
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccessing unusual heterocycles: ring expansion of benzoborirenes by formal cycloaddition reactions†
Marvin
Sindlinger
 a,
Markus
Ströbele
b,
Jörg
Grunenberg
*c and
Holger F.
Bettinger
*a
a,
Markus
Ströbele
b,
Jörg
Grunenberg
*c and
Holger F.
Bettinger
*a
aInstitut für Organische Chemie, Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany
bInstitut für Anorganische Chemie, Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany
cInstitut für Organische Chemie, Technische Universität (TU) Braunschweig, 38106 Braunschweig, Germany
First published on 13th September 2023
Abstract
Benzoborirenes are a very rare class of strained boron heterobicyclic systems. In this study a kinetically stabilized benzoborirene 1 is shown to react with multiple bonds of trimethylphosphine oxide, acetaldehyde, and tert-butyl isonitrile. The (2 + 2) cycloaddition product with trimethylphosphine oxide, benzo[c][1,2,5]oxaphosphaborole, has a long apical PO bond (194.0 pm) that must be considered on the border line between ionic and covalent according to the natural bond orbital, quantum theory of atoms in molecules, and compliance matrix approaches to the description of chemical bonding. The coordination compound between the benzoborirene and phosphine oxide was observed by NMR spectroscopy at 213 K. The Lewis acidity of 1 is similar to that of B(OCH2CF3)3 and B(C6F5)3 based on the 31P{1H} NMR chemical shift of the Lewis acid base complexes with trimethylphosphine oxide at 213 K. Benzoboriene 1 does not react with acetone, but forms a (2 + 2) cycloaddition product, an oxaborole, with acetaldehyde. In contrast, it undergoes a double (2 + 1) reaction with tert-butyl isonitrile to yield a boro-indane derivative under mild conditions. The observed reactivity of 1 is in agreement with computational analyses of the respective potential energy surfaces.
Introduction
Strained compounds are privileged in modern organic synthesis as the relief of ring strain provides ready synthetic access to complex molecular targets.1–12 An unusual scaffold in this context is cyclopropabenzene (benzocyclopropene). This highly strained bicyclic compound combines a three-membered ring fused to a benzene moiety.13–20 Its boron analogue, 1-boracyclopropabenzene, more commonly known as benzoborirene, has long escaped synthesis.21 Following identification in crossed molecular beam and matrix isolation experiments,22–25 successful solution-phase syntheses of benzoborirenes were eventually reported.26,27 We recently described the synthesis of the kinetically stabilized benzoborirene 1 and provided the first single crystal X-ray structure.28 The sterically demanding Trip2C6H3 (Trip = 2,4,6-iso-Pr3C6H2) substituent provides considerable stabilization to the boron centre in 1 as evidenced by high thermal stability as well as inertness towards a number of weak nucleophiles (acetonitrile, 2,6-lutidine, and 2-picoline) and weak electrophiles (trimethylchlorosilane and benzophenone).28 So far, only methanolysis and formation of a Lewis acid–base complex with pyridine have been observed for 1,28 while the other two known benzoborirenes react with the stronger Lewis base IiPrMe2 by opening the three-membered ring (Scheme 1a).26,27 The related carborane-fused boriranes undergo cycloaddition reactions with various substrates resulting in an expansion of the three-membered ring.29–31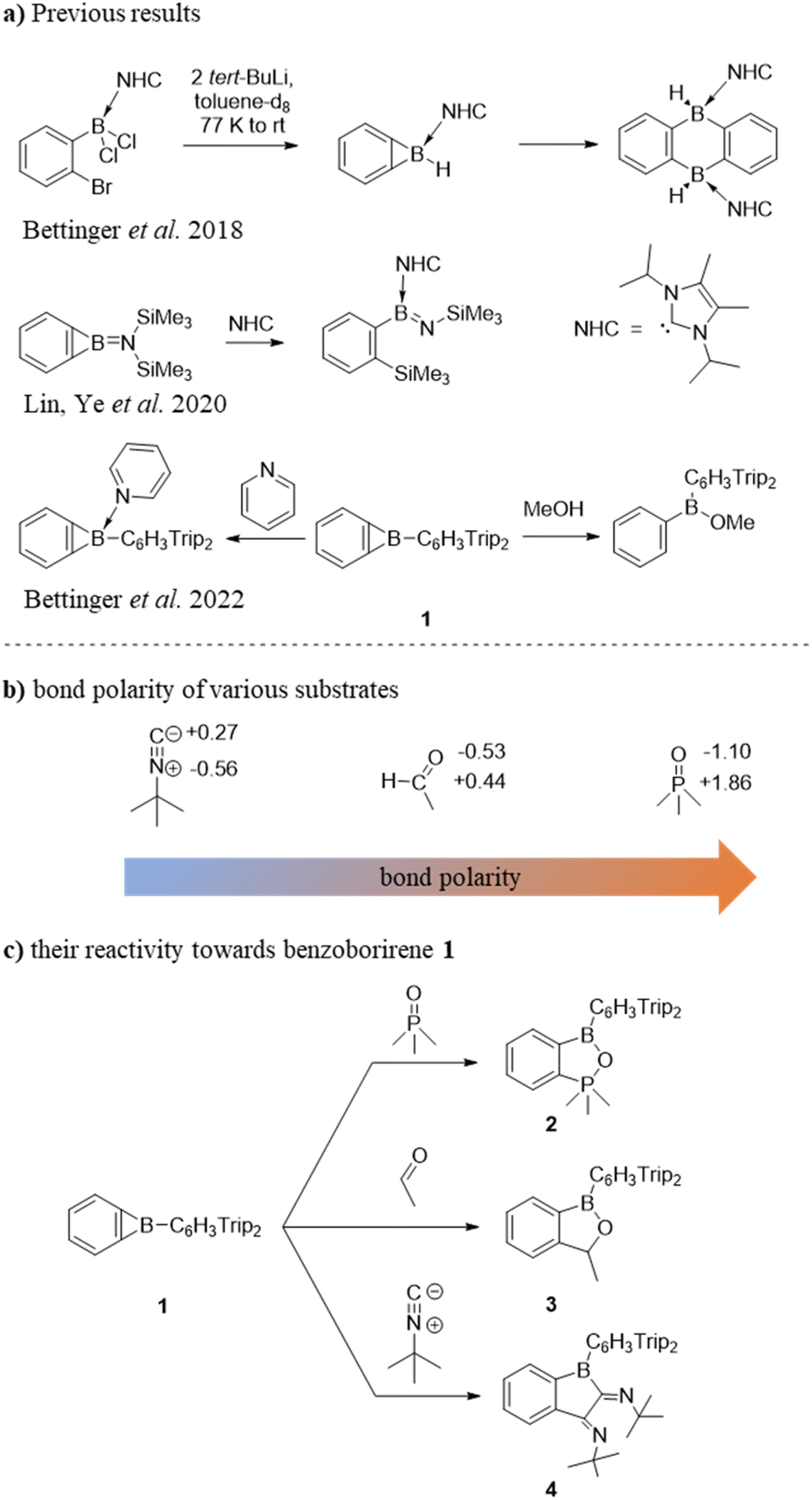 | ||
| Scheme 1 (a) Selected reactivity patterns of benzoborirenes prior to this work; (b) bond polarity of various substrates; NPA charges (e) were computed at the M0-62X/6-311+G** level of theory; (c) cycloaddition reactions of 1 with various substrates. | ||
Clearly, the readily accessible benzoborirene 1 offers the potential for unique reactivity as it combines an electrophilic boron centre31 and a strained three-membered as well as a distorted benzene ring. We here investigate this “ménage à trois” in its behaviour towards multiple bonds as provided by phosphine oxides R3PO, carbonyl compounds (R2CO and RCHO), and isonitriles RNC. The substrates were chosen to provide varying degrees of polarity of the Xδ+–Yδ– bond and philicities (nucleophilicity and electrophilicity) of their constituent atoms (Scheme 1b). We show that acetaldehyde (but not acetone) and trimethylphosphine oxide (but not triphenylphosphine oxide) undergo (2 + 2) cycloaddition reactions, while tert-butyl isocyanide undergoes twofold (2 + 1) cycloaddition with the strained C–B bond of 1 to give five-membered heterocycles (Scheme 1c).
Results and discussion
Reaction with Me3PO
Earlier investigations with pyridine and its derivatives have shown that the steric demand of the Trip2C6H3 moiety limits access to the boron centre.28 We thus employed the smaller Me3PO instead of the standard Gutmann–Beckett reagent Et3PO to afford qualitative information about the electrophilicity of benzoborirene 1. However, when 1 was treated with one equivalent of Me3PO in toluene we did not observe the expected 31P{1H} NMR shift towards regions lower-field than 30.0 ppm of Me3PO. Instead, a new signal almost 100 ppm further high-field at −68.2 ppm emerged, while a new signal in the 11B{1H} NMR spectrum at 41.2 ppm was observed. Single crystals were grown from pentane at −30 °C and X-ray crystallography revealed formation of benzo[c][1,2,5]oxaphosphaborole 2 that crystallizes in the monoclinic space group P21/c with four molecules in the unit cell.The phosphorus atom is coordinated in a slightly distorted trigonal bipyramidal fashion with bond angles between the equatorial carbon atoms of 118.12(4)°, 118.57(4)° and 120.64(5)° and a bond angle between the apical carbon and oxygen atoms of 175.15(4)°. The central ring of the terphenyl moiety is tilted by 85.5° relative to the benzo[c][1,2,5]oxaphosphaborole plane.
The P–O bond distance is 194.0 pm and therefore about 10 pm longer relative to comparable benzo[c][1,2,5]oxaphospholes,32,33 but also about 11 pm shorter than the bond in a comparable carborane fused oxaphosphaborole.31 The latter product forms in a similar reaction of a carborane-fused borirane with Et3PO.31 In the closely related intramolecular B–O–P adducts with four-coordinate B and P atoms the P–O bond lengths are significantly shorter with 154.2–154.6 pm; the B–O bonds on the other hand are significantly longer than those in benzo[c][1,2,5]oxaphosphaborole 2 and range from 155.0–158.4 pm.34
We analysed the character of the B–O and P–O bonds by NBO analysis at the M06-2X/6-311+G** level of theory. However, with the default settings of the NBO analysis no Lewis bonds towards the oxygen atom were identified. Rather, the oxygen atom is considered a separate fragment with four lone pairs. When the P–O and B–O bonds are enforced as Lewis bonds, the two electrons of the B–O bond are localized on the oxygen at 82.0%. In the framework of the second order perturbation theory analysis of the Fock matrix in the NBO basis, the B(p) orbital strongly interacts (46.8 kcal mol−1) with an oxygen lone pair and with the P–O σ-bond (36.3 kcal mol−1). The P–O σ-bond, on the other hand, has an occupancy of only 1.8 electrons, localized at 93.6% on the oxygen. In the framework of the second order perturbation theory the anti-bonding P–O orbital interacts considerably with the opposing C–P bond (100.8 kcal mol−1) as well as with the three equatorial C–P bonds (36.3, 43.5, and 56.1 kcal mol−1). Additionally, it interacts (19.4 kcal mol−1) with a lone pair of oxygen.
In order to get a deeper understanding of the bonding forces and electronic structure of the surprisingly long P–O bond in the oxaphosphaborole 2, we computed all relaxed force constants in combination with an analysis of the electron density, applying modern DFT methods. We would like to point out that in contrast to traditional force constants, which can easily be misleading,35 the computation of relaxed force constants allows the unique quantification of bond stiffness (or softness).36,37 Applying a range-separated version of Becke's 97 functional including dispersion correction (ωB97XD/def2-TZVP), developed by the group of Head-Gordon,38 and a polarized triple-ζ basis, the P–C bond in trans position to oxygen seems to be a regular two-electron bond with a relaxed force constant of 2.42 N cm−1.
Nevertheless, our computed value for the P–O bond of only 0.6 N cm−1 (see Fig. 2) points indeed to a very soft non-covalent interaction. This low value is far away from that of any typical two-electron P–O sigma bond for example in phosphorus pentoxide (4.51 N cm; see the ESI†).
Since we calculated the force constants by applying the full compliance matrix approach,39 the coupling constants are available too, allowing the quantification of electronic coupling in general. Interestingly, the computed coupling force constant (for a detailed discussion of the coupling terms, see: Turner et al.,40 Grunenberg41 and Zhao et al.42) between the P–O interaction and the opposing P–C bond is synergetic and pronounced (−0.20 N cm−1, see Table 1). This means that even if there is no classical covalent P–O bond, the electrostatic interaction seems to be considerable (see Table 2). Additionally, the Löwdin and Mulliken bond orders both indicate a pronounced double bond character of the B–O bond (see Table 1) and provide an explanation for its relatively short length.
| Atom | Natural charge | Mulliken charge | Electrostatic charge |
|---|---|---|---|
| B | +0.98 | +0.29 | −0.12 |
| C(7) | −0.69 | −0.50 | −0.79 |
| O | −0.97 | −0.59 | −0.48 |
| P | +1.77 | +0.88 | +1.25 |
These findings are in line with our NBO analysis (see above). Since population analysis sometimes tends to produce contradictory results when it comes to the description of unusual electronic structures (see, for example, the heated discussions on the gallyne or the quadruple bond between two carbon atoms)43 we, in a last step, analysed the electron density of 2 in terms of Bader's quantum theory of atoms in molecules (QTAIM) approach.44 First of all, we indeed find a (3, −1) critical point between the phosphorus and oxygen atoms. The calculated density at this line critical point (we follow Shahbazian's definition)45 is nevertheless very tenuous with only 0.08 e bohr−3 and should not be equated with the existence of a Lewis bond (see Fig. 3).46,47 The value of the electron density for the opposing “classical” P–C bond, for example, is nearly twice as high (0.15 e bohr−3).
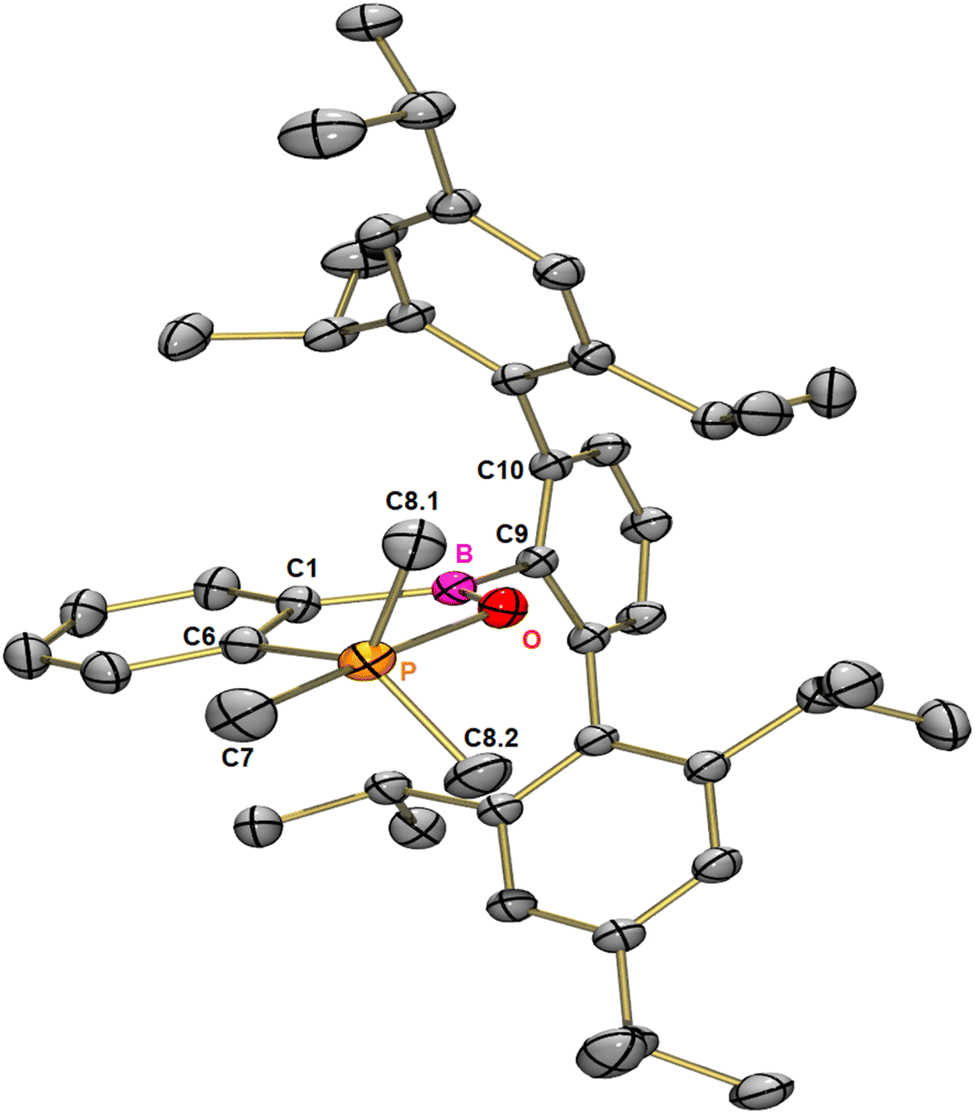 | ||
| Fig. 1 Molecular structure of 2 in the solid state. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected distances [pm] and angles [°] for 3: P–O 194.0, P–C6 183.2, P–C7 188.0, P–C8.1 182.0, P–C8.2 182.1, B–O 132.1, B–O–P 114.5, C1–B–O 112.3, O–P–C6 86.7, O–P–C7 175.2, O–P–C8.1 83.7, O–P–C8.2 83.3, C6–P–C8.1 118.6, C6–P–C8.2 118.1, C8.1–P–C8.2 120.6, and C1–B–C10–C11 85.5. | ||
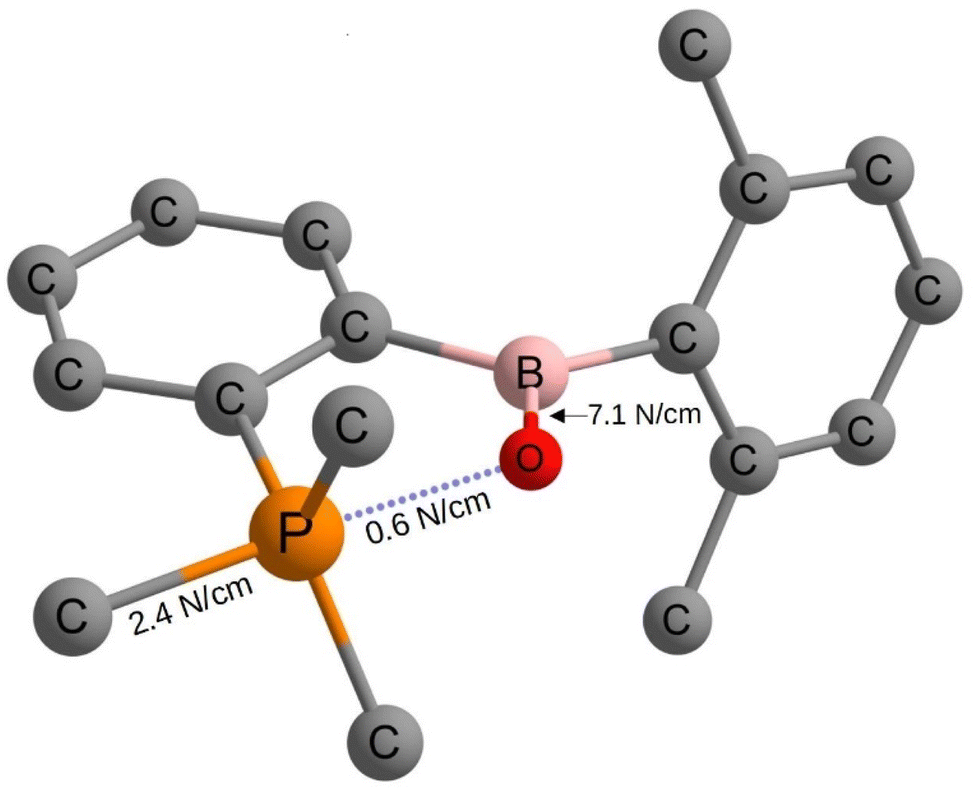 | ||
| Fig. 2 Selected relaxed force constants in 2 computed at the wB97XD/def2-tzvp level of theory. All hydrogen atoms as well as the 2,4,6-iso-Pr3C6H2 moieties are omitted for clarity. The coupling force constant between the P–O interaction and the opposing P–C bond is synergistic (note the negative sign) and pronounced at −0.20 N cm−1. | ||
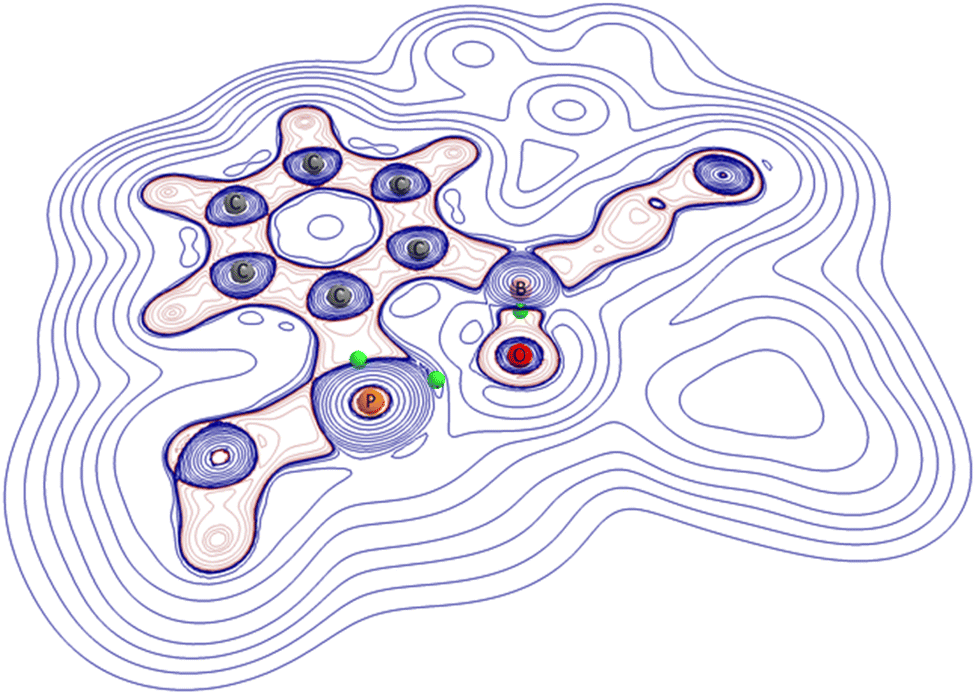 | ||
| Fig. 3 Computed positions of three selected (3, −1) critical points (green spheres) according to Bader's AIM analysis of 2 for the B–O, P–O and P–C interaction. The value of the electron density Rho for the very soft P–O interaction at its (3, −1) point is only 0.08 e bohr−3. The contour map depicts the Laplacian of the electron density: negative values are red; positive values are blue. The orientation of the molecule is comparable to that in Fig. 2. All hydrogen atoms as well as the 2,4,6-iso-Pr3C6H2 moieties are omitted for clarity. | ||
Finally, in order to further characterize the long P–O bond in 2 we computed the Laplacian of the electron density at the critical point. The slightly positive value of ∇2ρ (+0.01) for the P–O interaction is in line with our interpretation as an interaction, which is right at the border between electrostatics and covalency. Considering these findings, the electronic structure of compound 2 can be described by the resonance forms below (Scheme 2).
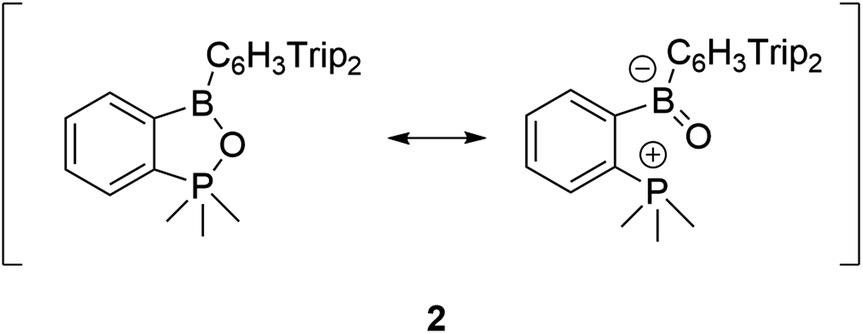 | ||
| Scheme 2 Resonance structures for compound 2. | ||
Utilizing DFT calculations (M06-2X/6-311+G**) for the reaction of the parent benzoborirene with trimethyl phosphine oxide (see ESI, Table S3 and Fig. S31†) we reasoned that the coordination compound between benzoborirene 1 and Me3PO (1·OPMe3) may be observable by NMR spectroscopy at low temperatures (Scheme 3). Indeed, the insertion reaction was slow at −60 °C, which allowed us to detect 1·OPMe3 using NOESY cross peaks between the methyl moieties of the phosphine oxide and the iso-propyl moieties of the terphenyl substituent (see ESI, Fig. S10†). Further support comes from a 11B{1H} NMR shift of −5.6 ppm, which is typical for tetra-coordinated boron centres. The observed 31P{1H} NMR shift was 63.0 ppm, 30.0 ppm further downfield than Me3PO at −60 °C. A survey of the 31P chemical shifts of coordination complexes of Me3PO with other Lewis acids at −60 °C (see Table 3) indicates a slightly higher electrophilicity of the boron centre of 1 than tris-(2,2,2-trifluoroethyl) borate (62.9 ppm) and a slightly lower Lewis acidity than B(C6F5)3. The bulkier Ph3PO did not react with 1, most likely due to steric hindrance.
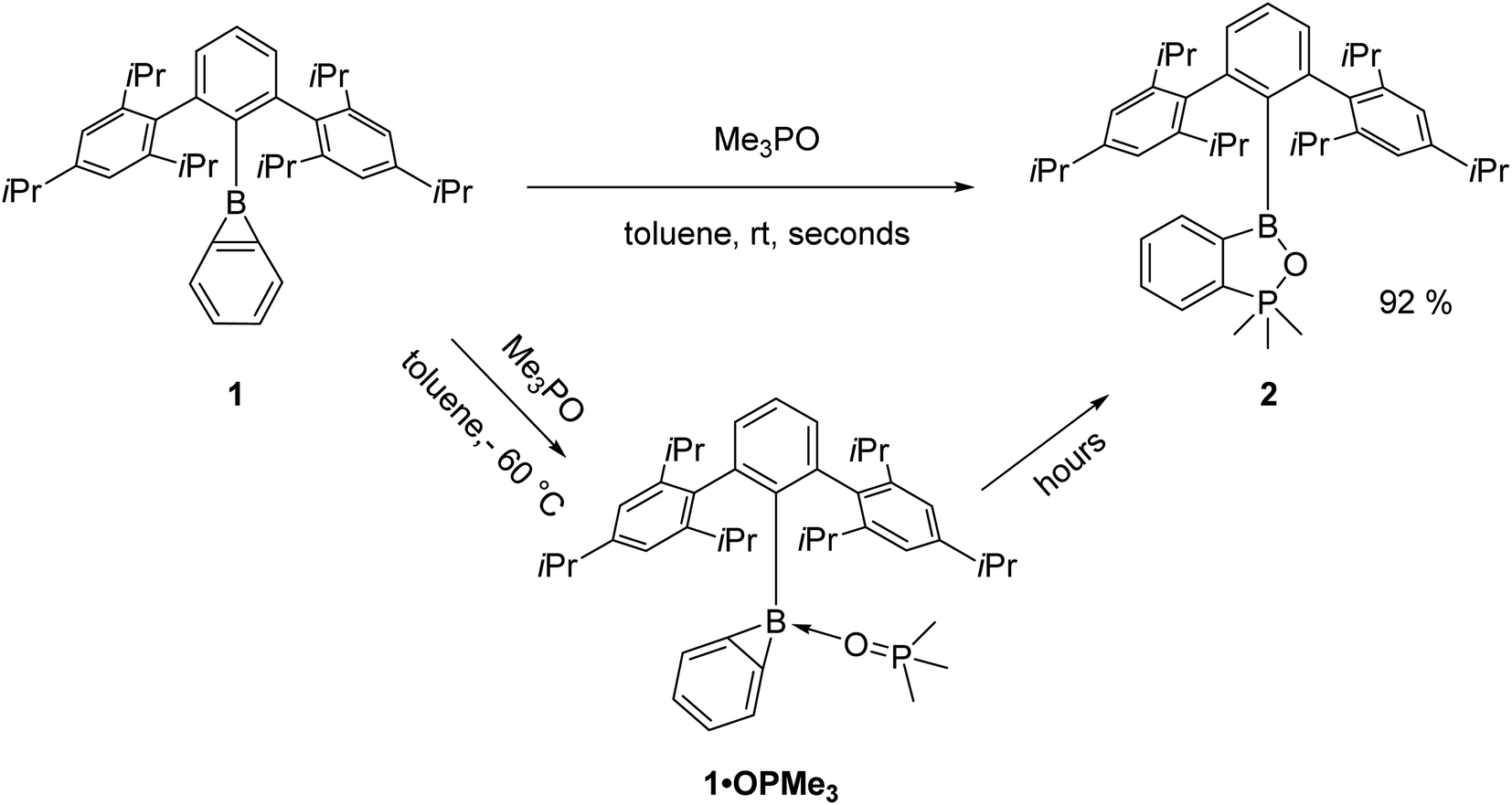 | ||
| Scheme 3 Reactivity of 1 towards trimethylphosphine oxide. | ||
| Lewis acid | Chemical shift |
|---|---|
| None | 33.0 ppm |
| B(NMe2)3 | 31.8 ppm |
| B(OMe)3 | 34.2 ppm |
| B(OCH2CF3)3 | 62.9 ppm |
| B(C6F5)3 | 65.8 ppm |
| 1 | 63.0 ppm |
Reaction with carbonyl compounds
Carbonyl compounds are less polar than phosphine oxides (Scheme 1b), and indeed we observed earlier that benzoborirene 1 does not react with benzophenone.28 To rule out steric hindrance as a cause for this we investigated the reactivity of 1 towards the smallest ketone, acetone. Remarkably, even when heated to 50 °C for hours no reaction takes place, proving that 1 is not Lewis acidic enough to react with simple aliphatic or aromatic ketones. We then focused our attention on acetaldehyde to reduce steric hindrance. Indeed, over the course of ten days the formation of a new compound with a 11B{1H} NMR shift of 49.3 ppm could be observed (Scheme 4).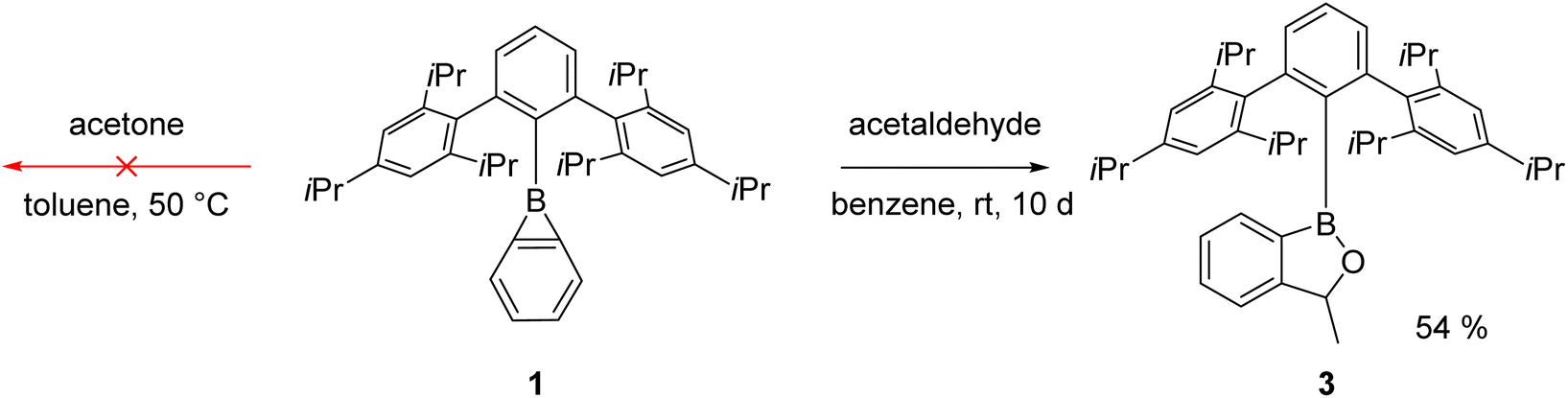 | ||
| Scheme 4 Reactivity of 1 towards acetone and acetaldehyde. | ||
After purification by column chromatography single crystals suitable for X-ray crystallography were grown by slow evaporation of toluene from a concentrated solution at reduced pressure. 1-(Trip2C6H3)-3-methyl-1,3-dihydrobenzo[c][1,2]oxaborole 3 is the product of the insertion of acetaldehyde into the B–C-bond of the three-membered ring and crystallizes in the monoclinic space group P21/n with four molecules in the unit cell (Fig. 4). The five-membered ring is almost planar with a sum of inner angles of 539.9° and tilted by 75.8° relative to the terphenyl moiety.
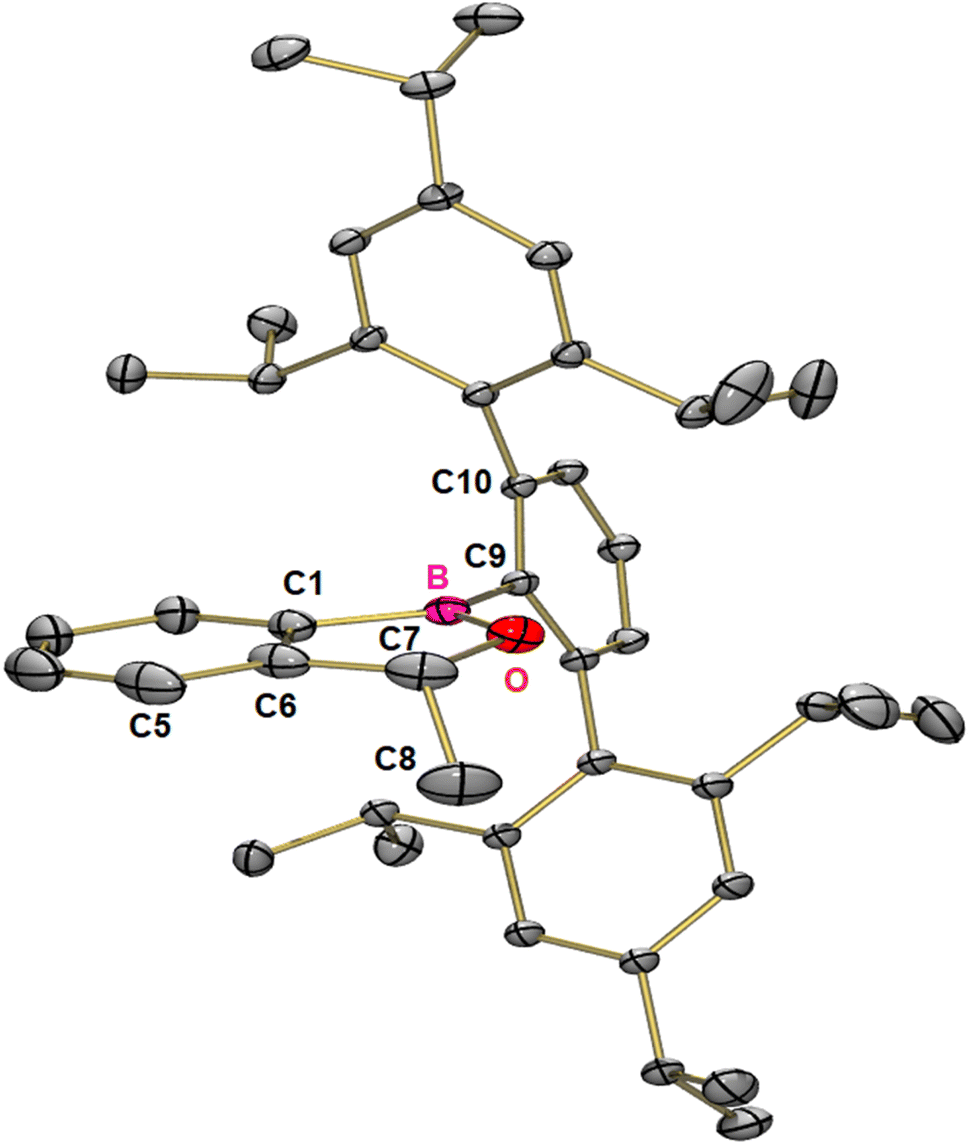 | ||
| Fig. 4 Molecular structure of 3 in the solid state. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected distances [pm] and angles [°] for 5: C1–B 155.5, B–C9 157.5, B–O 136.4, C1–C6 140.0, C6–C7 150.6, C7–C8 152.3, C7–O 145.0, C1–B–O 108.2, C1–B–C9 128.6, C9–B–O 123.0, C6–C1–B 105.1, B–O–C7 111.4, O–C7–C6 105.1, O–C7–C8 108.9, C6–C7–C8 113.2, C1–C6–C7 110.2, C1–B–C9–C10 75.8, C5–C6–C7–C8 63.4, and C1–C6–C7–C8 115.7. | ||
DFT computations (M06-2X/6-311+G**) show that the free energy of the coordination compound between the parent benzoborirene and acetaldehyde (M-1) is 4.7 kcal mol−1 higher in energy relative to the separated reactants (see ESI, Table S4 and Fig. S32†). The transition state for the formation of the five-membered ring lies 14.8 kcal mol−1 higher than M-1. This provides an explanation of the slow rate of the reaction of benzoborirene 1 with acetaldehyde as compared to trimethylphosphine oxide.
Reaction with tert-butyl isonitrile
Benzoborirene 1 does not react with acetonitrile.28 We here investigated the reactivity of benzoborirene 1 towards tert-butyl isonitrile, the least polar compound in the series of substrates studied in this investigation. In addition, its carbon centre has some degree of carbene character,48 which makes it an interesting reagent. Isocyanides are known to react twice in formal cycloaddition reactions.49–54When benzoborirene 1 is treated with one equivalent of tert-butyl isonitrile at room temperature the solution turns red instantly. The 1H NMR spectra showed the formation of a new compound, but also signals of unreacted 1 remained, resulting in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture. In fact, two equivalents of tert-butyl isonitrile are needed for a complete transformation. Red crystals suitable for X-ray crystallography, grown by slow evaporation of toluene from a concentrated solution at reduced pressure revealed the new compound to result from a double (2 + 1) cycloaddition of tert-butyl isonitrile into the B–C bond of the three-membered ring. The N,N′-di-tert-butyl-benzo[b]borole-2,3-diimine 4 crystallizes in the monoclinic space group P21/c with four molecules in the unit cell (Fig. 5). The five-membered ring has an envelope conformation with a sum of inner angles of 530.08°. All members of the five-membered ring are trigonal planar coordinated with angle sums of 358.96° (C8), 359.02° (C7), 359.57° (C1), 359.59° (C6) and 359.99° (B).
:50 mixture. In fact, two equivalents of tert-butyl isonitrile are needed for a complete transformation. Red crystals suitable for X-ray crystallography, grown by slow evaporation of toluene from a concentrated solution at reduced pressure revealed the new compound to result from a double (2 + 1) cycloaddition of tert-butyl isonitrile into the B–C bond of the three-membered ring. The N,N′-di-tert-butyl-benzo[b]borole-2,3-diimine 4 crystallizes in the monoclinic space group P21/c with four molecules in the unit cell (Fig. 5). The five-membered ring has an envelope conformation with a sum of inner angles of 530.08°. All members of the five-membered ring are trigonal planar coordinated with angle sums of 358.96° (C8), 359.02° (C7), 359.57° (C1), 359.59° (C6) and 359.99° (B).
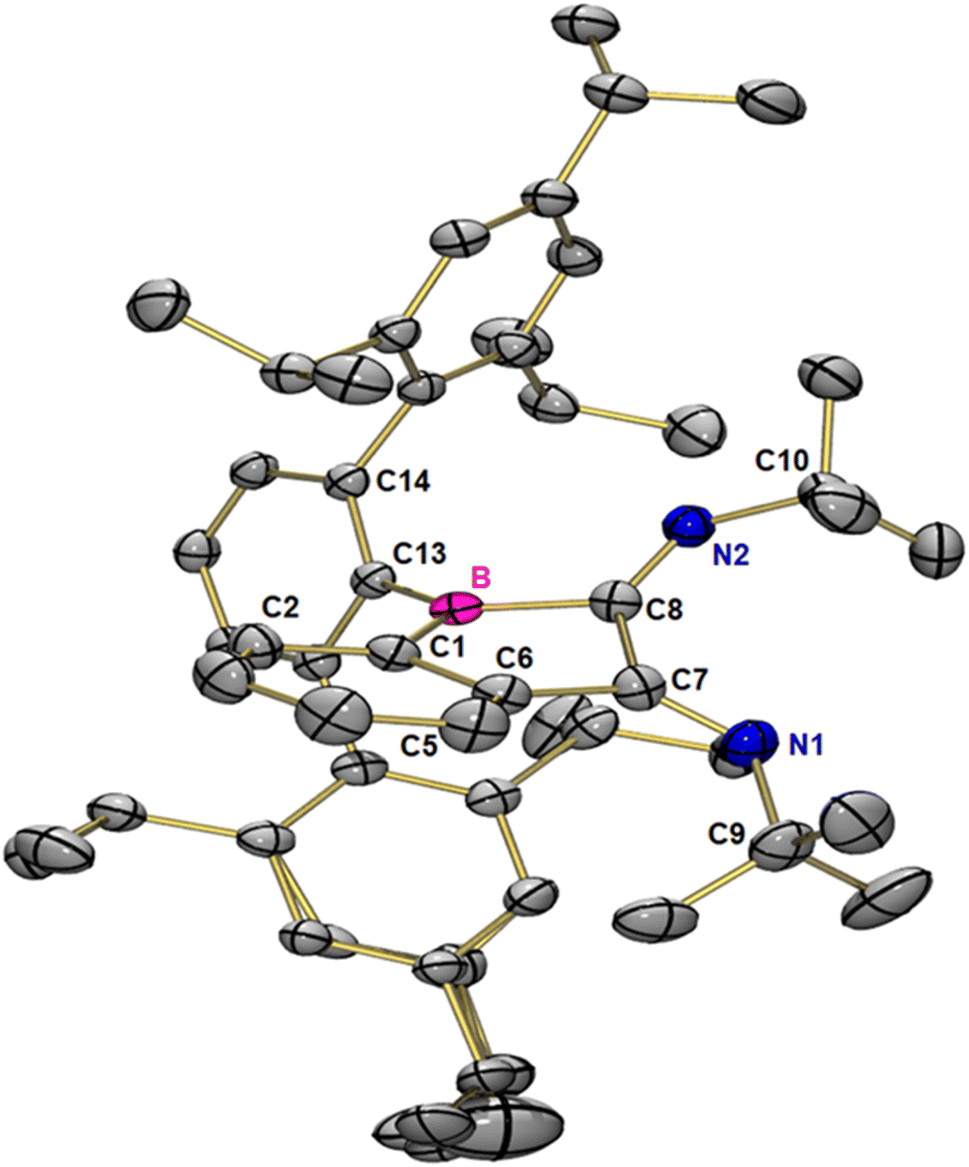 | ||
| Fig. 5 Molecular structure of 4 in the solid state. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms and a co-crystalized toluene molecule have been omitted for clarity. Selected distances [pm] and angles [°] for 4: B–C1 155.7, B–C8 158.1, C6–C7 150.0, C7–C8 152.6, N1–C7 127.3, N1–C9 148.1, N2–C8 127.9, N2–C10 147.9, C1–B–C8 101.5, B–C8–C7 104.4, C8–C7–C6 104.5, C7–N1–C9 127.2, C8–N2–C10 125.0, C6–C1–B–C8 15.2, C1–C6–C7–C8 22.6, C1–B–C13–C14 107.5, C1–C6–C7–N1 145.2, and N1–C7–C8–N2 53.0. | ||
The identity of 4 could also be derived from high resolution mass spectrometry as well as NMR spectroscopy at 70 °C. No signal was detected in the 11B{1H} NMR spectra in solution, but detection of the boron signal in the solid state was successful (see the ESI†) and arrived at an isotopic 11B NMR shift of 66.0 ppm. To the best of our knowledge borole diimines are completely unknown. The most closely related compounds in the literature are a set of 3,4-dimethylene borolanes reported by Herberich et al. in the early 1990s.55,56
The double (2 + 1) cycloaddition of isonitrile can be compared with the report by Paetzold et al. of a twofold tert-butyl isonitrile insertion into the B–C double bond of alkylidene aminoborane iPr2N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) BC(SiMe3)2 resulting in boretane a (Scheme 5a)Link "Scheme 5a" and its floatanchor here.49 Similar to these earlier observations, the reaction of a second isonitrile molecule with the primary 1:1 adduct is faster than the reaction between isonitrile and benzoborirene 1. The B–C bond of the three-membered ring thus behaves quite similarly as a BC bond in the acyclic compound (Scheme 5).
BC(SiMe3)2 resulting in boretane a (Scheme 5a)Link "Scheme 5a" and its floatanchor here.49 Similar to these earlier observations, the reaction of a second isonitrile molecule with the primary 1:1 adduct is faster than the reaction between isonitrile and benzoborirene 1. The B–C bond of the three-membered ring thus behaves quite similarly as a BC bond in the acyclic compound (Scheme 5).
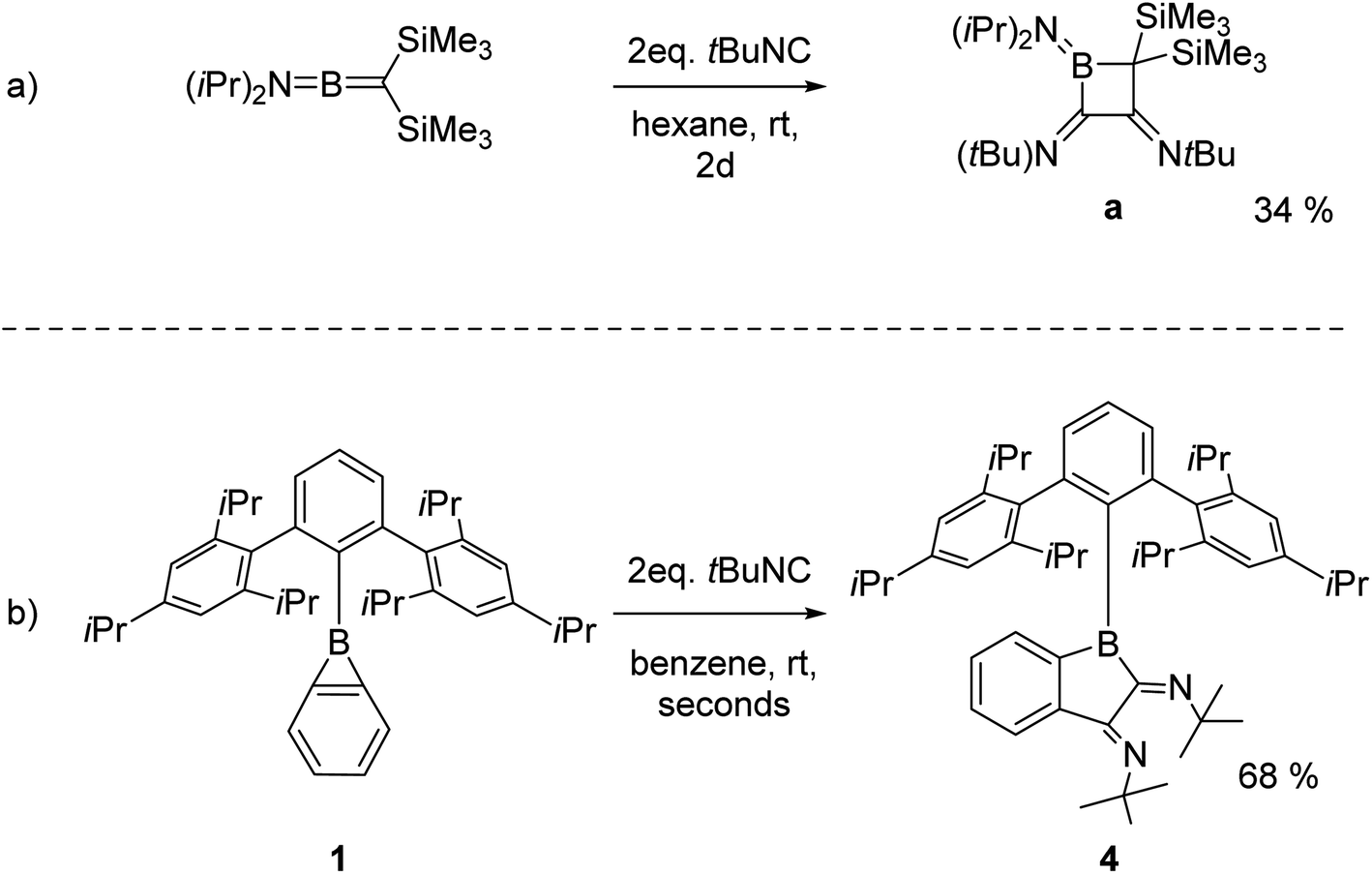 | ||
| Scheme 5 (a) Twofold tert-butyl isonitrile insertion into a BC bond reported by Paetzold et al.49 and (b) reactivity of 1 towards tert-butyl isonitrile. | ||
Addition of a third equivalent of tert-butyl isonitrile results in a yellow reaction mixture with a significantly reduced symmetry according to the 1H NMR spectrum, making unambiguous peak assignments increasingly difficult (see ESI, Fig. S15†). The 11B{1H} NMR shift of −12.6 ppm, however, indicates a tetra-coordinated boron centre instead of a third (2 + 1) cycloaddition (see ESI, Fig. S16†). Further purification and crystallization attempts proved unsuccessful.
DFT calculations (M06-2X/6-311+G**) for the parent benzoborirene/methyl isonitrile model system reveal the reactivity of the strained heterocyclic system towards methyl isonitrile (Fig. 6). The reaction with the first equivalent of methyl isonitrile is less exergonic and has a higher barrier (TS4vs.TS6) than the reaction with the second equivalent. The reaction with the third equivalent can only produce a coordination compound M9 as the insertion product M10 is about 5.4 kcal mol−1 higher in Gibbs free energy.
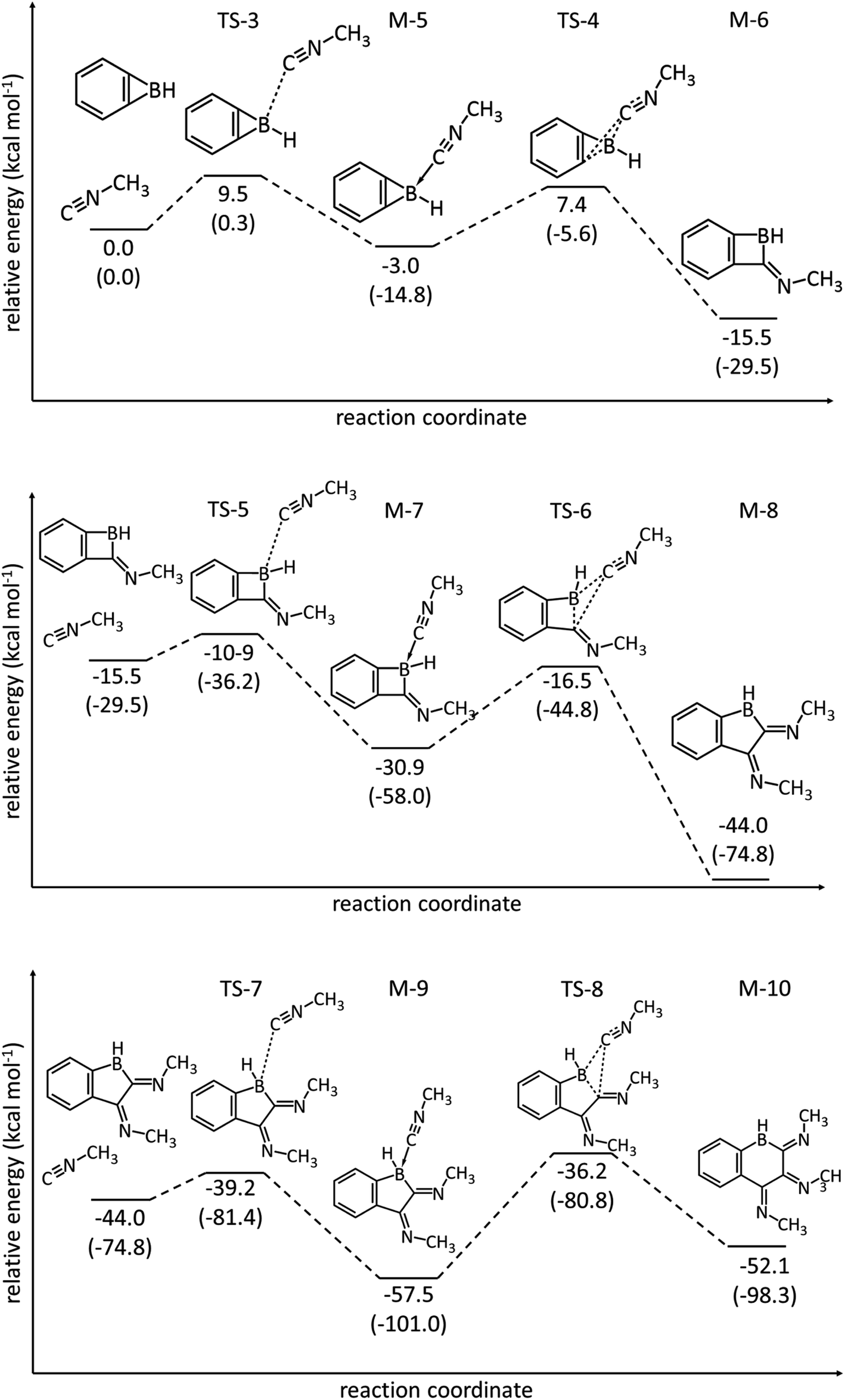 | ||
| Fig. 6 Energy profiles calculated (M06-2X/6-311+G**) for the cycloaddition of the parent benzoborirene (BBI) with the first (top), second (middle) and third (bottom) equivalents of methyl isonitrile. The relative Gibbs free energies (calculated at 298 K) and electronic energies (in parentheses) are given in kcal mol−1. | ||
Summary
In conclusion, the kinetically stabilized benzoborirene 1 allows formal (2 + 2) cycloadditions with acetaldehyde and trimethylphosphine oxide resulting in benzo[c][1,2,5]oxaphosphaborole 2 and 1,3-dihydrobenzo[c][1,2]oxaborole 3, respectively, by insertion into one of the B–C bonds. According to our analysis of the unusual bonding situation in benzo[c][1,2,5]oxaphosphaborole 2 by applying three different quantum-chemical descriptors (localized natural orbitals, the electron density, and relaxed force constants) the P–O bond in 2 seems to represent a bonding situation right at the borderline between shared and non-shared interactions in general. To the best of our knowledge, the softness of the P–O bond in 2 is unprecedented in the literature. The coordination compound of benzoborirene 1 and trimethylphosphine oxide was observed at low temperatures which allowed the estimation of the electrophilicity of the boron centre in 1 to be between that of B(C6F5)3 and B(OCH2CF3)3 at −60 °C.Furthermore, benzoborirene 1 reacts with tert-butyl isonitrile in a formal double (2 + 1) cycloaddition to form N,N′-di-tert-butyl benzo[b]borole-2,3-diimine 4, revealing the double bond nature of the B–C bond in the benzoborirene scaffold. Isonitrile in excess of two equivalents is likely only coordinated to the boron centre as a third insertion is energetically unfavourable according to DFT computations.
Data availability
The ESI† includes experimental details, NMR and MS spectra, details on X-ray crystallography, reference experiments for the 31P NMR shifts, computational details, and Cartesian coordinates.Author contributions
M. Sindlinger synthesized and characterized all compounds and grew single crystals. M. Ströbele conducted X-ray crystallography. J. G. carried out the bonding analysis of compound 2. M. Sindlinger carried out the mechanistic DFT computations. H. F. B. conceptualized and managed the project and acquired the funding. M. Sindlinger wrote the manuscript. All authors contributed to editing and reviewing the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are very grateful to the German Research Foundation (DFG) for the support of this work (BE 3183/5-3). The computations were performed on the BwForCluster JUSTUS2. The authors acknowledge support from the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/575-1 FUGG. We thank Dr Klaus Eichele at the Institute of Inorganic Chemistry at the University of Tübingen for the measurement of the solid-state NMR spectra.References
- J. Turkowska, J. Durka and D. Gryko, Strain release – an old tool for new transformations, Chem. Commun., 2020, 56, 5718–5734 RSC.
- B. Biletskyi, P. Colonna, K. Masson, J.-L. Parrain, L. Commeiras and G. Chouraqui, Small rings in the bigger picture: ring expansion of three- and four-membered rings to access larger all-carbon cyclic systems, Chem. Soc. Rev., 2021, 50, 7513–7538 RSC.
- A. Luque, J. Paternoga and T. Opatz, Strain Release Chemistry of Photogenerated Small-Ring Intermediates, Chem. –Eur. J., 2021, 27, 4500–4516 CrossRef CAS PubMed.
- M. Murakami and N. Ishida, Cleavage of Carbon–Carbon σ-Bonds of Four-Membered Rings, Chem. Rev., 2021, 121, 264–299 CrossRef CAS PubMed.
- V. Pirenne, B. Muriel and J. Waser, Catalytic Enantioselective Ring-Opening Reactions of Cyclopropanes, Chem. Rev., 2021, 121, 227–263 CrossRef CAS PubMed.
- R. Vicente, C–C Bond Cleavages of Cyclopropenes: Operating for Selective Ring-Opening Reactions, Chem. Rev., 2021, 121, 162–226 CrossRef CAS PubMed.
- C. B. Kelly, J. A. Milligan, L. J. Tilley and T. M. Sodano, Bicyclobutanes: from curiosities to versatile reagents and covalent warheads, Chem. Sci., 2022, 11721–11737 RSC.
- M. Golfmann and J. C. L. Walker, Bicyclobutanes as unusual building blocks for complexity generation in organic synthesis, Commun. Chem., 2023, 6, 9 CrossRef CAS PubMed.
- J. L. Tyler and V. K. Aggarwal, Synthesis and Applications of Bicyclo[1.1.0]butyl and Azabicyclo[1.1.0]butyl Organometallics, Chem. –Eur. J., 2023, e202300008 CrossRef CAS PubMed.
- W. Dai, S. J. Geib and D. P. Curran, Ring-Opening Reactions of NHC-Boriranes with In Situ Generated HCl: Synthesis of a New Class of NHC-Boralactones, J. Am. Chem. Soc., 2019, 141, 3623–3629 CrossRef CAS PubMed.
- A. S. Harmata, B. J. Roldan and C. R. J. Stephenson, Formal Cycloadditions Driven by the Homolytic Opening of Strained, Saturated Ring Systems, Angew. Chem., Int. Ed., 2023, 62, e202213003 CrossRef CAS PubMed.
- T. Yu, J. Yang, Z. Wang, Z. Ding, M. Xu, J. Wen, L. Xu and P. Li, Selective [2σ + 2σ] Cycloaddition Enabled by Boronyl Radical Catalysis: Synthesis of Highly Substituted Bicyclo[3.1.1]heptanes, J. Am. Chem. Soc., 2023, 145, 4304–4310 CrossRef CAS PubMed.
- R. Anet and F. Anet, Synthesis of a Benzocyclopropene Derivative, J. Am. Chem. Soc., 1964, 86, 525–526 CrossRef CAS.
- B. Halton, Benzocyclopropenes, Chem. Rev., 1973, 73, 113–126 CrossRef CAS.
- B. Halton, Developments in cycloproparene chemistry, Chem. Rev., 1989, 89, 1161–1185 CrossRef CAS.
- B. Halton, Cycloproparenes, Chem. Rev., 2003, 103, 1327–1370 CrossRef CAS PubMed.
- W. E. Billups, Synthesis and chemistry of benzocyclopropenes, Acc. Chem. Res., 1978, 11, 245–251 CrossRef CAS.
- W. Billups, W. A. Rodin and M. M. Haley, Cycloproparenes, Tetrahedron, 1988, 44, 1305–1338 CrossRef CAS.
- M. Khrapunovich, E. Zelenova, L. Seu, A. N. Sabo, A. Flaherty and D. C. Merrer, Regioselectivity and Mechanism of Dihalocarbene Addition to Benzocyclopropene, J. Org. Chem., 2007, 72, 7574–7580 CrossRef CAS PubMed.
- T. Matsuda, in Cleavage of Carbon–Carbon Single Bonds by Transition Metals, ed. M. Murakami and N. Chitani, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2015, ch. 2, pp. 35–88 Search PubMed.
- U. M. Dzhemilev, L. I. Khusainova, K. S. Ryazanov and L. O. Khafizova, Boron-containing small rings: synthesis, properties, and application prospects, Russ. Chem. Bull., 2021, 70, 1851–1892 CrossRef CAS.
- H. F. Bettinger, Phenylborylene: Direct Spectroscopic Characterization in Inert Gas Matrices, J. Am. Chem. Soc., 2006, 128, 2534–2535 CrossRef CAS PubMed.
- H. F. Bettinger and R. I. Kaiser, Reaction of Benzene and Boron Atom: Mechanism of Formation of Benzoborirene and Hydrogen Atom, J. Phys. Chem. A, 2004, 108, 4576–4586 CrossRef CAS.
- H. F. Bettinger, Generation of iodobenzoborirene, a boraaromatic cyclopropabenzene derivative, Chem. Commun., 2005, 21, 2756–2757 RSC.
- R. I. Kaiser and H. F. Bettinger, Gas-Phase Detection of the Elusive Benzoborirene Molecule, Angew. Chem., 2002, 114, 2456–2458 CrossRef.
- J. Hahn, C. Keck, C. Maichle-Mössmer, E. von Grotthuss, P. N. Ruth, A. Paesch, D. Stalke and H. F. Bettinger, Synthesis and Ring Strain of a Benzoborirene-N-Heterocyclic Carbene Adduct, Chem. –Eur. J., 2018, 24, 18634–18637 CrossRef CAS PubMed.
- H. Zhang, J. Wang, W. Yang, L. Xiang, W. Sun, W. Ming, Y. Li, Z. Lin and Q. Ye, Solution-Phase Synthesis of a Base-Free Benzoborirene and a Three-Dimensional Inorganic Analogue, J. Am. Chem. Soc., 2020, 142, 17243–17249 CrossRef CAS PubMed.
- M. Sindlinger, M. Ströbele, C. Maichle-Mössmer and H. F. Bettinger, Kinetic stabilization allows structural analysis of a benzoborirene, Chem. Commun., 2022, 2818–2821 RSC.
- H. Wang, J. Zhang and Z. Xie, Reversible Photothermal Isomerization of Carborane-Fused Azaborole to Borirane: Synthesis and Reactivity of Carbene-Stabilized Carborane-Fused Borirane, Angew. Chem., Int. Ed., 2017, 56, 9198–9201 CrossRef CAS PubMed.
- H. Wang, J. Zhang and Z. Xie, Ring-opening and ring-expansion reactions of carborane-fused borirane, Chem. Sci., 2021, 12, 13187–13192 RSC.
- Y. Wei, J. Wang, W. Yang, Z. Lin and Q. Ye, Boosting Ring Strain and Lewis Acidity of Borirane: Synthesis, Reactivity and Density Functional Theory Studies of an Uncoordinated Arylborirane Fused to o-Carborane, Chem. –Eur. J., 2023, 29, e202203265 CrossRef CAS PubMed.
- A. Bhunia, T. Kaicharla, D. Porwal, R. G. Gonnade and A. T. Biju, Multicomponent reactions involving phosphines, arynes and aldehydes, Chem. Commun., 2014, 50, 11389 RSC.
- A. Bhunia, T. Roy, R. G. Gonnade and A. T. Biju, Rapid Access to Benzoxaphospholes and Their Spiro Analogues by a Three-Component Coupling Involving Arynes, Phosphines, and Activated Ketones, Org. Lett., 2014, 16, 5132–5135 CrossRef CAS PubMed.
- J. M. Breunig, F. Lehmann, M. Bolte, H.-W. Lerner and M. Wagner, Synthesis and Reactivity of o-Phosphane Oxide Substituted Aryl(hydro)borates and Aryl(hydro)boranes, Organometallics, 2014, 33, 3163–3172 CrossRef CAS.
- J. Grunenberg, How Strong is a Reverse Dative Bond? Compliance Constants as Unique Bond Strength Descriptors, Inorg. Chem., 2022, 61, 20–22 CrossRef CAS PubMed.
- K. Brandhorst and J. Grunenberg, How strong is it? The interpretation of force and compliance constants as bond strength descriptors, Chem. Soc. Rev., 2008, 37, 1558–1567 RSC.
- K. Brandhorst and J. Grunenberg, Efficient computation of compliance matrices in redundant internal coordinates from Cartesian Hessians for nonstationary points, Chem. Phys., 2010, 132, 184101 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Systematic optimization of long-range corrected hybrid density functionals, Chem. Phys., 2008, 128, 084106 Search PubMed.
- W. J. Taylor and K. S. Pitzer, Vibrational frequencies of semirigid molecules: a general method and values for ethylbenzene, J. Res. Natl. Inst. Stand. Technol., 1947, 38, 1–17 CAS.
- J. J. Turner and J. A. Timney, Relaxed and local mode force constants: Linear ABC as a model system, J. Mol. Spectrosc., 2022, 387, 111662 CrossRef CAS.
- J. Grunenberg, III-defined concepts in chemistry: rigid force constants vs. compliance constants as bond strength descriptors for the triple bond in diboryne, Chem. Sci., 2015, 6, 4086–4088 RSC.
- L. Zhao, M. Zhi and G. Frenking, The strength of a chemical bond, Int. J. Quantum Chem., 2022, 122, e26773 CrossRef CAS.
- J. Grunenberg, Ill-defined chemical concepts: The problem of quantification, Int. J. Quantum Chem., 2017, 117, e25359 CrossRef.
- R. F. W. Bader, P. L. A. Popelier and T. A. Keith, Theoretical Definition of a Functional Group and the Molecular Orbital Paradigm, Angew. Chem., Int. Ed., 1994, 33, 620–631 CrossRef.
- C. Foroutan-Nejad, S. Shahbazian and R. Marek, Toward a Consistent Interpretation of the QTAIM: Tortuous Link between Chemical Bonds, Interactions, and Bond/Line Paths, Chem. –Eur. J., 2014, 20, 10140–10152 CrossRef CAS PubMed.
- C. R. Wick and T. Clark, On bond-critical points in QTAIM and weak interactions, J. Mol. Model., 2018, 24, 142 CrossRef PubMed.
- S. Shahbazian, Why Bond Critical Points Are Not “Bond” Critical Points, Chem. –Eur. J., 2018, 24, 5401–5405 CrossRef CAS PubMed.
- R. Ramozzi, N. Chéron, B. Braïda, P. C. Hiberty and P. Fleurat-Lessard, A valence bond view of isocyanides' electronic structure, New J. Chem., 2012, 36, 1137 RSC.
- A. Tapper, T. Schmitz and P. Paetzold, Reaktionen an der BC-Doppelbindung von iPr2N=B=C(SiMe3)2, Chem. Ber., 1989, 122, 595–601 CrossRef CAS.
- A. Ansorge, D. J. Brauer, S. Buchheim-Spiegel, H. Burger, T. Hagen and G. Pawelke, Novel heterocycles from alkylamino-bis(trifluoromethyl) borane, (CF3)2BNR2, and isocyanides. Crystal and molecular structure of (NC)(CF3)2B · NHMe2, J. Organomet. Chem., 1995, 501, 347–358 CrossRef CAS.
- N. Chatani, M. Oshita, M. Tobisu, Y. Ishii and S. Murai, A GaCl3-Catalyzed [4+1] Cycloaddition of α,β-Unsaturated Carbonyl Compounds and Isocyanides Leading to Unsaturated γ-Lactone Derivatives, J. Am. Chem. Soc., 2003, 125, 7812–7813 CrossRef CAS PubMed.
- M. Oshita, K. Yamashita, M. Tobisu and N. Chatani, Catalytic [4+1] Cycloaddition of α,β-Unsaturated Carbonyl Compounds with Isocyanides, J. Am. Chem. Soc., 2005, 127, 761–766 CrossRef CAS PubMed.
- A. Habibi, E. Sheikhhosseini and N. Taghipoor, A simple and efficient approach to the synthesis of furopyran derivatives: four-component reaction of isocyanides with arylidene-substituted Meldrum's acid, Chem. Heterocycl. Compd., 2013, 49, 968–973 CrossRef CAS.
- T. Kaur, P. Wadhwa, S. Bagchi and A. Sharma, Isocyanide based [4+1] cycloaddition reactions: an indispensable tool in multi-component reactions (MCRs), Chem. Commun., 2016, 52, 6958–6976 RSC.
- G. E. Herberich, U. Eigendorf and C. Ganter, Borylation of dicarbanions: syntheses of new five-and eight-membered boron-carbon rings, J. Organomet. Chem., 1991, 402, C17–C19 CrossRef CAS.
- G. E. Herberich, U. Eigendorf and U. Englert, 3,4-Dimethylenborolane und 3,7-Diborabicyclo[3.3.0]oct-1(5)-ene, Chem. Ber., 1994, 127, 1037–1039 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2172288, 2220111 and 2252479. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03433j |
| This journal is © The Royal Society of Chemistry 2023 |