
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn emissive charge-transfer excited-state at the well-defined hetero-nanostructure interface of an organic conjugated molecule and two-dimensional inorganic nanosheet†
Tomokazu
Umeyama
 *a,
Daizu
Mizutani
b,
Yuki
Ikeda
b,
W. Ryan
Osterloh
b,
Futa
Yamamoto
a,
Kosaku
Kato
c,
Akira
Yamakata
*c,
Masahiro
Higashi
*b,
Takumi
Urakami
b,
Hirofumi
Sato
b and
Hiroshi
Imahori
*bde
*a,
Daizu
Mizutani
b,
Yuki
Ikeda
b,
W. Ryan
Osterloh
b,
Futa
Yamamoto
a,
Kosaku
Kato
c,
Akira
Yamakata
*c,
Masahiro
Higashi
*b,
Takumi
Urakami
b,
Hirofumi
Sato
b and
Hiroshi
Imahori
*bde
aDepartment of Applied Chemistry, Graduate School of Engineering, University of Hyogo, Himeji, Hyogo 671-2280, Japan. E-mail: umeyama@eng.u-hyogo.ac.jp
bDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Kyoto 615-8510, Japan. E-mail: higashi@moleng.kyoto-u.ac.jp; imahori@scl.kyoto-u.ac.jp
cGraduate School of Natural Science and Technology, Okayama University, Okayama 700-8530, Japan. E-mail: yamakata@okayama-u.ac.jp
dInstitute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University, Kyoto 606-8501, Japan
eInstitute for Liberal Arts and Sciences (ILAS), Kyoto University, Kyoto 606-8501, Japan
First published on 17th October 2023
Abstract
Precise engineering of excited-state interactions between an organic conjugated molecule and a two-dimensional semiconducting inorganic nanosheet, specifically the manipulation of charge-transfer excited (CTE) states, still remains a challenge for state-of-the-art photochemistry. Herein, we report a long-lived, highly emissive CTE state at structurally well-defined hetero-nanostructure interfaces of photoactive pyrene and two-dimensional MoS2 nanosheets via an N-benzylsuccinimide bridge (Py-Bn-MoS2). Spectroscopic measurements reveal that no charge-transfer state is formed in the ground state, but the locally-excited (LE) state of pyrene in Py-Bn-MoS2 efficiently generates an unusual emissive CTE state. Theoretical studies elucidate the interaction of MoS2 vacant orbitals with the pyrene LE state to form a CTE state that shows a distinct solvent dependence of the emission energy. This is the first example of organic–inorganic 2D hetero-nanostructures displaying mixed luminescence properties by an accurate design of the bridge structure, and therefore represents an important step in their applications for energy conversion and optoelectronic devices and sensors.
Introduction
Manipulations of the excited-state interaction between the electron donor (D) and acceptor (A) are of crucial importance for high-performance energy conversion and optoelectronic devices and sensors based on organic materials.1 For instance, for light–electricity energy conversion in organic photovoltaic (OPV) devices, locally excited (LE), charge-transfer excited (CTE), and charge-separated (CS) states are known to be sequentially generated at the D–A interface.2 It is pointed out that the precise control of the elusive intermediate CTE state is of utmost importance to attain high power conversion efficiency (PCE).3 For electricity–light energy conversion in organic light-emitting diodes (OLEDs), conversion of the CS state to the LE state, which is an opposite process to that in OPVs, lies at the basis of device performance.4 Hence, sophisticated OLEDs with high efficiencies also require judicious engineering of the CTE state. In this context, intensive investigations on the CTE states formed at the interface between the organic D and A in intramolecular D–A linked systems and intermolecular D/A systems have been conducted.1,5,6 Not only the HOMO–LUMO energy levels but also the D–A distance, orientation, environment, and dynamic motion have been proved to exert a profound impact on the CTE state behaviors, i.e., quantum yields of the CTE state formation from the LE state, relaxation by radiative or non-radiative decay from the CTE state to the ground state, and the CS state formation from the CTE state.During the last couple of decades, hybrid D–A interfaces composed of organic π-conjugated compounds and inorganic nanostructures have also received considerable attention due to their potential applications in energy conversion devices.7 However, in contrast to the extensive studies on organic D–A interfaces, it is still a challenge to address CTE behaviors formed at hybrid organic–inorganic nanostructure D–A interfaces. Specifically, in spite of the excellent electronic and optical properties of two-dimensional (2D) transition metal dichalcogenide (TMD) nanosheets, the low chemical reactivity at the surface has hampered the preparation of structurally well-defined D–A linked systems based on organic molecules and TMD nanosheets.8 In this study, we designed a hetero-nanostructure interface between a photoactive organic π-conjugated molecule (pyrene, Py) and a semiconducting 2D inorganic nanosheet (mono- or few-layered MoS2) with a well-defined bridge at a molecular level. As the bridge linking Py to the MoS2 basal plane, we employed N-benzylsuccinimide (H-Bn-suc) to form Py-Bn-MoS2 (Scheme 1). Recently it has been shown that the H-Bn-suc groups can be introduced onto MoS2 basal planes at high densities by the reaction with N-benzylmaleimide (H-Bn-mal).9 The H-Bn-suc bridge has a relatively short, rigid structure that can define the separation distance between Py and MoS2 well (ca. 1 nm). The sole flexible part is a methylene, which enables the angle between Py and the MoS2 plane to be modulated, resulting in a stable conformation with an enhanced interaction between Py and MoS2. Therefore, one expects that the LE state of Py can rapidly interact with MoS2 to form CTE and/or CS states in Py-Bn-MoS2. Indeed, due to such an elaborate bridge design, the photoexcitation of Py on the MoS2 basal plane affords the CTE state efficiently, exhibiting long-lived, solvent polarity-dependent, strong steady-state emission. To the best of our knowledge, this is the first observation of unusually intense emission from the CTE state at the well-defined hetero-nanostructure interface of organic π-conjugated molecules and 2D inorganic nanosheets.
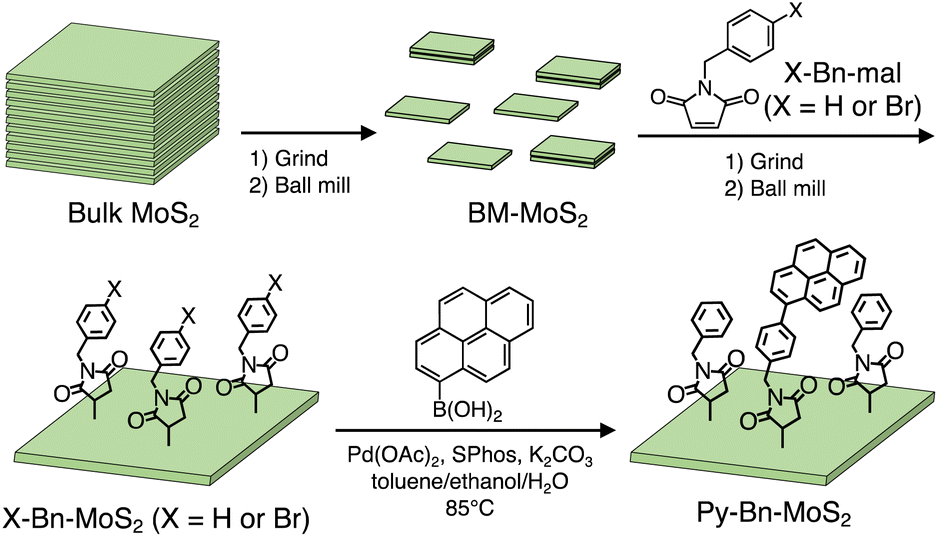 | ||
| Scheme 1 Synthetic scheme for Py-Bn-MoS2. | ||
Results and discussion
Exfoliation and covalent functionalization of MoS2 utilizing the ball-mill method
Exfoliation of bulk MoS2 was conducted with a planetary ball-mill system after grinding in an agate mortar (Scheme 1, see the ESI† for experimental details).10 Note here that ball-milling is a simple dry-phase process with an easy scale-up capability in contrast to the sonochemical method,11 and thereby recently has been utilized to exfoliate various 2D materials. The MoS2 nanosheets exfoliated by the ball-mill method are denoted as BM-MoS2. Atomic force microscopy (AFM) and field-emission scanning electron microscopy (FE-SEM) measurements imaged the nanosheet structures of BM-MoS2 (Fig. S1†). Statistical analysis of the AFM images showed that the average thickness and lateral size are 3.8 ± 0.9 nm and 197 ± 25 nm, respectively, supporting the exfoliated structures.Then, the planetary ball-mill method was also employed to perform the covalent functionalization of BM-MoS2. Although solid–state reactions using ball-mill systems have been applied to fullerenes and 2D materials,12 this technique is yet to be utilized for the covalent functionalization of MoS2 with organic molecules. As such, to evaluate the utility of the solid–state reaction, we first prepared H-Bn-suc-functionalized MoS2 nanosheets (denoted as H-Bn-MoS2) by the solid–state reaction of H-Bn-mal and BM-MoS2 (Scheme 1). Note here that H-Bn-MoS2 was previously synthesized by a solution-phase reaction.9 The brief procedure of the solid–state reaction and purification is as follows. A mixed powder of BM-MoS2 and H-Bn-mal with a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5 was treated with a planetary ball-mill system for 24 h under an argon atmosphere. Then, the resultant powder was dispersed in acetonitrile, and the dispersion was sonicated and filtered. The product was further washed with acetonitrile by repeated sonication and filtration to thoroughly remove unreacted H-Bn-mal (see the ESI† for further experimental details).
:2.5 was treated with a planetary ball-mill system for 24 h under an argon atmosphere. Then, the resultant powder was dispersed in acetonitrile, and the dispersion was sonicated and filtered. The product was further washed with acetonitrile by repeated sonication and filtration to thoroughly remove unreacted H-Bn-mal (see the ESI† for further experimental details).
The structure of H-Bn-MoS2 prepared by the solid–state reaction was characterized by Fourier transform infrared (FT-IR) and resonant Raman spectra, X-ray photoelectron spectroscopy (XPS), AFM, FE-SEM, and thermogravimetric analysis (TGA) as is the case for H-Bn-MoS2 synthesized by the solution reaction.9 In the FT-IR spectrum of H-Bn-MoS2, signals for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch and the S–C stretch appear, whereas that of alkene C–H bending mode is depleted (Fig. 1a and b). This confirms that CC of the maleimide unit was saturated to form C–S bonds on the MoS2 nanosheet of H-Bn-MoS2. In addition, the C–H and Ar–H vibration signals were observed in the FT-IR spectrum of H-Bn-MoS2, supporting the presence of –CH2– and phenylene groups (Fig. S2a†).
O stretch and the S–C stretch appear, whereas that of alkene C–H bending mode is depleted (Fig. 1a and b). This confirms that CC of the maleimide unit was saturated to form C–S bonds on the MoS2 nanosheet of H-Bn-MoS2. In addition, the C–H and Ar–H vibration signals were observed in the FT-IR spectrum of H-Bn-MoS2, supporting the presence of –CH2– and phenylene groups (Fig. S2a†).
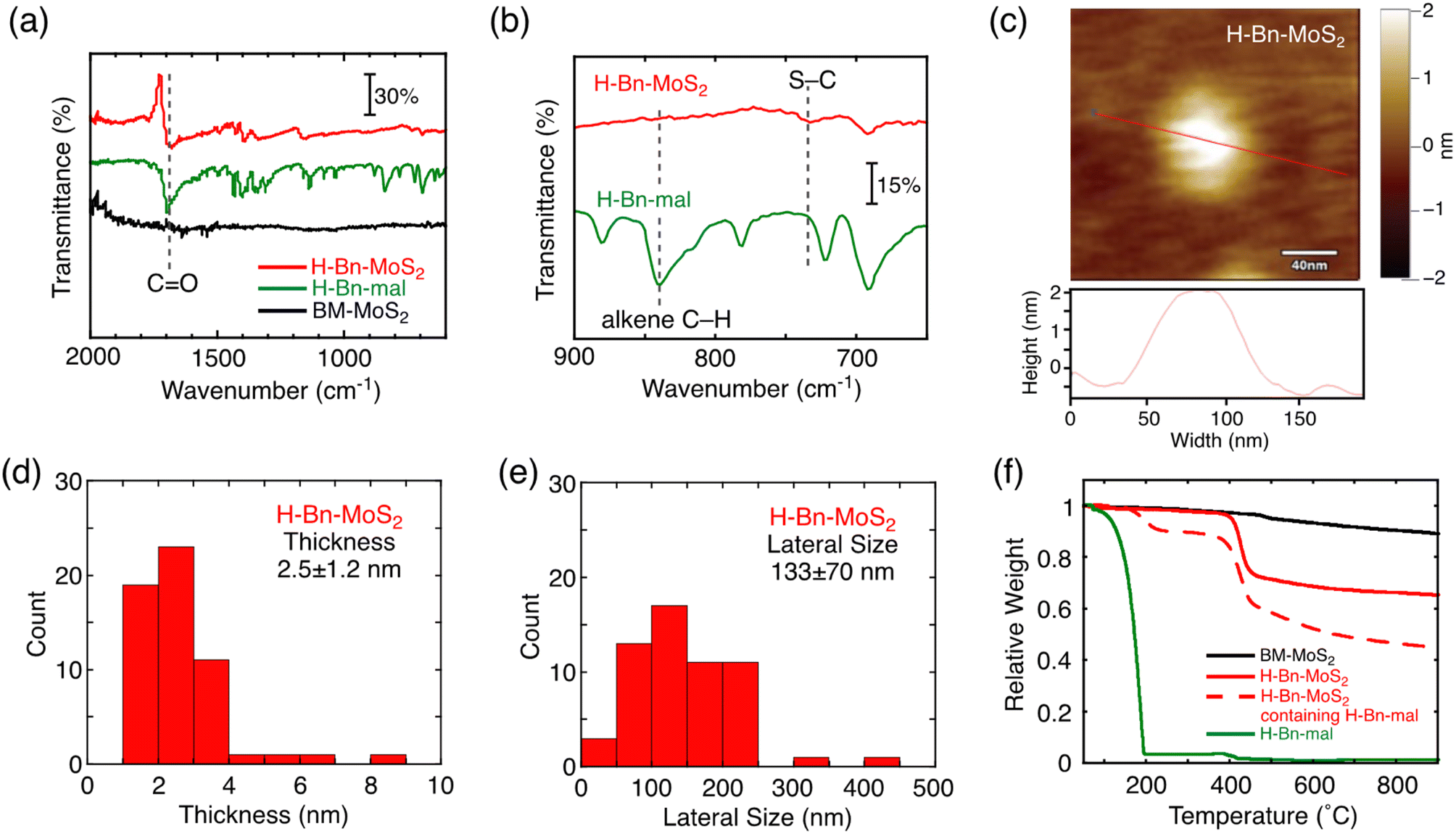 | ||
| Fig. 1 (a and b) FT-IR spectra of H-Bn-MoS2 (red), H-Bn-mal (green), and BM-MoS2 (black) in the regions of (a) 2000–600 cm−1 and (b) 900–650 cm−1. (c) AFM image with the section profile of H-Bn-MoS2 spin coated on mica from the NMP dispersion. The color scale represents the height topography, with light and dark representing the highest and lowest features, respectively. (d) Thickness and (e) lateral size distributions of H-Bn-MoS2 estimated by AFM measurements. 50 individual sheets were analyzed. (f) TGA of BM-MoS2 (black solid), H-Bn-MoS2 after thorough washing (red solid), H-Bn-MoS2 containing physisorbed H-Bn-mal (red dashed), and H-Bn-mal (green solid). The analyses were performed under nitrogen with a heating rate of 10 °C min−1. | ||
Resonant Raman spectra of H-Bn-MoS2 and BM-MoS2 excited at 532 nm exhibit characteristic bands of MoS2, including in-plane (E12g), out-of-plane (A1g), and S-vacancy-induced (2LA(M)) vibrations at around 380, 400, and 450 cm−1, respectively (Fig. S2b and c†).13 The absence of the J peak series (around 155, 225, and 330 cm−1) characteristic to metallic 1T MoS2 indicates the preservation of the semiconducting 2H nature of MoS2 after covalent functionalization.14 When the Raman spectrum of H-Bn-MoS2 with an excitation wavelength of 633 nm is compared with that of BM-MoS2, an increase in the relative intensity of the 2LA(M) mode is observed following functionalization (Fig. S2d–f†). The averaged intensity ratios IA1g/I2LA(M) of H-Bn-MoS2 and BM-MoS2 from the measurements of 255 points are 2.49 and 4.91, respectively. This result implies that the solid–state reaction using the ball-mill system increased the S-vacancy defects in the MoS2 nanosheet to some extent.13
Covalent functionalization by the solid–state reaction is also evidenced by comparing XPS patterns of H-Bn-MoS2 and BM-MoS2 (Fig. S3†). In the C region of H-Bn-MoS2, components for CO (288.4 eV), C–S (286.6 eV), and C–N (286.0 eV) are needed for appropriate fitting. In addition, the broad signal in the S 2p region of H-Bn-MoS2 also requires additional components that correspond to the S–C bond at 164.6 and 164.2 eV. While the XPS patterns of H-Bn-MoS2 and BM-MoS2 in the Mo region are similar, a characteristic peak for N 1s appears at 400.6 eV in the XPS pattern of H-Bn-MoS2, which can be unambiguously assigned to the succinimide fragment.
AFM (Fig. 1c) and FE-SEM (Fig. S4a†) measurements support the nanosheet structures of H-Bn-MoS2. Despite the introduction of H-Bn-suc units onto the MoS2 nanosheet surface, the average thickness (2.5 ± 1.2 nm) and lateral size (133 ± 70 nm) of H-Bn-MoS2 (Fig. 1d and e) became smaller than those of BM-MoS2 (3.8 ± 0.9 nm and 197 ± 25 nm, respectively, Fig. S1†), implying further exfoliation and fragmentation of the MoS2 nanosheets during the solid–state reaction. Considering the heights of single-layered MoS2 nanosheets (0.99 nm by AFM)15 and H-Bn-suc (0.89 nm, Fig. S4b†), H-Bn-MoS2 mainly contains mono-layered MoS2 nanosheets and tilted H-Bn-suc moieties on the MoS2 nanosheets.
TGA measurements were conducted to estimate the functionalization ratio (Fig. 1f). TGA of H-Bn-MoS2 after thorough washing revealed a sharp weight decrease at 400–450 °C (red solid line). Considering that TGA of BM-MoS2 showed no clear weight-loss steps (black solid line), this sharp weight loss is attributable to the thermal decomposition of organic components. Meanwhile, the H-Bn-MoS2 sample contains not only covalently-linked H-Bn-suc groups, but also physisorbed H-Bn-mal molecules on MoS2 nanosheets after washing only once, and exhibited sharp weight decreases at 170–220 °C as well as 400–450 °C (red dashed line). Furthermore, the pyrolysis of neat H-Bn-mal was completed below 200 °C (green solid line), which is similar to the former weight loss temperature of H-Bn-MoS2 containing physisorbed H-Bn-mal molecules (red dashed line). These results indicate that decomposition temperatures of physisorbed H-Bn-mal and covalently linked H-Bn-suc on MoS2 nanosheets are distinguishable, and the thermal detachment of the covalently-linked H-Bn-suc groups from MoS2 nanosheets occurs at 400–450 °C. From the difference in the relative weights of BM-MoS2 (black solid line) and H-Bn-MoS2 (red solid line) at 500 °C, the weight ratio of H-Bn-suc moieties in H-Bn-MoS2 is estimated to be 28 wt%. This corresponds to 1 H-Bn-suc unit every 6 S atoms. It is noteworthy that the feed ratio of H-Bn-mal to BM-MoS2 for the solid–state reaction (H-Bn-mal:BM-MoS2 = 2.5:1 in weight) is much lower than that for the reported solution reaction (typically, H-Bn-mal:exfoliated MoS2 = 47:1 in weight).9 Nevertheless, the solid–state reaction can successfully functionalize the MoS2 basal plane covalently with H-Bn-suc units at high densities as in the case of the solution reaction, demonstrating the enhanced reactivity in the solid–state reaction of MoS2 with N-benzylsuccinimide.
Synthesis of Py-Bn-MoS2
The covalent functionalization of BM-MoS2 by using a planetary ball-mill system was attempted using 4-(1-pyrenyl)-N-benzylmaleimide (Py-Bn-mal) instead of H-Bn-mal to directly obtain the pyrene-linked MoS2 nanosheet (Fig. S5a†). However, the functionalization reaction did not occur, which was evidenced by the absence of weight loss due to the thermal decomposition of the organic moieties in TGA (Fig. S5b†). The bulky Py group may decrease the contacts between the MoS2 nanosheet and the reactive maleimide moiety. In addition, the large Py group may also inhibit intercalation into the MoS2 nanosheets.Alternatively, a two-step reaction strategy was employed (Scheme 1). The first step is the modification of the MoS2 nanosheet by the solid–state reaction with less sterically-hindered N-p-bromobenzylmaleimide (Br-Bn-mal) to produce the N-p-bromobenzylsuccinimide (Br-Bn-suc)-functionalized MoS2 nanosheet (denoted as Br-Bn-MoS2). The synthesis and purification procedures of Br-Bn-MoS2 are the same as those of H-Bn-MoS2. The structure characterization of Br-Bn-MoS2 is described in the ESI (Fig. S6–S10†). TGA revealed the contents of organic moieties in Br-Bn-MoS2 to be 40 wt% (Fig. 2a), indicating that the functionalization ratio is 1 Br-Bn-suc unit in every 5 S atoms.
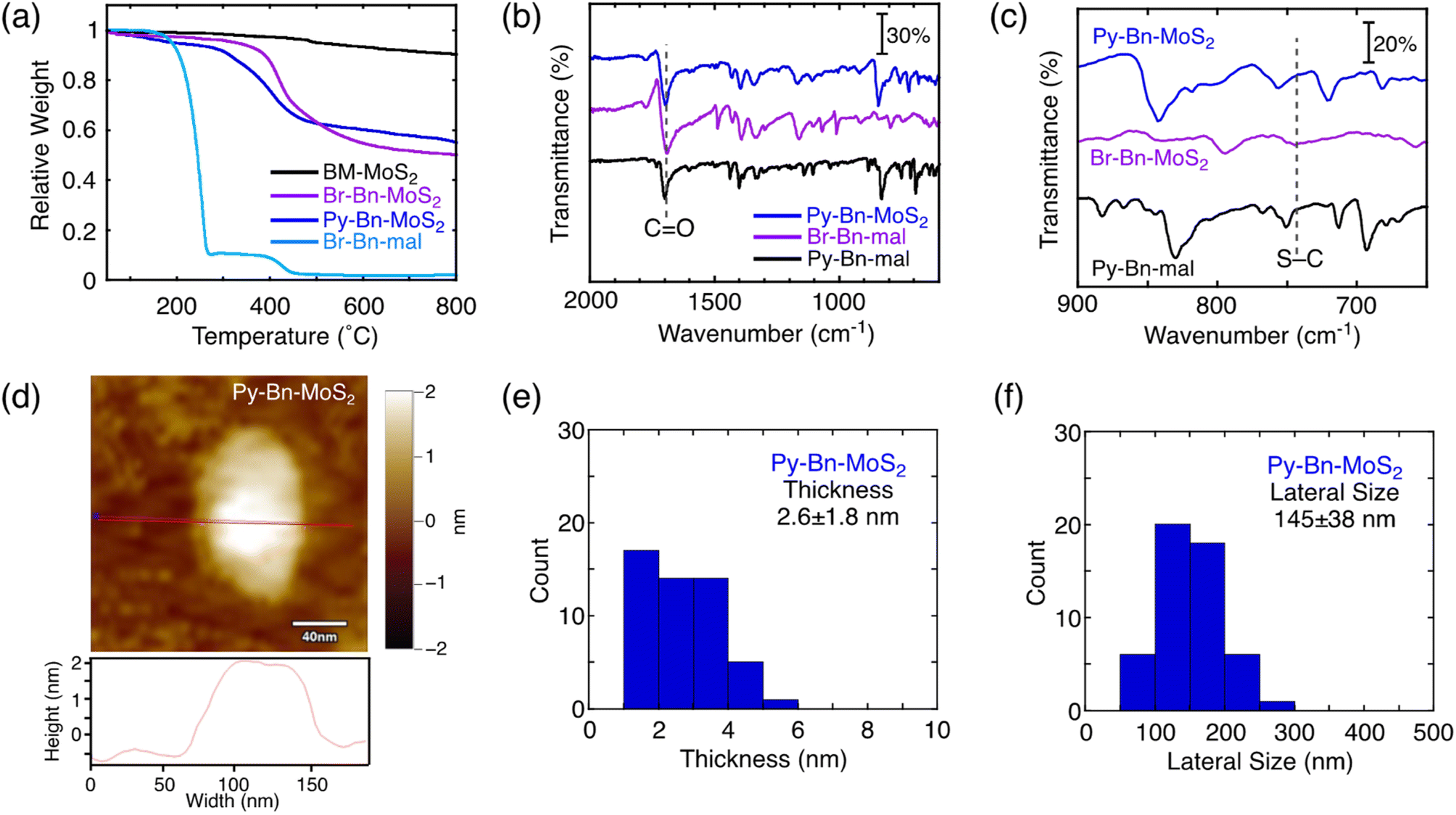 | ||
| Fig. 2 (a) TGA of BM-MoS2 (black), Br-Bn-MoS2 (purple), Py-Bn-MoS2 (blue), and Br-Bn-mal (light blue). The analyses were performed under nitrogen with a heating rate of 10 °C min−1. (b and c) FT-IR spectra of Py-Bn-MoS2 (blue), Br-Bn-mal (purple), and Py-Bn-mal (black) in the regions of (b) 2000–600 cm−1 and (c) 900–650 cm−1. (d) AFM image with the section profile of Py-Bn-MoS2 spin coated on mica from the NMP dispersion. The color scale represents the height topography, with light and dark representing the highest and lowest features, respectively. (e) Thickness and (f) lateral size distributions of Py-Bn-MoS2 estimated by AFM measurements. 50 individual sheets were analyzed. | ||
As the second step, a Suzuki coupling reaction between Br-Bn-MoS2 and pyrene boronic acid was conducted to obtain the N-p-pyrenylbenzylsuccinimide (Py-Bn-suc)-linked MoS2 nanosheet (denoted as Py-Bn-MoS2, Scheme 1). In the FT-IR spectrum of Py-Bn-MoS2, the signal for the CO stretch remains, suggesting the preservation of the succinimide structure after the Suzuki coupling reaction (Fig. 2b). Meanwhile, the S–C stretch signal is buried in the signals from the pyrene moiety (Fig. 2c). Resonant Raman spectra of Py-Bn-MoS2 excited at 532 nm showed E12g and A1g vibration bands, but the 2LA(M) signal was obscure (Fig. S11b†), likely due to the highly tilted Py groups that covered the MoS2 nanosheet and thereby decreased the signal-to-noise (S/N) ratio of the Raman signal from the MoS2 nanosheet. The low S/N ratio also makes it difficult to determine whether the metallic J series peaks existed or not. Meanwhile, the resonant Raman spectrum of Py-Bn-MoS2 at an excitation wavelength of 633 nm clearly exhibited the 2LA(M) signal (Fig. S11c and d†) and revealed a lower IA1g/I2LA(M) value (1.67) than those of BM-MoS2 (4.91) and H-Bn-MoS2 (2.49), indicating a further increase in the S-vacancy defects during the Suzuki coupling reaction to some extent.
TGA of Py-Bn-MoS2 exhibited a weight decrease at 250–450 °C (Fig. 2a), which corresponds to organic moieties covalently linked to the MoS2 nanosheet. From the difference in the relative weights of BM-MoS2 and Py-Bn-MoS2 at 600 °C, the weight ratio of the covalently linked organic moieties in Py-Bn-MoS2 is estimated to be 37 wt%, which is lower than that of Br-Bn-MoS2 (40 wt%). In addition, in the XPS spectrum of the Br core level for Py-Bn-MoS2, no signals were detected (Fig. S7e†). These results suggest that the Br atoms in Br-Bn-suc units were not only replaced by Py, but also consumed by the incomplete Suzuki coupling reaction during the second functionalization step. Considering that 1 Br-Bn-suc unit exists in every 5 S atoms in Br-Bn-MoS2, ca. 30% of Br-Bn-suc units in Br-Bn-MoS2 successfully reacted with 1-pyrene boronic acid. The other Br-Bn-suc units did not react with pyrene boronic acid and lost the bromo group. Therefore, it is estimated that Py-Bn-MoS2 has 1 Py-Bn-suc unit in every 17 S atoms, while one H-Bn-suc unit exists in every 7 S atoms.
The retainment of the nanosheet structures after the Suzuki coupling reaction was further confirmed by AFM and FE-SEM measurements (Fig. 2d and S12†). The average thickness and lateral size of Py-Bn-MoS2 are 2.6 ± 1.8 nm and 145 ± 38 nm, respectively (Fig. 2e and f). This result suggests that Py-Bn-MoS2 mainly contains mono-layered MoS2 nanosheets15 and tilted Py-Bn-suc moieties on the MoS2 nanosheets. In addition, the second functionalization reaction, i.e., stirring at 85 °C for several days, did not lead to fragmentation of the MoS2 nanosheets.
Steady-state absorption and photoluminescence spectra and emission lifetimes
The UV-vis absorption spectra of Py-Bn-MoS2, Br-Bn-MoS2, and the pyrene reference compound (1-phenylpyrene, Py-Ph) in DMF are displayed in Fig. 3a. Br-Bn-MoS2 exhibits A and B excitonic transitions at around 670 and 610 nm, respectively, accompanied with transitions from the valence band to the conduction one at around 390 and 440 nm. The spectrum of Py-Bn-MoS2 contains features reflecting both those of Br-Bn-MoS2 and Py-Ph spectra; that is, a pyrene π–π* band at 347 nm and broad absorption bands arising from the MoS2 moiety. The same π–π* peak positions of Py-Bn-MoS2 and Py-Ph indicate the negligible electronic interactions between the pyrene and MoS2 moieties of Py-Bn-MoS2, that is, no formation of the CT state in the ground state.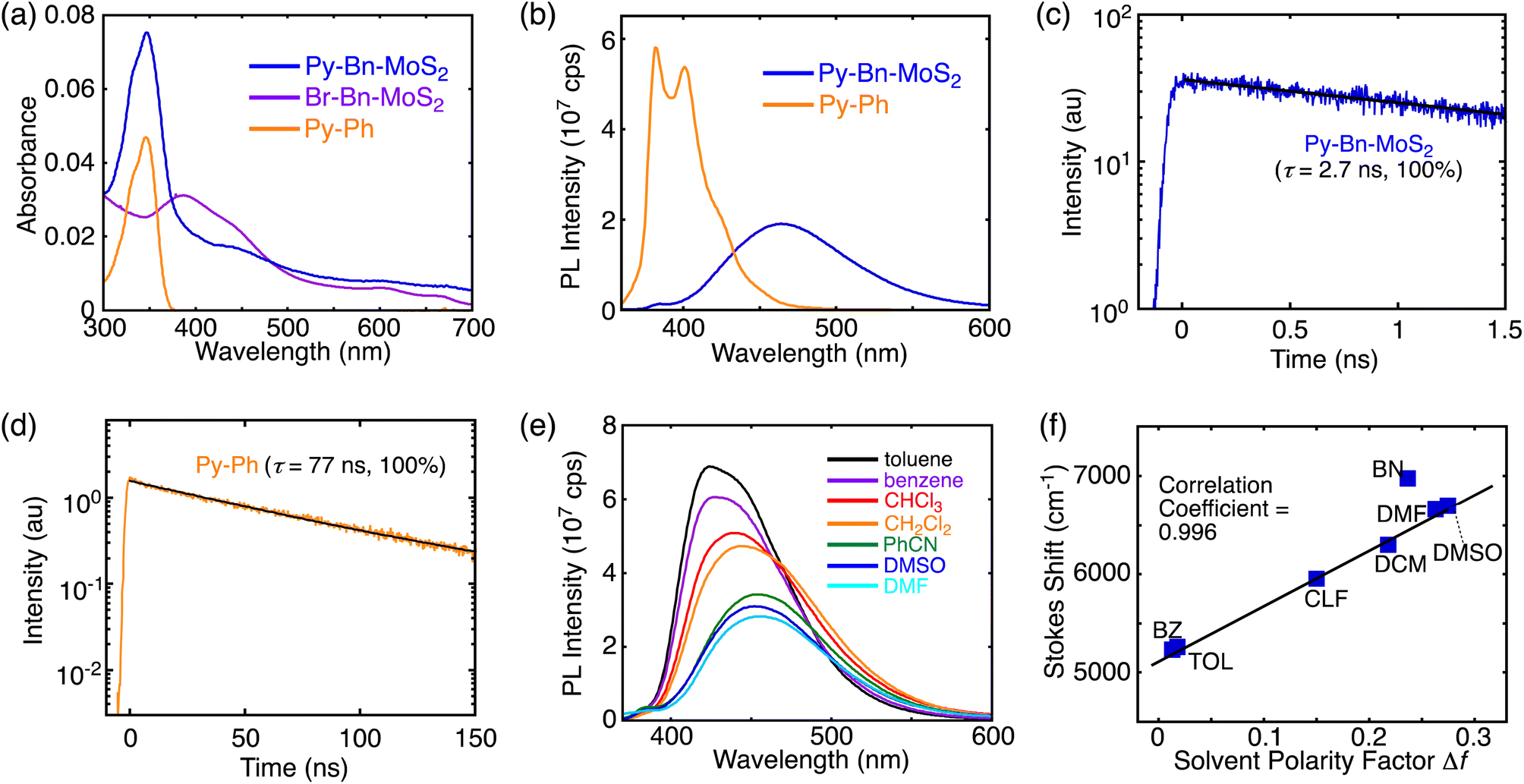 | ||
| Fig. 3 (a) UV-visible absorption spectra of Py-Bn-MoS2 (blue), Br-Bn-MoS2 (purple), and Py-Ph (orange) in DMF. (b) Photoluminescence spectra of Py-Bn-MoS2 (blue) and Py-Ph (orange) in DMF excited at 345 nm. At the excitation wavelength, the absorption intensities of pyrene moieties in Py-Bn-MoS2 and Py-Ph were normalized. Photoluminescence decays of (c) Py-Bn-MoS2 and (d) Py-Ph with fitting lines (black line) in DMF. The excitation wavelength was 345 nm. The emission was detected at 460 nm for Py-Bn-MoS2 and 400 nm for Py-Ph. The emission lifetimes (τ) and ratios are also shown in the figures. (e) Photoluminescence spectra of Py-Bn-MoS2 in various solvents excited at 345 nm. At the excitation wavelength, the absorption intensities were 0.05 in all solvents. (f) Lippert–Mataga plot for Py-Bn-MoS2 in benzene (BZ), toluene (TOL), chloroform (CLF), dichloromethane (DCM), benzonitrile (BN), DMSO, and DMF. The fitting line was made excluding the BN plot. | ||
The photoluminescence spectra of covalently-functionalized MoS2 samples and Py-Ph were also measured in DMF. The Py-Ph solution excited at 345 nm showed a strong emission with peaks at 382 and 402 nm (Fig. 3b), of which the photoluminescence quantum yield (PLQY) is 0.56. When the DMF dispersions of H-Bn-MoS2 and Br-Bn-MoS2 were irradiated at 600 nm at the B excitonic band, both of them showed no emission in the region of up to 850 nm, despite H-Bn-MoS2 and Br-Bn-MoS2 mainly containing mono-layered MoS2 nanosheets (see Fig. 1d and S8b† for estimated thickness).16 Structure defects in H-Bn-MoS2 and Br-Bn-MoS2 induced by the ball-mill treatments may significantly decrease the PLQYs of the MoS2 nanosheet. Then, to evaluate the electronic interactions in the excited state of Py with a MoS2 nanosheet, the photoluminescence spectrum of Py-Bn-MoS2 was measured with an excitation of the pyrene moiety (excitation wavelength: 345 nm, Fig. 3b). The pyrene emission is notably quenched and a broad and red-shifted intense emission emerges at 453 nm. Note here that no emission was observed in the 360–600 nm region by the excitation of Br-Bn-MoS2 at 345 nm. The emission decay profiles reveal that the photoluminescence lifetime of Py-Bn-MoS2 is as short as 2.7 ns,17 while Py-Ph exhibits a typical monoexponential decay of 77 ns,18 which is associated with the S1 state (Fig. 3c and d). Furthermore, the emission peak position of Py-Bn-MoS2 significantly differs from that of the excimer emission of Py-Ph in DMF (480 nm, Fig. S13†), ruling out the formation of pyrene excimers on the MoS2 nanosheet of Py-Bn-MoS2. These results indicate the generation of a new emissive species by the interaction between the MoS2 nanosheet and pyrene in the excited state.
A solvatochromism study was performed for Py-Bn-MoS2 in different solvents to evaluate the effect of solvent polarity on its optical properties. Py-Bn-MoS2 shows negligible changes in absorption profiles with peaks of the pyrene π–π* transition present at 346–349 nm on varying the polarity of the solvents (Table S1†). However, Py-Bn-MoS2 exhibits prominent positive solvatochromism in the photoluminescence spectra (Fig. 3e and Table S1†). This corroborates that Py-Bn-MoS2 is more polarized in the excited state than in the ground state. It can be assumed that the pyrene unit and the MoS2 nanosheet act as an electron donor and acceptor, respectively,11,19 and the CTE state is formed. The CTE state will be more stabilized by polar solvents, leading to a red-shift in emission. The solvatochromism data were analyzed by using the Lippert–Mataga plot (Fig. 3f),20 which illustrates a good linear correlation with a slope of +5560 cm−1. This result verifies the charge transfer character of Py-Bn-MoS2 in the excited state. In addition, consistent with photoluminescence intensities in various solvents (Fig. 3e), the PLQY of Py-Bn-MoS2 increases notably with decreasing solvent polarity (0.26, 0.42, and 0.68 in DMF, CHCl3, and toluene, respectively). It should be emphasized that, despite the larger Stokes shift of Py-Bn-MoS2 (5260 cm−1) than that of Py-Ph (2500 cm−1) in toluene, PLQY of Py-Bn-MoS2 (0.68) is significantly higher than that of Py-Ph (0.56) in toluene. Compared to the typical intermolecular CTE state (also regarded as an exciplex) between pyrene derivatives and N,N-dimethylaniline with PLQYs of 0.18–0.23 in even low polarity media,21 the PLQY of Py-Bn-MoS2 CTE state is unusually high.
Energy levels
From cyclic voltammetry (CV) and differential pulse voltammetry (DPV) measurements in DMF, the energy level of the bottom of the conduction band (CB) of Br-Bn-MoS2 was estimated to be −3.56 eV (Fig. S14a†). Here, the energy level of ferrocene/ferrocenium (Fc/Fc+) is assumed to be −4.80 eV below the vacuum level.22 The CB of the functionalized MoS2 nanosheet was found to be lower than the LUMO energy level of Py-Ph (−2.39 eV, Fig. S14b†). Because the electrochemical assessments did not exhibit any clear signals for the oxidation of Br-Bn-MoS2, the measurement of photoemission yield spectroscopy in air (PYSA) was conducted to determine the energy level of the valence band (VB) of Br-Bn-MoS2 (Fig. S14c†). The VB energy level of the functionalized MoS2 nanosheet was −5.70 eV, which is also lower than the HOMO energy level of Py-Ph (−5.60 eV, Fig. S13b†). These results support the formation of the CTE state of ((Py)δ+-Bn-(MoS2)δ−)*, that is, Py acting as an electron donor and MoS2 as an acceptor.Regarding Py-Bn-MoS2, its low dispersibility in DMF containing electrolyte (Bu4NPF6) led to aggregation during the electrochemical measurements, which prohibited evaluation of the redox potentials. Meanwhile, the PYSA suggested that the VB energy level of Py-Bn-MoS2 is −5.62 eV (Fig. S14d†). However, this value resulted from two components, the MoS2 nanosheet moiety (VB: −5.70 eV) and Py-Ph units (HOMO: −5.60 eV) on the MoS2 surface.
The optical gap of Py-Ph was determined to be 3.33 eV from the intersection of normalized absorption and fluorescence spectra (Fig. S15a†). In addition, the optical gap of Br-Bn-MoS2 was estimated to be 1.80 eV from the absorption edge of the A excitonic transition (Fig. S15b†). Although the electrochemical and optical gaps of Py-Ph (3.21 and 3.33 eV, respectively) are similar, the optical bandgap of the functionalized MoS2 nanosheet (1.80 eV) is significantly smaller than the bandgap estimated by using PYSA and electrochemistry (2.14 eV). This suggests that the interaction between electrons and holes may significantly stabilize the exciton in the functionalized MoS2 nanosheet.23
Transient absorption studies
To further elucidate the interaction in the excited states, the pump-probe transient absorption (TA) measurements were carried out (Fig. 4). The TA spectra of Br-Bn-MoS2 (Fig. 4b) resemble that of the non-functionalized MoS2 nanosheet,8b demonstrating the preservation of the 2H character of MoS2 nanosheets after the solid–state reaction. Immediately after the photoexcitation of Br-Bn-MoS2 at 345 nm, two minima at 663 and 609 nm and two maxima at 493 and 704 nm are observed. The photobleaching coincides well with the A and B excitonic transitions in the steady-state absorption spectrum (Fig. 3a). All these signals are gradually blue-shifted due to the cooling of hot excitons.8b Meanwhile, the TA spectra of Py-Ph (Fig. 4c) show an instantaneously formed transient species that has broad absorption bands in visible and NIR regions with two maxima at around 600 and 650 nm. This state is metastable and formation of another species follows, which is characterized by a positive signal at 490 nm and a long lifetime (>2 ns, beyond the time limit of the TA measurement system). These two species can be assigned to the second and first singlet excited states, respectively.24 The long decay lifetime of the second species (>2 ns) is consistent with the fluorescence lifetime of Py-Ph (77 ns, Fig. 3d).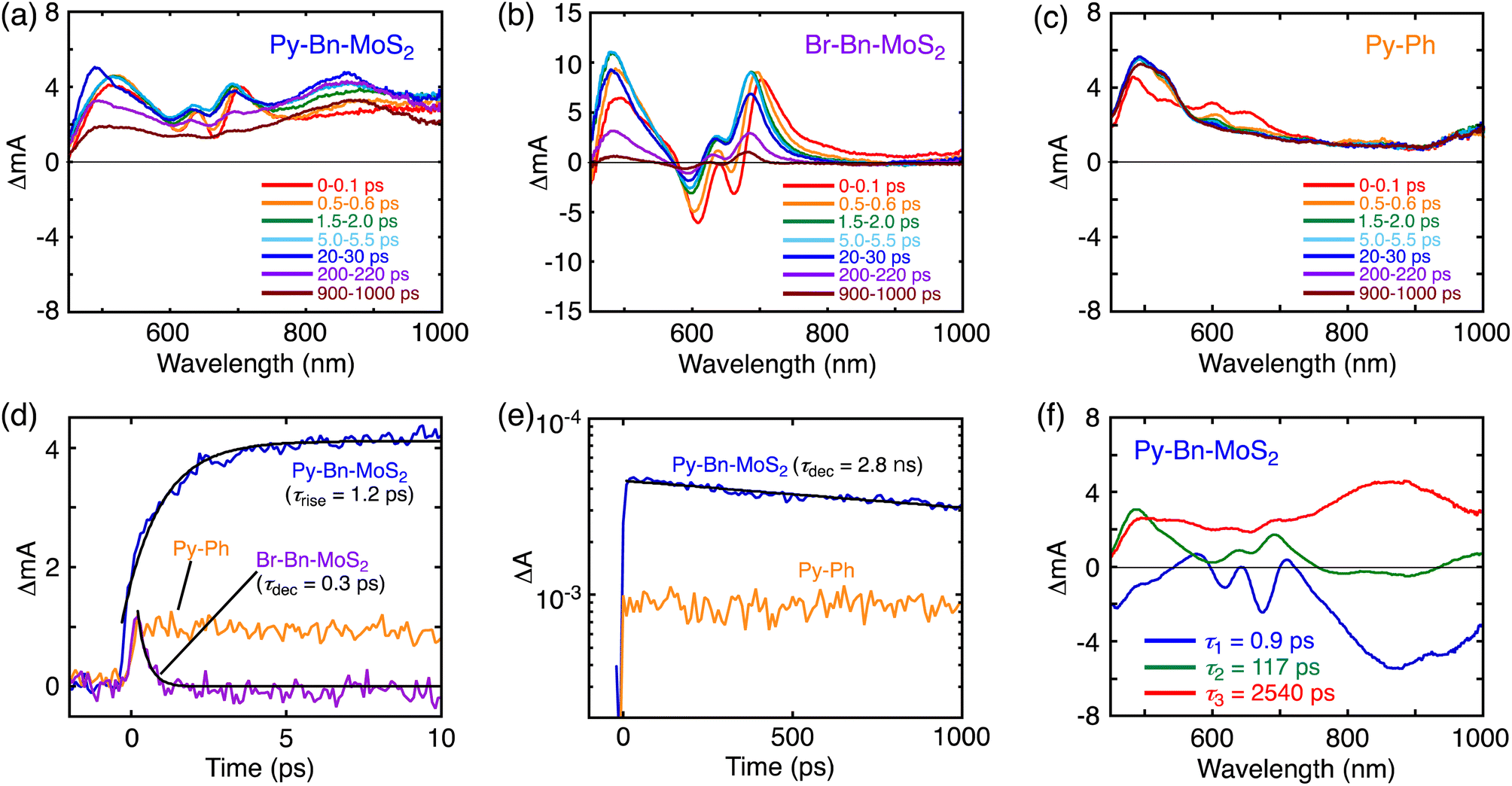 | ||
| Fig. 4 (a–c) Transient absorption spectra of (a) Py-Bn-MoS2, (b) Br-Bn-MoS2, and (c) Py-Ph in DMF. The excitation wavelength was 345 nm. (d and e) Transient absorption profiles of Py-Bn-MoS2 (blue), Br-Bn-MoS2 (purple), and Py-Ph (orange) with fitting curves (black line) in DMF with time ranges of up to (d) 10 ps and (e) 1000 ps. The excitation and detection wavelengths were 345 and 850 nm, respectively. The decay or rise lifetimes are given in parentheses. (f) Transient absorption decay component spectra of Py-Bn-MoS2 in DMF obtained with a global three-component fit of the data. The excitation wavelength was 345 nm. The lifetimes of the respective components are given in the figure. | ||
Fig. 4a depicts the TA spectra of Py-Bn-MoS2 excited at 345 nm, where the Py moiety mainly absorbs the excitation light. At 0–0.1 ps after photoexcitation, the spectrum largely coincides with the sum of the TA spectra of Br-Bn-MoS2 and Py-Ph (Fig. 4b and c), which reflects formations of excited states of both Br-Bn-MoS2 and Py-Ph. TA spectra of Py-Bn-MoS2 with time delays of up to 1000 ps display a characteristic positive signal in the NIR region with a peak at around 870 nm. This spectral feature suggests the evolution of a new transient species involving both pyrene and MoS2 moieties. The time profiles of the new species signal of Py-Bn-MoS2 detected at 850 nm are recorded as shown in Fig. 4d and e. The profile at <10 ps after the excitation clearly reveals the existence of a rise component with a time constant of 1.2 ps, whereas such a rise is not seen in the time profiles of Br-Bn-MoS2 and Py-Ph (Fig. 4d). These observations signify that a new transient species of Py-Bn-MoS2 is formed with a rate constant of 8.3 × 1011 s−1. In addition, the time profile of Py-Bn-MoS2 at <1000 ps discloses that the lifetime of the new transient species is 2.8 ns. This value matches well with the emission lifetime of Py-Bn-MoS2 (Fig. 3c), allowing the assignment of a new species to the emissive CTE state.
Multiexponential global fittings were applied to transient absorption decay curves of Py-Bn-MoS2 at different wavelengths. Three components with lifetimes of τ1 = 0.9 ps, τ2 = 117 ps, and τ3 = 2540 ps are rationally extracted, and the decaying component spectra are represented in Fig. 4f. The peak positions of positive and negative bands are summarized in Table S2.† Taking into account the broad negative signal in 700–1000 nm with a mirror image of the third component with a peak at around 875 nm, the first decay component can be attributed to the formation of the CTE state. The existence of negative peaks at 618 and 674 nm that originate from the initial photobleaching of the excitonic transitions of the MoS2 nanosheet indicates that the hot excitons in the MoS2 nanosheet also cause the formation of the CTE state to some extent. However, such a contribution should be minor due to the lower absorption intensity of the MoS2 moiety at the excitation wavelength (Fig. 3a) and the competition with the rapid cooling of the MoS2 hot exciton (Fig. 4b). Then, we can assign the second component to the decay of the cooled MoS2 exciton, which was formed by the direct excitation of MoS2 and the subsequent cooling or energy transfer from the excited Py, because the spectral shape of the second component coincides well with that of blue-shifted Br-Bn-MoS2 transient absorptions (Fig. 4b). This suggests that the cooled MoS2 exciton directly decays to the ground state without interacting with pyrene moieties. Finally, the third component that exhibits broad positive signals in visible and NIR regions with a peak at around 860 nm is attributable to the decay of the CTE state. The absence of an additional component with a longer lifetime implies that the CTE state decays to the ground state via photoemission and vibrational relaxation without forming a complete CS state. In addition, relaxation of the CTE state via the MoS2 cooled exciton is also energetically possible. The above relaxation pathways of Py-Bn-MoS2 are summarized in Fig. 5.
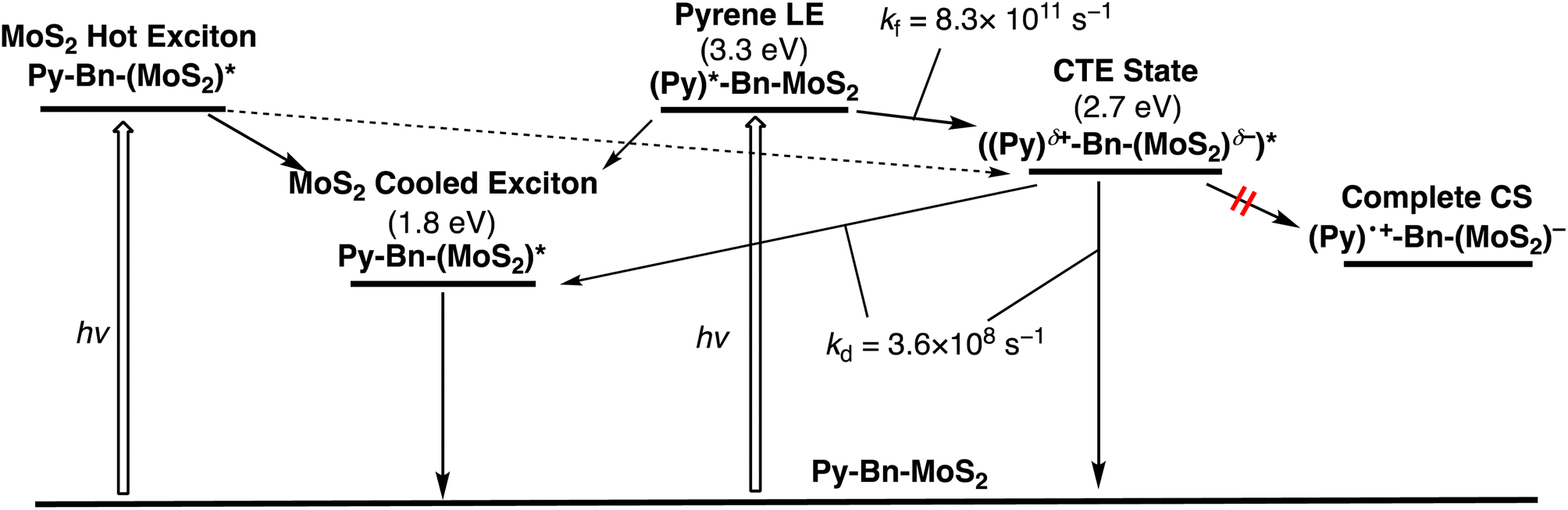 | ||
| Fig. 5 Relaxation pathways of Py-Bn-MoS2 in DMF. The energy level of the CTE state was determined by using the peak position of the emission. kf: the rate constant for the CTE state formation. kd: the rate constant for the CTE state decay. | ||
The signal intensity of the photobleaching in the third component was significantly lower than those in the first and second component spectra. Although the reason for the decrease in the photobleaching intensity in the third component is not clear at the current stage, it may be attributed to the difference in spatial extent of the CTE state and the exciton in the MoS2 nanosheet. In the CTE state of Py-Bn-MoS2, MoS2 has a partial negative charge, which interacts with the partial positive charge on the covalently linked pyrene moiety. Therefore, the CTE state is spatially confined compared to the freely movable exciton with a nanometer-scale size,25 resulting in a limited impact of the CTE state on the absorption behavior of the MoS2 nanosheet. In addition, the broad absorption of the CTE state in the region of 500–700 nm may also obscure the photobleaching signals.
The photoluminescence decay of Py-Bn-MoS2 and the transient absorption spectra of Py-Bn-MoS2, Br-Bn-MoS2, and Py-Ph were also recorded in toluene (Fig. S16†) to investigate photodynamics of Py-Bn-MoS2 in low polarity media. As is the case in DMF, these measurements exemplify the formation of a CTE state and its decay to the ground state without forming a complete CS state. However, the lifetime of the CTE state in toluene (1.8 ns) is significantly shorter than that in DMF (2.7 ns). This result is consistent with the highly polarized character of the CTE state, which is less stable and decays more rapidly to the ground state in a non-polar solvent than in a polar one.
Theoretical studies
Theoretical studies were also performed to elucidate the unprecedented formation of the highly emissive CTE state at the interface of the organic molecule–inorganic nanosheet. We prepared a Py-Bn-MoS2 model with a Mo28S63 nanosheet, in which one N-p-pyrenylbenzylsuccinimide unit and two N-benzylsuccinimide units are covalently bonded to sulfur atoms. The geometry optimization of the Py-Bn-MoS2 model suggests the nearly horizontal orientation of the Py moiety (Fig. S17†), which is consistent with the low thickness of the experimentally obtained Py-Bn-MoS2 (2.6 ± 1.8 nm, Fig. 2e). Moreover, the time-dependent density functional theory (TDDFT) and 3-dimensional reference interaction site model (3D-RISM)26 were employed to examine the emission behavior of Py-Bn-MoS2. Computational details are shown in the ESI.†First, we assessed molecular orbitals contributing to emission transitions of the Py-Bn-MoS2 model calculated at the cLR-SMD(DMF)-ωB97X-D/def2-SV(P) level (Fig. 6a, S18, and S19†). Despite the existence of neighboring N-benzylsuccinimide units, the LUMO of Py of the N-p-pyrenylbenzylsuccinimide unit is mixed with the MoS2 vacant orbitals, indicating CTE state formation by interaction between the MoS2 vacant orbitals and the pyrene excited state. The natural transition orbital (NTO) analysis also demonstrates the mixing of Py and MoS2 vacant orbitals (Fig. S20†). As a result, the charge transfer character in which the positive and negative charges are located at Py and MoS2, respectively, is included in emission transitions, and the difference in electronic dipole moment between the ground and excited states is large (11.0 Debye). This result sharply contrasts with the absence of dipole moment changes in the emission process of a Py molecule itself (Fig. 6b).
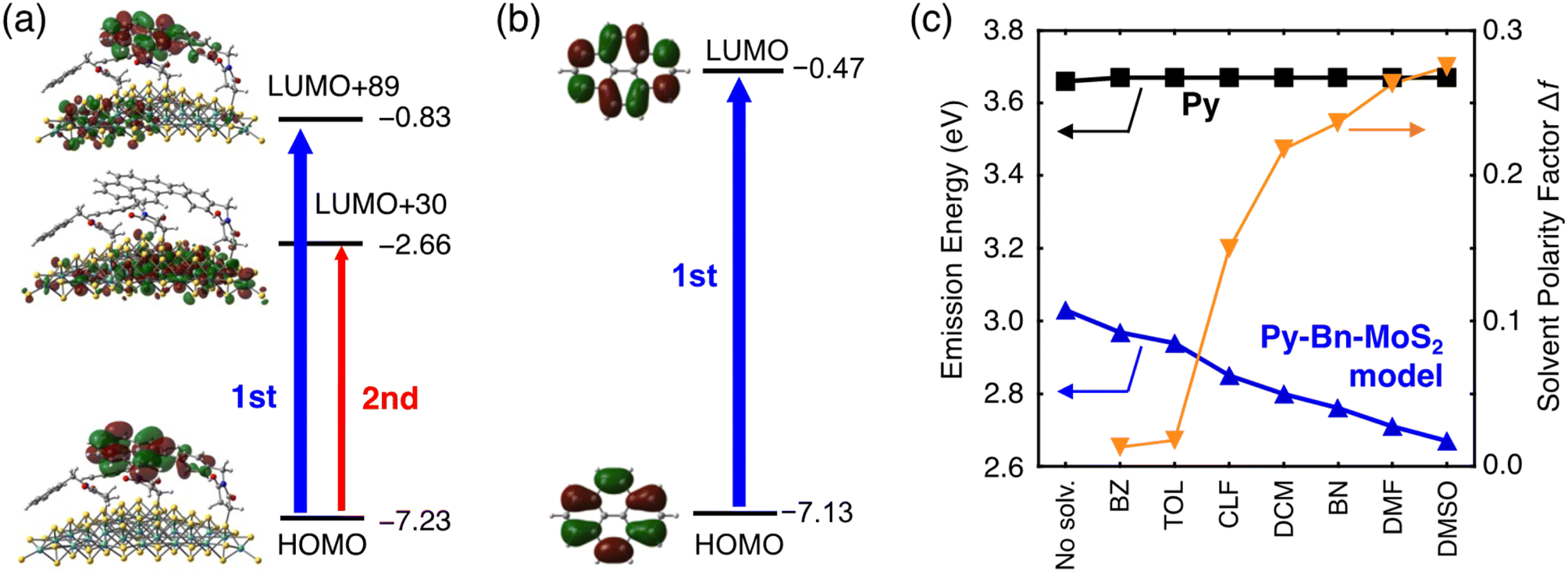 | ||
| Fig. 6 Molecular orbitals of (a) the Py-Bn-MoS2 model and (b) Py that mainly contribute to the emission transitions. Orbital energies are also shown in eV. LUMO+89 and LUMO+30 in (a) represent the 89th and 30th molecular orbitals above the LUMO. (c) Calculated emission energies of pyrene (Py, black square) and the Py-Bn-MoS2 model (blue triangle) in various solvents. Solvent polarity factors (orange inverted triangle) of the solvents are also shown. | ||
Next, solvent dependence of emission energy was evaluated with the 3D-RISM method based on the obtained charge distributions (Fig. 6c and Table S3†). No solvent dependence was found for the Py molecule. In contrast, fluorescence energies of the Py-Bn-MoS2 model become lower with increasing solvent polarity, which is consistent with the experimental results. A solvent spatial distribution obtained from 3D-RISM calculations reveals that the CTE state of the Py-Bn-MoS2 model is stabilized by strong solvation of the positively charged Py units with oxygen atoms of a polar solvent, DMSO (Fig. S21†). Overall, our theoretical calculations clearly confirm the formation of the CTE state, exhibiting the solvent dependency of emission energy.
Conclusions
In conclusion, the covalent linkage of pyrene carrying a structurally well-defined N-benzylsuccinimide bridge on MoS2 nanosheets was achieved utilizing a planetary ball-mill method. The most important new finding in this study is that the photoexcitation of Py in the Py–MoS2 covalently linked system effectively formed the long-lived, highly emissive CTE state that delocalizes over two heterogeneous materials, i.e., an organic π-conjugated unit (pyrene) and an inorganic semiconducting nanosheet (mono- or few-layered MoS2). The formation of an unusual emissive CTE state was fully demonstrated by the systematic steady-state, transient absorption and photoluminescence spectroscopies. Furthermore, theoretical studies elucidated the interaction of MoS2 vacant orbitals with the pyrene LE state to form the CTE state that shows clear solvent dependency of the emission energy. These results unambiguously demonstrate an unexplored potential of organic–inorganic 2D hetero-nanostructures with elaborated bridge structures for optoelectronic devices and sensors. In addition, the unique highly emissive CTE state may give us a hint to reduce the voltage loss derived from nonradiative charge recombination in OPV devices. As has been proposed by several groups,1,3d,e if the CTE state was more emissive, nonradiative vibrational charge recombination would be suppressed. Therefore, this study represents an important step for the applications of organic–inorganic 2D hetero-nanostructures in energy conversion and optoelectronic devices and sensors.Data availability
Additional experimental and computational details and data are already provided in the ESI.†Author contributions
T. Um. and H. I. conceived the concept and prepared the manuscript with feedback from the other authors. D. M., Y. I., and F. Y. conducted the syntheses, characterization studies, theoretical calculations, and optical measurements. W. R. O. conducted the electrochemical measurements. K. K. and A. Y. carried out the PL decay and TA measurements. M. H., T. Ur., and H. S. performed the theoretical calculations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (grants 20H02567, 20H05831, 20H05832, 20H05838, 20H05839, and 23H01808), the Foundation of Kinoshita Memorial Enterprise, Yashima Environment Technology Foundation, and The Asahi Glass Foundation. We thank Prof. K. Tanaka and Dr M. Gon (Kyoto University) for the measurements of photoluminescence quantum efficiencies. We also thank Prof. H. Ohkita, Dr H. D. Kim, and Mr J. Jeon (Kyoto University) for the PYSA measurements.Notes and references
- H. Imahori, Y. Kobori and H. Kaji, Acc. Mater. Res., 2021, 2, 501 CrossRef CAS.
- (a) G. Zhang, F. R. Lin, F. Qi, T. Heumuller, A. Distler, H.-J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K.-Y. Jen and H.-L. Yip, Chem. Rev., 2022, 122, 14180 CrossRef CAS PubMed; (b) D. He, F. Zhao, C. Wang and Y. Lin, Adv. Funct. Mater., 2022, 32, 2111855 CrossRef CAS; (c) T. Umeyama and H. Imahori, Acc. Chem. Res., 2019, 52, 2046 CrossRef CAS PubMed.
- (a) C. Piliego and M. A. Loi, J. Mater. Chem., 2012, 22, 4141 RSC; (b) T.-W. Ng, M.-F. Lo, M.-K. Fung, W.-J. Zhang and C.-S. Lee, Adv. Mater., 2014, 26, 5569 CrossRef CAS PubMed; (c) Z.-W. Zhao, Q.-Q. Pan, Y. Geng, Y. Wu, L. Zhao, M. Zhang and Z.-M. Su, ACS Sustainable Chem. Eng., 2019, 7, 19699 CrossRef CAS; (d) T. Saito, S.-I. Natsuda, K. Imakita, Y. Tamai and H. Ohkita, Sol. PPL, 2020, 4, 2000255 CAS; (e) H. Imahori, Bull. Chem. Soc. Jpn., 2023, 96, 339 CrossRef CAS.
- (a) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed; (b) Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915 RSC; (c) H. Kaji, H. Suzuki, T. Fukushima, K. Shizu, K. Suzuki, S. Kubo, T. Komino, H. Oiwa, F. Suzuki, A. Wakamiya, Y. Murata and C. Adachi, Nat. Commun., 2015, 6, 8476 CrossRef CAS PubMed; (d) Y. Wada, H. Nakagawa, S. Matsumoto, Y. Wakisaka and H. Kaji, Nat. Photonics, 2020, 14, 643 CrossRef CAS; (e) Y. Wada, Y. Wakisaka and H. Kaji, ChemPhysChem, 2021, 22, 625 CrossRef CAS PubMed.
- (a) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS; (b) D. M. Guldi, Phys. Chem. Chem. Phys., 2007, 9, 1400 RSC; (c) O. S. Wenger, Chem. Soc. Rev., 2011, 40, 3538 RSC.
- (a) T. Higashino, T. Yamada, M. Yamamoto, A. Furube, N. V. Tkachenko, T. Miura, Y. Kobori, R. Jono, K. Yamashita and H. Imahori, Angew. Chem., Int. Ed., 2016, 55, 629 CrossRef CAS PubMed; (b) T. Miura, R. Tao, S. Shibata, T. Umeyama, T. Tachikawa, H. Imahori and Y. Kobori, J. Am. Chem. Soc., 2016, 138, 5879 CrossRef CAS PubMed; (c) T. Umeyama and H. Imahori, Nanoscale Horiz., 2018, 3, 352 RSC.
- (a) V. M. Agranovich, Y. N. Gartstein and M. Litinskaya, Chem. Rev., 2011, 111, 5179 CrossRef CAS PubMed; (b) R. Otero, A. L. V. Parga and J. M. Gallego, Surf. Sci. Rep., 2017, 72, 105 CrossRef CAS; (c) H. Jin, J. Li, J. Iocozzia, X. Zeng, P.-C. Wei, C. Yang, N. Li, Z. Liu, J. H. He, T. Zhu, J. Wang, Z. Lin and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 15206 CrossRef CAS PubMed; (d) C. Zang, M. Xu, L. Zhang, S. Liu and W. Xie, J. Mater. Chem. C, 2021, 9, 1484 RSC.
- (a) T. R. Kafle, B. Kattel, P. Yao, P. Zereshki, H. Zhao and W.-L. Chan, J. Am. Chem. Soc., 2019, 141, 11328 CrossRef CAS; (b) R. Canton-Vitoria, H. B. Gobeze, V. M. Blas-Ferrando, J. Ortiz, Y. Jang, F. Fernandez-Lazaro, A. Sastre-Santos, Y. Nakanishi, H. Shinohara, F. D'Souza and N. Tagmatarchis, Angew. Chem., Int. Ed., 2019, 58, 5712 CrossRef CAS PubMed; (c) R. Canton-Vitoria, Y. Sayed-Ahmad-Baraza, M. Pelaez-Fernandez, R. Arenal, C. Bittencourt, C. P. Ewels and N. Tagmatarchis, npj 2D Mater. Appl., 2017, 1, 13 CrossRef.
- (a) M. Vera-Hidalgo, E. Giovanelli, C. Navío and E. M. Pérez, J. Am. Chem. Soc., 2019, 141, 3767 CrossRef CAS PubMed; (b) R. Quirós-Ovies, M. Vázquez Sulleiro, M. Vera-Hidalgo, J. Prieto, I. J. Gómez, V. Sebastián, J. Santamaría and E. M. Pérez, Chem.–Eur. J., 2020, 26, 6629 CrossRef PubMed.
- (a) G. Liu and N. Komatsu, ChemNanoMat, 2016, 2, 500 CrossRef CAS; (b) M. Mohamed Ismail, J. Vigneshwaran, S. Arunbalaji, D. Mani, M. Arivanandhan, S. P. Jose and R. Jayavel, Dalton Trans., 2020, 49, 13717 RSC; (c) T. Umeyama, H. Xu, T. Ohara, Y. Tsutsui, S. Seki and H. Imahori, J. Phys. Chem. C, 2021, 125, 13954 CrossRef CAS.
- J. Baek, T. Umeyama, W. Choi, Y. Tsutsui, H. Yamada, S. Seki and H. Imahori, Chem.–Eur. J., 2018, 24, 1561 CrossRef CAS PubMed.
- (a) K. Komatsu, Top. Curr. Chem., 2005, 254, 185 CrossRef CAS; (b) N. Rubio, K.-C. Mei, R. Klippstein, P. M. Costa, N. Hodgins, J. T.-W. Wang, F. Festy, V. Abbate, R. C. Hider, K. L. A Chan and K. T. Al-Jamal, ACS Appl. Mater. Interfaces, 2015, 7, 18920 CrossRef CAS PubMed; (c) X. Zhu, T. Zhang, D. Jiang, H. Duan, Z. Sun, M. Zhang, H. Jin, R. Guan, Y. Liu, M. Chen, H. Ji, P. Du, W. Yan, S. Wei, Y. Lu and S. Yang, Nat. Commun., 2018, 9, 4177 CrossRef PubMed.
- (a) S. Bae, N. Sugiyama, T. Matsuo, H. Raebiger, K. Shudo and K. Ohno, Phys. Rev. Appl., 2017, 7, 0240015 Search PubMed; (b) I. K. Sideri, Y. Jang, J. Garcés-Garcés, Á. Sastre-Santos, R. Canton-Vitoria, R. Kitaura, F. Fernández-Lázaro, F. D'Souza and N. Tagmatarchis, Angew. Chem., Int. Ed., 2021, 60, 9120 CrossRef CAS PubMed.
- I. Gómez-Muñoz, S. Laghouati, R. Torres-Cavanillas, M. Morant-Giner, N. V. Vassilyeva, A. Forment-Aliaga and M. Giménez-Marqués, ACS Appl. Mater. Interfaces, 2021, 13, 36475 CrossRef PubMed.
- Z. Zeng, Z. Yin, X. Huang, H. Li, Q. He, G. Lu, F. Boey and H. Zhang, Angew. Chem., Int. Ed., 2011, 50, 11093 CrossRef CAS PubMed.
- M. Amani, D.-H. Lien, D. Kiriya, J. Xiao, A. Azcatl, J. Noh, S. R. Madhvapathy, R. Addou, S. KC, M. Dubey, K. Cho, R. M. Wallace, S.-C. Lee, J.-H. He, J. W. Ager III, X. Zhang, E. Yablonovitch and A. Javey, Science, 2015, 350, 1065 CrossRef CAS PubMed.
- (a) A. Nakajima, Bull. Chem. Soc. Jpn., 1973, 46, 2602 CrossRef CAS; (b) N. V. Tkachenko, H. Lemmetyinen, J. Sonoda, K. Ohkubo, T. Sato, H. Imahori and S. Fukuzumi, J. Phys. Chem. A, 2003, 107, 8834 CrossRef CAS; (c) D. Das and D. N. Nath, J. Phys. Chem. B, 2007, 111, 11009 CrossRef CAS.
- (a) A. Wiessner, G. Hüttmann, W. Kühnle and H. Staerk, J. Phys. Chem., 1995, 99, 14923 CrossRef CAS; (b) W. Weigel, W. Rettig, M. Dekhtyar, C. Modrakowski, M. Beinhoff and A. D. Schlülter, J. Phys. Chem. A, 2003, 107, 5941 CrossRef CAS; (c) M. Fakis, J. S. Beckwith, K. Seintis, E. Martinou, C. Nançoz, N. Karakostas, I. Petsalakis, G. Pistolis and E. Vauthey, Phys. Chem. Chem. Phys., 2018, 20, 837 RSC; (d) M. Imran, A. A. Sukhanov, P. Maity, A. Elmali, J. Zhao, A. Karatay, O. F. Mohammed and V. K. Voronkova, J. Phys. Chem. B, 2021, 125, 9244 CrossRef CAS PubMed.
- T. Umeyama, J. Baek, Y. Sato, K. Suenaga, F. Abou-Chahine, N. V. Tkachenko, H. Lemmetyinen and H. Imahori, Nat. Commun., 2015, 6, 7732 CrossRef CAS PubMed.
- (a) N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465 CrossRef CAS; (b) J. P. Ceron-Carrasco, D. Jacquemin, C. Laurence, A. Planchat, C. Reichardt and K. Sraidi, J. Phys. Org. Chem., 2014, 27, 512 CrossRef CAS.
- C. D. Borsarelli, J. J. Cosa and C. M. Previtali, Langmuir, 1992, 8, 1070 CrossRef CAS.
- W. Hong, B. Sun, H. Aziz, W.-T. Park, Y.-Y. Nohd and Y. Li, Chem. Commun., 2012, 48, 8413 RSC.
- H. Yu, X. Cui, X. Xu and W. Yao, Natl. Sci. Rev., 2015, 2, 57 CrossRef CAS.
- N. P. Gritsan, E. A. Pritchina, I. L. Barabanov, G. T. Burdzinski and M. S. Platz, J. Phys. Chem. C, 2009, 113, 11579 CrossRef CAS.
- M. Goryca, J. Li, A. V. Stier, T. Taniguchi, K. Watanabe, E. Courtade, S. Shree, C. Robert, B. Urbaszek, X. Marie and S. A. Crooker, Nat. Commun., 2019, 10, 4172 CrossRef CAS.
- (a) D. Beglov and B. Roux, J. Phys. Chem. B, 1997, 101, 7821 CrossRef CAS; (b) A. Kovalenko and F. Hirata, Chem. Phys. Lett., 1998, 290, 237 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedures for measurements, theoretical calculations, and synthesis, characterization data, photophysical properties, and computational results. See DOI: https://doi.org/10.1039/d3sc03604a |
| This journal is © The Royal Society of Chemistry 2023 |