
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoordinating activation strategy enables 1,2-alkylamidation of alkynes†
Jing
Ren‡
,
Junhua
Xu‡
,
Xiangxiang
Kong
,
Jinlong
Li
and
Kaizhi
Li
 *
*
Biopharmaceutical Research Institute, West China Hospital of Sichuan University, 37 Guoxue Alley, Chengdu, 610041, P. R. China. E-mail: kzli@scu.edu.cn
First published on 30th September 2023
Abstract
The radical 1,2-difunctionalization reaction of alkynes has been evolved into a versatile approach for expeditiously increasing the complexity of the common feedstock alkyne. However, intermolecular 1,2-carboamidation with general alkyl groups is an unsolved problem. Herein, we show that a coordinating activation strategy could act as an efficient tool for enabling radical 1,2-alkylamidation of alkynes. With the employment of diacyl peroxides as both alkylating reagents and internal oxidants, a large library of β-alkylated enamides is constructed in a three-component manner from readily accessible amides and alkynes. This protocol exhibits broad substrate scope with good functional group compatibility and is amenable for late-stage functionalization of natural molecules and biologically compounds.
Introduction
The enamide is a privileged subunit in pharmaceuticals and biomolecules featuring excellent biological and physiological properties,1 as well as being a useful and versatile synthetic platform for further elaboration in organic synthesis.2 Several approaches for the preparation of enamides have been reported,3 including condensation of amides with carbonyls,3a carbofunctionalization of ynamides,3b,c transition-metal catalyzed N-alkenylation of amides,3d–f isomerization of allylamides,3g,h hydroamidation of alkynes,3i–l dehydrogenation of the corresponding amides,3m–p and others.3q–w However, one of the most straightforward approaches to access enamides is arguably the vicinal difunctional carboamidation of alkynes.The intermolecular radical 1,2-difunctionalization reaction of alkyne is a straightforward route for expeditiously increasing the complexity of the common feedstock alkyne starting materials for target molecule synthesis.4 This reaction typically proceeds through the addition of the radical species to the alkyne, followed by recombination of the formational vinyl radical intermediate with a metal catalyst or other radical species. Although it has been studied extensively in the past several decades, the intermolecular radical carboamination of alkyne remains a long-standing challenge (Fig. 1a). The difficulty in this transformation originates perhaps from either the strong Lewis basicity of the amines5 or the weak coordinating ability of the amides,6 interfering with the generation of the vinyl–metal–nitrogen intermediate. In 2015, the groups of Liu7a and Liang7b independently established an elegant synthetic route involving a copper-catalyzed trifluoromethylazidation of alkynes, while seminal work by Bao8 in 2019 identified carboazidated alkynes as attractive candidates for the synthesis of fluoroalkylated azirines. Very recently, Liu and co-workers reported another pioneering work on the efficient synthesis of vinylarene atropisomers through copper-catalyzed enantioselective trifluoromethylazidation of alkynes.7c These exceptions mainly rely on employing azide that serves as a reagent with good coordinating ability, and where coordination of the metal catalyst enables capture of the vinyl radical to form a vinyl–metal–N3 intermediate. Notwithstanding these significant breakthroughs, a general strategy that enables the intermolecular radical carboamination of alkynes utilizing common amine derivatives as the nitrogen source with general alkyl groups remains an unsolved problem.
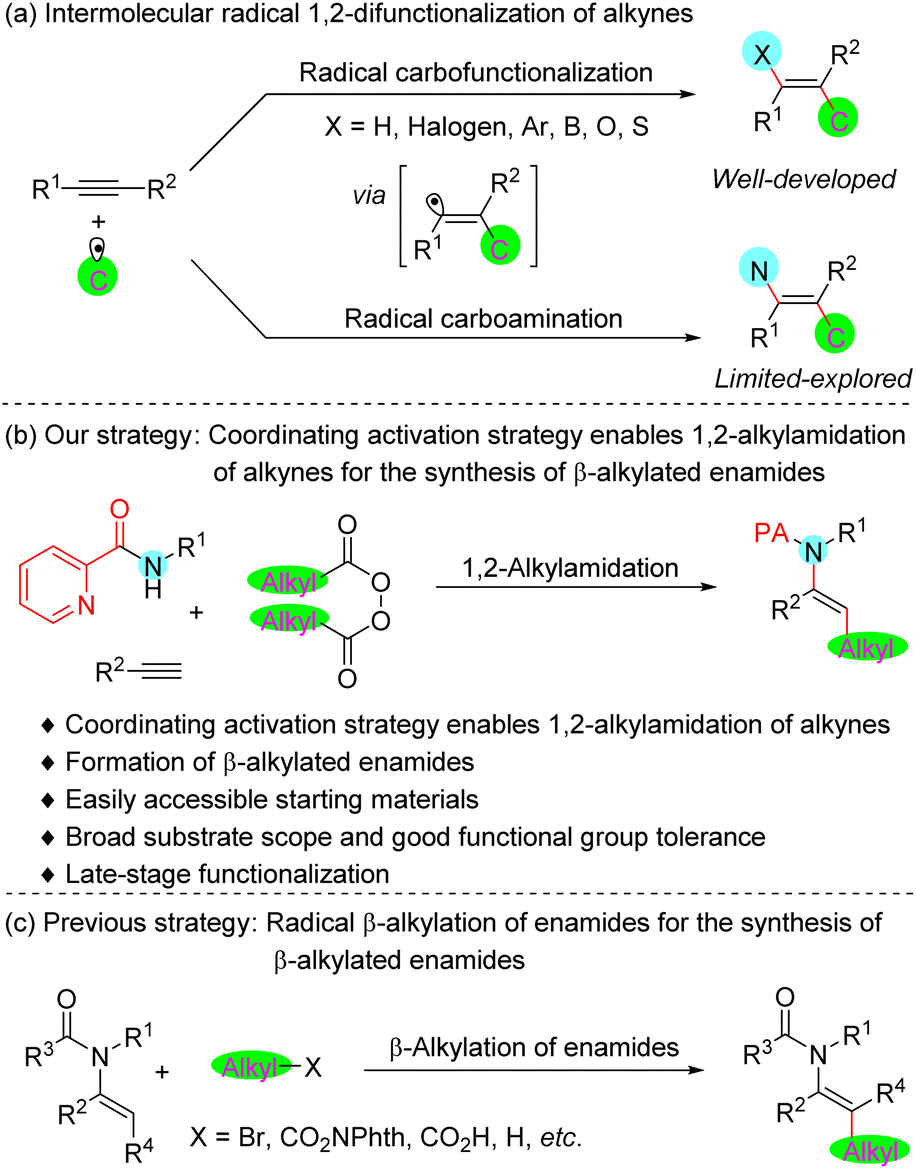 | ||
| Fig. 1 Background and project synopsis. | ||
In order to achieve this challenging catalytic process, we considered installing a 2-pyridylacyl group9 on the amine to serve as the coordinating group. We surmised that the metallacycle species, a coordination product formed from the reaction of the metal catalyst with a substrate bearing a bidentate auxiliary, could efficiently catch the vinyl radical intermediate, thus providing a straightforward strategy for the realization of difunctionalization carboamidation of alkynes. Herein, we report the general 1,2-difunctional alkylamidation of alkynes by utilizing this coordinating activation strategy for the synthesis of β-alkylated enamides (Fig. 1b). Recently, tremendous progress on radical β-alkylation of enamides has been demonstrated for the construction of such compounds (Fig. 1c).10 However, the representative known synthetic strategies typically start from prefunctionalized enamide substrates. Our current protocol is the first universal strategy that could straightforwardly build β-alkylated enamides from commercially available alkynes and readily accessible amides and diacyl peroxides. In addition, a variety of reactants derived from pharmaceuticals and natural products bearing proven biological activities can be adopted in this protocol and lead to the late-stage modifications of bioactive compounds.
Results and discussion
Optimization of the reaction conditions
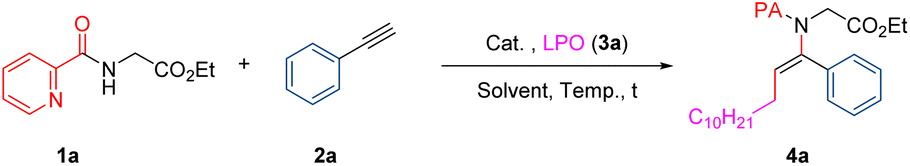
Based on our hypothesis, the bench-stable, commercially available chemical feedstock dilauroyl peroxide11 (LPO, 3a) was first used to serve as the alkylating and oxidizing agent simultaneously to illustrate the feasibility of the difunctional alkylamidation reaction. We initiated our studies by screening metal catalysts in the alkylamidation reaction of ethyl picolinoylglycinate (1a) with ethynylbenzene (2a) in the presence of LPO (3a). To our delight, Cu(acac)2 could lead to the desired alkylamidation product 4a in 36% yield in benzene at 120 °C (Table 1, entry 1). When the reaction temperature was increased to 130 °C, the yield of 4a was improved to 45% (entry 2). The copper catalyst examination suggested Cu(tfacac)2 was the better choice (entries 2–5). However, other copper salts, for instance CuCl, CuBr, CuI, Cu(OAc)2 and CuBr2, gave only trace amount of product (entry 6). A control experiment demonstrated copper anions played an important role in the catalytic activity (entry 7). Besides, some other metal catalysts, such as Fe(acac)2, Co(acac)2, Ni(acac)2, and Mn(acac)2, were proven to be inert for this reaction (entry 8). Gratifyingly, the yield could be improved by appropriately increasing the reaction time. In terms of the solvent effect, several solvents were assessed, and benzene turned out to be the best (entries 13–18). No reaction occurred in the absence of a metal catalyst (entry 19). Finally, further optimization of the reaction conditions revealed that the best yield was obtained with the help of 10 mol% Cu(tfacac)2 in benzene at 130 °C under a nitrogen atmosphere for 28 h (entry 11, for detailed optimization, see Table S1†).|
|
||||
|---|---|---|---|---|
| Entry | Cat. (equiv.) | Solvent | t (h) | Yieldb (%) |
| a Reaction conditions: 1a (0.10 mmol, 1.0 equiv.), 2a (0.20 mmol, 2.0 equiv.), LPO (3a, 0.30 mmol, 3.0 equiv.), cat. and solvent (1.0 mL) at 130 °C under N2 atmosphere. b Isolated yield after chromatographic purification. c The reaction at 120 °C. d CuCl, CuBr, CuI, Cu(OAc)2 or CuBr2 was used as the metal catalyst. e 2,2,6,6-Tetramethylheptane-3,5-dione (20 mol%) was added. f Fe(acac)2, Co(acac)2, Ni(acac)2 or Mn(acac)2 was used as the metal catalyst. g The reaction at 140 °C. Cu(hmacac)2 = copper(II) bis(2,2,6,6-tetramethyl-3,5-heptanedionate). Cu(tfacac)2 = copper(II) trifluoroacetylacetonate. Cu(hfacac)2 = copper(II) hexafluor-2,4-pentanedionate. n.d. = no product detected. PA = 2-pyridylacyl. | ||||
| 1c | Cu(acac)2 (0.2) | Benzene | 20 | 36 |
| 2 | Cu(acac)2 (0.2) | Benzene | 20 | 45 |
| 3 | Cu(hmacac)2 (0.2) | Benzene | 20 | 51 |
| 4 | Cu(tfacac)2 (0.2) | Benzene | 20 | 65 |
| 5 | Cu(hfacac)2 (0.2) | Benzene | 20 | 53 |
| 6d | Copper salt (0.2) | Benzene | 20 | Trace |
| 7e | Cu(OAc)2 (0.2) | Benzene | 20 | 44 |
| 8f | M(acac)2 (0.2) | Benzene | 20 | n.d. |
| 9 | Cu(tfacac)2 (0.1) | Benzene | 24 | 74 |
| 10g | Cu(tfacac)2 (0.1) | Benzene | 24 | 72 |
| 11 | Cu(tfacac)2 (0.1) | Benzene | 28 | 81 |
| 12 | Cu(tfacac)2 (0.1) | Benzene | 32 | 72 |
| 13 | Cu(tfacac)2 (0.1) | Cyclohexane | 28 | 43 |
| 14 | Cu(tfacac)2 (0.1) | Toluene | 28 | 67 |
| 15 | Cu(tfacac)2 (0.1) | PhCF3 | 28 | 75 |
| 16 | Cu(tfacac)2 (0.1) | PhCl | 28 | 73 |
| 17 | Cu(tfacac)2 (0.1) | MeOH | 28 | 48 |
| 18 | Cu(tfacac)2 (0.1) | HOAc | 28 | n.d. |
| 19 | — | Benzene | 28 | n.d. |
Investigation of reaction scope
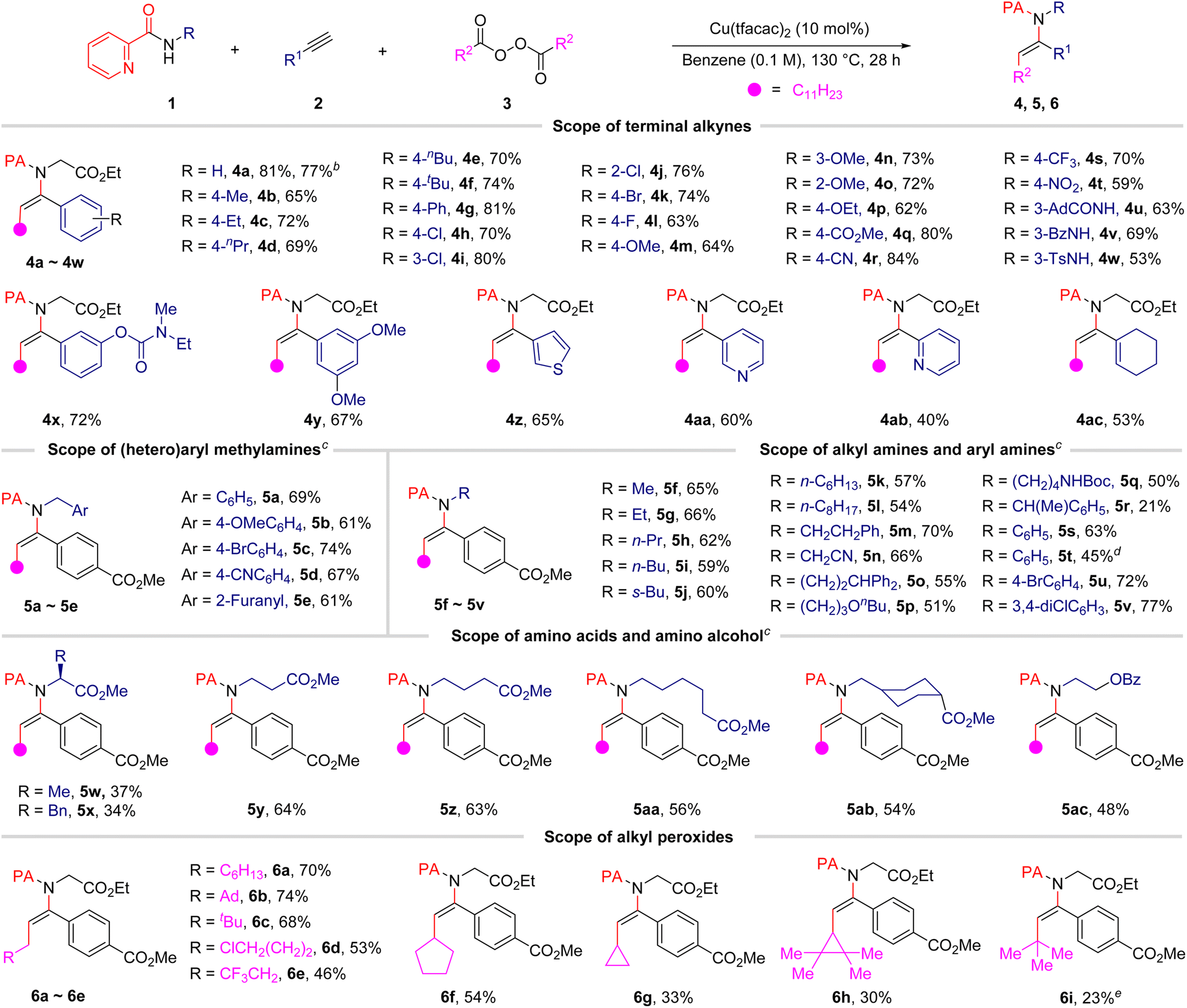
After having established the optimal reaction conditions, the substrate scope of the terminal alkynes was firstly examined and the results are summarized in Table 2. In general, the current protocol was applicable to terminal aryl alkynes bearing a series of substituents on the aromatic ring, including electron-withdrawing and electron-donating groups. In addition, various synthetically useful functional substituents, including halogen, ester, cyano, nitro, and amide groups, were tolerated with the reaction conditions, thus suggesting the suitability of the alkylamidation products for further structural elaboration. It is notable that an amide or sulfamide group is present at the 2-picolinamide moiety, C–N cross-coupling selectively took place at the 2-picolinamide–nitrogen, delivering the related products (4u–4w) in acceptable yields. This result not only establishes the importance of chelation for this transformation but also highlights the exquisite chemoselectivity of our strategy. Gratifyingly, heteroaryl substituted alkynes, such as 3-ethynylthiophene, 3-ethynylpyridine and 2-ethynylpyridine, could also proceed smoothly to furnish corresponding products in satisfactory yields. It is notable that the conjugated alkenyl acetylene 1-ethynylcyclohex-1-ene could also furnish the desired product 4ac with moderate yield. Unfortunately, no desired products were detected when internal and alkyl-substituted alkynes were utilized (see details in the ESI†). Notably, the reaction also proved to be scalable, with only a modest reduction in the yield of 4a from 81% to 77% observed when the reaction was performed on a 5.0 mmol scale in the presence of a lower catalyst loading (for details, see the ESI†).| a Reaction conditions: 1 (0.1 mmol, 1.0 equiv.), 2 (0.2 mmol, 2.0 equiv.), 3 (0.30 mmol, 3.0 equiv.), and Cu(tfacac)2 (0.01 mmol, 0.1 equiv.) in benzene (1.0 mL) under N2 atmosphere at 130 °C for 28 h; isolated yield is given. b The reaction was conducted on a 5.0 mmol scale. c 3a (0.40 mmol, 4.0 equiv.) was used. d Ethynylbenzene (2a) was used instead of methyl 4-ethynylbenzoate (2q) for 32 h. e tert-Butyl 2,2-dimethylpropaneperoxoate 3k (0.30 mmol, 3.0 equiv.) was used. |
|---|
|
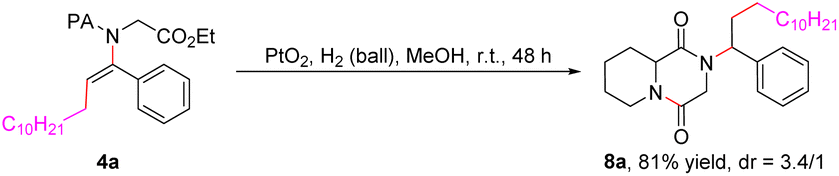
The scope with respect to 2-picolinamide derivatives was next evaluated, and the results are also summarized in Table 2. We were pleased to observe that a diverse range of 2-picolinamides were suitable for this difunctional alkylamidation reaction. The 2-picolinamides having (hetero)benzyl (5a–5e), aryl (5s–5v) and tethered aliphatic amines (5f–5q) exhibited good compatibility and produced the corresponding products in satisfactory yields. To our delight, the widespread natural and non-proteinogenic amino acids also proceeded smoothly to afford the desired products (5w–5ab). Notably, α,α-disubstituted 2-picolinamides did not turn out to be well-suited coupling partners for this transformation, possibly due to steric hindrance (5r, 5w, and 5x). The scope of the diacyl peroxides, which are readily prepared from carboxylic acids, as alkylating reagents was then studied under the optimized reaction conditions. The results showed that various primary alkyl radicals could be generated from the corresponding alkyl peroxides and afforded the alkylamidation products 6a–6e in good yields. A chlorine-substituted diacyl peroxide is compatible with the reaction conditions, providing the related product (6d) in 53% yield. In addition, products containing secondary and tertiary alkyl groups also could be obtained in relatively low yields from alkyl diacyl peroxides or alkyl peresters under the present reaction conditions (6f–6i). More importantly, the 2-pyridylacyl auxiliary group could be transformed (eqn (1)) or removed (eqn (2)) efficiently after achievement of the desired reactivity.
 | (1) |
 | (2) |
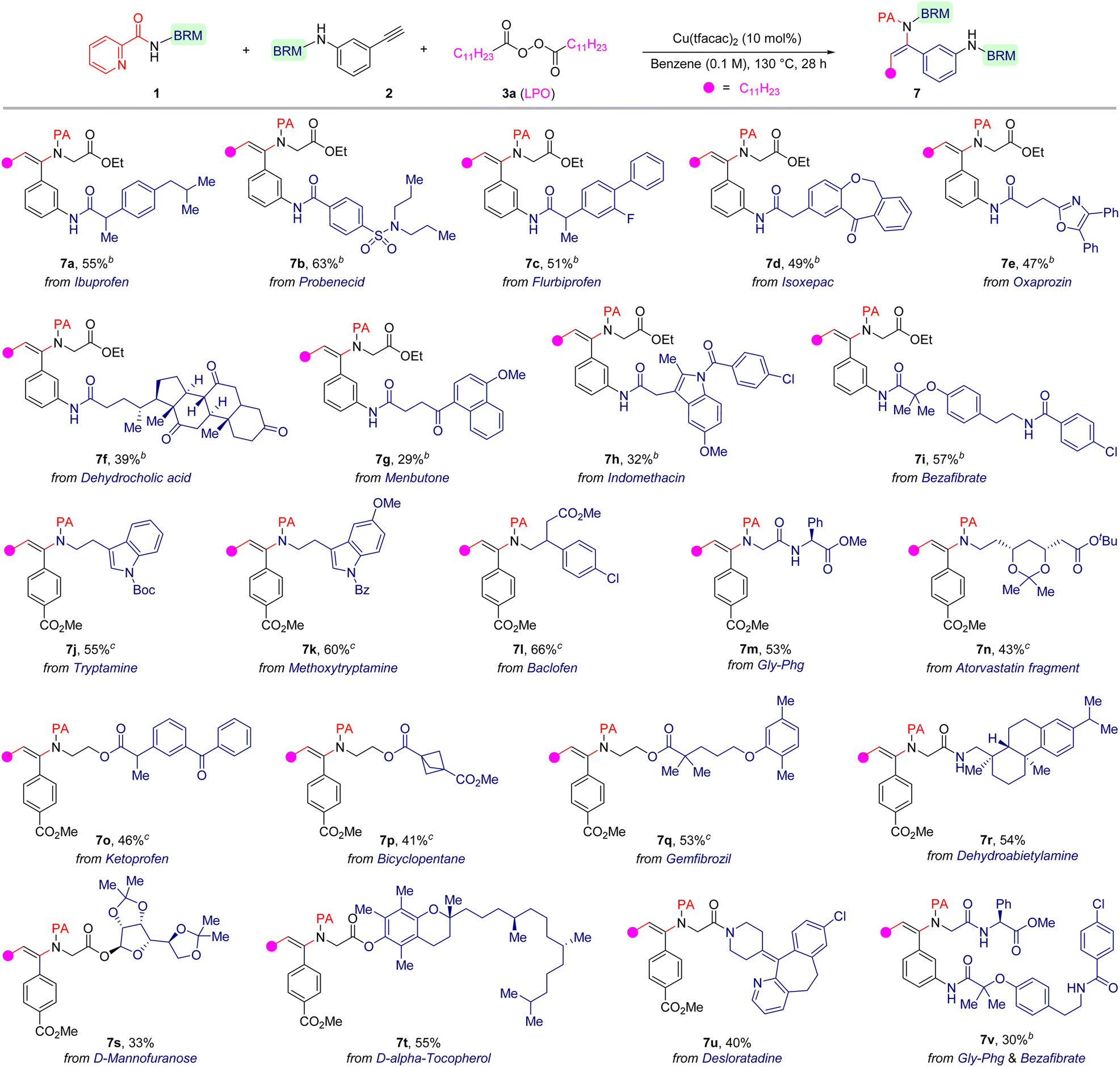
Because of the good functional group compatibility in this reaction, we evaluated its practicality for late-stage functionalization of multifunctionalized medicinally relevant molecules to show the generality of this method (Table 3). Pleasingly, the title reaction proved to be a reliable and robust methodology. We found that terminal alkynes derived from ibuprofen (7a), probenecid (7b), flurbiprofen (7c), isoxepac (7d), oxaprozin (7e), dehydrocholic acid (7f), menbutone (7g), indomethacin (7h) and bezafibrate (7i) could be used as substrates to deliver the corresponding β-alkylated enamides in acceptable yields (29–63%) with trans-selectivity. The current method was also applicable for sophisticated 2-picolinamides derived from bioactive compounds or drugs, such as tryptamine (7j), baclofen (7l), atorvastatin fragment (7n), ketoprofen (7o), gemfibrozil (7q), dehydroabietylamine (7r), D-mannofuranose (7s), D-α-tocopherol (7t) and desloratadine (7u), which deliver the corresponding products in moderate yields (33–66%). Significantly, a dipeptide could also serve as a suitable precursor and smoothly take part in this alkylamidation reaction, and the reaction only occurred at the 2-picolinamide-nitrogen with the other amide group intact (7m). Excitingly, 2-picolinamide derived from a peptide and alkyne derived from a drug scaffold could likewise be employed to couple together, resulting in an unprecedented peptide/drug conjugate (7v).
| a Reaction conditions: 1 (0.1 mmol, 1.0 equiv.), 2 (0.2 mmol, 2.0 equiv.), LPO (0.30 mmol, 3.0 equiv.), and Cu(tfacac)2 (0.01 mmol, 0.1 equiv.) in benzene (1.0 mL) under N2 atmosphere at 130 °C for 28 h; isolated yield is given. b 2 (0.15 mmol, 1.5 equiv.) was used. c 3a (0.4 mmol, 4.0 equiv.) was used. BRM = biologically relevant motif. |
|---|
|
Mechanistic study
To shed light on the mechanism, a series of mechanistic experiments were carried out (Fig. 2). First, a series of N-protecting groups on phenylmethanamine were examined under the optimized conditions (Fig. 2a). It was found that N-protecting groups with non-chelating properties, such as 3-pyridinecarbonyl, 4-pyridinecarbonyl, benzoyl, and p-toluenesulfonyl, were incapable of promoting the reaction. This further underlines the unique character of this coordinating activation strategy. Next, the reaction of 1a with deuterated alkyne [D]-2m proceeded smoothly to give the product with deuterium being partly incorporated into the product (81% deuterium content). Meanwhile, in the presence of deuterium oxide (D2O), the reaction of 1a with 2m afforded a deuterated product with 55% deuterium incorporation. This is possibly caused by the reversible H/D atom exchange of the terminal alkyne under the copper/heating conditions (Fig. 2b).12 Furthermore, intermolecular competition experiments with differently substituted aryl alkynes were carried out (Fig. 2c). The results showed that electron-withdrawing groups in the aryl alkyne gave a higher reactivity. In addition, competition experiments between differently N-substituted 2-picolinamides were examined as well, and the order of reactivity is CH2COOEt > Bn > Et. This trend presumably is due to the decreased acidity of the N–H bond of 2-picolinamide substrates.13 Notably, the rate of the reaction is sensitive to the amino group steric hindrance as shown by a similar competition experiment with N-benzyl-picolinamide 1b and N-(1-phenylethyl)-picolinamide 1s, which predominantly forms 5a (5a/5r > 20/1). Moreover, bis(N-phenylpicolinamido)copper(II) IM1a, which was prepared according to the literature14 and characterized using X-ray crystallographic analysis,15 along with an acetylacetone type ligand could be used to achieve this alkylamidation process (Fig. 2d). This result suggested that a ligand exchange of 2-picolinamide with Cu(tfacac)2 might occur initially in the mechanistic pathway. Additionally, when stoichiometric radical scavengers, 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and 1,1-diphenylethene (DPE), were added to the reaction of 1a and 2b under standard conditions, the formation of β-alkylated enamide 4a was suppressed (Fig. 2e). Instead, alkyl and alkylated vinyl radical intermediates were confirmed by the formation of radical scavengers trapped adducts that were detected using HRMS (High Resolution Mass Spectrometry), demonstrating the radical property of this process and the possible involvement of alkyl and alkylated vinyl radicals (for details, see the ESI†).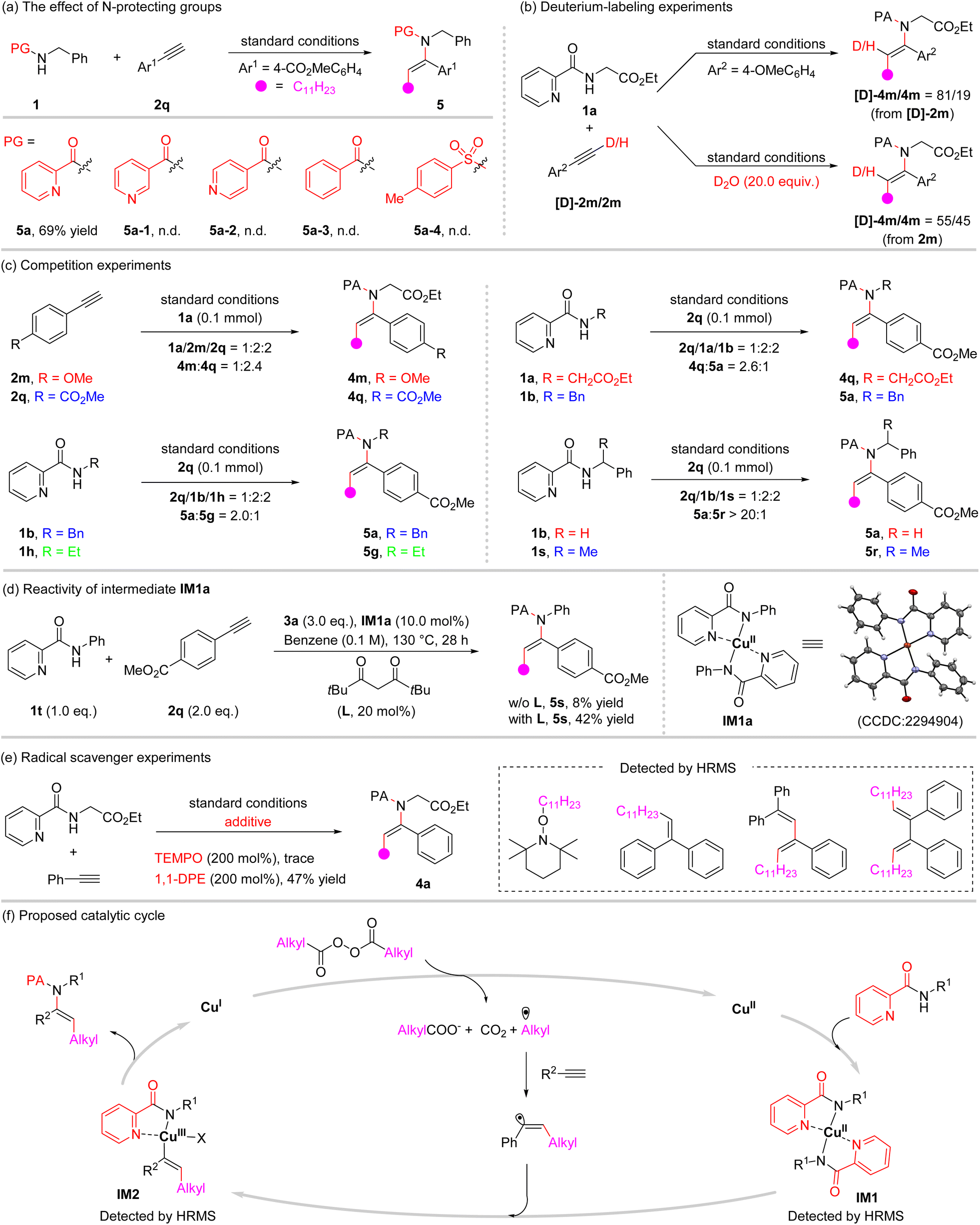 | ||
| Fig. 2 Mechanistic investigations. (a) The effect of N-protecting groups. (b) Deuterium-labeling experiments. (c) Competition experiments. (d) Reactivity of intermediate IM1a. (e) Radical scavenger experiments. (f) Proposed catalytic cycle. TEMPO = 2,2,6,6-tetramethylpiperidinyloxy. 1,1-DPE = 1,1-diphenylethylene. | ||
Based on the experimental results and literature precedence, a plausible reaction mechanism is proposed in Fig. 2f. Initially, an alkyl radical is generated from the diacyl peroxide with the aid of the copper catalyst and/or thermal decomposition,11 which subsequently undergoes regioselective addition to the alkyne to furnish the alkylated vinyl radical. Meanwhile, the 2-picolinamide acts as a ligand to coordinate with copper(II) to yield the intermediate IM1, and then the alkylated vinyl radical is trapped by this coordinated species to form a copper(III) intermediate IM2, followed by reductive elimination to furnish the difunctionalization product. The intermediates IM1 and IM2 could be detected based on HRMS analysis of the reaction mixture. Finally, the released low-valent metal catalyst Cu(I) is reoxidized by diacyl peroxides to fulfill a catalytic cycle. Diacyl peroxide serves simultaneously as the alkylating and oxidizing agent. The role of the 2-picolinamide group is assumed to involve the following aspects. First, the coordination of catalyst copper toward the 2-picolinamide group enables the N–H bond activation.9c–j Secondly, the 2-picolinamide group serving as a bidentate ligand gives access to the stabilization of the Cu(II) and Cu(III) intermediates, IM1 and IM2, and from the latter to the desired C–N bond formation.
Conclusions
In summary, we have developed a novel strategy to allow the radical difunctional alkylamidation of alkynes. With the employment of the diacyl peroxides as both alkylating reagents and internal oxidants, various β-alkylated enamides are constructed in a three-component manner from readily accessible amides and alkynes. This methodology exhibits broad substrate scope with good functional group compatibility and is amenable for late-stage functionalization of natural molecules and biologically relevant motifs. Additionally, various mechanistic experiments have been carried out to obtain a better understanding of the reaction process, such as competition experiments, H/D isotopic exchange experiments and radical trap experiments. We anticipate that this protocol will simplify the synthesis of a series of β-alkylated enamides, helping to fuel the design and discovery of novel biologically molecules. Though further improvements of the reaction are certainly needed, such as lowering the reaction temperature, reducing equivalents of diacyl peroxides, improving the yields of secondary and tertiary alkyl products, and replacement of benzene by eco-friendly solvents, and this will be explored in due course.Data availability
All experimental procedures, characterisation data, control experiments and NMR spectra for all compounds can be found in the ESI.†Author contributions
J. R. and J. X. contributed equally to this work. K. L. conceived the concept and prepared the manuscript. J. R. and J. X. performed and analyzed the data. X. K. and J. L. conducted preparation of starting materials. K. L. directed the project. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support for this work from the National Natural Science Foundation of China (22201190), the Department of Science and Technology of Sichuan Province (2023NSFSC1088, QR2021003) and the start-up grant from West China Hospital of Sichuan University. The authors thank Feijing Su and Qifeng Liu (Core Facilities of West China Hospital) for NMR recording and Qianlun Pu (Mass Spectrometry Center of West China Hospital) for HRMS measurements.Notes and references
- For selected references of pharmaceutical and natural molecules containing enamides and their derivatives, see: (a) R. Shen, C. T. Lin, E. J. Bowman, B. J. Bowman and J. A. Porco, J. Am. Chem. Soc., 2003, 125, 7889–7901 CrossRef CAS PubMed; (b) S. Lin, Z.-Q. Yang, B. H. B. Kwok, M. Koldobskiy, C. M. Crews and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 6347–6355 CrossRef CAS PubMed; (c) M. J. Martín, L. Coello, R. Fernández, F. Reyes, A. Rodríguez, C. Murcia, M. Garranzo, C. Mateo, F. Sánchez-Sancho, S. Bueno, C. de Eguilior, A. Francesch, S. Munt and C. Cuevas, J. Am. Chem. Soc., 2013, 135, 10164–10171 CrossRef PubMed; (d) T. Kuranaga, Y. Sesoko and M. Inoue, Nat. Prod. Rep., 2014, 31, 514–532 RSC.
- For selected reviews on application of enamides, see: (a) R. Matsubara and S. Kobayashi, Acc. Chem. Res., 2008, 41, 292–301 CrossRef CAS PubMed; (b) D. R. Carbery, Org. Biomol. Chem., 2008, 6, 3455–3460 RSC; (c) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS PubMed; (d) K. Gopalaiah and H. B. Kagan, Chem. Rev., 2011, 111, 4599–4657 CrossRef CAS PubMed; (e) N. Gigant, L. Chausset-Boissarie and I. Gillaizeau, Chem.–Eur. J., 2014, 20, 7548–7564 CrossRef CAS PubMed; (f) G. Bernadat and G. Masson, Synlett, 2014, 25, 2842–2867 CrossRef CAS; (g) T. Courant, G. Dagousset and G. Masson, Synthesis, 2015, 47, 1799–1826 CrossRef CAS; (h) M.-X. Wang, Chem. Commun., 2015, 51, 6039–6049 RSC; (i) T. Zhu, S. Xie, P. Rojsitthisak and J. Wu, Org. Biomol. Chem., 2020, 18, 1504–1521 RSC; (j) F. Beltrana and L. Miesch, Synthesis, 2020, 52, 2497–2511 CrossRef.
- For selected references for enamide synthesis, see: (a) J. Genovino, B. Lagu, Y. Wang and B. B. Touré, Chem. Commun., 2012, 48, 6735–6737 RSC; (b) B. Gourdet and H. W. Lam, J. Am. Chem. Soc., 2009, 131, 3802–3803 CrossRef CAS PubMed; (c) L. L. Baldassari, A. de la Torre, J. Li, D. S. Lüdtke and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 15723–15727 CrossRef CAS PubMed; (d) T. Kuranaga, Y. Sesoko and M. Inoue, Nat. Prod. Rep., 2014, 31, 514–532 RSC; (e) Y. Bolshan and R. Batey, Angew. Chem., Int. Ed., 2008, 47, 2109–2112 CrossRef CAS PubMed; (f) T. Kuranaga, Y. Sesoko, K. Sakata, N. Maeda, A. Hayata and M. Inoue, J. Am. Chem. Soc., 2013, 135, 5467–5474 CrossRef CAS PubMed; (g) B. M. Trost, J. J. Cregg and N. Quach, J. Am. Chem. Soc., 2017, 139, 5133–5139 CrossRef CAS PubMed; (h) L. Wang, C. Liu, R. Bai, Y. Pan and A. Lei, Chem. Commun., 2013, 49, 7923–7925 RSC; (i) S. S. Yudha, Y. Kuninobu and K. Takai, Org. Lett., 2007, 9, 5609–5611 CrossRef CAS PubMed; (j) N. Panda and R. Mothkuri, J. Org. Chem., 2012, 77, 9407–9412 CrossRef CAS PubMed; (k) C. Kuai, L. Wang, H. Cui, J. Shen, Y. Feng and X. Cui, ACS Catal., 2016, 6, 186–190 CrossRef CAS; (l) C. Yamamoto, K. Hirano and M. Miura, Chem. Lett., 2017, 46, 1048–1050 CrossRef CAS; (m) P. Chuentragool, M. Parasram, Y. Shi and V. Gevorgyan, J. Am. Chem. Soc., 2018, 140, 2465–2468 CrossRef CAS PubMed; (n) G. Li, P. A. Kates, A. K. Dilger, P. T. Cheng, W. R. Ewing and J. T. Groves, ACS Catal., 2019, 9, 9513–9517 CrossRef CAS; (o) N. Holmberg-Douglas, Y. Choi, B. Aquila, H. Huynh and D. A. Nicewicz, ACS Catal., 2021, 11, 3153–3158 CrossRef CAS PubMed; (p) P. Spieß, M. Berger, D. Kaiser and N. Maulide, J. Am. Chem. Soc., 2021, 143, 10524–10529 CrossRef PubMed; (q) X. Wang and J. A. Porco Jr, J. Org. Chem., 2001, 66, 8215–8221 CrossRef CAS PubMed; (r) M. J. V. Villa, S. M. Targett, J. C. Barnes, W. G. Whittingham and R. Marquez, Org. Lett., 2007, 9, 1631–1633 CrossRef CAS PubMed; (s) J. E. Mathieson, J. J. Crawford, M. Schmidtmann and R. Marquez, Org. Biomol. Chem., 2009, 7, 2170–2175 RSC; (t) H. Liu, Y. Zhou, X. Yan, C. Chen, Q. Liu and C. Xi, Org. Lett., 2013, 15, 5174–5177 CrossRef CAS PubMed; (u) M.-N. Zhao, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Org. Lett., 2014, 16, 608–611 CrossRef CAS PubMed; (v) X.-T. Min, D.-W. Ji, H. Zheng, B.-Z. Chen, Y.-C. Hu, B. Wan and Q.-A. Chen, Org. Lett., 2020, 22, 3386–3391 CrossRef CAS PubMed; (w) L. Kong, X. Han, P. Hu, F. Wang and X. Li, Chem. Commun., 2023, 59, 6690–6693 RSC.
- For selected reviews on radical difunctionalization of alkynes, see: (a) U. Wille, Chem. Rev., 2013, 113, 813–853 CrossRef CAS PubMed; (b) S. R. Chemler and M. T. Bovino, ACS Catal., 2013, 3, 1076–1091 CrossRef CAS PubMed; (c) Y. Shimizu and M. Kanai, Tetrahedron Lett., 2014, 55, 3727–3737 CrossRef CAS; (d) P. Gao, X.-R. Song, X.-Y. Liu and Y.-M. Liang, Chem.–Eur. J., 2015, 21, 7648–7661 CrossRef CAS PubMed; (e) V. P. Boyarskiy, D. S. Ryabukhin, N. A. Bokach and A. V. Vasilyev, Chem. Rev., 2016, 116, 5894–5986 CrossRef CAS PubMed; (f) H. Mei, Z. Yin, J. Liu, H. Sun and J. Han, Chin. J. Chem., 2019, 37, 292–301 CrossRef CAS; (g) C. Lin and L. Shen, ChemCatChem, 2019, 11, 961–968 CrossRef CAS; (h) X. Ren and Z. Lu, Chin. J. Catal., 2019, 40, 1003–1019 CrossRef CAS; (i) W. Liu and W. Kong, Org. Chem. Front., 2020, 7, 3941–3955 RSC; (j) D. Bag, S. Mahajan and S. D. Sawant, Adv. Synth. Catal., 2020, 362, 3948–3970 CrossRef CAS; (k) K. Keerthika, S. Nath and K. Geetharani, Catal. Sci. Technol., 2020, 10, 7142–7159 RSC; (l) D. Bag, H. Kour and S. D. Sawant, Org. Biomol. Chem., 2020, 18, 8278–8293 RSC; (m) H. Yao, W. Hu and W. Zhang, Molecules, 2021, 26, 105–136 CrossRef CAS PubMed; (n) S. Ghosh, R. Chakrabortty and V. Ganesh, ChemCatChem, 2021, 13, 4262–4298 CrossRef CAS; (o) J. Corpas, P. Mauleón, R. G. Arrayás and J. C. Carretero, ACS Catal., 2021, 11, 7513–7551 CrossRef CAS; (p) N. Chalotra, J. Kumar, T. Naqvic and B. A. Shah, Chem. Commun., 2021, 57, 11285–11300 RSC; (q) J.-B. Tang, J.-Q. Bian, Y.-S. Zhang, Y.-F. Cheng, H.-T. Wen, Z.-L. Yu, Z.-L. Li, Q.-S. Gu, G.-Q. Chen and X.-Y. Liu, Org. Lett., 2022, 24, 2536–2540 CrossRef CAS PubMed.
- (a) A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129–133 CrossRef CAS PubMed; (b) M.-L. Li, J.-H. Yu, Y.-H. Li, S.-F. Zhu and Q.-L. Zhou, Science, 2019, 366, 990–994 CrossRef CAS PubMed.
- H. Sigel and R. B. Martin, Chem. Rev., 1982, 82, 385–462 CrossRef CAS.
- (a) F. Wang, N. Zhu, P. Chen, J. Ye and G. Liu, Angew. Chem., Int. Ed., 2015, 54, 9356–9360 CrossRef CAS PubMed; (b) Y.-T. He, Q. Wang, J. Zhao, X.-Y. Liu, P.-F. Xu and Y.-M. Liang, Chem. Commun., 2015, 51, 13209–13212 RSC; (c) L. Fu, X. Chen, W. Fan, P. Chen and G. Liu, J. Am. Chem. Soc., 2023, 145, 13476–13483 CrossRef CAS PubMed.
- H. Xiong, N. Ramkumar, M.-F. Chiou, W. Jian, Y. Li, J.-H. Su, X. Zhang and H. Bao, Nat. Commun., 2019, 10, 122–128 CrossRef PubMed.
- For selected reviews, see: (a) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed; (b) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS PubMed. For selected examples on application of bidentate directing groups in N–H bond functionalization, see: (c) X. Li, B. Li, J. You and J. Lan, Org. Biomol. Chem., 2013, 11, 1925–1928 RSC; (d) N. Xu, J. Liu, D. Lia and L. Wang, Org. Biomol. Chem., 2016, 14, 4749–4757 RSC; (e) H. Sahoo, S. Mukherjee, G. S. Grandhi, J. Selvakumar and M. Baidya, J. Org. Chem., 2017, 82, 2764–2771 CrossRef CAS PubMed; (f) Q. Yue, Z. Xiao, Z. Kuang, Z. Su, Q. Zhang and D. Li, Adv. Synth. Catal., 2018, 360, 1193–1198 CrossRef CAS; (g) Z. Xiao, S. Shu, Y. Lin, Q. Zhang, P. Ren and D. Li, Asian J. Org. Chem., 2018, 7, 2053–2056 CrossRef CAS; (h) S. Bera, S. Roy, S. C. Pal, A. Anoop and R. Samanta, ACS Catal., 2021, 11, 10847–10854 CrossRef CAS; (i) H. Ji, H. Zhan, S. Chen, Z. Fang, Q. Zhang and D. Li, Asian J. Org. Chem., 2022, 11, e202200454 CrossRef CAS; (j) J. Ren, K. Liu, N. Wang, X. Kong, J. Li and K. Li, ACS Catal., 2023, 13, 11001–11011 CrossRef CAS.
- For selected references on radical β-alkylation of enamides, see: (a) H. Jiang, C. Huang, J. Guo, C. Zeng, Y. Zhang and S. Yu, Chem.–Eur. J., 2012, 18, 15158–15166 CrossRef CAS PubMed; (b) P. Li, J. Zhao, C. Xia and F. Li, Org. Lett., 2014, 16, 5992–5995 CrossRef CAS PubMed; (c) R. Ding, Z.-D. Huang, Z.-L. Liu, T.-X. Wang, Y.-H. Xu and T.-P. Loh, Chem. Commun., 2016, 52, 5617–5620 RSC; (d) R. Ding, W.-G. Lu, H. Ci, Y.-Y. Mao and L. Liu, ChemistrySelect, 2019, 4, 6954–6957 CrossRef CAS; (e) J.-Y. Guo, T. Guan, J.-Y. Tao, K. Zhao and T.-P. Loh, Org. Lett., 2019, 21, 8395–8399 CrossRef CAS PubMed; (f) J.-Y. Guo, Z.-Y. Zhang, T. Guan, L.-W. Mao, Q. Ban, K. Zhao and T.-P. Loh, Chem. Sci., 2019, 10, 8792–8798 RSC; (g) S. Bertho, I. Dondasse, P. Retailleau, C. Nicolas and I. Gillaizeau, New J. Chem., 2020, 44, 7129–7141 RSC; (h) S. Bertho, R. Maazaoui, D. Torun, I. Dondasse, R. Abderrahim, C. Nicolas and I. Gillaizeau, New J. Chem., 2021, 45, 17475–17482 RSC; (i) J.-X. Wang, Y.-T. Wang, H. Zhang and M.-C. Fu, Org. Chem. Front., 2021, 8, 4466–4472 RSC; (j) X.-D. Su, B.-B. Zhang, Q. Liu, J.-T. Cheng, Z.-X. Wang and X.-Y. Chen, Org. Lett., 2021, 23, 8262–8266 CrossRef CAS PubMed; (k) X. Zhang, T.-M. Yang, L.-M. Hu and X.-H. Hu, Org. Lett., 2022, 24, 8677–8682 CrossRef CAS PubMed; (l) T. Guan, J.-Y. Guo, Q.-H. Xu, X.-W. Zhang, X.-Y. Yu, Y. Zhang and K. Zhao, Green Chem., 2022, 24, 6524–6530 RSC; (m) J.-Y. Tao, Y.-X. Wang, Q.-H. Zhang, K. Ni, T.-H. Zhu and K. Zhao, Green Chem., 2022, 24, 4004–4011 RSC; (n) K. Minami, K. Ohmatsu and T. Ooi, ACS Catal., 2022, 12, 1971–1976 CrossRef CAS; (o) X.-Y. Lu, J.-C. Wang, X.-M. Sun, M.-T. Gao, W.-J. Ying, M.-Y. Ge, Z.-H. Wei, Z. Liu and X.-K. Chen, J. Org. Chem., 2023, 88, 513–524 CrossRef CAS PubMed; (p) H. Liu, Y. Wu, L. Liu, J.-T. Yu and C. Pan, Chem. Commun., 2023, 59, 8556–8559 RSC.
- (a) H. Liu, J.-T. Yu and C. Pan, Chem. Commun., 2021, 57, 6707–6724 RSC; (b) S. Kawamura, S. Mukherjee and M. Sodeoka, Org. Biomol. Chem., 2021, 19, 2096–2109 RSC.
- A. Adeleke, A. P. N. Brown, L.-J. Cheng, K. A. M. Mosleh and C. J. Cordier, Synthesis, 2017, 49, 790–801 CrossRef CAS.
- The pKa values of the N–H bond of the substrates (1a, 1b, and 1h) were calculated using theoretical calculations, and the values are 44.4, 45.3, and 45.8, respectively (see the ESI† for computational details).
- C. Sambiagio, R. H. Munday, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem.–Eur. J., 2014, 20, 17606–17615 CrossRef CAS PubMed.
- The crystal data of IM1a has been deposited in the Cambridge Crystallographic Data Centre as CCDC 2294904†.
Footnotes |
| † Electronic supplementary information (ESI) available: General information, experimental section, compound characterization, and NMR spectra. CCDC 2294904. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03786j |
| ‡ These authors contributed equally: Jing Ren and Junhua Xu. |
| This journal is © The Royal Society of Chemistry 2023 |