
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn-DNA hydroalkylation of N-vinyl heterocycles via photoinduced EDA-complex activation†
Mohammed
Sharique‡
a,
Bianca
Matsuo‡
 a,
Albert
Granados‡
a,
Saegun
Kim
a,
Mahwish
Arshad
a,
Hyunjung
Oh
a,
Victoria E.
Wu
b,
Minxue
Huang
b,
Adam
Csakai
b,
Lisa A.
Marcaurelle
b and
Gary A.
Molander
*a
a,
Albert
Granados‡
a,
Saegun
Kim
a,
Mahwish
Arshad
a,
Hyunjung
Oh
a,
Victoria E.
Wu
b,
Minxue
Huang
b,
Adam
Csakai
b,
Lisa A.
Marcaurelle
b and
Gary A.
Molander
*a
aRoy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, Pennsylvania 19104-6323, USA. E-mail: gmolandr@sas.upenn.edu
bEncoded Library Technologies/NCE Molecular Discovery, R&D Medicinal Science and Technology, GSK, 200 Cambridge Park Drive, Cambridge, MA 02140, USA
First published on 14th September 2023
Abstract
The emergence of DNA-encoded library (DEL) technology has provided a considerable advantage to the pharmaceutical industry in the pursuit of discovering novel therapeutic candidates for their drug development initiatives. This combinatorial technique not only offers a more economical, spatially efficient, and time-saving alternative to the existing ligand discovery methods, but also enables the exploration of additional chemical space by utilizing novel DNA-compatible synthetic transformations to leverage multifunctional building blocks from readily available substructures. In this report, a decarboxylative-based hydroalkylation of DNA-conjugated N-vinyl heterocycles enabled by single-electron transfer (SET) and subsequent hydrogen atom transfer through electron-donor/electron-acceptor (EDA) complex activation is detailed. The simplicity and robustness of this method permits inclusion of a broad array of alkyl radical precursors and DNA-tethered nitrogenous heterocyles to generate medicinally relevant substituted heterocycles with pendant functional groups. Moreover, a successful telescoped route provides the opportunity to access a broad range of intricate structural scaffolds by employing basic carboxylic acid feedstocks.
Introduction
The journey of drug discovery to marketing a potential drug in the pharmaceutical industry is a multifaceted process that necessitates substantial investment and includes various stages. A pivotal step in this process is known as hit identification, which involves identifying small molecules from a large pool of compounds that can bind to a specific target and elicit a desired biological effect, such as inhibiting the activity of a disease-causing protein.1–4 There are several conventional methods for hit identification,5–8 but DNA-encoded library (DEL) screening technology has gained significant attention in recent years, both in academic and pharmaceutical industry settings.9–14 This technique involves encoding numerous small molecules with unique DNA tags and exposing them to the target protein, allowing the identification of molecules that selectively bind to the protein by sequencing their DNA tags (Fig. 1).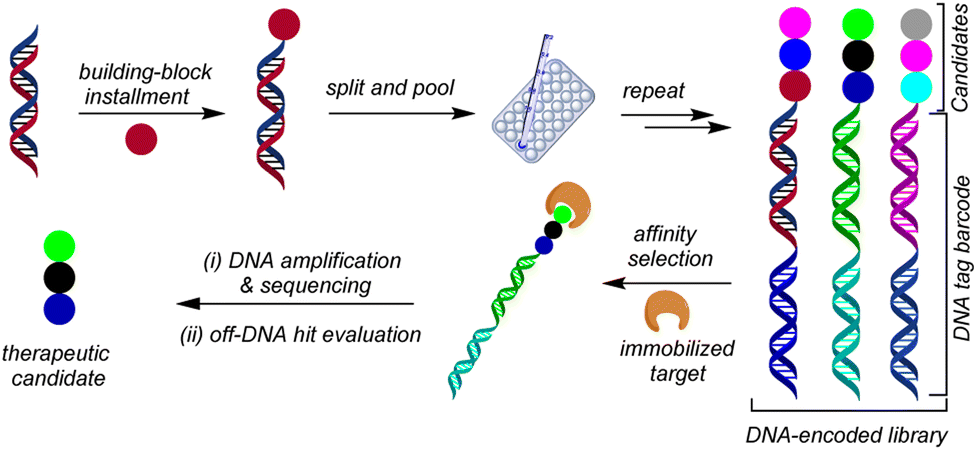 | ||
| Fig. 1 Schematic representation of DEL workflow. | ||
Conceptualized by Brenner and Lerner in 1992,15 DEL technology offers several advantages over other hit identification methods because of its ability to screen large libraries of compounds (>106 to 1012 discrete members) in a relatively short period of time, because the library of compounds can be screened against a biological target in a single step, rather than requiring the screening of individual compounds.16 This leads to significant time and cost savings, particularly in the early stages of drug discovery. Another advantage of DEL synthesis is its ability to identify hits with high binding affinity to a target protein. Thus, compounds in the library are covalently linked to a DNA barcode, which enables the screening of billions of compounds simultaneously. This allows the identification of compounds that bind specifically and tightly to the target protein, which can be important for the development of effective drugs.17
To generate structurally valuable DEL libraries, it is essential to explore and translate innovative synthetic transformations to this platform.
Compared to traditional synthetic reactions, DEL reactions are often viewed as unconventional because they involve simultaneously manipulating the small molecule building blocks while preserving the valuable DNA tags. As a result, these reactions present unique challenges and often require optimal tuning of reaction parameters. Additionally, the reaction must exhibit high chemo-selectivity, scalability, and versatility toward different functional groups.18 In this context, photoredox chemistry is an appealing and powerful synthetic tool for conducting DEL reactions. This is primarily because of its ability to operate under extremely mild conditions, faster reaction times, and compatibility with dilute aqueous media.19,20
Among various transformations developed in the realm of DEL-chemistry, C(sp3)–C(sp3) bond formation has garnered significant attention.19–22 In particular, photo-induced (hydro)alkylation reactions have proven to be valuable tools for incorporating pharmaceutically relevant alkyl scaffolds. These reactions utilize alkyl radicals derived from readily available radical precursors (RP) through photoinduced SET, which are subsequently reacted with DNA-tethered electrophilic Giese-type acceptors, making them an efficient way to introduce these scaffolds (Scheme 1A).23 Relying on this strategy, Flanagan and co-workers developed a decarboxylative alkylation approach to introduce stabilized α-amino- or α-oxy radicals onto DNA-tagged styrenes, acrylamides, and vinyl benzamides.24 Subsequently, the Liu (2021)25 and Lu (2021)26 research groups independently reported similar methods for adding α-amino radicals to on-DNA acylamides via the hydrogen atom transfer pathway in the presence of an iridium-based photocatalyst, yielding amino-alkylated products. However, these approaches are largely limited to stabilized alkyl radicals, with the exception of a 2020 report by Mendoza et al., where they employed a redox-active ester (RAE) to incorporate unstabilized alkyl radicals into on-DNA acrylates/acylamides through an EDA-complex route using NADH as an electron donor species.27
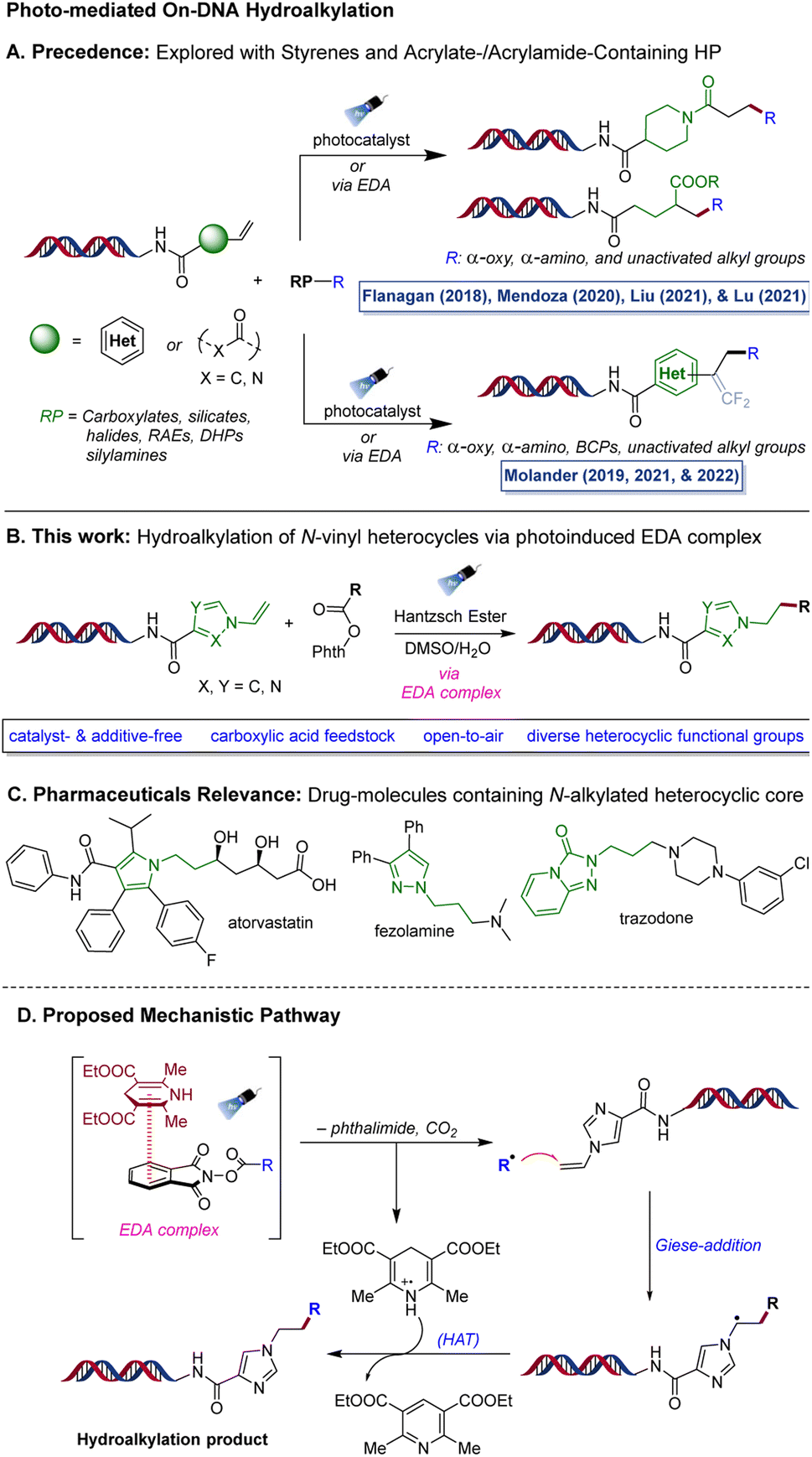 | ||
| Scheme 1 Photo-mediated on-DNA hydroalkylation. (A) Previous reports of DNA hydroalkylation (B) hydroalkylation of N-vinyl heterocycles via EDA complex activation (C) medicinal drugs containing N-alkylated heterocyclic core structure. (D) Proposed mechanism. | ||
As part of a research program focused on creating innovative synthetic methods to broaden the chemical space in DEL, our laboratory has been particularly involved in developing (hydro)alkylation reactions on various functionalized DNA headpieces (HPs) using photoredox approaches. In 2019, we reported a defluorinative alkylation method that utilizes photoredox radical-polar crossover pathway to synthesize on-DNA gem-difluoroalkenes from trifluoromethylated alkenes.28 We also developed a metal-free hydroalkylation approach that was used to introduce alkyl groups on DNA trifluoromethylated styrene conjugates, enabling access to complex trifluoromethylated benzylic scaffolds via a decarboxylative EDA complex activation mechanism.29 Very recently, in 2022, our laboratory reported an organo-photocatalyzed method for incorporating medicinally relevant bicyclo[1.1.1]pentanes (BCPs), which are arene bioisosteres, into functionalized on-DNA styrenes via a photocatalyzed halogen transfer pathway.30
Although significant progress has been made in the field of on-DNA hydroalkylation, the existing methods are limited to a few types of Giese acceptors such as styrenes and acrylates/acrylamides, indicating the need to explore new chemical methods to broaden the hydroalkylation space. To complement these efforts, we became interested in pushing the boundaries of photochemical paradigms to access on-DNA N-alkylated heterocyclic scaffolds via hydroalkylation of N-vinyl heterocyclic precursors (Scheme 1B). Heterocycles play a vital role in medicinal chemistry research and are a fundamental component of numerous pharmaceutical drugs.31–33 In particular, N-alkylated heterocycles are found in essential medicinal drugs such as atorvastatin/Lipitor® (for treating hypercholesterolemia) as well as fezolamine34,35 and trazodone (both antidepressants) (Scheme 1C). This underscores the significance of developing new chemical reactions focused on N-containing heterocycles. Off-DNA examples have successfully adopted the strategy of photoactivation of EDA complexes of RAEs and Hantzsch ester (HE) in a metal-free setting to promote the Giese addition of alkyl radicals to activated olefins, as demonstrated initially by Shang36 and later by Allred and Overman.37
Therefore, we envisioned a metal-free protocol that leverages the electron-donor/electron-acceptor (EDA)38–41 activation pathway to generate alkyl radicals from redox active esters, which are subsequently added into the N-vinyl heterocyclic HPs in a Giese-addition manner followed by hydrogen atom transfer, allowing the synthesis of on-DNA N-alkylated heterocyclic products (Scheme 1D).
Although the proposed transformation seems promising, there were challenges to overcome. For instance, N-vinyl heterocycles are electron-rich systems, which can result in a polarity mismatch of reactivity with the alkyl radicals that are also electron-rich species. To the best of our knowledge, no report on this specific hydroalkylation of N-vinyl heterocycles has been disclosed either on- or off-DNA, which made this transformation particularly appealing to investigate.
Discussion
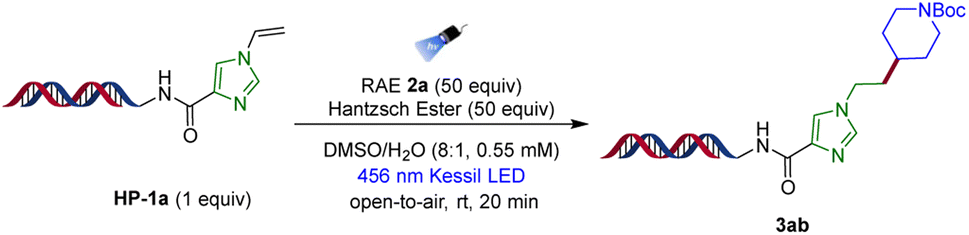
To validate the feasibility of the proposed hydroalkylation of electron-rich vinyl heterocyclic assemblies in light of the aforementioned challenges, we began the optimization study using N-vinyl imidazole DNA headpiece HP-1a and an unactivated redox-active ester (RAE) 2a as model substrates (Table 1). The reaction was performed under blue Kessil irradiation (λmax = 456 nm), and efficient conversion (95%) to the desired hydroalkylation product 3ab was observed using 50 equiv. of the radical precursor and 50 equiv. of Hantzsch ester (HE) under ambient reaction conditions within minutes of illumination (entry 1). Unlike radical-mediated alkylation processes that use metal reductants or external photoredox catalysts,42 this EDA paradigm operates in an open-to-air environment, making it highly practical for implementation in high-throughput settings, concomitantly circumventing the side reactions caused by singlet oxygen generation through triplet-energy transfer.43 Further, the effect of stoichiometric variation of EDA coupling partners on the reaction yield was investigated. Reducing the amount of RAE 2a and HE from 50 equiv. to 25 equiv. and 12 equiv., respectively, resulted in decreased reactivity, with the desired reaction conversion of 42% and 18%, respectively (entries 2 and 3). When only 6 equivalents of both RAE 2a and HE were used, only trace amounts of the desired product were observed (entry 4). Furthermore, when the reaction was carried out in the absence of either the HE or the light source, no product was detected, confirming the dependence of this hydroalkylation on photo-mediated charge-transfer complex formation (entries 5 and 6).|
|
||
|---|---|---|
| Entry | Deviation from std conditions | % conversionb |
a Reaction conditions: N-vinyl imidazole HP-1a (1.0 equiv., 25 nmol), RAE 2a (50 equiv., 1.25 μmol), Hantzsch ester (50 equiv., 1.25 μmol), 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMSO/H2O (0.55 mM), 20 min irradiation with Kessil lamps (λmax = 456 nm, 40 W).
b Conversion to 3ab was determined by LC/MS (see ESI). :1 DMSO/H2O (0.55 mM), 20 min irradiation with Kessil lamps (λmax = 456 nm, 40 W).
b Conversion to 3ab was determined by LC/MS (see ESI).
|
||
| 1 | None | 95 |
| 2 | RAE (25 equiv.), HE (25 equiv.) | 42 |
| 3 | RAE (12 equiv.), HE (12 equiv.) | 18 |
| 4 | RAE (6 equiv.), HE (6 equiv.) | Trace |
| 5 | No HE | 0 |
| 6 | No light | 0 |

|
||
After establishing suitable conditions, the generality of the on-DNA hydroalkylation reaction was assessed (Table 2). Initially, the scope of redox-active esters was investigated by utilizing primary-, secondary-, and tertiary radical precursors and reacting them with DNA-tagged pyrazole HP-1b. Notably, the method is effective with primary radicals that are relatively unstable and feature complex functional groups. The methyl radical 3a exhibited favorable conversion (49%). Redox-active esters with phenyl- and phenacyl on the side chain generated the desired products in good conversions (3b–3c). The reaction showed good tolerance toward some other primary radicals as well, including Boc-protected amine 3d, the bulkier methyl adamantyl group 3e, and the thiyl radical 3f, all of which provided modest to good conversions.
|
a Conversion was determined by LC/MS analysis (see ESI). Reaction conditions: DNA-N-vinyl pyrazole HP-1b (1.0 equiv., 25 nmol), RAE 2 (50 equiv., 1.25 μmol), Hantzsch ester (50 equiv., 1.25 μmol), 8:1 DMSO/H2O (0.55 mM), 20 min irradiation with Kessil lamp (λmax = 456 nm, 40 W).
|
|---|

|
We then proceeded to investigate the feasibility of using secondary- and tertiary alkyl-substituted redox-active esters in the reaction. We examined a range of secondary radical precursors that contained protected nitrogen groups. Piperidine 3g, pyrrolidine 3h, and azetidine 3i, all of which were Boc-protected, showed mostly excellent conversions. Similarly, other substructures, such as pyrimidine-containing 3j and benzyl ester 3k, also displayed good reactivity. In addition, cyclohexyl 3l, difluorocyclohexyl 3m, and difluorocyclobutyl (3n) radicals all exhibited good conversion. Moreover, compounds containing both internal and external alkene functional groups (3o–3p) also displayed excellent tolerance to the reaction, expanding the potential to attach further building blocks, an important aspect in DEL synthesis. In addition, the developed conditions were found to be effective for α-amino 3q, α-oxy 3r, and sugar-containing radicals (3s–3t). However, the bis-benzylic radical 3u exhibited low conversion under the same conditions.
To expand the scope of the process, structurally diverse tertiary radicals were examined in the reaction. N-Boc-protected cyclobutyl- and cyclohexylamines (3v–3w) exhibited excellent conversion. 4-Methylpentene 3x and cyclopropylbenzene 3y were also accommodated. Additionally, gemfibrozil 3z, which is a lipid-regulating medication, demonstrated good compatibility and conversion in the reaction, furnishing the desired compound with 72% conversion.
Next, we sought to evaluate the scope of this transformation with various heterocyclic HPs using a set of primary-, secondary-, and tertiary redox-active esters (Table 3). HP-1a, which contains an imidazole moiety, exhibited moderate reactivity with primary radical 3aa, but excellent reactivity with secondary- and tertiary radicals (3ab–3ad). Surprisingly, 2-bromo-imidazole HP-1c, which contains a carbon–halogen bond that is prone to photochemical cleavage, also exhibited good tolerance in the reaction and provided good to excellent conversions with all three types of RAEs (3ae–3aj). The scope of the investigation was broadened to encompass additional important heterocycles, such as pyrazoles and pyrroles. These heterocycles have been extensively studied and utilized in the design and development of various therapeutic agents because of their wide range of biological activities, structural diversity, synthetic accessibility, and favourable drug-like properties.44 In this regard, 5-methyl pyrazole HP-1d and 5-methyl pyrrole HP-1e demonstrated a notable ability to undergo reaction under the experimental conditions. Specifically, 5-methyl pyrazole HP-1d demonstrated moderate conversions with un-activated radicals (3ak–3am), but excellent results with more stabilized radicals (3an–3ap). On the other hand, 5-methyl pyrrole HP-1e displayed only moderate conversions (below 60%) when reacted with various alkyl radicals (3aq–3av). Furthermore, two nitrogen-rich heterocycles, triazole HP-1f and benzotriazole HP-1g were incorporated, which are easily obtained through Click reactions and hold significance in drug discovery.45,46 These heterocycles also showed excellent compatibility in the reaction and exhibited good to excellent reactivity (3aw–3bh), showcasing the robustness and versatility of the developed method, which can accommodate numerous heterocyclic functional groups crucial for medicinal chemistry research.
|
a Conversion was determined by LC/MS analysis (see ESI). Reaction conditions: DNA-N-vinyl Het (1.0 equiv., 25 nmol), RAE 2 (50 equiv., 1.25 μmol), Hantzsch ester (50 equiv., 1.25 μmol), 8:1 DMSO/H2O (0.55 mM), 20 min irradiation with Kessil lamp (λmax = 456 nm, 40 W).
|
|---|

|
The structural diversity and commercial availability of carboxylic acids make them a desirable choice for multifunctional building blocks in DEL. To demonstrate the modularity of this approach, a one-pot photoinduced decarboxylative alkylation protocol was devised by forming aliphatic RAEs in situ using N-hydroxyphthalimide tetramethyluronium hexafluorophosphate (HITU), which is a stable solid that can be easily synthesized in large quantities.40 The carboxylic acid 4, DIPEA, and HITU were microdosed in DMSO under air, followed by 3 hours of activation time. The RAE 2a formed in situ was then directly subjected to the developed conditions with a solution of HE and the DNA headpiece HP-1b. Gratifyingly, the reaction attains 80% yield to provide product 3g, which is similar to the yield obtained when preformed RAE was used (81%) (Scheme 2).
 | ||
| Scheme 2 On-DNA photoinduced decarboxylative alkylation: in situ activation of RAEs with N-hydroxyphthalimide tetramethyluronium hexafluorophosphate (HITU). | ||
DNA damage assessment
The practical application of the developed protocol for DEL preparation is intrinsically correlated with the capability of preserving the DNA tag upon chemical transformation, because the candidate building blocks of higher affinity to a biological target would be identified via qPCR, PCR, and sequencing analysis of the DNA barcode associated with them. Confirming the mildness of the photoinduced on-DNA hydroalkylation reaction reported herein, a 4-cycle tag mimic sequence was submitted to the standard reaction conditions under blue light irradiation. The samples were then elongated by ligation to introduce the essential PCR primers and then quantified by qPCR. The results showed that the reaction conditions do not impact the amount of amplifiable DNA present in a significant way, which demonstrates the valuable application of the on-DNA hydroalkylation reaction in preparing diversified and more elaborated N-vinyl heterocycles for rapid screening of drug candidates in medical chemistry research.Conclusions
In summary, a straightforward yet highly reliable method for the hydroalkylation of N-vinyl heterocycles on-DNA has been developed using an EDA complex activation pathway. This method eliminates the need for costly metal-based photocatalysts and can be carried out under mild, open-air reaction conditions, allowing the synthesis of biologically relevant N-alkylated heterocycles from readily available or easily synthesized redox-active esters and N-vinyl heterocycles. The simplicity and robustness of this method allow the incorporation of a wide range of alkyl-radical precursors and DNA-tethered nitrogenous heterocycles, which results in substituted heterocycles with pendant functional groups that are medicinally significant. Additionally, a successful telescoped route allows the creation of complex structural scaffolds using a simple carboxylic acid feedstock. Finally, the practical applicability of this method for DELT was successfully confirmed with a DNA integrity assessment, highlighting the mildness of the visible-light-mediated protocols as an extraordinary tool for diversifying on-DNA libraries.Data availability
General considerations, preparation of headpieces, general procedure for the photochemical hydroalkylation reaction, and characterization of all the synthesized compounds are available in the ESI.†Author contributions
Mohammed Sharique, Bianca Matsuo, and Albert Granados conceived the topic, designed the experiments and wrote the manuscript with input from Adam Csakai, Lisa A. Marcaurelle, and Prof. Gary A. Molander. Saegun Kim, Mahwish Arshad, and Hyunjung Oh contributed to the method scope development. Victoria E. Wu and Minxue Huang performed the UPLC/MS yield analysis and DNA damage assessment. Mohammed Sharique, Bianca Matsuo, and Albert Granados contributed equally as co-first authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support provided by NIGMS (R35 GM 131680 to G. M.) and GSK. The NSF Major Research Instrumentation Program (award NSF CHE-1827457), the NIH supplement awards 3R01GM118510-03S1 and 3R01GM087605-06S1, as well as the Vagelos Institute for Energy Science and Technology supported the purchase of NMRs used in this study. We thank Dr Charles W. Ross, III (UPenn) for mass spectral data. Kessil is thanked for the donation of lamps.Notes and references
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed.
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhoti, Nat. Rev. Drug Discovery, 2016, 15, 605–619 CrossRef CAS PubMed.
- M. Schenone, V. Dančík, B. K. Wagner and P. A. Clemons, Nat. Chem. Biol., 2013, 9, 232–240 CrossRef CAS PubMed.
- B. R. Stockwell, Nature, 2004, 432, 846–854 CrossRef CAS PubMed.
- D. A. Erlanson, B. J. Davis and W. Jahnke, Cell Chem. Biol., 2019, 26, 9–15 CrossRef CAS PubMed.
- P. J. Hajduk and J. Greer, Nat. Rev. Drug Discovery, 2007, 6, 211–219 CrossRef CAS PubMed.
- D. B. Kitchen, H. Decornez, J. R. Furr and J. Bajorath, Nat. Rev. Drug Discovery, 2004, 3, 935–949 CrossRef CAS PubMed.
- R. Macarron, M. N. Banks, D. Bojanic, D. J. Burns, D. A. Cirovic, T. Garyantes, D. V. S. Green, R. P. Hertzberg, W. P. Janzen, J. W. Paslay, U. Schopfer and G. S. Sittampalam, Nat. Rev. Drug Discovery, 2011, 10, 188–195 CrossRef CAS PubMed.
- A. Gironda-Martínez, E. J. Donckele, F. Samain and D. Neri, ACS Pharmacol. Transl. Sci., 2021, 4, 1265–1279 CrossRef PubMed.
- R. A. Goodnow, C. E. Dumelin and A. D. Keefe, Nat. Rev. Drug Discovery, 2017, 16, 131–147 CrossRef CAS PubMed.
- M. Song and G. T. Hwang, J. Med. Chem., 2020, 63, 6578–6599 CrossRef CAS PubMed.
- S. Melkko, J. Scheuermann, C. E. Dumelin and D. Neri, Nat. Biotechnol., 2004, 22, 568–574 CrossRef CAS PubMed.
- D. Neri and R. A. Lerner, Annu. Rev. Biochem., 2018, 87, 479–502 CrossRef CAS PubMed.
- J. Scheuermann, C. E. Dumelin, S. Melkko and D. Neri, J. Biotechnol., 2006, 126, 568–581 CrossRef CAS PubMed.
- S. Brenner and R. A. Lerner, Proc. Natl. Acad. Sci. U.S.A., 1992, 89, 5381–5383 CrossRef CAS PubMed.
- D. C. Swinney and J. Anthony, Nat. Rev. Drug Discovery, 2011, 10, 507–519 CrossRef CAS PubMed.
- M. A. Clark, R. A. Acharya, C. C. Arico-Muendel, S. L. Belyanskaya, D. R. Benjamin, N. R. Carlson, P. A. Centrella, C. H. Chiu, S. P. Creaser, J. W. Cuozzo, C. P. Davie, Y. Ding, G. J. Franklin, K. D. Franzen, M. L. Gefter, S. P. Hale, N. J. V. Hansen, D. I. Israel, J. Jiang, M. J. Kavarana, M. S. Kelley, C. S. Kollmann, F. Li, K. Lind, S. Mataruse, P. F. Medeiros, J. A. Messer, P. Myers, H. O'Keefe, M. C. Oliff, C. E. Rise, A. L. Satz, S. R. Skinner, J. L. Svendsen, L. Tang, K. van Vloten, R. W. Wagner, G. Yao, B. Zhao and B. A. Morgan, Nat. Chem. Biol., 2009, 5, 647–654 CrossRef CAS PubMed.
- M. L. Malone and B. M. Paegel, ACS Comb. Sci., 2016, 18, 182–187 CrossRef CAS PubMed.
- S. Patel, S. O. Badir and G. A. Molander, Trends Chem., 2021, 3, 161–175 CrossRef CAS PubMed.
- B. Matsuo, A. Granados, G. Levitre and G. A. Molander, Acc. Chem. Res., 2023, 56, 385–401 CrossRef CAS PubMed.
- R. J. Fair, R. T. Walsh and C. D. Hupp, Bioorg. Med. Chem. Lett., 2021, 51, 128339 CrossRef CAS PubMed.
- V. M. Lechner, M. Nappi, P. J. Deneny, S. Folliet, J. C. K. Chu and M. J. Gaunt, Chem. Rev., 2022, 122, 1752–1829 CrossRef CAS PubMed.
- A. L. Gant Kanegusuku and J. L. Roizen, Angew. Chem., Int. Ed., 2021, 60, 21116–21149 CrossRef CAS PubMed.
- D. K. Kölmel, R. P. Loach, T. Knauber and M. E. Flanagan, ChemMedChem, 2018, 13, 2159–2165 CrossRef PubMed.
- R. Wu, T. Du, W. Sun, A. Shaginian, S. Gao, J. Li, J. Wan and G. Liu, Org. Lett., 2021, 23, 3486–3490 CrossRef CAS PubMed.
- J. Shan, X. Ling, J. Liu, X. Wang and X. Lu, Bioorg. Med. Chem., 2021, 42, 116234 CrossRef CAS PubMed.
- R. Chowdhury, Z. Yu, M. L. Tong, S. V. Kohlhepp, X. Yin and A. Mendoza, J. Am. Chem. Soc., 2020, 142, 20143–20151 CrossRef CAS PubMed.
- J. P. Phelan, S. B. Lang, J. Sim, S. Berritt, A. J. Peat, K. Billings, L. Fan and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 3723–3732 CrossRef CAS PubMed.
- S. O. Badir, A. Lipp, M. Krumb, M. J. Cabrera-Afonso, L. M. Kammer, V. E. Wu, M. Huang, A. Csakai, L. A. Marcaurelle and G. A. Molander, Chem. Sci., 2021, 12, 12036–12045 RSC.
- E. Yen-Pon, L. Li, G. Levitre, J. Majhi, E. J. McClain, E. A. Voight, E. A. Crane and G. A. Molander, J. Am. Chem. Soc., 2022, 144, 12184–12191 CrossRef CAS PubMed.
- A. Gomtsyan, Chem. Heterocycl. Compd., 2012, 48, 7–10 CrossRef CAS.
- M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247–44311 RSC.
- D. P. Rotella, in Advances in Heterocyclic Chemistry, ed. N. A. Meanwell and M. L. Lolli, Academic Press, 2021, vol. 134, pp. 149–183 Search PubMed.
- D. M. Bailey, P. E. Hansen, A. G. Hlavac, E. R. Baizman, J. Pearl, A. F. DeFelice and M. E. Feigenson, J. Med. Chem., 1985, 28, 256–260 CrossRef CAS PubMed.
- S. Zisook, J. Mendels, D. Janowsky, J. Feighner, J. C. M. Lee and A. Fritz, Neuropsychobiology, 2008, 17, 133–138 CrossRef PubMed.
- C. Zheng, G.-Z. Wang and R. Shang, Adv. Synth. Catal., 2019, 361, 4500–4505 CrossRef CAS.
- S. P. Pitre, T. K. Allred and L. E. Overman, Org. Lett., 2021, 23, 1103–1106 CrossRef CAS PubMed.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- C. G. S. Lima, T. de M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS.
- L. Zheng, L. Cai, K. Tao, Z. Xie, Y.-L. Lai and W. Guo, Asian J. Org. Chem., 2021, 10, 711–748 CrossRef CAS.
- M. Sharique, J. Majhi, R. K. Dhungana, L. M. Kammer, M. Krumb, A. Lipp, E. Romero and G. A. Molander, Chem. Sci., 2022, 13, 5701–5706 RSC.
- J. Wang, H. Lundberg, S. Asai, P. Martín-Acosta, J. S. Chen, S. Brown, W. Farrell, R. G. Dushin, C. J. O'Donnell, A. S. Ratnayake, P. Richardson, Z. Liu, T. Qin, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, E6404–E6410 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, Molecules, 2020, 25, 1909 CrossRef CAS PubMed.
- A. Rajasekaran and K. A. Rajagopal, Acta Pharm., 2009, 59, 355–364 CAS.
- I. Briguglio, S. Piras, P. Corona, E. Gavini, M. Nieddu, G. Boatto and A. Carta, Eur. J. Med. Chem., 2015, 97, 612–648 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and mechanistic studies details, as well as spectral data. See DOI: https://doi.org/10.1039/d3sc03731b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |