
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An overlooked oxidation mechanism of toluene: computational predictions and experimental validations†
Zihao
Fu‡
a,
Fangfang
Ma‡
a,
Yuliang
Liu
 b,
Chao
Yan
b,
Dandan
Huang
c,
Jingwen
Chen
a,
Jonas
Elm
d,
Yuanyuan
Li
b,
Aijun
Ding
b,
Lukas
Pichelstorfer
ef,
Hong-Bin
Xie
*a,
Wei
Nie
*b,
Joseph S.
Francisco
*g and
Putian
Zhou
*e
b,
Chao
Yan
b,
Dandan
Huang
c,
Jingwen
Chen
a,
Jonas
Elm
d,
Yuanyuan
Li
b,
Aijun
Ding
b,
Lukas
Pichelstorfer
ef,
Hong-Bin
Xie
*a,
Wei
Nie
*b,
Joseph S.
Francisco
*g and
Putian
Zhou
*e
aKey Laboratory of Industrial Ecology and Environmental Engineering (Ministry of Education), School of Environmental Science and Technology, Dalian University of Technology, Dalian 116024, China. E-mail: hbxie@dlut.edu.cn
bJoint International Research Laboratory of Atmospheric and Earth System Sciences, School of Atmospheric Sciences, Nanjing University, Nanjing 210023, China. E-mail: niewei@nju.edu.cn
cState Environmental Protection Key Laboratory of Formation and Prevention of Urban Air Pollution Complex, Shanghai Academy of Environmental Sciences, Shanghai, China
dDepartment of Chemistry, iClimate, Aarhus University, Langelandsgade 140, DK-8000 Aarhus C, Denmark
eInstitute for Atmospheric and Earth System Research/Physics, Faculty of Science, University of Helsinki, P. O. Box 64, FIN-00014 Helsinki, Finland. E-mail: putian.zhou@helsinki.fi
fpi-numerics, Wallbachsiedlung 28, 5202 Neumarkt am W., Austria
gDepartment of Earth and Environmental Science, University of Pennsylvania, Philadelphia, PA, USA 19104-6316. E-mail: frjoseph@sas.upenn.edu
First published on 27th October 2023
Abstract
Secondary organic aerosols (SOAs) influence the Earth's climate and threaten human health. Aromatic hydrocarbons (AHs) are major precursors for SOA formation in the urban atmosphere. However, the revealed oxidation mechanism dramatically underestimates the contribution of AHs to SOA formation, strongly suggesting the importance of seeking additional oxidation pathways for SOA formation. Using toluene, the most abundant AHs, as a model system and the combination of quantum chemical method and field observations based on advanced mass spectrometry, we herein demonstrate that the second-generation oxidation of AHs can form novel epoxides (TEPOX) with high yield. Such TEPOX can further react with H2SO4 or HNO3 in the aerosol phase to form less-volatile compounds including novel non-aromatic and ring-retaining organosulfates or organonitrates through reactive uptakes, providing new candidates of AH-derived organosulfates or organonitrates for future ambient observation. With the newly revealed mechanism, the chemistry-aerosol box modeling revealed that the SOA yield of toluene oxidation can reach up to 0.35, much higher than 0.088 based on the original mechanism under the conditions of pH = 2 and 0.1 ppbv NO. This study opens a route for the formation of reactive uptake SOA precursors from AHs and significantly fills the current knowledge gap for SOA formation in the urban atmosphere.
Introduction
Secondary organic aerosols (SOAs) represent a major constituent of atmospheric aerosols,1 and impact human health and global climate.2,3 Gaseous organic compounds are potential SOA precursors, especially volatile organic compounds (VOCs). In the past 70 years, gas-phase oxidation of VOCs followed by condensation has been suggested to dominate SOA formation.1 Great efforts have been made to reveal the oxidation mechanism of VOCs, and identify the SOA precursors to build a quantitative relationship of gaseous organic compounds with SOA formation.4,5 However, atmospheric models based on current information consistently underestimate the global SOA budget.6–8 This underestimation of SOAs strongly suggests the importance of seeking additional pathways leading to SOA formation.Increasing evidence suggests that multiphase chemistry caused by the formation of reactive uptake precursors (RUPs) in the oxidation of VOCs is an important pathway for SOA formation. For example, the reactive uptake of isoprene-derived epoxydiols (IEPOX) has been demonstrated to be a significant source of atmospheric SOAs globally.9–12 Most recently, it was suggested that even in polluted urban environments, RUPs of anthropogenic origin significantly contributed to SOA formation locally.13–16 However, the underlying sources and chemical identities of these anthropogenic RUPs remain unclear. Therefore, the identification of the mechanisms of RUP formation from anthropogenic VOC precursors is of great interest.
Aromatic hydrocarbons (AHs) comprise a significant fraction (up to 60%) of total VOCs in the urban atmosphere.17 The oxidation of AHs can significantly contribute to SOA formation with up to 50% in the urban atmosphere in eastern China.18 The most abundant AHs are monocyclic aromatic hydrocarbons (MAHs) such as benzene and toluene.17 The first-generation and multi-generation oxidation mechanisms of MAHs have previously been investigated.19–29 Particularly, the autoxidation mechanism which leads to the formation of highly oxygenated organic molecules (HOMs) has been identified for alkylbenzene.22,23 With the revealed mechanism, the condensation of the low-volatility HOMs and multiphase chemistry of glyoxal and methylglyoxal were found to be the main processes for SOA formation of MAH oxidation. However, considering these processes still underestimated the SOA yield,30–33 indicates the existence of missing MAH oxidation mechanisms and possible unidentified RUPs.
Here, we demonstrate that the second-generation oxidation of MAHs can produce a significant yield of epoxides, enhancing SOA production through reactive uptakes under low pH conditions, similar to the case of isoprene.34,35 We selected toluene (T) as the representative compound as it is the most abundant AH in the urban atmosphere.20,36 Specifically, the modeled system started with hydroperoxide T-ROOH and organonitrate T-RONO2, which are important first-generation products of toluene upon oxidation by OH.19 The formation of epoxides is revealed by quantum chemical calculations and kinetics modeling, which are supported by field observations. With the chemistry-aerosol SOSAA-Box model, the SOA yield is shown to increase substantially when the reactive uptake of epoxides is considered. This study presents a new route for RUP formation from AHs, guides the detection of novel AH-derived SOA precursors, and fills the current knowledge gap in SOA formation in the urban atmosphere.
Materials and methods
Global minimum search
The global minimum of T-ROOH and T-RONO2 was selected as the initial conformations for the study of the multi-generation oxidation mechanism. A similar scheme for the global minimum search has been employed in our previous studies.37,38 Briefly, ab initio molecular dynamics (AIMD) within the TURBOMOLE 6.5 program package39 was first performed to produce a range of conformations of T-ROOH and T-RONO2. Selected conformations from the AIMD run were then further optimized at the M06-2X/6-31+G(d,p) level of theory, followed by ROCBS-QB3 single-point energy calculations. The conformation with the lowest Gibbs free energy was identified as the global minimum of T-ROOH and T-RONO2 (see their structures in Fig. S1†).Ab initio electronic structure calculations
All electronic structure and energy calculations were performed using the GAUSSIAN 09 program package.40 The geometry optimizations and harmonic vibrational frequency calculations for reactants (R), pre-complexes (RCs), post-complexes (PCs), intermediates (IMs), transition states (TSs) and products involved in all reaction pathways were performed at the M06-2X/6-31+G(d,p) level of theory,41 followed by a higher level ROCBS-QB3 single-point energy calculation.42 The combination of M06-2X functional with the ROCBS-QB3 scheme has previously been used in studying the oxidation of AHs.22,24,43–46 Since reaction pathways with high reaction barriers contribute negligibly to the reaction kinetics, only low level energies of the species involved in the pathways were provided (shown in the ESI†) considering the computational costs. Values of T1 diagnostics for the TSs in all reaction pathways were less than the threshold value (0.045) for the open-shell systems,47 indicating that single-reference methods are well suited to describe the target systems. To check the wavefunction stability of RC, the keyword “stable” was used. When considering reactions in the aqueous phase, the SMD solvation model was employed to account for the water solvent effect.48 In addition, the proportion of different dissociation forms of the reactants involved in the aqueous phase under different pH conditions was calculated based on the pKa values. Intrinsic reaction coordinate (IRC) calculations were performed to confirm the connection of each TS between designated local minima.Kinetics calculations
Reaction rate constants for the unimolecular reactions with a well-defined transition state as well as the competition between the uni- and biomolecular reactions were modeled using Rice–Ramsperger–Kassel–Marcus (RRKM)-master equation (ME) theory in the MESMER program.49 For ·OH-initiated reactions, RCs involved in the ·OH-addition reaction pathways were considered for all the kinetic calculations,50–54 since the ·OH-addition reaction is the dominant pathway. Reaction rate constants for the barrierless bimolecular reactions from R to RC in the OH-initiated reaction were calculated by combining the use of long-range transition state theory with a dispersion force potential and the inverse Laplace transformation (ILT) method.49,55 For the bimolecular reaction (alkyl radicals + O2), a constant value of 6.0 × 10−12 cm3 per molecule per s was used, which is similar to previous studies.22,56–58 N2 was used as the buffer gas. The average collisional activation/deactivation energy transfer of all the molecules is set to 200 cm−1 (ΔEd) per collision and the grain size is 50 cm−1. To explore the effects of ΔEd and grain size on the results, we additionally run the simulations at other ΔEd (150, 250 and 300 cm−1) and grain size (25 cm−1). The empirical method proposed by Gilbert and Smith was applied to estimate the Lennard-Jones parameters of intermediates (Table S2†).59 The theory for calculating the fractional yields of the main intermediates is presented in the ESI.† A one-dimensional unsymmetrical Eckart barrier was used to account for the tunneling effects in all the reaction rate constant calculations involving H-shift or H-abstraction.60Field observations
Ambient data of toluene and oxygenated organic molecules (OOMs) from aromatic oxidation were collected during the summer in Nanjing, a megacity in eastern China.61 Detailed description of this data has been presented in our previous study.61 Briefly, toluene was measured using a PTR-TOF-MS (Ionicon Analytik, TOF 1000 ultra),62 while OOMs were measured by using a nitrate-ion-based chemical ionization atmospheric pressure interface time-of-flight mass spectrometer (nitrate CI-APi-TOF), with a mass resolution of 8000–12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Th Th−1 (Th denotes Thomsons).63,64 The concentrations of OOMs were estimated via65,66
000 Th Th−1 (Th denotes Thomsons).63,64 The concentrations of OOMs were estimated via65,66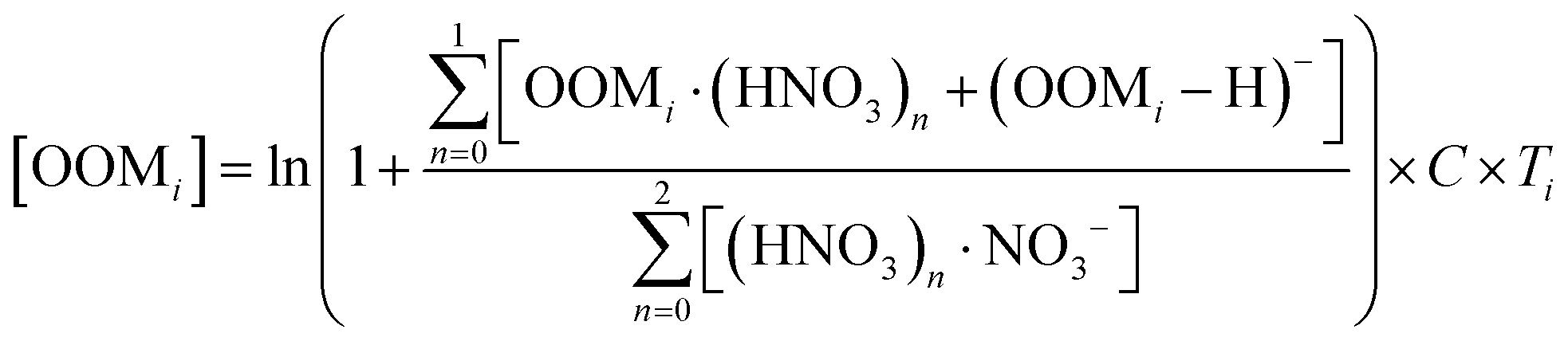 | (1) |
The primary RO2· (PC7-Aro-RO2) is calculated as kOH × [toluene] × [·OH], where kOH is the reaction rate constant (5.0 × 10−13 cm3 per molecule per s) of toluene with ·OH,18 and [toluene] and [·OH] are the concentrations of toluene and ·OH, respectively. [·OH] was estimated from the concentration of SA ([SA]) (viaeqn (2)).68
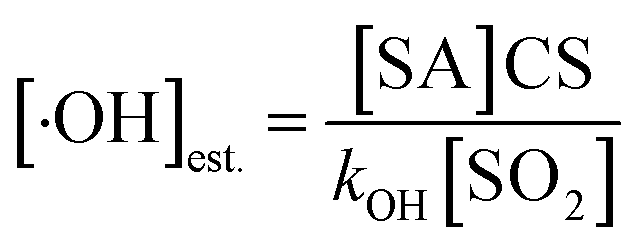 | (2) |
Box modeling
The box model SOSAA-Box69 (model to simulate organic vapours, sulphuric acid and aerosols) was applied to simulate the effect of the new oxidation pathways of toluene on SOA mass yields. The chemistry scheme was first generated with the MCM v3.3.1 (Master Chemical Mechanism version 3.3.1)70 by selecting the following species: toluene and CH4. The reaction rates of the oxidation of SO2 by stabilized Criegee intermediate (sCI) radicals were increased to 7.0 × 10−13 cm3 per molecule per s from 7.0 × 10−14 cm3 per molecule per s as suggested in Boy et al.71 All considered reaction pathways are presented in the ESI.† The background particle size distributions (PSDs) representing the environmental conditions in typical cities refer to the data collected in Wu and Boor,72 in which all the measured PSDs have been fitted with three lognormal modes. For example, one sample of PSDs in Beijing measured by Massling et al.73 has been fitted to three modes in the range of 3 nm to 800 nm with geometric mean diameters of 5.7 nm, 32.8 nm, and 114.5 nm, geometric standard deviations of 1.33, 2.61, and 1.55, and the peak number concentrations of 615 molecules cm−3, 31702 molecules cm−3, and 614 molecules cm−3, respectively (see Table S3† in Wu and Boor72). These PSDs were also applied and kept constant in this study to simulate the background aerosol environment. Therefore, nucleation and coagulation were not considered in the simulations.
In order to quantify how the oxidation products can contribute to SOA formation under different conditions and chemistry mechanisms, the condensation/evaporation processes of condensable organic vapors were simulated with the analytical predictor of condensation (APC) scheme modified from Jacobson.74 All the condensed organic compounds were considered to be well-mixed in the liquid phase. In this study, we have focused on the contribution of organic products, so the condensation of inorganic species is not considered. Each particle size was assumed to be internally mixed. The saturation vapor pressure (SVP) of the chemical species over a flat pure compound surface was obtained from the database in ARCA-Box,75 the SVP values of additional species in the new oxidation pathway were calculated using the SIMPOL method76 or via the EPI suite software (US EPA, 2012).77 The Raoult effect and Kelvin effect were included when calculating the SVP values over the particle surface. The activity coefficients were assumed to be one for all condensable vapors. Moreover, the method from eqn (17) in Jacobson78 was applied at each time step to constrain the mass of condensed vapors to not exceed the total available amount. The aqueous phase chemical reactions were calculated explicitly after condensation/evaporation processes when needed in the simulation cases. Other details for the model setup are presented in the ESI.†
Results and discussion
Formation of epoxides in the reactions of T-ROOH and T-RONO2 with ·OH
By carefully considering all possible reaction pathways (Fig. S2–S5†) for the ·OH-initiated oxidation of T-ROOH and T-RONO2, we identify the reaction pathways for forming novel epoxides (Fig. 1), which can potentially contribute to SOA formation through multiphase reactions similar to IEPOX.9–12 Two types of epoxides are identified including ring-opening (here the ring refers to a six-membered ring) dicarbonyl epoxides (PTH-1-1-2 and PTN-1-1-1) and ring-retaining epoxides (TEPOX). Ring-opening epoxides are formed in a multi-step reaction mechanism that proceeds via a C-centered radical intermediate formed by ·OH addition to the α-site C-atom of the –COOH/CONO2 group. The formation mechanism of the ring-opening epoxides for the reaction of T-ROOH is slightly different from that for the reactions of T-RONO2. For the reactions of T-ROOH, the formed RO· from C-centered radicals intermediately dissociates to form PTH-1-1-2, but not IMTH-1-1-1via the lower reaction energy barriers (Ea) (see detailed analysis in the ESI). However, the formed RO· from C-centered radical intermediates needs multiple steps to finally form PTN-1-1-1 for the reactions of T-RONO2. Differing from the ring-opening epoxides, ring-retaining TEPOX is formed via a two-step reaction mechanism that proceeds via ·OH addition to the β-site C-atom of the –COOH/CONO2 group, followed by a concerted O–O/O–N bond rupture and C–O–C cyclization.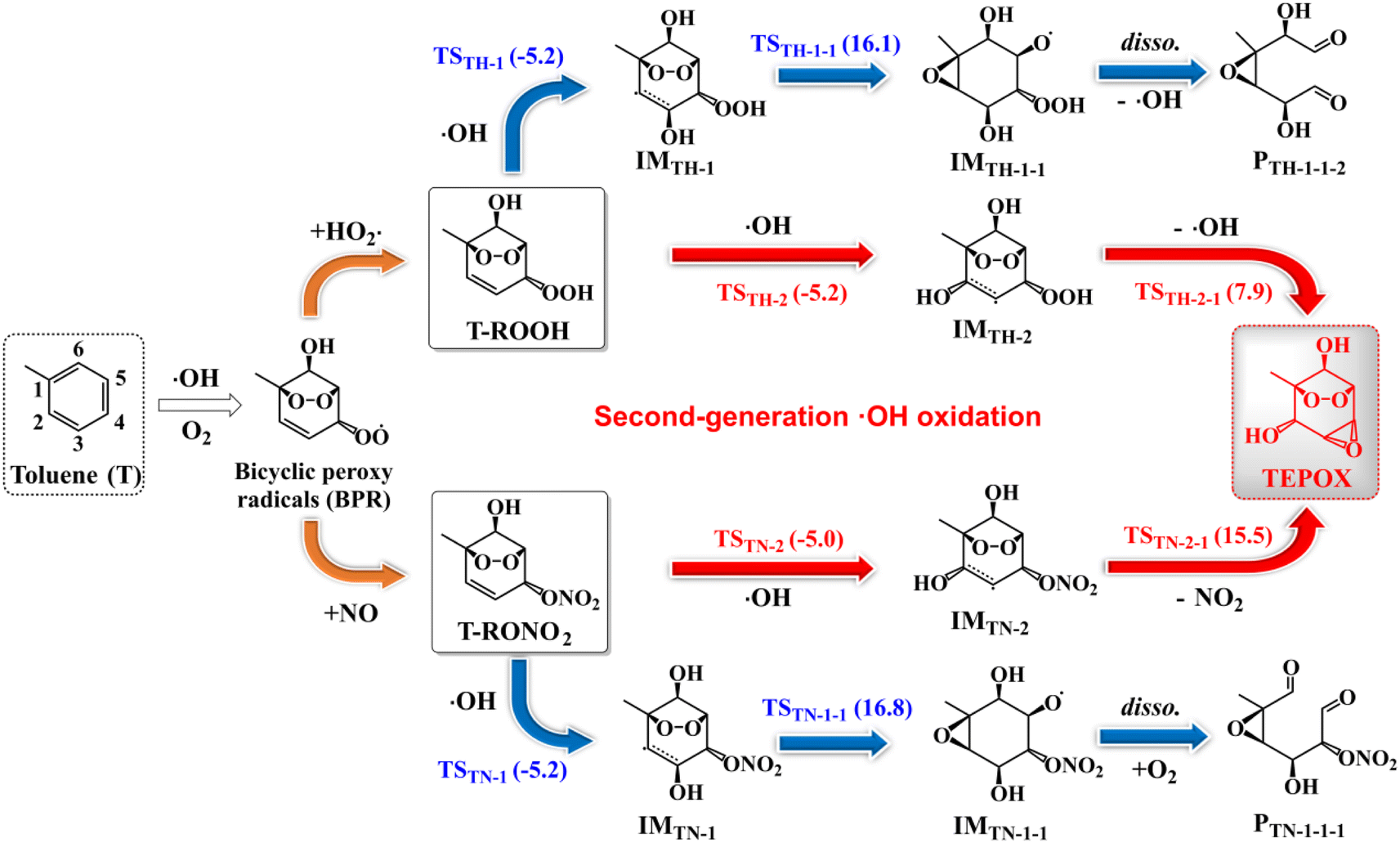 | ||
| Fig. 1 Reaction pathways of forming epoxides for reactions of T-ROOH and T-RONO2 with ·OH starting from toluene (T). The numbers (in kcal mol−1) near the arrows are zero-point corrected reaction energy barriers for the corresponding reactions at the ROCBS-QB3//M06-2X/6-31+G(d,p) level of theory. The labels TSTH/TN-m, IMTH/TN-m and PTH/TN-m represent the transition states, intermediates and products, respectively, where subscripts TH/TN were used to differentiate the reactions starting from T-ROOH/T-RONO2 + ·OH, respectively, and m presents different species. | ||
The formation mechanism for ring-retaining TEPOX from T-ROOH and T-RONO2 is similar to that of IEPOX from organic hydroperoxide ISOPOOH and organonitrate ISOPONO2 formed from the oxidation of isoprene.9,79 In view of the molecular structure, the similar reaction mechanism should result from the fact that they contain similar ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–C(–OOH/ONO2)
CH–C(–OOH/ONO2)![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) structural units, which act as the reactive core for forming the epoxides. It is noteworthy that the reaction energy barriers (Ea) for the formation of TEPOX from T-ROOH are much lower than that from T-RONO2, which resembles the formation of IEPOX from ISOPOOH and ISOPONO2.79
structural units, which act as the reactive core for forming the epoxides. It is noteworthy that the reaction energy barriers (Ea) for the formation of TEPOX from T-ROOH are much lower than that from T-RONO2, which resembles the formation of IEPOX from ISOPOOH and ISOPONO2.79
Similar to a previous study,80 by considering all possible competitive reaction pathways (T-ROOH + ·OH → IMTH-1/IMTH-2 → IMTH-1-1/PTH2-1 and IMTH-1/IMTH-2 + O2 → IMTH-1-O2/IMTH-2-O2), the fractional yields of ring-opening epoxides (PTH-1-1-2) and ring-retaining TEPOX (PTH2-1) are calculated to be 1.44% and 56.1% for the reaction of T-ROOH with ·OH, respectively (Fig. 2, details in Fig. S6†). Therefore, epoxides, mainly consisting of ring-retaining TEPOX, are important products for the reactions of T-ROOH with ·OH. We noted that previous studies found that the yields of epoxides are low for the reactions of alkoxy radicals produced in the first-generation oxidation of AHs.44,45,81,82 To the best of our knowledge, this is the first time to illustrate that ring-retaining epoxides (TEPOX) can be formed in high yields in the second-generation oxidation of toluene, similar to that of isoprene oxidation. In addition, the ring-opening epoxides also have a considerable yield (1.44%), presenting a novel mechanism for the formation of ring-opening epoxides in the atmosphere. Different from the reactions of T-ROOH with ·OH, the calculated fractional yield of ring-retaining TEPOX (22.4%) from the reaction of T-RONO2 with ·OH (Fig. 2) is low based on the favorable reaction pathways (T-RONO2 + OH → IMTN-1/IMTN-2 → IMTN-1-1/PTN-2-1 and IMTN-1/IMTN-2 + O2 → IMTN-1-O2/IMTN-2-O2). The lower yield of TEPOX results from its corresponding high unimolecular reaction energy barrier (15.5 kcal mol−1). A previous study on the oxidation of isoprene found that the yield of IEPOX from the reaction of ISOPOOH with ·OH is much higher than that from the reaction of ISOPONO2 with ·OH.79 This is consistent with our findings for toluene here. In addition, the yield (0.200%) of ring-opening epoxides from the T-RONO2 with ·OH is also lower than that (1.44%) of the corresponding reactions of T-ROOH with ·OH.
 | ||
| Fig. 2 Calculated fractional yields of main intermediates and products in the reactions of T-ROOH (a) and T-RONO2 (b) initiated by ·OH at 298 K and 1 atm. The labels IMTH/TN-m-O2 and PTH/TN-m represent the peroxy radicals and products, respectively, where subscripts TH/TN were used to differentiate the reactions starting from T-ROOH/T-RONO2 + ·OH, respectively, and m presents different species. | ||
Besides TEPOX, peroxy radicals also have high yields in the reactions of T-ROOH and T-RONO2 initiated by ·OH, presenting another main oxidation pathway. For the reaction of T-ROOH, peroxy radicals are mainly formed from the reactions of C-centered IMTH-1 radicals with O2. The yield of the formed peroxy radicals IMTH-1-O2 is 42.4%. For the reaction of T-RONO2, peroxy radicals are formed from the C-centered radicals IMTN-1 and IMTN-2. The yields of the formed IMTN-1-O2 and IMTN-2-O2 are 56.0% and 21.4%, respectively. These peroxy radicals can subsequently react with NO or HO2· to form organonitrates, hydroperoxides and alkoxy radicals. The formed alkoxy radicals eventually produce a range of dicarbonyl products including C3H4O3, C4H6O3, C4H6O5, C4H6NO5, C7H9NO8 and methylglyoxal (see details in Fig. S3 and S5†). In addition, we found that the selection of ΔEd (from 150 to 300 cm−1) and grain size have little effect on the yields of the important species mentioned above (Table S3†).
Comparison with recent laboratory studies
The main atmospheric oxidation pathways and products of the ·OH-initiated reactions of T-ROOH and T-RONO2 are summarized in Fig. S7.† Overall, the main products include C7H10O5, C3H4O3, C4H6O3, C4H6NO5, C7H9NO8 and methylglyoxal, some of which (C7H10O5, C4H6O3 and C7H9NO8) have been detected in the chamber experiments of toluene oxidation performed by Zaytsev et al.26 More importantly, Zaytsev et al.26 suggested that C7H10O5 is a mixture of first- and second-generation oxidation products of toluene, consistent with our finding that the molecular formula corresponds to T-ROOH (first-generation products) and ring-retaining TEPOX (second-generation products). The evidence from these experiments further corroborates our mechanistic findings.Supporting evidence from field observations
We further conducted field observations at the Station for Observing Regional Processes of the Earth System (SORPES)83 during the summer of 2019 in Nanjing, China. A nitrate CI-Api-TOF was employed to detect the oxidation products of toluene, especially ring-retaining TEPOX in the real atmosphere. Most molecules identified in our revealed mechanism can be observed in the real atmosphere, including both the key oxidation products of C7H9NO6, C7H10O5 and C7H9NO8, and the fragmentation products (C4H5NO6, C4H6O5, C4H6O3 and C3H4O3). The product molecules C7H9NO6, C7H10O5 and C7H9NO8 that are not fragmented have a double-bond-equivalent (DBE) of 3, suggesting they were formed via ·OH attacking the benzene ring of toluene at daytime.18As shown in Fig. 3a, the observed C7H9NO6 correlates with the primary RO2· (PC7-Aro-RO2) from the ·OH-initiated oxidation of toluene. Therefore, C7H9NO6 should correspond to T-RONO2 and is probably a first-generation product of toluene oxidation. However, we cannot determine whether C7H10O5 is T-ROOH, ring-retaining TEPOX or both directly from its elemental formula, since the mass spectrometry observations cannot distinguish molecular structures. As proposed above, the ring-retaining TEPOX molecule is a second-generation product, while T-ROOH is a first-generation product. Therefore, we infer the attribution of C7H10O5 by their distinctive diurnal variation patterns. As shown in Fig. 3b, there is no correlation between C7H10O5 and PC7-Aro-RO2. More importantly, the daytime peak of C7H10O5 was around 14:00-15:00, well after the possible first-generation product C7H9NO6 (10:00–11:00) (Fig. 3c). Therefore, it is more likely that C7H10O5 is mainly composed of second-generation products (i.e. ring-retaining IEPOX), although some first-generation products may also be present in the morning. This is consistent with the previous lab study that C7H10O5 is a mixture of first- and second-generation products for the oxidation of toluene.26 Overall, the field observations suggest that a significant amount of ring-retaining TEPOX exists in this suburban environment.
 | ||
| Fig. 3 Ambient observation of toluene oxidation products of C7H10O5 and C7H9NO6. (a) Correlation of C7H9NO6 with a production rate of RO2 (PC7 Aro-RO2) from ·OH-initiated oxidation of toluene during daytime, (b) correlation of C7H10O5 with the PC7-Aro-RO2 during daytime, and (c) variation of C7H10O5, C7H9NO6, NO and temperature at daytime during the field observation campaign. | ||
Box modelling
Implementing this new mechanism of T-ROOH and T-RONO2 initiated by ·OH, a SOSAA-Box model69 simulation shows that SOA yield significantly increases by 0.26 and 0.080 at pH = 2 and pH = 4 (Fig. 4a), respectively, when low NO concentration (e.g., 0.1 ppbv) is considered. Even under the conditions of high NO concentration (e.g., 5 ppbv), the SOA yield can increase by 0.023 and 0.018 at pH = 2 and pH = 4 (Fig. 4b), respectively. By analyzing the contribution of species to SOA, the TEPOX takes a high percentage (51.92%) at the condition of pH = 2 and 0.1 ppbv NO (see details in the ‘Analysis of sensitivity simulations’ part and Fig. S10 in the ESI†). This is consistent with its high fractional yields (56.1% for T-ROOH and 22.4% for T-RONO2) from the kinetic calculations. With increasing the pH and NO concentration, the contribution of TEPOX to SOA formation decreases, similar to the case of IEPOX.34,35 In addition, high SOA yield in low pH should mainly result from a high reaction rate of TEPOX (see box modeling details and sensitivity analysis in the ESI†). Therefore, this study uncovers a new mechanism for the formation of reactive uptake precursors that eventually connects gas-phase toluene oxidation to the SOA formation in an urban atmosphere, especially at low pH and low NO concentration.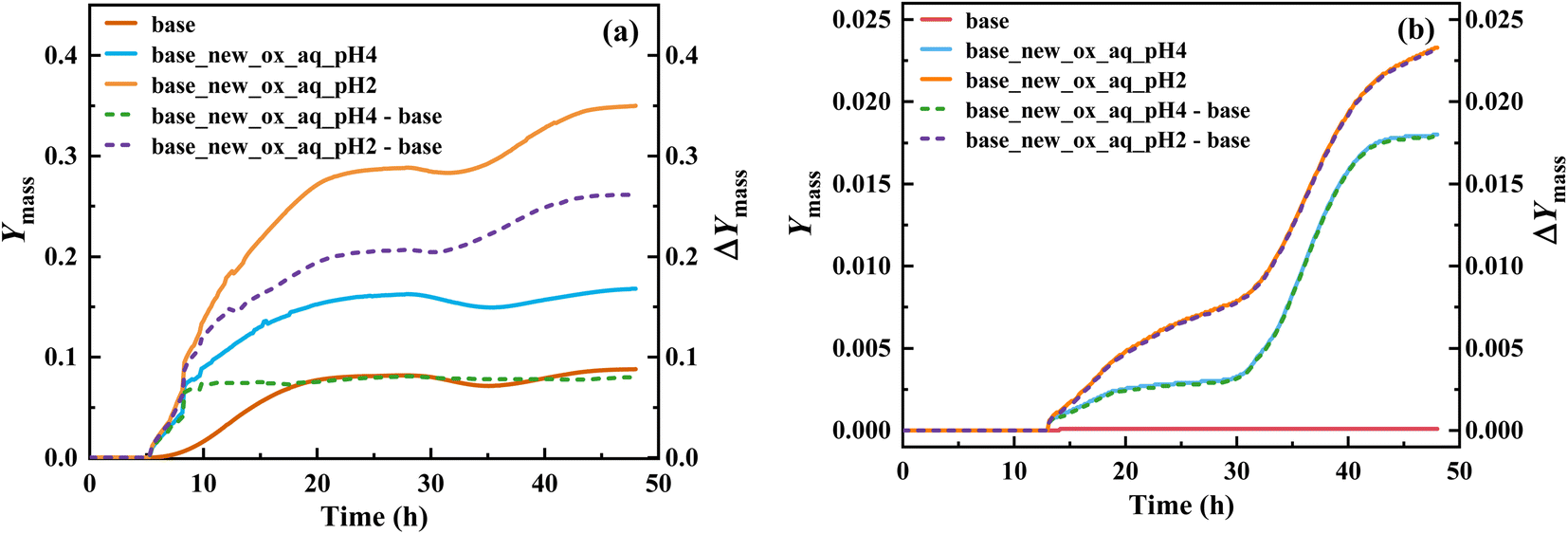 | ||
| Fig. 4 SOA mass yields (Ymass) for toluene oxidation based on the original mechanism (base) and new mechanism (base-new) and improved SOA mass yields (ΔYmass) caused by the consideration of the new mechanism at pH = 2 and pH = 4 as a function of reaction time, under low NO concentration (0.1 ppbv) (a) and high NO concentration (5 ppbv) (b) conditions. | ||
Atmospheric implication and conclusions
Our theoretical study and field observation reveal that ·OH-initiated oxidation of T-ROOH and T-RONO2, an important second-generation oxidation process of toluene, lead to the formation of ring-retaining TEPOX, and a range of dicarbonyl products. The formation of ring-retaining TEPOX, which resembles the formation of IEPOX from isoprene,9,10,79 has not been previously recognized. The formed TEPOX can form ring-retaining and non-aromatic organosulfates, organonitrates or polyols via acid-catalyzed ring-opening reactions once partitioned into the aerosol phase (see details in Figure S8†-9), similar to the heterogeneous reactions of the well-characterized IEPOX.11,35,84 Therefore, the identified TEPOX is a novel reactive uptake precursor for SOA formation. It should be noted that only aromatic organosulfates have been detected in the ambient particles via target analysis.85–87 This study suggests the existence of non-aromatic and ring-retaining AHs-derived organosulfates and organonitrates, which should be further investigated in future atmospheric measurements.The revealed mechanism can significantly lead to the SOA increase for toluene oxidation, filling the SOA gap between experiment and model prediction under the conditions of low pH and low NO concentration, especially since the frequency of low-NO conditions has increased significantly in recent years due to NOx emission controls.88 Additionally, It is known that other AHs, especially MAHs, can form AHs-derived hydroperoxides and organonitrates in atmospheric oxidation.26 Accordingly, the oxidation of other AHs could also lead to the formation of epoxides through a similar pathway as toluene oxidation, which could significantly enhance SOA formation via reactive uptakes. More importantly, the present findings fill a gap in mechanistic chemical insight between measured and simulated SOA for AH oxidation, thereby, warranting future studies on the global contribution of this new mechanism to SOA formation.
Data availability
The ESI† contains the details of box modelling; computational details for fractional yield calculation; comparison of the formation of PTH-1-1-2 and IMTH-1-1-1; discussion about the energies of RCs and TSs; all considered reaction pathways for the reactions of T-ROOH/RONO2 + ·OH; Lennard-Jones parameters of intermediates used in the MESMER simulations; effects of selection of ΔEd and grain size on the yields of important species, proportion of different dissociation forms of TEPOX as a function of pH; cartesian coordinates and electronic energies.Author contributions
H. B. X. and J. S. F. designed the study. W. N., Y. L. L. and Y. Y. L. performed the field observation and analyzed the data. P. T. Z. and L. P. performed box model simulation. Z. H. F. and F. F. M. performed the quantum chemical calculation. H. B. X., J. S. F., C. Y., D. D. H. J. W. C., J. E., A. J. D., W. N., P. T. Z., Z. H. F. and F. F. M. wrote the manuscript. All coauthors participated in relevant scientific discussions and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22176022, 22236044, 21876024, 22206020, and 22306002), the China Postdoctoral Science Foundation (2022M720640 and 2023M730054), EU H2020 project FORCeS (821205), the ACCC Flagship funded by the Academy of Finland (337549), the Autumn 2020 Arctic Avenue (spearhead research project between the University of Helsinki and Stockholm University) and Independent Research Fond Denmark (IRFD) (grant number 9064-00001B). We thank the Supercomputing Center of Dalian University of Technology and CSC-IT Center for Science, Finland for computational resources.Notes and references
- M. Glasius and A. H. Goldstein, Environ. Sci. Technol., 2016, 50, 2754–2764 CrossRef CAS PubMed.
- M. Shiraiwa, K. Ueda, A. Pozzer, G. Lammel, C. J. Kampf, A. Fushimi, S. Enami, A. M. Arangio, J. Fröhlich-Nowoisky, Y. Fujitani, A. Furuyama, P. S. J. Lakey, J. Lelieveld, K. Lucas, Y. Morino, U. Pöschl, S. Takaharna, A. Takami, H. J. Tong, B. Weber, A. Yoshino and K. Sato, Environ. Sci. Technol., 2017, 51, 13545–13567 CrossRef CAS PubMed.
- M. Shrivastava, C. D. Cappa, J. W. Fan, A. H. Goldstein, A. B. Guenther, J. L. Jimenez, C. Kuang, A. Laskin, S. T. Martin, N. L. Ng, T. Petaja, J. R. Pierce, P. J. Rasch, P. Roldin, J. H. Seinfeld, J. Shilling, J. N. Smith, J. A. Thornton, R. Volkamer, J. Wang, D. R. Worsnop, R. A. Zaveri, A. Zelenyuk and Q. Zhang, Rev. Geophys., 2017, 55, 509–559 CrossRef.
- M. Ehn, J. A. Thornton, E. Kleist, M. Sipilä, H. Junninen, I. Pullinen, M. Springer, F. Rubach, R. Tillmann, B. Lee, F. Lopez-Hilfiker, S. Andres, I. H. Acir, M. Rissanen, T. Jokinen, S. Schobesberger, J. Kangasluoma, J. Kontkanen, T. Nieminen, T. Kurtén, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard, M. Canagaratna, M. D. Maso, T. Berndt, T. Petäjä, A. Wahner, V. M. Kerminen, M. Kulmala, D. R. Worsnop, J. Wildt and T. F. Mentel, Nature, 2014, 506, 476–479 CrossRef CAS PubMed.
- G. Isaacman-VanWertz, P. Massoli, R. O'Brien, C. Lim, J. P. Franklin, J. A. Moss, J. F. Hunter, J. B. Nowak, M. R. Canagaratna, P. K. Misztal, C. Arata, J. R. Roscioli, S. T. Herndon, T. B. Onasch, A. T. Lambe, J. T. Jayne, L. P. Su, D. A. Knopf, A. H. Goldstein, D. R. Worsnop and J. H. Kroll, Nat. Chem., 2018, 10, 462–468 CrossRef CAS PubMed.
- J. H. Kroll and J. H. Seinfeld, Atmos. Environ., 2008, 42, 3593–3624 CrossRef CAS.
- A. H. Goldstein and I. E. Galbally, Environ. Sci. Technol., 2007, 41, 1514–1521 CrossRef CAS PubMed.
- C. L. Heald and J. H. Kroll, Sci. Adv., 2020, 6, eaay8967 CrossRef CAS PubMed.
- F. Paulot, J. D. Crounse, H. G. Kjaergaard, A. Kürten, J. M. St Clair, J. H. Seinfeld and P. O. Wennberg, Science, 2009, 325, 730–733 CrossRef CAS PubMed.
- Y. H. Lin, H. F. Zhang, H. O. T. Pye, Z. F. Zhang, W. J. Marth, S. Park, M. Arashiro, T. Q. Cui, H. Budisulistiorini, K. G. Sexton, W. Vizuete, Y. Xie, D. J. Luecken, I. R. Piletic, E. O. Edney, L. J. Bartolotti, A. Gold and J. D. Surratt, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6718–6723 CrossRef CAS PubMed.
- J. D. Surratt, A. W. H. Chan, N. C. Eddingsaas, M. N. Chan, C. L. Loza, A. J. Kwan, S. P. Hersey, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6640–6645 CrossRef CAS PubMed.
- L. Xu, H. Y. Guo, C. M. Boyd, M. Klein, A. Bougiatioti, K. M. Cerully, J. R. Hite, G. Isaacman-VanWertz, N. M. Kreisberg, C. Knote, K. Olson, A. Koss, A. H. Goldstein, S. V. Hering, J. de Gouw, K. Baumann, S. H. Lee, A. Nenes, R. J. Weber and N. L. Ng, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 37–42 CrossRef CAS PubMed.
- G. I. Gkatzelis, D. K. Papanastasiou, V. A. Karydis, T. Hohaus, Y. Liu, S. H. Schmitt, P. Schlag, H. Fuchs, A. Novelli, Q. Chen, X. Cheng, S. Broch, H. Dong, F. Holland, X. Li, Y. H. Liu, X. F. Ma, D. Reimer, F. Rohrer, M. Shao, Z. Tan, D. Taraborrelli, R. Tillmann, H. C. Wang, Y. Wang, Y. S. Wu, Z. J. Wu, L. M. Zeng, J. Zheng, M. Hu, K. D. Lu, A. Hofzumahaus, Y. H. Zhang, A. Wahner and A. Kiendler-Scharr, Geophys. Res. Lett., 2021, 48, e2020GL091351 CrossRef.
- S. Guo, M. Hu, Q. F. Guo, X. Zhang, M. Zheng, J. Zheng, C. C. Chang, J. J. Schauer and R. Y. Zhang, Environ. Sci. Technol., 2012, 46, 9846–9853 CrossRef CAS PubMed.
- R. Z. Tang, Z. P. Wu, X. Li, Y. J. Wang, D. J. Shang, Y. Xiao, M. R. Li, L. M. Zeng, Z. J. Wu, M. Hallquist, M. Hu and S. Guo, Atmos. Chem. Phys., 2018, 18, 4055–4068 CrossRef CAS.
- S. Yang, F. K. Duan, Y. L. Ma, H. Li, J. L. Wang, Z. Y. Du, Y. Z. Xu, T. Zhang, L. D. Zhu, T. Huang, T. Kimoto, L. F. Zhang and K. B. He, Sci. Total Environ., 2020, 746, 141209 CrossRef CAS PubMed.
- D. Cabrera-Perez, D. Taraborrelli, R. Sander and A. Pozzer, Atmos. Chem. Phys., 2016, 16, 6931–6947 CrossRef CAS.
- W. Nie, C. Yan, D. D. Huang, Z. Wang, Y. L. Liu, X. H. Qiao, Y. S. Guo, L. H. Tian, P. G. Zheng, Z. N. Xu, Y. Y. Li, Z. Xu, X. M. Qi, P. Sun, J. P. Wang, F. X. Zheng, X. X. Li, R. J. Yin, K. R. Dallenbach, F. Bianchi, T. Petäjä, Y. J. Zhang, M. Y. Wang, M. Schervish, S. N. Wang, L. P. Qiao, Q. Wang, M. Zhou, H. L. Wang, C. Yu, D. W. Yao, H. Guo, P. L. Ye, S. C. Lee, Y. J. Li, Y. C. Liu, X. G. Chi, V. M. Kerminen, M. Ehn, N. M. Donahue, T. Wang, C. Huang, M. Kulmala, D. Worsnop, J. K. Jiang and A. J. Ding, Nat. Geosci., 2022, 15, 255–261 CrossRef CAS.
- C. Bloss, V. Wagner, M. E. Jenkin, R. Volkamer, W. J. Bloss, J. D. Lee, D. E. Heard, K. Wirtz, M. Martin-Reviejo, G. Rea, J. C. Wenger and M. J. Pilling, Atmos. Chem. Phys., 2005, 5, 641–664 CrossRef CAS.
- I. Suh, R. Zhang, L. T. Molina and M. J. Molina, J. Am. Chem. Soc., 2003, 125, 12655–12665 CrossRef CAS PubMed.
- Y. M. Ji, J. Zhao, H. Terazono, K. Misawa, N. P. Levitt, Y. X. Li, Y. Lin, J. F. Peng, Y. Wang, L. Duan, B. W. Pan, F. Zhang, X. D. Feng, T. C. An, W. Marrero-Ortiz, J. Secrest, A. L. Zhang, K. Shibuya, M. J. Molina and R. Y. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8169–8174 CrossRef CAS PubMed.
- S. N. Wang, R. R. Wu, T. Berndt, M. Ehn and L. M. Wang, Environ. Sci. Technol., 2017, 51, 8442–8449 CrossRef CAS PubMed.
- U. Molteni, F. Bianchi, F. Klein, I. El Haddad, C. Frege, M. J. Rossi, J. Dommen and U. Baltensperger, Atmos. Chem. Phys., 2018, 18, 1909–1921 CrossRef CAS.
- S. N. Wang, M. J. Newland, W. Deng, A. R. Rickard, J. F. Hamilton, A. Muñoz, M. Ródenas, M. M. Vázquez, L. M. Wang and X. M. Wang, Environ. Sci. Technol., 2020, 54, 7798–7806 CrossRef CAS PubMed.
- O. Garmash, M. P. Rissanen, I. Pullinen, S. Schmitt, O. Kausiala, R. Tillmann, D. F. Zhao, C. Percival, T. J. Bannan, M. Priestley, Å. M. Hallquist, E. Kleist, A. Kiendler-Scharr, M. Hallquist, T. Berndt, G. McFiggans, J. Wildt, T. F. Mentel and M. Ehn, Atmos. Chem. Phys., 2020, 20, 515–537 CrossRef CAS.
- A. Zaytsev, A. R. Koss, M. Breitenlechner, J. E. Krechmer, K. J. Nihill, C. Y. Lim, J. C. Rowe, J. L. Cox, J. Moss, J. R. Roscioli, M. R. Canagaratna, D. R. Worsnop, J. H. Kroll and F. N. Keutsch, Atmos. Chem. Phys., 2019, 19, 15117–15129 CrossRef CAS PubMed.
- R. H. Schwantes, K. A. Schilling, R. C. McVay, H. Lignell, M. M. Coggon, X. Zhang, P. O. Wennberg and J. H. Seinfeld, Atmos. Chem. Phys., 2017, 17, 3453–3474 CrossRef CAS.
- M. Y. Wang, D. X. Chen, M. Xiao, Q. Ye, D. Stolzenburg, V. Hofbauer, P. L. Ye, A. L. Vogel, R. L. Mauldin, A. Amorim, A. Baccarini, B. Baumgartner, S. Brilke, L. Dada, A. Dias, J. Duplissy, H. Finkenzeller, O. Garmash, X. C. He, C. R. Hoyle, C. Kim, A. Kvashnin, K. Lehtipalo, L. Fischer, U. Molteni, T. Petäjä, V. Pospisilova, L. L. J. Quéléver, M. Rissanen, M. Simon, C. Tauber, A. Tomé, A. C. Wagner, L. Weitz, R. Volkamer, P. M. Winkler, J. Kirkby, D. R. Worsnop, M. Kulmala, U. Baltensperger, J. Dommen, I. El Haddad and N. M. Donahue, Environ. Sci. Technol., 2020, 54, 7911–7921 CrossRef CAS PubMed.
- Y. X. Li, J. Y. Zhao, Y. Wang, J. H. Seinfeld and R. Y. Zhang, Environ. Sci. Technol., 2021, 55, 8592–8603 CrossRef CAS PubMed.
- J. L. Xu, R. J. Griffin, Y. Liu, S. Nakao and D. R. Cocker, Atmos. Environ., 2015, 101, 217–225 CrossRef CAS.
- Q. Chen, T. M. Fu, J. L. Hu, Q. Ying and L. Zhang, Nati. Sci. Rev., 2017, 4, 806–809 CrossRef CAS.
- L. Xing, J. R. Wu, M. Elser, S. R. Tong, S. X. Liu, X. Li, L. Liu, J. J. Cao, J. M. Zhou, I. El-Haddad, R. J. Huang, M. F. Ge, X. X. Tie, A. S. H. Prévôt and G. H. Li, Atmos. Chem. Phys., 2019, 19, 2343–2359 CrossRef CAS.
- J. L. Hu, P. Wang, Q. Ying, H. L. Zhang, J. J. Chen, X. L. Ge, X. H. Li, J. K. Jiang, S. X. Wang, J. Zhang, Y. Zhao and Y. Y. Zhang, Atmos. Chem. Phys., 2017, 17, 77–92 CrossRef CAS.
- M. I. Jacobs, A. I. Darer and M. J. Elrod, Environ. Sci. Technol., 2013, 47, 12868–12876 CrossRef CAS PubMed.
- N. C. Eddingsaas, D. G. VanderVelde and P. O. Wennberg, J. Phys. Chem. A, 2010, 114, 8106–8113 CrossRef CAS PubMed.
- Y. Gao, H. Wang, X. Zhang, S. Jing, Y. Peng, L. Qiao, M. Zhou, D. D. Huang, Q. Wang, X. Li, L. Li, J. Feng, Y. Ma and Y. Li, Environ. Sci. Technol., 2019, 53, 8664–8671 CrossRef CAS PubMed.
- F. F. Ma, Z. Z. Ding, J. Elm, H. B. Xie, Q. Yu, C. Liu, C. Li, Z. Q. Fu, L. L. Zhang and J. W. Chen, Environ. Sci. Technol., 2018, 52, 9801–9809 CrossRef CAS PubMed.
- Q. Yu, H. B. Xie and J. W. Chen, Sci. Total Environ., 2016, 571, 1105–1114 CrossRef CAS PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- M. J. T. Frisch, G. W. Schlegel, H. B. Scuseria, G. E. Robb, M. A. Cheeseman, J. R. Scalmani, G. Barone, V. Mennucci, B. Petersson, G. A. Nakatsuji, H. Caricato, M. Li, X. Hratchian, H. P. Izmaylov, A. F. Bloino, J. Zheng, G. Sonnenberg, J. L. Hada, M. Ehara, M. Toyota, K. Fukuda, R. Hasegawa, J. Ishida, M. Nakajima, T. Honda, Y. Kitao, O. Nakai, H. Vreven, T. Montgomery, J. A. Peralta Jr, J. E. Ogliaro, F. Bearpark, M. Heyd, J. J. Brothers, E. Kudin, K. N. Staroverov, V. N. Kobayashi, R. Normand, J. Raghavachari, K. Rendell, A. Burant, J. C. Iyengar, S. S. Tomasi, J. Cossi, M. Rega, N. Millam, J. M. Klene, M. Knox, J. E. Cross, J. B. Bakken, V. Adamo, C. Jaramillo, J. Gomperts, R. Stratmann, R. E. Yazyev, O. Austin, A. J. Cammi, R. Pomelli, C. Ochterski, J. W. Martin, R. L. Morokuma, K. Zakrzewski, V. G. Voth, G. A. Salvador, P. Dannenberg, J. J. Dapprich, S. Daniels, A. D. Farkas, O. Foresman, J. B. Ortiz, J. V. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 Search PubMed.
- G. P. F. Wood, L. Radom, G. A. Petersson, E. C. Barnes, M. J. Frisch and J. A. Montgomery, J. Chem. Phys., 2006, 125, 094106 CrossRef PubMed.
- X. C. Zhao and L. M. Wang, J. Phys. Chem. A, 2017, 121, 3247–3253 CrossRef CAS PubMed.
- L. M. Wang, R. R. Wu and C. Xu, J. Phys. Chem. A, 2013, 117, 14163–14168 CrossRef CAS PubMed.
- R. R. Wu, S. S. Pan, Y. Li and L. M. Wang, J. Phys. Chem. A, 2014, 118, 4533–4547 CrossRef CAS PubMed.
- Z. Z. Ding, Y. Y. Yi, W. X. Wang and Q. Z. Zhang, Chemosphere, 2021, 263 Search PubMed.
- J. C. Rienstra-Kiracofe, W. D. Allen and H. F. Schaefer, J. Phys. Chem. A, 2000, 104, 9823–9840 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- D. R. Glowacki, C. H. Liang, C. Morley, M. J. Pilling and S. H. Robertson, J. Phys. Chem. A, 2012, 116, 9545–9560 CrossRef CAS PubMed.
- À. González-Lafont and J. M. Lluch, J. Mol. Struct.: THEOCHEM, 2004, 709, 35–43 CrossRef.
- J. R. Alvarez-Idaboy, N. Mora-Diez, R. J. Boyd and A. Vivier-Bunge, J. Am. Chem. Soc., 2001, 123, 2018–2024 CrossRef CAS PubMed.
- N. Mora-Diez, J. R. Alvarez-Idaboy and R. J. Boyd, J. Phys. Chem. A, 2001, 105, 9034–9039 CrossRef CAS.
- A. Galano, J. R. Alvarez-Idaboy, M. E. Ruiz-Santoyo and A. Vivier-Bunge, J. Phys. Chem. A, 2002, 106, 9520–9528 CrossRef.
- V. H. Uc, J. R. Alvarez-Idaboy, A. Galano, I. García-Cruz and A. Vivier-Bunge, J. Phys. Chem. A, 2006, 110, 10155–10162 CrossRef CAS PubMed.
- Y. Georgievskii and S. J. Klippenstein, J. Chem. Phys., 2005, 122, 194103 CrossRef PubMed.
- S. N. Wang and L. M. Wang, Phys. Chem. Chem. Phys., 2016, 18, 7707–7714 RSC.
- F. Ma, X. Guo, D. Xia, H. B. Xie, Y. Wang, J. Elm, J. Chen and J. Niu, Environ. Sci. Technol., 2021, 55, 4399–4409 CrossRef CAS PubMed.
- F. Ma, H.-B. Xie, M. Li, S. Wang, R. Zhang and J. Chen, Chemosphere, 2021, 273, 129207 CrossRef CAS PubMed.
- R. G. Gilbert and S. C. Smith, Theory of Unimolecular and Recombination Reactions, Blackwell Scientific, 1990, pp. 319 Search PubMed.
- C. Eckart, Phys. Rev., 1930, 35, 1303–1309 CrossRef CAS.
- Y. L. Liu, W. Nie, Y. Y. Li, D. F. Ge, C. Liu, Z. N. Xu, L. D. Chen, T. Y. Wang, L. Wang, P. Sun, X. M. Qi, J. P. Wang, Z. Xu, J. Yuan, C. Yan, Y. J. Zhang, D. D. Huang, Z. Wang, N. M. Donahue, D. Worsnop, X. G. Chi, M. Ehn and A. J. Ding, Atmos. Chem. Phys., 2021, 21, 14789–14814 CrossRef CAS.
- Z. N. Xu, W. Nie, Y. L. Liu, P. Sun, D. D. Huang, C. Yan, J. Krechmer, P. L. Ye, Z. Xu, X. M. Qi, C. J. Zhu, Y. Y. Li, T. Y. Wang, L. Wang, X. Huang, R. Z. Tang, S. Guo, G. L. Xiu, Q. Y. Fu, D. Worsnop, X. G. Chi and A. J. Ding, Geophys. Res. Lett., 2021, 48, e2020GL089276 CrossRef CAS.
- H. Junninen, M. Ehn, T. Petaja, L. Luosujarvi, T. Kotiaho, R. Kostiainen, U. Rohner, M. Gonin, K. Fuhrer, M. Kulmala and D. R. Worsnop, Atmos. Meas. Techn., 2010, 3, 1039–1053 CrossRef CAS.
- T. Jokinen, M. Sipilä, H. Junninen, M. Ehn, G. Lonn, J. Hakala, T. Petäjä, R. L. Mauldin, M. Kulmala and D. R. Worsnop, Atmos. Chem. Phys., 2012, 12, 4117–4125 CrossRef CAS.
- J. Tröstl, W. K. Chuang, H. Gordon, M. Heinritzi, C. Yan, U. Molteni, L. Ahlm, C. Frege, F. Bianchi, R. Wagner, M. Simon, K. Lehtipalo, C. Williamson, J. S. Craven, J. Duplissy, A. Adamov, J. Almeida, A. K. Bernhammer, M. Breitenlechner, S. Brilke, A. Dias, S. Ehrhart, R. C. Flagan, A. Franchin, C. Fuchs, R. Guida, M. Gysel, A. Hansel, C. R. Hoyle, T. Jokinen, H. Junninen, J. Kangasluoma, H. Keskinen, J. Kim, M. Krapf, A. Kürten, A. Laaksonen, M. Lawler, M. Leiminger, S. Mathot, O. Möhler, T. Nieminen, A. Onnela, T. Petäjä, F. M. Piel, P. Miettinen, M. P. Rissanen, L. Rondo, N. Sarnela, S. Schobesberger, K. Sengupta, M. Sipilä, J. N. Smith, G. Steiner, A. Tomè, A. Virtanen, A. C. Wagner, E. Weingartner, D. Wimmer, P. M. Winkler, P. L. Ye, K. S. Carslaw, J. Curtius, J. Dommen, J. Kirkby, M. Kulmala, I. Riipinen, D. R. Worsnop, N. M. Donahue and U. Baltensperger, Nature, 2016, 533, 527–531 CrossRef PubMed.
- D. Stolzenburg, L. Fischer, A. L. Vogel, M. Heinritzi, M. Schervish, M. Simon, A. C. Wagner, L. Dada, L. R. Ahonen, A. Amorim, A. Baccarini, P. S. Bauer, B. Baumgartner, A. Bergen, F. Bianchi, M. Breitenlechner, S. Brilke, S. B. Mazon, D. X. Chen, A. Dias, D. C. Draper, J. Duplissy, I. Haddad, H. Finkenzeller, C. Frege, C. Fuchs, O. Garmash, H. Gordon, X. C. He, J. Helm, V. Hofbauer, C. R. Hoyle, C. Kim, J. Kirkby, J. Kontkanen, A. Kuerten, J. Lampilahti, M. Lawler, K. Lehtipalo, M. Leiminger, H. Mai, S. Mathot, B. Mentler, U. Molteni, W. Nie, T. Nieminen, J. B. Nowak, A. Ojdanic, A. Onnela, M. Passananti, T. Petäjä, L. L. J. Quéléver, M. P. Rissanen, N. Sarnela, S. Schallhart, C. Tauber, A. Tome, R. Wagner, M. Y. Wang, L. Weitz, D. Wimmer, M. Xiao, C. Yan, P. L. Ye, Q. Z. Zha, U. Baltensperger, J. Curtius, J. Dommen, R. C. Flagan, M. Kulmala, J. N. Smith, D. R. Worsnop, A. Hansel, N. M. Donahue and P. M. Winkler, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9122–9127 CrossRef CAS PubMed.
- M. Heinritzi, M. Simon, G. Steiner, A. C. Wagner, A. Kürten, A. Hansel and J. Curtius, Atmos. Meas. Techn., 2016, 9, 1449–1460 CrossRef CAS.
- L. W. Yang, W. Nie, Y. L. Liu, Z. N. Xu, M. Xiao, X. M. Qi, Y. Y. Li, R. X. Wang, J. Zou, P. Paasonen, C. Yan, Z. Xu, J. P. Wang, C. Zhou, J. Yuan, J. N. Sun, X. G. Chi, V. M. Kerminen, M. Kulmala and A. J. Ding, Environ. Sci. Technol., 2021, 55, 6665–6676 CrossRef CAS PubMed.
- D. Chen, C. Xavier, P. Clusius, T. Nieminen, P. Roldin, X. Qi, L. Pichelstorfer, M. Kulmala, P. Rantala, J. Aalto, N. Sarnela, P. Kolari, P. Keronen, M. P. Rissanen, D. Taipale, B. Foreback, M. Baykara, P. Zhou and M. Boy, Environ. Sci.: Atmos., 2021, 1, 449–472 CAS.
- M. E. Jenkin, S. M. Saunders, V. Wagner and M. J. Pilling, Atmos. Chem. Phys., 2003, 3, 181–193 CrossRef CAS.
- M. Boy, D. Mogensen, S. Smolander, L. Zhou, T. Nieminen, P. Paasonen, C. Plass-Dülmer, M. Sipilä, T. Petäjä, L. Mauldin, H. Berresheim and M. Kulmala, Atmos. Chem. Phys., 2013, 13, 3865–3879 CrossRef.
- T. R. Wu and B. E. Boor, Atmos. Chem. Phys., 2021, 21, 8883–8914 CrossRef CAS.
- A. Massling, M. Stock, B. Wehner, Z. J. Wu, M. Hu, E. Brüggemann, T. Gnauk, H. Herrmann and A. Wiedensohler, Atmos. Environ., 2009, 43, 1578–1589 CrossRef CAS.
- M. Z. Jacobson, Aerosol Sci. Technol., 1997, 27, 491–498 CrossRef CAS.
- P. Clusius, C. Xavier, L. Pichelstorfer, P. T. Zhou, T. Olenius, P. Roldin and M. Boy, Geosci. Model Dev., 2022, 15, 7257–7286 CrossRef CAS.
- J. F. Pankow and W. E. Asher, Atmos. Chem. Phys., 2008, 8, 2773–2796 CrossRef CAS.
- U. EPA, Estimation Programs Interface Suite™ for Microsoft® Windows, v 4.11, United States Environmental Protection Agency, Washington, DC, USA, 2012 Search PubMed.
- M. Z. Jacobson, J. Geophys. Res.: Atmos., 2002, 107, D19 Search PubMed.
- M. I. Jacobs, W. J. Burke and M. J. Elrod, Atmos. Chem. Phys., 2014, 14, 8933–8946 CrossRef.
- J. M. St Clair, J. C. Rivera-Rios, J. D. Crounse, H. C. Knap, K. H. Bates, A. P. Teng, S. Jørgensen, H. G. Kjaergaard, F. N. Keutsch and P. O. Wennberg, Phys. Chem. A, 2016, 120, 1441–1451 CrossRef CAS PubMed.
- S. S. Pan and L. M. Wang, J. Phys. Chem. A, 2014, 118, 10778–10787 CrossRef CAS PubMed.
- D. R. Glowacki, L. M. Wang and M. J. Pilling, J. Phys. Chem. A, 2009, 113, 5385–5396 CrossRef CAS PubMed.
- A. J. Ding, W. Nie, X. Huang, X. G. Chi, J. N. Sun, V.-M. Kerminen, Z. Xu, W. D. Guo, T. Petäjä, X. Q. Yang, M. Kulmala and C. B. Fu, Front. Environ. Sci. Eng., 2016, 10, 15 CrossRef.
- I. R. Piletic, E. O. Edney and L. J. Bartolotti, Phys. Chem. Chem. Phys., 2013, 15, 18065–18076 RSC.
- S. Kundu, T. A. Quraishi, G. Yu, C. Suarez, F. N. Keutsch and E. A. Stone, Atmos. Chem. Phys., 2013, 13, 4865–4875 CrossRef.
- S. Staudt, S. Kundu, H. J. Lehmler, X. R. He, T. Q. Cui, Y. H. Lin, K. Kristensen, M. Glasius, X. L. Zhang, R. J. Weber, J. D. Surratt and E. A. Stone, Atmos. Environ., 2014, 94, 366–373 CrossRef CAS PubMed.
- J. Z. Song, M. J. Li, B. Jiang, S. Y. Wei, X. J. Fan and P. A. Peng, Environ. Sci. Technol., 2018, 52, 2575–2585 CrossRef CAS PubMed.
- W. Nie, C. Yan, L. W. Yang, P. Roldin, Y. L. Liu, A. L. Vogel, U. Molteni, D. Stolzenburg, H. Finkenzeller, A. Amorim, F. Bianchi, J. Curtius, L. Dada, D. C. Draper, J. Duplissy, A. Hansel, X. C. He, V. Hofbauer, T. Jokinen, C. Kim, K. Lehtipalo, L. Nichman, R. L. Mauldin, V. Makhmutov, B. Mentler, A. Mizelli-Ojdanic, T. Petäjä, L. L. J. Quééver, S. Schallhart, M. Simon, C. Tauber, A. Tomé, R. Volkamer, A. C. Wagner, R. Wagner, M. Y. Wang, P. L. Ye, H. Y. Li, W. Huang, X. M. Qi, S. J. Lou, T. Y. Liu, X. G. Chi, J. Dommen, U. Baltensperger, I. El Haddad, J. Kirkby, D. Worsnop, M. Kulmala, N. M. Donahue, M. Ehn and A. J. Ding, Nat. Commun., 2023, 14, 3347 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03638c |
| ‡ Z. H. F. and F. F. M. contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |