
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite selective gold(I)-catalysed benzylic C–H amination via an intermolecular hydride transfer to triazolinediones†
Kevin
Bevernaege
a,
Nikolaos V.
Tzouras
 b,
Albert
Poater
c,
Luigi
Cavallo
d,
Steven P.
Nolan
*e,
Fady
Nahra
*be and
Johan M.
Winne
*a
b,
Albert
Poater
c,
Luigi
Cavallo
d,
Steven P.
Nolan
*e,
Fady
Nahra
*be and
Johan M.
Winne
*a
aDepartment of Organic and Macromolecular Chemistry, Ghent University, Krijgslaan 281-S4, B-9000 Ghent, Belgium. E-mail: johan.winne@ugent.be
bDepartment of Chemistry and Center for Sustainable Chemistry, Ghent University, Krijgslaan 281-S3, B-9000 Ghent, Belgium
cDepartament de Química, Institut de Química Computacional i Catàlisi, Universitat de Girona, C/Maria Aurèlia Capmany 69, 17003 Girona, Spain
dKAUST Catalysis Center, Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology, Thuwal, 23955, Saudi Arabia
eSeparation and Conversion Technology, VITO (Flemish Institute for Technological Research), Boeretang 200, 2400 Mol, Belgium
First published on 24th August 2023
Abstract
Triazolinediones are known as highly reactive dienophiles that can also act as electrophilic amination reagents towards enolisable C–H bonds (ionic pathway) or weak C–H bonds (free radical pathway). Here, we report that this C–H amination reactivity can be significantly extended and enhanced via gold(I)-catalysis. Under mild conditions, several alkyl-substituted aryls successfully undergo benzylic C–H aminations at room temperature. The remarkable site selectivity that is observed points towards strong electronic activation and deactivation effects, that go beyond a simple weakening of the C–H bond. The observed catalytic C–H aminations do not follow the expected trends for a free radical-type C–H amination and show complementarity to existing methods. Density functional theory (DFT) calculations and distinct experimental trends provide a clear mechanistic rationale for observed selectivity patterns, postulating a novel pathway for triazolinedione-induced aminations via a carbon-to-nitrogen hydride transfer.
Introduction
Cookson-type reagents, or 4-substituted-1,2,4-triazoline-3,5-diones (TADs) have a remarkable reactivity towards dienes and olefins in classical Diels–Alder and Alder–ene reactions, displaying extremely rapid reaction kinetics compared to more classical dienophiles and enophiles.1 The Alder–ene reaction is formally an insertion of a urazole moiety into an allylic C–H bond, with concomitant displacement of the olefinic bond (Scheme 1a).2 Direct C–H amination reactivity with triazolinediones is also known in electrophilic aromatic substitutions,3a–f and for ‘enolisable’ C–H bonds.3g Direct insertions of triazolinediones into weak C–H bonds, such as thio-ether α-hydrogens, are known to proceed via a free radical substitution chain reaction involving hydrogen atom abstraction by a nitrogen-centred radical species.4 For these processes, radical initiators or photochemical conditions are required, and azodicarboxylates are often used as oxidative traps for the nitrogenation of alkyl radicals (Scheme 1b).5,6 Free radical benzylic aminations with azodicarboxylate esters are unselective between different benzylic positions, but several catalytic systems for more selective C–H aminations have been developed in recent years, including one that is quite selective for the sterically less hindered benzylic positions.6 TADs are not known as general amination reagents for benzylic positions, and have been reported to more readily lead to electrophilic aromatic substitution in the presence of Brønsted acids or under the influence of UV-light.7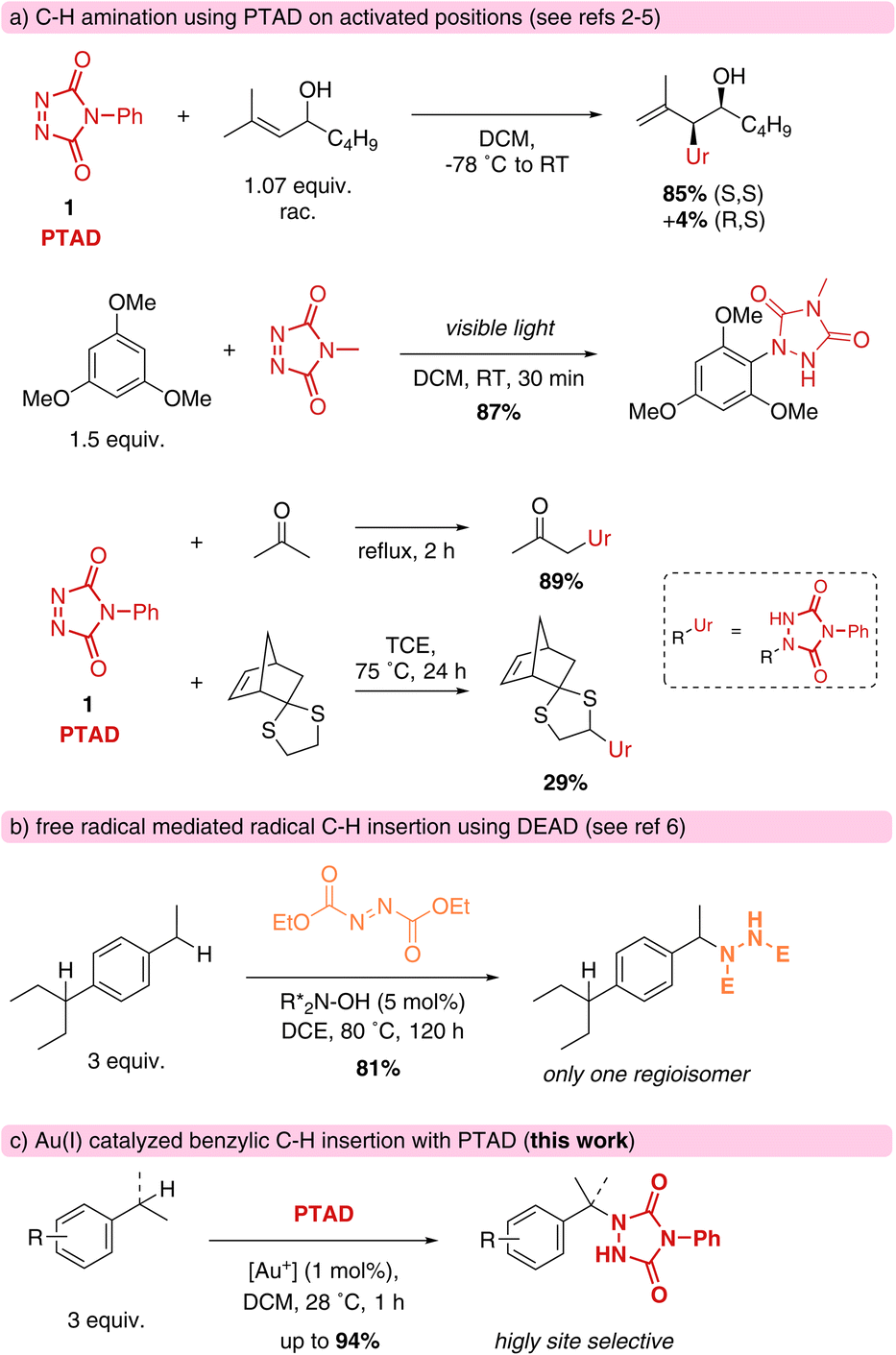 | ||
| Scheme 1 Rationale and positioning of the reported work. | ||
We now report on our investigations of catalytic C–H amination of benzylic substrates using triazolinediones as the electrophilic amination reagent. These high energy reagents have so far been ignored in catalytic C–H aminations, although their remarkable reactivity should create opportunities for various substrate types. In fact, our study has uncovered a previously unappreciated reactivity mode of TADs via a novel reaction pathway involving an unexpected intermolecular hydride transfer. This new mechanistic pathway exhibits a selectivity profile that is strongly influenced by electronic effects of relatively remote substituents, which in turn leads to remarkably site-selective aminations that discriminate between different benzylic positions.
Results and discussion
For our initial benzylic amination study using TAD reagents, we selected dialkylbenzene 2a as a model substrate, since this gave the closest 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 selectivity in free radical benzylic amination reactions.6a This would allow us to probe efficiency as well as regioselectivity, based on the work by Kato and Maruoka in this area.6 Reacting the commercially available 4-phenyl-triazolinedione (PTAD) reagent 1 with 2a, in the absence of any catalyst, results in a slow consumption of PTAD over 5 days, giving the C–H inserted products 2b and 2c in moderate yield, albeit with clear selectivity for one regioisomer (Table 1, entries 1 and 2). Heating the reaction leads to shorter reaction times, but the overall amination efficiency remains low (Table 1, entry 3). The addition of the radical initiator N-hydroxyl-phthalimide (NHPI, entry 4), had no effect on the reaction rate compared to the purely thermal reaction of PTAD and 2a (entry 2), which rules out a competitive free radical hydrogen transfer mechanism. Surprisingly, however, a slightly higher yield was obtained after full conversion of the PTAD reagent. This indicates that NHPI, rather than accelerate the C–H amination reaction, acts as an inhibitor of the background decomposition reaction of the TAD reagent, thus favouring the amination reaction. Initial catalyst screening (see ESI†) revealed that combining [AuCl(IPr)] with AgSbF6 showed good activity towards promoting the C–H amination.8
:1 selectivity in free radical benzylic amination reactions.6a This would allow us to probe efficiency as well as regioselectivity, based on the work by Kato and Maruoka in this area.6 Reacting the commercially available 4-phenyl-triazolinedione (PTAD) reagent 1 with 2a, in the absence of any catalyst, results in a slow consumption of PTAD over 5 days, giving the C–H inserted products 2b and 2c in moderate yield, albeit with clear selectivity for one regioisomer (Table 1, entries 1 and 2). Heating the reaction leads to shorter reaction times, but the overall amination efficiency remains low (Table 1, entry 3). The addition of the radical initiator N-hydroxyl-phthalimide (NHPI, entry 4), had no effect on the reaction rate compared to the purely thermal reaction of PTAD and 2a (entry 2), which rules out a competitive free radical hydrogen transfer mechanism. Surprisingly, however, a slightly higher yield was obtained after full conversion of the PTAD reagent. This indicates that NHPI, rather than accelerate the C–H amination reaction, acts as an inhibitor of the background decomposition reaction of the TAD reagent, thus favouring the amination reaction. Initial catalyst screening (see ESI†) revealed that combining [AuCl(IPr)] with AgSbF6 showed good activity towards promoting the C–H amination.8
|
|
||||
|---|---|---|---|---|
| Entry | Cat. (1 mol%) | Timeb | Yieldc (%) | 2b:2cd |
| a Reaction conditions: 0.5 mmol PTAD (1 equiv.), 1.5 mmol 1-ethyl-4-isopropylbenzene (3 equiv.), PhCF3 (0.1 M), shielded from light at room temperature. b Reaction was stirred until the red colour of the PTAD disappeared. c Mean NMR yield of three repetitions vs. internal standard (1,3,5-trimethoxybenzene). d Mean NMR ratio of three repetitions. e Only performed once. f Reaction stopped before the colour of the PTAD disappeared as to have the same timeframe as entry 5. g Conversion was too low to determine a reliable ratio. h Was heated to 80 °C. i Isolated yield. j DCM was used as solvent. k DCE was used as solvent. l 0.1 mol% of catalyst was used. | ||||
| 1e,f | — | 30 min | 2 | —g |
| 2 | — | 5 days | 47 | 85:15 |
| 3e,h | — | 2.5 hc | 46 | 81:19 |
| 4e | NHPI (10 mol%) | 5 days | 66 | 84:16 |
| 5 | [AuCl(IPr)] + AgSbF6 | 30 min | 72 | 76:24 |
| 6e,f | [AuCl(IPr)] | 30 min | 2 | —g |
| 7e,f | AgSbF6 | 30 min | 2 | —g |
| 8 | [AuCl(IPr)] + NaBArF | 30 min | 80 | 80:20 |
| 9 | [Au(IPr)(MeCN)]BF 4 | 2 h | 82 |
80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :20 :20
|
| 10 | [Au(NTf2)(IPr)] | 5 h | 77 | 80:20 |
| 11 | [{Au(IPr)}2(μ-OH)]BF4 | 2 h | 82 | 80:20 |
| 12 | [Au(NTf2)(PPh3)] | 3 days | 36 | 72:28 |
| 13 | [Au(JohnPhos)(MeCN)]SbF6 | 4 h | 53 | 67:33 |
| 14 | [Au(NTf2)(IMes)] | 2.5 h | 67 | 81:19 |
| 15 | [AuCl(IAd)] + NaBArF | 2.5 h | 70 | 80:20 |
| 16 | [AuCl(IPrMe)] + NaBArF | 30 min | 78 | 78:22 |
| 17 | [Au(IPrCl)(MeCN)]BF4 | 5 h | 73 | 78:22 |
| 18 | [AuCl(SIPr)] + NaBArF | 30 min | 77 | 76:24 |
| 19 | [Au(OTf)(IPr*)] | 2 h | 73 | 62:38 |
| 20 | [Au(IPr)(MeCN)]BF4 | 2 h | 88i | 81:19 |
| 21 | [Au(IPr)(MeCN)]BF 4 | 30 min | 94 |
80:20
|
| 22k | [Au(IPr)(MeCN)]BF4 | 30 min | 94i | 79:21 |
| 23j,l | [Au(IPr)(MeCN)]BF4l | 2 days | 80i | 88:12 |
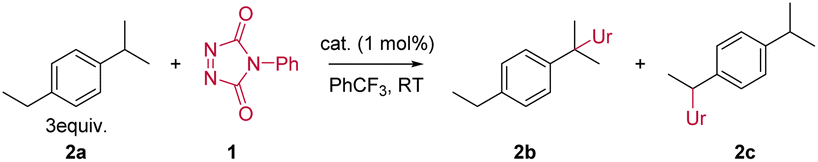
Using 1 mol% loading of both the [AuCl(IPr)] and AgSbF6 afforded full conversion after only 30 minutes with an NMR yield of 72% and a 76:24 ratio of regioisomers 2b and 2c (Table 1, entry 5). To evaluate if both metal complexes were necessary, the gold and silver complexes were tested independently (Table 1 entries 6 and 7) under the same conditions as entry 5. Thus, it was confirmed that the combination of both gold and silver complexes to form the cationic gold active species was necessary for the C–H-amination to proceed smoothly.
Even though using AgSbF6 to generate cationic gold is quite straightforward, this method still has several drawbacks, including so-called silver-effects and light sensitivity of silver reagents.9 Therefore, other methods to generate the cationic gold species were explored. The alternative halogen abstractor NaBArF (sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate) gave improved results in the same reaction time (Table 1, entry 8). More importantly, these reactions proved to be more reproducible. To avoid halogen abstractors altogether and reduce the complexity of the system, the use of well-defined cationic gold(I) species was investigated.10 In this context, we assayed several well-defined cationic gold species (Table 1, entries 9–11). All three complexes gave similar results with slightly increased reaction times compared to when either AgSbF6 or NaBArF were used. From these experiments we could conclude that the counterion plays a minor role in the outcome of the reaction.
Next, the effect of the NHC ligand was assessed. To that end, NHCs with different electronic and steric properties were explored, with a focus on the effect of steric bulk and ligand electron donating capabilities. The results are summarized in Table 1 (entries 12–19). For comparison, two well-defined cationic gold(I) phosphine complexes were also used (Table 1, entries 12 and 13), but these gave inferior results. When looking at the increase in the catalyst's steric bulk, highlighted by the increase of %Vbur (ref. 11) of the corresponding NHC (%Vbur: IMes < IAd < IPr < IPrMe < IPrCl < SIPr < IPr*), a general and logical trend is observed (Table 1, entries 12–19): bulkier ligands shift the selectivity towards the least hindered position. Smaller ligands are therefore better suited to promote the C–H amination at the more hindered position. The benchmark [Au(IPr)(MeCN)]BF4 (ref. 10) (Table 1, entry 9) provided the best balance between yield and selectivity.
Several solvents were screened (Table 1, entries 20–22). The initial choice of solvent (trifluoromethylbenzene) was mainly governed by compatibility issues where only solvents that would not give background reactivity towards PTAD could be tested. From the solvent screening (see ESI†) PhCF3, DCM and DCE emerged as good solvents (see Table 1, entries 20–22), although reactions in PhCF3 were slower and provided slightly lower yields. DCM was then adopted as solvent of choice for testing the substrate scope, while dichloroethane (DCE) was found to be most practical for higher temperature reactions of the slower reacting substrates.
Finally, and interestingly, a low catalyst loading (0.1 mol%, entry 23) still gave a high isolated yield, even though the reaction time required for full conversion of PTAD increased from 30 minutes to two days. The lower yield can be attributed to the slow degradation of PTAD over time.
Having identified a suitable catalytic system and reaction conditions to activate PTAD towards C–H amination, we next examined different substrates in order to investigate general reactivity and selectivity patterns (Scheme 2). As expected, simple cumene derivatives that only possess one benzylic hydrogen all gave good yields of the aminated products (Scheme 2a, 3b–6b) within reasonable reaction times. Remarkably, we found that there is a significant and unusual difference in reactivity between mono-alkyl and di-alkyl substituted aryls, even though the reactive C–H-bonds in these substrates should have very similar bond strengths. For example, our model substrate 2a reacted with PTAD reaching full conversion at room temperature within minutes, whereas the corresponding cumene, lacking a para-ethyl group, reacted more sluggishly under the same conditions to yield 3b in 10 h (Scheme 2a). Thus, not only the direct substitution pattern of the reactive C–H bond is important in this amination reaction (isopropyl vs. ethyl as in substrate 2a), but also the substitution pattern on the aromatic ring appears to be critical. The same can be seen when cumene and 4-tert-butylcumene are compared, where the latter again reacts much faster than the former yielding 4b in excellent yield after 30 minutes.
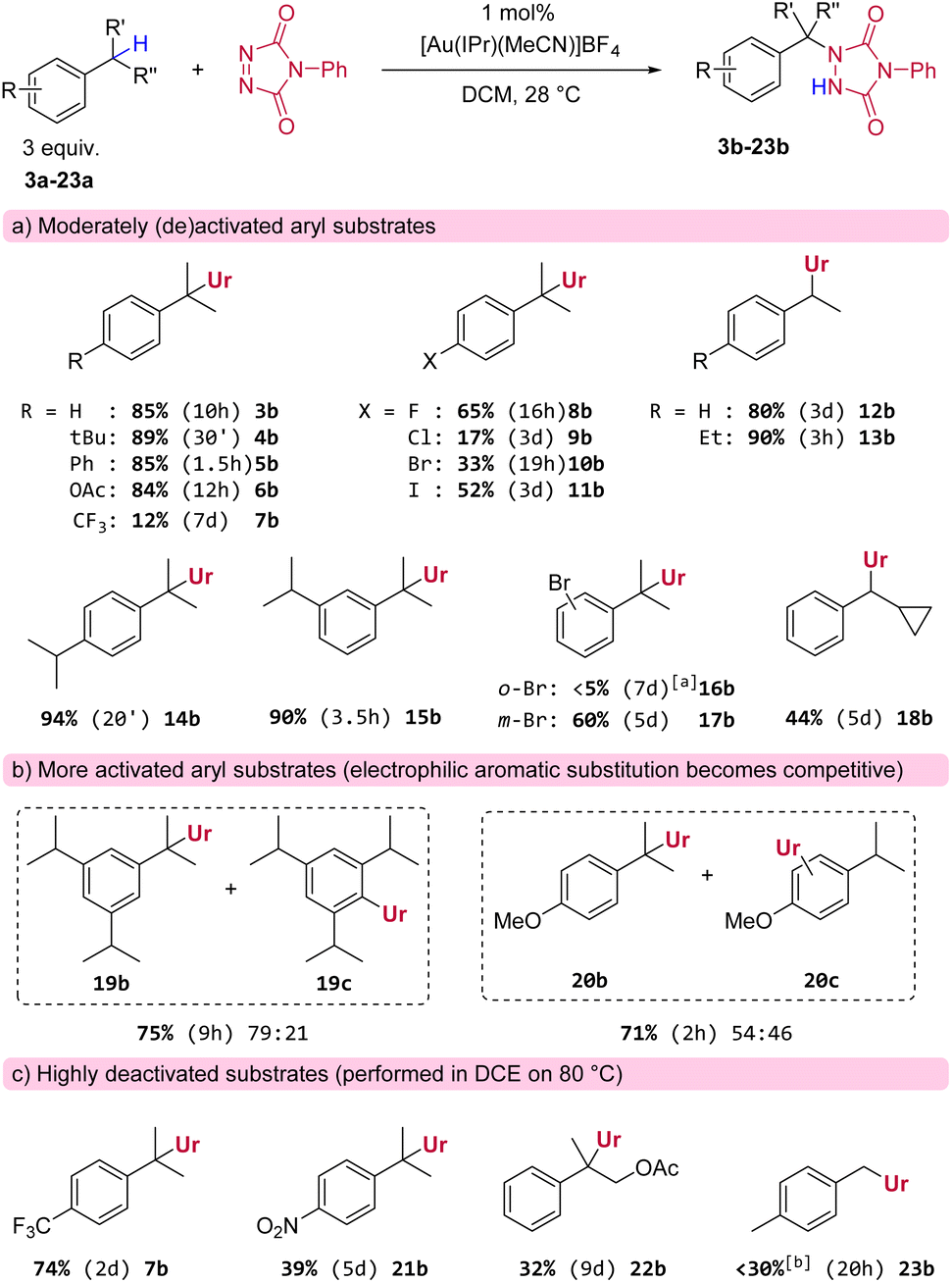 | ||
| Scheme 2 Substrate scope of gold-catalysed benzylic amination. Reaction conditions: 0.5 mmol PTAD (1 equiv.), 1.5 mmol substrate (3 equiv.), [Au(IPr)(MeCN)]BF4 (1 mol%), DCM (0.1 M) at 28 °C. Reported reaction times refer to the time it took for the red colour of the PTAD to disappear. Yields are isolated, isomer ratios are based on NMR integration. aReaction was stopped after 7 days (90% conversion of PTAD (1)). bBased on NMR analysis using an internal standard. | ||
Furthermore, halogenated cumenes reacted more slowly and also gave lower yields (8b–11b). Amination of ethylbenzene with PTAD affords 12b in similar yield as for 3b, but the reaction requires a significantly longer time (3 days for 12bvs. 10 h for 3b). Conversely, the reaction of 1,4-diethylbenzene with PTAD happens more swiftly (within only 3 h at rt), and the amination product 13b is obtained in excellent yield (90%). Here again, a strong effect of para-alkyl substitution can be seen (13bvs.12b). Surprisingly, in this reaction only traces of bis-adducts derived from 13a were observed, indicating that the ethyl group in the aminated product 13b is deactivated compared to the initial ethyl groups of the starting material 13a (vide infra).
The relative position of alkyl groups on the aryl ring was also of interest, as 1,4-diisopropylbenzene (yielding 14b after 20 minutes) reacted faster than 1,3-diisopropylbenzene (yielding 15b after 3.5 h). Again, only traces of bis-adducts were observed for both dialkylsubstrates 14a or 15a. The effect of ortho- and meta-substitution was also examined for the electron withdrawing bromo-substituent (16a and 17a). The meta-positioning was clearly better tolerated, while the ortho-position proved to be detrimental for the reaction. The reaction of 1,3,5-triisopropylbenzene was more complex. Here, reactions were slow and bis-adducts were again not observed, but now the electrophilic aromatic amination product 19c is formed as a major side product (see Scheme 2b). Electrophilic aromatic substitution (EAS) is a known reactivity mode of PTAD with highly activated aryls.3a–f It should also be noted here that the reaction rate for 1,3,5-triisopropylbenzene is slower than that for 1,3-diisopropylbenzene (respectively 9 h compared to 3.5 h for complete conversion of PTAD). The same competitive EAS was noted for 4-isopropylanisole. This substrate gave the C–H aminated product 20b as the major product, along with aromatic substitution products (20c). Thus, for very electron rich aryls, competitive aromatic amination becomes an important side reaction. For electron poor aryl substrates, almost no reaction was observed under standard conditions (e.g.7b, Scheme 2a). However, heating the reaction at 80 °C gave the expected amination products in shorter reaction times and higher yields (e.g.7b, Scheme 2c), even for strongly deactivated systems such as p-nitro cumene (see Scheme 2c), although 21b was obtained in poor yield and only after 5 days of heating. Similarly, electron-withdrawing groups near the benzylic position also greatly prolong reaction times, as the aminated product 22b was only formed in 32% yield after reacting 2-phenylpropyl acetate (22a) for 9 days at 80 °C. Finally, a general finding was that benzylic methyls mostly lacked reactivity towards PTAD in this gold(I)-catalyzed amination protocol. For example, we found that PTAD resisted reaction with an excess of para-xylene (23a) for over a week at room temperature. Heating this reaction to 80 °C did give complete consumption of PTAD in 20 h, but only a small amount was converted to the expected mono-adduct 23b (∼25%), next to various inseparable degradation products of PTAD that could not be characterized here.
In order to further profile this novel reactivity mode of TADs, and because of the observed strong reactivity trends induced by substitution patterns, we turned to intra- and intermolecular competition experiments. In Scheme 3, the relative ratios of observed regioisomers can be seen for various substrates (Scheme 3a) and their combinations in intermolecular competitions (Scheme 3b). These isomer ratios can be expected to reflect the relative rate of the different benzylic positions under our standard gold(I)-catalysed reaction conditions.
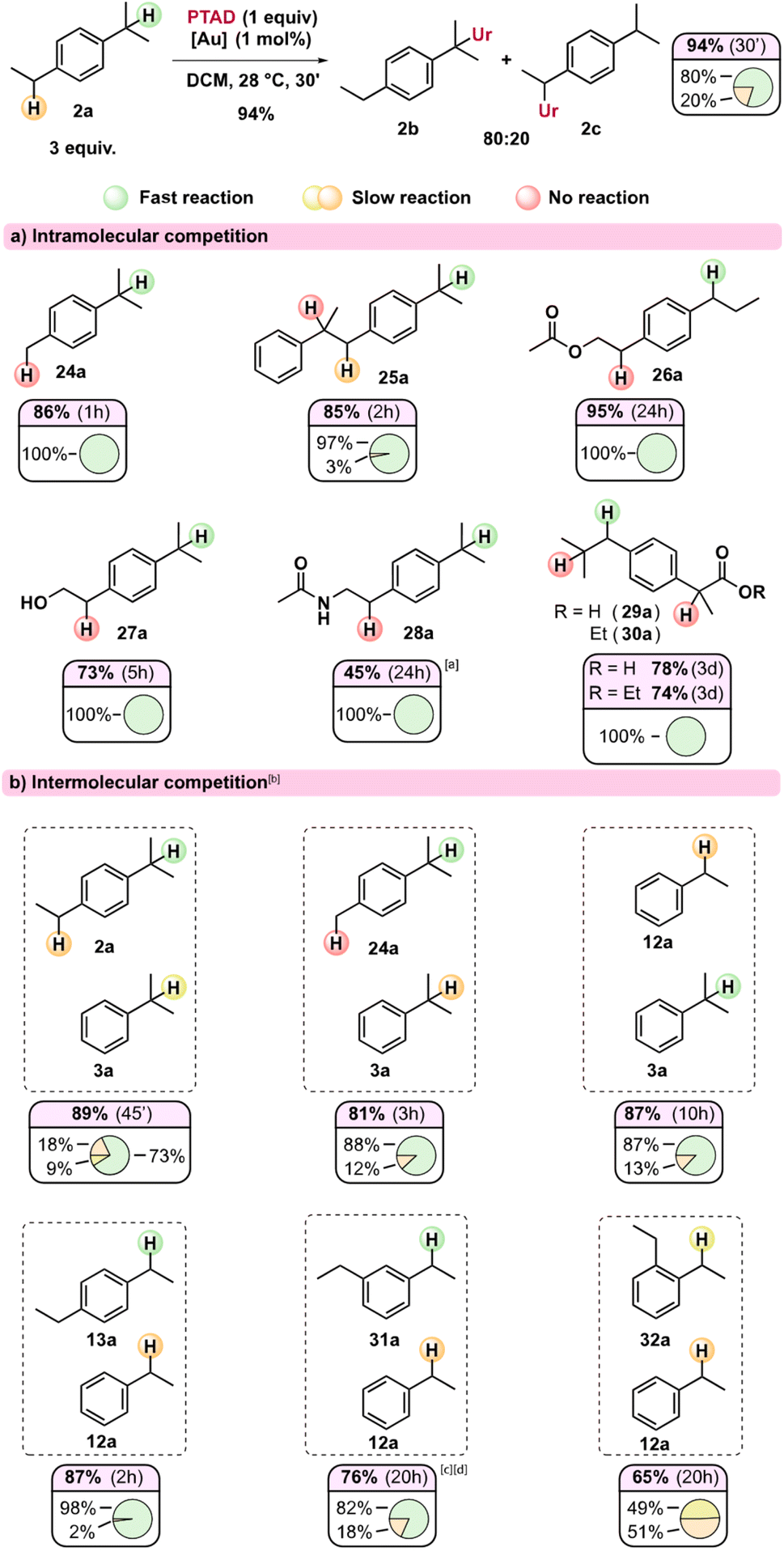 | ||
| Scheme 3 Intra- and intermolecular competition experiments for the C–H amination method. Reaction conditions: 0.5 mmol PTAD (1 equiv.), 1.5 mmol substrate (3 equiv.), [Au(IPr)(MeCN)]BF4 (1 mol%), DCM (0.1 M) at 28 °C. Reaction was stirred at least until the red colour of the PTAD disappeared (mostly longer). Yields are isolated yields; isomer ratios are based on NMR integration. aLower yield can be attributed to low conversion (95% yield brsm). Although traces of undetermined side-products were present, none were of the benzylic regioisomer. bIn case of intermolecular competition reactions, both substrates were used in 1.5 mmol (thus both 3 equiv.). cEAS products were also observed. dRatio determined by deconvolution of the NMR spectrum. | ||
Initially, it could be seen that the tertiary position of p-cymene 24a reacts exclusively, as the methyl remains unaffected, in line with our previous observations (viz.23b). Substrate 25a has three different benzylic positions but shows an almost exclusive reaction on one of the isopropyl substituents. The internal positions here are both deactivated with the aryl group acting as an inductive electron-withdrawing group, while the internal tertiary benzylic C–H also lacks a para-alkyl substituent. Likewise, when an electron-withdrawing group is near one of the benzylic positions (Scheme 3a, 26a–30a), the reaction proceeds exclusively at the benzylic position furthest away from this electron withdrawing group. The reaction shows relatively broad functional group tolerance, as esters, amides, free hydroxyls and even carboxylic acids are all tolerated (Scheme 3a, 26a–30a). As expected, substrates with a basic amine do not result in C–H amination (see ESI†) as PTAD is known to degrade rapidly under the influence of amines.12 Moreover, amines are known to strongly coordinate cationic Au(I) catalysts. However, ammonium salts are useful substrates in the reaction (see ESI†). The acetamide (28a) shows delayed reactivity, which could be rationalized due to some catalyst poisoning by the more coordinating amide function. Interestingly, both ibuprofen (29a) as its ethyl ester (30a), which is a popular substrate in C–H activation studies,13 react exclusively at the secondary benzylic position, giving complete selectivity over the tertiary position and the benzylic position alpha to the carbonyl. Such selectivity is usually not seen in free radical benzylic C–H activations, and is preserved for radical-polar cross-over reactions.14
Intermolecular competition experiments reveal further distinct selectivity trends (Scheme 3b). Interestingly, in the reaction of an equimolar mixture of 2a and 3a with PTAD, even the secondary position in dialkylbenzene 2a appears to react faster than the tertiary position in monoalkylbenzene 3a. This indicates that a para-alkyl substitution (isopropyl in 2avs. H in 3a) is more important than direct alkyl substitution of the benzylic position itself (secondary in 2avs. tertiary in 3a). The competition experiment of 24avs.3a confirms this effect. In competitions between mono- and diethyl benzenes, 1,4-diethylbenzene (13a) reacted almost exclusively with PTAD in a head-to-head competition with simple ethylbenzene (12a). When using 1,3-diethylbenzene (31a), this selectivity dropped to 82:18 ratio in favour of the diethyl substrate. In line with previous observations, both substrates 13a and 31a did not produce detectable amounts of bis-PTAD-adducts. Finally, and remarkably, 1,2-ethylbenzene (32a) and ethylbenzene (12a) showed almost equal reactivity towards PTAD, giving a near 1:1 mixture of the corresponding amination products (32b and 12b). This effect can be explained as ortho-substitution can prevent coplanarity of the aryl ring, making it less stabilizing. Similar effects have been observed in benzylic oxidation reactions involving benzylic cation intermediates.15
As shown above, an electron withdrawing group reduces the reactivity of the substrate, and actually prevents a C–H insertion in nearby sp3-carbon atoms (Scheme 3, 25a–28a), and also slows down the general reactivity of a substrate (5 h for 27avs. 3 h for 13a and 24 h for 28avs. 30 min for 2a). This effect explains why only traces of bis-adducts can be observed on substrates like 12a–14a as the urazole acts as an electron withdrawing group after the first addition, slowing down the second addition. Even when only one equivalent of substrate 13a is used, less than 1% of bis-adduct can be observed (see Scheme 4). This reaction also shows that using 1 equivalent of substrate still gives good isolated yields, with just a slight increase in reaction time and some erosion in yield.
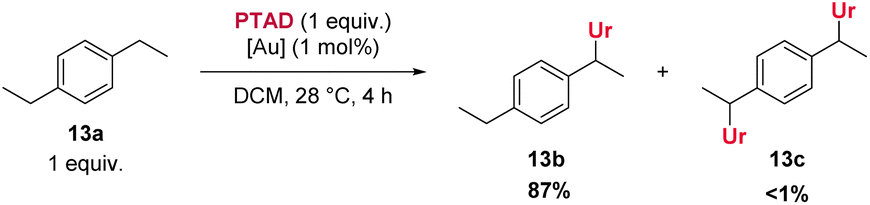 | ||
| Scheme 4 C–H amination with only one equivalent of substrate. | ||
An enantioselective C–H amination was attempted with 1,4-diethylbenzene (13a) as substrate and various chiral gold(I) complexes (see ESI, Table S8†). To our delight, modest to good enantioselectivities were immediately observed for all chiral ligands, the best result is given in Scheme 5. These results exclude a reaction pathway in which a free radical substitution of weak C–H-bonds is operating, and imply a close involvement of the gold catalyst with the bond forming process.
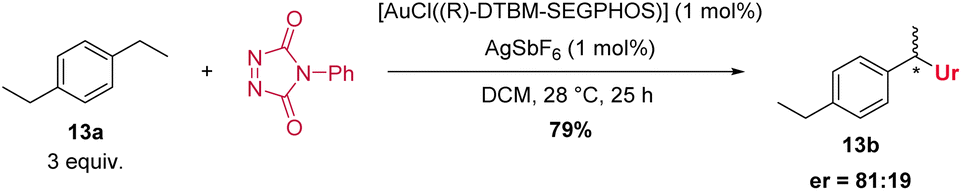 | ||
| Scheme 5 Enantioselective gold-catalysed C–H amination. | ||
Mechanistic investigation
To gain further insight into the mechanism, the possibility of a kinetic isotope effect (KIE) was investigated using a deuterated substrate (Scheme 6). The deuterated version of 33a showed a much slower reaction as judged by the conversion of PTAD in a stoichiometric experiment, indicating a C–H-bond being broken during the rate determining step (RDS). Analysis of the isotope ratios before and after reaction showed a strong KIE of 4.79 based on NMR integrations.16 | ||
| Scheme 6 Kinetic isotope effect reaction. | ||
To rationalize the reaction outcomes and the observed remarkable selectivity patterns, DFT calculations were performed for the non-catalysed and Au(I) catalysed reactions of PTAD with ethylbenzene (Scheme 7). These calculations showed that in both cases the reaction operates via a rate-determining hydride transfer from the benzylic carbon to the PTAD (1) nitrogen atom (TSI). From the calculated energy profiles, a sharp accelerative effect of the gold coordination can be noted for the rate-determining hydride transfer transition state, which is lowered by 7.1 kcal mol−1 when comparing catalysed vs. non-catalysed pathways. This reaction step is essentially the same for the catalysed and non-catalysed pathways and represents an intermolecular hydride shift from the benzylic position to an azo-bonded nitrogen atom. Such hydride transfers are known for carbocationic reagents, but hydride transfer to nitrogens are rare.17 Moreover, well-controlled hydride transfer catalytic pathways are usually confined to intramolecular reactions,18 or to enzymatic reactions with hydride-accepting co-factors such as NAD+ or FAD.19 The difference in reaction rate between different substrates as observed here can be quite readily rationalized according to this carbocationic pathway, and the DFT-calculated energies for different substrates are in complete agreement with experiments (see ESI†).
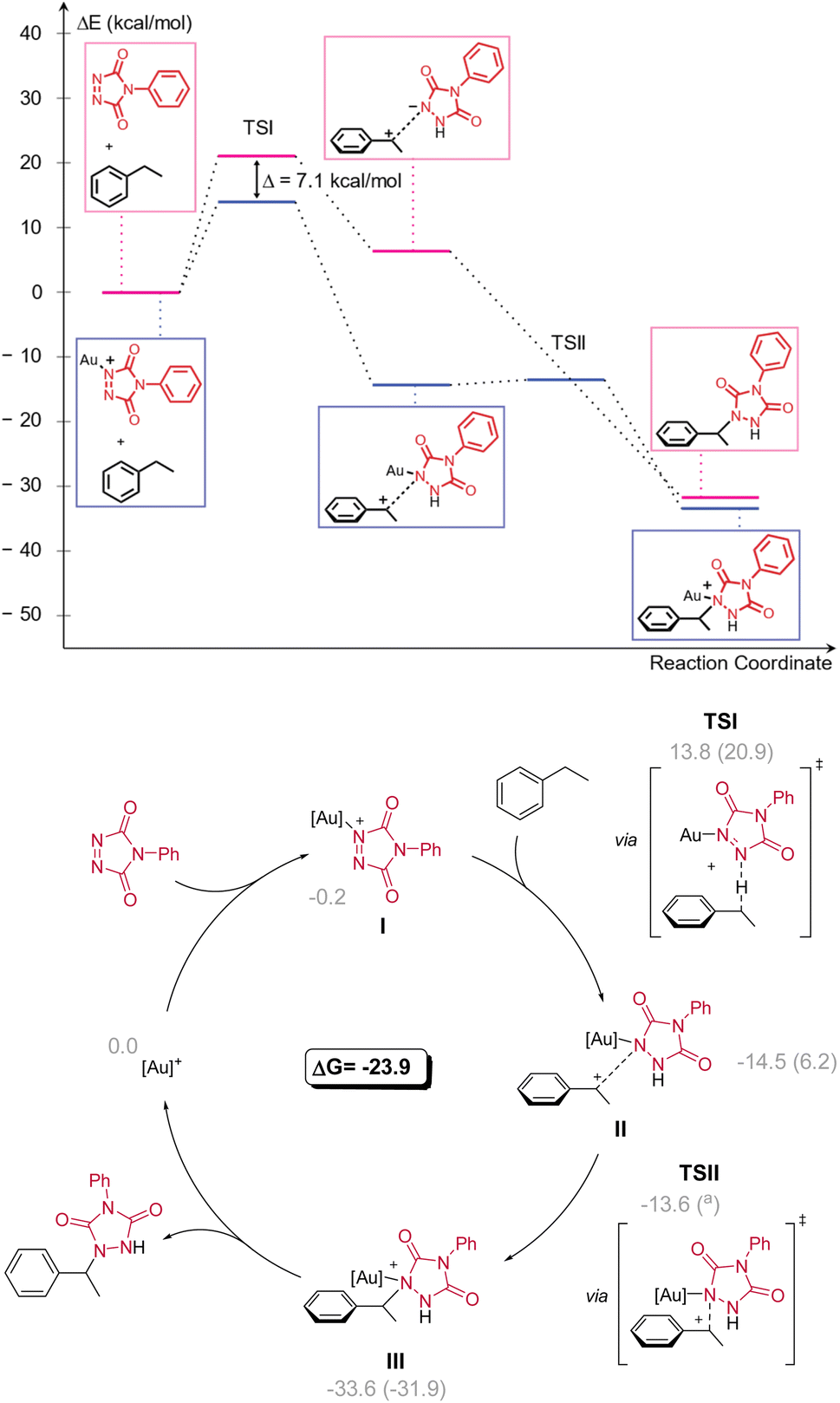 | ||
| Scheme 7 Catalytic cycle calculated at the M06/Def2-TZVP∼sdd(SMD-DCM)//BP86/Def2-SVP∼sdd level of theory (Gibbs energies in kcal mol−1 referenced to the cationic complex Au(IPr)+ (=[Au]+), in parentheses the corresponding values without the metal catalyst, a = not located). | ||
The calculated catalytic cycle (Scheme 7) explains the observed strong electronic substituent effects, as they can easily make substrates significantly more/less hydride donating. To further confirm our mechanistic rationale, which is a deviation from what is generally expected in gold catalysis and C–H activation pathways, effort was undertaken to provide experimental support for intermediate I. After generating cationic gold (by reacting [AuCl(IPr)] and AgSbF6) and adding a stoichiometric amount of PTAD (1) to the mixture (see Scheme 8), clear shifts in the 13C{1H}-NMR peaks of both PTAD (1) and the gold moiety were observed. Indicating the formation of a single new gold complex, intermediate I. The same complex was also observed when the reaction was conducted in the presence of ethylbenzene (12a), strongly indicating that the coordination of gold to PTAD (1) is more favourable than that of gold to the alkyl-substituted aryl substrates. Upon a closer look at the 13C{1H}-NMR peaks after PTAD (1) addition to the cationic gold, a desymmetrisation of PTAD (1) can be observed, giving further evidence of gold coordinating to the 1-N and not the 4-N, the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond or the aryl of PTAD (1). Calculations also showed that the coordination of the cationic [Au(IPr)+] is favoured on the N instead of the O by a difference of 10.6 kcal mol−1. Interestingly, when [Au(IPr)(MeCN)]BF4 was used, practically no complexation of the PTAD to the gold was observed. This led us to believe that MeCN is binding more strongly to the gold than PTAD (1). This was then confirmed by adding MeCN to the complex formed between PTAD and cationic gold. Visually, one can already see the colour of the solution changing from dark red to bright red upon adding MeCN, indicating the release of PTAD (1) from the complex. The formation of [Au(IPr)(MeCN)]SbF6 was then confirmed by NMR and purification of this complex.
N bond or the aryl of PTAD (1). Calculations also showed that the coordination of the cationic [Au(IPr)+] is favoured on the N instead of the O by a difference of 10.6 kcal mol−1. Interestingly, when [Au(IPr)(MeCN)]BF4 was used, practically no complexation of the PTAD to the gold was observed. This led us to believe that MeCN is binding more strongly to the gold than PTAD (1). This was then confirmed by adding MeCN to the complex formed between PTAD and cationic gold. Visually, one can already see the colour of the solution changing from dark red to bright red upon adding MeCN, indicating the release of PTAD (1) from the complex. The formation of [Au(IPr)(MeCN)]SbF6 was then confirmed by NMR and purification of this complex.
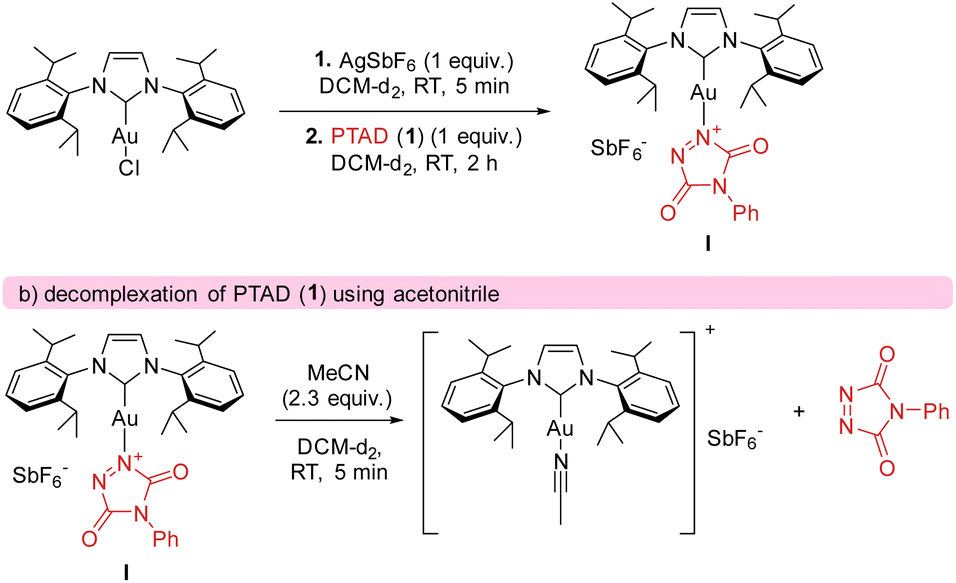 | ||
| Scheme 8 Complexation of PTAD (1) with gold. | ||
The fact that acetonitrile shows a stronger interaction with the gold cation than PTAD (1), also explains some of our experimental observations. Substrates bearing a functional group that could be a competitive ligand for the gold centre, lead to longer reaction times. For example, this explains the slower reaction of 20a (compared to 24a) and 6a (compared to 3a) even though they are bearing more electron donating groups in the para position that should in theory lead to a faster activation of the hydride transfer.
Synthetic applications
Although the newly found C–H amination reactivity of TADs is interesting from a mechanistic point of view, and also shows a relatively wide substrate scope for such a reaction type, the formed products should also find good use in synthesis and other applications. Urazole derivatives have for example shown to possess interesting biological activities.20 Moreover, 1-alkyl urazoles have recently emerged as a valuable tool to label and crosslink tyrosine residues on native proteins.21 Furthermore, urazoles can be transformed into other useful functional groups. Reductive cleavage of the N–N bond can be achieved by use of Raney-Ni19a or addition of an alkylmagnesiumbromide reagent in excess19b yielding simple (protected) amine products. Conversion of 1,4-disubstituted-urazoles to a primary amine is thus feasible in a few steps.22c–e The group of Sarlah has made great progress in the synthetic applications of TAD reagents in recent years, showing that quite a few functional group transformations are possible even beyond amines.22e For a benzylic substrate, urazoles can be regarded as mild leaving groups, comparable in nucleofugality to acetates. Indeed, as simple SN1-type hydrolysis affords the tertiary alcohol in excellent yield (see Scheme 9).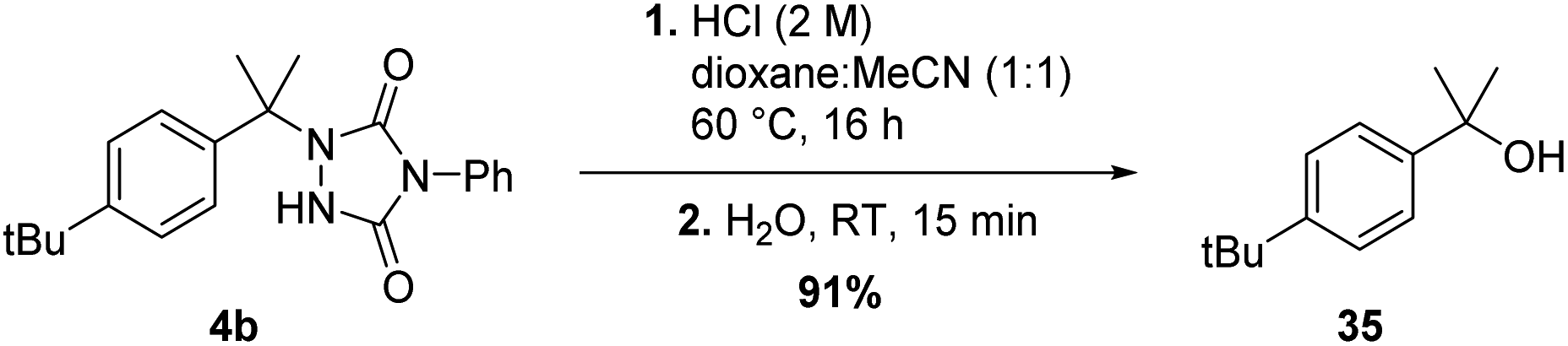 | ||
| Scheme 9 Hydrolysis of the formed N-alkylurazole. | ||
Conclusion
In conclusion, we have developed a site-selective benzylic C–H amination method which critically depends on the substitution pattern of the benzylic carbon atom, but also on the substitution pattern of the aryl group. This selectivity contrasts the normally observed reactivity patterns for such reactions and is not governed primarily by sterics or C–H-bond strengths but can be readily rationalized by a rate-limiting intermolecular hydride transfer to give a benzylic carbocation intermediate, which is strongly affected by both electron donating and electron-withdrawing groups. This peculiar mechanism can be regarded as an SN1-type hydride displacement at carbon and appears to be an overlooked intrinsic reactivity mode of TADs, that can be envisioned as a synthetically useful method. Further studies examining synthetic applications of this Au(I)-accelerated C–H activation reaction and its intriguing mechanism are currently underway in our laboratories.Data availability
All detailed procedures, characterization data, and spectra are available in the ESI.† Data not included there (e.g. IR and HRMS spectra) can be provided on request.Author contributions
The draft manuscript was written by K. B., and reviewed by all authors. All authors contributed to revising the manuscript. The study was conceptualized by J. M. W. and K. B. with important input of F. D. and S. P. N. Funding for the experimental synthetic work was acquired by K. B., S. P. N. and J. M. W. All the C–H amination reactions, the synthesis of the substrates and chiral NHC–Au complexes, complexation tests and synthetic data analysis were performed by K. B. A. P. and L. C. performed and acquired funding for the DFT calculations. All gold catalysts, except those containing chiral NHCs, were synthesised by N. V. T.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the The Research Foundation – Flanders (FWO) and the BOF (starting and senior grants to SPN) as well as the iBOF C3 project for financial support. The FWO is also acknowledged for Fundamental Research PhD fellowships to KB (11E1122N) and NVT (11I6921N). The FWO is also acknowledged by JMW for financial support (G005720N). A. P. thanks the Spanish Ministerio de Ciencia e Innovación for project PID2021-127423NB-I00. A. P. is a Serra Húnter Fellow and ICREA Academia Prize 2019.References
- (a) R. C. Cookson, S. S. H. Gilani and I. D. R. Stevens, Tetrahedron Lett., 1962, 3, 615–618 CrossRef; (b) R. C. Cookson, S. S. H. Gilani and I. D. R. Stevens, J. Chem. Soc. C, 1967, 1905–1909 RSC; (c) K. De Bruycker, S. Billiet, H. A. Houck, S. Chattopadhyay, J. M. Winne and F. E. Du Prez, Chem. Rev., 2016, 116, 3919–3974 CrossRef CAS PubMed.
- (a) K. De Bruycker, S. Billiet, H. A. Houck, S. Chattopadhyay, J. M. Winne and F. E. Du Prez, Chem. Rev., 2016, 116, 3919–3974 CrossRef CAS PubMed; (b) M. Squillacote, M. Mooney and J. De Felippis, J. Am. Chem. Soc., 2002, 112, 5364–5365 CrossRef; (c) A. H. Gau, G. L. Lin, B. J. Uang, F. L. Liao and S. L. Wang, J. Org. Chem., 1999, 64, 2194–2201 CrossRef CAS; (d) Y. Kuroda, Org. Lett., 2022, 24, 6224–6229 CrossRef CAS PubMed.
- (a) R. C. Cookson, S. S. H. Gilani and I. D. R. Stevens, J. Chem. Soc. C, 1967, 1905–1909 RSC; (b) G. W. Breton, Tetrahedron Lett., 2011, 52, 733–735 CrossRef CAS; (c) G. W. Breton and K. R. Hoke, J. Org. Chem., 2013, 78, 4697–4707 CrossRef CAS PubMed; (d) E. H. Southgate, J. Pospech, J. Fu, D. R. Holycross and D. Sarlah, Nat. Chem., 2016, 8, 922–928 CrossRef CAS PubMed; (e) D. Kaiser, J. M. Winne, M. E. Ortiz-Soto, J. Seibel, T. A. Le and B. Engels, J. Org. Chem., 2018, 83, 10248–10260 CrossRef CAS PubMed; (f) K. W. Decoene, K. Unal, A. Staes, O. Zwaenepoel, J. Gettemans, K. Gevaert, J. M. Winne and A. Madder, Chem. Sci., 2022, 13, 5390–5397 RSC; (g) R. Marshall Wilson, A. C. Hengge, A. Ataei and N. Chantarasiri, J. Org. Chem., 2002, 55, 193–197 CrossRef.
- (a) W. Ando, K. Ito and T. Takata, Tetrahedron Lett., 1982, 23, 3909–3912 CrossRef CAS; (b) W. Adam, O. De Lucchi, K. Hill, E. -M Peters, K. Peters and H. G. von Schnering, Chem. Ber., 1985, 118, 3070–3088 CrossRef CAS.
- (a) M. Shibuya, T. Orihashi, Y. Li and Y. Yamamoto, Chem. Commun., 2021, 57, 8742–8745 RSC; (b) Y. Jin, Q. Zhang, L. Wang, X. Wang, C. Meng and C. Duan, Green Chem., 2021, 23, 6984–6989 RSC; (c) Q. An, Z. Wang, Y. Chen, X. Wang, K. Zhang, H. Pan, W. Liu and Z. Zuo, J. Am. Chem. Soc., 2020, 142, 6216–6226 CrossRef CAS PubMed.
- (a) T. Kato and K. Maruoka, Angew. Chem., Int. Ed., 2020, 59, 14261–14264 CrossRef CAS PubMed; (b) T. Kato and K. Maruoka, Chem. Commun., 2022, 58, 1021–1024 RSC.
- (a) H. Wamhoff and K. Wald, Chem. Ber., 1977, 110, 1699–1715 CrossRef CAS; (b) D. W. Borhani and F. D. Greene, J. Org. Chem., 1986, 51, 1563–1570 CrossRef CAS.
- For reviews of other gold-catalysed C–H activation methods, see: (a) Q. Zhao, G. Meng, S. P. Nolan and M. Szostak, Chem. Rev., 2020, 120, 1981–2048 CrossRef CAS PubMed; (b) G. Meera, K. R. Rohit, G. S. S. Treesa and G. Anilkumar, Asian J. Org. Chem., 2020, 9, 144–161 CrossRef CAS; (c) J. Xie, C. Pan, A. Abdukader and C. Zhu, Chem. Soc. Rev., 2014, 43, 5245–5256 RSC; (d) S. Gaillard, C. S. J. Cazin and S. P. Nolan, Acc. Chem. Res., 2012, 45, 778–787 CrossRef CAS PubMed; (e) T. Deharo and C. Nevado, Synthesis, 2011, 2011, 2530–2539 CrossRef; (f) T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910–1925 RSC; (g) P. Lu, T. C. Boorman, A. M. Z. Slawin and I. Larrosa, J. Am. Chem. Soc., 2010, 132, 5580–5581 CrossRef CAS PubMed; (h) A. S. K. Hashmi, R. Salathé, T. M. Frost, L. Schwarz and J. H. Choi, Appl. Catal., A, 2005, 291, 238–246 CrossRef CAS.
- (a) M. Veguillas, G. M. Rosair, M. W. P. Bebbington and A. L. Lee, ACS Catal., 2019, 9, 2552–2557 CrossRef CAS; (b) D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. L. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012–9019 CrossRef CAS PubMed.
- P. De Frémont, E. D. Stevens, M. R. Fructos, M. Mar Díaz-Requejo, P. J. Pérez and S. P. Nolan, Chem. Commun., 2006, 2045–2047 RSC.
- (a) A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759–1766 CrossRef CAS; (b) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC.
- (a) Q. Y. Hu, M. Allan, R. Adamo, D. Quinn, H. Zhai, G. Wu, K. Clark, J. Zhou, S. Ortiz, B. Wang, E. Danieli, S. Crotti, M. Tontini, G. Brogioni and F. Berti, Chem. Sci., 2013, 4, 3827–3832 RSC; (b) L. H. Dao and D. Mackay, Can. J. Chem., 1979, 57, 2727–2733 CrossRef CAS.
- Y. Zhang, T. Zhang and S. Das, Chem, 2022, 8, 3175–3201 CAS.
- (a) S. E. Suh, S. J. Chen, M. Mandal, I. A. Guzei, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2020, 142, 11388–11393 CrossRef CAS PubMed; (b) I. N. M. Leibler, M. A. Tekle-Smith and A. G. Doyle, Nat. Commun., 2021, 12, 1–10 CrossRef PubMed.
- G. A. Olah and N. Friedman, J. Am. Chem. Soc., 1966, 88, 5330–5331 CrossRef CAS.
- D. A. Singleton and A. A. Thomas, J. Am. Chem. Soc., 1995, 117, 9357–9358 CrossRef CAS.
- (a) N. P. van Leest, L. Grooten, J. I. van der Vlugt and B. de Bruin, Chem. –Eur. J., 2019, 25, 5987–5993 CrossRef CAS PubMed; (b) M. E. Rotella, R. M. B. Dyer, M. K. Hilinski and O. Gutierrez, ACS Catal., 2019, 10, 897–906 CrossRef PubMed; (c) T. G. Jo and J. E. M. N. Klein, ChemCatChem, 2021, 13, 4087–4091 CrossRef CAS.
- (a) M. C. Haibach and D. Seidel, Angew. Chem., Int. Ed., 2014, 53, 5010–5036 CrossRef CAS PubMed; (b) X. Wen, X. Li, X. Luo, W. Wang, S. Song and N. Jiao, Chem. Sci., 2020, 11, 4482–4487 RSC; (c) X. De An and J. Xiao, Org. Chem. Front., 2021, 8, 1364–1383 RSC; (d) H. Zhang, M. C. Sun, L. M. Yin, D. Wei, M. P. Song, D. Yang and J. L. Niu, Org. Chem. Front., 2021, 8, 6888–6894 RSC; (e) J. L. Miller, J. M. I. A. Lawrence, F. O. Rodriguez del Rey and P. E. Floreancig, Chem. Soc. Rev., 2022, 51, 5660–5690 RSC.
- (a) J. M. Berrisford and L. A. Sazanov, J. Biol. Chem., 2009, 284, 29773–29783 CrossRef CAS PubMed; (b) M. W. Fraaije and A. Mattevi, Trends Biochem. Sci., 2000, 25, 126–132 CrossRef CAS PubMed.
- (a) S. Gupta, P. Saluja and J. M. Khurana, Tetrahedron, 2016, 72, 3986–3993 CrossRef CAS; (b) T. Tang, C. Hartig, Q. Chen, W. Zhao, A. Kaiser, X. Zhang, H. Zhang, H. Qu, C. Yi, L. Ma, S. Han, Q. Zhao, A. G. Beck-Sickinger and B. Wu, Nat. Commun., 2021, 12(1), 1–9 CrossRef PubMed.
- (a) Q. Yan, M. Li, Y. Zhang, H. Liu, F. Liu, W. Liao, Y. Wang, H. Duan and Z. Wei, Talanta, 2023, 258, 124421 CrossRef CAS PubMed; (b) E. Llabani, R. W. Hicklin, H. Y. Lee, S. E. Motika, L. A. Crawford, E. Weerapana and P. J. Hergenrother, Nat. Chem., 2019, 11(6), 521–532 CrossRef CAS PubMed; (c) S. Sato, K. Hatano, M. Tsushima and H. Nakamura, Chem. Commun., 2018, 54, 5871–5874 RSC.
- (a) J. Yang, J. W. Zhang, W. Bao, S. Q. Qiu, S. Li, S. H. Xiang, J. Song, J. Zhang and B. Tan, J. Am. Chem. Soc., 2021, 143, 12924–12929 CrossRef CAS PubMed; (b) Y. Kuroda, Org. Lett., 2022, 24, 6224–6229 CrossRef CAS PubMed; (c) W. Adam, A. Pastor and T. Wirth, Org. Lett., 2000, 2, 1295–1297 CrossRef CAS PubMed; (d) T. W. Bingham, L. W. Hernandez, D. G. Olson, R. L. Svec, P. J. Hergenrother and D. Sarlah, J. Am. Chem. Soc., 2019, 141, 657–670 CrossRef CAS PubMed; (e) M. Okumura, A. S. Shved and D. Sarlah, J. Am. Chem. Soc., 2017, 139, 17787–17790 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03683a |
| This journal is © The Royal Society of Chemistry 2023 |