
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering a molecular ruthenium catalyst into three-dimensional metal covalent organic frameworks for efficient water oxidation†
Wang-Kang
Han
a,
Yong
Liu
a,
Jing-Dong
Feng
a,
Xiaodong
Yan
 a,
Huan
Pang
b and
Zhi-Guo
Gu
*a
a,
Huan
Pang
b and
Zhi-Guo
Gu
*a
aKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, China. E-mail: zhiguogu@jiangnan.edu.cn
bSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou 225002, China
First published on 5th October 2023
Abstract
The water oxidation reaction plays an important role in clean energy conversion, utilization, and storage, but mimicking the oxygen-evolving complex of photosystem II for designing active and stable water oxidation catalysts (WOCs) is still an appealing challenge. Here, we innovatively engineered a molecular ruthenium WOC as a metal complex building unit to construct a series of three-dimensional metal covalent organic frameworks (3D MCOFs) for realizing efficient oxidation catalysis. The resultant MCOFs possessed rare 3D interlocking structures with inclined interpenetration of two-dimensional covalent rhombic nets, and the Ru sites were periodically arranged in the crystalline porous frameworks. Impressively, these MCOFs showed excellent performance towards water oxidation (the O2 evolution rate is as high as 2830 nmol g−1 s−1) via the water nucleophilic attack pathway. Besides, the MCOFs were also reactive for oxidizing organic substrates. This work highlights the potential of MCOFs as a designable platform in integrating molecular catalysts for various applications.
Introduction
Water oxidation is the primary and key step of both natural and artificial photosynthesis.1–3 It is also the half reaction in schemes of solar fuel production, since water oxidation supplies electrons and protons for carrying out H+ reduction to hydrogen or CO2 reduction to a reduced form of carbon.4–6 However, the oxidation of water to molecular oxygen requires the transfer of 4e−/4H+ along with the cleavage of the O–H bond and the formation of the O–O bond, leading to high energy barriers and sluggish kinetics.7,8 It is thus highly desired to explore efficient water oxidation catalysts (WOCs) that are able to perform this reaction at low-energy pathways. Of note is that, in the natural photosystem II (PS II), the water oxidation occurs through first storing four oxidizing equivalents into the oxygen-evolving complex (OEC) featuring the Mn4CaO5 reaction center and then catalyzing the O–O bond formation.9–11 Inspired by the OEC in PS II, great efforts have been devoted to the investigation of molecular and supramolecular WOCs being structurally and functionally analogous to the Mn4CaO5 cluster.12–16In particular, a significant number of Ru coordination complexes have been identified as effective WOCs, some of which are comparable to that of nature.17–21 Unfortunately, homogeneous molecular Ru-WOCs often suffer from instability and difficulty in recycling. In OEC-PS II, the above challenge was elegantly solved by embedding the Mn4CaO5 cluster into proteinic environments for effectively protecting the oxygen-evolving complex from degradation.11 Hence, it is believed that the combination of active molecular WOCs and stable supports could help enhance the durability of the catalysts. To achieve this goal, two main strategies have been explored. One is based on the post-synthetic immobilization of molecular WOCs onto a heterogeneous support such as the metal organic frameworks.22,23 Another is a host–guest approach to encapsulate molecular WOCs in a porous host.24–26 However, in the aforementioned integrated system, the amount and position of the catalytic centers cannot be precisely controlled, and the active sites are often poorly accessible. In this context, the development of newly heterogeneous materials for effective water oxidation is strongly desired.
To design comprehensively heterogeneous WOCs, three key elements should be considered: (1) stable framework, (2) H2O capture, and (3) chemical conversion of H2O (Scheme 1a). In this regard, metal covalent organic frameworks (MCOFs) with a tailorable structure, permanent porosity and modular functionality are attractive candidates, which have recently emerged as a new class of crystalline functional materials for a variety of applications.27–29 Conceivably, upon engineering molecular WOCs, such as Ru-based polypyridine complexes, as the building units for MCOFs, the active WOC modules can be integrated into crystalline frameworks for combining the following advantages (Scheme 1b): (i) structural controllability: the active modules can be predictably arranged into the architecture; (ii) increased stability: the catalytic units are stabilized in the covalent rigid skeleton, and prevent disadvantageous aggregation; (iii) abundant porosity: the interconnected channels contribute to the exposure of active sites as well as for better diffusion of substrates; (iv) effective recyclability: the heterogeneous nature allows for catalyst separation and recycling. To realize this appealing strategy, the functionality, stability, geometry and connectivity of the engineered building unit have to be considered together, which is still fraught with challenges and never been realized in MCOFs.
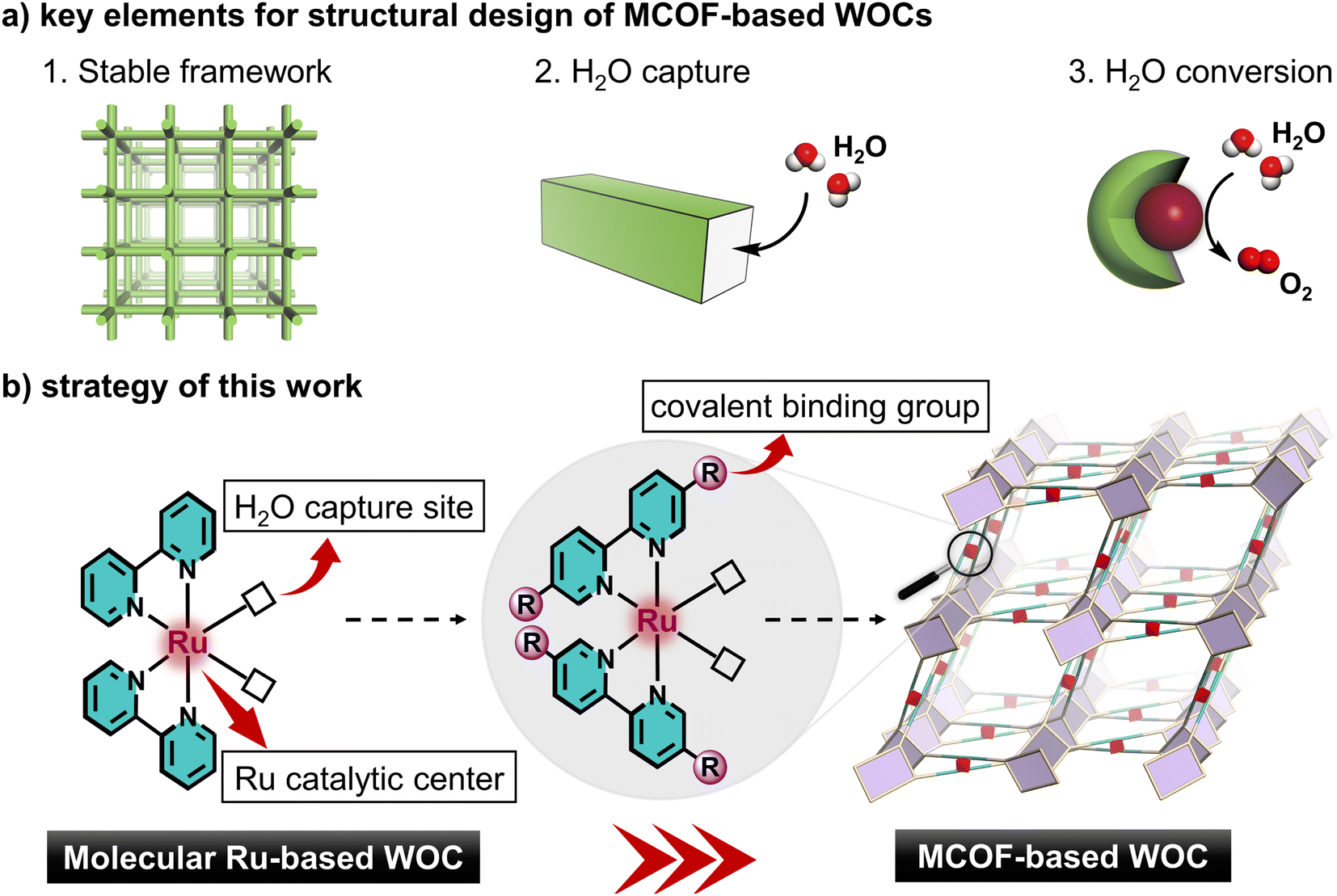 | ||
| Scheme 1 Illustration of (a) key elements for structural design of MCOF-based WOCs and (b) the strategy of this work for engineering a molecular ruthenium WOC as a metal complex building unit to construct MCOF-based WOCs. | ||
Results and discussion
Synthesis and characterization
With this in mind, we synthesized an aldehyde-functionalized Ru(II) complex, namely Ru(bpy-CHO)2Cl2, as a 4-connected metal complex building unit to construct MCOFs (Fig. 1 and S1–S5, ESI†). The single crystal X-ray structure of Ru(bpy-CHO)2Cl2 showed that the two linear bipyridine derived ligands were posited in two approximately parallel geometrical planes. In a top view, the intersection angle between the two linear bipyridine derived units is 60.3° with a distorted rectangular shape (Fig. S6, ESI†). The model reaction of condensing Ru(bpy-CHO)2Cl2 with aniline yielded imine product Ru(bpy-Ph)2Cl2, which was confirmed by ESI-MS (Fig. S7 and S8, ESI†) and single crystal X-ray diffraction (Fig. 1 and S9, ESI†). This model reaction also suggested that the Ru(bpy-CHO)2Cl2 building unit is stable under solvothermal conditions for the synthesis of MCOFs. Ru(bpy-CHO)2Cl2 was then reacted with amine linkers ETTA and ETTBA, respectively, in a solvent mixture of 1,4-dioxane, mesitylene, acetonitrile and acetic acid at 120 °C for 5 days, to form two crystalline products, namely RuCOF-100 and RuCOF-101 (Fig. 1).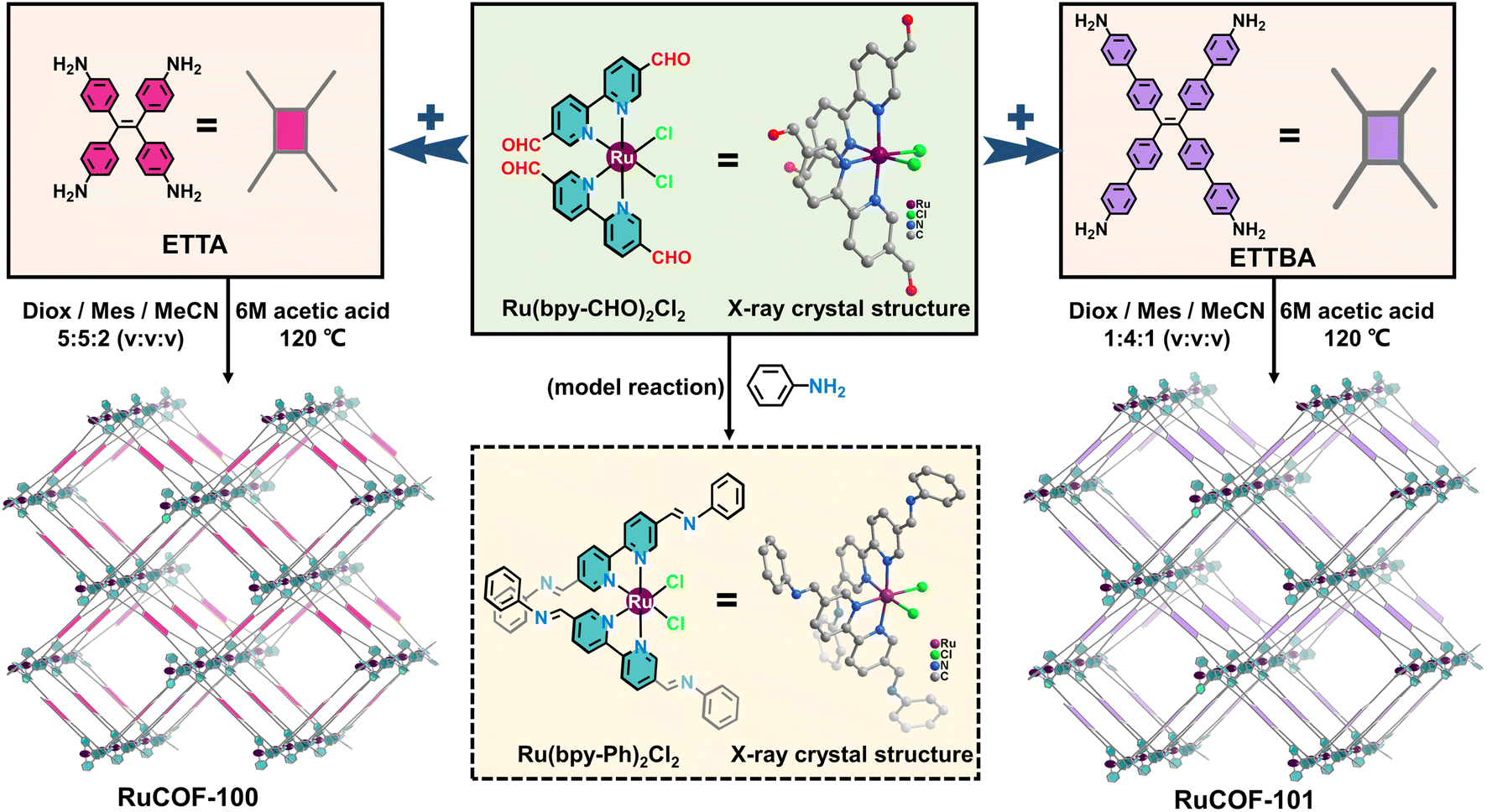 | ||
| Fig. 1 The synthetic procedures for model compounds Ru(bpy-Ph)2Cl2, RuCOF-100 and RuCOF-101, respectively. The corresponding X-ray single crystal structures of metal complex building unit Ru(bpy-CHO)2Cl2 and model compound Ru(bpy-Ph)2Cl2 are shown, in which all H atoms and solvent molecules have been removed for clarity. | ||
Fourier transform infrared and 13C cross-polarization magic-angle-spinning NMR spectroscopies confirmed the formation of imine linkages (Fig. S10–S13, ESI†). The resultant RuCOFs were stable in acid and base solutions, and common organic solvents (Fig. S14 and S15, ESI†). Thermogravimetric analysis showed a weight loss of less than 5% below 350 °C under a N2 atmosphere (Fig. S16, ESI†). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) displayed uniform nanoparticles of the RuCOFs (Fig. 2a and b). High-resolution TEM images revealed their crystalline feature with clear lattice fringes (Fig. S17 and S18, ESI†). Elemental mapping images and X-ray photoelectron spectroscopies confirmed the elemental compositions of C, N, Ru and Cl in the RuCOFs (Fig. 2a, b, S19 and S20, ESI†). The ICP analysis showed that the Ru contents were 10.87 and 8.01 wt% for RuCOF-100 and RuCOF-101, respectively, which were close to their theoretical values of 11.02 and 8.27 wt%.
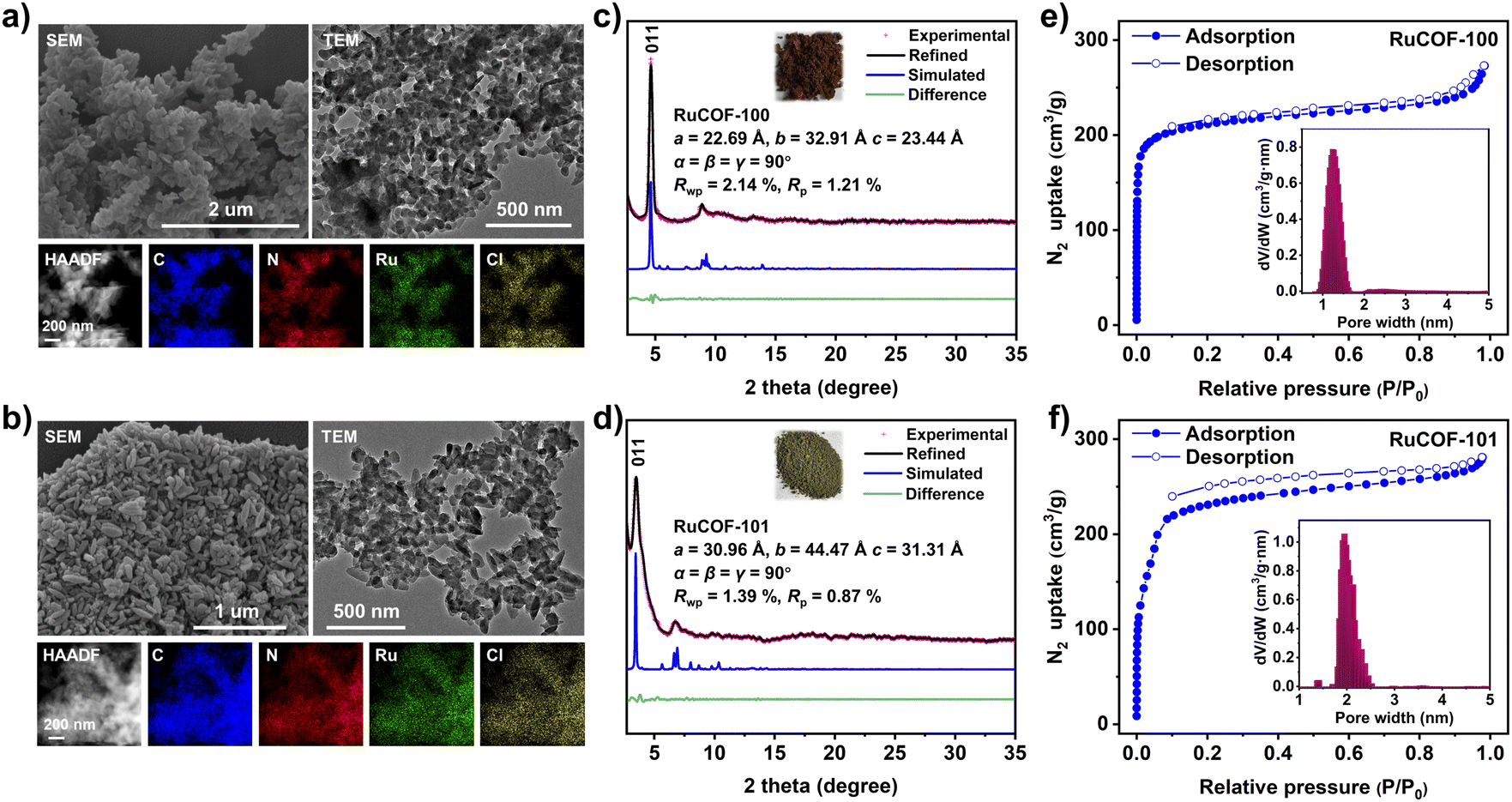 | ||
| Fig. 2 The SEM, TEM and HAADF-STEM with corresponding elemental mapping images of (a) RuCOF-100 and (b) RuCOF-101. The PXRD plots of (c) RuCOF-100 and (d) RuCOF-101 (Insets: the powder samples and unit cell parameters of corresponding RuCOFs). N2 adsorption/desorption isotherms with the pore size distributions of (e) RuCOF-100 and (f) RuCOF-101. | ||
The crystalline structures of RuCOFs were explored by powder X-ray diffraction (PXRD) analysis together with theoretical simulation. The experimental diffraction patterns exhibited intense main (011) Bragg diffraction peaks of RuCOF-100 and RuCOF-101 at 2θ = 4.64° and 3.45° (Fig. 2c and d), respectively, followed by a series of less intense peaks, which differed greatly from that of the starting monomers (Fig. S21 and S22, ESI†), indicative of the highly crystalline nature of the newly formed frameworks. Subsequently, several possible two-dimensional (2D) and three-dimensional (3D) structural models were built on the basis of the geometry and connectivity of the building units (Fig. S23–S28, ESI†). It was found that the experimental PXRD results matched well with the simulated patterns obtained from the lvt topology. Both Pawley and Rietveld refinements were carried out against the experimental PXRD data to refine the structures in the Pnc2 space group, respectively, providing the unit cell parameters with good agreement factors (Fig. 2c, d, S29 and S30, ESI†). Moreover, the structural regularities were also supported by small-angle X-ray scattering (SAXS) experiments, in which the SAXS patterns were consistent with the PXRD results (Fig. S31 and S32, ESI†).
According to the structural model, the two isostructural RuCOFs were formed by covalently stitching together Ru(bpy-CHO)2Cl2 and quadrilateral tetramine linkers with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N linkages (Fig. 3 and S33, ESI†). RuCOF-101 was selected as a representative to illustrate the unique structure. The Ru(II) center is surrounded by two linear bipyridine derived ligands and can be regarded as a 4-connected node, while the ETTBA unit can be considered as another 4-connected node (Fig. 3a). Every Ru(II) building unit is linked with four ETTBA units, and every ETTBA unit is also linked to four Ru(II) units, forming a 3D metalated framework with an overall lvt topology. The rhombic one-dimensional (1D) channels along the a-axis with dimensions of 21 × 21 Å2 are formed (Fig. 3b). Interestingly, the covalent components of RuCOF-101 have a rare interlocking structure with inclined interpenetration of 2D nets. The 2D covalent rhombic grids with sql topology are formed by stitching together linear bipyridine derived units and quadrilateral ETTBA linkers. Meanwhile, another set of chemically equivalent 2D sql layers thread through the wide opening of the rhombic windows, resulting in the two set of layers entangled together (Fig. 3c and e). The dihedral angle between the two sets of interdigitated rhombic grids is around 64° (Fig. 3d). The Ru(II) ions serve as points of registry to direct the mechanical interdigitation (Fig. 3e). Finally, the 3D framework with an inclined interpenetrated network is formed (Fig. 3d and f). Notably, the well-constructed porous framework provides facile pathways for substrate diffusion. Meanwhile, all the Ru sites are placed in a beneficial manner with geometrical orientation to the inside of channels, making the Ru sites easily accessible for substrate binding.
N linkages (Fig. 3 and S33, ESI†). RuCOF-101 was selected as a representative to illustrate the unique structure. The Ru(II) center is surrounded by two linear bipyridine derived ligands and can be regarded as a 4-connected node, while the ETTBA unit can be considered as another 4-connected node (Fig. 3a). Every Ru(II) building unit is linked with four ETTBA units, and every ETTBA unit is also linked to four Ru(II) units, forming a 3D metalated framework with an overall lvt topology. The rhombic one-dimensional (1D) channels along the a-axis with dimensions of 21 × 21 Å2 are formed (Fig. 3b). Interestingly, the covalent components of RuCOF-101 have a rare interlocking structure with inclined interpenetration of 2D nets. The 2D covalent rhombic grids with sql topology are formed by stitching together linear bipyridine derived units and quadrilateral ETTBA linkers. Meanwhile, another set of chemically equivalent 2D sql layers thread through the wide opening of the rhombic windows, resulting in the two set of layers entangled together (Fig. 3c and e). The dihedral angle between the two sets of interdigitated rhombic grids is around 64° (Fig. 3d). The Ru(II) ions serve as points of registry to direct the mechanical interdigitation (Fig. 3e). Finally, the 3D framework with an inclined interpenetrated network is formed (Fig. 3d and f). Notably, the well-constructed porous framework provides facile pathways for substrate diffusion. Meanwhile, all the Ru sites are placed in a beneficial manner with geometrical orientation to the inside of channels, making the Ru sites easily accessible for substrate binding.
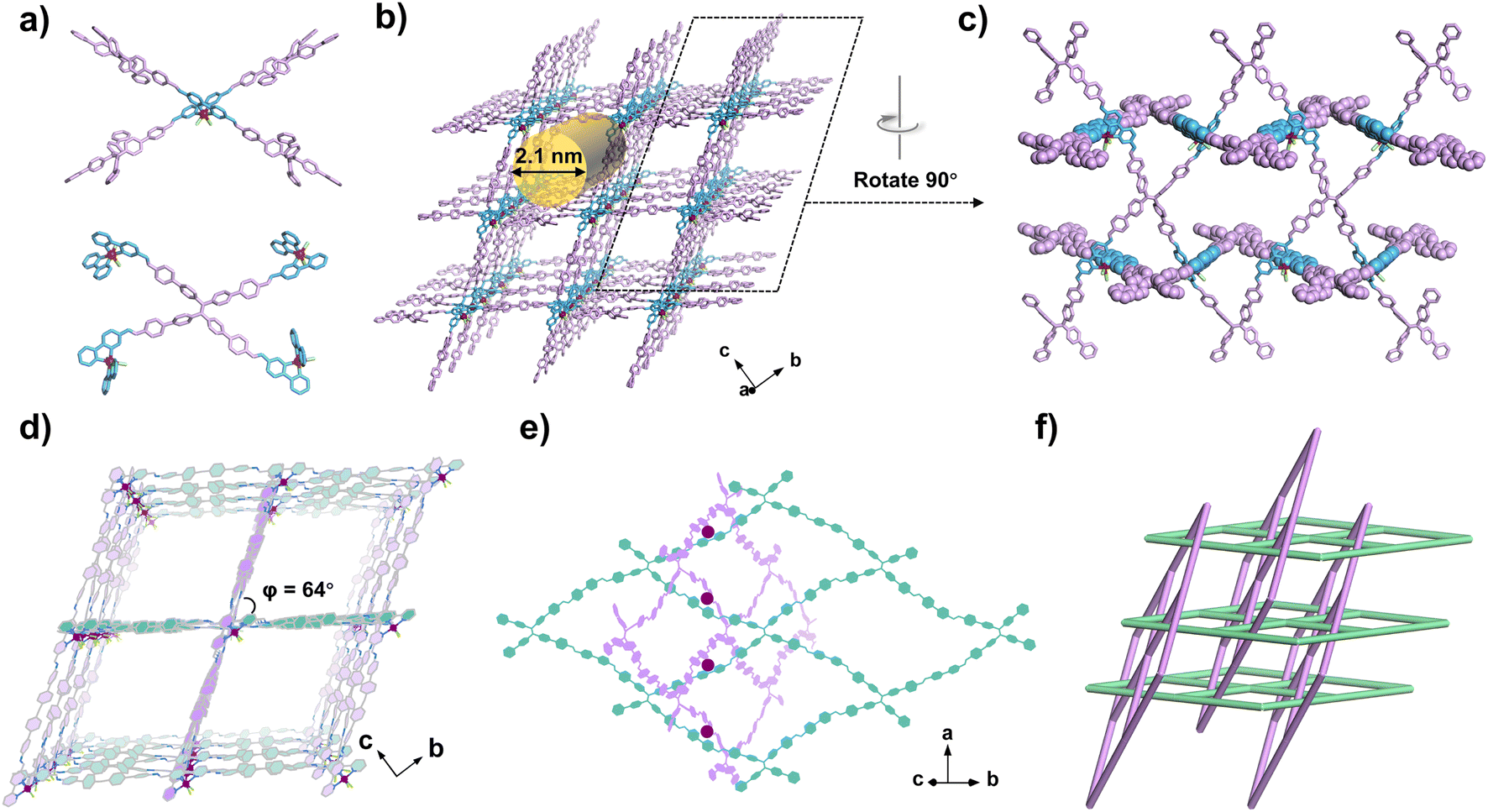 | ||
| Fig. 3 The structure of RuCOF-101: (a) Linking Ru(bpy-CHO)2Cl2 and ETTBA with a 4,4-connected pattern, (b) affording the formation of a 3D framework. (c) Views showing the inclined interpenetration of the covalent components. (d) Overall 3D framework showing the interlocking of 2D networks, where the 2D networks are represented in green and pink, respectively. (e) Structural representation of two 2D sql layers threaded through the wide opening of the rhombic windows with the Ru(II) ions as templates. (f) The corresponding 3D topology, highlighting the interdigitation of 2D networks. | ||
The porosity of the RuCOFs was then assessed by N2 adsorption–desorption measurements, showing that the Brunauer–Emmett–Teller (BET) surface areas of RuCOF-100 and RuCOF-101 were 825.38 and 973.79 m2 g−1, respectively (Fig. 2e and f). Nonlocal density functional theory calculation provided pore sizes of about 1.28 nm for RuCOF-100 and 1.96 nm for RuCOF-101, which are consistent with their theoretical pore diameters (Fig. S34 and S35, ESI†).
Catalytic water oxidation reaction
Encouraged by the highly crystalline nature, accessible pores, and abundant Ru sites, we evaluated the catalytic performance of RuCOFs towards the water oxidation reaction. The O2 evolution was first assessed by using the Ce(IV) oxidant at different concentration of RuCOFs. Unfortunately, RuCOF-100 and RuCOF-101 showed negligible catalytic activities. It was speculated that the coordinated Cl was sluggish to water molecule substitution, which restricted the availability of water molecules around Ru centers. In order to improve the interaction of the Ru center with water molecules, the Cl ligands were then removed to produce RuCOF-100′ and RuCOF-101′. PXRD analysis confirmed the retention of the crystallinity after the substitution of Cl ligands (Fig. S36 and S37, ESI†). The water contact tests showed that both of RuCOF-100′ and RuCOF-101′ have enhanced hydrophilicities compared to the Cl coordinated counterparts, promoting their good dispersibility in water (Fig. S38, ESI†). Significantly enhanced oxygen-releasing activities were then observed for both of RuCOF-100′ and RuCOF-101′ (Fig. 4a and b). The plots of initial rates versus RuCOF concentrations revealed first-order kinetics of 2305 nmol g−1 s−1 for RuCOF-100′ and 2830 nmol g−1 s−1 for RuCOF-101′ (Fig. 4c), suggesting that the water oxidation follows a water nucleophilic attack (WNA) mechanism.17 And the H/D kinetic isotope effect (KIE) experiments further provided evidence for this WNA pathway, which showed the reduced catalytic performance in heavy water with the KIE quotient around 2.5 (Fig. 4d, S39 and S40, ESI†).17,19 The turnover frequency (TOF) of RuCOF-100′ and RuCOF-101′ was estimated to be 0.0021 s−1 and 0.0034 s−1, respectively, comparable to that of some molecular Ru catalysts (Fig. S41 and Table S3, ESI†). Moreover, the catalytic activity and structural integrity of RuCOFs were well retained after five consecutive cycles (Fig. S43 and S44, ESI†).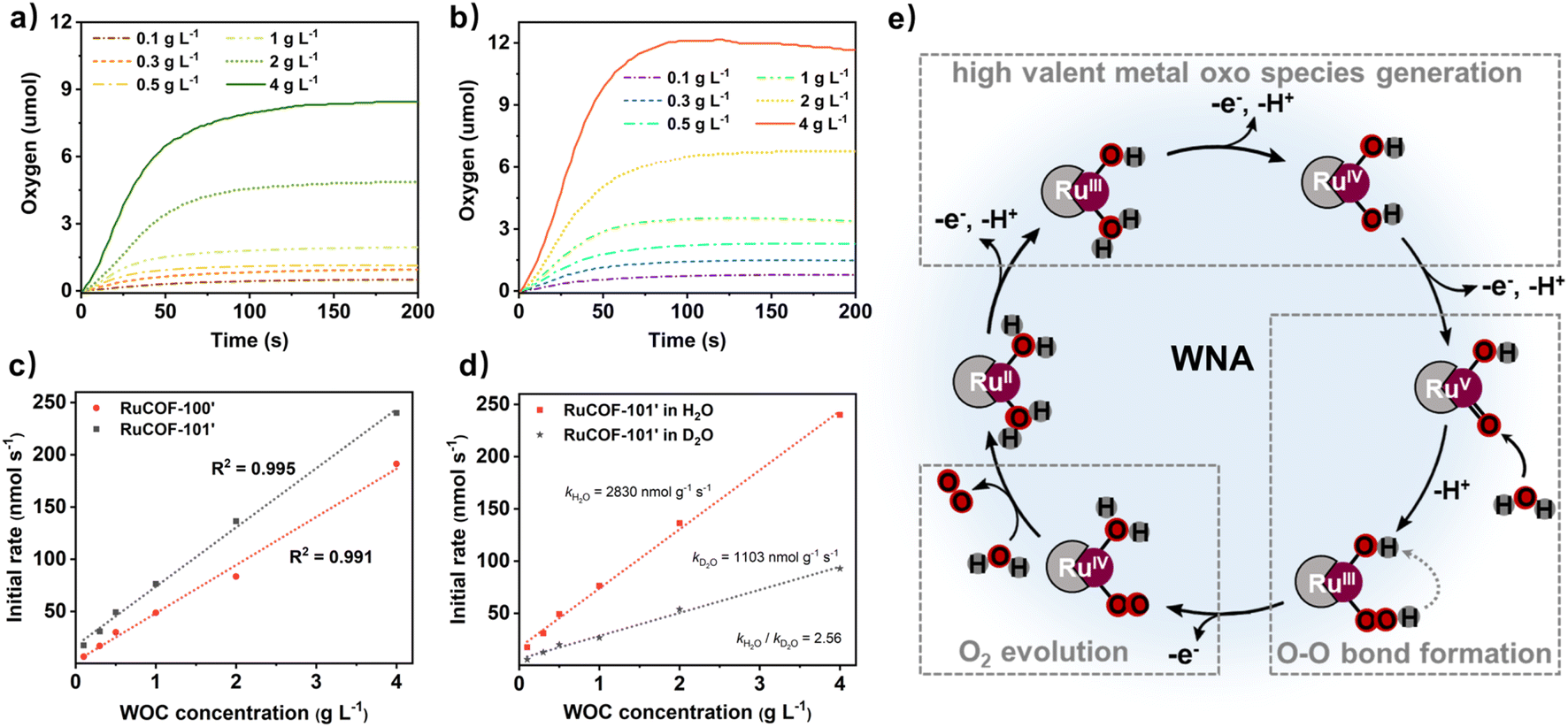 | ||
| Fig. 4 Chemical water oxidation catalysis: the time-dependent oxygen evolution of (a) RuCOF-100′ and (b) RuCOF-101′ with varying catalyst concentrations in 20 mL aqueous pH = 1 solution. (c) Plot of initial catalytic rates (obtained by linear fitting between 10 and 25 s of the oxygen evolution curve) versus RuCOF concentration with corresponding linear regression fit. (d) The kinetic isotope effect experiments for RuCOF-101′ in H2O and D2O, respectively, showing the plot of initial catalytic rates versus RuCOF concentration with corresponding linear regression fit. (e) The proposed mechanism for water oxidation with RuCOF-based WOCs. | ||
Based on our experimental results and the related literature,17–21 a possible WNA reaction pathway was proposed (Fig. 4e, S45 and S46, ESI†). Catalysis initially starts with Ru(II) that is stepwise oxidized to a high valent Ru(V)O intermediate. The electrochemical properties can verify this redox process. Three consecutive oxidation peaks versus a normal hydrogen electrode (NHE) at +0.60, +0.95, and +1.20 V and +0.61, +0.97, and +1.26 V were measured for RuCOF-100′ and RuCOF-101′, respectively, which are assignable to the formal oxidation of Ru2+/3+, Ru3+/4+, and Ru4+/5+ (Fig. S45, ESI†).20,21 Then, the nucleophilic attack of a water molecule on the reactive Ru(V)O species occurs, forming the ruthenium hydroperoxo intermediate Ru(III)–O–OH that further undergoes a proton-coupled electron transfer process to form Ru(IV)–OO. This subsequently releases dioxygen, and coordinates substrate water molecules, consequently subjecting to the next catalytic cycle. Furthermore, the density functional theory (DFT) calculations were performed to deeply understand the catalytic mechanism. The DFT calculation predicted a Ru–O distance of 1.753 Å for the proposed intermediate, which is consistent with the bond distance of Ru(V)O reported in the literature (about 1.7 Å).30 Besides, the calculated result suggested that the O–O bond formation is the rate-determining step of the reaction, as judged by its largest energy barrier of 66.38 kcal mol−1 (Fig. S47, S48 and Table S4†).
As a more sustainable alternative, the RuCOFs were then applied to photocatalytic water oxidation in the presence of Ru(bpy)32+ as a photosensitizer (PS) and Na2S2O8 as a sacrificial electron acceptor (Fig. 5a). The photocatalytic activities of the RuCOFs were observed even at a very low catalyst loading of 0.05 g L−1 (Fig. 5b and c). Again, the linear dependency of oxygen production on catalyst concentration was obtained, providing evidence for first-order kinetics with reaction rate constants of 315 and 425 nmol g−1 s−1 for RuCOF-100′ and RuCOF-101′, respectively (Fig. 5d). These findings point out that the photocatalytic water oxidation follows a similar WNA mechanism for the O–O bond formation. Notably, the photocatalytic water oxidation rates of RuCOF-100′ and RuCOF-101′ outperform most of the reported COF-based photocatalysts to date (Fig. 5e and Table S5, ESI†), such as BtB-COF31 (185 nmol g−1 s−1), Pt@TpBpy-NS32 (17.8 nmol g−1 s−1) and g-C54N6-COF33 (14.2 nmol g−1 s−1). Control experiments showed no oxygen release in the absence of either RuCOF catalysts or light or PS. Besides, the photocatalytic activity of the RuCOFs shows only a slight change after five runs (Fig. S49 and S50, ESI†).
 | ||
Fig. 5 Photocatalytic water oxidation: (a) scheme of the photocatalytic cycle of water oxidation. The time-dependent photocatalytic oxygen evolution of (b) RuCOF-100′ and (c) RuCOF-101′ with varying catalyst concentrations in 20 mL 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 CH3CN/aqueous phosphate buffer solution (pH = 7). Irradiation started at t = 30 s. (d) Plot of initial catalytic rates (obtained by linear fitting between 90 and 110 s of the oxygen evolution curve) versus RuCOF concentration with corresponding linear regression fit. (e) The comparison of photocatalytic O2 evolution activity of RuCOF-based WOCs in this work with reported representative COF-based WOCs. :7 CH3CN/aqueous phosphate buffer solution (pH = 7). Irradiation started at t = 30 s. (d) Plot of initial catalytic rates (obtained by linear fitting between 90 and 110 s of the oxygen evolution curve) versus RuCOF concentration with corresponding linear regression fit. (e) The comparison of photocatalytic O2 evolution activity of RuCOF-based WOCs in this work with reported representative COF-based WOCs. | ||
Additionally, to demonstrate the general applicability of our strategy for integrating a molecular ruthenium catalyst into MCOFs, the electrochemical water oxidation was performed (Fig. S51, ESI†). Moreover, the catalytic oxidation of organic substrates was also investigated. The C–H bond is one of the most common bonds in organic compounds, and the activation of saturated C–H bonds is challenging.34,35 Herein, it was found that the as-synthesized RuCOFs were robust catalysts toward selective oxidation of sp3 C–H bonds to carbonyls with considerable activities (Section 5, ESI†).
Conclusions
In conclusion, engineering a molecular Ru catalyst as an integral subcomponent for the construction of functional MCOFs was illustrated, and the type of metal complex building unit for structural enriching of the MCOF family was further expanded. The resultant Ru-based MCOFs were shown to be catalytically active and recyclable heterogeneous catalysts for water oxidation as well as organic transformation. Importantly, the modular and designable nature of MCOFs should allow the integration of other molecular catalysts for various applications.Data availability
Essential data are provided in the main text and the ESI.† Data can be available from the corresponding author upon reasonable request.Author contributions
Z.-G. G. and H. P supervised the project. Z.-G. G. and W.-K. H. conceived and designed the project. W.-K. H., Y. L. and J.-D. F. conducted the experiments. W.-K. H. X. D. Y. and Z.-G. G. analysed the data and wrote the paper. All the authors discussed the results and contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22075108 and 21905116), the Natural Science Foundation of Jiangsu Province (BK20190614), and the Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (2022B01).References
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- D. K. Dogutan and D. G. Nocera, Acc. Chem. Res., 2019, 52, 3143–3148 CrossRef CAS PubMed.
- E. A. Reyes Cruz, D. Nishiori, B. L. Wadsworth, N. P. Nguyen, L. K. Hensleigh, D. Khusnutdinova, A. M. Beiler and G. F. Moore, Chem. Rev., 2022, 122, 16051–16109 CrossRef CAS PubMed.
- X. Tao, Y. Zhao, S. Wang, C. Li and R. Li, Chem. Soc. Rev., 2022, 51, 3561–3608 RSC.
- S. L. Meng, C. Ye, X. B. Li, C. H. Tung and L. Z. Wu, J. Am. Chem. Soc., 2022, 144, 16219–16231 CrossRef CAS PubMed.
- J. H. Kim, D. Hansora, P. Sharma, J. W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- Y. Fang, Y. Hou, X. Fu and X. Wang, Chem. Rev., 2022, 122, 4204–4256 CrossRef CAS PubMed.
- J. Lloret-Fillol and M. Costas, Nat. Energy, 2016, 1, 16023 CrossRef CAS.
- P. Greife, M. Schonborn, M. Capone, R. Assuncao, D. Narzi, L. Guidoni and H. Dau, Nature, 2023, 617, 623–628 CrossRef CAS PubMed.
- A. Bhowmick, R. Hussein, I. Bogacz, P. S. Simon, M. Ibrahim, R. Chatterjee, M. D. Doyle, M. H. Cheah, T. Fransson, P. Chernev, I. S. Kim, H. Makita, M. Dasgupta, C. J. Kaminsky, M. Zhang, J. Gatcke, S. Haupt, I. I. Nangca, S. M. Keable, A. O. Aydin, K. Tono, S. Owada, L. B. Gee, F. D. Fuller, A. Batyuk, R. Alonso-Mori, J. M. Holton, D. W. Paley, N. W. Moriarty, F. Mamedov, P. D. Adams, A. S. Brewster, H. Dobbek, N. K. Sauter, U. Bergmann, A. Zouni, J. Messinger, J. Kern, J. Yano and V. K. Yachandra, Nature, 2023, 617, 629–636 CrossRef CAS PubMed.
- J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175–4205 CrossRef CAS PubMed.
- M. D. Karkas, O. Verho, E. V. Johnston and B. Akermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed.
- R. Matheu, P. Garrido-Barros, M. Gil-Sepulcre, M. Z. Ertem, X. Sala, C. Gimbert-Surinach and A. Llobet, Nat. Rev. Chem, 2019, 3, 331–341 CrossRef CAS.
- M. Kondo, H. Tatewaki and S. Masaoka, Chem. Soc. Rev., 2021, 50, 6790–6831 RSC.
- M. Gil-Sepulcre and A. Llobet, Nat. Catal., 2022, 5, 79–82 CrossRef CAS.
- N. Vereshchuk, M. Gil-Sepulcre, A. Ghaderian, J. Holub, C. Gimbert-Surinach and A. Llobet, Chem. Soc. Rev., 2023, 52, 196–211 RSC.
- C. Casadevall, V. Martin-Diaconescu, W. R. Browne, S. Fernandez, F. Franco, N. Cabello, J. Benet-Buchholz, B. Lassalle-Kaiser and J. Lloret-Fillol, Nat. Chem., 2021, 13, 800–804 CrossRef CAS PubMed.
- B. Zhang and L. Sun, J. Am. Chem. Soc., 2019, 141, 5565–5580 CrossRef CAS PubMed.
- M. Schulze, V. Kunz, P. D. Frischmann and F. Wurthner, Nat. Chem., 2016, 8, 576–583 CrossRef CAS PubMed.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- N. F. Suremann, B. D. McCarthy, W. Gschwind, A. Kumar, B. A. Johnson, L. Hammarstrom and S. Ott, Chem. Rev., 2023, 123, 6545–6611 CrossRef CAS PubMed.
- R. Ezhov, A. Karbakhsh Ravari, A. Page and Y. Pushkar, ACS Catal., 2020, 10, 5299–5308 CrossRef CAS.
- G. Paille, M. Gomez-Mingot, C. Roch-Marchal, B. Lassalle-Kaiser, P. Mialane, M. Fontecave, C. Mellot-Draznieks and A. Dolbecq, J. Am. Chem. Soc., 2018, 140, 3613–3618 CrossRef CAS PubMed.
- F. Yu, D. Poole 3rd, S. Mathew, N. Yan, J. Hessels, N. Orth, I. Ivanovic-Burmazovic and J. N. H. Reek, Angew. Chem., Int. Ed., 2018, 57, 11247–11251 CrossRef CAS PubMed.
- B. Li, F. Li, S. Bai, Z. Wang, L. Sun, Q. Yang and C. Li, Energy Environ. Sci., 2012, 5, 8229–8233 RSC.
- Q. Guan, L. L. Zhou and Y. B. Dong, Chem. Soc. Rev., 2022, 51, 6307–6416 RSC.
- J. Dong, X. Han, Y. Liu, H. Li and Y. Cui, Angew. Chem., Int. Ed., 2020, 59, 13722–13733 CrossRef CAS PubMed.
- W.-K. Han, Y. Liu, X. Yan and Z.-G. Gu, Mater. Chem. Front., 2023, 7, 2995–3010 RSC.
- D. Moonshiram, J. W. Jurss, J. J. Concepcion, T. Zakharova, I. Alperovich, T. J. Meyer and Y. Pushkar, J. Am. Chem. Soc., 2012, 134, 4625–4636 CrossRef CAS PubMed.
- Y. He, G. Liu, Z. Liu, J. Bi, Y. Yu and L. Li, ACS Energy Lett., 2023, 8, 1857–1863 CrossRef CAS.
- Y. Yang, X. Chu, H. Y. Zhang, R. Zhang, Y. H. Liu, F. M. Zhang, M. Lu, Z. D. Yang and Y. Q. Lan, Nat. Commun., 2023, 14, 593 CrossRef CAS PubMed.
- J. Xu, C. Yang, S. Bi, W. Wang, Y. He, D. Wu, Q. Liang, X. Wang and F. Zhang, Angew. Chem., Int. Ed., 2020, 59, 23845–23853 CrossRef CAS PubMed.
- C. Tang, X. Qiu, Z. Cheng and N. Jiao, Chem. Soc. Rev., 2021, 50, 8067–8101 RSC.
- X. Xiao, Z. Ruan, Q. Li, L. Zhang, H. Meng, Q. Zhang, H. Bao, B. Jiang, J. Zhou, C. Guo, X. Wang and H. Fu, Adv. Mater., 2022, 34, e2200612 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthetic procedures, characterization and catalytic experiments. CCDC 2270378 and 2270379. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03681b |
| This journal is © The Royal Society of Chemistry 2023 |