
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment and application of decatungstate catalyzed C–H 18F- and 19F-fluorination, fluoroalkylation and beyond
Zheliang
Yuan
 *ab and
Robert
Britton
*a
*ab and
Robert
Britton
*a
aDepartment of Chemistry, Simon Fraser University, Burnaby, British Columbia V5A 1S2, Canada
bKey Laboratory of the Ministry of Education for Advanced Catalysis Materials, Department of Chemistry, Zhejiang Normal University, Jinhua, Zhejiang 321004, China. E-mail: zyuan88@zjnu.edu.cn; rbritton@sfu.ca
First published on 1st November 2023
Abstract
Over the past few decades, photocatalytic C–H functionalization reactions have received increasing attention due to the often mild reaction conditions and complementary selectivities to conventional functionalization processes. Now, photocatalytic C–H functionalization is a widely employed tool, supporting activities ranging from complex molecule synthesis to late-stage structure–activity relationship studies. In this perspective, we will discuss our efforts in developing a photocatalytic decatungstate catalyzed C–H fluorination reaction as well as its practical application realized through collaborations with industry partners at Hoffmann–La Roche and Merck, and extension to radiofluorination with radiopharmaceutical chemists and imaging experts at TRIUMF and the BC Cancer Agency. Importantly, we feel that our efforts address a question of utility posed by Professor Tobias Ritter in “Late-Stage Fluorination: Fancy Novelty or Useful Tool?” (ACIE, 2015, 54, 3216). In addition, we will discuss decatungstate catalyzed C–H fluoroalkylation and the interesting electrostatic effects observed in decatungstate-catalyzed C–H functionalization. We hope this perspective will inspire other researchers to explore the use of decatungstate for the purposes of photocatalytic C–H functionalization and further advance the exploitation of electrostatic effects for both rate acceleration and directing effects in these reactions.
 Zheliang Yuan | Zheliang Yuan received his B.S. degree in Chemistry from Zhejiang Normal University (ZJNU) in 2011. In 2016, he received his PhD degree under the supervision of Prof. Guosheng Liu in organic chemistry from Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Sciences (CAS). After his postdoctoral research at TRIUMF and Simon Fraser University (SFU) with Prof. Paul Schaffer and Prof. Robert A. Britton, he started his independent academic career at ZJNU in 2019. He currently is a visiting scholar at SFU supported by China Scholarship Council (CSC). His research interests are the development of new trifluoromethyl chalcogen reagents, radical difunctionalization and fluorination. |
 Robert Britton | Robert Britton obtained his B. Sc. from the University of Waterloo in 1996 working with Professor Victor Snieckus. He then earned a PhD. in natural product isolation, structural elucidation, and total synthesis from the University of British Columbia in 2002 with Professors Edward Piers and Raymond Anderson. He spent two years at the University of Cambridge with Professor Ian Paterson as an NSERC PDF then joined the Process Research Group at Merck as a Senior Scientist in 2004. In 2005, he started an independent academic career at Simon Fraser University and was promoted to Professor in 2015. He is a Michael Smith Foundation for Health Research Career Scholar. Professor Britton has broad research interests that include natural product drug discovery, medicinal chemistry and radiotracer synthesis. Research in these areas have led to 18F-labelled amino acids and peptides for PET imaging in oncology, tools for late-stage modification of drug leads, processes for improving the manufacture of drugs, and strategies for synthesizing structurally complex natural products. |
1 Introduction
Synthetic organic chemistry continues to play a key role in both the pharmaceutical and agrochemical sectors. Here, a constant stream of innovation has revolutionized how we approach molecular design, the construction of complex molecules and the practice of drug discovery. These innovations have significantly expanded the boundaries of accessible chemical space, improved process efficiencies, and facilitated the adoption of safer, environmentally conscious and cost-effective processes.1–3 Undoubtedly, the development of new synthetic methods has enabled the discovery and development of medicines aimed at treating a wide variety of diseases, ultimately improving the quality of life worldwide.1–31.1 Fluorination in medicinal chemistry
An area of particular interest to drug discovery involves the incorporation of a fluorine atom or fluorinated substituent into a lead candidate. This seemingly simple modification can impart unique and sometimes unexpected effects on the physicochemical properties and biological activities of organic molecules.4–7 The strong electron-withdrawing nature of the fluorine atom or small fluorinated functional groups can modulate the pKa of proximal amines and enhance metabolic stability, resulting in an overall improved bioavailability and extended half-life.4–7 Moreover, fluorine atoms or fluorinated groups in drug molecules can modulate the binding affinity and selectivity of drugs by affecting conformation8 and interactions with target proteins or enzymes.4,9,10 The lipophilic nature of fluorine substitutions can also be used to tune the lipophilicity of drug molecules, influencing distribution and partitioning within biological systems. As a result, the modification of drug candidates with fluorine or fluorinated groups can have a profound impact on their pharmacokinetic and pharmacodynamic properties. It is noteworthy that approximately 20% of pharmaceuticals and 50% of agrochemicals currently available on the market contain at least one fluorine atom.11,12In addition to these features relevant to medicinal chemistry, 18F is a widely used radioisotope for positron emission tomography (PET) owing to its convenient half-life (t1/2 = 109.8 min), high positron decay ratio (97%), and low positron energy (0.64 Mev), which leads to images with high spatial resolution.6,13,14 Thus, the development of 18F-labelled radiotracers continues to attract considerable attention,14,15 and PET imaging agents such as 2-deoxy-2-[18F]fluoro-D-glucose ([18F]FDG) have become essential tools for the diagnoses and treatment monitoring of various diseases. Further, in vivo pharmacological imaging using 18F-labelled radiotracers supports biodistribution and drug–target engagement studies, which are crucial in guiding drug development decisions and optimizing therapeutic strategies.
Despite the clear importance of fluorination to (radio)pharmaceutical discovery and development, the incorporation of fluorine into drug-like molecules is often constrained by limitations in synthetic chemistry. Consequently, the development of convenient methods for the introduction of fluorine and fluorine-containing groups into drug-like molecules continues to attract attention.
1.2 Photocatalytic C–H fluorination
Over the past few decades, photochemistry has transformed how chemists contemplate complex molecule synthesis.1,16–21 This broad family of transformations have attracted mounting attention due to their often-mild reaction conditions and unique functional group compatibilities. The application of photoredox catalysis in late-stage functionalization (LSF)22–25 has proven particularly advantageous and often involves activation of nonconventional C–H bonds in drug-like molecules. Here, remarkable site and chemo-selectivity make photoredox catalysis a go-to tool for the precise installation of functional groups in complex molecules.Considering the clear importance of fluorination to medicinal chemistry, much effort has been directed towards LSF via photocatalytic C–H fluorination.26,27 Notably, by avoiding often laborious pre-functionalization steps, late-stage fluorination can support rapid evaluation of structure–activity relationships (SARs). Additionally, the tendency for photocatalytic C–H fluorination to target weak C–H bonds that are also sites for metabolism, provides unique opportunities to rapidly probe metabolic hypothesis. Finally, many of these strategies support the introduction of 18F into complex bioactive molecules, further facilitating discovery efforts in radiopharmaceutical chemistry.
1.3 Decatungstate chemistry
The decatungstate anion belongs to the polyoxometalate family and is often used as the tetrabutylammonium (TBADT, (nBu4N)4[W10O32], Scheme 1a) or sodium salt (NaDT, Na4[W10O32]). Both species have demonstrated outstanding photochemical activity and have attracted considerable attention for the purposes of photocatalytic C–H functionalization.28–34 In the 1980s the use of decatungstate and other polyoxometalates as photocatalysts was reviewed by Papaconstantino35 and dectungstate in particular was further developed through pioneering work by Hill and co-workers,36 and later by Albini, Fagnoni, Ravelli, Ryu and other groups.28–33 The absorption spectrum of the decatungstate anion shows a broad band centered at 324 nm, and the absorption signal gradually diminishes as the wavelength of incident light increases, with absorption disappearing around 420 nm.37 Based on mechanistic studies by Hill,36,37 Tanielian33 and others,28–32 it is proposed that upon photoexcitation, an excited state ([W10O32]4−*) species formed by intramolecular O-to-W ligand to metal charge transfer (LMCT) rapidly (∼30 ps) decays to a reactive state termed wO (lifetime: 55 ± 20 ns) with a quantum yield of around 0.6 (Scheme 1b).38 The wO species is highly electrophilic with partial radical character on the oxygen center and has a predicted redox potential E (wO/[W10O32]5−) around +2.44 V vs. saturated calomel electrode (SCE). wO is proposed to be the species that interacts with organic substrates.39 Depending on the redox properties of the substrate, a single-electron transfer (SET) event may take place when the substrate has an oxidation potential <+2.44 V, leading to a radical cation. Alternatively, hydrogen-atom transfer (HAT) can occur when the substrate has an oxidation potential greater than +2.44 V, resulting in the generation of a radical intermediate (Scheme 1b). SET reactions occur mainly with substrates (such as aromatic amines and aromatic hydrocarbons) that are easy to oxidize and lack a weak C–H bond.31,40 HAT reactions occur in a polarity matched manner with substrates (such as alkanes, phenols, aliphatic alcohols, aliphatic amines, and halogenated hydrocarbons) with typical rate constants in the 107–108 M−1 s−1 range.31,41 Regardless of the pathway (SET or HAT), oxidation then leads to the formation of a reduced decatungstate ([W10O32]5−), either protonated or not, which has a characteristic deep blue color [W10O32]5− can then undergo back hydrogen donation or oxidation to regenerate the catalyst ([W10O32]4−). Alternatively, [W10O32]5− can undergo disproportionation, leading to the formation of a further reduced [W10O32]6− and an oxidized [W10O32]4−.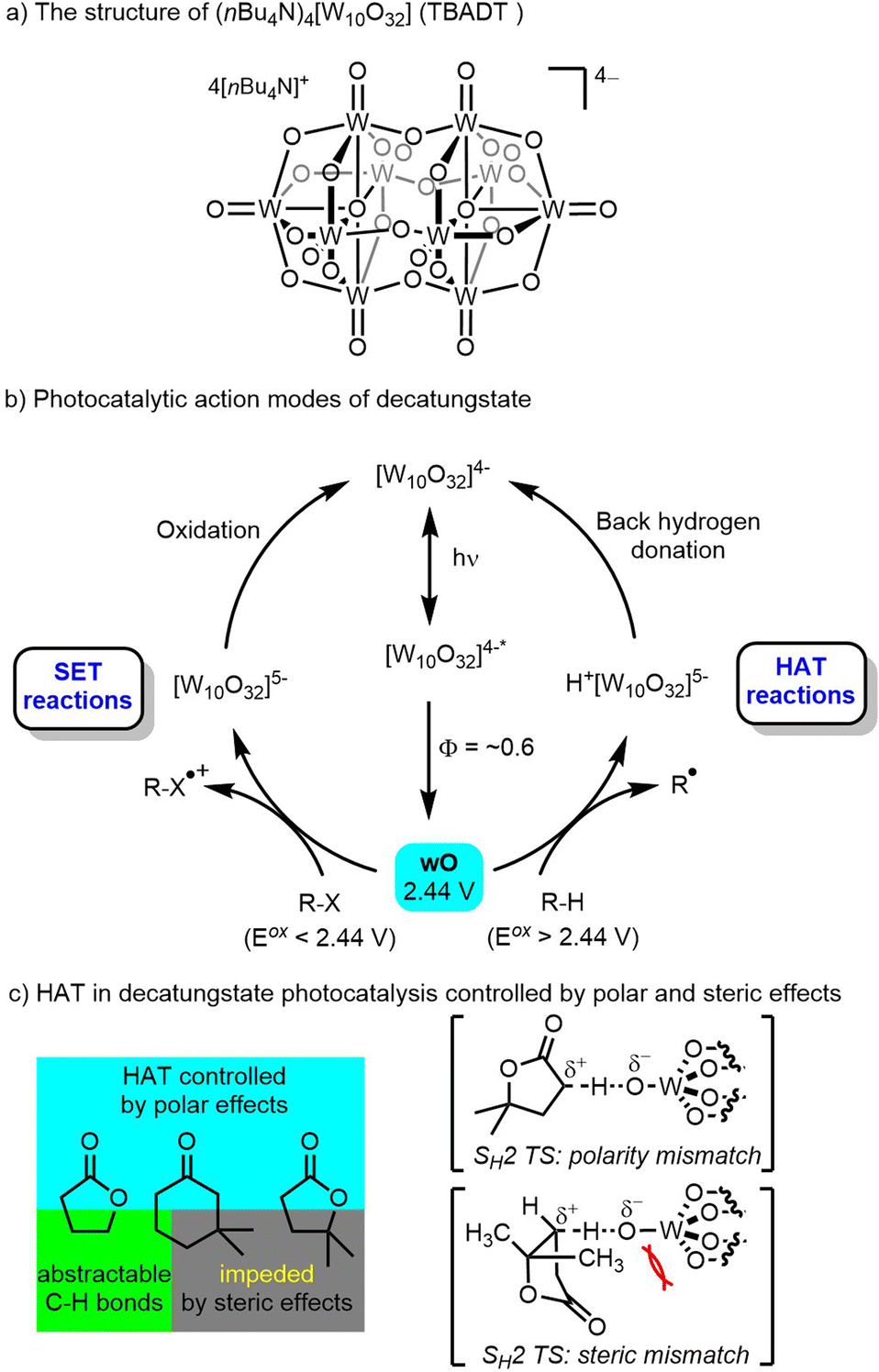 | ||
| Scheme 1 The structure of decatungstate, photocatalytic activation of decatungstate and HAT process controlled by polar and steric effects. (SH2: homolytic bimolecular substitution; TS: transition state). | ||
Decatungstate photocatalysis has found wide application in C–H bond transformation, such as alkane dehydrogenation,36,42 epimerization,43 oxidation28,44 and C–C,45 C–N,46 C–F,47 C–S,48 C–D49 bond formation. Moreover, it has been applied in the asymmetric synthesis by Melchiorre,50a Wang50b and Wang.50c MacMillan and co-workers have successfully developed efficient protocols for the direct C(sp3)–H transformations through a combination of decatungstate photocatalysis and transition-metal catalysis,51 while Noël and co-workers explored C(sp3)–H functionalization reactions using decatungstate photocatalysis in flow.19,45a,46b Notably, the HAT process in decatungstate catalysis typically shows good site-selectivity with synergistic control by both polar and steric effects due to the highly electrophilic oxygen center and steric bulk of the decatungstate anion (Scheme 1c). These effects were comprehensively described by Fagnoni, Ryu and co-workers based on extensive investigations.30
Based on our experiences with decatungstate-catalyzed C–H fluorination, here we will discuss the discovery of this unique reaction as well as its practical application in drug synthesis and radiofluorination through collaborations with scientists at Hoffmann–La Roche, Merck, TRIUMF (TRI University Meson Facility) and the BC (British Columbia) Cancer Agency. For more comprehensive summaries of decatungstate-catalyzed C–H transformations, one should refer to reviews by Hill, Fagnoni, Ryu and others.28–34 In addition, we will showcase advances in decatungstate-catalyzed C–H fluoroalkylation and the interesting role that electrostatic effects play in decatungstate chemistry. We hope this perspective will inspire other researchers to use decatungstate for photocatalytic C–H functionalization and further advance the exploitation of electrostatic effects for both rate acceleration and directing effects in these reactions.
2 Decatungstate catalyzed C–H 18F- and 19F-fluorination
2.1 19F-fluorination of small molecules
At an early stage during the emergence of radical C–H fluorination, we initiated a research program focused on decatungstate catalyzed fluorination. Of particular inspiration to our work, in 2012 Groves and co-workers disclosed that a manganese porphyrin complex can catalyze radical C–H fluorination using fluoride ion as the fluorine source and iodosylbenzene as an oxidant.52 Around this time, Sammis and Paquin reported the homolytic N–F bond dissociation energy (BDE) of common electrophilic fluorinating reagents, such as N-fluorobenzenesulfonimide (NFSI; BDENF = 63.1 kcal mol−1 in MeCN), Selectfluor (BDENF = 60.9 kcal mol−1 in MeCN) and N-fluoropyridinium salts (BDENF = 75.1 kcal mol−1 in MeCN).53 The relatively low N–F BDEs of NFSI and Selectfluor suggested their potential use as fluorine atom transfer reagents and these groups demonstrated that alkyl radicals, generated through decarboxylative processes, could undergo radical fluorine transfer with NFSI to afford the corresponding fluorinated products.53 This seminal study by Sammis and Paquin prompted a renaissance in radical fluorination using NFSI and Selectfluor.42–45Inspired by the above discoveries and the rich history of decatungstate catalyzed C–H transformations via alkyl radical formation,52–57 in collaboration with Rainer Martin at Hoffmann–La Roche, Shira Halperin and Hope Fan in our group at Simon Fraser University (SFU) developed the first decatungstate catalyzed direct fluorination of unactivated C–H bonds using NFSI as the fluorine atom source (Scheme 2).47a This process proved to be exceptionally mild (e.g. room temperature, wet solvents), and supported the synthesis of a variety of fluorinated compounds (e.g., 2a–2d), including fluorinated natural products, acyl fluorides, and fluorinated amino acid derivatives. Distinct from contemporary radical C–H fluorination protocols, including pioneering by Lectka,55 which employed Selectfluor as the fluorine atom transfer reagent,54,57 NFSI proved to be far superior in the decatungstate reaction. Additionally, when compared to many reports using decatungstate-catalyzed C–H functionalization that required C–H substrates to be used as solvents or in large excess,28,31 our method proceeded well with C–H substrates as the limiting reagent, an important caveat for medicinal chemistry purposes. Our proposed mechanism for this process is presented in Scheme 2 (bottom). Here, following excitation, the decatungstate anion (W10O324−) produces the short-lived excited state which rapidly decays to a long-lived reactive intermediate (wO). The electrophilic wO then abstracts a hydrogen atom from the substrate with preference for a more sterically accessible hydrogen that also generates a more stable carbon-centered radical (e.g.4A). Fluorine atom transfer from NFSI to 4A then delivers the desired product as well as the N-centered radical 4B, which abstracts a hydrogen from the H+[W10O32]5− to regenerate the decatungstate catalyst as well as NHSI. Notably, an alternative pathway involving a single-electron transfer from the radical species 4A to NFSI to generate a carbocation, followed by fluoride transfer53 is also feasible and was partially supported by the formation of compound 2a′. This work laid a solid foundation for efforts focused on C–H fluorination for both drug and radiopharmaceutical discovery purposes.
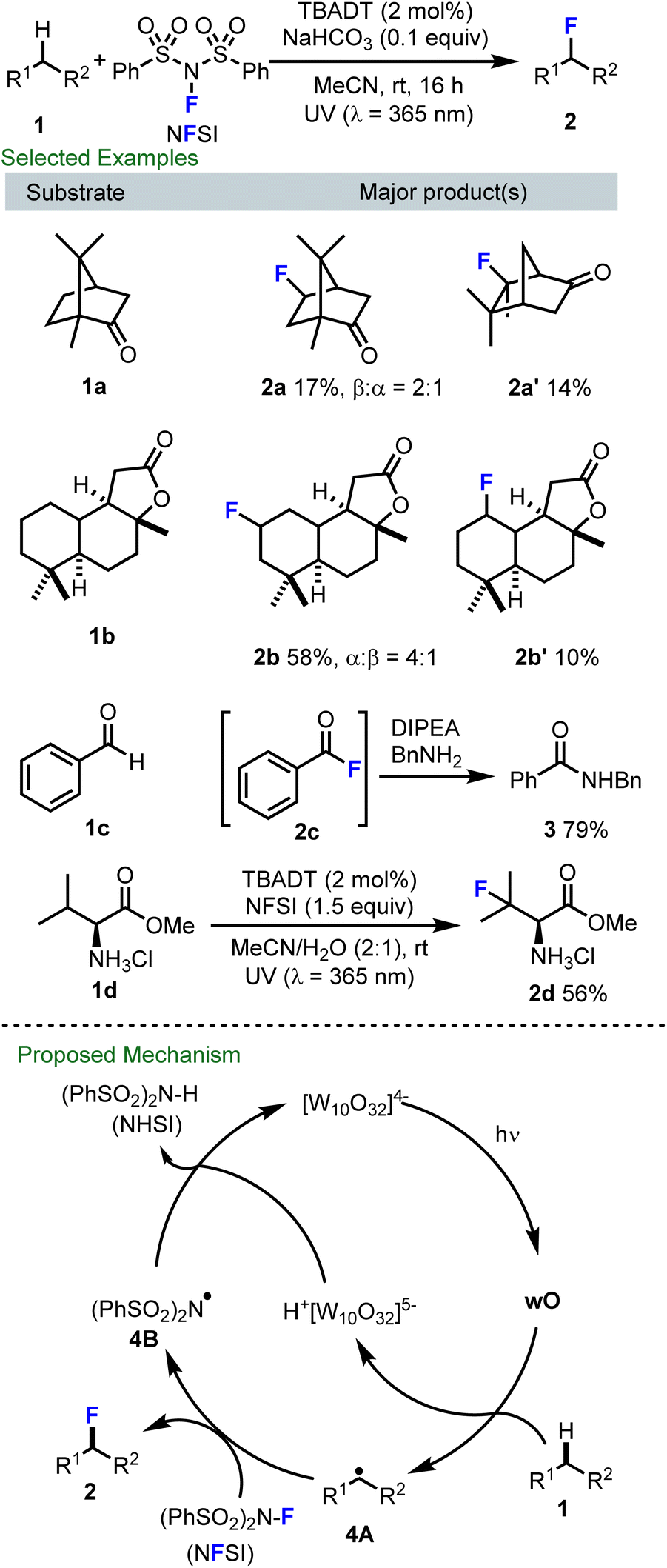 | ||
| Scheme 2 The initial work of decatungstate catalyzed radical fluorination of unactivated C–H bonds. | ||
In a further collaboration with scientists at Hoffmann–La Roche, Matthew Nodwell and Abhimanyu Bagai in our group extended the decatungstate process to include radical fluorination of benzylic C–H bonds and identified a complimentary strategy that relies on azobisisobutyronitrile (AIBN)-initiation (Scheme 3a).58 In benzylic C–F fluorination using NFSI, the addition of base (NaHCO3 or Li2CO3) is required to avoid the formation of Ritter-type acetamide byproducts. Here, the acidic NHSI acts as a Brønsted acid and catalyzes fluoride displacement by solvent (MeCN). This relatively mild process tolerates a wide range of functional groups and provides benzyl fluoride products in generally good yield (6a–6f). Notably, decatungstate-catalyzed fluorination showed better reactivity and generality than the equivalent reaction initiated by AIBN. In order to further showcase the utility of decatungstate-catalyzed benzylic fluorination, we demonstrated the fluorination of ibuprofen methyl ester (5f) in flow, which reduced the reaction time from 24 h (batch) to 5 h (bottom right, Scheme 3a) without impacting the yield. Evaluation of the pharmacokinetic properties of ibuprofen and fluorinated ibuprofen 6f indicated improved metabolic stability for the latter compound in both human and rat microsomes. For example, fluorination decreased the clearance from 19 to 12 mg min−1 mg−1 protein in human microsomes, and 71 to 39 mg min−1 mg−1 protein in rat microsomes, highlighting the utility of late-stage fluorination for medicinal chemistry purposes. Michael Meanwell, Johannes Lehmann from our Group and Marc Eichenberger and Rainer Martin at F. Hoffman–La Roche also explored decatungstate-catalyzed C(sp2)–H fluorination of aromatic and aliphatic aldehydes 7 to generate the acyl fluorides 8via acyl radical intermediates (Scheme 3b).59 Due to the instability of resulting acyl fluorides, these products were generally treated with benzylamine and N,N-diisopropylethylamine (DIPEA) in situ to obtain the corresponding amides 9. Furthermore, benzylic and aliphatic alcohols 10 could be converted directly into N-benzyl amides 9via decatungstate catalysis using an excess of NFSI (2.5 equiv.) in a one-pot fashion (Scheme 3b, bottom).
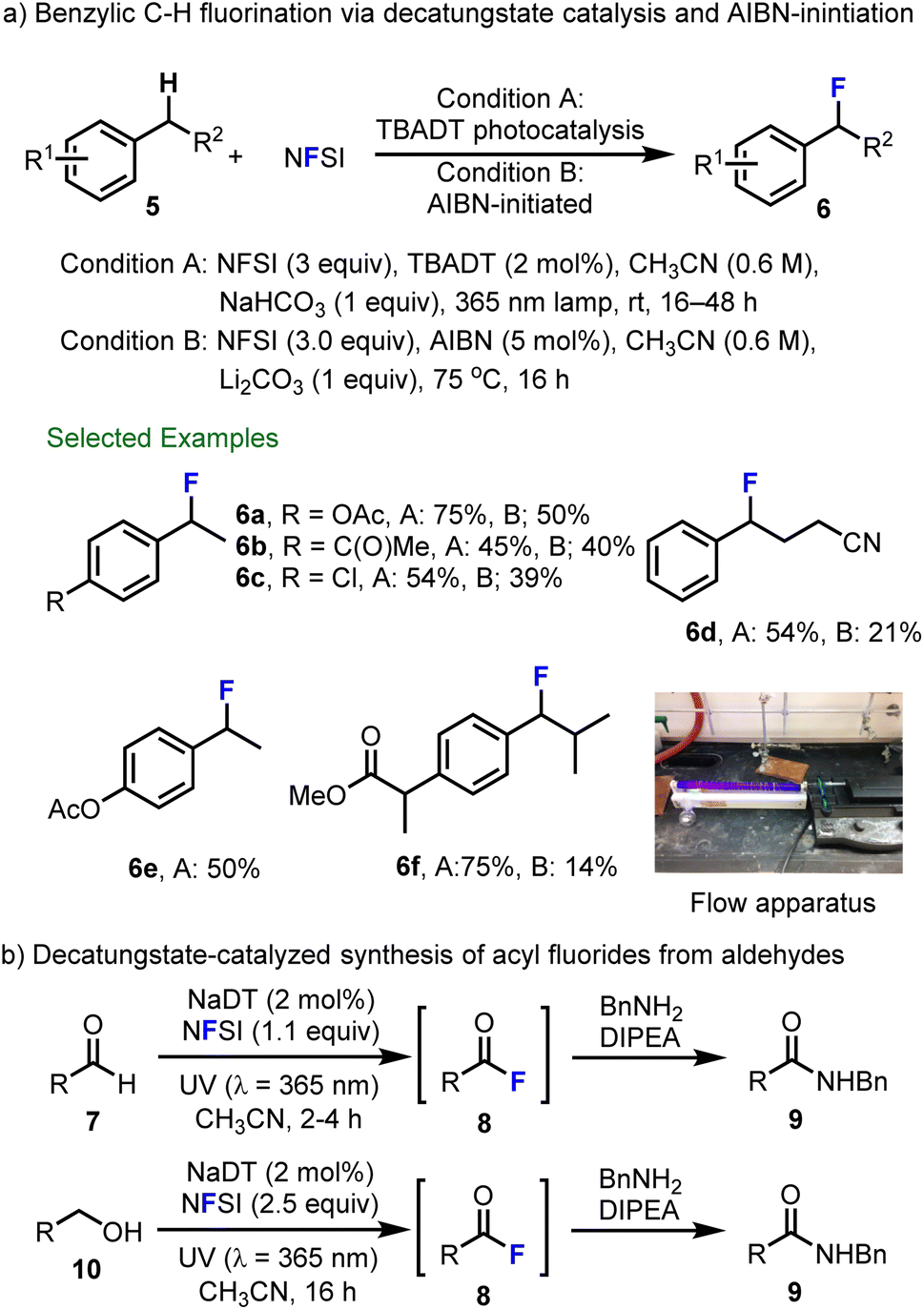 | ||
| Scheme 3 (a) Decatungstate-catalyzed and AIBN-initiated fluorination of benzylic C–H bonds. (b) Decatungstate-catalyzed synthesis of acyl fluorides from aldehydes. Image reproduced with permission from ref. 58, copyright 2015 the Royal Society of Chemistry. | ||
Considering the importance of tuning both the metabolism and pKa of heteroaromatics,60 in collaboration with scientists at Hoffmann–La Roche, we aimed to expand decatungstate catalyzed benzylic fluorination47,58 to include pyridylic fluorination, which at the time had been less studied. After considerable effort, Michael Meanwell in our group at SFU found that the decatungstate catalyst was not necessary, and catalyst free late-stage C–H monofluorination of alkylpyridines with NFSI alone was developed (Scheme 4a).61 This process showed good compatibility with 2- and 4-alkyl pyridines, and annulated pyridines, and provides pyridylic fluoride products in good to excellent yields (12a–12f). Notably, 3-alkyl-pyridines were not competent substrates for this reaction (e.g., 12g). This transformation was also applied to the late-stage fluorination of aldosterone synthase (CYP11B2) inhibitors62 (12f), demonstrating the utility of this method for LSF. Importantly, the decatungstate-free process provides complimentary selectivity to our photocatalytic decatungstate C–H fluorination reaction.47,58 For instance, compound 11h underwent selective fluorination at the pyridylic (a) or benzylic (b) position to provide 12h or 12h′ using the appropriate method. Mechanistically, a non-radical process was proposed for pyridylic fluorination and was supported by the absence of radical rearrangement products using 11c. As highlighted in Scheme 4a, sulfonylation of pyridine by NFSI leads to an N-sulfonylpyridinium salt 13A and intermediate 13B. Intermediate 13B can decompose to release fluoride and nitrene 13D. As a result of the increased acidity of the pyridylic protons in intermediate 13A, deprotonation by mild base then delivers the alkylidene dihydropyridine 13C, which subsequently reacts with the electrophilic fluorinating reagent NFSI to provide the fluorinated product 12. By increasing the equivalents of NFSI, it was found that heterobenzylic difluorination of quinoline (15a–15b), pyridine (15c), quinazolie (15d) and purine (15e) could be realized in acceptable yields (Scheme 4b).63
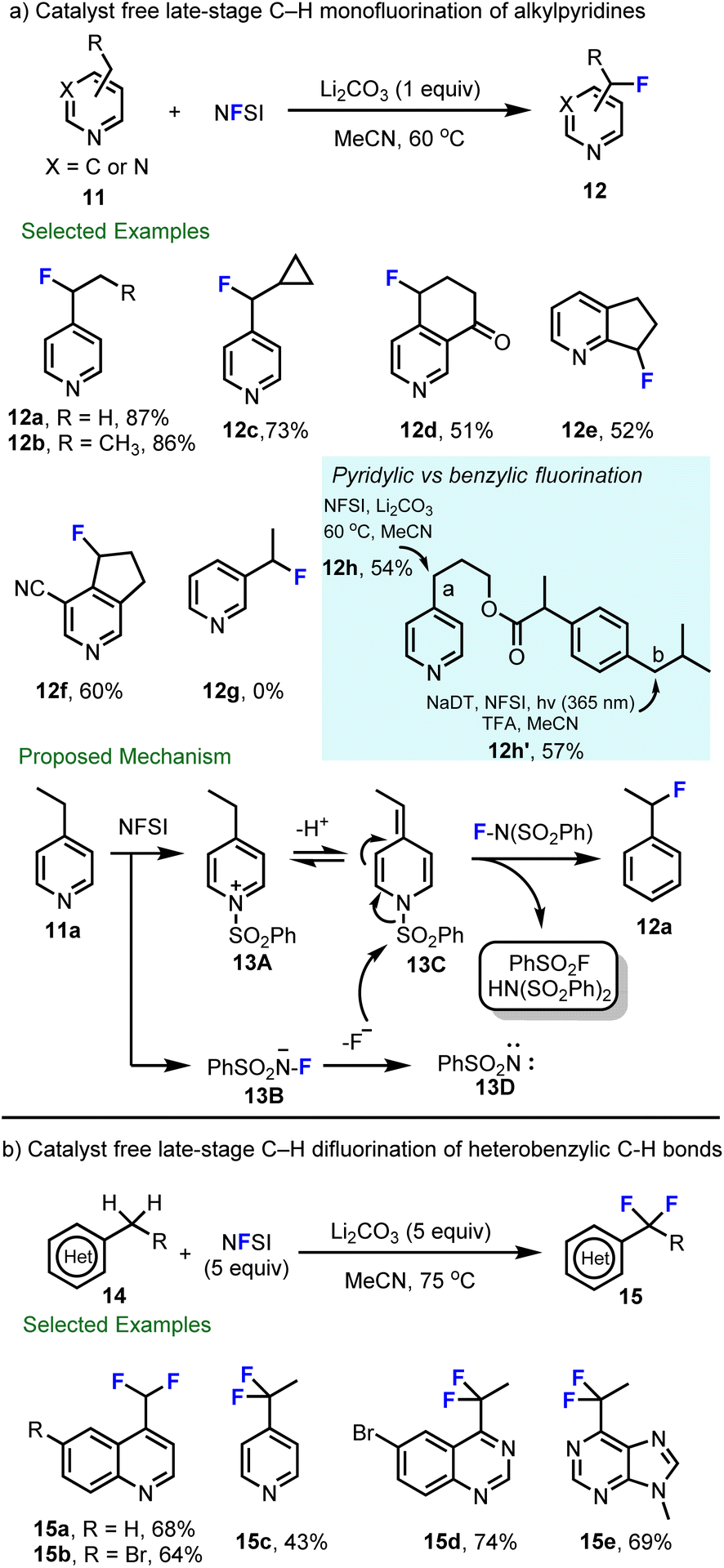 | ||
| Scheme 4 Catalyst free late-stage C–H monofluorination and difluorination of heterobenzylic C–H bonds. | ||
Armed with this suite of tools for mono and difluorination via radical and polar pathways, we next sought to demonstrate the practical applications of these chemistries. In the essay entitled “Late-Stage Fluorination: Fancy Novelty or Useful Tool?” published in 2015, it is stated that “recent advances in late-stage fluorination have not yet had a substantial impact on the synthesis of bulk chemicals, 18F positron emission tomography (PET) tracers, or materials”.64 Based on our experiences described above, and opportunistic collaborations with medicinal and process research chemists at Hoffmann–La Roche and Merck, and radiopharmaceutical chemists and PET imaging experts at both TRIUMF and the BC Cancer Agency, we felt that we were uniquely positioned to address these limitations.
2.2 Scalable 19F-fluorination for drug development
First, in collaboration with Erik Regalado, Daniel DiRocco and Louis-Charles Campeau from the Process Chemistry group at Merck we developed a concise and scalable synthesis of (2S)-γ-fluoroleucine methyl ester (17a) using our decatungstate-catalyzed C–H fluorination process (Scheme 5).47b (2S)-γ-fluoroleucine ethyl ester (17b) was a key intermediate required for the synthesis of the clinical candidate odanacatib. At the time, odanacatib was undergoing a phase III clinical trial for the treatment of osteoporosis.65 Although scientists at Merck had developed five unique and scalable synthetic routes (from starting materials 18–21) to this important intermediate (Scheme 5, bottom), the length of these syntheses and requirement for a stereocontrolled introduction and preservation of the α-chiral amine and tertiary alkyl fluoride functions detracted from their utility.66 After additional optimization carried out at Merck using high-throughput experimentation (HTE), it was found that sodium decatungstate (NaDT) showed better reactivity than tetrabutylammonium decatungstate (TBADT) in this process. Further, the counterion, MeCN–H2O ratio and temperature were optimized to ultimately enable production of 44.7 g (90% yield) of (2S)-γ-fluoroleucine methyl ester bisulfate (17a) in a one-step flow photocatalytic synthesis. Here a custom built 365 nm flow photoreactor (Scheme 5, middle right) was utilized and showcased the practical utility of this LSF for process research purposes.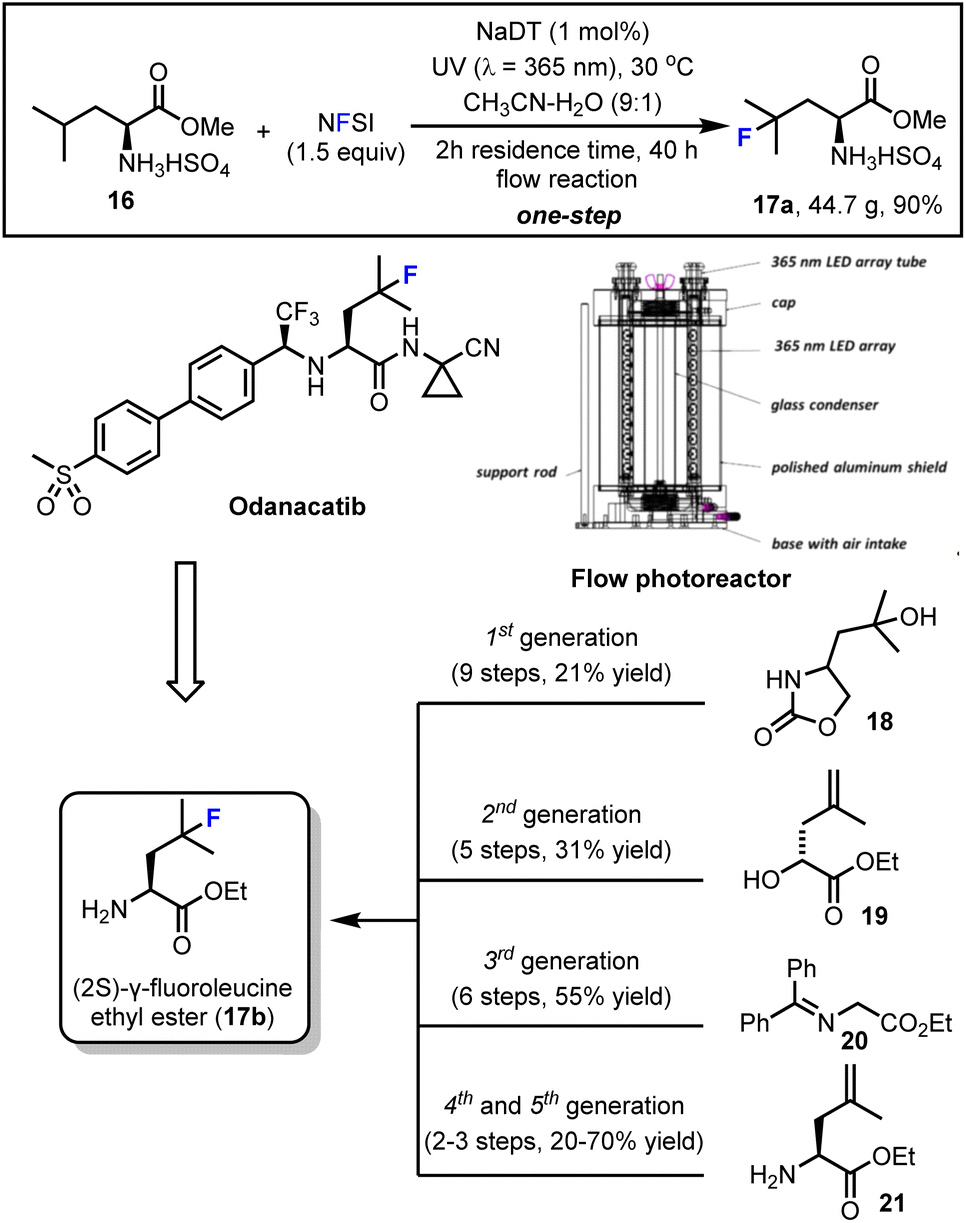 | ||
| Scheme 5 Decatungstate-catalyzed scalable synthesis of an odanacatib precursor in flow. Image reproduced with permission from ref. 47b, copyright 2015 American Chemical Society. | ||
2.3 Rapid 18F-fluorination for radiotracer synthesis
A major hallmark of tumor cells is sustained proliferation, which requires increased nutrients for rapid growth and division.67 Apart from glucose and the well-studied Warburg effect,68 amino acids are also essential for rapid and sustained proliferation,69 leading to overexpression of L-type AA transporters (LATs) in many cancers.70 Specifically, LAT1 mediates the transport of large, neutral, branched amino acids (e.g., leucine, isoleucine, phenylalanine).70 Thus, LAT1 is a promising target for cancer imaging via positron emission tomography (PET).71–73 Several amino acid radiotracers have been developed that exploit LAT transport and accumulate in cancer cells, enabling differentiation from healthy cells and tissues. For example, 6-[18F]-fluoro-3,4-dihydroxy-L-phenylalanine ([18F]-FDOPA) has been used in the clinical management of neuroendocrine tumors.74 Additionally, trans-1-amino-3-[18F]-fluoro cyclobutane carboxylic-acid (anti-[18F]-FACBC), a leucine analogue, has been approved for imaging of recurrent prostate cancer (PCa)75 and is a promising PET imaging agent for multiple myeloma.76 Building on our previous results and aware of the importance of leucine in cancer biology, we sought to develop an 18F-labelled leucine radiotracer. Below are some of the key considerations for the development of new PET radiochemical reactions:14,15,77• Reaction compatibility with micro or nanoscale.
• Short reaction and purification times consistent with the half life of PET radionuclides. As general rule, the duration between the end of bombardment (EOB) and the injection of the tracer should be shorter than three isotope half-lives.15,77
• High radiochemical yield (RCY).
• Depending on application, a high level of molar activity (>1 Ci μmol−1 or >37 GBq μmol−1) may be required.
• Execution of radiochemistry should be straightforward and readily translatable to other PET centers for use by non-experts.
Towards these goals, Matthew Nodwell spearheaded our efforts to prepare [18F]-4-fluoroleucine in collaboration with Paul Schaffer and Hua Yang at TRIUMF and Milena Čolović, Helen Merkens and François Bénard at the BC Cancer Agency and Rainer Martin at F. Hoffman La Roche (Scheme 6).78 First, we took advantage of a process reported by Gouverneur and Luthra for the generation of [18F]NFSI.79 Here, with some optimization of the reported purification of [18F]NFSI, a solution of [18F]NFSI in H2O/CH3CN could be obtained using C18 SepPak purification step in 5 min. Further, we modified the decatungstate process to meet the requirements for 18F-labelling. Specifically, after evaluating several reaction configurations, we found that a PTFE tube reactor was optimal and consisted of a narrow-bore PTFE tube wrapped a BLB (blacklight blue) lamp (Scheme 6, upper right). Using this apparatus, direct 18F-fluorination of the unprotected amino acid leucine could be realized with ∼60% conversion to 4-fluoroleucine after only 40 min. Considering that leucine is an endogenous amino acid, separation from 4-fluoroleucine ([18F]23) was not necessary as residual leucine 22 would not interfere with the performance of the tracer.80 Thus a straightforward purification procedure was developed for the [18F]-4-fluoroleucine/leucine mixture that simply required filtering through a strong cation exchange resin (no HPLC) and provided material suitable for direct injection and imaging. Further, owing to the requirement for water in the fluorination step, our modified production of [18F]NFSI avoided the typical azeotropic distillation steps required for 18F-labelling. As such, the entire process required only 50 min and proceeded with a radiochemical yield (RCY) for isolated 4-[18F]fluoroleucine (4-[18F]FL, 23) of 23 ± 3.3% (n = 4, decay corrected) and a molar activity of 7.1 ± 1.9 MBq μmol−1. The uptake of 4-[18F]FL (23) in several cancer cell lines was then investigated and correlated well with the expression levels of LAT1.81 However, a biodistribution study of 4-[18F]FL (23) showed high bone uptake, suggestive of in vivo defluorination, and halted further development. Interestingly, we had also observed the instability of 19F-23 during chromatographic purification and found that this amino acid tends to lactonize via fluoride displacement. In an effort to decrease the rate of lactonization, we prepared the homologue 5-[18F]fluorohomoleucine (5-[18F]FHL, 24), which proved to be stable in vivo and showed very little bone uptake (∼2% vs. ∼12% for 23). Further, in imaging studies, 5-[18F]FHL showed high tumor accumulation in mice bearing human glioma (U87) and prostate cancer (PC3) xenografts, which provided excellent tumor visualization (Scheme 6b). It is notable that although this method uses low molar activity (MA) [18F]F2 gas (∼1 GBq μmol−1) resulting in MAs of labelled amino acids of ∼7 MBq μmol−1, clinically relevant doses of several amino acid radiotracers have been manufactured from [18F]F2.82
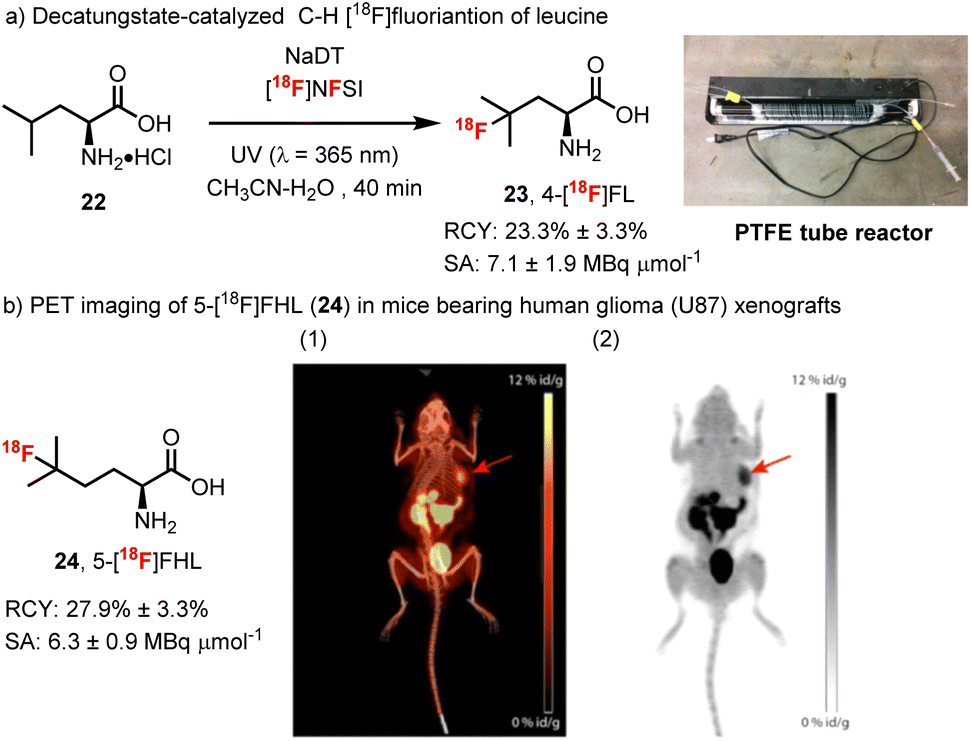 | ||
| Scheme 6 (a) Decatungstate-catalyzed C–H 18F-fluorination of leucine. (b) PET imaging of 5-[18F]FHL in mice bearing human glioma (U87) xenografts. Maximum intensity projection images overlaid on CT (a) and standalone PET images (b) of the biodistribution of 5-[18F]FHL (24) at 60 min show high accumulation of 5-[18F]FHL (24) in the tumor (red arrow). Images reproduced with permission from ref. 78, copyright 2017 American Chemical Society. | ||
Considering the potential application of our method for high throughput production of radiotracers, our team then prepared a series of 11 fluorinated branched-chain amino acids using a modified apparatus, which involved a microreactor glass chip placed on a transilluminator (365 nm) and examined the affinity of each new fluorinated amino acid for LAT (Scheme 7).83 From this study, (S)-5-[18F]fluorohomoleucine ((S)-5-[18F]FHL, 24) proved to be ideal, showing rapid tumor uptake and optimal tumor visualization, which plateaued at around 10 min and remained stable for at least 30 min in vivo. The similar tumor and brain uptake of (S)-5-[18F]FHL (24) (Scheme 7b) and the clinical LAT radiotracer O-(2-[18F]-flurorethyl-L-tyrosine) ([18F]-FET)84 suggested that (S)-5-[18F]FHL (24) has the potential for further development as an oncologic PET imaging agent.
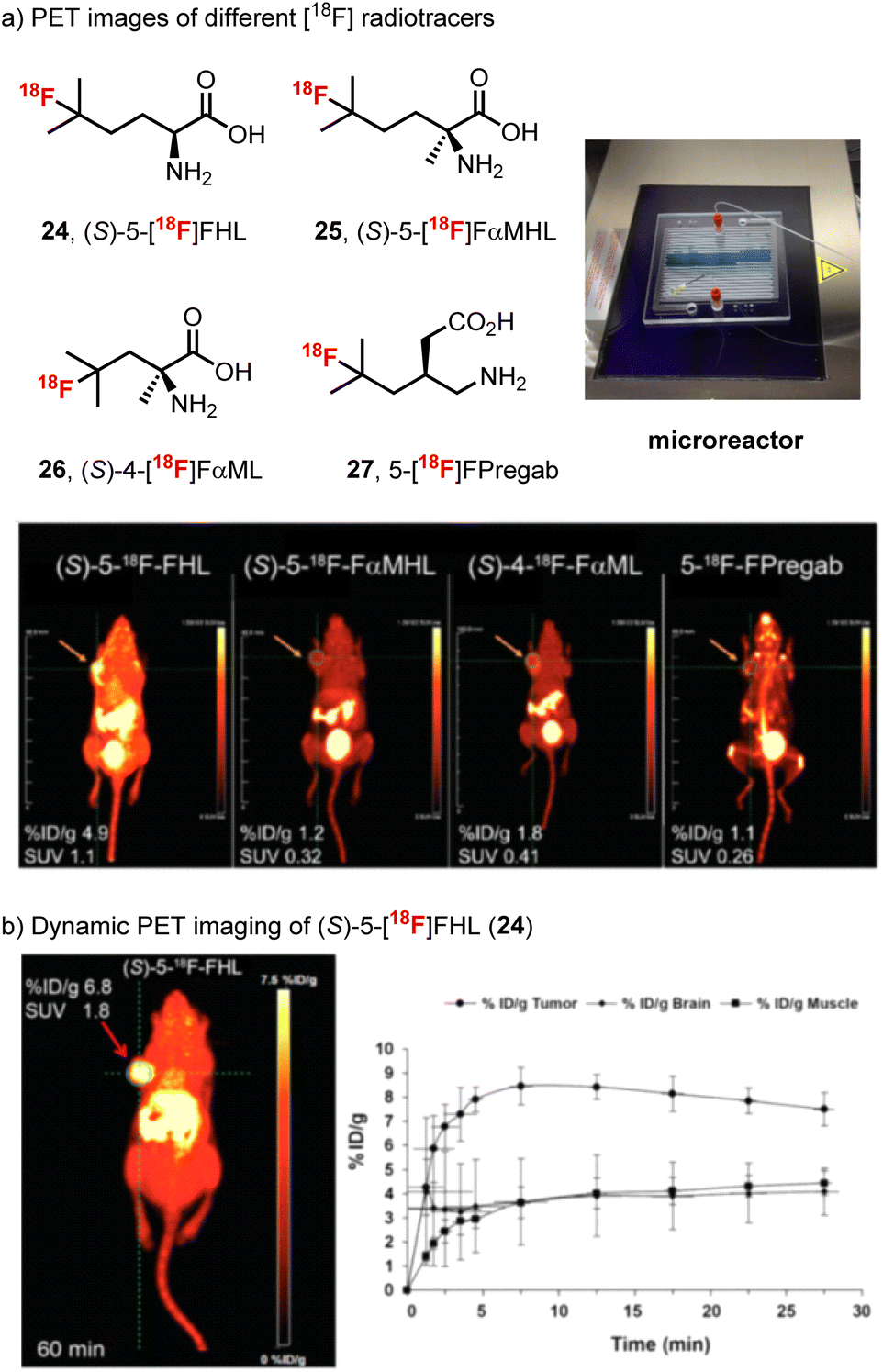 | ||
| Scheme 7 (a) PET images at 10 min for U-87 xenograft-bearing mice with (S)-5-[18F]-FHL (24), (S)-5-[18F]-FαMHL (25), (S)-4-[18F]-FαML (26), and 5-[18F]-FPregab (27). The orange arrow indicates the site of tumor. (b) In vivo dynamic PET imaging of (S)-5-[18F]-FHL in NSG mouse bearing U-87 xenograft tumor at 60 min after injection and time–activity curve of (S)-5-[18F]-FHL (24) in tumor, brain, and muscle over 30 min after injection, n = 3. Images reproduced with permission from ref. 83, copyright 2019 the Society of Nuclear Medicine and Molecular Imaging. | ||
2.4 Selective 18F-fluorination of unprotected peptides
The recognition that branched aliphatic amino acids such as leucine and homoleucine undergo rapid and clean fluorination using decatungstate catalysis inspired us to explore the selective fluorination of leucine residues in unprotected peptides. Through further collaboration with scientists at Hoffmann–La Roche, TRIUMF and the BC Cancer Agency, we examined the decatungstate catalyzed C–H fluorination of leucine-containing peptides (Scheme 8).85 Initial studies on all 20 leucine-containing natural dipeptides indicated that this mild fluorination process was compatible with most natural amino acids. The exceptions included the relatively rare and easily oxidized amino acids methionine, tryptophan and cysteine. Moreover, in a series of leucine-containing tetrapeptides, and bioactive peptides (FALGEA-NH2 (ref. 86) and ZJ-43 (ref. 87)), selective fluorination at the branched position of leucine could be achieved. By employing larger surface area reactors and higher intensity light sources PTFE tube reactor (Scheme 6) or a microreactor (Scheme 7) to improve the reaction efficiency, rapid 18F-fluorination of peptides could be realized (28–31). This protocol has certain advantages over methods involving 18F-fluorination of prosthetic groups attached to a peptide,88 where additional radiochemistry steps are often required and the prosthetic group can alter the physical properties of the bioactive peptide. Notably, this work culminated in the direct 18F-labelling of ZJ-43, a potent ligand for the prostate-specific membrane antigen (PSMA).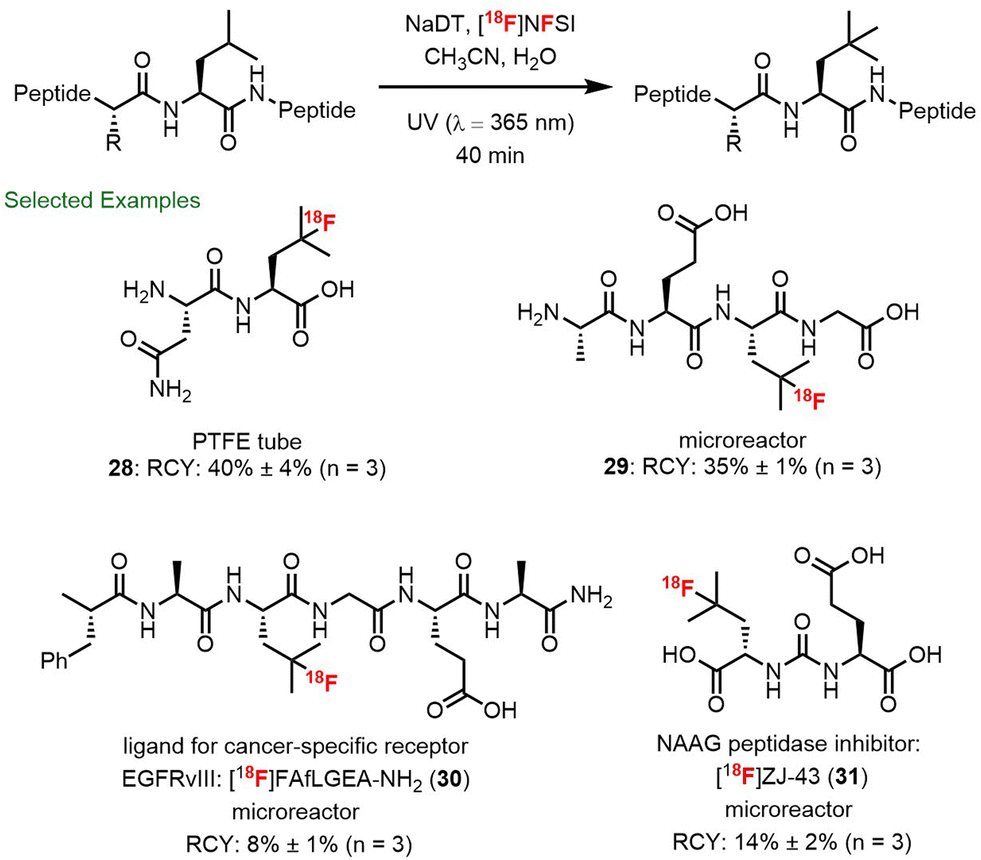 | ||
| Scheme 8 Direct site-selective 18F-fluorination of unprotected peptides. | ||
During these studies, we observed and defined an important electrostatic effect in decatungstate catalysis (see Section 4.1 for additional details), where cationic ammonium groups in substrates promote the formation of a precursor complex with decatungstate anion (DT−) that ultimately increases the rate of C–H abstraction (Scheme 9).89 This rate accelerating electrostatic effect proved to be generally useful for the C–H fluorination of small molecules (i.e. amino acids, heterocycles and pseudopeptides). Most importantly, we were able to exploit this effect and accelerate the fluorination of ZJ-43 and Glu-U-HLeu using cationic derivatives (32–34) and identified Glu-U-FHLeu (35) as a potent PSMA binding ligand. Exploiting these findings we showed that the PSMA-binding ligand [18F]Glu-U-FHLeu (37) could be readily prepared by decatungstate catalyzed radiofluorination and hydrolysis.
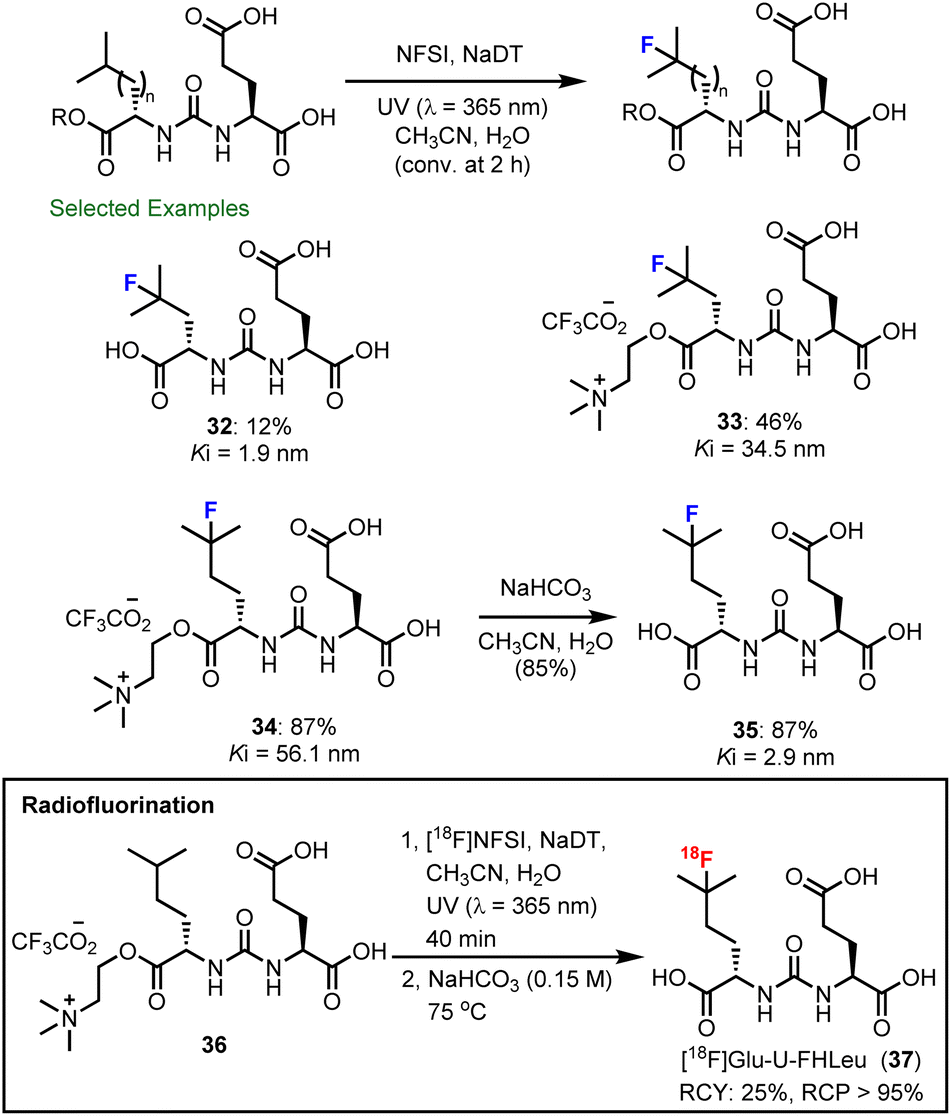 | ||
| Scheme 9 Accelerated fluorination of choline esters of ZJ-43 and Glu-U-HLeu. PSMA binding of each ligand (Ki) was measured by competition assays (n ≥ 3) using [18F]DCFPyL and determined by applying a one-site Fit Ki model using GraphPad Prism (7.04). | ||
3 Decatungstate catalyzed C–H fluoroalkylation
Besides C–H fluorination, decatungstate-catalyzed C–H functionalization has additionally been applied to trifluoromethylation, difluoromethylthiolation and trifluoromethylthiolation. Further, in 2020, MacMillan and co-workers described the first merger of decatungstate photocatalysis with copper catalysis and realized the direct trifluoromethylation of both strong aliphatic (39a–39f) and benzylic (39g–39h) C–H bonds using Togni's II reagent as the trifluoromethylating agent (Scheme 10).90 This procedure enables the trifluoromethylation of pharmaceuticals and natural products including sclareolide, celecoxib and others (39i–39j). In some cases, the addition of sodium chloride was required to minimize oxidation by the copper intermediate through chloride coordination. As established prior,44b acidic conditions were required to avoid the reactivity of unprotected amines and deactivate C–H bonds adjacent to the nitrogen atom, which in turn enhances site-selectivity by a polarity matched effect during HAT process (39a–39d). Based on mechanistic studies, it was proposed that electrophilic excited decatungstate anion site-selectively abstracts an electron-rich and sterically accessible C–H bond to generate the alkyl radical 40A. The alkyl radical 40A is then trapped by a CuII–CF3 species to afford the Cu(III) intermediate 40B, which undergoes reductive elimination to afford the desired trifluoromethylated products 39 and regenerate the Cu(I) catalyst 40C. Formal reduction of an electrophilic CF3 source by the reduced decatungstate (H+[W10O32]5–) and Cu(I) catalyst 40C would then afford a CuII–CF3 intermediate 40D and regenerate the decatungstate anion ([W10O32]4−)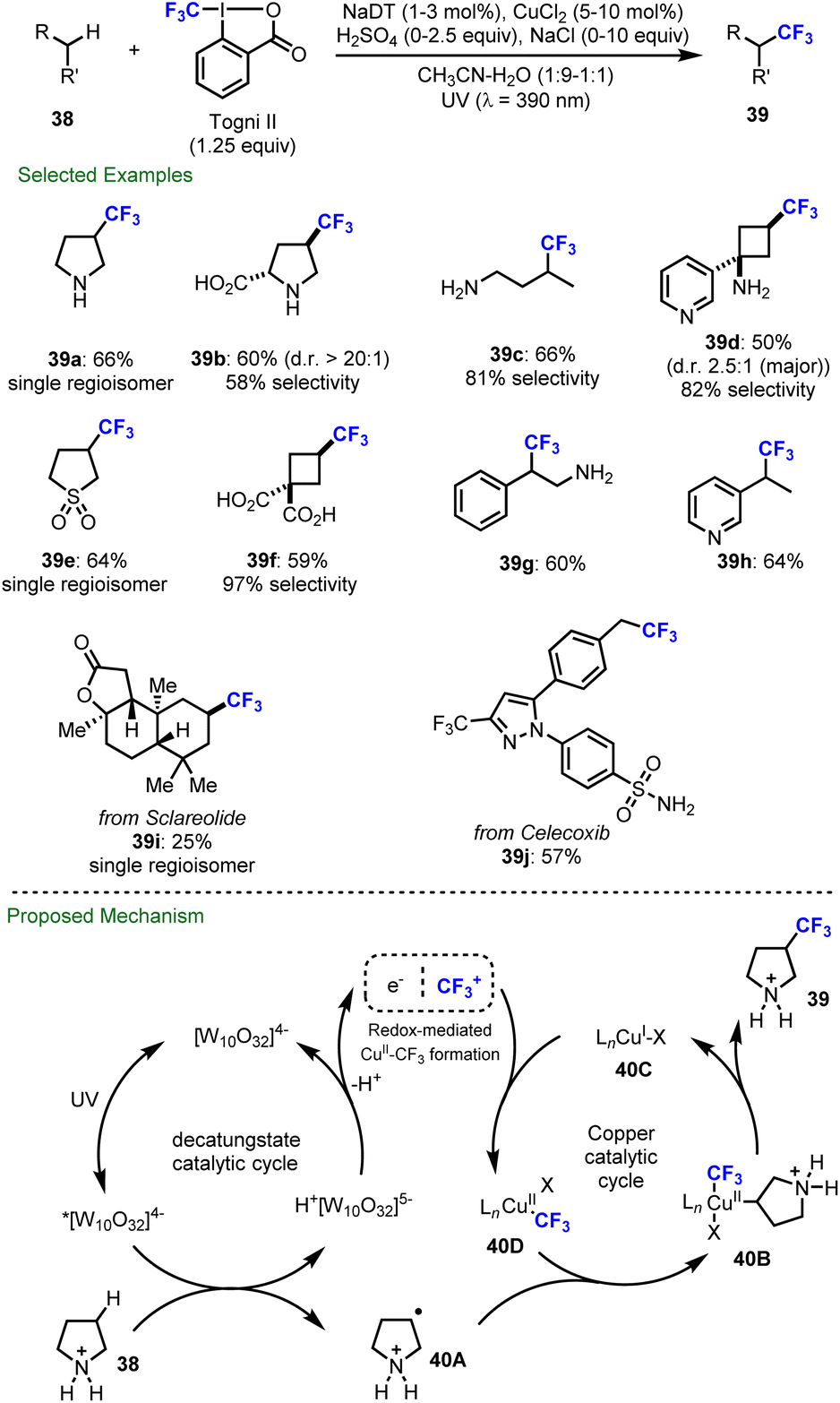 | ||
| Scheme 10 Direct C(sp3)–H trifluoromethylation via decatungstate and copper catalysis. | ||
Apart from fluorination and trifluoromethylation, decatungstate chemistry has also been applied to C–H fluoroalkylthiolation, including difluoromethylthiolation and trifluoromethylthiolation. As for the mechanism, similar to fluorination (Scheme 2, bottom), the reaction initiates with the HAT between excited decatungstate and C–H substrates to generate the radical intermediate (alkyl or acyl radical), which subsequently undergoes fluoroalkylthio group transfer to afford the final products (Scheme 11a). In 2020, Wang and co-workers developed a decatungstate catalyzed C–H difluoromethylthiolation of aldehydes using PhSO2SCF2H (Scheme 11b(1)).48a More recently, they have reported the decatungstate catalyzed C–H trifluoromethylthiolation of aldehydes with PhthSCF3 (Scheme 11b(2)).48b The difluoromethylthiolation and trifluoromethylthiolation use aldehydes as the limiting reagents, and require a high loading of fluoroalkylthiolation reagents. Around the same time, König and co-workers reported a decatungstate catalyzed trifluoromethylthiolation of alkyl C(sp3)–H bonds and formyl C(sp2)–H bonds with PhthSCF3 (Scheme 11b(3)),91 while we also reported a trifluoromethylthiolation of aldehydes using NaDT as the catalyst and trifluoromethylthiosaccharin as the CF3S source (Scheme 11b(4)).92 In contrast to Wang's work, we found that the reaction was more efficient with trifluoromethylthiosaccharin used as the limiting reagent, though the separation of products from the unreacted starting materials proved problematic in certain cases. Together, this suite of redox-neutral decatungstate catalyzed C–H fluoroalkylthiolation protocols provide new opportunities for LSF in drug discovery.
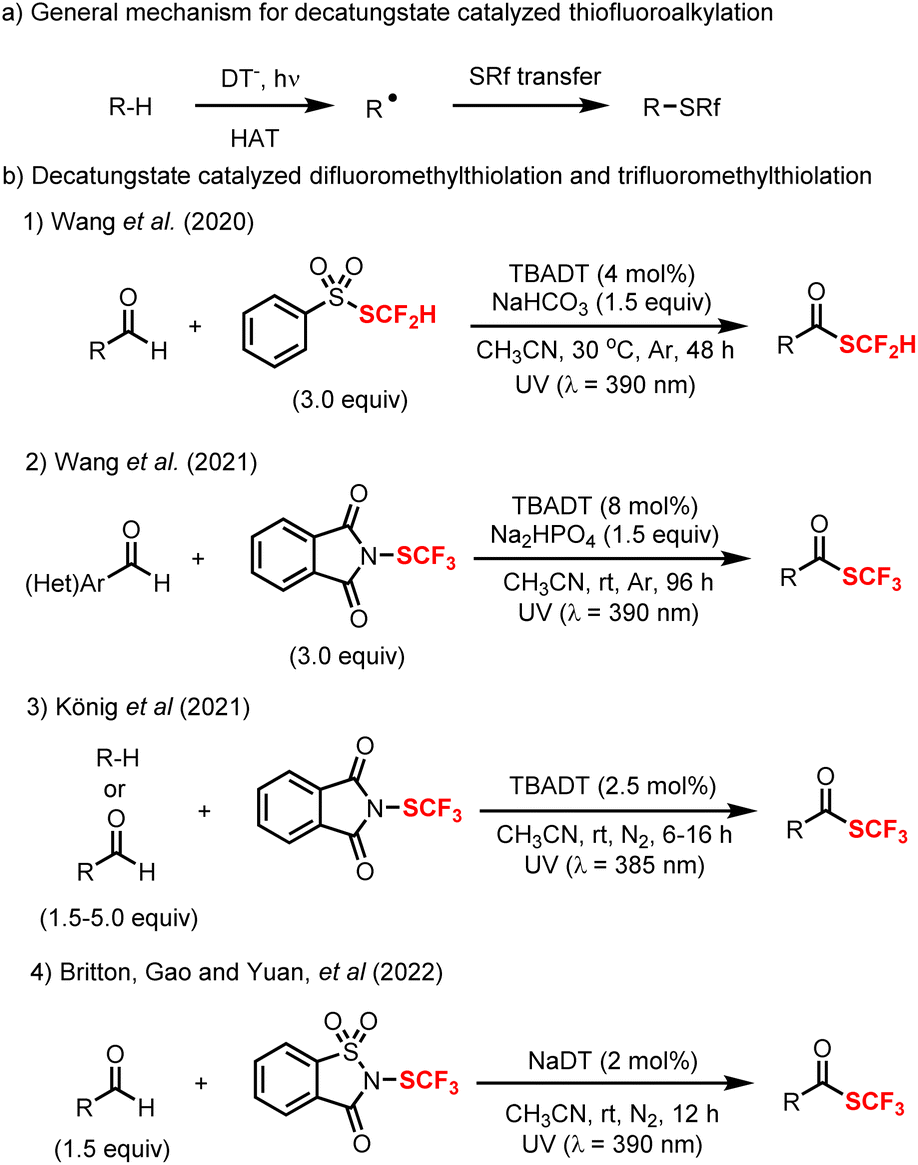 | ||
| Scheme 11 Decatungstate catalyzed C–H fluoroalkylthiolation. | ||
4 The electrostatic effects in decatungstate catalysis
The electrostatic effect, also referred to as an ion-pairing interaction, is an attraction between two groups of opposite charge and is a fundamentally important noncovalent interaction.93–96 Although this interaction is not as widely exploited as hydrogen bonding in organic catalysis, recent developments have shown that electrostatic effects offer unique opportunities to control site-selectivity and even enantioselectivity.97,98 In this part, two types of electrostatic effects in decatungstate-catalyzed C–H functionalization will be discussed: (i) a rate accelerating effect in fluorination, and (ii) site-selectivity in the HAT step.4.1 Rate enhancement via electrostatic effects
Generally, the selectivity of decatungstate catalyzed C–H functionalization is governed by C–H bond strength, polar and steric effects that have been described by Fagnoni and Ryu.30 During our own studies on decatungstate catalyzed C–H fluorination,47,85 similar trends were observed. Here, weak, sterically accessible tertiary C–H bonds were generally selected for HAT. This is particularly evident in our work on peptide fluorination, where the branched position of a leucine hexapeptide could be selected for over ∼20 other distinct C–H bonds.85 However, during these studies several confusing phenomena were observed. For example, the rate and yield for leucine fluorination varied significantly based on the position of a leucine residue in a peptide85 and the rate for C–H fluorination of branched aliphatic amino acids was far greater than that for benzylic fluorination despite lower BDE for benzylic C–H bonds.58 These and other observations led us to probe the impact simple substituents have on the rate of leucine fluorination (e.g., ester, amide, sulfonamide) and ultimately identify a rate enhancement that originates from electrostatic interactions between the cationic ammonium group and the decatungstate anion (Scheme 12).89 For example, unprotected but protonated leucine works well in this reaction and there was only a modest impact on reaction outcome when the counter ion was changed (Scheme 12a, entries 1 and 2). However, the fluorination yield dropped significantly when the amine group was acylated (entries 3 and 4) or sulfonylated (not shown). Here, using a bulky t-butyl amide, the reaction was further impeded in accordance with known steric effects (entry 4). In contrast, the reaction outcome was little changed when a methyl ester of leucine was employed (entry 5). Again, N-acylation of leucine methyl ester (entry 6) led to a decrease in yield. Interestingly, the yield was not significantly impacted when an N-acyl group containing a cationic ammonium function was incorporated (entry 7). These results demonstrate the importance of the ammonium function for rapid fluorination. Furthermore, Jeffrey Warren, a colleague at SFU, and David Weber examined these reactions using transient absorption spectroscopy to determine the individual rate constants for HAT by excited decatungstate. Here, HAT rates paralleled the reaction yields, with leucine derivatives containing cationic ammonium groups displaying larger HAT rate constants (entries 1, 5, and 7). These results also support our contention that HAT is the rate-determining step in the fluorination process.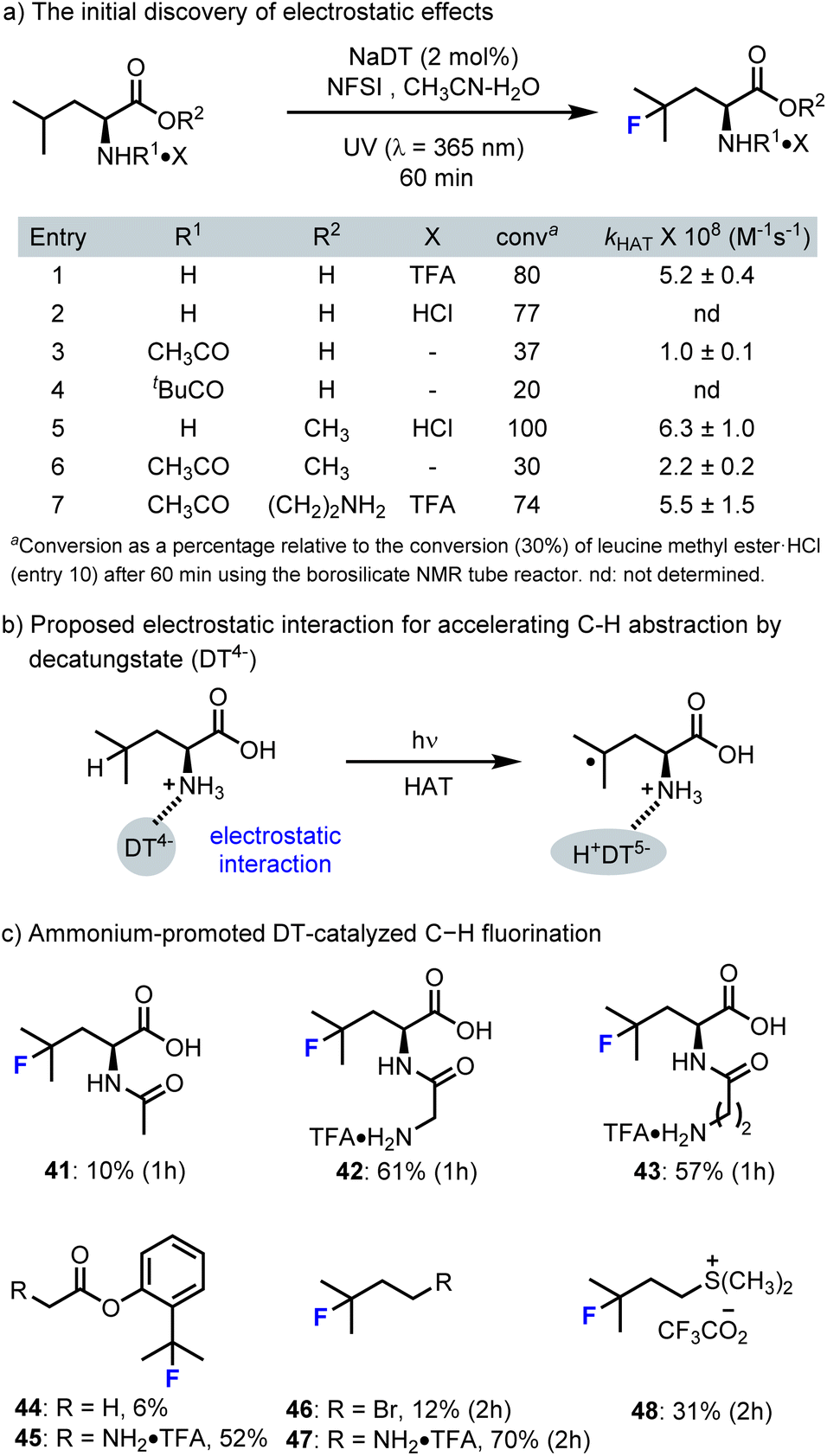 | ||
| Scheme 12 The cationic ammonium groups accelerate DT-catalyzed C–H fluorination by electrostatic effects. | ||
To further validate the rate-accelerating electrostatic effects (Scheme 12c), we examined the fluorination of several N-acyl leucine derivatives with attached ammonium functionalities. Here, we were pleased to find that the reaction rate increased significantly with the re-introduction of cationic ammonium groups (41–43). This rate accelerating electrostatic effect also extended to other aliphatic C–H fluorination reactions, such as N-glycyl·TFA derivatives of 2-aminobutane (44vs.45, 46vs.47). Further demonstration of rate acceleration for C–H fluorination by other cationic groups, such as dimethylsulfonium (46vs.48), highlights the potential generality of this approach to accelerating dectungstate catalysis. Exploiting these findings, we also demonstrated the rapid production of [18F]Glu-U-FHLeu (37), an 18F-labelled prostate specific membrane antigen (PSMA)-binding ligand (Scheme 9). Notably, our own attempts to realize site-selective C–H fluorination by strategic incorporation of ammonium functionalities were largely unsuccessful.
4.2 Controlling the site-selectivity via electrostatic effects
In 2017, Schultz and co-workers from Merck reported a decatungstate catalyzed C–H oxyfunctionalization of remote C–H bonds in cyclic amines to provide a series of important oxo-heterocycles using either H2O2 or O2 as the terminal oxidant (Scheme 13).44b Here, protonation deactivates the adjacent position to the amine function to HAT. For example, pyrrolidine undergoes selective oxidation to give pyrrolidin-3-one (51a), while piperidine was also oxidized to piperidines, formed as a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of γ:β keto products (50b). In contrast, only γ C–H oxygenation of azepane (51c) was observed despite the presence of multiple reactive sites. From this study, it was proposed that site-selectivity derived from an electrostatic interaction between protonated azepane and decatungstate anion, leading to site-selective HAT. Although no additional studies were presented to validate this hypothesis, this work demonstrated that electrostatic effects could be exploited to promote site-selective C–H functionalization using decatungstate photocatalysis.
:1 mixture of γ:β keto products (50b). In contrast, only γ C–H oxygenation of azepane (51c) was observed despite the presence of multiple reactive sites. From this study, it was proposed that site-selectivity derived from an electrostatic interaction between protonated azepane and decatungstate anion, leading to site-selective HAT. Although no additional studies were presented to validate this hypothesis, this work demonstrated that electrostatic effects could be exploited to promote site-selective C–H functionalization using decatungstate photocatalysis.
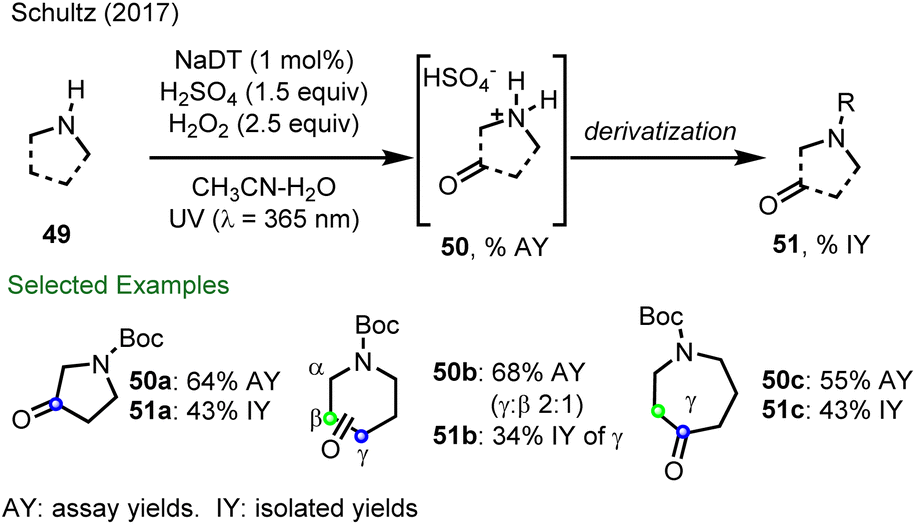 | ||
| Scheme 13 Decatungstate catalyzed C–H oxyfunctionalization of the remote C–H bonds of aliphatic amines and the observation of site-selective C–H oxidation. AY: assay yields by 1H NMR analysis; IY: yields of isolated products. | ||
More recently, Torigoe, Kuninobu and co-workers reported that electrostatic interactions enable site-selective C–H alkylation using decatungstate photocatalysis (Scheme 14a).99 Here, electrostatic interactions between the anionic decatungstate and cationic ammonium group in several anilines were shown to direct HAT to the benzylic position proximal to the ammonium group. A series of 2-methylaniliniums containing multiple C(sp3)–H bonds with similar properties were then selectively functionalized at the C-2 position by coupling the intermediate radical with electron-deficient alkenes (53a–53c). In contrast, the noncationic substrate, 1-chloro-2,4-dimethylbenzene (54), preferred to react at the distal methyl group (55b:55a–9:1), with selectivity based on both electronic and steric factors (Scheme 14b). In addition, although electronically deactivated for HAT by the cationic ammonium group, in a competition reaction between toluene (56) and the TFA salt of o-toluidine 57, the major product was that derived from C(sp3)–H alkylation of 57 to afford 58b (selectivity ∼4:1) (Scheme 14c). These results highlight the importance of electrostatic interactions between decatungstate and ammonium functionalities for directing and accelerating HAT (Scheme 14d).
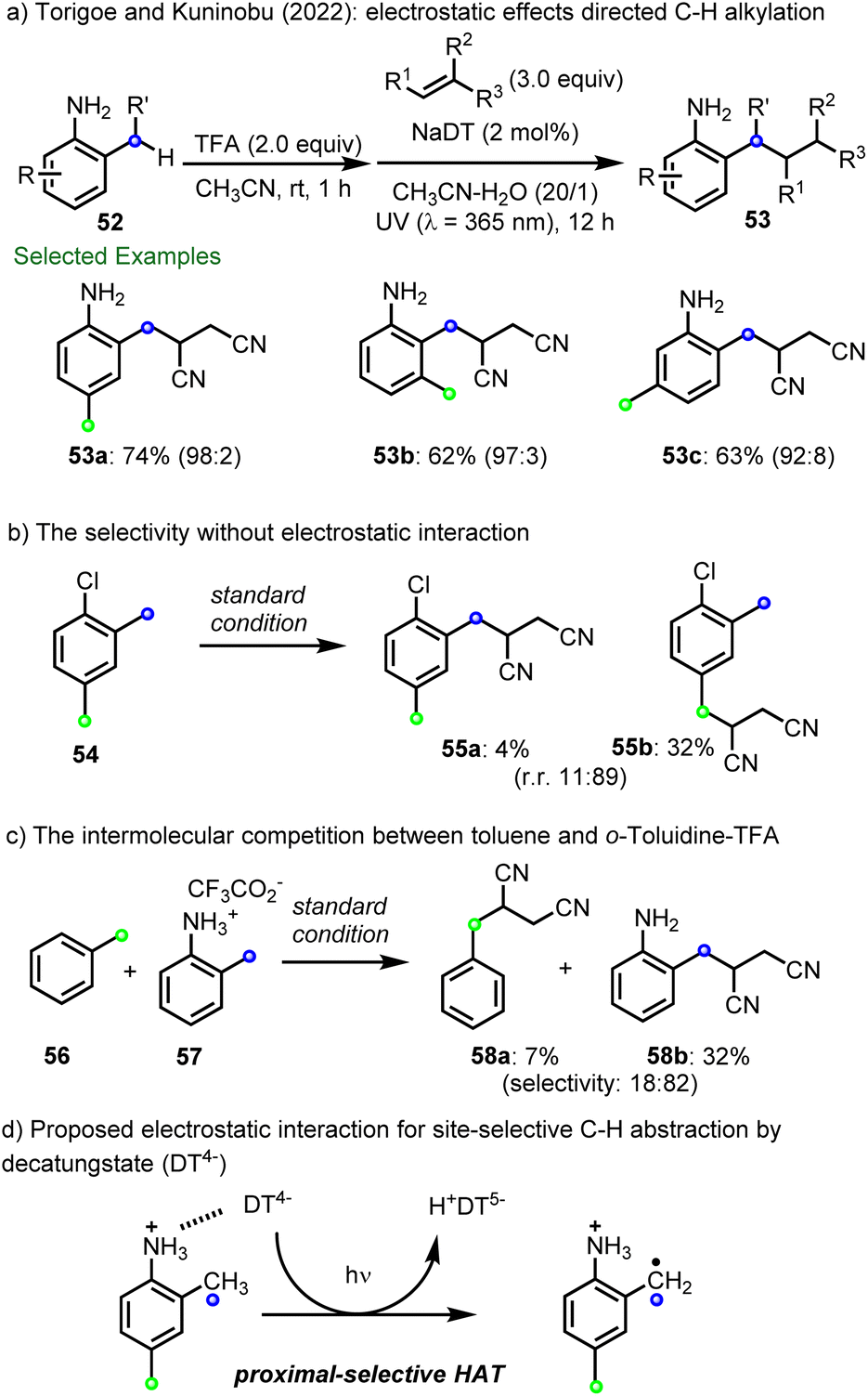 | ||
| Scheme 14 Decatungstate-catalyzed site-selective C–H alkylation via electrostatic effects. | ||
In further explorations of site-selective C(sp3)–H functionalization, Torigoe, Kuninobu and co-workers have reported a decatungstate C(sp3)–H alkylation of valine derivatives 60 with electron-deficient alkenes.100 Here, the initially alkylated product 61 undergoes hydrolysis and cyclization to afford δ-lactam 62 (Scheme 15a). Notably, and similar to our findings with leucine,89 the valine derivatives, H–Val–OMe (63), Ac–Val–OMe (64) and methyl isovalerate (65) did not react under the same conditions, highlighting the importance of electrostatic interactions in accelerating the HAT step (Scheme 15b). The C(sp3)–H alkylation of dipeptides containing a valine residue (such as Ala–Val, 66) at the C-terminus could also be realized. Furthermore, the Val–Val dipeptide 68 was selectively alkylated at the N-terminus to provide 69 along with traces of the dialkylated product 70 (Scheme 15c). This work represents the state of the art in combining LSF with both cationic rate acceleration and directing effects in decatungstate catalysis and should inspire further efforts in each of these areas.
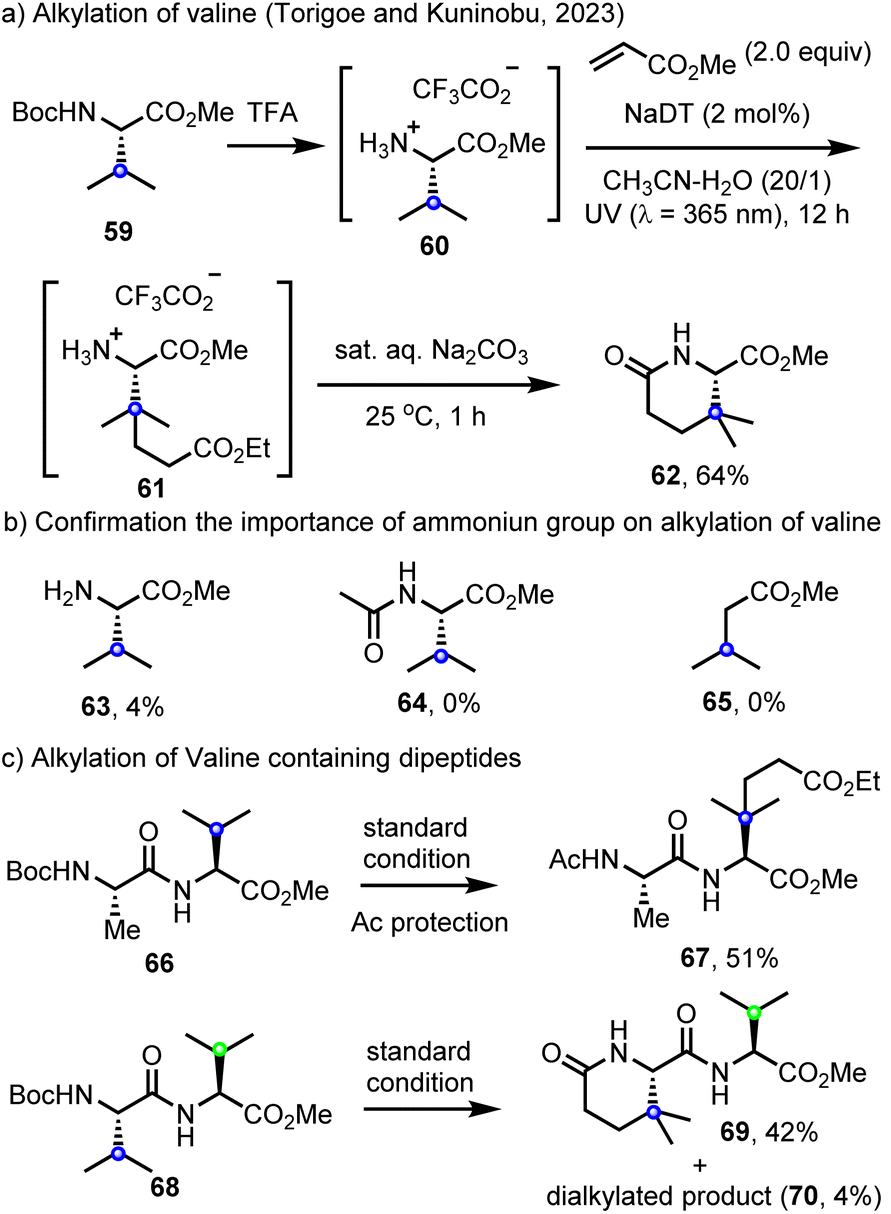 | ||
| Scheme 15 Decatungstate-catalyzed site-selective C–H alkylation valine residue. | ||
5 Conclusions and outlook
In this perspective, we have summarized our efforts and those of others in C–H (radio)fluorination/fluoroalkylation chemistry using decatungstate photocatalysis. We hope our experiences in developing both practical and scalable processes will inspire wider uptake and highlight the benefits of industry-academia collaborations to support basic reaction discovery that can often translate in unexpected ways. Further, we highlight our efforts in 18F-radiolabelling of amino acids and peptides that led to the identification of electrostatic effects in dectungstate chemistry that were also exploited to accelerate C–H (radio)fluorination. These electrostatic effects have now been more widely developed and, in particular, the work of Torigoe and Kuninobu has highlighted how this effect can be exploited for site-selective C(sp3)–H alkylation.With regards to decatungstate-catalyzed C–H (radio)fluorination, through collaboration with leaders in medicinal chemistry and process research as well as radiochemists and cancer biologists (Hoffmann–La Roche, Merck, TRIUMF and BC Cancer Agency), we summarize our modifications to reaction parameters, including a flow apparatus for large scale C–H fluorination and the development of microscale reactors for radiofluorination, that have supported both the development of industry-relevant processes and the discovery of new radiotracers for PET imaging. For example, with scientists at Merck, a one-step high yielding synthesis of (2S)-γ-fluoroleucine, a key intermediate in the “synthesis of the” drug candidate odanacatib, demonstrated the utility of our method for process research. Further, with scientists at Hoffmann–La Roche, TRIUMF and BC Cancer Agency, we succeeded in the development of decatungstate-catalyzed C–H 18F-fluorination and identified (S)-5-[18F]FHL (24) for further development as an oncologic PET imaging agent. While the relatively low molar activity of (S)-5-[18F]FHL is sufficient for imaging due to the accumulatory uptake mechanism for this radiotracer, widespread use of dectungstate catalysis for generating 18F-labelled compounds will require the generation of 18F-labelled fluorine atom transfer reagents (e.g., NFSI) from [18F]fluoride, a major challenge for the field. We hope that our efforts summarized here will inspire further developments in this area that may eventually translate to clinical PET imaging.
With respect to decatungstate-catalyzed C–H fluoroalkylation, these transformations are limited to decatungstate-catalyzed C–H trifluoromethylation difluoromethylthiolation and trifluoromethylthiolation. However, the merger of decatungstate with transition metal catalysis (i.e. Cu, Ni, Co etc.) developed by the MacMillan group, now provides unique opportunities for further exploration of C–H fluoroalkylation and other functionalizations, including asymmetric induction.
Finally, we highlight our efforts that resulted in the identification of electrostatic effects in decatungstate photocatalysis. These findings offer real potential to accelerate LSF reactions and, thanks to the work of others, control the site-selectivity in HAT processes. These works should inspire the exploitation of electrostatic effects in decatungstate photocatalyzed C–H functionalization chemistry. Further, through the rational design of cationic co-catalysts that also interact with substrate functionalities (e.g., carbonyl), it may prove possible to greatly expand the utility of decatungstate photochemistry.
Author contributions
Z. Y. and R. B. conceived the topic and structure of the manuscript. Both authors contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Natural Sciences and Engineering Research Council (NSERC) of Canada (Discovery grant: 2019-06368) was received by R. B. R. B. also acknowledges support from F. Hoffman-La Roche. Z. Y. gratefully acknowledge financial support from China Scholarship Council (CSC) (202107260030) and National Natural Science Foundation of China (22001233).Notes and references
- L. Candish, K. D. Collins, G. C. Cook, J. J. Douglas, A. Gómez-Suárez, A. Jolit and S. Keess, Chem. Rev., 2022, 122, 2907–2980 CrossRef CAS PubMed.
- K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef CAS PubMed.
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305–321 CrossRef CAS.
- (a) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC; (b) R. Mondal, M. Agbaria and Z. Nairoukh, Chem.–Eur. J., 2021, 27, 7193–7213 CrossRef CAS.
- C. Y. Kim, P. P. Chandra, A. Jain and D. W. Christianson, J. Am. Chem. Soc., 2001, 123, 9620–9627 CrossRef CAS PubMed.
- F. Hof, D. M. Scofield, W. B. Schweizer and F. Diederich, Angew. Chem., Int. Ed., 2004, 43, 5056–5059 CrossRef CAS PubMed.
- R. Britton, V. Gouverneur, J.-H. Lin, M. Meanwell, C. Ni, G. Pupo, J.-C. Xiao and J. Hu, Nat. Rev. Methods Primers, 2021, 1, 47 CrossRef CAS.
- F.-L. Qing, X.-Y. Liu, J.-A. Ma, Q. Shen, Q. Song and P. Tang, CCS Chem., 2022, 4, 2518–2549 CrossRef CAS.
- O. Jacobson, D. O. Kiesewetter and X. Chen, Bioconjugate Chem., 2015, 26, 1–18 CrossRef CAS PubMed.
- A. F. Brooks, J. J. Topczewski, N. Ichiishi, M. S. Sanford and P. J. H. Scott, Chem. Sci., 2014, 5, 4545–4553 RSC.
- R. Halder and T. Ritter, J. Org. Chem., 2021, 86, 13873–13884 CrossRef CAS PubMed.
- M. H. Shaw, J. Twilton and D. W. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS PubMed.
- L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem., 2021, 5, 522–545 CrossRef CAS.
- D. A. DiRocco, K. Dykstra, S. Krska, P. Vachal, D. V. Conway and M. Tudge, Angew. Chem., Int. Ed., 2014, 53, 4802–4806 CrossRef CAS PubMed.
- L. Zhang and T. Ritter, J. Am. Chem. Soc., 2022, 144, 2399–2414 CrossRef CAS PubMed.
- R. Szpera, D. F. J. Moseley, L. B. Smith, A. J. Sterling and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 14824–14848 CrossRef CAS.
- P. Bellotti, H.-M. Huang, T. Faber and F. Glorius, Chem. Rev., 2023, 123, 4237–4352 CrossRef CAS PubMed.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS.
- K. Suzuki, N. Mizuno and K. Yamaguchi, ACS Catal., 2018, 8, 10809–10825 CrossRef CAS.
- D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa and I. Ryu, ACS Catal., 2018, 8, 701–713 CrossRef CAS.
- D. Ravelli, S. Protti and M. Fagnoni, Acc. Chem. Res., 2016, 49, 2232–2242 CrossRef CAS PubMed.
- M. D. Tzirakis, I. N. Lykakis and M. Orfanopoulos, Chem. Soc. Rev., 2009, 38, 2609–2621 RSC.
- C. Tanielian, Coord. Chem. Rev., 1998, 178–180, 1165–1181 CrossRef CAS.
- X. Yuan, G. Yang and B. Yu, Chinese J. Org. Chem., 2020, 40, 3620–3632 CrossRef CAS.
- E. Papaconstantinou, Chem. Soc. Rev., 1989, 18, 1–31 RSC.
- C. L. Hill, Synlett, 1995, 1995, 127–132 CrossRef.
- D. C. Duncan, T. L. Netzel and C. L. Hill, Inorg. Chem., 1995, 34, 4640–4646 CrossRef CAS.
- C. Tanielian, K. Duffy and A. Jones, J. Phys. Chem. B, 1997, 101, 4276–4282 CrossRef CAS.
- V. De Waele, O. Poizat, M. Fagnoni, A. Bagno and D. Ravelli, ACS Catal., 2016, 6, 7174–7182 CrossRef.
- I. Texier, J. A. Delaire and C. Giannotti, Phys. Chem. Chem. Phys., 2000, 2, 1205–1212 RSC.
- D. C. Duncan and M. A. Fox, J. Phys. Chem. A, 1998, 102, 4559–4567 CrossRef CAS.
- For selected examples, see: (a) R. F. Renneke, M. Pasquali and C. L. Hill, J. Am. Chem. Soc., 1990, 112, 6585–6594 CrossRef CAS; (b) J. G. West, D. Huang and E. J. Sorensen, Nat. Commun., 2015, 6, 10093 CrossRef PubMed.
- For selected examples, see: (a) L. A. Combs-Walker and C. L. Hill, J. Am. Chem. Soc., 1992, 114, 938–946 CrossRef CAS; (b) H. M. Carder, Y. Wang and A. E. Wendlandt, J. Am. Chem. Soc., 2022, 144, 11870–11877 CrossRef CAS PubMed; (c) Y.-A. Zhang, V. Palani, A. E. Seim, Y. Wang, K. J. Wang and A. E. Wendlandt, Science, 2022, 378, 383–390 CrossRef CAS PubMed.
- For selected examples, see: (a) I. N. Lykakis, C. Tanielian, R. Seghrouchni and M. Orfanopoulos, J. Mol. Catal. A, 2007, 262, 176–184 CrossRef CAS; (b) D. M. Schultz, F. Lévesque, D. A. DiRocco, M. Reibarkh, Y. Ji, L. A. Joyce, J. F. Dropinski, H. Sheng, B. D. Sherry and I. W. Davies, Angew. Chem., Int. Ed., 2017, 56, 15274–15278 CrossRef CAS PubMed.
- For selected examples, see: Csp3–Csp3 bond formation, (a) G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92–96 CrossRef CAS; (b) S. Angioni, D. Ravelli, D. Emma, D. Dondi, M. Fagnoni and A. Albini, Adv. Synth. Catal., 2008, 350, 2209–2214 CrossRef CAS . Csp3–Csp2 bond formation,; (c) H. Cao, Y. Kuang, X. Shi, K. L. Wong, B. B. Tan, J. M. C. Kwan, X. Liu and J. Wu, Nat. Commun., 2020, 11, 1956 CrossRef CAS; (d) I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. C. MacMillan, Nature, 2018, 560, 70–75 CrossRef CAS PubMed . Csp3–Csp bond formation,; (e) L. Capaldo and D. Ravelli, Org. Lett., 2021, 23, 2243–2247 CrossRef CAS PubMed . Csp3–Csp bond formation,; (f) K. Chen, Q. Zeng, L. Xie, Z. Xue, J. Wang and Y. Xu, Nature, 2023, 620, 1007–1012 CrossRef CAS PubMed . Csp2–Csp3 bond formation,; (g) S. Esposti, D. Dondi, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2007, 46, 2531–2534 CrossRef CAS; (h) L. Capaldo, M. Fagnoni and D. Ravelli, Chem.–Eur. J., 2017, 23, 6527–6530 CrossRef CAS PubMed . Csp2–Csp2 bond formation,; (i) P. Fan, C. Zhang, L. Zhang and C. Wang, Org. Lett., 2020, 22, 3875–3878 CrossRef CAS PubMed; (j) L. Wang, T. Wang, G.-J. Cheng, X. Li, J.-J. Wei, B. Guo, G. Zheng, G. Chen, C. Ran and C. Zheng, ACS Catal., 2020, 10, 7543–7551 CrossRef CAS . Csp2–Csp bond formation, ref. 45e..
- For selected examples, see: (a) I. Ryu, A. Tani, T. Fukuyama, D. Ravelli, S. Montanaro and M. Fagnoni, Org. Lett., 2013, 15, 2554–2557 CrossRef CAS PubMed; (b) T. Wan, L. Capaldo, G. Laudadio, A. V. Nyuchev, J. A. Rincón, P. García-Losada, C. Mateos, M. O. Frederick, M. Nuño and T. Noël, Angew. Chem., Int. Ed., 2021, 60, 17893–17897 CrossRef CAS PubMed.
- For selected examples, see: (a) S. D. Halperin, H. Fan, S. Chang, R. E. Martin and R. Britton, Angew. Chem., Int. Ed., 2014, 53, 4690–4693 CrossRef CAS PubMed; (b) S. D. Halperin, D. Kwon, M. Holmes, E. L. Regalado, L. C. Campeau, D. A. Dirocco and R. Britton, Org. Lett., 2015, 17, 5200–5203 CrossRef CAS PubMed.
- For selected examples, see: (a) J. Dong, F. Yue, X. Wang, H. Song, Y. Liu and Q. Wang, Org. Lett., 2020, 22, 8272–8277 CrossRef CAS PubMed; (b) X. Wang, J. Dong, Y. Liu, H. Song and Q. Wang, Chin. Chem. Lett., 2021, 32, 3027–3030 CrossRef CAS.
- J. Dong, X. Wang, Z. Wang, H. Song, Y. Liu and Q. Wang, Chem. Sci., 2020, 11, 1026–1031 RSC.
- For selected examples, see: (a) J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218–222 CrossRef CAS; (b) Z.-Y. Dai, Z.-S. Nong and P.-S. Wang, ACS Catal., 2020, 10, 4786–4790 CrossRef CAS; (c) P. Fan, Y. Lan, C. Zhang and C. Wang, J. Am. Chem. Soc., 2020, 142, 2180–2186 CrossRef CAS PubMed.
- I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. C. MacMillan, Nature, 2018, 560, 70–75 CrossRef CAS PubMed.
- W. Liu, X. Huang, M.-J. Cheng, R. J. Nielson, W. A. Goddard III and J. T. Groves, Science, 2012, 337, 1322–1325 CrossRef CAS PubMed.
- M. Rueda-Becerril, C. Chatalova Sazepin, J. C. T. Leung, T. Okbinoglu, P. Kennepohl, J.-F. Paquin and G. M. Sammis, J. Am. Chem. Soc., 2012, 134, 4026–4029 CrossRef CAS PubMed.
- S. Bloom, C. R. Pitts, D. Curtin Miller, N. Haselton, M. G. Holl, E. Urheim and T. Lectka, Angew. Chem., Int. Ed., 2012, 51, 10580–10583 CrossRef CAS.
- Y. Amaoka, M. Nagatomo and M. Inoue, Org. Lett., 2013, 15, 2160–2163 CrossRef CAS PubMed.
- F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401–10404 CrossRef CAS PubMed.
- R. Szpera, D. F. J. Moseley, L. B. Smith, A. J. Sterling and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 14824–14848 CrossRef CAS PubMed.
- M. B. Nodwell, A. Bagai, S. D. Halperin, R. E. Martin, H. Knust and R. Britton, Chem. Commun., 2015, 51, 11783–11786 RSC.
- M. Meanwell, J. Lehmann, M. Eichenberger, R. E. Martinand and R. Britton, Chem. Commun., 2018, 54, 9985–9988 RSC.
- M. Meanwell and R. Britton, Synthesis, 2018, 50, 1228–1236 CrossRef CAS.
- M. Meanwell, M. B. Nodwell, R. E. Martin and R. Britton, Angew. Chem., Int. Ed., 2016, 55, 13244–13248 CrossRef CAS PubMed.
- R. E. Martin, J. Lehmann, T. Alzieu, M. Lenz, M. A. Carnero Corrales, J. D. Aebi, H. P. M-rki, B. Kuhn, K. Amrein, A. V. Mayweg and R. Britton, Org. Biomol. Chem., 2016, 14, 5922–5927 RSC.
- M. Meanwell, B. S. Adluri, Z. Yuan, J. Newton, P. Prevost, M. B. Nodwell, C. M. Friesen, P. Schaffer, R. E. Martin and R. Britton, Chem. Sci., 2018, 9, 5608–5613 RSC.
- C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3216–3221 CrossRef CAS PubMed.
- (a) B. Langdahl, N. Binkley, H. Bone, N. Gilchrist, H. Resch, J. Rodriguez Portales, A. Denker, A. Lombardi, C. Le Bailly De Tilleghem, C. DaSilva, E. Rosenberg and A. Leung, J. Bone Miner. Res., 2012, 27, 2251–2258 CrossRef CAS PubMed; (b) K. Brixen, R. Chapurlat, A. M. Cheung, T. M. Keaveny, T. Fuerst, K. Engelke, R. Recker, B. Dardzinski, N. Verbruggen, S. Ather, E. Rosenberg and A. E. de Papp, J. Clin. Endocrinol. Metab., 2013, 98, 571–580 CrossRef CAS.
- (a) V. L. Truong, J. Y. Gauthier, M. Boyd, B. Roy and J. Scheigetz, Synlett, 2005, 1279–1290 CrossRef CAS; (b) C. Nadeau, F. Gosselin, P. D. O'Shea, I. W. Davies and R. P. Volante, Synlett, 2006, 291–295 CrossRef CAS; (c) G. Humphrey, C. K. Chung, N. R. Rivera and K. Belyk, PCT Patent Application, WO 2013/148550 A1, 2013; (d) J. Limanto, A. Shafiee, P. N. Devine, V. Upadhyay, R. A. Desmond, B. R. Foster, D. R. Gauthier Jr., R. A. Reamer and R. P. Volante, J. Org. Chem., 2005, 70, 2372–2375 CrossRef CAS; (e) M. D. Truppo and G. Hughes, Org. Process Res. Dev., 2011, 15, 1033–1035 CrossRef CAS; (f) N. Rivera, Y. R. Pendri, S. Mende and B. N. Reddy, Patent Application, WO2013/148554 A1, 2013.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- M. V. Liberti and J. W. Locasale, Trends Biochem. Sci., 2016, 41, 211–218 CrossRef CAS PubMed.
- A. M. Hosios, V. C. Hecht, L. V. Danai, M. O. Johnson, J. C. Rathmell, M. L. Steinhauser, S. R. Manalis and M. G. Vander Heiden, Dev. Cell, 2016, 36, 540–549 CrossRef CAS PubMed.
- (a) Q. Wang and J. Holst, Am. J. Cancer Res., 2015, 5, 1281–1294 Search PubMed; (b) P. Häfliger and R.-P. Charles, Int. J. Mol. Sci., 2019, 20, 2428 CrossRef PubMed.
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed.
- P. M. Matthews, E. A. Rabiner, J. Passchier and R. N. Gunn, Br. J. Clin. Pharmacol., 2012, 73, 175–186 CrossRef CAS PubMed.
- M. E. Phelps, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9226–9233 CrossRef CAS PubMed.
- W. Chen, D. H. S. Silverman, S. Delaloye, J. Czernin, N. Kamdar, W. Pope, N. Satyamurthy, C. Schiepers and T. Cloughesy, J. Nucl. Med., 2006, 47, 904–911 CAS.
- D. M. Schuster, C. Nanni, S. Fanti, S. Oka, H. Okudaira, Y. Inoue, J. Sörensen, R. Owenius, P. Choyke, B. Turkbey, T. V. Bogsrud, T. Bach-Gansmo, R. K. Halkar, J. A. Nye, O. A. Odewole, B. Savir-Baruch and M. M. Goodman, J. Nucl. Med., 2014, 55, 1986–1992 CrossRef CAS PubMed.
- V. Morath, M. Heider, M. Mittelhäuser, H. Rolbieski, J. Stroh, J. Calais, M. Eiber, F. Bassermann and W. A. Weber, EJNMMI Res., 2022, 12, 4 CrossRef CAS PubMed.
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed.
- M. B. Nodwell, H. Yang, M. Čolović, Z. Yuan, H. Merkens, R. E. Martin, F. Bénard, P. Schaffer and R. Britton, J. Am. Chem. Soc., 2017, 139, 3595–3598 CrossRef CAS PubMed.
- H. Teare, E. G. Robins, E. Arstad, S. K. Luthra and V. Gouverneur, Chem. Commun., 2007, 2330–2332 RSC.
- J. Demling, K. Langer, M. Wörthmüller and V. Yusufu, Amino Acids, 1993, 5, 253–262 CrossRef CAS.
- (a) Q. Wang and J. Holst, Am. J. Cancer Res., 2015, 5, 1281–1294 Search PubMed; (b) B. C. Fuchs and B. P. S. Bode, Cancer Biol., 2005, 15, 254–266 CrossRef CAS; (c) H. Uchino, Y. Kanai, D. K. Kim, M. F. Wempe, A. Chairoungdua, E. Morimoto, M. W. Anders and H. Endou, Mol. Pharmacol., 2002, 61, 729–737 CrossRef CAS PubMed.
- (a) H. Teare, E. G. Robins, A. Kirjavainen, S. Forsback, G. Sandford, O. Solin, S. K. Luthra and V. Gouverneur, Angew. Chem., Int. Ed., 2010, 49, 6821–6824 CrossRef CAS PubMed; (b) N. Turjanski, G. V. Sawle, E. D. Playford, R. Weeks, A. A. Lammerstma, A. J. Lees and D. J. Brooks, J. Neurol., Neurosurg. Psychiatry, 1994, 57, 688–692 CrossRef CAS PubMed.
- M. B. Nodwell, H. Yang, H. Merkens, N. Malik, M. Colovic, B. Wagner, R. E. Martin, F. Bénard, P. Schaffer and R. Britton, J. Nucl. Med., 2019, 60, 1003–1009 CrossRef CAS PubMed.
- H. J. Wester, M. Herz, W. Weber, P. Heiss, R. Senekowitsch-Schmidtke, M. Schwaiger and G. Stöcklin, J. Nucl. Med., 1999, 40, 205–212 CAS.
- Z. Yuan, M. B. Nodwell, H. Yang, N. Malik, H. Merkens, F. Bénard, R. E. Martin, P. Schaffer and R. Britton, Angew. Chem., Int. Ed., 2018, 57, 12733–12736 CrossRef CAS PubMed.
- C. L. Denholt, P. R. Hansen, N. Pedersen, H. S. Poulsen, N. Gillings and A. Kjær, Biopolymers, 2009, 91, 201–206 CrossRef CAS PubMed.
- R. T. Olszewski, N. Bukhari, J. Zhou, A. P. Kozikowski, J. T. Wroblewski, S. Shammi-Noori, B. Wroblewska, T. Bzdega, S. Vicini, F. B. Barton and J. H. Neale, J. Neurochem., 2004, 89, 876–885 CrossRef CAS PubMed.
- (a) F. Wuest, L. Kçhler, M. Berndt and J. Pietsch, Amino Acids, 2009, 36, 283–295 CrossRef CAS PubMed; (b) G. Vaidyanathan and M. R. Zalutsky, Nat. Protoc., 2006, 1, 1655–1661 CrossRef CAS PubMed.
- Z. Yuan, H. Yang, N. Malik, M. Čolović, D. S. Weber, D. Wilson, F. Bénard, R. E. Martin, J. J. Warren, P. Schaffer and R. Britton, ACS Catal., 2019, 9, 8276–8284 CrossRef CAS.
- P. J. Sarver, V. Bacauanu, D. M. Schultz, D. A. DiRocco, Y.-h. Lam, E. C. Sherer and D. W. C. MacMillan, Nat. Chem., 2020, 12, 459–467 CrossRef CAS.
- T. E. Schirmer, A. B. Rolka, T. A. Karl, F. Holzhausen and B. König, Org. Lett., 2021, 23, 5729–5733 CrossRef CAS PubMed.
- Z. Ye, Z. Lei, X. Ye, L. Zhou, Y. Wang, Z. Yuan, F. Gao and R. Britton, J. Org. Chem., 2022, 87, 765–775 CrossRef CAS PubMed.
- H. J. Davis and R. J. Phipps, Chem. Sci., 2017, 8, 864–877 RSC.
- Y. Kuninobu and T. Torigoe, Org. Biomol. Chem., 2020, 18, 4126–4134 RSC.
- H. Schneider, J. Phys. Org. Chem., 2022, 35, e4340 CrossRef CAS.
- S. E. Wheeler, T. J. Seguin, Y. Guan and A. C. Doney, Acc. Chem. Res., 2016, 49, 1061–1069 CrossRef CAS PubMed.
- J. E. Gillespie, A. Fanourakis and R. J. Phipps, J. Am. Chem. Soc., 2022, 144, 18195–18211 CrossRef CAS.
- M. Mihai and R. Phipps, Synlett, 2017, 28, 1011–1017 CrossRef CAS.
- J. Zeng, T. Torigoe and Y. Kuninobu, ACS Catal., 2022, 12, 3058–3062 CrossRef CAS.
- J. Song, T. Torigoe and Y. Kuninobu, Org. Lett., 2023, 25, 3708–3712 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |