
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAza-dicyclopenta[a,g]naphthalenes: controllable seesaw-like emissive behavior and narrowband AIEgens†
Jinbiao
Li
,
Jiaxin
Lao
 and
Hongbin
Zou
*
and
Hongbin
Zou
*
College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, 310058, P. R. China. E-mail: zouhb@zju.edu.cn
First published on 26th September 2023
Abstract
Molecular motions significantly influence the emissive behavior and properties of organic fluorescent molecules. However, achieving controllable emission remains a major challenge for fluorophores. In the case of aggregation-induced emission luminogens (AIEgens), the desired properties of aggregated emission and narrowband spectrum demand molecular motion patterns that inherently oppose each other. A nitrogen-containing dicyclopenta[a,g]naphthalene scaffold was discovered as a controllable luminogenic structure through a highly efficient one-step intermolecular cascade reaction. By carefully balancing molecular motions and introducing additional nitrogen atoms into the skeleton, pyrrole-conjugated dicyclopenta[a,g]naphthalenes with aggregation-caused quenching (ACQ) could be transformed into dual-state emission luminogens (DSEgens). This transformation was achieved by incorporating an additional weak H-bond “lock.” Furthermore, the DSEgens could be converted into AIEgens with an exciting narrow full-width-at-half-maximum (FWHM, <50 nm) by methylation. This unprecedented discovery is attributed to the contribution of the weak H-bond “lock,” which overcomes the limitations of broad band emission in AIEgens caused by restrictions of intramolecular motion. Specific organelle probes were developed by replacing the methyl group of the onium product with different positioning groups. This study emphasizes the delicate balance of molecular motions in controlling luminescence and demonstrates a successful approach to designing organic luminogens with controllable emission and narrowband AIEgens.
Introduction
Intramolecular motion plays a vital role in the emission behavior of fluorescent molecules and has a significant impact on their applications in bioimaging and optoelectronic devices.1 Traditional organic luminescent materials typically have large conjugation planes and exhibit strong emission in solution but become non-emissive when aggregated. This phenomenon, known as aggregation-caused quenching (ACQ), results from the intermolecular π–π stacking of aromatic rings in the aggregated state (Fig. 1A).2 Since 2001, the development of aggregation-induced emission (AIE) fluorophores with opposite optical properties has generated considerable interest from theoretical and applied perspectives.3 These fluorophores are non-emissive in dilute solution but exhibit strong emissions when aggregated. Adequate molecular motions in AIE molecules enhance nonradiative decay rates in solution, while the twisted structure prevents detrimental close plane packing, enabling radiative decay in the aggregated state.4 As a result, ACQ luminogens (ACQgens) are limited to use in the dilute solution state, whereas AIE luminogens (AIEgens) can be used in the aggregated state. Recently, a few fluorophores that exhibit effective emission in both solution and aggregation states have been discovered, and they are referred to as dual-state emission (DSE) fluorophores.5 DSE emission relies on achieving a delicate balance of intramolecular motions within a complex molecular structure. Despite their unique optical properties and promising potential applications, the design and construction of these novel DSE luminogens (DSEgens) remain a significant challenge due to the delicate balance required for molecular motion.6 These emission behaviors demand further highlighting of the significance and challenge of precise control of intramolecular motion, especially for switchable luminogens with the same or similar chemical skeleton. | ||
| Fig. 1 Effect of intramolecular motion on the emissive behavior (A) and properties (B) of fluorescent molecules, and the discovery of controllable ideal aza-dicyclopenta[a,g]naphthalene luminogens via delicate balance of intramolecular motions (C). | ||
Molecular motions also influence the width of the emission peak, and a narrow bandwidth emission spectrum is preferred due to its pure fluorescence color and a wider range of applications.7 As shown in Fig. 1B, the peak width of fluorescence emission is determined by the number of vibrational transitions (0–0, 0–1, 0–2…). Structural changes caused by charge redistribution in the excited state lead to movement of the excited state potential energy surface, resulting in an increase in the number of vibrational transitions. Without any structural changes in the excited state relative to the ground state, an extremely narrowband emission (0–0) can be expected according to the Franck–Condon principle.8 Thus, viable methods for reducing spectral widening include suppressing excited-state structural deformation and minimizing vibronic coupling.9 Generally, coplanar molecules with a rigid structure exhibit narrowed emission, often accompanied by a small Stokes shift and ACQ effect resulting from π–π stacking of a large conjugate system. In contrast, AIEgens with an intrinsic twisted structure offers the advantage of a large Stokes shift (>100 nm). However, the broadened emission with a full-width-at-half-maximum (FWHM) greater than 100 nm of AIEgens is often caused by the variety of excitation energies associated with diverse molecular conformations, which can limit their practical applications. Despite the development of several powerful strategies to narrow the FWHM of ACQgens in recent years,10 it seems contradictory to produce narrowband AIEgens while maintaining their properties through a diverse range of molecular conformations resulting from naturally occurring molecular motions (Fig. 1B). Although some pioneering work has reported narrow emission in AIE materials through a light-harvesting strategy,11 it is still a significant challenge to obtain AIEgens with narrowband emission by effectively controlling inherent molecular motion.
Polycyclic aromatic hydrocarbons (PAHs) are an integral part of organic fluorescent molecules, and the introduction of heteroatoms into PAHs can significantly alter their photophysical properties.12 Given the influence of intramolecular motion on fluorescent emissive behavior and properties, we anticipate that the identification of core nitrogen-containing PAHs with a delicate balance of molecular motions could yield fluorophores with superior properties. As such, aza-dicyclopenta[a,g]naphthalenes, namely 4,11-diaryl substituted pyrrolo[1′,2′:4,5]pyrazino[2,3-f]indolizines (PPI) and 4,11-diaryl substituted imidazo[1′,2′:1,6]pyrido[3,4-e]imidazo[1,2-a]pyrazines (IPIP), were synthesized through a simple intermolecular cascade condensation of N-substituted pyrroles and imidazoles, respectively (Fig. 1C). The PPI compound, with a large and rigid plane, exhibits the ACQ effect, and the 11-aryl group is positioned perpendicular to the main plane due to steric hindrance from the hydrogen of the core structure. However, the resulting highly planarized structure poses challenges in effectively preventing the π–π stacking of aromatic rings in the aggregated state. By introducing additional nitrogen atoms into the core plane of PPI, IPIP is created, with a weak H-bond “lock” between the hydrogen on the 11-aryl group and the nitrogen atom.13 Moreover, the twisted structure of the molecule partially inhibits the intermolecular stacking of the aromatic rings. Consequently, IPIP exhibits emission in both solution and solid states, indicating that it belongs to the family of DSEgens. Subsequent substitution at the N5 position allows IPIP-ND for potential restriction of intramolecular motion (RIM) spatially. Further experiments demonstrated that the delicate balance between the weak H-bond “lock” and RIM in IPIP-ND resulted in an AIE molecule with a narrow FWHM (<50 nm). To our knowledge, this marks the first instance of a small organic fluorescent molecule exhibiting the AIE effect with such a narrow FWHM. Additionally, the incorporation of organelle-specific groups on IPIP enabled the discovery of different organelle probes.
Results and discussion
Pyrrole-conjugated PAHs are crucial organic fluorescent molecules due to their diverse photophysical properties and wide range of applications.14 The synthesis of these molecules typically involves multiple steps.15 In previous studies, we successfully developed pyrrole-conjugated AIE systems, such as pyrrolo[1,2-b]isoquinolines and aza-dicyclopenta[a,f]naphthalenes, through intra- or intermolecular cascade reactions of versatile N-substituted 2-ketopyrrole synthons (Scheme 1A).16 Building upon these synthetic strategies, we employed similar N-substituted 2-ketopyrroles in a one-step intermolecular reaction transformation to synthesize novel aza-dicyclopenta[a,g]naphthalenes called PPI. N-substituted 2-ketoimidazoles were utilized for the efficient construction of aza-dicyclopenta[a,g]naphthalenes, leading to the formation of IPIP. The process also helped introduce a potential weak H-bond “lock” into PPI.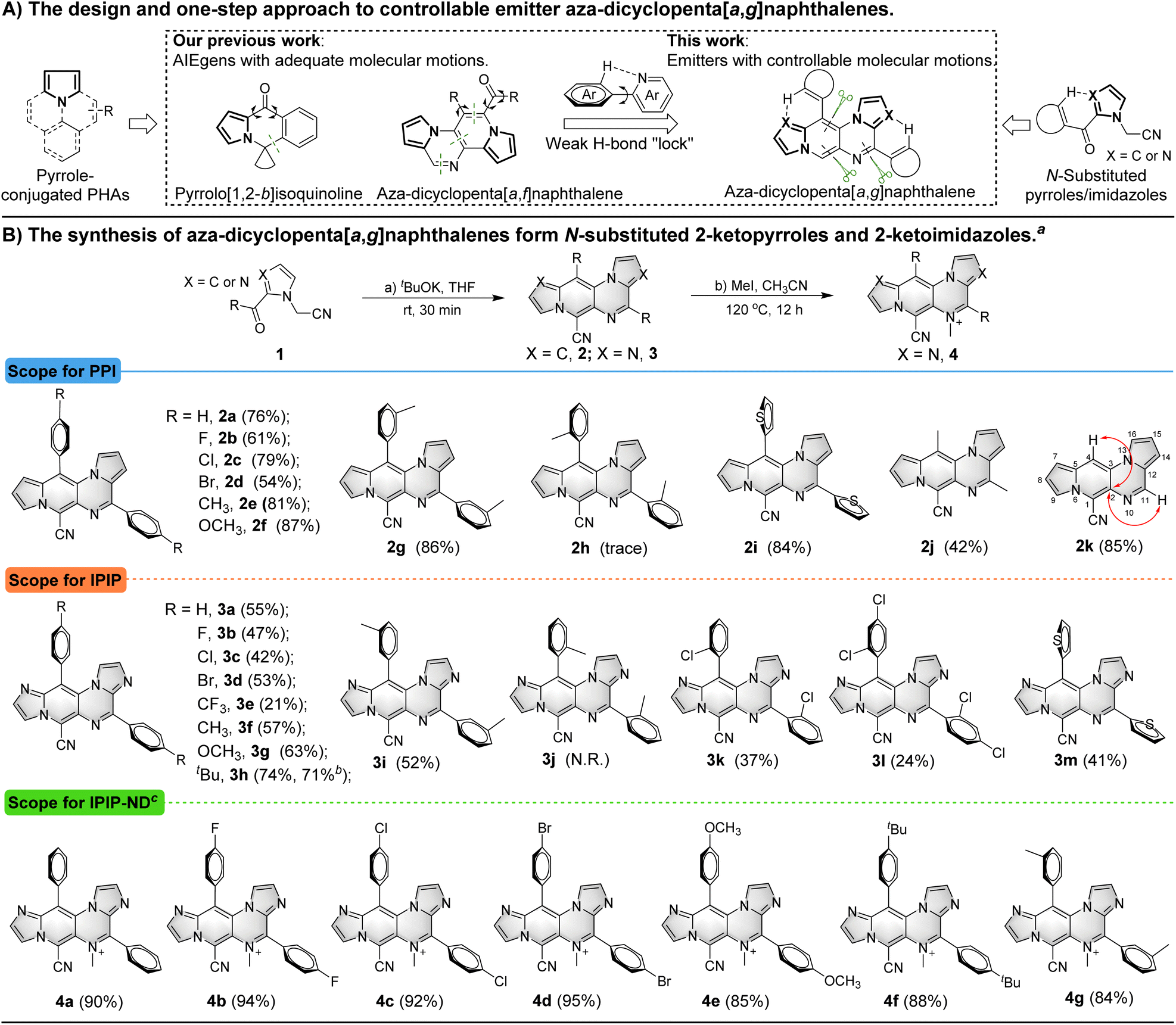 | ||
| Scheme 1 (A) Design of and one-step approach to the emitter aza-dicyclopenta[a,g]naphthalenes; (B) the synthesis of aza-dicyclopenta[a,g]naphthalenes from N-substituted 2-ketopyrroles and 2-ketoimidazoles, and scope for PPI, IPIP, and IPIP-ND. Reaction conditions: (a) 1 (0.2 mmol), tBuOK (0.4 mmol), THF (2 mL), room temperature, 30 minutes, under N2; (b) 2 (0.1 mmol), MeI (0.2 mmol), CH3CN (2 mL), 120 °C, 12 h; aisolated yield; bgram-scale; ciodide. | ||
The reaction was initiated by treating 2-(2-benzoyl-1H-pyrrol-1-yl)acetonitrile (1a) with tBuOK, resulting in the observation of aza-dicyclopenta[a,g]naphthalene 2a with three nitrogens in the skeleton (Table S1 in the ESI†).
We then screened critical reaction factors, including the base, solvents, temperature, and time, to determine the most successful reaction conditions for the production of 2a from 1a. The optimal conditions involved tBuOK in THF at room temperature for 30 min. With these efficient one-step reaction conditions established, we investigated the scope of the reaction for aza-dicyclopenta[a,g]naphthalene (PPI) formation (Scheme 1B). Various pyrrole derivatives, including electron-rich and electron-deficient substituents on the phenyl ring's para-position of 2-(2-benzoyl-1H-pyrrol-1-yl)acetonitrile, were found to be compatible with this transformation (2a–2f). Products with electron-withdrawing groups yielded relatively lower yields (2b–2d, 54–79%) than those with electron-donating groups (81% and 87% for 2e and 2f, respectively). The impact of substituted positions on the phenyl ring was also examined using methyl as an example. While high yields of 2e (81%) and 2g (86%) were obtained with para- and meta-methyl substitution, only a trace amount of ortho-methyl substituted product 2h was obtained due to steric hindrance. Moreover, pyrrole derivatives with thiophenyl, acetyl, and formyl substituents successfully delivered the corresponding products in moderate to good yields (2i–2k, 42–85%).
We introduced two additional nitrogen atoms to the pyrrolo[1′,2′:4,5]pyrazino[2,3-f]indolizine skeleton using imidazole derivatives as synthons to control the delicate balance of molecular motions. The intermolecular cascade reactions of 1l–1x (Scheme S1, ESI†) resulted in the formation of corresponding imidazo[1′,2′:1,6]pyrido[3,4-e]imidazo[1,2-a]pyrazine derivatives (3a–3m) in moderate yields (21–74%). A similar trend was observed for the formation of aza-dicyclopenta[a,g]naphthalenes 3a–3h with electrophilic and nucleophilic groups on the imidazole derivatives. Likewise, the steric hindrance effect of the methyl group on the phenyl ring from imidazole derivative synthons was also observed. The para- and meta-methyl derivatives gave corresponding products 3f (57%) and 3i (52%) in moderate yields, whereas the ortho-methyl analogue failed to produce any corresponding products. However, the smaller ortho-chloro substituted imidazole derivative was tolerated and the corresponding product 3k was generated in a low yield of 37%. The imidazole derivative with the 2,4-dichloro group on the phenyl ring also yielded 3l, albeit with a relatively lower yield of 24%. The thiopheneyl substituted imidazole derivative resulted in the production of 3m with a moderate yield (41%), while acetyl or other acyl substituents failed to achieve the transformation. A gram-scale reaction (3.0 mmol) of 1s was also conducted to afford product 3h with isolated yields of 71%, demonstrating the synthetic utility of this approach. Encouraged by the results and our previous research on methylation of aromatic N-heterocyclics to AIEgens,17 we attempted to incorporate a methyl group to PPI or IPIP using iodomethane as the methylation reagent. We tried different methylation reagents and reaction conditions and could not introduce a methyl group to the PPI structure. However, it succeeded in the methylation of IPIP derivatives to give corresponding IPIP-ND products (4a–4g) in good to excellent yields (84–95%) by simply refluxing with 2.0 equivalents of iodomethane in a sealed tube for 12 h. All of these structures were confirmed by HRMS and NMR, and the molecular structures of 2 were revealed by 2D NMR using 2k as an example (see the ESI† for details).
In spite of sharing a similar core scaffold, PPI, IPIP, and IPIP-ND exhibit distinct emissive behavior when excited by UV light. PPI shows fluorescence only in solution. IPIP exhibits bright emission in solution and solid states, while IPIP-ND only exhibits emission in aggregated states. Compounds 2, 3, and 4 were subjected to photoluminescence (PL) testing to investigate these property differences further. The detailed excitation (λex), emission (λem), Stokes shift, full-width-at-half-maximum (FWHM), quantum yield (Φf), and αAIE data are summarized in the ESI (Tables S2–S4†). The maximum absorption wavelength of these compounds is ∼380 nm, attributed to the π–π* absorption of the conjugated coplanar structure. Tables S2 and S3† show that the fluorescence of PPI ranges from 474 nm (2k) to 531 nm (2f), while IPIP exhibits a maximum emission wavelength of ∼550 nm. IPIP-ND, with different substituents, displays bright green emission in the aggregated state and has maximum emission wavelengths of ∼520 nm. PPI exhibits a large Stokes shift (>100 nm) and a medium FWHM (82–106 nm) in the presence of the ACQ effect. Interestingly, IPIP demonstrates DSE properties with a narrower FWHM (∼70 nm). In the aggregated state, IPIP-ND exhibits narrowband emission (FWHM < 50 nm) and a maximum emission wavelength of ∼520 nm (Table S4†).
The fluorescence performance of these compounds is represented by examples 2c, 3c, and 4c (Fig. 2 and Table S5†). As shown in Fig. 2A, product 2c displays blue fluorescence in solution, product 4c exhibits bright green emission in the aggregated state, while 3c shows strong emission in solution, aggregation, solid, and crystal states upon UV excitation. The PL spectra of 2c, 3c, and 4c in solution show absorption maxima at 378, 373, and 370 nm and emission maxima at 496, 550, and 512 nm, respectively (Fig. 2B–D). Products 3c and 4c exhibit shoulder absorption bands ∼440 nm, and all compounds display large Stokes shifts (>100 nm). Furthermore, a slight blue shift is observed for 2c in the aggregated state, with a peak at 484 nm and FWHM of 86 nm (Fig. 2B). Compound 3c shows emission maxima at 551 nm, with an acromion-emission at 581 nm in the aggregated state and red-shifted wavelengths of 567 and 584 nm in the solid and crystal states, respectively (Fig. 2C). Compared to the aggregated state of 2c, that of 3c showed a narrow FWHM of 73 nm. Notably, the fluorescence of 4c exhibits a slight red shift in maximum emission wavelengths to 524 nm in the aggregated state, with an unexpectedly narrow FWHM of 47 nm (Fig. 2D). Table S5† confirms the ACQ effect of 2c, with a quantum yield of 11.4% in solution and 0.87% in the aggregated state. The quantum yields of 3c in the solution, aggregation, solid, and crystal states are measured to be 14.9%, 8.3%, 7.2%, and 6.1%, respectively, indicating exciting DSE properties. The quantum yield of 4c in solution and aggregation was measured to be 0.46% and 18.8%, respectively, confirming its AIE features. However, the quantum yield of 4c in the crystal state was measured to be 0.58%, illustrating the emission quenching of 4c in a tightly arranged state. The emission lifetimes of 2c, 3c, and 4c in different states were detected to be several nanoseconds, and the results are shown in the ESI.† Markedly, the designed additional two nitrogens of ACQgens PPI (2c) led to the successful production of IPIP (3c) with DSE properties, while further small structure manipulation resulted in IPIP-ND (4c), thus exhibiting AIE effects with an unprecedented narrow FWHM (<50 nm). Interestingly, all of these products also retained their large Stokes shifts (>100 nm).
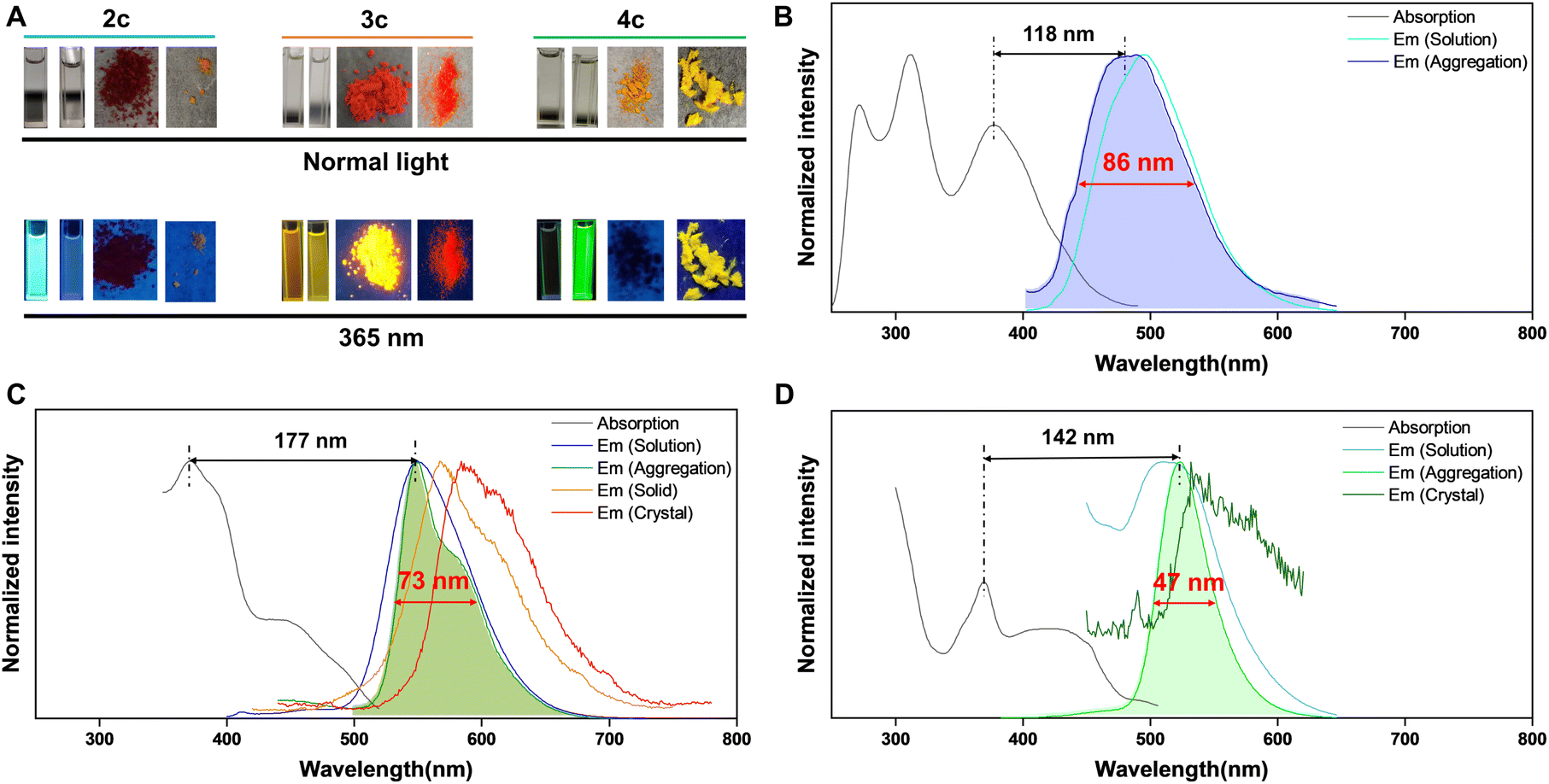 | ||
| Fig. 2 Luminescence properties of 2c, 3c, and 4c. (A) Photos of compounds 2c, 3c, and 4c (from left to right: solution, aggregation, solid, and crystal state) under normal light (up) and 365 nm UV light (down). Normalized absorption and PL spectra of 2c (B), 3c (C), and 4c (D) in different states. Concentration: 10 μM. | ||
The fluorescence properties of 2c, 3c, 3h, 4c, and 4f were examined in different solvents and water fractions to explore the emissive behavior of the compounds further. The absorption bands of these compounds remained stable ∼380 nm in various solvents, but the fluorescence intensity and emission maxima showed significant changes. Compound 2c exhibited solvatochromism in different solvents and the maximum emission shifts from 470 nm in toluene to 496 nm in DMSO (Fig. S1†). However, 2c presents a fluorescence spectrum with two peaks centered at 497 and 583 nm in petroleum (Fig. S1C, ESI†), which could be ascribed to the twisted intramolecular charge transfer (TICT) effect.18 Similar solvatochromism was observed for 3c (Fig. S2†) and 3h (Fig. S3†), with identical core structures. A strong intensity was observed with green-light emissions ∼520 nm in low polar solvents such as petroleum ether, toluene, tetrahydrofuran, chloroform, and dioxane. However, in high polar solvents such as acetone, acetonitrile, methanol, and dimethyl sulfoxide, the emission peak red-shifted to ∼540 nm, resulting in yellow to orange-light emissions. Compared to 2c with a broad FWHM of ∼90 nm, 3c and 3h, with the introduction of nitrogen atoms, resulted in a narrow full width at half maximum (FWHM) of ∼70 nm in different solvents. Compounds 4c and 4f exhibited blue-light emissions in tetrahydrofuran and dioxane among the low polarity solvents, with emission maxima at ∼470 nm, and green-light emissions in acetone, acetonitrile and methanol with a peak of ∼520 nm. Notably, a narrow FWHM of ∼50 nm was observed in 4c and 4f in high-polarity solvents such as acetone, acetonitrile, and methanol. These results indicate that solvents play a crucial role in defining the luminescence properties of these compounds. The emission spectra were measured by adding water (a poor solvent) to the solution of the products in dimethyl sulfoxide (DMSO), a good solvent (Fig. 3), to investigate the emission behavior of these compounds in an aggregated state. The solution of 2c displayed bright blue fluorescence with an emission spectrum centered at 496 nm. Further increasing the water fraction to 95% resulted in a gradual decrease in intensity to 0.08 times, demonstrating that product 2c exhibited ACQ properties (Fig. 3A and F). The solution of 3c exhibited strong orange fluorescence with an emission spectrum centered at 551 nm. Increasing the water fraction caused a decrease in fluorescence intensity at 551 nm and the appearance of a peak at 581 nm. Further increase in the water fraction resulted in a gradual decrease in intensity at 551 nm and 581 nm. Although there was a 0.35-fold decrease in emission intensity at 556 nm in the 95% water fraction, different trends were observed on the tert-butyl substituted 3h due to its large steric hindrance. Increasing the water fraction from 0% to 60% led to its gradual decrease in intensity, which indicates that water can effectively quench the emission of 3h. However, further increase of the water fraction does not significantly affect the emission intensity. Interestingly, the tert-butyl group on the benzene ring appears to be more effective at preventing intermolecular stacking than the chloro group (as shown in Fig. 3C and F). Notably, 3c and 3h exhibit strong fluorescence in solution, as well as in the aggregated, solid, and crystalline states, which is consistent with their DSE properties. The addition of water to the DMSO solution of 4c resulted in a significant increase in emission intensity, with a 46.17-fold enhancement observed as the water fraction increased from 0% to 95%. Similar AIE effects were observed for the tert-butyl substituted 4f, with a 14.00-fold increase in emission intensity at 532 nm from 0% to 80% water fraction. However, a further increase in the water fraction led to a slight decrease in intensity. Both 4c and 4f exhibited pure green emission with a narrow FWHM of 47 nm and 43 nm, respectively, in their aggregated states. These results highlight the influence of solvents and water fraction on the fluorescence properties of the compounds, including color changes, intensity variations, and AIE effects in aggregated states.
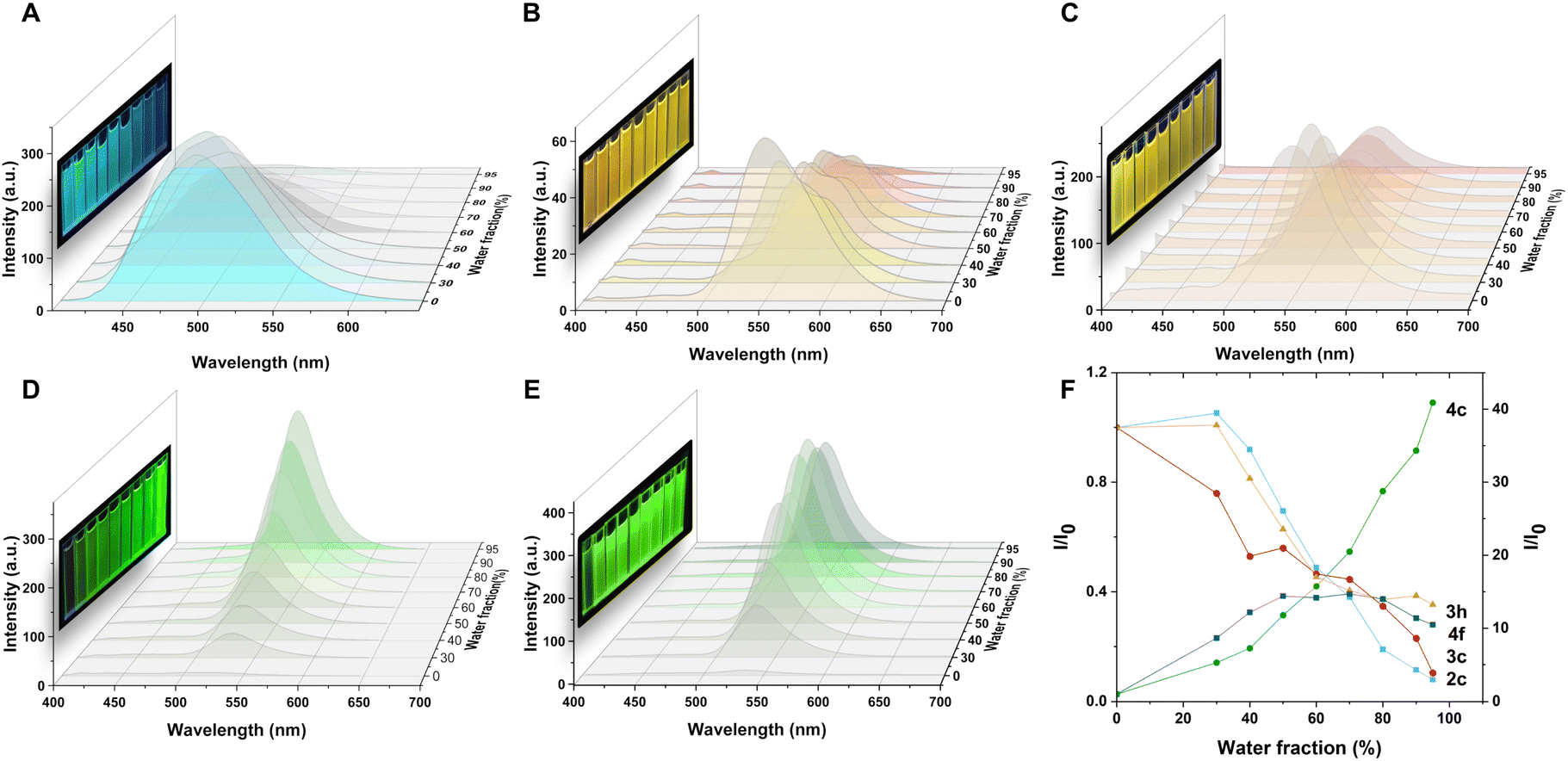 | ||
| Fig. 3 Emission spectra of 2c (A), 3c (B), 3h (C), 4c (D), 4f (E), and (F) plots of emission intensity I/I0 of 2c (496 nm, blue line), 3c (550 nm, red line), 3h (551 nm, orange line), 4c (524 nm, light green line), and 4f (533 nm, deep green line) in different water fractions (inset: photos of 2c, 3c, 3h, 4c, and 4f in various water fractions under 365 nm UV light). Concentration: 10 μM. | ||
To gain insight into the effect of molecular motion during the aggregation process, we measured the particle sizes of products 2c, 3c, and 4c using a diameter analyzer across a range of water fractions. As the water fraction was gradually increased, the average hydrodynamic diameters of these products also increased, reaching ∼100 nm in a 95% water fraction, as measured by dynamic light scattering (DLS, Fig. 4A–C and S8–S10 in the ESI†). Similarly, transmission electron microscopy (TEM) images of 2c and 4c in a 95% water fraction revealed the formation of spherical nanoparticles with sizes in the 100 nm range (Fig. 4D and E). Interestingly, we observed two different shapes of 3c in the aggregated state, as shown in Fig. 4F. By slowly dropping the DMSO solution into water, we observed the formation of both crystalline structures and spherical nanoparticles. This observation is consistent with the emissive behavior of 3c, which exhibits two peaks at 551 and 581 nm in the aggregated state (Fig. 3B). These results sparked our interest in investigating the relationship between the emissive behavior of 3c and its various states. We also utilized powder X-ray diffraction (PXRD) to investigate the changes of 3c between its crystal and solid states, as depicted in Fig. 4G. The pristine crystal of 3c displayed a bright red emission and strong diffraction peaks in the PXRD pattern. Upon gentle grinding, the emission color and diffraction peaks remained largely unchanged, indicating the stability of the crystal phase and the subtle nature of molecular motions. However, complete grinding led to broadened and weakened diffraction peaks in the PXRD pattern, a change in emission color to orange, and emission behavior more closely resembling the solid state of 3c. This significant change suggests that the crystal phase of 3c transformed from the crystalline state into the amorphous state. These results emphasize the profound impact of molecular motion on the emission behavior of fluorescent molecules.
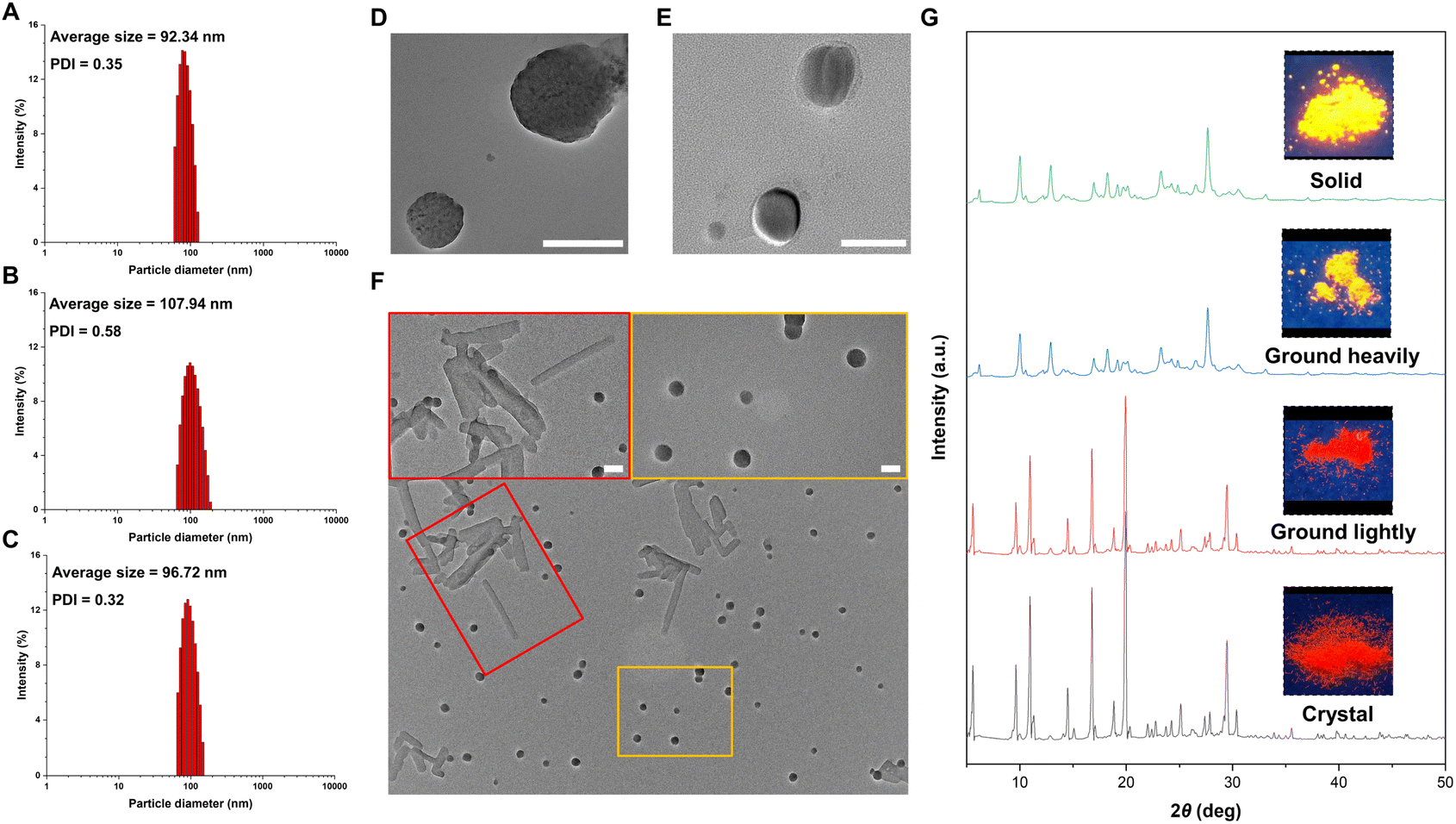 | ||
| Fig. 4 DLS size distribution of 2c (A), 3c (B), and 4c (C) in 95% water/5% DMSO; TEM of 2c (D), 4c (E), and 3c (F) in 95% water/5% DMSO; powder X-ray diffraction patterns of 3c (G) in different states (scale bar: 100 nm). | ||
Fluorescent molecules are frequently designed with specific functional groups for various applications, such as bioimaging, ion sensing, and biological diagnosis.19 However, in many cases, these fluorescent probes are synthesized using a linear approach with multiple steps, resulting in a significant waste of raw materials, time, and cost.20 In this study, the one-step transformation of DSEgens IPIP to easily methylated IPIP-ND led to AIEgens with a fascinating narrow FWHM, as illustrated in Scheme 1 and Fig. 2. This approach inspired us to explore the transformable potential of the protocol by combining IPIP with various functional groups to synthesize different types of potential fluorescent probes, as shown in Scheme 2. Using 3h as a starting material with excellent DSEgen properties, we first introduced ethyl and propyl groups to the skeleton by utilizing iodoethane or iodopropane as nucleophiles, resulting in the corresponding products 5a and 5b with good yields of 89% and 91%, respectively. Functionalizable groups containing alkenyl, alkynyl, hydroxyl, and sulfonic acid (5c–5g, 88–94%) were well-tolerated, providing potential reactive groups. Similarly, ethyl 1-bromopropan-2-one and ethyl 2-bromoacetate successfully delivered the corresponding products with good yields of 85% and 87% (5h and 5i). Due to its high transformability, we chose to use 3h as a starting material to produce various specific organelle probes. Fragments with localization effects for membranes, mitochondria, and lysosomes were successfully incorporated into the structure of IPIP, resulting in the corresponding potential organelle probes (5j–5l, 53–86%). Fluorescence tests confirmed that these molecules exhibited good AIE properties with a narrow FWHM (Table S4, ESI†).
 | ||
| Scheme 2 Scope for IPIP-ND derivatives with different functional groups. Reaction conditions: 3h (0.1 mmol), RI or RBr (0.2 mmol), CH3CN (2 mL), 120 °C, 12 h. | ||
The highly reactive groups present in IPIP-ND derivatives (Scheme 2) enabled us to investigate further the relationship between fluorescence and their molecular motions (Fig. 5). Through a KOH-catalyzed substitution reaction between the cyano and aliphatic hydroxyl groups of 5f, we successfully synthesized product 6 with a planar benzoquinone structure (Fig. 5A). By introducing a double-bond “lock” to the rotatable 4-phenyl ring in 5f, a planar segment without the RIM effect was formed in product 6. As shown in Fig. 5B, the emissive behavior of product 6 was the opposite of that exhibited by 5f. We observed a significant increase in emission intensity at 531 nm as the water fraction gradually increased from 0% to 95%. Compound 5f displayed pure green emission in its aggregated state with a narrow FWHM of 43 nm (Fig. 5B and S6†). In contrast, the solution of product 6 exhibited bright fluorescence with a blue-shifted emission peak at 496 nm. As the water fraction further increased to 95%, we observed a gradual decrease in intensity. These findings indicated that product 6 exhibited ACQ properties, characterized by a broader FWHM of 60 nm (Fig. 5B and S7†). The remarkable results obtained here demonstrate promising potential for utilizing molecular motions in the rational design to balance coplanar and rotating structures, concurrently controlling the emission behavior and properties in different states.
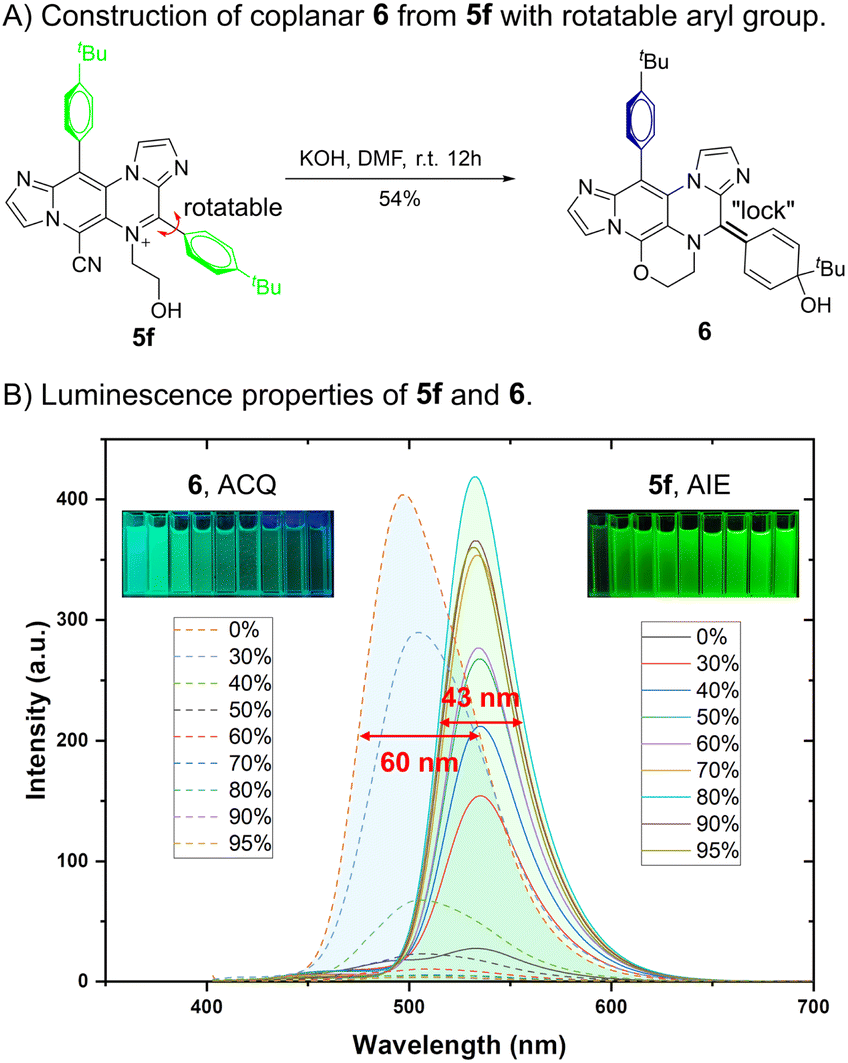 | ||
| Fig. 5 Further showcase of controllable fluorescence via delicate molecular motion balance. (A) Construction of coplanar 6 from 5f with a rotatable aryl group. (B) Luminescence properties of 5f (solid line) and 6 (dashed line) in different water fractions (inset: photos of 5f and 6 in various water fractions under 365 nm UV light). Concentration: 10 μM. | ||
Specific organelle probes play a crucial role in cell research due to their excellent photophysical characteristics, high sensitivity, rapid response, low cost, non-invasiveness, and ability to enable real-time imaging. To investigate the potential of the designed molecules (3h, 5j, 5k, and 5l, as shown in Schemes 1 and 2) for organelle imaging in living cells, we first tested their cytotoxicities against HeLa cells (see the ESI, Fig. S11†). All of the tested compounds exhibited uniform 100% cell viability, even at a high concentration of 50 μM in an aqueous solution. We obtained cell image data by irradiating HeLa cells co-stained with 3h or the commercially available lipid droplet-specific tracker (Nile red) at 405 nm and 550 nm, respectively. The emission spectra of 3h and Nile red, measured in the ranges of 520–580 nm and 580–640 nm, respectively, exhibited a high correlation, as indicated by a Pearson's coefficient of 0.90. In parallel, we observed that the emission spectra of 5j in the range of 480–540 nm overlapped with the commercial membrane-specific tracker (DiI) emission spectra (560–580 nm), with a high Pearson coefficient of 0.93. Furthermore, co-staining experiments of 5k and 5l with commercially available mitochondria-specific tracker (Mit-tracker Red) and lysosome-specific tracker (Lyso-tracker Red) allowed specific labeling of mitochondria or lysosomes in living cells, with Pearson's coefficients of 0.82 and 0.76, respectively. These results demonstrate the potential utility of these probes for organelle imaging in living cells (Fig. 6).
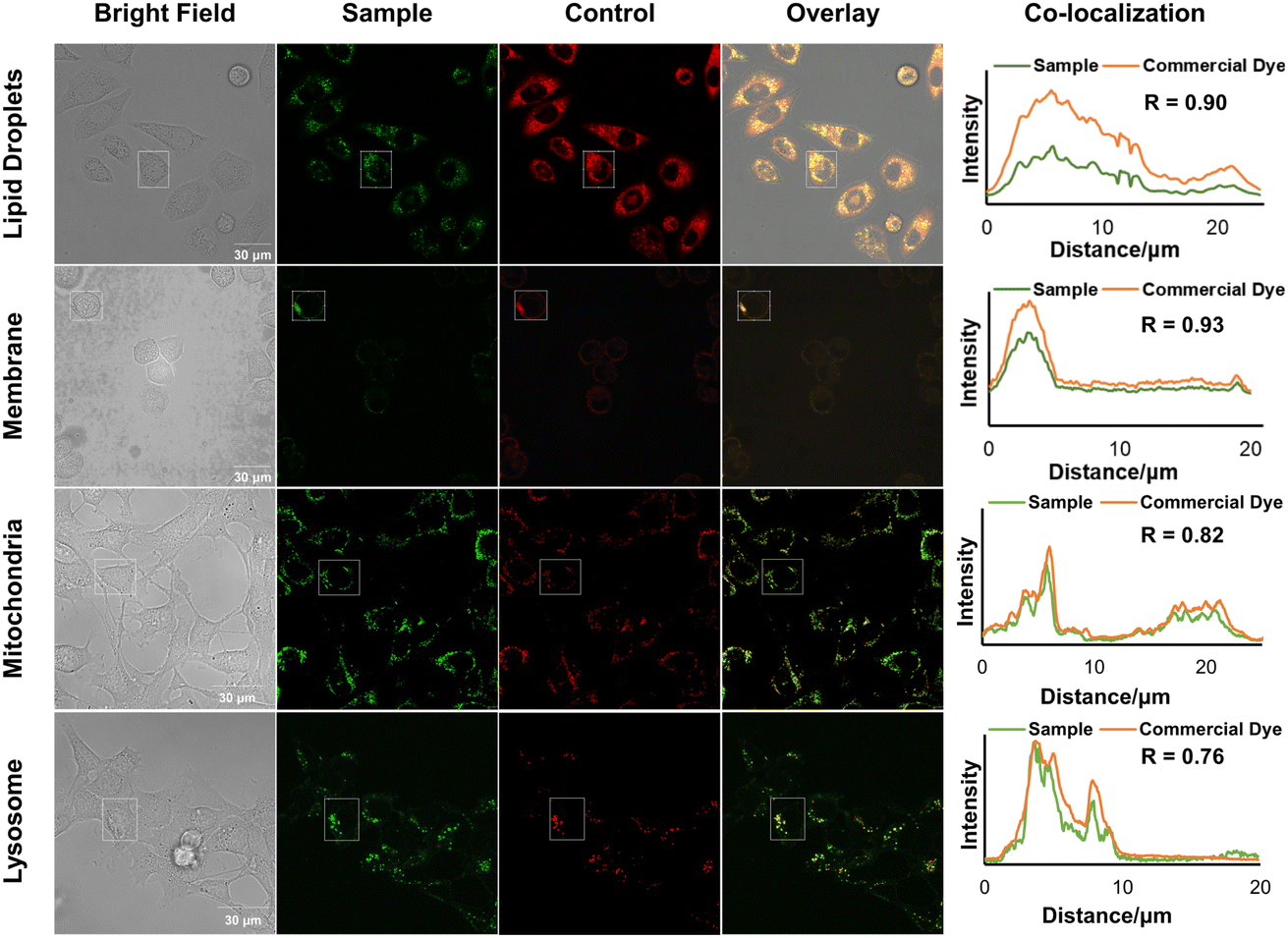 | ||
| Fig. 6 Co-localization images of HeLa cells stained with products 3h, 5j, 5k, and 5l (10 μM, irradiation at 405 nm) and commercially available organelle-probes. 3h (λem = 520–580 nm), 5j (λem = 480–540 nm), 5k (λem = 480–540 nm), 5l (λem = 480–540 nm), Nile red (λex = 550 nm, λem = 580–640 nm), DiI (λex = 550 nm, λem = 560–580 nm), Mito-tracker Red (λex = 580 nm, λem = 590–650 nm), Lyso-tracker Red (λex = 580 nm, λem = 590–650 nm) measured by confocal lasing scanning microscopy (CLSM). Scale bar: 30 μm. | ||
Conclusions
In summary, we have successfully developed aza-dicyclopenta[a,g]naphthalenes that exhibit controllable emissive properties by carefully balancing molecular motions. The introduction of nitrogen atoms has transformed the traditional ACQ molecules, such as PPI, into IPIP with a distinct DSE effect, achieved through the formation of weak H-bond “locks”. Furthermore, methylation of the aromatic nitrogen in IPIP generates its onium derivative, IPIP-ND, which exhibits excellent AIE properties. Notably, when nanoparticle aggregates of IPIP-ND are formed, they exhibit a strong emission with an unprecedented narrow FWHM of less than 50 nm. This breakthrough demonstrates that the unique property of AIE luminogens can be achieved, although it poses significant challenges, through the delicate balance of RIM and rigid coplanarity. We have successfully developed specific organelle probes by functionalizing IPIP with different groups following a simple one-step reaction. These significant findings hold enormous potential for the design of advanced organic luminogens with exceptional optical performance through the precise control of molecular motion balance.Data availability
The data supporting the findings of this study are available within the article and in the ESI.†Author contributions
J. Li conceived the project, carried out the experiments, analyzed the results and wrote the paper. J. Lao performed the cell experiments and provided assistance for the synthesis. H. Zou supervised and directed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (No. 2017YFE0102200), the National Natural Science Foundation of China (No. 21472170) and the Key Projects of Natural Science Foundation of Zhejiang Province (LZ21B020001).Notes and references
- (a) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081 CrossRef CAS; (b) D. Roke, S. J. Wezenberg and B. L. Feringa, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9423 CrossRef CAS PubMed; (c) B. He, J. Huang, J. Zhang, H. H. Y. Sung, J. W. Y. Lam, Z. Zhang, S. Yan, D. Wang, J. Zhang and B. Z. Tang, Angew. Chem., Int. Ed., 2022, 61, e202117709 CrossRef CAS PubMed; (d) D. Tu, J. Zhang, Y. Zhang, H. H. Y. Sung, L. Liu, R. T. K. Kwok, J. W. Y. Lam, I. D. Williams, H. Yan and B. Z. Tang, J. Am. Chem. Soc., 2021, 143, 11820 CrossRef CAS PubMed.
- K. Li, T. B. Ren, S. Huan, L. Yuan and X. B. Zhang, J. Am. Chem. Soc., 2021, 143, 21143 CrossRef CAS PubMed.
- (a) F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 14192 CrossRef PubMed; (b) Q. Wan, Y. Li, K. Ding, Y. Xie, J. Fan, J. Tong, Z. Zeng, Y. Li, C. Zhao, Z. Wang and B. Z. Tang, J. Am. Chem. Soc., 2023, 145, 1607 CrossRef CAS; (c) X. Cai and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 9868 CrossRef CAS; (d) Q. Wu, J. Liu, Y. Li, M. M. S. Lee, L. Hu, Y. Li, P. Zhou, D. Wang and B. Z. Tang, Matter, 2021, 4, 3286 CrossRef CAS; (e) S. S. Chen, H. Wang, B. Wu, Q. Li, J. Gong, Y. L. Zhao, Y. Zhao, X. Xiao, J. W. Y. Lam, Z. Zhao, X. D. Luo and B. Z. Tang, ACS Cent. Sci., 2023, 9, 883 CrossRef CAS PubMed; (f) J. Dai, X. Wu, S. Ding, X. Lou, F. Xia, S. Wang and Y. Hong, J. Med. Chem., 2020, 63, 1996 CrossRef CAS.
- (a) T. Brixner, R. Hildner, J. Köhler, C. Lambert and F. Würthner, Adv. Energy Mater., 2017, 7, 1700236 CrossRef; (b) H. Zhang, Z. Zhao, A. T. Turley, L. Wang, P. R. McGonigal, Y. Tu, Y. Li, Z. Wang, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Adv. Mater., 2020, 32, 2001457 CrossRef CAS; (c) J. Liu, H. Zhang, L. Hu, J. Wang, J. W. Y. Lam, L. Blancafort and B. Z. Tang, J. Am. Chem. Soc., 2022, 144, 7901 CrossRef CAS.
- (a) S. Bhuin, P. Sharma, P. Chakraborty, O. P. Kulkarni and M. Chakravarty, J. Mater. Chem. B, 2023, 11, 188 RSC; (b) Y. Liu, L. Teng, C. Xu, T. Ren, S. Xu, X. Lou, L. Yuan and X. Zhang, CCS Chem., 2021, 3, 2156 Search PubMed; (c) Y. Zheng, Y. Zhou, S. Jiang, X. Xie, G. Du, X. Shen, X. Zhao and Z. Yu, Org. Chem. Front., 2023, 10, 1495 RSC; (d) Q. Liu, M. Zhang, Y. Fu, S. Shen and L. Zhu, Chin. Chem. Lett., 2023, 34, 107612 CrossRef CAS; (e) L. A. Rodríguez-Cortés, F. J. Hernández, M. Rodríguez, R. A. Toscano, A. Jiménez-Sánchez, R. Crespo-Otero and B. Rodríguez-Molina, Matter, 2023, 6, 1140 CrossRef; (f) G. Chen, W. Li, T. Zhou, Q. Peng, D. Zhai, H. Li, W. Z. Yuan, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 4496 CrossRef CAS PubMed.
- (a) L. A. Rodríguez-Cortés, A. Navarro-Huerta and B. Rodríguez-Molina, Matter, 2021, 4, 2582 CrossRef; (b) J. L. Belmonte-Vázquez, Y. A. Amador-Sánchez, L. A. Rodríguez-Cortés and B. Rodríguez-Molina, Chem. Mater., 2021, 33, 7160 CrossRef; (c) H. Wu, Z. Chen, W. Chi, A. K. Bindra, L. Gu, C. Qian, B. Wu, B. Yue, G. Liu, G. Yang, L. Zhu and Y. Zhao, Angew. Chem., Int. Ed., 2019, 58, 11419 CrossRef CAS PubMed; (d) Y. Ni, L. Yang, L. Kong, C. Wang, Q. Zhang and J. Yang, Mater. Chem. Front., 2022, 6, 3522 RSC.
- M. Yang, I. S. Park and T. Yasuda, J. Am. Chem. Soc., 2020, 142, 19468 CrossRef CAS.
- X. Qiu, G. Tian, C. Lin, Y. Pan, X. Ye, B. Wang, D. Ma, D. Hu, Y. Luo and Y. Ma, Adv. Opt. Mater., 2021, 9, 2001845 CrossRef CAS.
- Y. Zhang, G. Li, L. Wang, T. Huang, J. Wei, G. Meng, X. Wang, X. Zeng, D. Zhang and L. Duan, Angew. Chem., Int. Ed., 2022, 61, e202202380 CrossRef CAS.
- (a) X. C. Fan, K. Wang, Y. Z. Shi, Y. C. Cheng, Y. T. Lee, J. Yu, X. K. Chen, C. Adachi and X. H. Zhang, Nat. Photonics, 2023, 17, 280 CrossRef CAS; (b) S. Oda, T. Sugitani, H. Tanaka, K. Tabata, R. Kawasumi and T. Hatakeyama, Adv. Mater., 2022, 34, 2201778 CrossRef CAS; (c) D. H. Ahn, S. W. Kim, H. Lee, I. J. Ko, D. Karthik, J. Y. Lee and J. H. Kwon, Nat. Photonics, 2019, 13, 540 CrossRef CAS; (d) S. Jiang, Y. Yu, D. Li, Z. Chen, Y. He, M. Li, G. X. Yang, W. Qiu, Z. Yang, Y. Gan, J. Lin, Y. Ma and S. J. Su, Angew. Chem., Int. Ed., 2023, 62, e202218892 CrossRef CAS; (e) T. Fan, Y. Zhang, L. Wang, Q. Wang, C. Yin, M. Du, X. Jia, G. Li and L. Duan, Angew. Chem., Int. Ed., 2023, 61, e202213585 CrossRef PubMed; (f) G. Meng, D. Zhang, J. Wei, Y. Zhang, T. Huang, Z. Liu, C. Yin, X. Hong, X. Wang, X. Zeng, D. Yang, D. Ma, G. Li and L. Duan, Chem. Sci., 2022, 13, 5622 RSC; (g) J. Liu, Y. Zhu, T. Tsuboi, C. Deng, W. Lou, D. Wang, T. Liu and Q. Zhang, Nat. Commun., 2022, 13, 4876 CrossRef CAS PubMed.
- (a) X. Zhu, J. X. Wang, L. Y. Niu and Q. Z. Yang, Chem. Mater., 2019, 31, 3573 CrossRef CAS; (b) T. Xiao, H. Qian, Y. Shen, C. Wei, D. Ren, L. Zhang, Z. Y. Li, L. Wang and X. Q. Sun, Mater. Today Chem., 2022, 24, 100833 CrossRef CAS; (c) X. Zhou, L. Zhao, K. Zhang, C. Yang, S. Li, X. Kang, G. Li, Q. Wang, H. Ji, M. Wu, J. Liu, Y. Qin and L. Wu, Chem. Sci., 2023, 14, 113 RSC; (d) Y. Xia, J. Li, X. Chen, A. Li, K. Guo, F. Chen, B. Zhao, Z. Chen and H. Wang, Chem.–Eur. J., 2022, 28, e202202434 CrossRef CAS PubMed.
- (a) M. Farinone, M. Kijewska, J. Cybińska, M. Siczek and M. Pawlicki, Chem. Commun., 2022, 58, 7269 RSC; (b) Y. Huang, J. Xing, Q. Gong, L. C. Chen, G. Liu, C. Yao, Z. Wang, H. L. Zhang, Z. Chen and Q. Zhang, Nat. Commun., 2019, 10, 169 CrossRef PubMed; (c) M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291 CrossRef CAS PubMed; (d) Z. Zhang and Q. Zhang, Mater. Chem. Front., 2020, 4, 3419 RSC; (e) M. S. S. Maier, M. S. N. Hippchen, F. Jester, M. Dodds, M. Weber, L. Skarjan, F. Rominger, J. Freudenberg and U. H. F. Bunz, Angew. Chem., Int. Ed., 2023, 62, e202214031 CrossRef PubMed; (f) Y. Kou, G. Li, Y. Han, M. Li, T. Wang, Z. Qu and Y. Chen, Chem. Sci., 2023, 14, 668 RSC.
- (a) Y. Zou, M. Yu, J. Miao, T. Huang, S. Liao, X. Cao and C. Yang, Chem. Sci., 2023, 14, 3326 RSC; (b) X. He, J. Lou, B. Li, H. Wang, X. Peng, G. Li, L. Liu, Y. Huang, N. Zheng, L. Xing, Y. Huo, D. Yang, D. Ma, Z. Zhao, Z. Wang and B. Z. Tang, Angew. Chem., Int. Ed., 2022, 61, e202209425 CrossRef CAS PubMed.
- (a) Y. Li, X. Li and Y. Xu, Sol. Energy, 2020, 196, 146 CrossRef CAS; (b) J. H. Delcamp, A. Yella, T. W. Holcombe, M. K. Nazeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2013, 52, 376 CrossRef CAS PubMed; (c) J. Hager, S. Kang, P. J. Chmielewski, T. Lis, D. Kim and M. Stępień, Org. Chem. Front., 2022, 9, 3179 RSC.
- (a) O. Dumele, L. Đorđević, H. Sai, T. J. Cotey, M. H. Sangji, K. Sato, A. J. Dannenhoffer and S. I. Stupp, J. Am. Chem. Soc., 2022, 144, 3127 CrossRef CAS PubMed; (b) Y. Hu, Y. Jia, Z. Tuo and W. Zhou, Org. Lett., 2023, 25, 1845 CrossRef CAS PubMed; (c) Q. Li, Y. Zhang, Z. Xie, Y. Zhen, W. Hu and H. Dong, J. Mater. Chem. C, 2022, 10, 2411 RSC.
- (a) J. Li, S. Zhang, J. Lao and H. Zou, ChemSusChem, 2021, 14, 3208 CrossRef CAS PubMed; (b) J. Li, S. Zhang and H. Zou, Org. Chem. Front., 2020, 7, 1218 RSC.
- J. Lao, J. Li and H. Zou, Chem.–Eur. J., 2022, 28, e202202179 CrossRef CAS PubMed.
- C. Wang, W. Chi, Q. Qiao, D. Tan, Z. Xu and X. Liu, Chem. Soc. Rev., 2021, 50, 12656 RSC.
- (a) M. K. Goshisht, N. Tripathi, G. K. Patra and M. Chaskar, Chem. Sci., 2023, 14, 5842 RSC; (b) J. Qian and B. Z. Tang, Chem, 2017, 3, 56 CrossRef CAS.
- (a) Y. Zhou, J. Hua, B. Z. Tang and Y. Tang, Sci. China: Chem., 2019, 62, 1312 CrossRef CAS; (b) W. Ma, L. Zhang, Y. Shi, Y. Ran, Y. Liu and J. You, Adv. Funct. Mater., 2020, 30, 2004511 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03921h |
| This journal is © The Royal Society of Chemistry 2023 |