
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular donor-stabilized tetra-coordinated germanium(IV) di-cations and their Lewis acidic properties†
Balakrishna
Peddi
a,
Souvik
Khan
a,
Rajesh G.
Gonnade
 b,
Cem B.
Yildiz
*c and
Moumita
Majumdar
*a
b,
Cem B.
Yildiz
*c and
Moumita
Majumdar
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research, Pune, Dr. Homi Bhabha Road, Pashan, Pune-411008, Maharashtra, India. E-mail: moumitam@iiserpune.ac.in
bPhysical and Materials Chemistry Division, CSIR-National Chemical Laboratory, Pune-411008, Maharashtra, India
cDepartment of Aromatic and Medicinal Plants, Aksaray University, 68100 Aksaray, Türkiye
First published on 16th November 2023
Abstract
We report the first examples of intramolecular phosphine-stabilized tetra-coordinated germanium(IV) di-cationic compounds: [LiPr2Ge][CF3SO3]23iPr and [LPh2Ge][CF3SO3]23Ph (LiPr = 6-(diisopropylphosphanyl)-1,2-dihydroacenaphthylene-5-ide; LPh = 6-(diphenylphosphanyl)-1,2-dihydroacenaphthylene-5-ide). The step wise synthetic strategy involves the isolation of neutral and mono-cationic Ge(IV) precursors: [LiPr2GeCl][X] (X = GeCl31iPr, OTf 2iPr), [LPh2GeCl2] 1Ph and [LPh2GeCl][OTf] 2Ph. Both 3iPr and 3Ph exhibit constrained spiro-geometry. DFT studies reveal the dispersion of di-cationic charges over P–Ge–P sites. Anion or Lewis base binding occurs at the Ge site resulting in relaxed distorted trigonal bipyramidal/tetrahedral geometry. 3iPr and 3Ph activate the Si–H bond initially at the P-site. The hydride ultimately migrates to the Ge-site rapidly giving [LPh2GeH][CF3SO3] 3PhH, while sluggishly forming [LiPr2GeH][CF3SO3] 3iPrH. Compounds 3iPr and 3Ph were tested as catalysts for the hydrosilylation of aromatic aldehydes. While catalytic hydrosilylation proceeded via the initial Et3Si–H bond activation in the case of 3iPr, compound 3Ph as a catalyst showed a masked Frustrated Lewis Pair (FLP) type reactivity in the catalytic cycle.
Introduction
Germanium(II) di-cations stabilized by neutral Lewis bases have been known for long,1 possessing inherent electrophilicity2 and rarely exhibiting nucleophilic behaviour.3 In contrast there are only very few examples of Ge(IV) di-cations known, which are stabilized by hyper-coordination involving neutral donors: [LGeF2]2+ (L = tris(1-ethyl-benzoimidazol-2-ylmethyl)amine) (Fig. 1A),4 [GeF2(OTf)2] (OTf = CF3SO3) coordinated by two mono-dentate phosphine donors or bi-dentate chelating phosphine donors (Fig. 1B),5 and [Me2Ge(OTf)2] coordinated by two 4-N,N′-dimethylaminopyridine (DMAP) units or a 2,2′-bipyridine (Fig. 1C).6 Alongside in silicon chemistry, terpyridine-stabilized silicon(IV) di-cations (R2Si2+), tri-cations (RSi3+),7 and silicon tetrakis(trifluoromethanesulfonate)8 have been established as Lewis superacids9 in recent times. The uses of Ge(IV) di-cations as Lewis acids hold immense potential; however they are yet to be unfolded. In general, while scheming Lewis acidic Ge(IV) polycations, an optimal balance between the stabilization of the polycationic species by neutral donors and accessibility of vacant orbitals is necessary. Therefore, focused investigations on the stabilization of Ge(IV) polycations using suitable donor groups that circumvent hyper-coordination are imperative for their potential Lewis acidity. Furthermore, isolation of such species has remained a worthwhile synthetic target on fundamental grounds.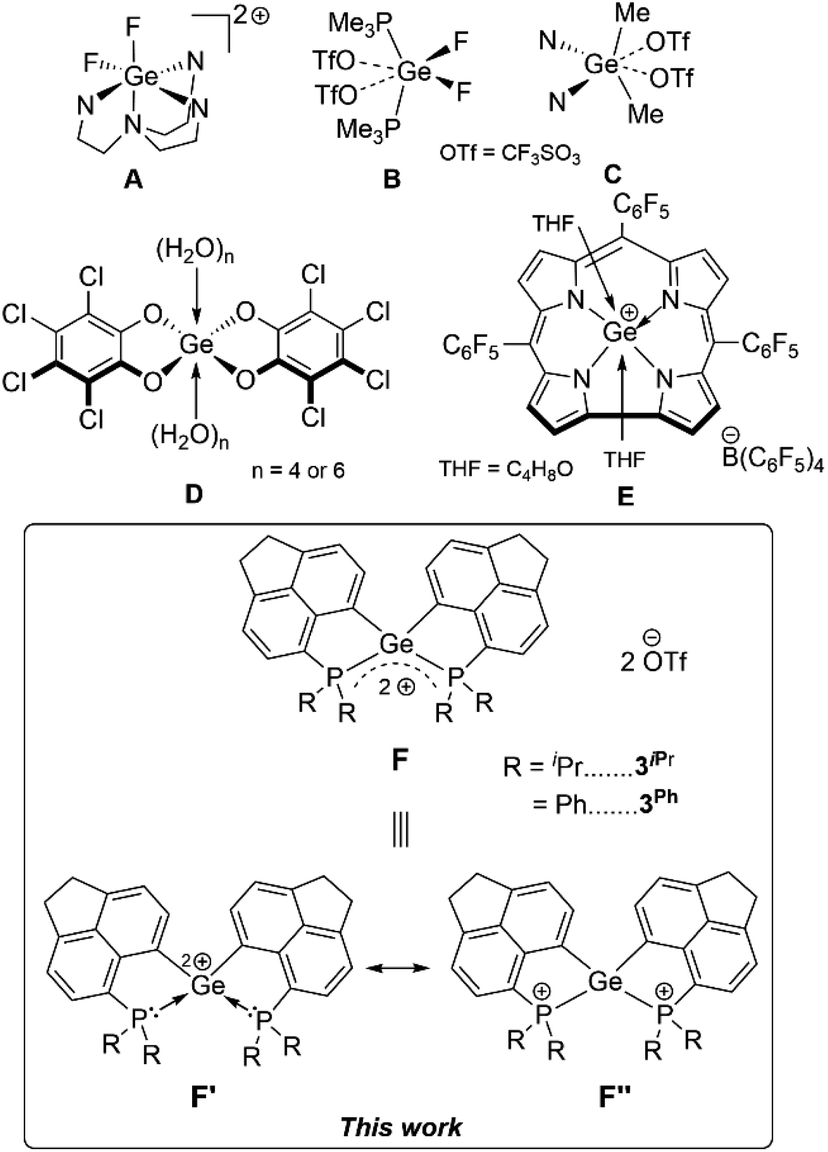 | ||
| Fig. 1 Reported examples of Ge(IV) di-cations (A) (N4 = tris(1-ethyl-benzoimidazol-2-ylmethyl)amine) and (B) and (C) (N = 4-N,N′-dimethylaminopyridine); examples of Ge(IV)-based Lewis acids (D) and (E) and the intramolecular phosphine-stabilized Ge(IV) di-cations (F, this work). | ||
As a matter of fact, compared to silicon analogues,10 there are scarce reports on Ge-based Lewis acids11 both in their neutral or cationic forms. Greb et al. have pioneered neutral bis(perchlorocatecholato)germane as both hard and soft Lewis superacids (Fig. 1D).12 Bis(catecholato)germane derivatives have been successfully implemented as Lewis acid catalysts.13 A significant example of the cationic form of Ge(IV) Lewis acid is [(TPFC)Ge(THF)2]+ (TPFC = tris(pentafluorophenyl)corrole, THF = tetrahydrofuran) (Fig. 1E).14 Recently, a Ge(II) mono-cationic σ-donor ligand towards Ni(0) has been reported with simultaneous Lewis superacidity.15 It is worth mentioning that Gabbaï et al. have demonstrated a formally cationic Ge(IV) σ-acceptor site in a dinuclear Pt–Ge(IV) complex.16
Contemporary research in the field of main-group Lewis acids17 has introduced the concepts of geometrically constrained Lewis acids,18 main group Lewis acid/ligand assisted cooperative bond activation,19 hidden frustrated Lewis pair (FLP) chemistry20 in intra-molecular Lewis base stabilized Lewis acidic fragments.21 Taking advantage of the enforced proximity in peri-substituted acenaphthenes,22 herein we have established the unprecedented intramolecularly phosphine-stabilized tetra-coordinated di-cationic Ge(IV) compounds. The aptitude of these geometrically constrained Ge(IV) di-cationic species as Lewis acids has been assessed based on experimental findings and computational analyses. Non-innocent roles of the phosphines have been observed. The proclivity of Ge(IV) di-cationic compounds for Et3Si–H bond activation coupled with FLP-type reactivity has been realized in a proof-of-concept catalytic hydrosilylation of p-methyl benzaldehyde.
Results and discussion
Syntheses and characterization of intramolecular phosphine-stabilized Ge(IV) mono- and di-cationic compounds
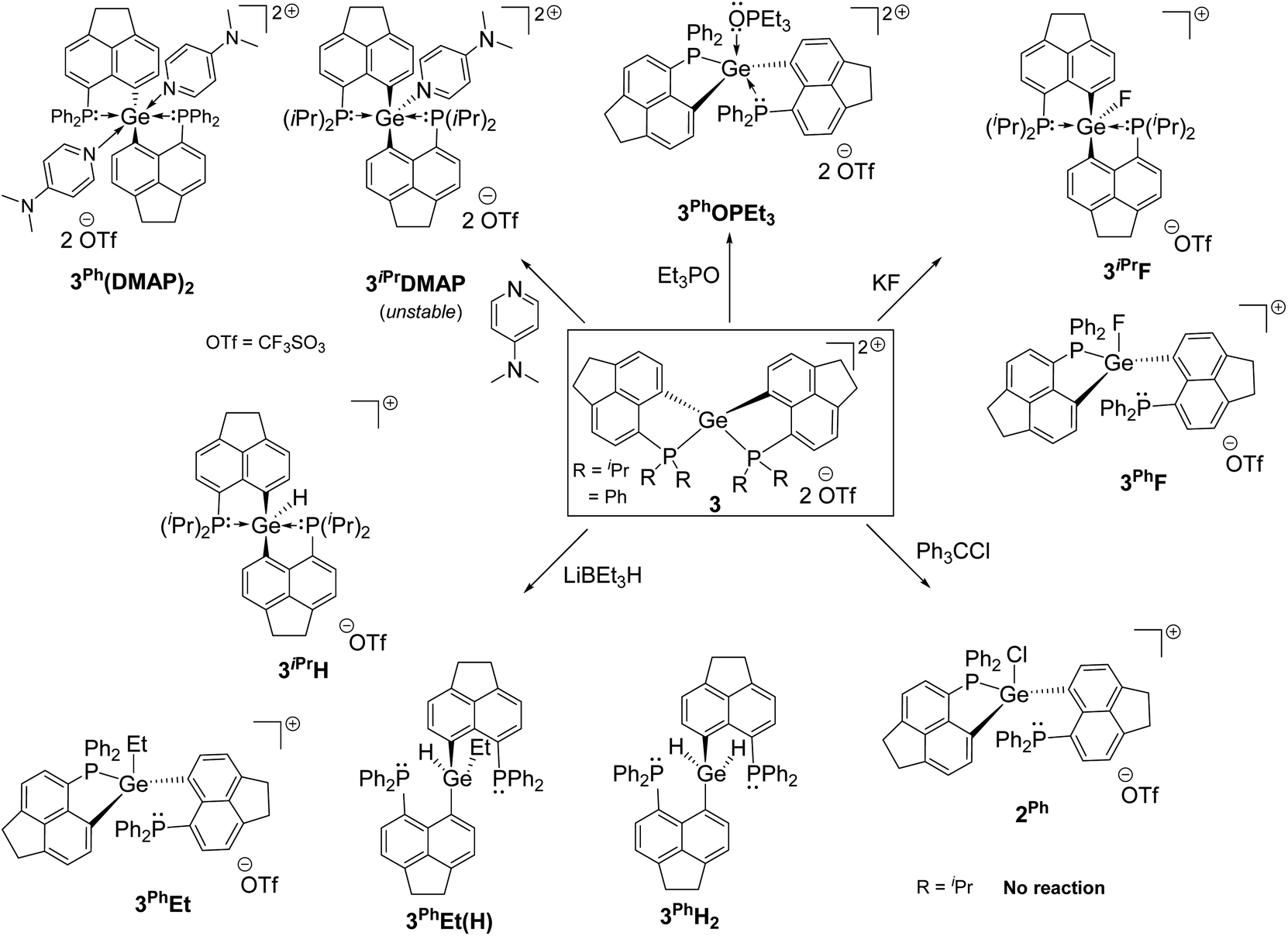
Intramolecular phosphine-stabilized Ge(IV) di-cationic compounds 3iPr and 3Ph were obtained in a step-wise manner as shown in Scheme 1. Initially, LiPr–Br and LPh–Br were synthesized and characterized (see the ESI† for Experimental procedures). Table 1 summarizes the NMR data for all compounds. The reaction between 1.5 equivalents of lithiated LiPr–Br and GeCl4 at −78 °C in a tetrahydrofuran (THF) medium, followed by extraction with dichloromethane (DCM), gave 1iPr (Scheme 1a). Structural elucidation of the single crystals of 1iPr obtained from acetonitrile (MeCN) reveals the formation of [LiPr2GeCl]+, interestingly possessing GeCl3 as a counter anion (Fig. S95, ESI†). The formation of the GeCl3 anion stems from the reducing ability of phosphines.23 The mono-cationic compound 1iPr was characterized by the 31P{1H} NMR chemical shift at −11.1 ppm. An additional peak at −19.5 ppm was observed in the 31P{1H} NMR spectrum of the crude reaction mixture arising from the corresponding P(V) oxidized product (Fig. S7, ESI†).24 Using straightforward lithiated LiPr–Br and GeCl4 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio also led to the formation of 1iPr. An anion exchange reaction of 1iPr with one equivalent of trimethylsilyl trifluoromethane sulfonate (TMSOTf) followed by crystallization in DCM resulted in the isolation of 2iPr (Scheme 1a). 2iPr was characterized in the solution state using NMR spectroscopy displaying the characteristic 31P{1H} NMR chemical shift at −10.8 ppm.
:1 ratio also led to the formation of 1iPr. An anion exchange reaction of 1iPr with one equivalent of trimethylsilyl trifluoromethane sulfonate (TMSOTf) followed by crystallization in DCM resulted in the isolation of 2iPr (Scheme 1a). 2iPr was characterized in the solution state using NMR spectroscopy displaying the characteristic 31P{1H} NMR chemical shift at −10.8 ppm.
 | ||
| Scheme 1 Syntheses of the Ge(IV) mono- and di-cationic compounds (see the ESI† for the detailed spectroscopic and experimental procedures of each reaction). | ||
| Compounds | 31P{1H} in ppm (deuterated solvent) | 19F{1H} in ppm (deuterated solvent) | J values (Hz) | Compounds | 31P{1H} in ppm (deuterated solvent) | 19F{1H} in ppm (deuterated solvent) | J values (Hz) |
|---|---|---|---|---|---|---|---|
| a Chemical shift values at room temperature. b Chemical shift values at low temperature. c In situ NMR. d Measured after 2 days. e Measured after one day. | |||||||
| LiPrBr | −1.58a (CDCl3) | 3iPrDMAP | +28.37 (3iPr), −13.19 (PiPr2)b (CD2Cl2) | ||||
| LPhBr | −9.40a (CDCl3) | 3Ph(DMAP)2 | −29.16b (CDCl3) | −79.21a (CDCl3) | |||
| 1iPr | −11.06a (CDCl3) | 3iPrF | −10.23(d)a (CDCl3) | −78.12(OTf), −141.03(t)a (CDCl3) | 2 J P–F = 184.9 | ||
| 1Ph | −34.79a (CDCl3) | 3PhF | −17.54(br.)a, −10.27(m), −23.66(m) (CDCl3) | −150.32(t)a (CDCl3) | 2 J P–F = 100.6a, JP–P = 119.8, JP–F = 98.9b | ||
| 2iPr | −10.77a (CDCl3) | −78.08a (CDCl3) | 3Ph + Ph3CClc | −6.31(3Ph), −13.46(2Ph)a (CDCl3) | |||
| 2Ph | −13.20a, −3.0(d), −20.5(d)b (CD2Cl2) | −78.84a (CD2Cl2) | 2 J P–P = 110b | 3iPrH | −5.15a (CD2Cl2) | −77.01a (CD2Cl2) | |
| 3iPr | +28.37a (CD2Cl2), +26.95a (CD3CN) | −78.84a (CD2Cl2) | 3Ph + LiBEt3Hc | −19.07, −19.82, −20.06a (CDCl3) | −78.35, −78.87a (CDCl3) | 1H NMR: 6.40(t) (J = 20.2, Ge–H2) | |
| 3Ph | −6.32a (CDCl3) | −79.74a (CDCl3) | 3iPr + Et3SiHc,d | +28.51 (3iPr), +20.28 (P–H), +12.16 (PiPr2)a (CD2Cl2) | 1 J P–H = 476 | ||
| 3iPrOPEt3 | +73.4 (bound Et3PO), −12.0 (PiPr2)a (CD2Cl2) | 3Ph + Et3SiHc,e | −19.0a (CDCl3) | −77.43a (CDCl3) | 2 J P–H = 30.4 | ||
| 3PhOPEt3 | +71.59 (bound Et3PO), −6.30 (3Ph), −13.38 (PPh2)a (CDCl3) | 3Ph + aldehydec | 36.02–32.08, −19.26a (CDCl3) | ||||
The molecular structure25 of 1iPr shows that the cationic part has an overall distorted trigonal bipyramidal (TBP) geometry with two pendant iPr2P groups occupying axial sites. Two Ge–C and one Ge–Cl bond constitute the trigonal plane with the sum of angles at the Ge atom close to 360°. The Ge1–P1 and Ge1–P2 bond lengths are 2.544(1) and 2.654(1) Å, respectively, which are longer than those of [GeF3(Ph2PCH2CH2PPh2)(OTf)] (avg. Ge–P = 2.43 Å)5 and diaminodiphosphine stabilized bis(chlorogermyliumylidene) (avg. Ge–P = 2.44–2.50 Å).26 The structural parameters of the cationic part in 2iPr (Fig. 2a) are analogous to 1iPr. The triflate counter anion in 2iPr is located far away from the Ge cationic site (closest Ge–O contact being 5.7 Å).
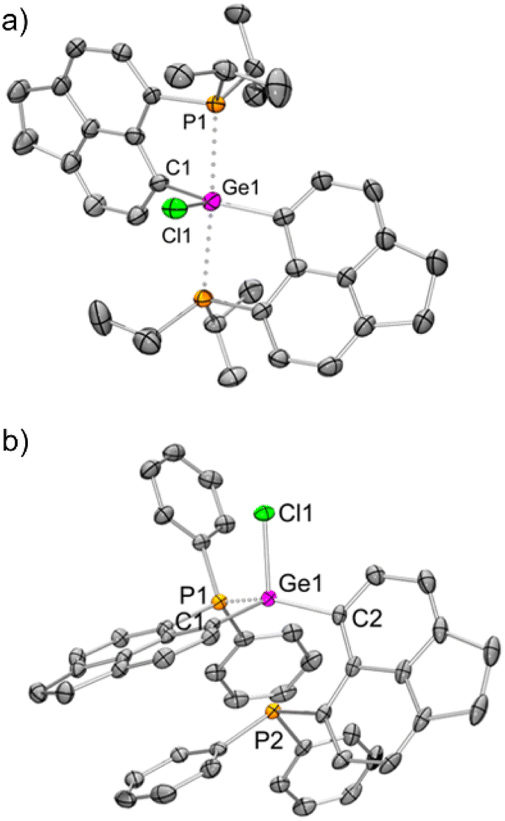 | ||
| Fig. 2 (a) Molecular structures of (a) 2iPr and (b) 2Ph (H atoms, solvent molecule and triflate anion have been removed for clarity; thermal ellipsoid 50%). Selected bond lengths [Å] and angles [°]: (a) Ge1–Cl1 = 2.228(2), Ge1–C1 = 1.988(4), Ge1–P1 = 2.643(1); C1–Ge1–C1 = 147.9(2), Cl1–Ge1–C1 = 106.1(1), Cl1–Ge1–P1 = 86.9(1), P1–Ge1–P1 = 173.9(1); (b) Ge1–C1 = 1.922(3), Ge1–C2 = 1.926(3), Ge1–Cl1 = 2.215(1), Ge1–P1 = 2.357(1); C1–Ge1–C2 120.0(1), C1–Ge1–Cl1 = 104.8(1), C2–Ge1–Cl1 = 104.2(1), C1–Ge1–P1 = 91.1(1), C2–Ge1–P1 = 135.5(1), Cl1–Ge1–P1 = 96.4(1). | ||
The formation of 3iPr can either proceed as a one-pot reaction between 1iPr and two equivalents of TMSOTf in DCM or stepwise chloride abstraction from 1iPrvia the intermediacy of 2iPr (Scheme 1a). Colourless crystals of 3iPr were obtained from the concentrated THF solution. The characteristic 31P{1H} NMR chemical shift values obtained for 3iPr in CD3CN (+27.0 ppm) and CD2Cl2 (+28.4 ppm) are similar, which invalidates possible solvent coordination.
The solid-state structure of 3iPr (Fig. 3a) shows a spirocyclic geometry19a with the Ge atom at the nexus and having two non-coordinating triflate counter anions (closest Ge–O contact being 5.8 Å). Due to the presence of dipositive charges in 3iPr, the Ge–C (avg. 1.92 Å) and Ge–P (avg. 2.36 Å) bond lengths are significantly shorter compared to those of 2iPr. The strongly electron-donating nature of the two intramolecular iPr2P groups adequately stabilizes the Ge(IV) di-cation in 3iPr in an overall tetra-coordinated environment.
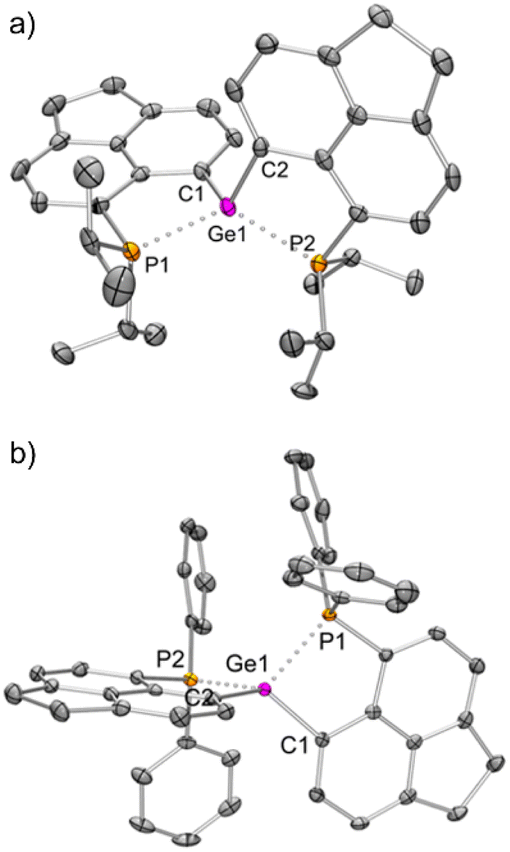 | ||
| Fig. 3 Molecular structures of (a) 3iPr and (b) 3Ph (H atoms, solvent molecules and triflate anions have been removed for clarity; thermal ellipsoid 50%). Selected bond lengths [Å] and angles [°]: (a) Ge1–C1 = 1.923(5), Ge1–C2 = 1.923(5), Ge1–P1 = 2.350(1), Ge1–P2 = 2.363(1); C1–Ge1–C2 = 112.5(2), P1–Ge1–P2 = 122.4(1), C1–Ge1–P1 = 90.9(2), C2–Ge1–P2 = 91.4(2), C1–Ge1–P2 = 120.8(2), C2–Ge1–P1 = 121.2(2); (b) Ge1–C1 = 1.924(3), Ge1–C2 = 1.920(3), P1–Ge1 = 2.344(1), Ge1–P2 = 2.338(1); C2–Ge1–C1 = 126.8(1), C2–Ge1–P2 = 91.5(1), C1–Ge1–P2 = 116.1(1), C2–Ge1–P1 = 115.7(1), C1–Ge1–P1 = 91.2(1), P2–Ge1–P1 = 118.1(1). | ||
In the case of the diphenylphosphanyl donor, the neutral compound [LPh2GeCl2] 1Ph could be obtained from the reaction between lithiated LPh–Br and GeCl4 taken in a 2:1 ratio in a THF medium (Scheme 1b and Fig. S96, see the ESI† for Experimental procedures). Chloride abstractions from 1Ph using one and two equivalents of TMSOTf in DCM led to the formation of mono-cationic 2Ph and di-cationic 3Ph compounds, respectively (Scheme 1b). The characteristic peaks in 31P{1H} NMR spectra for 2Ph and 3Ph appear at −13.2 and −6.3 ppm respectively.
Unlike 2iPr, the molecular structure of 2Ph depicts a distorted tetrahedral geometry (Fig. 2b). Compared to 2iPr, the Ge1–P1 bond length has noticeably reduced in 2Ph (2.357(1) Å). Another Ge1⋯P2 bond distance in 2Ph is 3.012(1) Å. The triflate anion is far away from the Ge atom (closest Ge–O contact being 7.2 Å). The low temperature 31P{1H} NMR for 2Ph echoes the presence of two inequivalent phosphines exhibiting chemical shift values at −3.0 and −20.5 ppm.
The solid-state structure of 3Ph exhibits a spiro-geometry with similar bond parameters to those in 3iPr (Fig. 3b). There are two non-coordinated triflate anions (closest Ge–O contact being 4.2 Å) present in the asymmetric unit. Both 3iPr and 3Ph in the solid-state show no sign of decomposition when exposed to air for a day.
The optimized geometries of 2oiPr, 2oPh, 3oiPr and 3oPh at the TPSS-D3(BJ)/def2-TZVPP level of theory are in close agreement with the X-ray parameters (see the ESI† for the detailed theoretical part). On par with the experimental findings, Natural Bond Orbital (NBO) analyses for 2oiPr suggest significant donor–acceptor (D–A) interactions (avg. 125 kcal mol−1) for 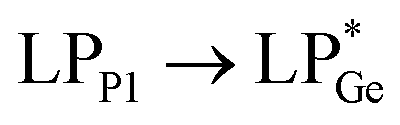 and
and 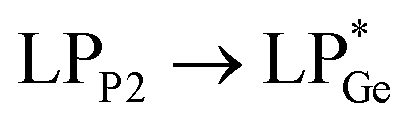 . However, dissimilar bonding situations in 2oPh between Ge1–P1 (covalent) and Ge1–P2 (D–A interaction) were confirmed by NBO and AIM (Atoms in Molecules) analyses. The calculated Wiberg bond index (WBI) of Ge1–P2 in 2oPh possesses dative nature with a value of 0.183 (0.712 for Ge1–P1). Additionally, no bonding orbital for Ge1–P2 was observed from the NBO analyses. Instead, only one D–A interaction
. However, dissimilar bonding situations in 2oPh between Ge1–P1 (covalent) and Ge1–P2 (D–A interaction) were confirmed by NBO and AIM (Atoms in Molecules) analyses. The calculated Wiberg bond index (WBI) of Ge1–P2 in 2oPh possesses dative nature with a value of 0.183 (0.712 for Ge1–P1). Additionally, no bonding orbital for Ge1–P2 was observed from the NBO analyses. Instead, only one D–A interaction 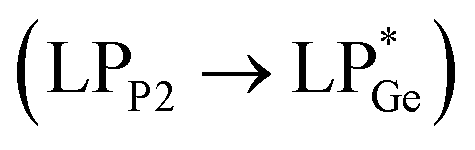 is determined in 2oPh with a stabilization energy of 32 kcal mol−1. Furthermore, we have performed AIM analyses. A negative Laplacian value at the BCPs (bond critical points) is associated with shared interactions, indicating covalent bonds, and positive Laplacian values reflect closed shell interactions such as ionic or dative bonds. In the case of 2oPh, the higher positive ∇2ρ(r) value on Ge–P2 shows dative bonding character.
is determined in 2oPh with a stabilization energy of 32 kcal mol−1. Furthermore, we have performed AIM analyses. A negative Laplacian value at the BCPs (bond critical points) is associated with shared interactions, indicating covalent bonds, and positive Laplacian values reflect closed shell interactions such as ionic or dative bonds. In the case of 2oPh, the higher positive ∇2ρ(r) value on Ge–P2 shows dative bonding character.
The frontier molecular orbitals (FMOs) of 3oiPr and 3oPh reflect similar contributions in terms of Ge–C and Ge–P σ* orbitals (Fig. 4a and b). The orbital scenario is analogous to those observed in the catecholato phosphonium ion19a or stibonium salt.27 There is no pure vacant p-orbital available28 on the central Ge atom in both cases. WBI calculations predict the values of 0.804 (3oiPr) and 0.770 (3oPh) for Ge–P bonds, displaying a mostly covalent bonding situation. The dispersion of positive charges among the P (+1.161, +1.145 for 3oiPr; +1.174, +1.174 for 3oPh) and Ge (+1.136 for 3oiPr; +1.157 for 3oPh) sites is evident from NBO partial charges and the electrostatic potential map analyses (Fig. 4c and d). Canonical forms describing the dispersion of positive charges in 3oiPr and 3oPh (F) are depicted in Fig. 1.
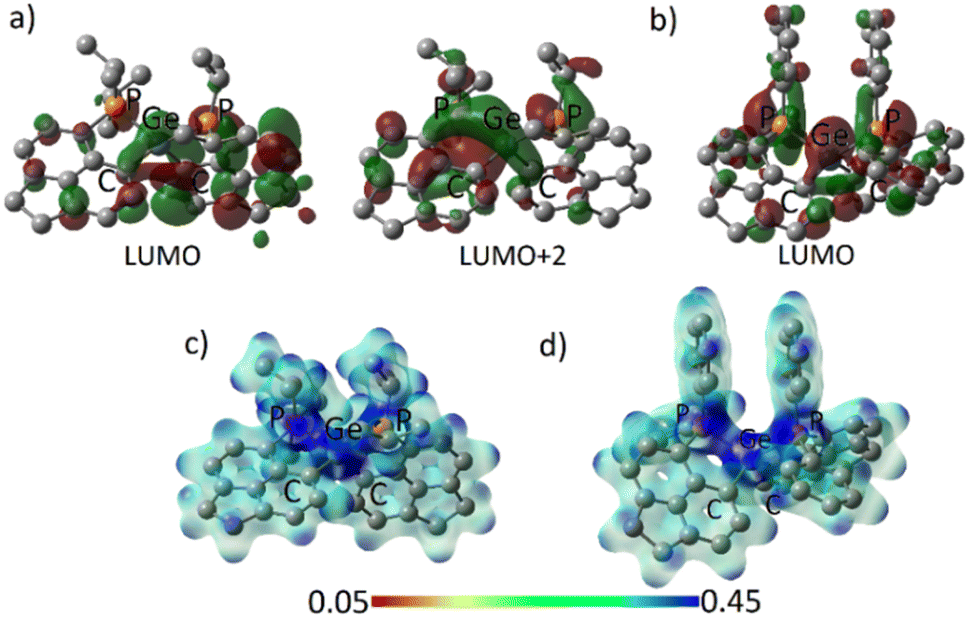 | ||
| Fig. 4 (a) Acceptor orbitals in 3oiPr; (b) LUMO in 3oPh; electrostatic potential map for (c) 3oiPr and (d) 3oPh. | ||
Lewis acidic properties of Ge(IV) di-cationic compounds
We have studied the effective Lewis acidity of both 3iPr and 3Ph from induced 31P NMR shift of triethylphosphine oxide (Et3PO) in the Gutmann–Beckett (GB) method (see the ESI† for details).29 Et3PO adducts were targeted by employing varying equivalents of Et3PO (0.2 to 3.0 equivalents) in deuterated solvents. The addition of 0.2 equivalents of Et3PO to 3iPr gave numerous peaks at +75.7, +74.2, −12.3 and −18.2 ppm. The complexity of the 31P{1H} NMR spectra obtained at room temperature impeded the Lewis acidity scaling of 3iPr by the GB method. However, we have observed peaks appearing at +73.4 and −12.0 ppm in the 31P{1H} NMR spectrum, when a 1:1 mixture of 3iPr and Et3PO was prepared in CD2Cl2 at −78 °C and subsequently raised to room temperature. This gave an induced 31P{1H} NMR chemical shift of Et3PO for 3iPr (Δ31P = 21.3 ppm) with respect to the free Et3PO (δ for free Et3PO in the same solvent is 52.1 ppm).
In the case of 3Ph, we have noticed the induced 31P{1H} NMR chemical shift from the addition of 0.2 equivalents of Et3PO in CDCl3 (Δ31P = 21.6 ppm, δ for free Et3PO in the same solvent is 50.0 ppm); without any complexity observed in the NMR spectrum at room temperature. These Δ31P values are close to the values reported for the Si(IV) di-cations (Δ31P = 23.5 ppm for [Ph2Si(terpy)]2+).7 Both 3iPr and 3Ph exhibit similar effective Lewis acidity as obtained following the GB method. The peak at +71.6 ppm in the 31P{1H} NMR spectrum assigned to the bound Et3PO is shifted up-field upon increasing the amount of Et3PO. This obvious shift is due to the expected weak acceptor ability upon further Et3PO coordination at the cationic site.10f We have crystallized the mono-adduct 3PhOPEt3 (Fig. S97†) under low temperature conditions from the reaction between 3Ph and Et3PO. The molecular structure of 3PhOPEt3 shows a penta-coordinated Ge site. Interestingly, the solution-state NMR study of 3PhOPEt3 shows characteristic 31P{1H} NMR chemical shifts at +68.5 and −20.5 ppm along with a peak at −6.3 ppm assigned to the in situ generated free 3Ph. This observation reflects the presence of the equilibrium in the solution-state.
Despite the dispersion of dipositive charges over the P–Ge–P sites in 3iPr and 3Ph, we have observed the preferential binding of Lewis bases such as Et3PO and 4-N,N′-dimethylaminopyridine (DMAP) at the Ge site (Scheme 2). The reaction between 3iPr and DMAP taken in a 1:1 ratio gave 3iPrDMAP, which was crystallized in small amounts from DCM/hexane layering at room temperature (Scheme 2). Single crystals of dimethylamino pyridium triflate were obtained in large amounts, which were separated from the 3iPrDMAP crystals. The molecular structure of 3iPrDMAP from X-ray analysis reveals an overall TBP geometry with the DMAP coordination at one of the trigonal planar equatorial sites of the Ge atom (Fig. S98†). However, 3iPrDMAP is unstable under room temperature conditions in the solution state. The 31P{1H} NMR of in situ generated 3iPrDMAP at −40 °C in CD2Cl2 shows a peak at −13.2 ppm in addition to the uncoordinated 3iPr peak (+28.4 ppm) present in the reaction medium. These peaks gradually disappeared upon warming to room temperature, ultimately giving new peaks at +75.7 ppm and −18.2 ppm (vide supra). We have observed a peak at +12.9 ppm in the 1H NMR spectrum corresponding to the formation of the conjugate acid dimethylamino pyridium triflate. Similar side-reactions might be involved in the reaction between 3iPr and donors (Et3PO and DMAP), as was reported earlier by Stephan et al.30
 | ||
| Scheme 2 Anion/Lewis base binding and bond activation by the Ge(IV) di-cationic compounds (see the ESI† for detailed spectroscopic and experimental procedures of each reaction). | ||
A dynamic process13a between mono- and bis-adduct formation was observed upon reacting 3Ph with one equivalent of DMAP at room temperature. The NMR scale reaction in CDCl3 shows a peak at −18.5 ppm and a broad peak centered at −27.0 ppm in the 31P{1H} NMR spectrum. The gradual decrease in temperature up to −50 °C resulted in a complete disappearance of the peak at −18.5 ppm. Concurrently two close sharp peaks at ∼−29 ppm appeared along with the generation of the peak for 3Ph at −6.3 ppm. Inferring from this NMR study, a bis-adduct was isolated (corresponding peak at ∼−29 ppm) from the reaction between DMAP and 3Ph at −35 °C. The crystallization from DCM/pentane layering at the same temperature gave single crystals of trans bis-adduct 3Ph(DMAP)2 (Fig. S99†). As a matter of fact, in this case we find the spontaneous formation of the more favourable hexa-coordinated bis-adduct over the mono-adduct at lower temperatures.11d The octahedral coordination type at the Ge atom of 3Ph(DMAP)2 comprises two LPh in the equatorial plane and two DMAPs occupying the axial sites. Thus, dynamic processes were observed for the adducts formed between 3Ph and donors.
The preferential binding of donor groups at the Ge site is associated with the relief in constraint on going from a strained spiro-geometry in 3iPr and 3Ph to the relaxed TBP (3iPrDMAP) and distorted tetrahedral geometry (3PhOPEt3) in the adducts, respectively. Notably, this results in a handful of penta-coordinated Ge compounds obtained having TBP geometry which are uncommon in Ge chemistry.11d The viable expansion of the peri-distances22 in the two intramolecular P–Ge bonds in 3iPr and 3Ph allows smooth structural rearrangements with the addition of nucleophiles.
We have validated the above understanding from the deformation energy9,29 values obtained in the spiro- to TBP geometric transformation. We have prepared 3iPrF from the addition of one equivalent of KF to 3iPr (Scheme 2), which exhibits a distorted TBP geometry (Fig. 5a) like 2iPr in the solid state. DFT calculations reveal that the fluoride ion binding to 3iPr involves a low deformation energy (Edef = 126 kJ mol−1), falling within the range reported in the literature.10g,19a,293iPrF was characterised by NMR spectroscopy in the solution state showing a characteristic triplet at −141.0 ppm (2JF–P = 185 Hz) in the 19F{1H} NMR spectrum and a corresponding doublet in the 31P{1H} NMR spectrum (−10.2 ppm).
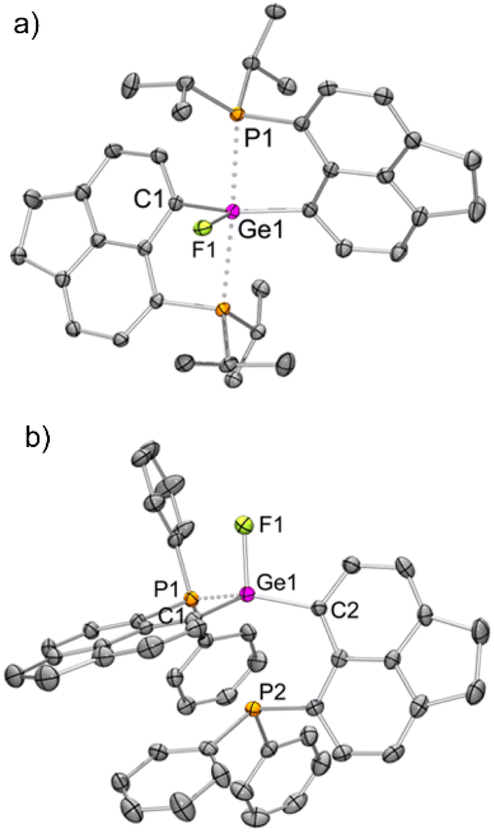 | ||
| Fig. 5 Molecular structures of (a) 3iPrF and (b) 3PhF (H atoms, solvent molecule and triflate anion have been removed for clarity; thermal ellipsoid 50%). Selected bond lengths [Å] and angles [°]: (a) Ge1–C1 = 1.951(2), Ge1–F1 = 1.785(2), Ge1–P1 = 2.565(1); C1–Ge1–C1 = 145.1(1), F1–Ge1–C1 = 107.5(1), P1–Ge1–P1 = 172.8(1), P1–Ge1–F1 = 86.4(1); (b) Ge1–F1 = 1.797(2), Ge1–C2 = 1.920(3), Ge1–C1 = 1.927(3), Ge1–P1 = 2.368(1), Ge1–P2 = 3.043(1); F1–Ge1–C2 = 102.1(1), F1–Ge1–C1 = 102.8(1), C2–Ge1–C1 = 120.6(1), F1–Ge1–P1 = 97.3(1), C2–Ge1–P1 = 137.3(1), C1–Ge1–P1 = 91.2(1). | ||
Like 3iPr, the addition of one equivalent of KF led to the formation of 3PhF (Scheme 2) showing similar characteristic peaks in NMR spectra (δ19F{1H}: −150.3 ppm, triplet, and 2JP–F = 100 Hz; δ31P{1H}: −17.5 ppm). The molecular structure of 3PhF (Fig. 5b) resembles that of 2Ph. The low temperature 31P{1H} NMR spectrum reveals the presence of inequivalent phosphines in 3PhF. In this case, the release of the structural constraint occurs on going from spirocyclic 3Ph to the relaxed geometry in 3PhF involving a deformation energy value of 148 kJ mol−1.10g,19a,29
The calculations on the gas-phase fluoride ion affinity (FIA)31 at the Ge sites give very high values of 873 kJ mol−1 and 859 kJ mol−1 for 3iPr and 3Ph respectively (gas-phase FIA for reference SbF5 is 497 kJmol−1) (see the ESI† for details). However, the calculated FIA values in DCM solvated models decreased significantly giving the values of 194 kJ mol−1 and 190 kJ mol−1 for 3iPr and 3Ph, respectively (solvent corrected FIA for reference SbF5 is 331 kJmol−1). This is a common phenomenon observed due to solvation damping, being more pronounced with a cationic Lewis acid.7,19a Both 3iPr and 3Ph did not abstract the fluoride anion when reacted with [PPh4][SbF6] and hence are not Lewis superacids. The chloride ion affinity study using trityl chloride shows that only 3Ph abstracts chloride with characteristic coloration10g and the NMR data reveal the formation of 2Ph (Scheme 2 and Fig. S62, ESI†).
Gas-phase hydride ion affinity (HIA)31 calculations also give very high values of 949 kJ mol−1 and 928 kJ mol−1 for 3iPr and 3Ph, respectively (gas-phase HIA for reference B(C6F5)3 is 517 kJ mol−1) (see the ESI† for details). Reasonable HIA values are obtained after considering the solvation in DCM: 254 kJ mol−1 and 269 kJ mol−1 for 3iPr and 3Ph, respectively (solvent corrected HIA for reference B(C6F5)3 is 270 kJmol−1). Thus, both 3iPr and 3Ph are hydrophilic in nature. 3iPr efficiently abstracts the hydride from LiBEt3H at room temperature leading to the formation of 3iPrH (Scheme 2). The 1H NMR spectrum of 3iPrH reveals the Ge–H resonance as a triplet (2JP–H = 36 Hz) at +7.2 ppm; the chemical shift value falls within the range of reported Ge-hydrides.15 The molecular structure of 3iPrH attains a distorted TBP geometry (Fig. S100†) analogous to 2iPr. The Ge–H bond length of 1.540 Å in the optimized geometry 3oiPrH agrees well with that found in the molecular structure of 3iPrH (1.54(3) Å) (see the ESI† for details). The reaction of 3Ph with two equivalents of LiBEt3H gave the neutral 3PhH2 (Scheme 2). The molecular structure of 3PhH2 is shown in Fig. S101.† The Ge⋯P distances in 3PhH2 are close to 3 Å; however they fall within the sum of van der Waal's radii ΣvdW(Ge–P) = 3.91 Å.32 The 1H NMR spectrum of 3PhH2 reveals a Ge–H2 resonance at 6.4 ppm as a triplet (JP–H = 20 Hz). The reaction between 3Ph and one equivalent of LiBEt3H followed by crystallization from different solvent combinations gives the single crystals of 3PhEt and 3PhH(Et) (Scheme 2 and Fig. S102, S103, ESI†), confirming B–C bond activation.33 Thus, preliminary reactivity studies identify 3Ph as a highly reactive di-cation.
Catalytic hydrosilylation of an aromatic aldehyde by the Ge(IV) di-cationic compounds
Given the hydrophilicity of the soft Ge-based Lewis acids 3iPr and 3Ph, we targeted the activation of the Si–H bond in Et3SiH (see the ESI† for details).34 Monitoring the reaction between 3iPr and excess Et3SiH at room temperature using 31P NMR displays the formation of two new peaks at +20.3 ppm and +12.2 ppm along with retention of 3iPr. The peak at +20.3 ppm showed a coupling of JPH ≈ 476 Hz. Furthermore, 1H–31P 1D and 2D Heteronuclear Single Quantum Coherence (HSQC) NMR measurements (CNST2 optimized at 480 Hz) confirm that 1JPH = 476 Hz.35 Thus, the Si–H bond activation and hydride binding occur preferentially at the P-site over the Ge-site giving 3iPr–PH. This is in contrast to 3iPrH formed by hydride abstraction from LiBEt3H (vide supra). Notably, although Si–H bond activation occurs at the P-site of 3iPr, standing of the reaction mixture for 14 days ultimately led to Ge–H bond formation (t, 2JP–H = 36 Hz at +7.2 ppm) giving 3iPrH.In contrast, 3Ph activates the Si–H bond in Et3SiH giving 3PhH (Scheme 3a) as the product within 24 hours (see the ESI† for details). However, the time dependent 31P NMR monitoring of the initial reaction mixture displays a minor peak at +10.0 ppm with a coupling of 1JPH = 534 Hz corresponding to the P–H formation, which subsequently disappeared giving 3PhH within 24 hours. The 1H NMR spectrum of 3PhH discloses the Ge–H resonance at +7.0 ppm as a triplet (2JP–H = 30 Hz) and a corresponding doublet at −19.0 ppm in the 31P{1H} NMR spectrum. The 29Si{1H} NMR of the reaction mixture shows the corresponding formation of Et3SiOTf (δ = +45.0 ppm).
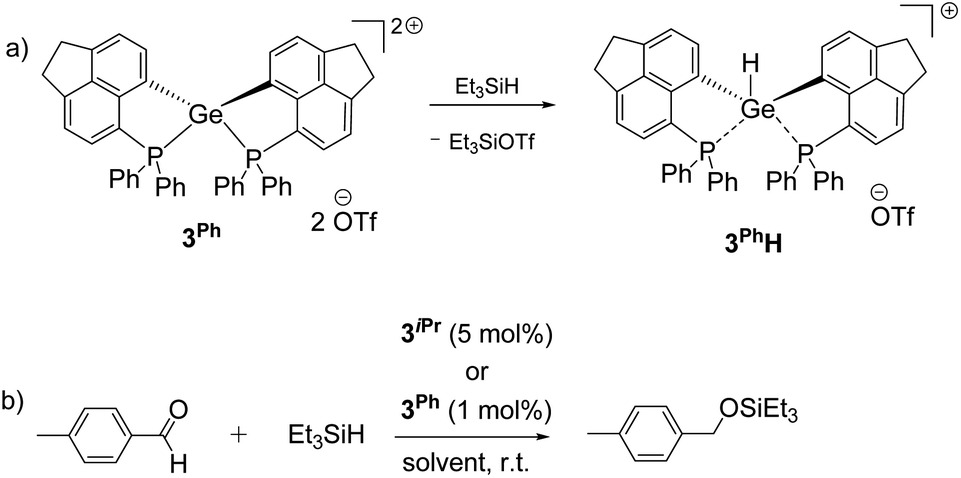 | ||
| Scheme 3 (a) Si–H bond activation by 3Ph leading to 3PhH within 24 hours; (b) hydrosilylation of p-methyl benzaldehyde using 3iPr or 3Ph as the catalyst (see the ESI† for the detailed spectroscopic and experimental procedures of each reaction). | ||
The above-mentioned observations suggest that the hydride binding initially occurs at the P-site due to the dispersion of dipositive charges over P–Ge–P units in these di-cationic species. Computational investigations done on the hydride migration from P to Ge for both 3iPr and 3Ph reveal the involvement of a relatively higher transition state energy barrier in the case of the former (Fig. S112, ESI†). The hydride migration mechanism proposed based on the DFT calculations performed on the cationic part of the compounds proves only the trends of the hydride shifts from P to Ge. Nonetheless, the very low energy barriers obtained are inadequate to support the longer time requirements for hydride migrations observed experimentally. Probably, the inclusions of the solvent models and counter anions to the systems would be able to resolve the disagreement at an associated extremely high computational cost. Thus, the hydride migration mechanism proposed herein is rather based on the experimentally observed data. Additionally, the strong electron donating nature of iPr2P compared to Ph2P pushes electron density towards the Ge-site in 3iPr thereby making the P-site appropriate for hydride. The ultimate formation of 3iPrH or 3PhH stems from the geometric constraint empowered Lewis acidity at the Ge site of 3iPr or 3Ph respectively (vide supra).
Despite the differences observed in the reaction with Et3SiH, the intrinsic hydrophilicity of 3iPr and 3Ph prompted us to perform a test reaction on the catalytic hydrosilylation36 of p-methyl benzaldehyde with Et3SiH at room temperature (Scheme 3b, see the ESI† for details). Using 5 mol% of 3iPr led to the complete conversion to the silyl ether within 24 hours. It is observed from the 1H NMR study that the aldehyde does not bind to the catalyst, unlike most Lewis acid catalysts.10e Thus, the reaction is likely to proceed via silane activation by 3iPr (Scheme 4).37 The NMR investigations of the catalytic reaction mixture show P-mediated Si–H bond activation (1H NMR = +8.5 ppm; 31P NMR = +20.2 ppm). The 29Si{1H} NMR spectrum shows the formation of a hydrosilylated product and the presence of Et3SiH with the corresponding peaks at +8.9 ppm and +0.3 ppm, respectively. The 29Si{1H} NMR spectrum did not show the presence of Et3SiOTf which is a competent catalyst. Nonetheless, the latter's presence in trace amounts and participation in the catalytic cycle cannot be completely neglected. The control experiment shows no hydrosilylated product being formed, as confirmed from the 1H NMR study of the reaction mixture containing p-methyl benzaldehyde and Et3SiH (Fig. S94, ESI†).
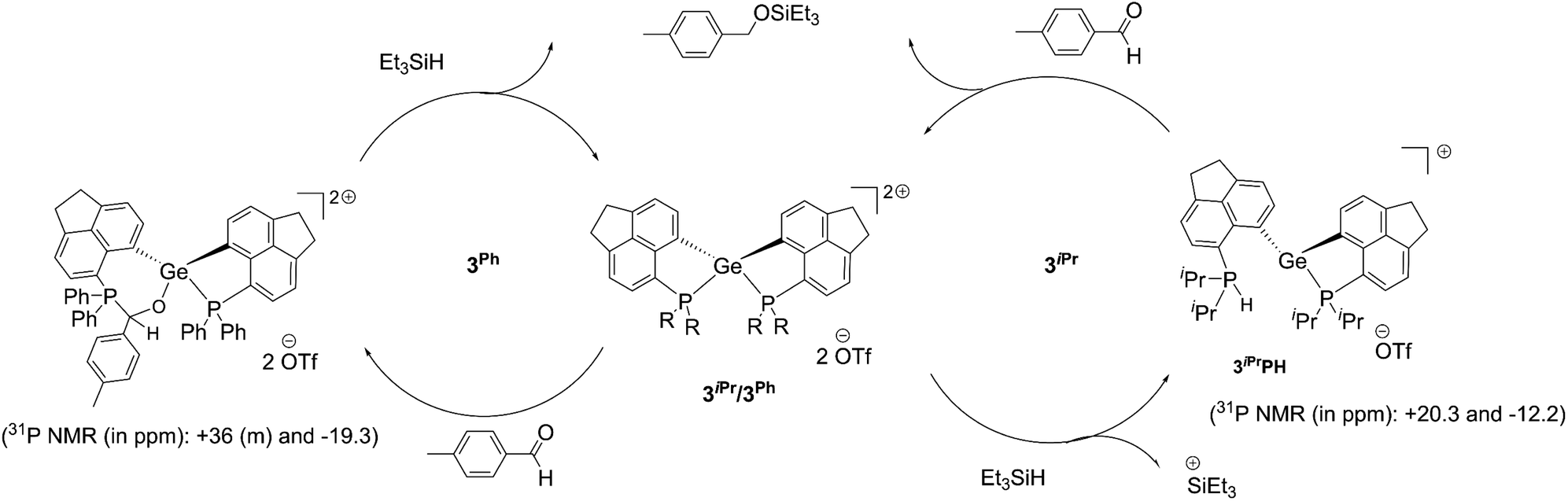 | ||
| Scheme 4 Proposed catalytic cycle for the hydrosilylation of aromatic aldehydes using 3iPr and 3Ph as catalysts. | ||
Full conversion of p-methyl benzaldehyde to the silyl ether was achieved within 24 hours under room temperature conditions by using a lower catalyst loading to 1 mol% of 3Ph (see the ESI† for details). Importantly, the aldehyde (in the absence of any other reactant) binds to 3Ph as evident from the NMR study. The 31P{1H} NMR spectrum depicts a peak at −19.3 ppm and multiple peaks at around +36 ppm in addition to the peak for free 3Ph (−6.3 ppm). Correspondingly, we have obtained single crystals from a THF solution of 3Ph and excess aldehyde. The crystal structure shows the insertion of an aldehyde into the P–Ge bond in 3Ph (Fig. S104, ESI†). Thus, a masked frustrated Lewis pair (FLP) type behavior21,38 of 3Ph was observed, which is unsurprising due to the adaptability of the P–Ge bond in peri-systems. The NMR spectrum recorded immediately after the addition of 3Ph to the reaction mixture of the aldehyde and Et3SiH also displays similar chemical shift values (−19.3 and around +36 ppm in the 31P{1H} NMR study). The NMR study did not show any peak arising from Si–H bond activation by 3Ph. This suggests the formation of the carbonyl-FLP insertion product taking place at the initial stage of the catalytic cycle. Notably, no Et3SiOTf formation is detected from the 29Si{1H} NMR monitoring (Fig. S93, ESI†). Thus, the catalytic hydrosilylation is likely to proceed via the carbonyl insertion followed by the R3Si–H addition across the carbonyl functional group (Scheme 4). The catalyst 3Ph is clearly observed after complete consumption of the reactants.
Thus, both 3iPr and 3Ph show different pathways catalysing the hydrosilylation of the aldehyde. Based on our experimental findings, while the former proceeds through the Si–H bond activation at the Lewis acidic site, the latter utilizes the masked FLP for the aldehyde insertion followed by Et3Si–H addition to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond.
O bond.
Conclusion
In this study, we have established the first examples of tetra-coordinated Ge(IV) di-cationic compounds 3iPr and 3Ph. This was achieved through the intramolecular stabilization of the Ge di-cationic site using a phosphine donor in peri-substituted acenaphthene. The strong P–Ge bonds led to the dispersion of di-cationic charges over Ge and P sites. The simple anion binding or Lewis base coordination occurs exclusively at the Ge site owing to the release of the geometric constraint on going from the spiro-geometry of the di-cation to the distorted TBP/tetrahedral geometry of the resultant species. Although the Si–H bond activation occurs initially at the P site for both the di-cationic species due to their dispersed di-positive charges, the migration of hydrides to the Ge site occurs promptly in the case of 3Ph compared to 3iPr. Preliminary tests on the catalytic hydrosilylation of the benzaldehyde demonstrate the involvement of two different catalytic pathways for the two catalysts 3iPr and 3Ph. In the case of 3iPr, the catalytic hydrosilylation was initiated with Et3Si–H bond activation. On the other hand, masked Frustrated Lewis Pair (FLP) type reactivity led to hydrosilylated product formation for 3Ph as the catalyst. Fine tuning the donor group properties in such charged peri species is being explored in our group for their applicability as catalysts in diverse organic transformations.Data availability
All data associated with this publication are provided in the ESI.†Author contributions
Investigation, methodology and formal analysis: Balakrishna Peddi and Souvik Khan. X-ray crystallography: Rajesh G. Gonnade. Computational study, comment and editing draft: Cem B. Yildiz. Conceptualization, funding acquisition, supervision, and writing-original draft: Moumita Majumdar.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank SERB India (SPF/2022/000046, CRG/2022/000673) for their financial support. The numerical calculations reported in this paper were partially performed at the TUBITAK ULAKBIM, High Performance and Grid Computing Center (TRUBA resources) and Param Brahma supercomputing facility at IISER Pune. The authors acknowledge the ‘Chemical Biology and Molecular Imaging Group, Department of Chemical Sciences, Tata Institute of Fundamental Sciences’ along with Prof. Ankona Datta and Ms. Deepika Rao for elemental analyses. The authors thank Mr Hritwik Haldar, a 5th year BS–MS student, for assisting in computational studies. The authors thank all the reviewers for their valuable comments in improving the manuscript.References
- Selected references: (a) P. A. Rupar, V. N. Staroverov, P. J. Ragogna and K. M. Baines, J. Am. Chem. Soc., 2007, 129, 15138 CrossRef CAS PubMed; (b) P. A. Rupar, V. N. Staroverov and K. M. Baines, Science, 2008, 322, 1360 CrossRef CAS PubMed; (c) Y. Xiong, T. Szilvási, S. Yao, G. Tan and M. Driess, J. Am. Chem. Soc., 2014, 136, 11300 CrossRef CAS PubMed; (d) P. A. Rupar, R. Bandyopadhyay, B. F. T. Cooper, M. R. Stinchcombe, P. J. Ragogna, C. L. B. Macdonald and K. M. Baines, Angew. Chem., Int. Ed., 2009, 48, 5155 CrossRef CAS; (e) F. Cheng, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Angew. Chem., Int. Ed., 2009, 48, 5152 CrossRef CAS; (f) T. A. Engesser, M. R. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789 RSC; (g) V. S. V. S. N. Swamy, S. Pal, S. Khan and S. S. Sen, Dalton Trans., 2015, 44, 12903 RSC; (h) S. Biswas, N. Patel, R. Deb and M. Majumdar, Chem. Rec., 2022, 22, e202200003 CrossRef CAS PubMed.
- R. Bandyopadhyay, J. H. Nguyen, A. Swidan and C. L. B. Macdonald, Angew. Chem., Int. Ed., 2013, 52, 3469 CrossRef CAS PubMed.
- R. K. Raut and M. Majumdar, Chem. Commun., 2017, 53, 1467 RSC.
- R. Suter, A. Swidan, C. L. B. Macdonald and N. Burford, Chem. Commun., 2018, 54, 4140 RSC.
- R. P. King, W. Levason and G. Reid, Dalton Trans., 2021, 50, 17751 RSC.
- A. P. M. Robertson, J. N. Fiedmann, H. A. Jenkins and N. Burford, Chem. Commun., 2014, 50, 7979 RSC.
- A. Hermannsdorfer and M. Driess, Angew. Chem., Int. Ed., 2020, 59, 23132 CrossRef CAS PubMed.
- A. Hermannsdorfer and M. Driess, Angew. Chem., Int. Ed., 2021, 60, 13656 CrossRef CAS.
- (a) L. Greb, Chem.–Eur. J., 2018, 24, 17881 CrossRef CAS PubMed; (b) L. O. Müller, D. Himmel, J. Stauffer, G. Steinfeld, J. Slattery, G. Santiso-Quiñones, V. Brecht and I. Krossing, Angew. Chem., Int. Ed., 2008, 47, 7659 CrossRef PubMed; (c) T. Thorwart and L. Greb, Lewis Superacids, in Encyclopedia of Inorganic and Bioinorganic Chemistry, 2021, DOI:10.1002/9781119951438.eibc2758.
- (a) H. F. T. Klare and M. Oestreich, Dalton Trans., 2010, 39, 9176 RSC; (b) H. F. T. Klare, L. Albers, L. Süsse, S. Keess, T. Müller and M. Oestreich, Chem. Rev., 2021, 121, 5889 CrossRef CAS PubMed; (c) T. Müller, in Functional Molecular Silicon Compounds I, ed. D. Scheschkewitz, Structure and Bonding, Springer, 2013, ch. 3 Search PubMed; (d) T. Stahl, H. F. T. Klare and M. Oestreich, ACS Catal., 2013, 3, 1578 CrossRef CAS; (e) A. L. L. Martin, R. G. Bergman and T. D. Tilley, J. Am. Chem. Soc., 2015, 137, 5328 CrossRef PubMed; (f) R. Maskey, M. Schadler, C. Legler and L. Greb, Angew. Chem., Int. Ed., 2018, 57, 1717 CrossRef CAS PubMed; (g) D. Hartmann, M. Schädler and L. Greb, Chem. Sci., 2019, 10, 7379 RSC; (h) F. S. Tschernuth, T. Thorwart, L. Greb, F. Hanusch and S. Inoue, Angew. Chem., Int. Ed., 2021, 60, 25799 CrossRef CAS PubMed.
- (a) H. C. Clark and C. J. Willis, J. Am. Chem. Soc., 1962, 84, 898 CrossRef CAS; (b) D. P. Graddon and B. A. Rana, J. Organomet. Chem., 1979, 165, 157 CrossRef CAS; (c) D. J. Brauer, H. Bürger and R. Eujen, Angew Chem. Int. Ed. Engl., 1980, 19, 836 CrossRef; (d) S. E. Denmark, R. T. Jacobs, G. Dai-Ho and S. Wilson, Organometallics, 1990, 9, 3015 CrossRef CAS; (e) S. Pelzer, B. Neumann, H.-G. Stammler, N. Ignat’ev and B. Hoge, Chem.–Eur. J., 2016, 22, 3327 CrossRef CAS PubMed; (f) S. Pelzer, B. Neumann, H.-G. Stammler, N. Ignat’ev and B. Hoge, Chem.–Eur. J., 2016, 22, 16460 CrossRef CAS PubMed; (g) T. A. Kinder, R. Pior, S. Blomeyer, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem.–Eur. J., 2019, 25, 5899 CrossRef CAS PubMed.
- D. Roth, H. Wadepohl and L. Greb, Angew. Chem., Int. Ed., 2020, 59, 20930 CrossRef CAS PubMed.
- (a) A. T. Henry, T. P. L. Cosby, P. D. Boyle and K. M. Baines, Dalton Trans., 2021, 50, 15906 RSC; (b) D. Basu and H. P. Nayek, Dalton Trans., 2022, 51, 10587 RSC.
- H. Fang, H. Jing, A. Zhang, H. Ge, Z. Yao, P. J. Brothers and X. Fu, J. Am. Chem. Soc., 2016, 138, 7705 CrossRef CAS PubMed.
- P. M. Keil and T. J. Hadlington, Angew. Chem., Int. Ed., 2022, 61, e202114143 CrossRef CAS PubMed.
- M. Karimi, E. S. Tabei, R. Fayad, M. R. Saber, E. O. Danilov, C. Jones, F. N. Castellano and F. P. Gabbai, Angew. Chem., Int. Ed., 2021, 60, 22352 CrossRef CAS PubMed.
- Selected reviews on Group 13 Lewis acids: (a) E. I. Davydova, T. N. Sevastianova and A. Y. Timoshkin, Coord. Chem. Rev., 2015, 297, 91 CrossRef; (b) I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270–271, 75 CrossRef CAS; Selected reviews on Group 14 Lewis acids: (c) H. Yamamoto, Silicon(IV) Lewis Acids, Wiley-VCH, Weinheim, 2000 Search PubMed; (d) T. Müller, Adv. Organomet. Chem., 2005, 53, 155 CrossRef; (e) S. A. Weicker and D. W. Stephan, Bull. Chem. Soc. Jpn., 2015, 88, 1003 CrossRef CAS; (f) D. J. Scott, N. A. Phillips, J. S. Sapsford, A. C. Deacy, M. J. Fuchter and A. E. Ashley, Angew. Chem., 2016, 128, 14958 CrossRef; Selected reviews on Group 15 Lewis acids: (g) C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374 CrossRef CAS PubMed; (h) J. M. Bayne and D. W. Stephan, Chem. Soc. Rev., 2016, 45, 765 RSC; (i) D. W. Stephan, Angew. Chem., Int. Ed., 2017, 56, 5984 CrossRef CAS PubMed; (j) J. M. Bayne and D. W. Stephan, Chem.–Eur. J., 2019, 25, 9350 CrossRef CAS PubMed; (k) C. Lichtenberg, Chem. Commun., 2021, 57, 4483 RSC; (l) M. Yang, D. Tofan, C.-H. Chen, K. M. Jack and F. P. Gabbai, Angew. Chem., Int. Ed., 2018, 57, 13868 CrossRef CAS PubMed.
- (a) K. Chulsky, I. Malahov, D. Bawari and R. Dobrovetsky, J. Am. Chem. Soc., 2023, 145, 3786 CrossRef CAS PubMed; (b) S. Volodarsky, D. Bawari and R. Dobrovetsky, Angew. Chem., Int. Ed., 2022, 61, e202208401 CrossRef CAS PubMed; (c) A. Ben Saida, A. Chardon, A. Osi, N. Tumanov, J. Wouters, A. I. Adjieufack, B. Champagne and G. Berionni, Angew. Chem., Int. Ed., 2019, 58, 16889 CrossRef CAS PubMed; (d) J. C. Gilhula and A. T. Radosevich, Chem. Sci., 2019, 10, 7177 RSC; (e) K. M. Marczenko, J. A. Zurakowski, M. B. Kindervater, S. Jee, T. Hynes, N. Roberts, S. Park, U. Werner-Zwanziger, M. Lumsden, D. N. Langelaan and S. S. Chitnis, Chem.–Eur. J., 2019, 25, 16414 CrossRef CAS PubMed.
- (a) D. Roth, J. Stirn, D. W. Stephan and L. Greb, J. Am. Chem. Soc., 2021, 143, 15845 CrossRef CAS PubMed; (b) F. Ebner, L. M. Sigmund and L. Greb, Angew. Chem., Int. Ed., 2020, 59, 17118 CrossRef CAS PubMed.
- (a) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018 CrossRef CAS PubMed; (b) D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed; (c) D. W. Stephan, Chemistry, 2020, 6, 1520 CrossRef CAS; (d) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS PubMed; (e) D. J. Scott, M. J. Fuchter and A. E. Ashley, Chem. Soc. Rev., 2017, 46, 5689 RSC.
- (a) D. Hartmann, S. Braner and L. Greb, Chem. Commun., 2021, 57, 8572 RSC; (b) I. Mallov, A. J. Ruddy, H. Zhu, S. Grimme and D. W. Stephan, Chem.–Eur. J., 2017, 23, 17692 CrossRef CAS PubMed; (c) T. Thorwart, D. Hartmann and L. Greb, Chem.–Eur. J., 2022, 28, e202202273 CrossRef CAS PubMed; (d) S. Freitag, J. Henning, H. Schubert and L. Wesemann, Angew. Chem., Int. Ed., 2013, 52, 5640 CrossRef CAS PubMed; (e) J. Schneider, K. M. Krebs, S. Freitag, K. Eichele, H. Schubert and L. Wesemann, Chem.–Eur. J., 2016, 22, 9812 CrossRef CAS PubMed; (f) J. Schneider, C. P. Sindlinger, S. M. Freitag, H. Schebert and L. Wesemann, Angew. Chem., Int. Ed., 2017, 56, 333 CrossRef CAS PubMed.
- (a) E. Hupf, E. Lork, S. Mebs and J. Beckmann, Organometallics, 2014, 33, 2409 CrossRef CAS; (b) J. Beckmann, E. Hupf, E. Lork and S. Mebs, Inorg. Chem., 2013, 52, 11881 CrossRef CAS PubMed; (c) P. Kilian, F. R. Knight and J. D. Woollins, Chem.–Eur. J., 2011, 17, 2302 CrossRef CAS PubMed; (d) Y. V. Vishnevskiy, A. A. Otlyotov, J.-H. Lamm, H.-G. Stammler, G. V. Girichev and N. W. Mitzel, Phys. Chem. Chem. Phys., 2023, 25, 11464 RSC.
- (a) V. Kumar, R. G. Gonnade, C. B. Yildiz and M. Majumdar, Angew. Chem., Int. Ed., 2021, 60, 25522 CrossRef CAS PubMed; (b) S. S. Chitnis, A. P. Robertson, N. Burford, J. J. Weigand and R. Fischer, Chem. Sci., 2015, 6, 2559 RSC.
- N. C. Gonnella, C. Busacca, S. Campbell, M. Eriksson, N. Grinberg, T. Bartholomeyzik, S. Ma and D. L. Norwood, Magn. Reson. Chem., 2009, 47, 461 CrossRef CAS PubMed.
- CCDC numbers: 2223776 (1iPr), 2223777 (2iPr), 2223780–2223782 (3iPr, 3iPrH, 3iPrF), 2223784 (3iPrDMAP), 2256454–2256458 (1Ph, 2Ph, 3Ph, 3Ph(DMAP)2, 3PhEt3PO), 2256463 (3PhF), 2256466–2256468 (3PhH2, 3PhEt, 3PhH(Et)), 2256567 (3PhOald).
- P. Sahoo, R. K. Raut, D. Maurya, V. Kumar, P. Rani, R. G. Gonnade and M. Majumdar, Dalton Trans., 2019, 48, 7344 RSC.
- B. Pan and F. P. Gabbai, J. Am. Chem. Soc., 2014, 136, 9564 CrossRef CAS PubMed.
- F. Ebner, H. Wadepohl and L. Greb, J. Am. Chem. Soc., 2019, 141, 18009 CrossRef CAS PubMed.
- P. Erdmann and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202114550 CrossRef CAS PubMed.
- M. H. Holthausen, R. R. Hiranandani and D. W. Stephan, Chem. Sci., 2015, 6, 2016 RSC.
- (a) P. Erdmann, J. Leitner, J. Schwarz and L. Greb, ChemPhysChem, 2020, 21, 987 CrossRef CAS PubMed; (b) P. Erdmann and L. Greb, ChemPhysChem, 2021, 22, 935 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- D. Carmichael, P. L. Floch, H. G. Trauner and F. Matthey, Chem. Commun., 1996, 971 RSC.
- (a) G. I. Nikonov, S. F. Vyboishchikov and O. G. Shirobokov, J. Am. Chem. Soc., 2012, 134, 5488 CrossRef CAS PubMed; (b) A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983 CrossRef CAS PubMed.
- M. H. Holthausen, J. M. Bayne, I. Mallov, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 7298 CrossRef CAS PubMed.
- (a) D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440 CrossRef CAS; (b) W. E. Piers and T. Chivers, Chem. Soc. Rev., 1997, 26, 345 RSC.
- D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090 CrossRef CAS PubMed.
- A. Dajnak, E. Maerten, N. Saffon-Merceron, A. Baceiredo and T. Kato, Organometallics, 2020, 39, 3403 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic analyses and theoretical details of all compounds. CCDC 2223776, 2223777, 2223780–2223782, 2223784, 2256454–2256458, 2256463, 2256466–2256468 and 2256567. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03717g |
| This journal is © The Royal Society of Chemistry 2023 |