
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insights into an NH4OAc-promoted imine dance in Rh-catalysed multicomponent double C–H annulations through an N-retention/exchange dual channel†
Shiqing
Li‡
 *ab,
Shihai
Lv‡
a,
Yanyan
Yang‡
a,
Peiyan
Zhu
a,
Dongbing
Zhao
*b and
Ming-Hua
Zeng
*ac
*ab,
Shihai
Lv‡
a,
Yanyan
Yang‡
a,
Peiyan
Zhu
a,
Dongbing
Zhao
*b and
Ming-Hua
Zeng
*ac
aGuangxi Key Laboratory of Electrochemical and Magneto-Chemical Functional Materials, College of Chemistry and Bioengineering, Guilin University of Technology, Guilin 541004, China. E-mail: lisq@glut.edu.cn
bState Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: dongbing.chem@nankai.edu.cn
cState Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, School of Chemistry and Pharmaceutical Sciences, Guangxi Normal University, 15 Yu Cai Road, Guilin, 541004, China. E-mail: zmh@mailbox.gxnu.edu.cn
First published on 27th October 2023
Abstract
Developing new and understanding multicomponent reactions (MCRs) is an appealing but challenging task. Herein, Rh(III)-catalyzed multicomponent double C–H annulations of cyclic diimines (or diketones and acetone), alkynes, and ammonium acetate to assemble functionalized 1,1′-biisoquinolines and C-bridged 1,1′-bisisoquinolines with controllable 14N/15N editing in one shot has been developed. Through a combination of isotopic-labeling (2H, 18O, and 15N) experiments, crystallography, and time-dependent ESI-MS, the reaction process was studied in detail. Ammonium acetate accounts for two rounds of Hofmann elimination and iminization, thus leading to an unprecedented imine dance, cyclic imine → N-alkenyl imine → NH imine. The N-alkenyl imine can immediately guide a C–H annulation (N-retention channel), and some of it is converted into NH-imine to trigger another annulation (N-exchange channel). The channels and 15N ratios can be regulated by the reaction mode and acidity. Moreover, the resulting 1,1′-biisoquinolines are a privileged ligand scaffold which is exemplified herein by a hydrazine–iodine exchange reaction to form drug-like benzo[c]cinnolines.
Introduction
Catalytic multicomponent reactions (MCRs) serve as an efficient and powerful strategy for the rapid editing of molecular complexity in a single operation, thus expanding the synthetic toolbox for assembling natural products, drugs, and materials.1 Hence, developing new highly ordered catalytic MCRs, especially from simple/commercially available raw materials, is being actively pursued. The tracking and understanding of MCRs are even more appealing yet challenging tasks. Fortunately, a combination of techniques, like crystallography and ESI-MS, has been cooperatively applied to study the processes of complex reactions.2,3 For example, time-dependent ESI-MS (TD ESI-MS) by sampling at multiple time points can offer more information than simple HRMS at a single time.4 However, using these combined techniques in organic MCRs is rare.5On the one hand, Rh-catalyzed C–H annulation has been developed as an efficient and frequently-used method to access N-heterocycles;6 but multicomponent cyclization via Rh catalysis is underdeveloped,7 especially for four or more component reactions. Recently, Li et al. realized a C–H activation/dual-directing group (DDG) strategy for multi-component C–H annulations to construct 1,1′-biisoquinolines (1,1′-BIQs)8 which cannot be obtained from the known C–H activation/1,3-diyne strategy.9–12 These linear DDGs may form bidentate ligands to poison the metal catalyst; and other unwanted Z/E isomers may also be generated in their preparation. Besides, the mechanism was not studied in detail and access to C-bridged counterparts has not been documented. Consequently, we turn our attention to cyclic DDGs which can perfectly avoid these problems. Readily available 4,5-diaryl 2H-imidazole is an ideal cyclic DDG platform that may undergo doubly directed C–H annulations at the two aryls. Nonetheless, only two-component mono [4 + 2]13 and [3 + 2]14 annulations with alkynes/alkenes to obtain isoquinolines and spiroimidazole-indenes have been reported (Scheme 1a), and the mechanism of such transformations remains ambiguous. Dong assumed a direct process of Int-B→Int-C′via reductive elimination (RE, Scheme 1b, path A),13a but the vital middle Int-C was not mentioned/identified. In addition, the annulation of N-protected imines with alkynes has been mechanistically problematic for a long time, because the true paths of the two processes (path A via reductive elimination; path B via concerted C–N formation and C–N cleavage) are not yet clear (Scheme 1b).15 Obviously, isolation/determination of the cationic Int-C can provide direct evidence.16 Encouraged by this pioneering work, we would like to develop novel MCRs based on cyclic DDGs and to fill in the blank in the bundled mechanism.
 | ||
| Scheme 1 C–H activation of 2H-imidazoles and our design. | ||
On the other hand, NH4OAc is a useful inorganic reagent in Rh-catalyzed three-component reactions to form isoquinolines,7 in which it just plays a single role of NH-imine formation, and 15N-labelled products are not involved. Herein, we disclose novel four/six-component reactions to assemble 1,1′-BIQs and C-bridged 1,1′-bisisoquinolines, containing dual N-retention and N-exchange channels, via a C–H activation/DDG strategy (Scheme 1c). The mechanism is studied in detail by a combination of isotopic labeling, crystallography, and TD ESI-MS, demonstrating NH4OAc as a multifunctional reagent to promote two rounds of Hofmann elimination and iminization, leading to an unprecedented imine dance. 15N incorporation can be controllably edited by the two channels and is affected by reaction mode and acidity. Moreover, the current 1,1′-BIQ ligand platform shows powerful catalytic activity in Cu-catalyzed diarylation to form benzo[c]cinnolines.
Results and discussion
Initially, 4,5-diphenyl-2H-imidazole 1a and two molecules of diphenylacetylene 2a were subjected to a simple Rh(III)/Cu catalytic system (Scheme 2a), but none of the complex transformations, of double annulated compound (di-3a), 1,1′-BIQ 3a, acyl isoquinoline 3a′ or spiral product 3a′′ occurred. In contrast, the reaction happened to stop at the first annulation, giving mono-cationic product Int-C in a high yield, as determined by X-ray diffraction (CCDC 2266530).17 Interestingly, 1,1′-BIQ 3a was obtained solely in 89% yield when 2 equiv. of NH4OAc was added. Optimization of the reaction parameters, such as N-sources, catalysts, oxidants, and solvents, led to give 3a in the highest yield of 93% in the presence of NH4OAc (2 equiv.), [Cp*RhCl2]2 (2.5 mol%) and Cu(OAc)2·H2O (1 equiv.) in TFE in air at 120 °C for 12 hours (see Table S1 in the ESI†). With treatment of Int-C with a single additive, such as [Cp*RhCl2]2, Cu(OAc)2, or NH4OAc, under thermodynamic conditions, only NH4OAc successfully gave ring-opening product 3a′ in 92% yield, showing that NH4OAc is the true promoter of Hofmann elimination (Scheme 2b). Both Int-C and 3a′ can transform to 3a in the presence of alkyne 2a and NH4OAc (Scheme 2c), hinting that they are key intermediates in this double C–H activation. These results lead to the hypothesis of an N-exchange mechanism (channel 1, Scheme 2d): first annulation → Hofmann elimination → hydrolysis → iminization → second annulation. Undoubtedly, reductive elimination process/Hofmann elimination (path A) is favored while the concerted process (path B) is not, because intermediate Int-C is isolated in high yield.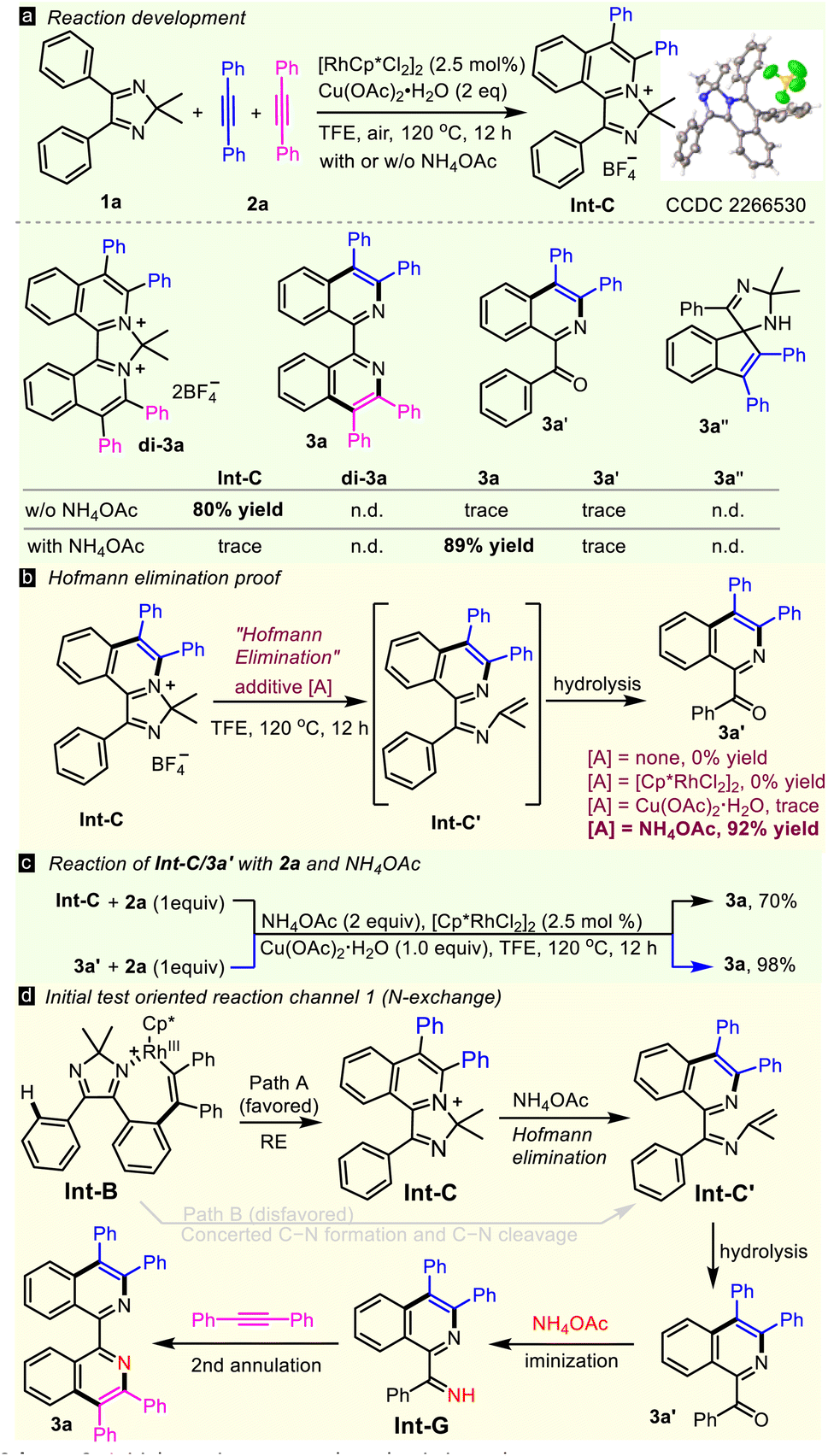 | ||
| Scheme 2 Initial reaction tests and mechanistic study. | ||
To prove the above reaction mechanism, comprehensive isotopic labeling experiments (D, 18O, and 15N) were conducted (Scheme 3 and ESI†). The reaction of 1a using deuterated methanol as a co-solvent furnished [D4]-1a with 28% D-incorporation (see ESI†). While no H/D-exchange was observed from 3a′, revealing that the first C–H activation was reversible, but the second was not. Then we performed detailed kinetic studies. For the first annulation reaction (Int-C), the KIE value was observed to be 2.3. A negligible KIE of 1.0 was observed in the second C–H activation. The KIE value of the di-annulation reaction was found to be 3.0. These kinetic studies implied that cleavage of the first C–H bond may be involved in the rate-determining step. Furthermore, the oxygen atom in 3a′ was proved to come from water in the Hofmann elimination-hydrolysis cascade of Int-C by adding 10 equiv. of heavy-oxygen water (Scheme 3a), which supported the proposed hydrolytic process. Notably, some unexpected results were received when using 15NH4OAc as the nitrogen source (Scheme 3b). For example, the reaction of 1a and 2a with 15NH4OAc gave mix-15N-3a (a mixture of 14N,15N- and 14N,14N-3a) with only about 39% rather than 100% 15N incorporation for one N atom of the two (Scheme 3c). Also, differentiated 15N ratios were observed for 3b (30%), 3c (9%), 3d (16%), 3j (67%), 3k (28%), and 3m (30%), respectively. These results revealed that another reaction route (the N-retention channel) must be included. Generally, the electron-rich substrate shows a higher 15N ratio, suggesting it favors the N-exchange channel over its electron-deficient counterpart. More interestingly, the 15N ratio of mix-15N-3a did not change (∼40%) throughout the entire reaction time (Scheme 3d), implying that the rate-determining step may be involved in an early stage and the value of the rate constant ratio k(N-exchange)/k(N-retention) is ∼0.4. Thus, an N-retention process (channel 2) in the second annulation should be included, where N-alkenyl imine Int-C′ guides the second C–H activation directly (Scheme 3e).
 | ||
| Scheme 3 Isotopic labeling experiments. | ||
To collect more process information on this dual-channel reaction, time-dependent electrospray ionization mass spectrometry (TD ESI-MS) was conducted (Fig. 1). First, at room temperature, besides substrate 1a, fragments at 485.14, 663.22, 425.20, and 385.17 corresponding to Int-A [M]+, Int-B [M]+, Int-C [M]+/Int-C′ [M + H]+, and Int-G [M + H]+ were detected. Then at 120 °C, the signal of Int-B disappeared after 0.5 h because it is not very stable and it quickly takes part in the subsequent transformation. The abundances of Int-A, Int-B, and Int-G were low, which could be ascribed to its low concentration/stability. Initially, the abundance of Int-C/Int-C′ increased, but after 2 h, they began to decrease. The abundances of Int-F and 3a increased gradually during the course of the reaction. On the other hand, all the 15N-labelled signals of these intermediates were observed when using 15NH4OAc as the N-source (see ESI†).
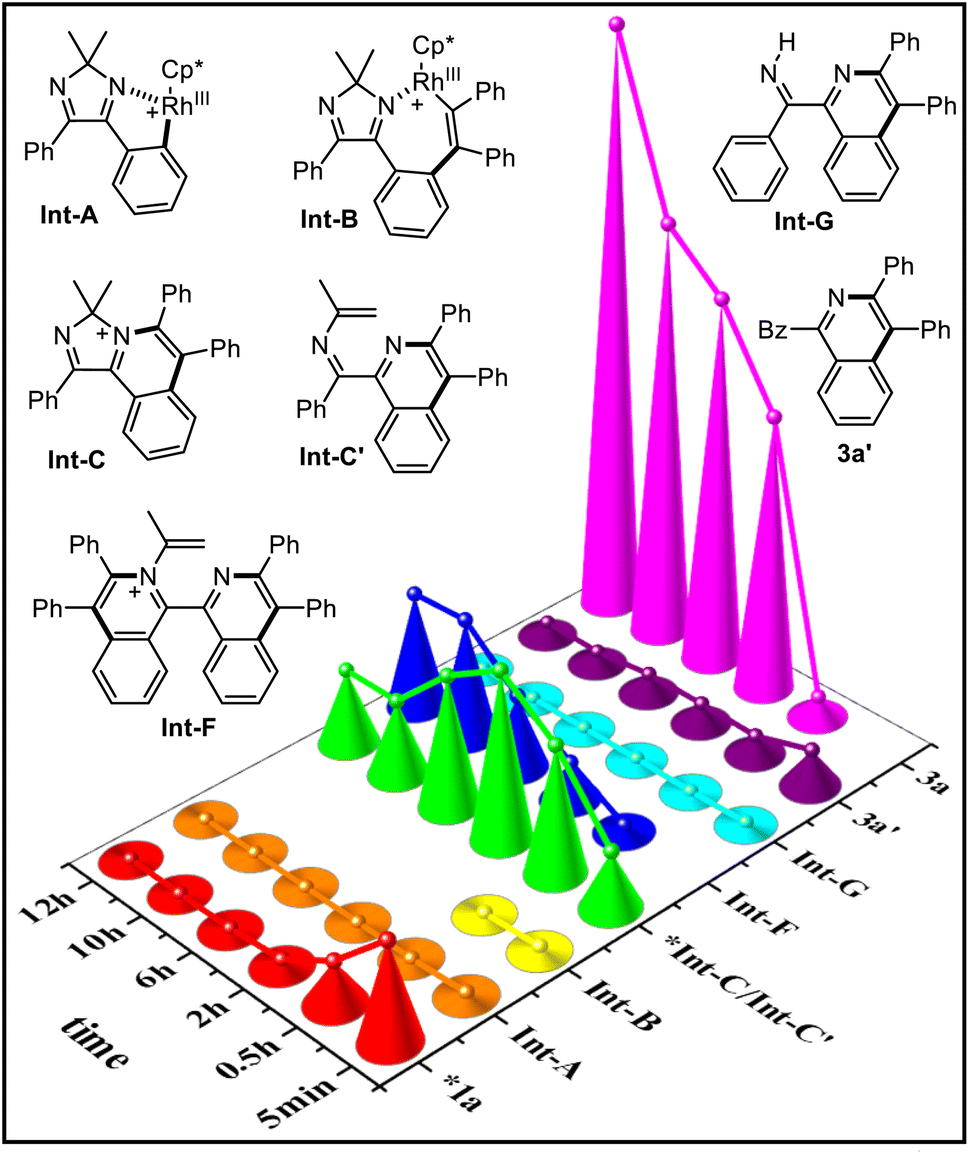 | ||
| Fig. 1 Relative intensities of the reaction system at different points in time (room temperature: 5 min; 120 °C: 0.5, 2, 6, 10, 12 h) under standard conditions (*intensity/100). | ||
Based on the above experiments, especially the isotopic experiments, ESI-MS analysis, and literature precedence,13,14 a dual-channel mechanism is proposed (Fig. 2). The cationic Cp*(OAc)Rh+ detected by ESI-MS is formed first, with the aid of Cu(OAc)2. Cyclic diimine 1a induces the first C–H activation with Cp*(OAc)Rh+ to form the first rhodacycle Int-A. Subsequent coordination and insertion with 2a give seven-membered complex Int-B, which undergoes reductive elimination to produce cationic compound Int-C, realizing the first C–H annulation. Then, ammonium acetate-mediated Hofmann elimination occurs on Int-C to furnish N-alkenyl imine Int-C′, starting-up the second C–H annulation, which divides into two channels. On the one hand there is the direct C–H activation of Int-C′ with Cp*Rh(III) species to afford the second imine-Rh species Int-D, which is followed by alkynyl insertion to form Int-E. Subsequent reductive elimination releases Cp*Rh(I) and Int-F, which proceeds to undergo the second Hofmann elimination to obtain final product 3a with N-retention. On the other hand, hydrolysis of imine Int-C′ yields 1-benzoyl isoquinoline 3a′, followed by iminization with NH4OAc to provide NH-imine Int-G. C–H activation of Int-G produces the third imine-Rh species Int-H. Subsequent insertion with alkyne and reductive elimination delivers Cp*Rh(I) species and 3a with N-exchange. Cp*Rh(III) is regenerated from Cp*Rh(I) through oxidation with Cu(II) and air to guarantee the catalytic cycle.
 | ||
| Fig. 2 A total dual-channel mechanism. | ||
With the optimal reaction conditions and plausible mechanism in hand, the reaction scope was then defined (Scheme 4). A number of 2H-imidazoles were subjected to reaction with NH4OAc and two molecules of alkyne 2a. 2H-Imidazole 1b with methyl at the para-position of phenyl reacted with 2a and NH4OAc to afford 3b in 64% yield. para-Chloro (1c) or -fluoro (1d) substituted imidazole gave 3c and 3d in 77% and 58% yields, respectively. Imidazole 1e with a meta-methyl group gave 3e in 80% yield with excellent regioselectivity. Thienyl-fused bipyridine 3f was successfully obtained for the first time with acceptable yield. Unsymmetric imidazole 1g was well tolerated to obtain unsymmetric 1,1′-BIQ 3g in a good yield of 75%. Thus, treatment of ortho-OMe-substituted alkyne 2b with 1a afforded 3h in a good yield of 69%. The reaction of meta-Br diphenylacetylene 2c and 1a generated 3i in 50% yield. para-OMe (2d), –Cl (2e), –Br (2f), and –CF3 (2g) substituted diphenylacetylenes smoothly underwent two-fold C–H activation and annulation and provided 1,1′-BIQs 3j–m in good to high yields. Notably, thienyl-incorporated 1,1′-BIQs (3f and 3n) were furnished with ∼40% yield. Hex-3-yne underwent this reaction smoothly to give 3o in 81% yield. Upon treatment of unsymmetric alkyl–aryl alkynes and with 1a, two types of regioisomer were generated (3p). Moreover, the title products could also be obtained through a six-component reaction of benzils, acetone, alkynes (×2), and ammonium acetate (×2). For selected products (3a, 3c, 3g, 3j, and 3m) were formed in 46–59% yields entirely from commercially available raw materials. In addition, treatment of 3a′ with one molecule of alkynes 2 in the presence of NH4OAc gave non-symmetric 1,1′-BIQs 4ai-4ao in excellent yields (all over 90%), while they were obtained in low yields in a one-pot, two-step manner—albeit with high chemo-selectivity (ESI†).
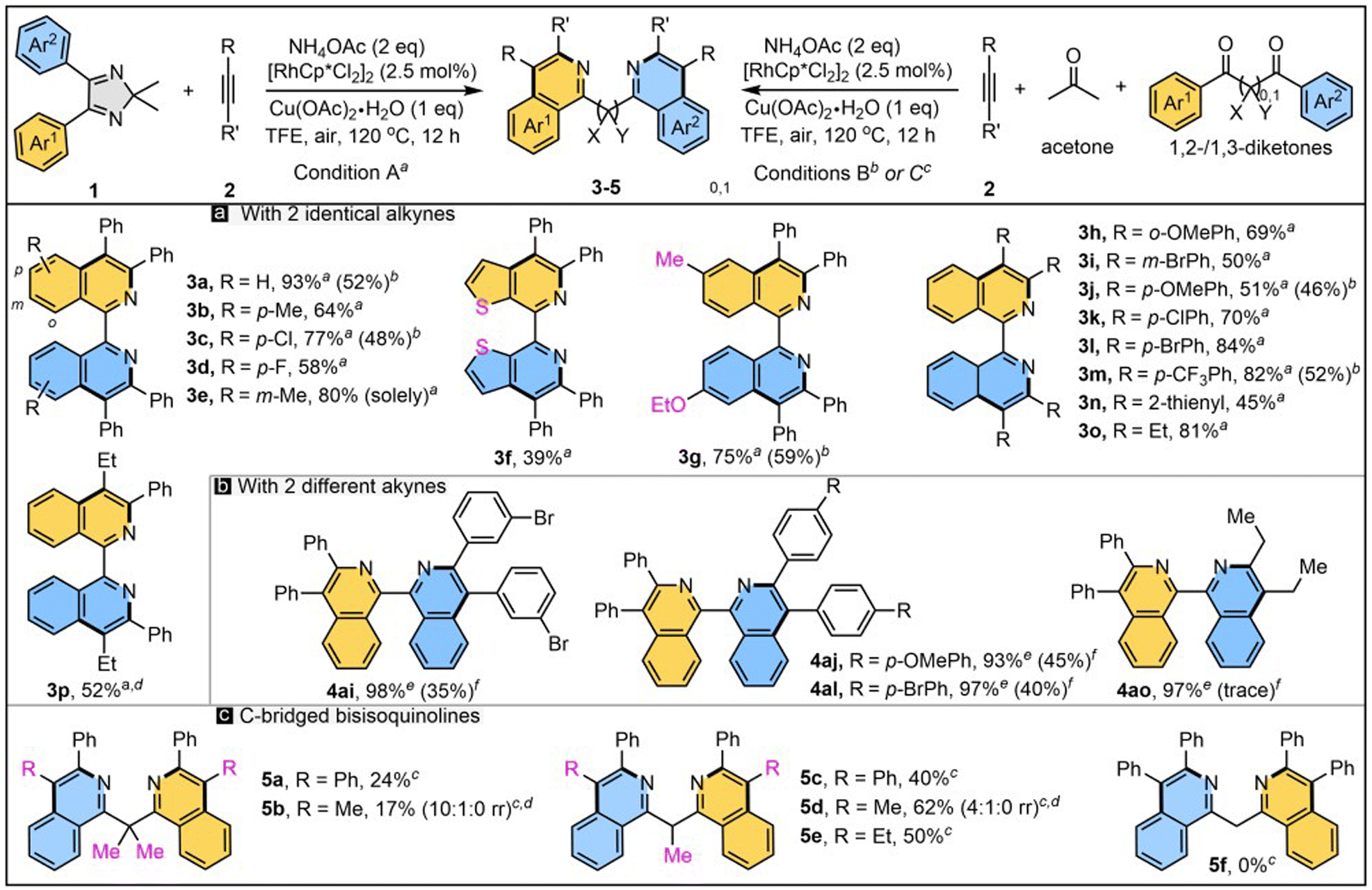 | ||
| Scheme 4 Scope of Substrates. aReaction conditions A: 1 (0.1 mmol), 2 (0.2 mmol), [RhCp*Cl2]2 (2.5 mol%), Cu(OAc)2·H2O (1 equiv.) and NH4OAc (2 equiv.) in TFE (2 mL) at 120 °C for 12 h under air, isolated yield. bReaction conditions B: benzil (0.1 mmol), acetone (0.15 mmol), 2 (0.2 mmol), [RhCp*Cl2]2 (2.5 mol%), Cu(OAc)2·H2O (1 equiv.) and NH4OAc (2 equiv.) in TFE (2 mL) at 120 °C for 12 h under air. cReaction conditions C: 1,3-diketone (0.1 mmol), 2 (0.2 mmol), [RhCp*Cl2]2 (2.5 mol%), Cu(OAc)2·H2O (1 equiv.) and NH4OAc (7 equiv.) in TFE (2 mL) at 120 °C for 12 h under air. dOnly the main isomer is shown. ePrepared from the reaction of 3a′ with alkynes. fPrepared in a one-pot, two-step manner. | ||
With success in preparing non-bridged 1,1′-BIQs from 1,2-diketones, we turned our attention to 1,3-diketones, which may offer interesting C-bridged N-heterobiaryls (Scheme 4c). However, the synthesis of simple methylene-bridged 1,1′-bisisoquinoline 5f was a failure, which may be due to its strong enol tautomerism. Conversely, enol tautomerism-blocked diketone was prepared and subjected to the double C–H annulation, to our delight giving gem-dimethyl-bridged product 5a in 24% yield. The 1,3-diketone with only one methyl-block afforded 5c in a higher yield of 40%. Similarly, the treatment of 1,3-diketones with non-symmetric aryl–alkyl acetylenes generated two of the three isomers. For example, symmetric 5b and non-symmetric isomer 5b′ were obtained as a mixture in a ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Bisisoquinoline 5d was afforded in good yield (62%) with an acceptable regioselectivity ratio (r.r. = 4:1). The reaction of Ph–Et alkyne with diketone gave 5e (50%) and 5e′ (16%) as two isolable isomers. To the best of our knowledge, this is the first example of constructing C-bridged 1,1′-bisisoquinolines through a transition-metal-catalyzed C–H activation strategy.18
:1. Bisisoquinoline 5d was afforded in good yield (62%) with an acceptable regioselectivity ratio (r.r. = 4:1). The reaction of Ph–Et alkyne with diketone gave 5e (50%) and 5e′ (16%) as two isolable isomers. To the best of our knowledge, this is the first example of constructing C-bridged 1,1′-bisisoquinolines through a transition-metal-catalyzed C–H activation strategy.18
Furthermore, the versatile functions of this protocol were explored (Scheme 5). First, an excellent yield (88%, 0.451 g) of product Int-C was obtained in a 1 mmol scale with low catalyst loading (1 mol% Rh) under air (Scheme 5a). It can smoothly convert into 3a′ with a 90% yield through Hofmann elimination and hydrolysis. When using aniline 6 as the N-source instead of NH4OAc, some new 1,1′-biisoquinolinium salts 7a–7c were obtained in good yields (Scheme 5b). Besides, treatment of 3a′ with hydroxylamine gave 1-oximido isoquinoline, which annulated with acrylaldehyde to form 3,4,4′-trisubstituted 1,1′-biisoquinoline 8 in 46% yield in two steps (Scheme 5c), which represents a novel type of 1,1′-BIQ. Other cyclic diimines could also undergo ring-opening double C–H annulation to form 3a. For instance, 5-membered furoxan 9 reacted with 2a to deliver 3a in a high yield of 84% (Scheme 5d). Six-membered diimine 10 afforded 3a with 30% yield through two rounds of Hofmann elimination; as expected, the di-annulation did not take place in the absence of NH4OAc. It is noted that with both N atoms fully 15N-labelled, product di-15N-3a was easily obtained in 50% yield through a one-pot six-component reaction with the aid of 15NH4OAc (Scheme 5e, left). Also, mono-15N-3a with only one N atom fully 15N-labelled was achieved in 95% yield from the reaction of 3a′, 2a, and 15NH4OAc (Scheme 5e, middle). As hydrolysis of Int-C′ is vital in the switch to the N-exchange channel, 15N-labeling can be handled by the pH, i.e., it is enhanced from 39% to 58% when HOAc is added, but decreases to 27% in the presence of NaOAc (Scheme 5e, right). Hence, a library of controllably 15N labelled 1,1′-BIQs could be facilely assembled, for the first time, by adjusting the reaction mode and acidity. Finally, the catalytic activity of this class of newly prepared bidentate ligand was surveyed in an Ullmann-type reaction to synthesize drug-like benzo[c]cinnolines.19 Phthalic hydrazide (11) and cyclic diaryliodonium triflate (12) were treated in the presence of CuI (5 mol%), K2CO3 (2 equiv.), DMF, and a ligand (Fig. 3). Compared to well-known N,N-ligands, like 2,2′-bipyridine (Bipy), 1,10-phenanthroline (Phen), or N,N′-dimethylethylenediamine (DMEDA), 1,1′-BIQ 3a showed higher activity and gave benzo[c]cinnoline (13a) in the highest yield (99%). Especially for the cyano-substituted cyclic iodonium, 3a exhibited much greater efficiency with 99% yield compared to 35% for Bipy. Other 1,1′-BIQs with examples of 3e, 3j, 3m, and 3o, all gave 13b in over 75% yield.
 | ||
| Scheme 5 Synthetic applications. | ||
 | ||
| Fig. 3 BIQ as ligand in double N-arylation to benzo[c]cinnolines. | ||
Conclusions
In conclusion, we have developed Rh(III)-catalyzed novel multi-component reactions to assemble axial 1,1′-biisoquinolines and C-bridged 1,1′-bisisoquinolines through a C–H activation/DDG strategy with controllable 15N editing. The combinations of techniques, crystallography, TD ESI-MS, and isotopic experiments prove that the current transformations include double channels of N-retention and N-exchange. NH4OAc plays irreplaceable multiple roles of two rounds of Hofmann elimination and iminization, leading to an unprecedented imine dance, cyclic imine → N-alkenyl imine → NH imine. The newly in situ formed alkenyl imine can directly guide a C–H annulation with N-retention; or it changes to NH imine via hydrolysis and reiminization, initiating another C–H annulation with N-exchange. The 15N distribution can be predicted and further adjusted by reaction mode and acidity. In this protocol, N-aryl 1,1′-biisoquinoliniums and 3,3′,4′-trisubstituted 1,1′-biisoquinoline have been synthesized for the first time. Moreover, the resultant 1,1′-BIQs are proved to be privileged N,N-ligands with powerful catalytic activity in Cu-catalyzed diarylation to form drug-like benzo[c]cinnolines. Over ten (15N labelled) intermediates have been characterized by the combined techniques, confirming reductive elimination/Hofmann elimination rather than a concerted process of Rh-catalyzed C–N breaking N-annulation, and thus enriching the evidence in Rh catalysis.Data availability
All detailed procedures, characterization data and NMR spectra are available in the ESI.†Author contributions
S. Li conceived the concept, directed the project, performed key experiments, and wrote the paper. S. Lv and Y. Yang carried out most of the experiments. P. Zhu conducted the ESI-MS. D. Zhao conceived the concept and helped edit the manuscript. M. Zeng analysed ESI-MS data and helped edit the manuscript. S. Li, S. Lv and Y. Yang contributed equally to this work.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (22001049, 22261013, 21525101 and 22171075), Guangxi Natural Science Foundation (2020GXNSFBA297003 and 2018GXNSFAA29404), the BAGUI talent program (no. 2019AC26001).Notes and references
- (a) Multicomponent Reactions, ed. J. Zhu and H. Bienaymé, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) Multicomponent Reactions, ed. T. J. J. Müller, Science of Synthesis, Thieme, Stuttgart, 2014, vol. 1, p. 2 Search PubMed; (c) Multicomponent Reactions in Organic Synthesis, ed. J. Zhu, Q. Wang and M.-X. Wang, Wiley-VCH, Weinheim, 2015 Search PubMed.
- For selected reviews, see: (a) S. Datta, M. L. Saha and P. J. Stang, Acc. Chem. Res., 2018, 51, 2047–2063 CrossRef CAS PubMed; (b) D. J. Tranchemontagne, J. L. Mendoza-Cortes, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38(5), 1257–1283 RSC.
- (a) J.-P. Zhong, B. Liu, T. Yang, Y.-J. Liu, Z.-H. Zhu, B.-F. Shi, M. Kurmoo and M.-H. Zeng, Inorg. Chem., 2017, 56, 10123–10126 CrossRef CAS PubMed; (b) M.-H. Zeng, Z. Yin, Z.-H. Liu, H.-B. Xu, Y.-H. Feng, Y.-Q. Hu, L.-X. Chang, Y.-X. Zhang, J. Huang and M. Kurmoo, Angew. Chem., Int. Ed., 2016, 55, 11407–11411 CrossRef CAS PubMed; (c) Y.-Q. Hu, M.-H. Zeng, K. Zhang, S. Hu, F.-F. Zhou and M. Kurmoo, J. Am. Chem. Soc., 2013, 135, 7901–7908 CrossRef CAS PubMed; (d) H.-L. Zheng, X.-L. Chen, T. Li, Z. Yin, Y. Zhang, M. Kurmoo and M.-H. Zeng, Chem.–Eur. J., 2018, 24, 7906–7912 CrossRef CAS PubMed.
- B. Liu, F. Yu, M. Tu, Z.-H. Zhu, Y. Zhang, Z.-W. Ouyang, Z. Wang and M.-H. Zeng, Angew. Chem., Int. Ed., 2019, 58, 3748–3753 CrossRef CAS PubMed.
- (a) R. Xie, Y. Xiao, Y. Wang, Z.-W. Xu, N. Tian, S. Li and M.-H. Zeng, Org. Lett., 2023, 25, 2415–2419 CrossRef CAS PubMed; (b) V. M. Williams, J. R. Kong, B. J. Ko, Y. Mantri, J. S. Brodbelt, M.-H. Baik and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 16054–16062 CrossRef CAS PubMed; (c) N. L. Bell, C. Xu, J. W. B. Fyfe, J. C. Vantourout, J. Brals, S. Chabbra, B. E. Bode, D. B. Cordes, A. M. Z. Slawin, T. M. McGuire and A. J. B. Watson, Angew. Chem., Int. Ed., 2021, 60, 7935–7940 CrossRef CAS PubMed.
- (a) L. Zhao and R. Hua, Chem. Rec., 2018, 18, 556–569 CrossRef PubMed; (b) V. P. Boyarskiy, D. S. Ryabukhin, N. A. Bokach and A. V. Vasilyev, Chem. Rev., 2016, 116, 5894–5986 CrossRef CAS PubMed; (c) J. C. Lewis, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2008, 41, 1013–1025 CrossRef CAS PubMed; (d) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed.
- For the examples of isoquinoline syntheses, see: (a) L. Zheng, J. Ju, Y. Bin and R. Hua, J. Org. Chem., 2012, 77, 5794–5800 CrossRef CAS PubMed; (b) J. Zhang, H. Qian, Z. Liu, C. Xiong and Y. Zhang, Eur. J. Org Chem., 2014, 2014, 8110–8118 CrossRef CAS; (c) H. Lee, Y.-K. Sim, J.-W. Park and C.-H. Jun, Chem.–Eur. J., 2014, 20, 323–333 CrossRef CAS PubMed; (d) X. Tan, X. Hou, T. Rogge and L. Ackermann, Angew. Chem., Int. Ed., 2021, 60, 4619–4624 CrossRef CAS PubMed.
- (a) S. Li, H. Lv, R. Xie, Y. Yu, X. Ye and X. Kong, Org. Lett., 2020, 22, 4207–4212 CrossRef CAS PubMed; (b) S. Lv, Y.-N. Tian, Y. Yang, C. Wen and S. Li, J. Org. Chem., 2022, 87, 16019–16025 CrossRef CAS PubMed; (c) C. Wen, Y. Wang, Y. Yang, Y.-N. Tian, J. Wang and S. Li, Org. Lett., 2023, 25, 2616–2621 CrossRef CAS PubMed; (d) Y.-N. Tian, S. Lv, L. Huang, C. Wen, Y. Yang, X. Kong, Q. Zhu and S. Li, Org. Chem. Front., 2023, 10, 83–91 RSC.
- D.-G. Yu, F. de Azambuja, T. Gensch, C. G. Gensch and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 9650–9654 CrossRef CAS PubMed.
- R. Feng, H. Ning, H. Su, Y. Gao, H. Yin, Y. Wang, Z. Yang and C. Qi, J. Org. Chem., 2017, 82, 10408–10417 CrossRef CAS PubMed.
- S. Kumar, A. M. Nair and C. M. R. Volla, Org. Lett., 2020, 22, 2141–2146 CrossRef CAS PubMed.
- A. Dey and C. M. R. Volla, Org. Lett., 2020, 22, 7480–7485 CrossRef CAS PubMed.
- (a) X.-L. Wu and L. Dong, Org. Lett., 2018, 20, 6990–6993 CrossRef CAS PubMed; (b) X.-L. Wu and L. Dong, Asian J. Org. Chem., 2018, 7, 2422–2426 CrossRef CAS.
- (a) Y. Luo, H. Liu, J. Zhang, M. Liu and L. Dong, Org. Lett., 2020, 22, 7604–7608 CrossRef CAS PubMed; (b) Z. Song, Z. Yang, P. Wang, Z. Shi, T. Li and X. Cui, Org. Lett., 2020, 22, 6272–6276 CrossRef CAS PubMed; (c) Y. Luo, W. Pu, Y.-J. Xu and L. Dong, Org. Chem. Front., 2021, 8, 4549–4553 RSC; (d) Y. Luo, H.-Y. Zhou, Y.-C. Gang and L. Dong, Org. Lett., 2022, 24, 6940–6944 CrossRef CAS PubMed.
- (a) N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908–6909 CrossRef CAS PubMed; (b) S.-C. Chuang, P. Gandeepan and C.-H. Cheng, Org. Lett., 2013, 15, 5750–5753 CrossRef CAS PubMed.
- Z. Yu, Y. Zhang, J. Tang, L. Zhang, Q. Liu, Q. Li, G. Gao and J. You, ACS Catal., 2020, 10, 203–209 CrossRef CAS.
- CCDC 2266530 (Int-C) contains the supplementary crystallographic data for this paper..
- D. E. Stephens, V. T. Nguyen, B. Chhetri, E. R. Clark, H. D. Arman and O. V. Larionov, Org. Lett., 2016, 18, 5808–5811 CrossRef CAS PubMed.
- A. L. Ruchelman, S. K. Singh, A. Ray, X. Wu, J.-M. Yang, N. Zhou, A. Liu, L. F. Liu and E. J. LaVoie, Bioorg. Med. Chem., 2004, 12, 795–906 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR data and crystallographic data. CCDC 2266530. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03861k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |