
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistically remodulating H+/Ca2+ gradients to induce mitochondrial depolarization for enhanced synergistic cancer therapy†
Xiaoni
Wang
a,
Xiyang
Ge
a,
Xiaowen
Guan
a,
Jin
Ouyang
 b and
Na
Na
*a
b and
Na
Na
*a
aKey Laboratory of Radiopharmaceuticals, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: nana@bnu.edu.cn
bDepartment of Chemistry, College of Arts and Sciences, Beijing Normal University at Zhuhai, Zhuhai City, Guangdong Province 519087, China
First published on 3rd October 2023
Abstract
The remodulation of H+/Ca2+ gradients in the mitochondria matrix could be effective to induce mitochondria depolarization for the enhancement of cancer therapy. However, it is still challenged by H+ homeostasis, insufficient Ca2+, uncoordinated regulations, and inefficient loading/delivery strategies. Herein, a supramolecular DNA nanocomplex (Ca@DNA–MF) was prepared to synergistically remodulate H+/Ca2+ gradients for mitochondrial depolarization. Upon targeted functionalization and TME-triggered delivery, multiple reagents were released in cancer cells for synergistic three-channel mitochondrial depolarization: the gene reagent of siMCT4 blocked the LA metabolism to induce mitochondrial acidification by downregulating monocarboxylate transporter 4 (MCT4); released Ca2+ disrupted Ca2+ homeostasis to facilitate Ca2+-based mitochondrial depolarization; specifically, TME-activated glutathione (GSH) depletion facilitated efficient generation of hydroxyl radicals (˙OH), further enhancing the mitochondrial depolarization. The remodulation not only triggered apoptosis but also led to ferroptosis to generate abundant ROS for efficient LPO-based apoptosis, providing a synergistic strategy for enhanced synergistic cancer therapy.
Introduction
Rational regulation of organelles, to administer cellular behaviour and fate, has attracted great attention for the intervention and treatment of diseases.1–3 As powerhouses and energy providers, mitochondria have been reported to play an important role in the occurrence of many major diseases and their regulation can effectively induce cancer cell death. Generally, the activity of mitochondria depends on the proton (H+) gradient of the inner mitochondrial membrane. To form the proton gradient, ATP is normally generated and provides energy to maintain cell metabolism and survival.4–6 Meanwhile, Ca2+ homeostasis in mitochondria also contributes to the regulation of H+ gradients for maintaining the normal function of mitochondria.7–9 Accordingly, modulating the H+ gradient and Ca2+ homeostasis would lead to mitochondrial damage and cell apoptosis via opening of mitochondrial permeability transition pores. This would facilitate the release of cytochrome C for the subsequent lysis of caspase-3 protein.10,11 Therefore, it can be imaged that the synergistic regulation of H+/Ca2+ influx to the mitochondrial matrix will lead to efficient mitochondrial depolarization for enhanced cancer therapy.Nevertheless, the mitochondrial depolarization could be a complex process, which was normally hindered in the heterogeneous and dynamic tumor microenvironment (TME).12–14 Firstly, TME homeostasis of tumor cells dramatically hinders the mitochondrial depolarization. For example, pH homeostasis could be maintained by the effusion of intracellular by-products of glycolysis metabolite and lactic acid (LA) metabolism in the TME.15,16 Besides, cytoplasm and mitochondria can serve as important Ca2+ storage hubs to maintain intracellular Ca2+ homeostasis, which greatly limited the mitochondrial depolarization.17,18 Furthermore, the toxic reactive oxygen species (ROS), a pivotal agent for mitochondrial membrane permeability, is easily depleted by abundant endogenous antioxidant glutathione (GSH), leading to poor therapeutic effectiveness.19–22 Therefore, multichannel synergistic regulations to balance TME homeostasis for sensitized mitochondrial depolarization are required.
Meanwhile, the effective loading and delivery of therapeutic agents as well as TME-responsive drug release are limited,23–25 which encounter undesirable leakage of “off-target” toxicity and adverse side effects.26–28 Therefore, the meticulous loading of multiple reagents into nanocarriers is essential for designing an ideal collaborative system. Fortunately, upon programmable assembly, deoxyribonucleic acid (DNA)-based materials exhibited stimulus-responsiveness (such as pH, enzymes, and metal ions), controlled network structures and convenient modifications, which showed great potential for cancer therapy.29–34 Therefore, a multifunctional DNA nanoplatform is expected to induce cell apoptosis upon efficient delivery of multiple reagents for safe and synergistic mitochondrial depolarization.
Herein, a supramolecular DNA nanocomplex (Ca@DNA–MF) was fabricated upon the dynamic assembly of multiple mitochondrial depolarization reagents into a DNA nanonetwork. Via the hybridization of designed DNA primers, the tiny “seeds” of Fe/Mn–polyphenol coordination polymers were packed by siRNA (siMCT4)-integrated DNA nanonetworks. The obtained spherical nanocomplex was further biomineralized with Ca2+ in the presence of hyaluronic acid (HA) to obtain Ca@DNA–MF for targeting tumor cells. Upon targeting functionalization with HA, Ca@DNA–MF accumulated at tumor sites. This was followed by efficient Ca2+ release to break Ca2+ homeostasis, leading to Ca2+-based mitochondrial depolarization. Simultaneously, ATP-triggered siMCT4 downregulated MCT4 expression on the tumor cell membrane to restrain lactate acid (LA) efflux and induce TME acidification. This can further accelerate the overloading of Ca2+ in the mitochondrial matrix. Specifically, the drastic cytoplasmic acidification also enhanced the Fenton reaction to generate adequate ROS. This further increased the mitochondrial membrane permeability and dissipated the proton gradient. This was achieved by the synergistic consume of excessive endogenous glutathione (GSH) by redox systems of Fe3+/Fe2+ and Mn2+/Mnx+. Consequently, via metabolic reprogramming and cancer cell disturbance, synergistic remodulation of H+/Ca2+ gradients was obtained for efficient mitochondrial depolarization. Upon triggering apoptosis and ferroptosis to enhance ROS generation, the remodulation was efficient for enhanced synergistic cancer therapy with low toxicity.
Results and discussion
Design of the Ca@DNA–MF nanocomplex
The present nanocomplex of Ca@DNA–MF was prepared upon multiple procedures of dynamic self-assembly. As shown in Scheme 1A, the primers of N1, H1-dimer and ATP aptamer-integrated H2-siRNA hairpins were designed. In the presence of N1, H1-dimer hybridized with H2 hairpins to form a DNA network under the hybridization chain reaction (C-HCR).35,36 Both the ATP aptamer and a specific MCT4 inhibitor of siMCT4 were integrated into H2 hairpins. This would facilitate ATP-driven dissociation for gene-mediated metabolic reprogramming, which depended on LA levels in high glycolytic cancer cells. Simultaneously, the “seeds” of Fe/Mn–polyphenol polymers (MF) were prepared by thermal decomposition of Fe3+ and Mn2+ in NH3·H2O, which then accumulated into “seeds” in the presence of citric acid. Thereafter, the “seeds” were added into the aforementioned DNA nanocomplex to obtain spherical MF-embedded DNA networks (DNA–MF) upon electrostatic interactions. Finally, the spherical nanocomplex was biomineralized by Ca2+ in the presence of HA. Therefore, Ca@DNA–MF was obtained for the controllable release of therapy reagents, avoiding reagent leaking in the vivo fluid circulation and eliminating side effects toward normal tissues.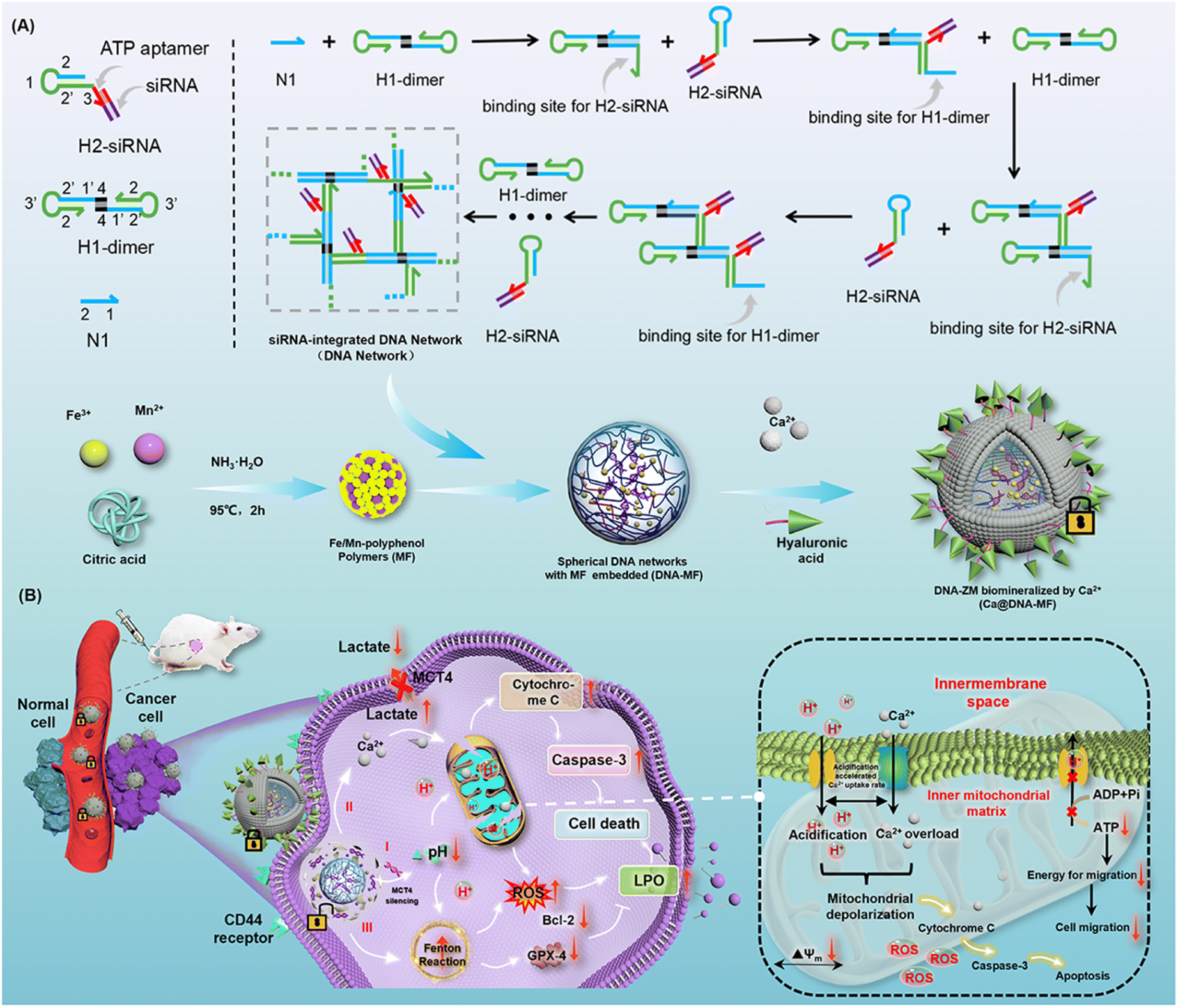 | ||
| Scheme 1 Synthesis and working procedures of Ca@DNA–MF. (A) The schematic illustration for the synthesis. (B) The working procedures for the synergistic multichannel depolarization of the mitochondrial. (I) The channel of MCT4 silencing for acidosis. (II) The channel of Ca2+ overload. (III) The channel of Fenton-like oxidative stress. | ||
In the vivo fluid circulation (Scheme 1B), Ca@DNA–MF site-specifically targeted tumor sites, because the surface HA specifically recognized highly expressed CD44 membrane proteins on the surface of cancer cells.37–39 Then, upon endocytosis, the Ca-biomineralized cover was on-demand “unlocked” in the slightly acidic TME. This initiated the subsequent synergistic multichannel mitochondrial depolarization. Firstly (channel I), the siMCT4 in the DNA networks was released upon the interaction between the integrated ATP-aptamer and the cytoplasmic ATP in cancer cells. The ATP-driven siMCT4 could downregulate the MCT4 expression on the tumor cell membrane to initiate intracellular retention of LA. This resulted in drastic acidification of cytoplasm and further induced mitochondria matrix acidification for mitochondrial depolarization. Simultaneously, the acidification of the mitochondria matrix would synergistically promote the Ca2+ overloading upon the Ca2+ release from the cover of the DNA nanocomplex, being another channel for mitochondrial depolarization (channel II). Thirdly, along with the decomposition of Ca@DNA–MF, the seeds of Fe/Mn–polyphenol polymers were exposed to release Fe3+, which immediately in situ reduced to Fe2+ by endogenous GSH. Subsequently, promoted by Mn2+, Fe2+ participated in the efficient Fenton-like catalytic reduction of endogenous H2O2 to cytotoxic ˙OH.40–43 This would further facilitate the mitochondrial depolarization upon the increase of mitochondrial membrane permeability for penetrating more H+/Ca2+ into the mitochondrion (channel III). Additionally, the acidosis in channel I would also enhance the Fenton-like ˙OH generation to facilitate the potential of apoptosis with LA metabolic modulation. Consequently, upon the synergistic “network” of multichannel mitochondrial depolarization, an increase of ROS can be eventually obtained. This would exhibit increased expression of both cytochrome C and caspase-3 (apoptotic-pathway-related proteins), along with the decrease of Bcl-2 and GPX-4 protein.
Characterization of the Ca@DNA–MF nanocomplex
To confirm the successful preparation of the Ca@DNA–MF nanocomplex, a series of characterization methods have been carried out. As demonstrated by transmission electron microscopy (TEM) characterization, the seeds of MF nanoparticles exhibited a tiny spherical morphology at a monodisperse size of 6 nm (Fig. 1A-MF). Meanwhile after being embedded into 100 nm of the DNA networks (Fig. S1†), larger spherical DNA–MF particles of approximately 120 nm diameter were obtained (Fig. 1A DNA–MF). Subsequently, after biomineralization with Ca2+ and HA, the size of the Ca@DNA–MF nanocomplex reached about 140 nm. Demonstrated by high-angle annular dark field-scanning TEM (HAADF-STEM) and elemental mapping images, the uniform element distribution of both DNA–MF (Fig. S2†) and Ca@DNA–MF (Fig. 1B) was revealed, verifying the successful assembly-based preparation. Furthermore, the stepped increase of particle size during the preparation was further confirmed by dynamic laser scattering (DLS) analysis (Fig. 1C). Besides, zeta potential characterization revealed the successful integration of MF (+18.64 mV) with DNA networks (−21.25 mV), which formed DNA–MF (−6.54 mV). This was followed by successful biomineralization by Ca2+, exhibiting the increased zeta potential of Ca@DNA–MF (−4.76 mV) (Fig. 1D).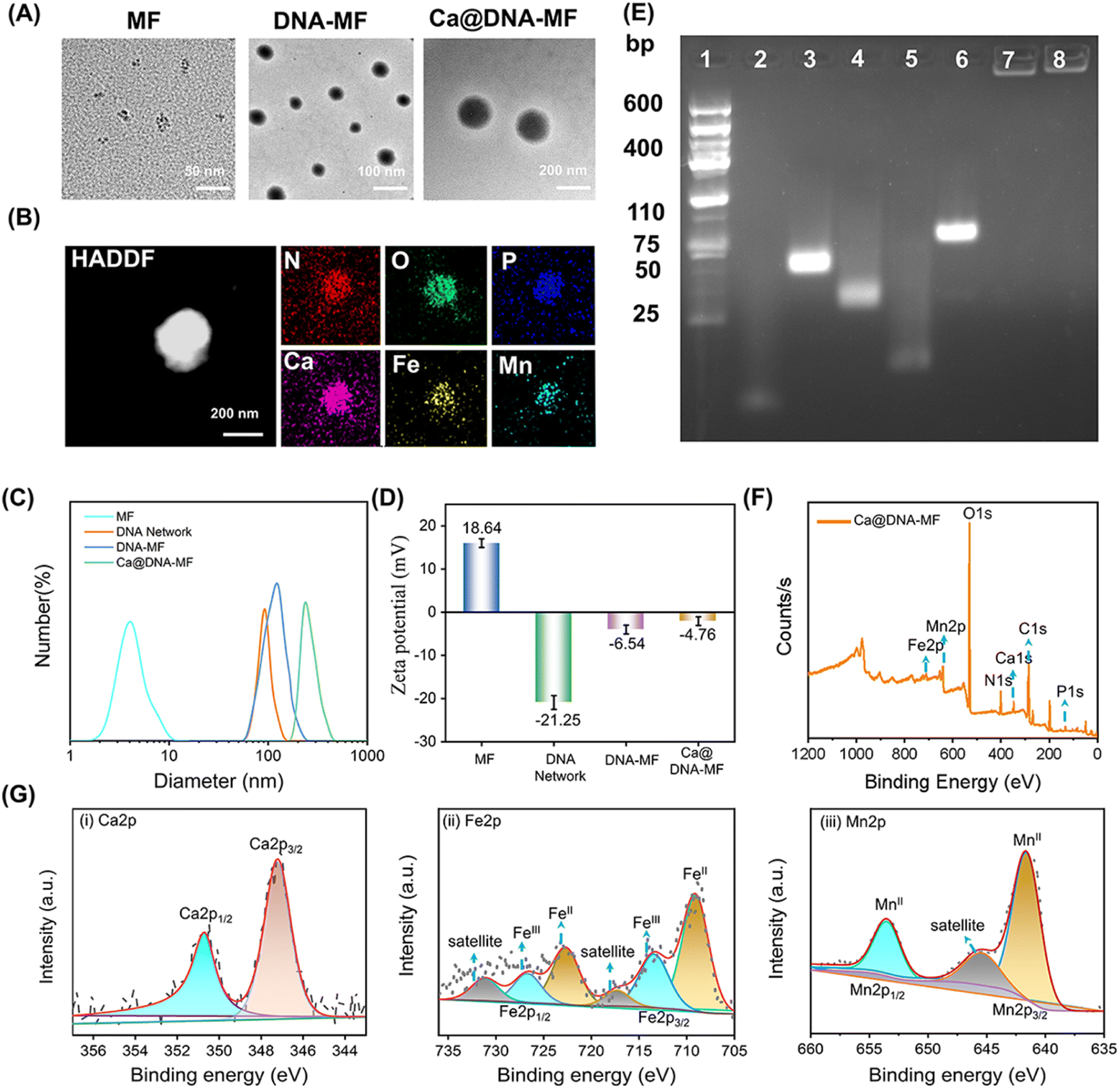 | ||
| Fig. 1 Characterization of the Ca@DNA–MF nanocomplex. (A) TEM images of MF, DNA–MF and Ca@DNA–MF. (B) TEM elemental mapping images of N, O, P, Ca, Fe and Mn. (C) DLS analysis. (D) Zeta potentials. (E) Polyacrylamide gel electrophoresis images of stepped products in C-HCR-based preparation of Ca@DNA–MF. (1) Ladder, (2) N1, (3) H1-dimer, (4) H2, (5) ssDNA, (6) H2-ssDNA, (7) DNA network, and (8) Ca@DNA–MF. (F) XPS spectrum of Ca@DNA–MF. (G) XPS high-resolution spectrum of Ca 2p (i), Fe 2p (ii) and Mn 2p (iii). | ||
The stepped-synthesis of the DNA network-based Ca@DNA–MF was also verified by polyacrylamide gel electrophoresis (PAGE). As shown in Fig. 1E, the C-HCR primers of N1 (lane 2), H1-dimer (lane 3), H2 (lane 4), and the siRNA mimic of a ssDNA (lane 5) exhibited relatively faster migration due to the different molecular weights. When the ssDNA was hybridized to H2 upon the complementation of the 5′ sequence of ssDNA to the 3′ end of H2, the obtained product of H2-ssDNA migrated much slower (lane 6). This indicated that the siRNA could be efficiently tethered to H2 at the 3′ end with the ATP aptamer overhung, which would facilitate the ATP-initiate release of siRNA. Meanwhile the C-HCR product of the DNA network (lane 7) and the final Ca@DNA–MF (lane 8) were trapped without any separation, verifying the successful synthesis of DNA networks via C-HCR. In addition, Ca@DNA–MF was quite stable in complicated biological environments, demonstrated by the fact that no DNA fragment was recorded after incubating with Ca@DNA–MF in 10% fetal bovine serum (FBS)-containing culture medium (Fig. S3†). The DLS results also demonstrated the good stability of Ca@DNA–MF under different physiological conditions (DI water, PBS, and RPMI 1640), which facilitated biological applications (Fig. S4†).
Furthermore, the related chemical composition and element valence state of the stepped synthesis products were verified by X-ray photoelectron spectroscopy (XPS). The co-existence of Ca, Fe and Mn was well recorded in the XPS spectra of MF, DNA–MF and the Ca@DNA–MF nanocomplex (Fig. S5† and 1F). In the XPS high-resolution spectra of Ca@DNA–MF, the Ca2+ peaks (Ca 2p3/2 at 346.5 and Ca 2p1/2 at 346.5 eV) (Ca 2p spectrum, Fig. 1G(i)), peaks of Fe2+ (709.7 and 724.08 eV) and Fe3+ (711.7 and 726.12 eV) (Fe 2p spectrum, Fig. 1G(ii)), and Mn2+ peaks (Mn 2p3/2 at 641.14 and Mn 2p1/2 at 653.07 eV) (Mn 2p spectrum, Fig. 1G(iii)) were well recorded. It should be noted that the important Fenton-like regent of Fe species exhibits the coexistence of Fe2+ and Fe3+, demonstrated by the Fe 2p1/2 and Fe 2p3/2 peaks. Consequently, the successful synthesis of the Ca@DNA–MF nanocomplex was confirmed.
The acidity-activated ROS generation and GSH consumption by Ca@DNA–MF
As designed, the mitochondrial depolarization could be facilitated by siMCT4-initiated acidosis (channel I in Scheme 1) and Ca2+ (channel II). Meanwhile, the acidity would further activate GSH depletion and hydroxyl radical (˙OH) generation by a Fenton-like reaction (channel III). This was a synergistic process, in which the acidification of channel I also facilitated the other two channels. To verify the synergistic roles of acidification for the other two mitochondrial dysfunction channels, the characterization of ion release, ˙OH generation and GSH consumption was carried out. Herein, the ion release (channel II) was evaluated by coupled plasma optical emission spectrometry (ICP-OES) tests. The generation of ˙OH and the depletion of GSH in channel III were evaluated by monitoring the decreased absorption of 3,3′,5,5′-tetramethylbenzidine (TMB) and 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), respectively. The redox recycling between Fe3+/Fe2+ and Mn2+/Mnx+ facilitated the GSH depletion and ˙OH generation (Fig. 2A).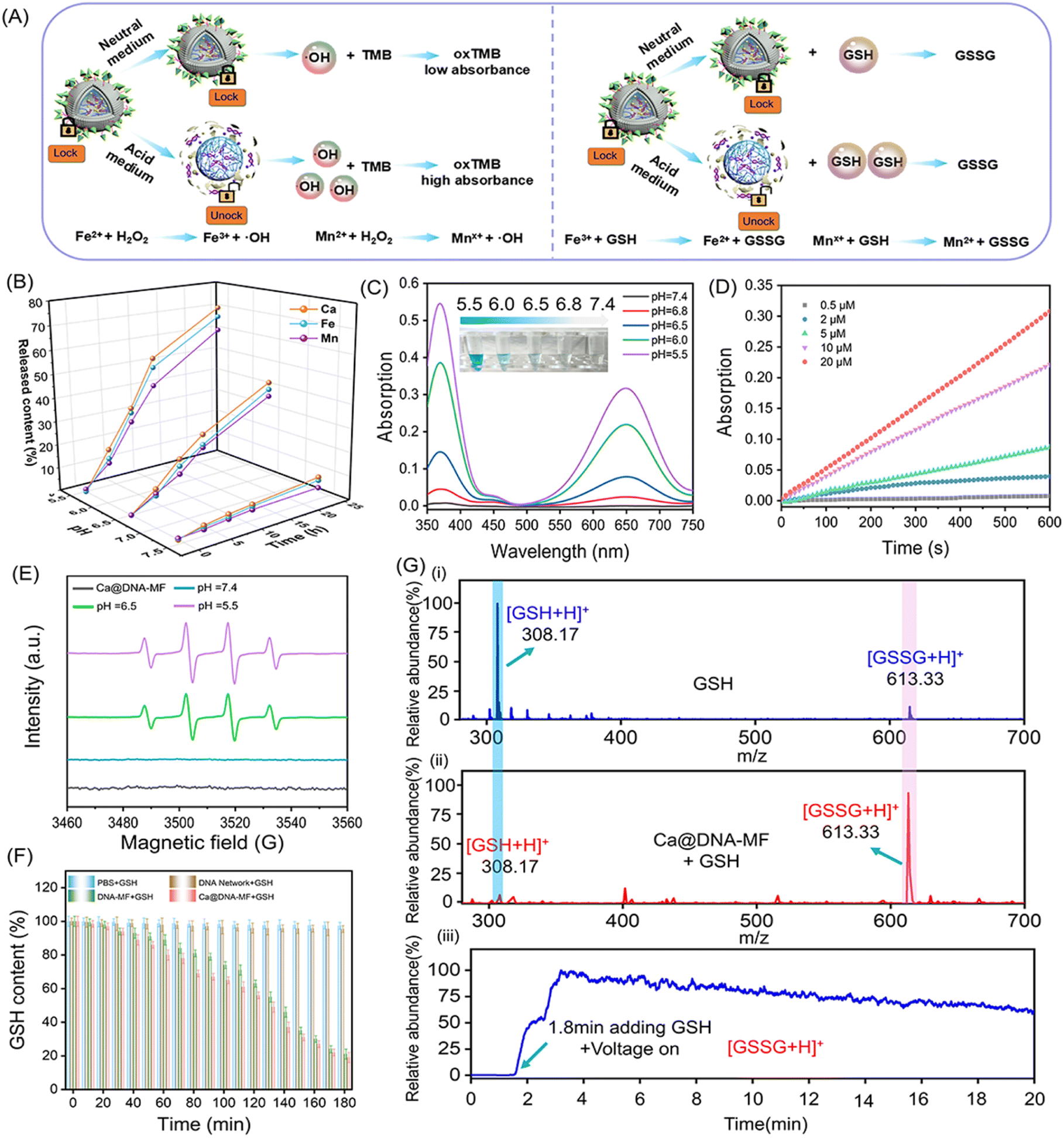 | ||
| Fig. 2 Evaluation of the acidosis-based synergistic multichannel mitochondrial dysfunctions by Ca@DNA–MF. (A) Schematic illustration for the catalytic ˙OH generation, which facilitated the oxidation of TMB (colorless) to oxTMB (blue) or oxidation of GSH to GSSG. (B) Profiles of the release of Ca2+, Fe3+ and Mn2+ during the incubation of Ca@DNA–MF in HEPES under different pH conditions. (C) TMB oxidation by 100 μM H2O2 in the presence of Ca@DNA–MF under different conditions. (D) Time-dependent absorbance of oxTMB by different amounts of H2O2 in HEPES buffer (pH = 6.5) with Ca@DNA–MF presented. (E) ESR spectra under different pH conditions using 5,5-dimethyl-1-pyrroline N-oxidation (DMPO) as the ˙OH scavenger. (F) Absorption of GSH after the depletion by different nanomaterials. (G) GSH residual concentration in PBS (i), Ca@DNA–MF (ii) and Ca@DNA–MF at different times (HEPES pH 6.5) (iii). | ||
As demonstrated by ICP-OES analysis (Fig. 2B), the release of Ca, Fe and Mn was inefficient under physiological conditions at pH 7.4. This indicated the good stability and low systemic toxicity of Ca@DNA–MF, preventing the leakage of reagents into blood circulations. While a relatively faster ion release was observed under acidic conditions (pH 5.5 and pH 6.5), and the highest release was exhibited at pH 5.5. Therefore, the TME (acidic condition)-initiated collapse of Ca@DNA–MF facilitated the subsequent mitochondrial dysfunction by Ca2+ (channel II) and by Fenton-like therapy (channel III). In addition, TME responsiveness would minimize the premature leakage-induced systematic toxicity and concurrently facilitate precise spatiotemporal therapy at tumor sites.
Besides, the Fenton-like therapy also exhibited acid-dependent enhancements. For the Fenton-like therapy, the delivered Fe3+ was firstly in situ reduced to Fe2+ to effectively consume GSH. This was confirmed by the increased absorption of the Fe2+ probe (1,10-phenanthroline) at 510 nm. As a result, the obtaining of Fe2+ was enhanced under acidic conditions, indicated by the higher absorption at pH 5.5 than at pH 6.5 and 7.4 (ESI Fig. S6†). This was consistent with the pH-responsive morphological variations in TEM and DLS characterization (Fig. S7†). Therefore, acidosis can facilitate the release of the mitochondrial dysfunction reagents, which would initiate the subsequent multichannel mitochondrial depolarization.
Inspired by the reduction of Fe3+ to Fe2+, the toxic hydroxyl radical (˙OH) was generated via a Fenton-like reaction to decompose H2O2. This was evaluated by 3,3′,5,5′-tetramethylbenzidine (TMB) oxidation. As demonstrated (Fig. 2C and S8†), the absorption of oxTMB (generated by ˙OH oxidation) increased with the decrease of the pH value, exhibiting increased blue colors (the inset image). Meanwhile poor catalytic activity was observed at pH 7.4, which further confirmed the activation of catalytic activity for generating ˙OH under acidic conditions. This was in accordance with the decreased H2O2 signals (with titanium sulfate as the probe, Fig. S9†) upon H2O2 consumption. Besides, the steady-state kinetics of the catalytic generation of ˙OH by peroxidase-like coversion were also examined. The kinetic curves of the catalytic conversion of different concentrations of H2O2 to ˙OH were recorded in a slightly acidic environment (Fig. 2D and S10†), which resulted in the maximum initial velocity (Vmax) and a Michaelis–Menten constant (Km) of 4.37 × 10−8 M s−1 and 15.06 μM, respectively. This ˙OH generation was also confirmed by the increased FL signals of terephthalic acid (TA) upon the oxidation of TA by ˙OH (Fig. S11†). In addition, the acidic-dependent generation of ˙OH was also confirmed by electron spin resonance (ESR) spectroscopic analysis, which exhibited increased characteristic ˙OH signals (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1) with the decrease of pH values (Fig. 2E). Besides, no obvious O2˙− signal was observed in the Ca@DNA–MF system and the groups in the presence of air or H2O2 at pH 5.5, 6.5 and 7.4 (Fig. S12†). Therefore, the good catalytic capability of Ca@DNA–MF was confirmed, which can generate efficient ˙OH for the subsequent mitochondrial dysfunction in channel III.
:2:2:1) with the decrease of pH values (Fig. 2E). Besides, no obvious O2˙− signal was observed in the Ca@DNA–MF system and the groups in the presence of air or H2O2 at pH 5.5, 6.5 and 7.4 (Fig. S12†). Therefore, the good catalytic capability of Ca@DNA–MF was confirmed, which can generate efficient ˙OH for the subsequent mitochondrial dysfunction in channel III.
Furthermore, the depletion of GSH in channel III was evaluated by comparison of ˙OH and GSH under different conditions. Moreover, the depletion of intracellular GSH to GSSG is beneficial for improving the efficiency of Fenton-like reactions. As demonstrated, with adding GSH into the H2O2–Ca@DNA–MF system, the amount of ˙OH increased to a certain extent (0–9 mM), indicated by the TMB degradation test (Fig. S13A†). This is in accordance with the depletion of GSH (Fig. 2F) for converting Fe3+ into Fe2+, which facilitated the ˙OH generation by the Fenton-like process (Fig. S13C†). Meanwhile with too much GSH added (higher than 15 nM), ˙OH obviously decreased due to the consumption of ˙OH by excessive GSH (Fig. S13B†). Simultaneously, the depletion of GSH was also confirmed by the decreased absorption of 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) during the incubation (Fig. S14†). Furthermore, the GSH consumption was monitored by ambient mass spectrometry, which directly extracted and ionized samples without any pre-treatment (Section 4.4 in the ESI†). As demonstrated by both off-line (Fig. 2G(i) and (ii)) and on-line examinations (Fig. 2G(iii)), with Ca@DNA–MF treatment, the characteristic GSH ion of [GSH + H]+ (at m/z 308) dramatically decreased along with the significant increase of [GSSG + H]+. Therefore, the depletion of endogenous GSH (lower than 10 mM of GSH expression in the TME) could facilitate the generation of ˙OH for synergetic Fenton-like therapy (channel III).44
The ATP-triggered intracellular delivery of mitochondrial dysfunction reagents
To explore the on-demand rapid ATP-triggered release and gene knockdown in cells, siRNA expression was evaluated for examining the release behavior. For visualized monitoring, DNA networks were labelled with red FL dye of TAMRA, whose FL signals were quenched by BHQ2 linked to the siRNAs upon fluorescence resonance energy transfer (FRET). As illustrated in Fig. 3A, in the presence of ATP, the siRNA was released along with the turn-on red signals due to the breaking of the FRET system. This process was initiated by ATP in the TME, which was demonstrated by the highest FL signals in the group of Ca@DNA–MF (functionalized with the ATP aptamer), relative to the negligible fluorescence signal in the nATP-Ca@DNA–MF group (without the ATP aptamer) (Fig. 3B, S15A and B†). In addition, this release rate was a time-dependent process, which was illustrated by PAGE analysis of H2-siRNA products after incubation for different times. As shown in Fig. 3C, the amount of H2-siRNA decreased during the incubation and satisfactory release can be achieved within 0.5 h. Meanwhile, no significant dissociation was recorded for the ssDNA complex with a scramble sequence of the ATP aptamer (donated as nATP-H2) even after incubation for 3 h (Fig. S15C†). Therefore, the ATP-triggered siRNA reagent release has been successfully designed, which would facilitate intracellular delivery and therapy applications. | ||
| Fig. 3 Examination of the ATP-triggered delivery of mitochondrial dysfunction reagents. (A) Schematic illustration of ATP-triggered release of siRNAs, which emitted “turn-on” FRET-based red FL signals of TAMRA that was labelled on siRNAs. (B) The increase of FL signals versus time in the presence of Ca@DNA–MF with or without the ATP-aptamer linked, respectively. (C) PAGE image for products of ATP-triggered collapse of H2-ssDNA (2 μM), after incubation at 37 °C for different times. Intercellular ATP was 5–10 mM. (D) Cell imaging of 4T1 cells after being incubated with Ca@DNA–MF with (i) and without (ii) the ATP-aptamer and the nuclear dye Hoechst (blue). Red signal of TAMRA indicated the location of siRNA. Scale bars: 50 μm. (E) Mean FL of flow cytometry data with (i) and without the ATP aptamer (ii). (F) CLSM Z-stack scanning of 3D multicellular spheroids of 4T1 cells treatment with different nanomaterials. Scale bars: 500 μm. | ||
Thereafter, the delivery of the mitochondrial dysfunction reagents was examined by confocal laser scanning microscopy (CLSM) imaging of 4T1 cell lines. As a result, a significantly increased red signal of TAMRA (indicating the release of siRNAs) was recorded during the incubation of the 4T1 cells with ATP aptamer-functionalized Ca@DNA–MF, even exhibiting obvious red signals at 4 h (Fig. 3D(i)). Meanwhile without the ATP aptamer (nATP-Ca@DNA–MF), a quite weak signal of TAMRA was observed even after 24 h of incubation (Fig. 3D(ii)). The important role of the ATP aptamer in the siRNA delivery was further demonstrated by flow cytometry analysis. As a result (Fig. 3E), a dramatically higher uptake of the reagents than that without the ATP aptamer was exhibited, implying effective cellular uptake for therapeutic applications. Time-dependent cell internalization via endocytosis was also demonstrated according to subcellular localization experiments. Indicated by Pearson's correlations (Fig. S16A†), the initial overlapped signals of Ca@DNA–MF and lysosomal were separated with time prolonging, suggesting efficient endosomal escape of Ca@DNA–MF. Meanwhile without the Ca cover, the DNA network group was stably trapped in lysosomes (Fig. S16B†). This was generated from the rapid decomposition of the Ca cover in acidic lysosomes, which facilitated the production of ROS to improve lysosomal escape for the efficient delivery of multiple reagents.45
Besides, the modification with HA was also important for tumor-targeted uptake, verified by the 3 times weaker cellular uptake of DNA–MF (without HA modified) than that of Ca@DNA–MF (Fig. S17 and S18†). The tumor-targeted intracellular delivery was further confirmed by the imaging comparison of different cell lines, including cancer cells (MCF-7, A549 and HeLa) and normal cells (HEK-293T, with low CD44 antigen expression). As a result, 20 times stronger cellular uptake was exhibited for the cancer cells than the normal cells, attributed to low expression of the CD44 antigen that selectively bound to HA on Ca@DNA–MF (Fig. S19A and B†). The role of HA in the selective targeting of tumor cells was also confirmed by flow cytometry analysis (Fig. S19C†). The study on the cellular uptake pathway demonstrated that the entrance of Ca@DNA–MF into 4T1 cells was inhibited by specific endocytic inhibitors and at low temperature. This indicated that the cellular uptake of Ca@DNA–MF was an energy-dependent endocytosis pathway (Fig. S20†). Additionally, the tumor infiltration properties were also evaluated by the imaging of 4T1 cell-based multicellular spheroids (MTSs) after being treated with different TAMRA-labeled nanonetworks. As demonstrated in Fig. 3F, no obvious red signal was observed in each depth group without the ATP-aptamer (nATP-DNA network). Alternatively, the strongest FL signals of tumor spheroids were recorded after being incubated with Ca@DNA–MF, indicating the best infiltration and targeted properties with both the ATP aptamer and HA functionalized on Ca@DNA–MF. In addition, the acidic conditions facilitated the reagent delivery, demonstrated by the much obvious red signals (Fig. S21A and B†) and higher cell uptakes (Fig. S21C†) at lower pH values. Therefore, the present Ca@DNA–MF exhibited the efficient tumor-targeted and safe delivery of mitochondrial dysfunction reagents with minimized off-target toxicity.
The performance of the mitochondrial dysfunction by remodulating of H+/Ca2+ gradients
The influence of Ca@DNA–MF on mitochondrial pH, morphological membrane potential (MMP), Ca2+ expression and ATP production in 4T1 cells was further evaluated. As illustrated in Fig. 4A, the level of free Ca2+ increased observably, owing to the disturbance of intramitochondrial Ca2+ homeostasis upon Ca@DNA–MF decomposition. The released siMCT4 initiated drastic intracellular acidification and facilitated the H+ influx/acidification in the mitochondrial matrix. This was further synergistically promoted by the overloaded Ca2+, which resulted in the dramatically increased penetration of H+/Ca2+ for depolarization in the internal mitochondrial matrix. Meanwhile, the mitochondrial depolarization would decrease the ATP expression, which induced low cell migration with low migration energy. More significantly, the efficient mitochondrial depolarization finally induced apoptosis along with the increase of apoptotic-pathway-related proteins such as cytochrome C and caspase-3.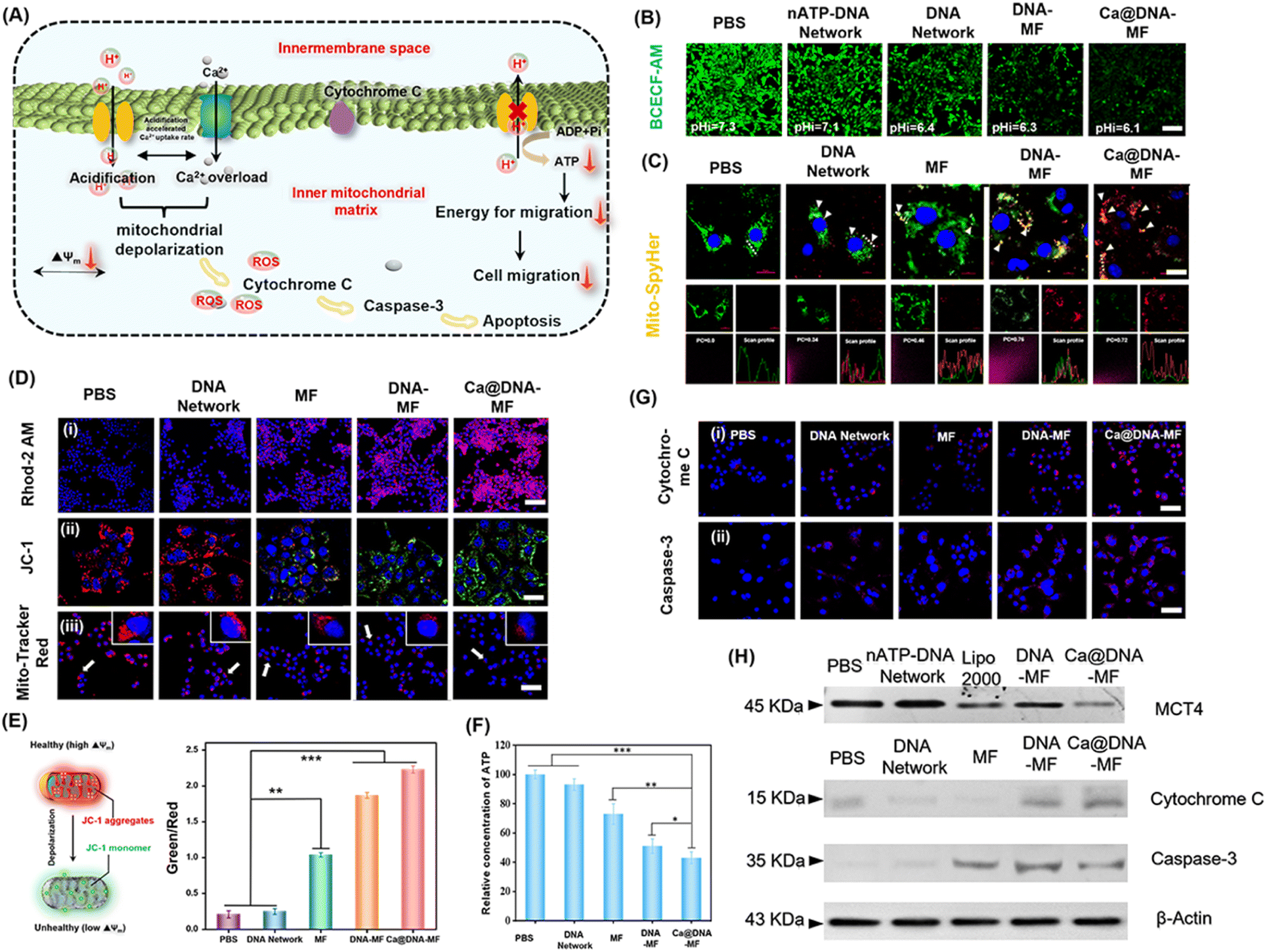 | ||
| Fig. 4 Examination of the mitochondrial dysfunction of 4T1 cells by different nano-networks. (A) Schematic illustration of mitochondria depolarization caused by remodulate H+/Ca2+ gradients. (B) Imaging of extracellular (PBS) and intracellular (cytoplasm) pH levels with DCECF-DA as the pH probe, which exhibited increased green signals at higher pH values. Scale bars: 100 μm. (C) Imaging of mitochondrial pH values in 4T1 cells transfected with pH-sensitive Mito-SypHer. The increased ratio of red (480 nm)/green (430 nm) indicated higher pH values. Scale bars: 10 μm. (D) Mitochondrial imaging for Ca2+ evaluation (showing purple signals with Rhod-2 AM as the probe). (i) Scale bars: 100 μm. Mitochondrial membrane potential measurement (with JC-1 as the probe, exhibiting the conversion of signals from red to green with mitochondrial dysfunction). (ii) Scale bars: 10 μm. The mitochondrial distribution with Mitochondria-Tracker Red as the probe to exhibit red signals (iii). Scale bars: 50 μm. (E) Corresponding quantitative analysis of (D). ***p < 0.001 and n.s.: no significance. Data presented as mean ± SD (n = 3). (F) Relative concentration of intracellular ATP. Bars represent mean ± SD (n = 3 independent samples) (*P < 0.05; ***P < 0.001, calculated using a two-tailed unpaired t test). (G) Expression of cytochrome C (i) and total caspase-3 (ii) in 4T1 cells after different treatments for 24 h. Scale bars: 50 μm. (H) Western blotting assay of MCT4 inhibition and the mitochondria apoptosis indicated by the contents of cytochrome C and caspase-3. Control: β-actin. | ||
To evaluate the performance of siMCT4-initiated intracellular acidification, the intracellular pH values were determined by cell imaging with BCECF-AM as the pH probe. With 4T1 cells as models (Fig. 4B), the intracellular pH was about 7.3 after being treated with PBS. Meanwhile the intracellular pH decreased and the lowest value was recorded after being treated with Ca@DNA–MF (pH 6.1). This confirmed the siMCT4-initiated cytosolic acidity via blocking of LA effusion (channel I). Furthermore, the highest intracellular LA level of Ca@DNA–MF further confirmed the successful MCT4 blockage (Fig. S22†). Besides, the pH values in the supernatant of Ca@DNA–MF further validated similar results to the above investigations (Fig. S23†). Therefore, the siMCT4-induced acidosis could be regulated by the LA metabolic content to remodel the tumor intracellular environment. To further examine the influx of H+ into the mitochondrial matrix, the mitochondrial pH values (pH-mito) were monitored with a pH-sensitive probe of mitochondrial FL protein Mito-SypHer. As a result (Fig. 4C), the significant H+ influx into mitochondria was recorded after being treated with Ca@DNA–MF, which was indicated by the obvious fluorescence emission-shift from green to red (white arrows). This would therefore facilitate the decrease of H+ gradient of the inner/outer mitochondria, for achieving mitochondrial depolarization. The mitochondrial dysfunction was further confirmed by the round-shaped 4T1 cells, accompanied by obvious matrix swelling and mitochondrial fission after being treated with Ca@DNA–MF (Fig. S24†).
In addition, the expression of Ca2+ in both cytoplasm and mitochondria was evaluated to support the Ca2+-based mitochondrial depolarization in channel II. From the cell imaging of cytosolic Ca2+, the strongest FL signal of Ca2+ (indicated by the green probe of Fluo-4 AM) was recorded in the group of Ca@DNA–MF (Fig. S25†), further confirming the pH-activated intracellular release of Ca2+. Besides, the intracellular acidification in channel I would accumulate H+ in the mitochondrial intermembrane. This would trigger the opening of the mitochondrial permeability transition pore, accelerating a significant influx of H+ and Ca2+ and resulting in mitochondrial dysfunction.46 Thereafter, the Ca2+ expression in mitochondria was evaluated by the mitochondrial Ca2+ probe of Rhod-2 AM (Fig. 4D(i)), which indicated the highest Ca2+ signal in the Ca@DNA–MF group. This Ca2+ expression was more significant than that in the DNA–MF group without Ca2+ coating, which further confirmed the good synergistic mitochondrial dysfunction of Ca2+ in channel II.
To evaluate the performance of the mitochondrial depolarization, a series of the characterization techniques have been employed. Firstly, the membrane potential (Δψ) was evaluated by using a JC-1 probe to indicate the proton gradient across the mitochondrial membrane. The increased ratio of the green/red signal indicated the decrease of Δψ upon the conversion of JC-1 aggregates (red) into monomers (green) via the mitochondrial depolarization (Fig. 4E). As a result (Fig. 4D(ii)), the Ca@DNA–MF group displayed the lowest Δψ, resulting in an ∼6.7-fold green/red ratio relative to the untreated one (Fig. 4E). This further confirmed the satisfactory mitochondrial depolarization by Ca@DNA–MF. This was also in accordance with the smallest number of mitochondria with severe mitochondrial damage by Ca@DNA–MF, which was demonstrated by the intracellular distribution of mitochondria staining with Mito-Tracker Deep Red (Fig. 4D(iii)). In addition, the most significant mitochondrial permeability transition pore (mPTP) activation upon mitochondrial depolarization with Ca@DNA–MF was confirmed by the calcein-AM loading/CoCl2 quenching strategy (Fig. S26†).47 Consequently, the significant mitochondrial depolarization by Ca@DNA–MF has been verified.
Next, intracellular adenosine triphosphate (ATP) was measured as a direct embodiment of LA metabolic reprogramming. As expected, the lowest ATP level was recorded in the Ca@DNA–MF group (Fig. 4F), which was attributed to the oxidative phosphorylation inside mitochondria. Given that ATP was defined as the cell “power plant” of mitochondria, the ATP-dependent cell migration was inhibited upon mitochondrial dysfunction and was demonstrated by the wound healing assay (Fig. S27†). In addition, the corresponding biomarkers of the mitochondria dysfunction (including the inhibition of MCT-4, the release of cytochrome C and the activation of the caspase family such as caspase-3) were also evaluated, upon mitochondria-related apoptotic cascades. As illustrated by imaging of 4T1 cells (Fig. 4G), the significantly increased signals of cytochrome C and caspase-3 were released from the mitochondria into the cytosol after being treated with Ca@DNA–MF. This was in accordance with the western blotting assay, which exhibited significantly decreased expression of MCT-4, and upregulated expression of cytochrome C and caspase-3 (Fig. 4H and S28†). Therefore, the significant mitochondrial dysfunction via the remodulating of H+/Ca2+ gradients by Ca@DNA–MF has been confirmed, which would facilitate the subsequent efficient apoptosis of cancer cells.
Generation of ROS for LPO-based cell apoptosis
Upon the synergistic multichannel mitochondrial dysfunction, the mitochondrial was damaged to facilitate the generation of ROS for LPO-based apoptosis. As illustrated in Fig. 5A, upon specific targeting of HA (on Ca@DNA–MF) to the CD44 receptor on the membrane of cancer cells, Ca@DNA–MF was endocytosed into cancer cells. This facilitated the release of multiple regents for mitochondrial dysfunctions in three channels, which induced the significant enhanced generation of ROS (i.e., H+/Ca2+ transport modulation and GSH depletion). Consequently, the high expression of ROS initiated the decrease of reductive protein of Bcl-2, which prevents apoptotic mitochondrial signaling by inhibiting the oligomerization and activation of proapoptotic factors. In addition, the increased ROS would initiate the apoptosis upon oxidation of R–OH to R–OOH, with the product of MDA as the significant biomarker. Besides, the oxidized GSH (GSSG) in channel III would also result in the decreased expression of reductive GPX-4. Considering that the reductive GPX-4 exhibited the capability of lipid reduction, the apoptosis would be further facilitated due to the inhibition of lipid reduction with the low expression of GPX-4.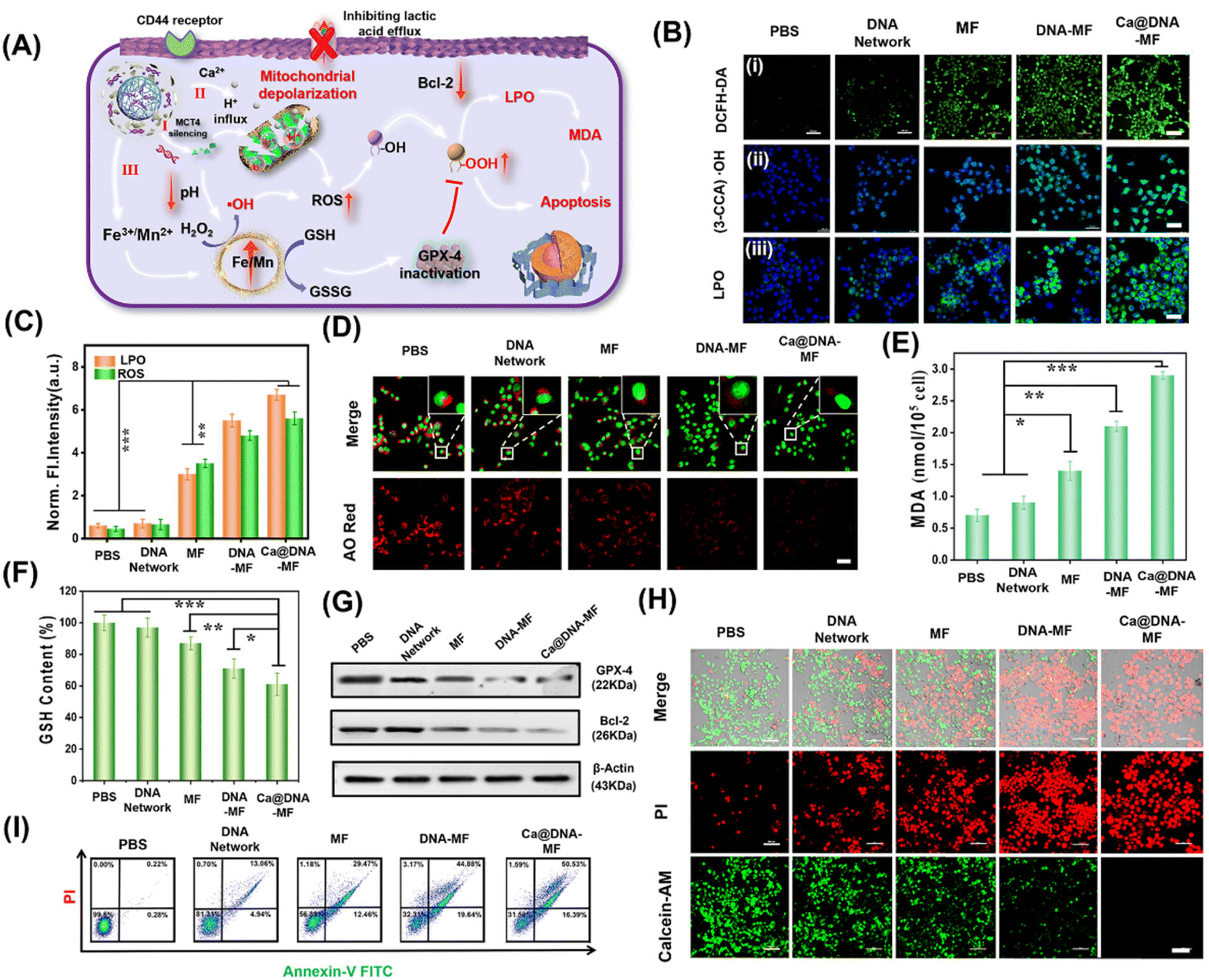 | ||
| Fig. 5 Evaluation of the LPO-based mitochondrial dysfunction after treating 4T1 cancer cells with different groups of PBS, DNA networks, MF, DNA–MF and Ca@DNA–MF. (A) Schematic illustration of ROS generation for the LPO-based mitochondrial dysfunction. (B) CLSM images. (i) Intracellular ROS levels with DCFH-DA as the indicator. Scale bars: 100 μm. (ii) ˙OH levels. Scale bars: 50 μm. (iii) LPO levels. Scale bars: 10 μm. The 4T1 cells were incubated with different nanonetworks for 24 h. Scale bars: 100 μm. (C) The fluorescence intensity of LPO and ˙OH in (B). (D) The imaging of the endosomal membrane content after 24 h of the treatments, based on red signals from AO staining. Scale bars: 10 μm. (E) Content of the LPO product of MDA after treating with different nanomaterials. The statistical analysis was performed in contrast to a control group (*p < 0.05, **p < 0.01, ***p < 0.001, and t-test). (F) GSH content. (G) Western blotting assay of intracellular GPX-4 and Bcl-2 proteins, indicating the therapy performance. (H) CLSM images of 4T1 cells upon different treatments and stained with calcein-AM/PI; scale bars: 100 μm; (I) flow cytometric apoptosis analysis of 4T1 cells stained with Annexin V-FITC/PI. | ||
Firstly, to examine the generation of ROS and level of LPO for the cell apoptosis, 4T1 cells were incubated with different nanomaterials for comparison. As demonstrated (Fig. 5B and C), the most significant ROS signals were recorded for 4T1 cells treated with Ca@DNA–MF, indicated by the ROS probe of dichlorofluorescein diacetate (DCFH-DA). This confirmed the accumulation of ROS by Ca@DNA–MF, which would elicit the mitochondrial depolarization and redox dyshomeostasis for the therapy. Besides, the highest fluorescence levels of ˙OH and LPO were observed in the Ca@DNA–MF group and no obvious signal was recorded in the control group (Fig. 5B). This indicated the tremendous upregulation of LPO by enhancing the ROS generation for efficient cell apoptosis, which was also consistent with the flow cytometry analysis of the LPO level (Fig. 5B(iii) and S29†). This was also confirmed by the obvious endosomal membrane rupture, which was indicated by dramatically decreased red signals of acridine orange (AO) (Fig. 5D).
Furthermore, some biomarkers or important therapy-related molecules were evaluated. Being a typical end product of LPO, the intracellular malondialdehyde (MDA) was highest when cells were treated with Ca@DNA–MF, in accordance with LPO imaging (Fig. 5E). Given that GSH depletion can inactive GPX-4 for cell apoptosis, GSH consumption activity was determined after various treatments. As expected, the most efficient depletion of GSH by Ca@DNA–MF was also confirmed (Fig. 5F), which was generated from the conversion of Fe3+ into Fe2+ for efficient Fenton-like therapy. The down-regulation expression of GPX-4 was determined by western blotting assay (Fig. 5G-GPX-4), which could be attributed to the oxidation of GSH to GSSG. In addition, the expression of Bcl-2 (the key anti-apoptosis protein) also decreased, which was generated from the modulation of metabolic phenotypes in the tumor cells (Fig. 5G-Bcl-2 and S30†). Therefore, efficient mitochondrial dysfunction-based apoptosis was obtained by Ca@DNA–MF, due to the provoking of excessive ROS by Ca@DNA–MF via multistage synergistic processes (channel I to channel III).
To investigate the in vitro therapeutic effects of Ca@DNA–MF, cell viability was evaluated using standard MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide) assay. As a result, Ca@DNA–MF exhibited dose-dependent cytotoxicity on 4T1 cells at a CD50 value of 128.4 μg mL−1 (ESI Fig. S31A†). In addition, negligible influence on the cell viability was observed for the HEK-293T cell (85.6%) compared to the 4T1 cancer cell (19.8%), which owe to the high express H2O2 and GSH levels in cancer cells. Furthermore, enhanced cytotoxicity was recorded at lower pH values (ESI Fig. S31C†), which was in accordance with the designed acidosis-based synergistic multichannel mitochondrial depolarization. The higher cytotoxicity under acidic conditions could be generated from the overloading of Ca2+ in the mitochondrial matrix upon the enhanced release of Ca2+ at lower pH values. More significantly, more ROS species would be generated due to the enhanced Fenton-like reactivity under acidic conditions, which consequently resulted in enhanced cytotoxicity. Furthermore, the performance of cellular apoptosis by Ca@DNA–MF was evaluated by calcein-AM and PI cell staining. As shown in Fig. 5H, the most obvious dead cells were obtained by Ca@DNA–MF after 24 h of incubation. Moreover, Annexin-V-FITC/PI-based flow cytometry analysis was also carried out (Fig. 5I). As a result, the apoptosis rates for the groups of PBS, DNA networks, MF, DNA–MF, and Ca@DNA–MF were 0.22%, 13.06%, 29.47%, 44.88%, and 50.53%, respectively. Therefore, the multichannel mitochondrial dysfunction behavior of Ca@DNA–MF for inducing admirable cancer cell apoptosis was confirmed.
In vivo examinations
Finally, the in vivo therapeutic performance of Ca@DNA–MF was assessed with 4T1 tumor-bearing mice as the models. The PBS, MF, DNA–MF, and Ca@DNA–MF were intravenously injected every three days by the tail vein, and the tumor volumes and body weights were monitored every 3 days for 14 days (Fig. 6A). As indicated by the tumor growth curves (Fig. 6B and C), the best tumor inhibition efficacy was obtained after being treated with the present Ca@DNA–MF, resulting the significant tumor suppression of 70.4% on the 14th day. This was in accordance with the smallest tumor for the group treated with Ca@DNA–MF (Fig. 6D). The distinctively higher inhibition efficiency of the Ca@DNA–MF group than that of the DNA–MF group indicated the significant permeability and targeted effect of the nano-reagents covered by Ca@HA. Besides, the accumulation and retention of Ca@DNA–MF at tumor sites were confirmed by the most significant FL signals (from the TAMRA-labeled Ca@DNA–MF) at the tumor, demonstrated by both FL images (Fig. 6E and S32†) and the quantitative data (Fig. 6F). Therefore, the present Ca@DNA–MF exhibited efficient therapeutic efficacy upon the efficient targeting of tumors.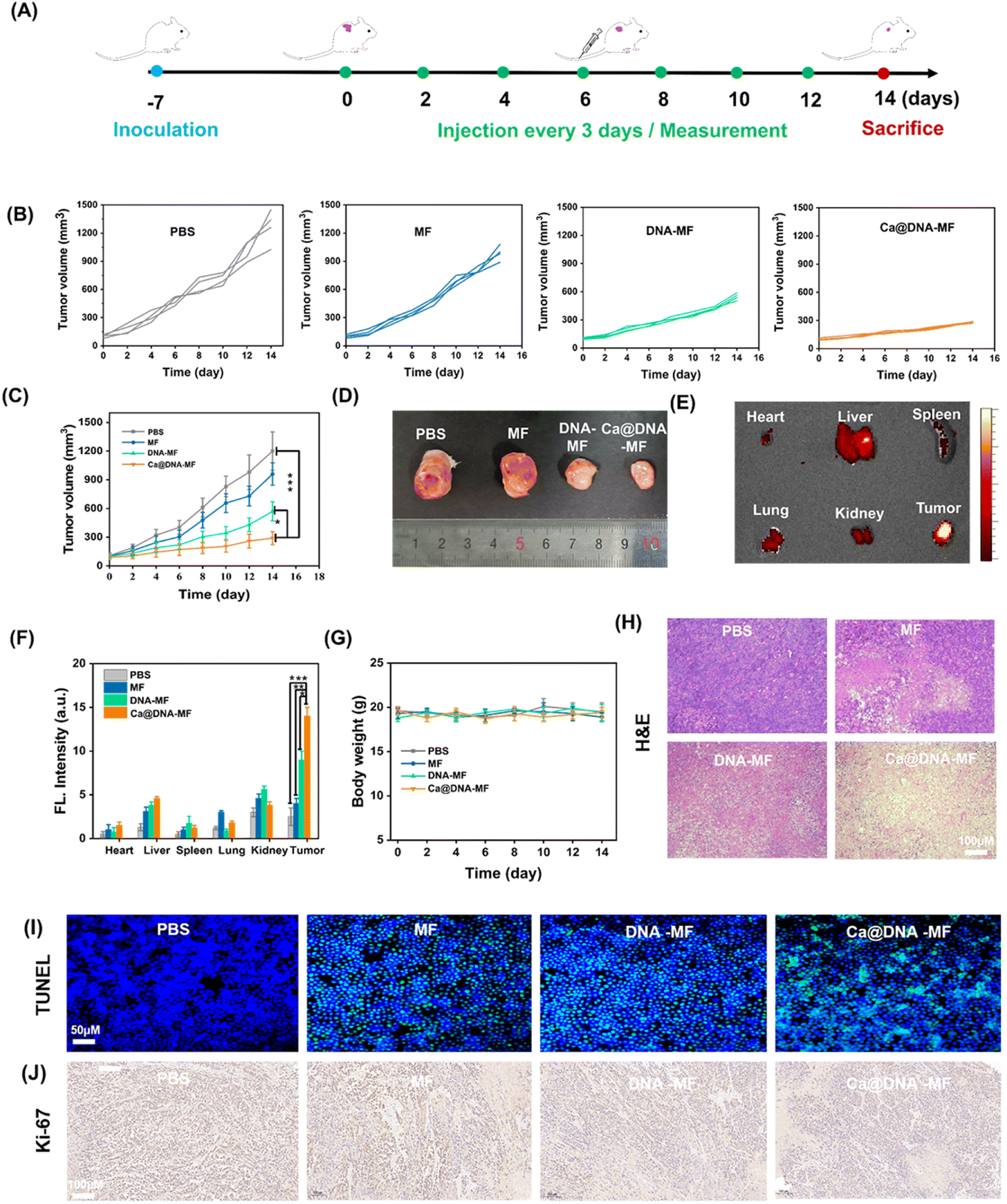 | ||
| Fig. 6 In vivo explorations on tumor targeting and antitumor activities. (A) Scheme of the establishment of the 4T1 tumor xenograft model and the treatment process. (B) Individual tumor growth curves and (C) relative growth curves of 4T1 tumors in different treatment groups (*p < 0.05, **p < 0.01, ***p < 0.001, and t-test). (D) Photographs of excised 4T1 tumors on the 14th day after different treatments. (E) FL images of tissue and (F) quantitative results of the average FL signals of the major organs (*p < 0.05, **p < 0.01, ***p < 0.001, and t-test). (G) Body-weight of 4T1 tumor-bearing female mice after different treatments. (H) Histochemical analyses (H&E) stained, (I) TUNEL-stained and (J) Ki67-stained of tumor tissues after different treatments for 14 days. | ||
Moreover, the biosafety of Ca@DNA–MF was evaluated by both hemolysis assay and in vivo experiments. As a result, no significant hemolysis was observed during incubating mice blood with even 200 μg mL−1 of Ca@DNA–MF (Fig. S33†). In addition, on obvious change of the bodyweight of the mice was observed during the in vivo treatment (Fig. 6G), also indicating the good biosafety of the formulations. Thereafter, the hematoxylin and eosin (H&E) staining of the major organs and tumor tissues was employed. As a result, severe fibrosis with an incomplete cellular morphology and many disintegrated nuclei was observed in the Ca@DNA–MF group, and no evident tissue necrosis was observed in the other groups (Fig. 6H). In addition, no obvious tissue necrosis was also recorded in other major organs after being treated with Ca@DNA–MF (Fig. S34†). Furthermore, the most significant cell apoptosis and the tumor cell proliferation inhibition by Ca@DNA–MF were confirmed via evaluating apoptosis-related proteins by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling (TUNEL) (Fig. 6I) and Ki67 staining (Fig. 6J). This was generated from the deactivation of Ca@DNA–MF in the normal tissues, without the activation species of high ATP expression or lower pH values. This can avoid the premature leakage of reagents ahead of arriving at the tumor, potentially reducing the off-target toxicity in normal tissues and achieving tumor-specific therapy.
Conclusion
A supramolecular DNA nanocomplex of Ca@DNA–MF was successfully prepared for the remodulation of H+/Ca2+ gradients to induce mitochondrial depolarization. The remodulation not only triggered apoptosis but also led to ferroptosis through the enhancement of ROS, which obtained the synergistical enhancement of cancer therapy. Upon targeted and TME-triggered intracellular delivery, multiple reagents including Ca2+, the MCT4 gene silencing inhibitor of siMCT4 and Fenton-like reagents were released into cancer cells. Subsequently, MCT4 gene silencing induced acidosis to enhance the conversion of endogenous H2O2 into highly toxic ˙OH. Simultaneously, GSH was oxidized to GSSG to facilitate ferroptosis upon the irreversible GPX-4 inactivation and lipid peroxidation process. Therefore, based on the three channels of the therapy, multiple and synergistic mitochondrial depolarization was obtained via the remodulating of H+/Ca2+ gradients. This dramatically initiated the significant generation of excessive ROS species by regulating the H+/Ca2+ gradient influx in the matrix, TME-activated GSH depletion and abundant ˙OH generation. Therefore, the intrinsic redox homeostasis was disturbed for LPO-based apoptosis. By both cell imaging and in vivo experiments, synergistic cancer therapy was achieved with low toxicity, providing an efficient strategy for enhanced cancer therapy.Ethical statement
All animal experiment protocols were reviewed and approved by the Animal Care and Use Committee of Institute of Beijing Normal University and complied with all relevant ethical regulations.Data availability
All relevant data is presented in the manuscript and ESI.† Raw data is available upon request by email to the corresponding author.Author contributions
X. Wang and N. Na conceived and designed the project. X. Wang performed the experiments. X.Ge, X. Guan supported figure preparations and J. Ouyang supported the characterizations. N. Na directed the whole research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (NNSFC, 22274012 and 21974010).Notes and references
- M. Fan, J. Zhang, C. Tsai, B. Orlando, M. Rodriguez, Y. Xu, M. Liao, M. F. Tsai and L. Feng, Nature, 2020, 582, 129–133 CrossRef CAS PubMed.
- K. Lee, S. Park, K. Lee, S. Kim, H. Kim, Y. Meroz, L. Mahadevan, K. Jung, T. K. Ahn, K. K. Parker and K. Shin, Nat. Biotechnol., 2018, 36, 530–535 CrossRef CAS PubMed.
- W. Li, S. Yin, Y. Shen, H. Li, L. Yuan and X. Zhang, J. Am. Chem. Soc., 2023, 145, 3736–3747 CrossRef CAS PubMed.
- S. Cassim, M. Vučetić, M. Ždralević and J. Pouyssegur, Cancers, 2020, 12, M1119 CrossRef PubMed.
- L. Cui, A. M. Gouw, E. L. LaGory, S. Guo, A. N. ttarwala, Y. Tang, J. Qi, Y. Chen, Z. Gao, K. M. Casey, A. A. Bazhin, M. Chen, L. Hu, J. Xie, M. Fang, C. Zhang, Q. Zhu, Z. Wang, A. J. Giaccia, S. S. Gambhir, W. Zhu, D. W. Felsher, M. D. Pegram, E. A. Goun, A. Le and J. Rao, Nat. Biotechnol., 2021, 39, 357–367 CrossRef CAS PubMed.
- E. Gouaux and R. MacKinnon, Science, 2005, 310, 1461–1465 CrossRef CAS PubMed.
- S. Matsuyama, J. Llopis, Q. L. Deveraux, R. Y. Tsien and J. C. Reed, Nat. Cell Biol., 2000, 2, 318–325 CrossRef CAS PubMed.
- P. Zheng, B. Ding, G. Zhu, C. Li and J. Lin, Angew. Chem., Int. Ed., 2022, 61, e202204904 CrossRef CAS PubMed.
- P. Zheng, B. Ding, R. Shi, Z. Jiang, W. Xu, G. Li, J. Ding and X. Chen, Adv. Mater., 2021, 33, 2007426 CrossRef CAS PubMed.
- C. Giorgi, S. Marchi and P. Pinton, Nat. Rev. Mol. Cell Biol., 2018, 19, 713–730 CrossRef CAS PubMed.
- Y. Mari, C. Katnik and J. Cuevas, Cell Calcium, 2010, 48, 70–82 CrossRef CAS PubMed.
- C. Xue, M. Li, C. Liu, Y. Li, Y. Fei, Y. Hu, K. Cai, Y. Zhao and Z. Luo, Angew. Chem., Int. Ed., 2021, 60, 8938–8947 CrossRef CAS PubMed.
- J. Cheng, Y. Zhu, X. Xing, J. Xiao, H. Chen, H. Zhang, D. Wang, Y. Zhang, G. Zhang, Z. Wu and Y. Liu, Theranostics, 2021, 11, 5418–5429 CrossRef CAS PubMed.
- C. Zhang, L. Chen, Q. Bai, L. Wang, S. Li, N. Sui, D. Yang and Z. Zhu, ACS Appl. Mater. Interfaces, 2022, 14, 27720–27732 CrossRef CAS PubMed.
- C. Yang, M. Wang, M. Chang, M. Yuan, W. Zhang, J. Tan, B. Ding, P. Ma and J. Lin, J. Am. Chem. Soc., 2023, 145, 7205–7217 CrossRef CAS PubMed.
- S. Puri and K. Juvale, Eur. J. Med. Chem., 2020, 199, 112393 CrossRef CAS PubMed.
- L. Xu, G. Tong, Q. Song, C. Zhu, H. Zhang, J. Shi and Z. Zhang, ACS Nano, 2018, 12, 6806–6818 CrossRef CAS PubMed.
- P. Zheng, B. Ding, R. Shi, Z. Jiang, W. Xu, G. Li, J. Ding and X. Chen, Adv. Mater., 2021, 33, e2007426 CrossRef PubMed.
- X. Meng, D. Li, L. Chen, H. He, Q. Wang, C. Hong, J. He, X. Gao, Y. Yang and B. Jiang, ACS Nano, 2021, 15, 5735–5751 CrossRef CAS PubMed.
- D. Wang, H. Wu, C. Wang, L. Gu, H. Chen, D. Jana, L. Feng, J. Liu, X. Wang and P. Xu, Angew. Chem., Int. Ed., 2021, 133, 3038–3044 CrossRef.
- S. Guo, Z. Li, J. Feng, W. Xiong, J. Yang, X. Lu, S. Yang, Y. Xu, A. Wu and Z. Shen, Nano Today, 2022, 47, 101663 CrossRef CAS.
- W. Wang, F. Fu, Z. Huang, W. Wang, M. Chen, X. Yue, J. Fu, X. Feng, Y. Huang, C. Wu and X. Pan, ACS Nano, 2022, 16, 8370–8387 CrossRef CAS PubMed.
- Y. Chen, Y. Liu, C. Guo, C. Yin, C. Xie and Q. Fan, Adv. Funct. Mater., 2023, 33, 2209927 CrossRef CAS.
- G. Guan, C. Zhang, H. Liu, Y. Wang, Z. Dong, C. Lu, B. Nan, R. Yue, X. Yin, X. Zhang and G. Song, Angew. Chem., Int. Ed., 2022, 61, e202117229 CrossRef CAS PubMed.
- C. Zhang, W. Bu, D. Ni, S. Zhang, Q. Li, Z. Yao, J. Zhang, H. Yao, Z. Wang and J. Shi, Angew. Chem., Int. Ed., 2016, 55, 2101–2106 CrossRef CAS PubMed.
- B. Yu, B. Choi, W. Li and D. Kim, Nat. Commun., 2020, 11, 3637 CrossRef CAS PubMed.
- B. Yang, Q. Liu, X. Yao, D. Zhang, Z. Dai, P. Cui, G. Zhang, X. Zheng and D. Yu, ACS Appl. Mater. Interfaces, 2019, 11, 38395–38404 CrossRef CAS PubMed.
- C. Yao, H. D. Qi, X. Jia, Y. Xu, Z. Tong, Z. Gu and D. Yang, Angew. Chem., Int. Ed., 2022, 61, e202113619 CrossRef CAS PubMed.
- Y. Dong, C. Yao, Y. Zhu, L. Yang, D. Luo and D. Yang, Chem. Rev., 2020, 120, 9420–9481 CrossRef CAS PubMed.
- X. Guo, F. Li, C. Liu, Y. Zhu, N. Xiao, Z. Gu, D. Luo, J. Jiang and D. Yang, Angew. Chem., Int. Ed., 2020, 59, 20651–20658 CrossRef CAS PubMed.
- X. Guan, F. Meng, H. Tan, X. Wang, J. Li, J. Wei, J. Ouyang and N. Na, Chem. Sci., 2022, 13, 8657–8666 RSC.
- P. Winterwerber, S. Harvey, D. Y. W. Ng and T. Weil, Angew. Chem., Int. Ed., 2020, 59, 6144–6149 CrossRef CAS PubMed.
- C. Whitfield, M. Zhang, P. Winterwerber, Y. Wu, D. Y. W. Ng and T. Weil, Chem. Rev., 2021, 121, 11030–11084 CrossRef CAS PubMed.
- H. Pei, N. Lu, Y. Wen, S. Song, Y. Liu, H. Yan and C. Fan, Adv. Mater., 2010, 22, 4754–4758 CrossRef CAS PubMed.
- J. Wang, J. Chao, H. Liu, S. Su, L. Wang, W. Huang, I. Willner and C. Fan, Angew. Chem., Int. Ed., 2017, 56, 2171–2175 CrossRef CAS PubMed.
- X. Shen, W. Xua, J. Ouyang and N. Na, Chin. Chem. Lett., 2022, 33, 4505–4516 CrossRef CAS.
- G. Nabil, R. Alzhrani, H. O. Alsaab, M. Atef, S. Sau, A. K. Iyer and H. E. Banna, Cancers, 2021, 13, 898–925 CrossRef CAS PubMed.
- W. S. Sun, J. X. Yang, M. F. Hou, S. W. Xie, L. Q. Xiong, B. Li and C. Zhang, Adv. Funct. Mater., 2021, 31, 2009116–2009132 CrossRef CAS.
- R. Wang, H. Yang, A. R. Khan, X. Yang, J. Xu, J. Ji and G. Zhai, J. Colloid Interface Sci., 2021, 598, 213–228 CrossRef CAS PubMed.
- Z. Sun, T. Wang, J. Wang, J. Xu, T. Shen, T. Zhang, B. Zhang, S. Gao, C. Zhao, M. Yang, F. Sheng, J. Yu and Y. Hou, J. Am. Chem. Soc., 2023, 145, 11019–11032 CrossRef CAS PubMed.
- Y. Zhu, P. Gong, J. Wang, J. Cheng, W. Wang, H. Cai, R. Ao, H. Huang, M. Yu, L. Lin and X. Chen, Angew. Chem., Int. Ed., 2023, 62, e202218407 CrossRef CAS PubMed.
- J. Wang, Z. Sun, S. Wang, C. Zhao, J. Xu, S. Gao, M. Yang, F. Sheng, S. Gao and Y. Hou, J. Am. Chem. Soc., 2022, 144, 19884–19895 CrossRef CAS PubMed.
- C. Cao, X. Wang, N. Yang, X. Song and X. Dong, Chem. Sci., 2022, 13, 863–889 RSC.
- Q. Sun, S. Zhang, X. Wei, T. Yang, J. Wang and M. Chen, Anal. Chim. Acta, 2022, 1221, 340100 CrossRef CAS PubMed.
- X. Wang, X. Shen, J. Li, X. Ge, J. Ouyang and N. Na, Anal. Chem., 2022, 94, 16803–16812 CrossRef CAS PubMed.
- F. Shen, S. Dai, N. Wong, S. Deng, A. Wong and D. Yang, J. Am. Chem. Soc., 2020, 142, 10769–10779 CrossRef CAS PubMed.
- P. He, F. Liu, M. Li, M. Ren, X. Wang, Y. Deng, X. Wu, Y. Li, S. Yang and J. Song, Adv. Healthcare Mater., 2023, 12, 2203106 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section; general information and additional data for material characterization, including the data of fluorescence spectra and TEM. Detailed DNA sequence, 4T1 cell imaging, colocalization experiments, flow cytometric analyses, ex vivo imaging of organs and tumor tissues, and H&E staining for major tissue sections. See DOI: https://doi.org/10.1039/d3sc03493c |
| This journal is © The Royal Society of Chemistry 2023 |