
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A cell-active cyclic peptide targeting the Nrf2/Keap1 protein–protein interaction†
Jessica
Iegre‡
 a,
Sona
Krajcovicova‡
ab,
Anders
Gunnarsson
c,
Lisa
Wissler
c,
Helena
Käck
c,
Anna
Luchniak
c,
Stefan
Tångefjord
d,
Frank
Narjes
*e and
David R.
Spring
*a
a,
Sona
Krajcovicova‡
ab,
Anders
Gunnarsson
c,
Lisa
Wissler
c,
Helena
Käck
c,
Anna
Luchniak
c,
Stefan
Tångefjord
d,
Frank
Narjes
*e and
David R.
Spring
*a
aYusuf Hamied Department of Chemistry, Lensfield Road, CB2 1EW, Cambridge, UK. E-mail: spring@ch.cam.ac.uk
bDepartment of Organic Chemistry, Palacky University Olomouc, Tr. 17. Listopadu 12, 77900, Olomouc, Czech Republic
cMechanistic and Structural Biology, Discovery Sciences, BioPharmaceuticals R&D, AstraZeneca, Pepparedsleden 1, 43183, Gothenburg, Sweden
dBioScience, Research & Early Development, Respiratory & Immunology, BioPharmaceuticals R&D, AstraZeneca, Pepparedsleden 1, 43183, Gothenburg, Sweden
eMedicinal Chemistry, Research & Early Development, Respiratory & Immunology, BioPharmaceuticals R&D, AstraZeneca, Pepparedsleden 1, 43183, Gothenburg, Sweden. E-mail: frank.narjes@astrazeneca.com
First published on 20th September 2023
Abstract
The disruption of the protein–protein interaction (PPI) between Nrf2 and Keap1 is an attractive strategy to counteract the oxidative stress that characterises a variety of severe diseases. Peptides represent a complementary approach to small molecules for the inhibition of this therapeutically important PPI. However, due to their polar nature and the negative net charge required for binding to Keap1, the peptides reported to date exhibit either mid-micromolar activity or are inactive in cells. Herein, we present a two-component peptide stapling strategy to rapidly access a variety of constrained and functionalised peptides that target the Nrf2/Keap1 PPI. The most promising peptide, P8-H containing a fatty acid tag, binds to Keap1 with nanomolar affinity and is effective at inducing transcription of ARE genes in a human lung epithelial cell line at sub-micromolar concentration. Furthermore, crystallography of the peptide in complex with Keap1 yielded a high resolution X-ray structure, adding to the toolbox of structures available to develop cell-permeable peptidomimetic inhibitors.
Introduction
The transcription factor Nuclear Factor Erythroid-2-related factor (Nrf2) protects the cells against oxidative stress by regulating the transcription of more than 200 genes containing the antioxidant response element (ARE). Nrf2 is negatively regulated by the protein–protein interaction (PPI) with the E3 ligase Kelch-like EH-associated protein 1 (Keap1), which as part of a Cullin3-RBX1 complex binds Nrf2 in the cytosol, promoting its ubiquitination and proteasomal degradation.1 Oxidative stress is recognised as a major driver in a variety of pathological conditions including cancer, neurodegenerative conditions, chronic obstructive pulmonary disease (COPD) and other inflammatory-driven diseases.2 Disruption of the Nrf2/Keap1 PPI is regarded as a promising strategy to upregulate Nrf2 levels and protect cells against oxidative stress and small molecule as well as peptide approaches have been pursued to achieve this goal.3,4 Nrf2 interacts with Keap1 via two conserved motifs in the Nrf2-ECH homology domain 2 (Neh2), namely DLG and ETGE. Unlike the DLG, the ETGE motif binds strongly to Keap1 (KD ∼ 5 nM) through H-bonds, electrostatic and van der Waals interactions.5,6 Of particular interest for targeting this PPI are peptide inhibitors since they resemble the structure of the epitope of Nrf2 interacting with Keap1. The hydrophilic binding site on Keap1 requires charged and polar sequences for high affinity interactions with Nrf2, making cell permeability an issue. A plethora of potent linear or cyclic peptides, with or without moieties to enhance cell permeability have been reported thus far; however, they either lack or show a significant drop-off in activity in cellular assays.7–23 The limited cellular activity hampers the use of Nrf2-like peptides as effective Keap1/Nrf2 inhibitors. On the other hand, small molecule Nrf2/Keap1 inhibitors can overcome the cell permeability issues.24 However, due to the plasticity of the Keap1 binding site, they can target different conformations of the Keap1 protein, and represent a complementary approach to Keap1/Nrf2 inhibition.25–30 Consequently, the development of a cell permeable Nrf2-like peptide that retains activity in the cellular environment would be a significant breakthrough. Herein, we describe a variety of Cys dependant stapling approaches of an Nrf2 peptide, of which the divinylpyrimidine (DVP)31 two-component peptide stapling (2C-PS) approach proved particularly useful to easily access a variety of peptides of different sizes carrying different functionalities. The most promising of the initial peptides, P3-F, was further optimised into a cell permeable peptide. To this end, we adopted different strategies and combinations thereof, namely peptide shortening, replacement of acidic residues with methyl esters, poly-arginine residues with the endosomal escape enhancer motif GWWG and incorporation of fatty acid tags (Fig. 1).32–35 Peptide P8-H (Fig. 2; shorter peptide containing a stearic tag) retained its binding affinity in the enzymatic assays alongside exhibiting a submicromolar EC50 value (0.75 μM) in a cellular assay. Peptide P8-H represents, to the best of our knowledge, the most potent Nrf2/Keap1 peptide inhibitor with cellular activity known to date. Moreover, we were able to obtain X-ray structures of Keap1 in complex with peptide P3-F and the shortened analogue P8H. The binding mode was found to maintain the hotspot interactions of the ETFG motif of the native epitope. These could present useful starting points for the design of peptidomimetics.36,37 | ||
| Fig. 1 (a) DVP peptide stapling methodology successfully used in this work; (b) strategies used in this work to aid cell permeability of Nrf-2 like peptides following 2C-PS with DVP staples. | ||
 | ||
| Fig. 2 Structures of peptides made in this work. Stapling positions on the different peptides are coloured in blue. Peptides were made using SPPS (ESI†) and cyclised as follows: cyclisation with staple A: linear peptide (1 eq.), H2O, TCEP·HCl (1.5 eq.), K2CO3 (3 eq.) then NEt3 (10 eq.), diiodomethane (8 eq.), THF, rt, 4 h; cyclisation with staple B: linear peptide (1 eq.), 50 mM NaPi buffer pH 8, TCEP·HCl (1.5 eq.), dichloroacetone (3 eq.), DMF, rt, 4 h,; cyclisation with staple C: linear peptide (1 eq.), TCEP·HCl (1.5 eq.), 50 mM NaPi pH 8, rt, 1 h, then 1,3-bisdibromomethylbenzene (3 eq.), DMF, rt, 4 h; cyclisation with DVP staples D–H: linear peptide (1 eq.), 50 mM NaPi buffer, pH 8, 5% DMF, staple D–H (1.1 eq.), rt, 1 h. | ||
Results and discussion
We based the design of our Nrf2-like cyclic peptides around the amino acid sequence of the ETGE motif (namely 12-mer 73QLQLDEETGEF84L) and SAR studies reported in the literature.9,10,12,14 The ETFE motif is known to adopt a beta-turn conformation upon binding to Keap1. Residues Q73-Q75 and L85 stabilise the bound conformation, L76-D77 and E79-G81 bind deep into the pocket with E79 and E82 forming electrostatic interactions with Keap1 and D77 and T80 stabilising the beta-turn via H-bonds with E78 not contributing to the binding. In particular, our peptides aim to retain the beta-turn conformation and featured the E78P mutation which has been reported to improve the binding affinity.9 Visual inspection of the X-ray structures of previously published peptides in complex with Keap1 and of the native Keap1/Nrf2 PPI (PDB: 4IFL,105WFV,382FLU6) suggested a combination of three sequences, P1–P3 and five different staples (A–E) to initiate our investigation (Fig. 2). In all three sequences, F83 was replaced with Cys or azido alanine (Aza), since it is well-established that this residue does not contribute to binding affinity.38 The ability of the cyclic peptides to bind to the Kelch domain of Keap1 was evaluated using surface plasma resonance (SPR), where the Kelch domain is immobilised onto a SPR sensor chip. Additionally, we assessed the ability of the peptides to inhibit the PPI between Nrf2 and the Kelch domain of Keap1 using a time-resolved FRET (TR-FRET) assay. Here, the IC50 for inhibition of the interaction between the human Keap1 Kelch domain and an Nrf2-ETGE peptide is measured.Compared to the 11-mer 74LQLDEETGEF84L peptide (entry 1, Table 1), which had a KD value of 52 nM and inhibited the TR-FRET assay with an IC50 of 5.3 nM, P1 and the P3 derived cycles showed approximately 2–3-fold improvement in binding affinity, nearly irrespective of the staple used.
| Entry | Sequence | SPR Kd (nM) | SPR kon (M−1 s−1) koff (s−1) | TR-FRET IC50 (nM) |
|---|---|---|---|---|
| a Tested twice. b Tested once. c Linear peptides P1, P2 and P3 containing free cysteines, were synthesised as starting material for the cyclised peptides. Their purity declined overtime, presumably due to intra- or intermolecular disulphide formation. The data for P1 and P3 were comparable over several testing occasions, whereas for b we were not able to obtain reproducible KD's or IC50's. | ||||
| 1 | 74LQLDPETGEF83L | 52 ± 1.4 | 1.7 × 105/7.9 × 10−3 | 5.3 ± 1.2 |
| 2 | P1 | 20 ± 1.2a | 8.6 × 104/1.4 × 10−3 | 4.9 ± 1.1 |
| 3 | P1-A | 19 ± 1.4 | 4.8 × 105/7.3 × 10−3 | 2.9 ± 1.7 |
| 4 | P1-B | 27 ± 1.3 | 4.1 × 105/8.7 × 10−3 | 4.4 ± 1.4 |
| 5 | P1-C | 17 ± 1.2a | 4.7 × 105/7.8 × 10−3 | 2.1b |
| 6 | P1-D | 25 ± 1.3 | 2.0 × 105/4.7 × 10−3 | 2.9 ± 1.4 |
| 7 | P2 | 75 | n.d.c | n.d. |
| 8 | P2-A | 70 ± 1.0 | 2.1 × 105/1.5 × 10−2 | 8.3 ± 1.1 |
| 9 | P2-B | 97 ± 1.4 | 3.5 × 105/2.8 × 10−2 | 9.8 ± 1.2 |
| 10 | P2-C | 36 ± 1.8a | 5.0 × 105/1.3 × 10−2 | 5.6 ± 1.1 |
| 11 | P2-D | 54 ± 1.2 | 1.6 × 105/8.3 × 10−3 | 4.8 ± 1.5 |
| 12 | P3 | 19 ± 1.8 | 1.8 × 105/4.9 × 10−3 | 5.0 ± 1.5 |
| 13 | P3Aza-E | 34 ± 1.3 | 1.7 × 105/5.4 × 10−3 | 4.0 ± 1.4 |
| 14 | P3-D | 22 ± 1.3 | 1.4 × 105/2.8 × 10−3 | 2.4 ± 1.3 |
| 15 | P3-F | 14 ± 1.4 | 2.4 × 105/3.2 × 10−3 | 1.9 ± 1.3 |
Cyclisation thus did not improve the affinity to a significant extent compared to the linear peptide, as previously reported. One reason for this is the preorganisation of the linear Keap1 peptide into a β-turn, which is even more pronounced by the presence of P78.39 On- and off rates were comparable across the two series. Pleasingly, in the TR-FRET assay, the P1 and P3 cyclic peptides showed nM enzymatic inhibition with IC50 ranging from 2 to 5 nM (Table 1). Conversely, the analogues in P2 series, having a smaller cycle size, had equal or less affinity compared to the parent peptide, which might be due to a faster off-rate for several analogues, when compared to the matched pairs in the P1 and P3 series. Among all the cyclic unmodified peptides, P3-D (KD = 22 nM and IC50 = 2.4 nM) was selected for further investigation due to the functional handle on its staple, that allows for facile elaboration.
To avoid the additional negative charge brought by the carboxylic acid on staple D, we changed the residue to an ethyl ester to yield P3-F (Fig. 2), which displayed similar activity displaying a KD of 12 ± 1 nM and an enzymatic IC50 of 2 ± 1.3 nM (Table 1).
An X-ray crystal structure of P3-F bound to Keap1 Kelch domain could be obtained and allowed to investigate its binding mode (Fig. 3). The peptide was found to bind to the Kelch domain of Keap1 and maintained the conformation of the backbone and the side-chains when compared to the structure of linear peptides (PDB: 5WFV and 2FLU).6,38 This includes the key interactions made by E79 with residues R415, R483 and S508 in Keap1 and of E82 with R380, N382 and S363, as well as the intramolecular hydrogen bonds established by T80 and D77 to stabilise the turn structure.6,38 The staple and the flanking residues are protruding from the binding site, lacking direct interactions with Keap1 and are consequently less ordered, as demonstrated by less defined electron density for this portion of P3-F (Fig. 3).
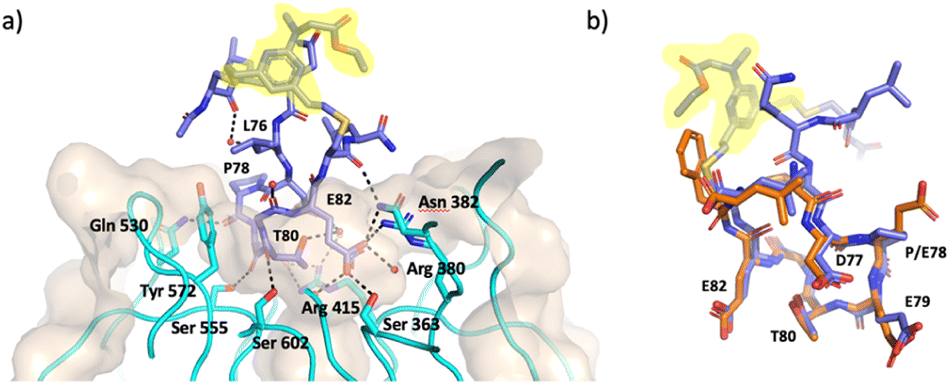 | ||
| Fig. 3 X-ray crystal structure of P3-F bound to Keap1 (PDB 8Q1Q, 1.4 Å). (a) Structure of P3-F (purple) in complex with Keap1 (cyan ribbons and beige surface) key interactions of P3-F hotspot with water molecules and Keap1 residues in the binding site are also shown. (b) Overlay of P3-F (purple) with the native peptide (orange, PDB: 5WFV). Staple highlighted in yellow. | ||
The effect of peptides on Nrf2-dependent gene expression was investigated in BEAS-2B cells, where we determined the upregulation of the mRNA of NAD(P)H quinone dehydrogenase (NQO1) by qPCR. The expression of NQO1 is Nrf2-dependent and it is one of the two major quinone reductases, which play a crucial role in protecting cells from oxidative stress.40§ Incubation of peptides P1-A, P1-D, P2-A, P2-D, P3-D, P3-F as well as the linear 11-mer peptide for 24 h did not lead to any induction of NQO1 mRNA at concentrations of up to 10 μM, whereas a small molecule inhibitor of the Keap1–Nrf2 interaction had an EC50 = 12 nM, matching the reported value.25 The lack of activity of the peptides is unsurprising considering the overall negative net charge, which will obstruct their cellular uptake. In order to improve the cellular uptake, we took of advantage of the 2C-PS moiety, which not only allows the easy synthesis of a variety of macrocycles of different ring sizes, but also permits the facile introduction of a diverse set of moieties, which could enhance cell permeability. We pursued a diverse set of strategies to make the Keap1 peptide more cell permeable, namely: masking of the acidic amino acids with methyl esters, introduction of polyarginine peptides, GWWG-polyarginine peptides or a long-chain fatty acid tag onto the staple, as well as peptide shortening and a combination of these. The rationale behind these strategies is explained herein, and details of their synthesis can be found in the ESI.†
Methyl esters to mask the acidic residues
It was hypothesised that by masking the Glu and Asp residues as methyl esters would allow peptides to overcome hydrophobic barriers more easily and hence facilitate cell penetration. Consequently, we synthesised four ester bearing peptides P4-F, P5-F, P6-F and P7-F, where different combinations of the carboxylates were masked as methyl esters. The Glu and Asp residues are deemed as critical to the binding, and hence, it was not surprising to see that the ester-bearing peptides showed reduced activity in both enzymatic assays (Table 2, entries 1 and 4). It was thought that upon cellular internalisation, intracellular carboxyl esterase would hydrolyse the methyl esters to the free carboxylic acids,41 leading to inhibition of the Keap1–Nrf2 interaction and expression of NQO1.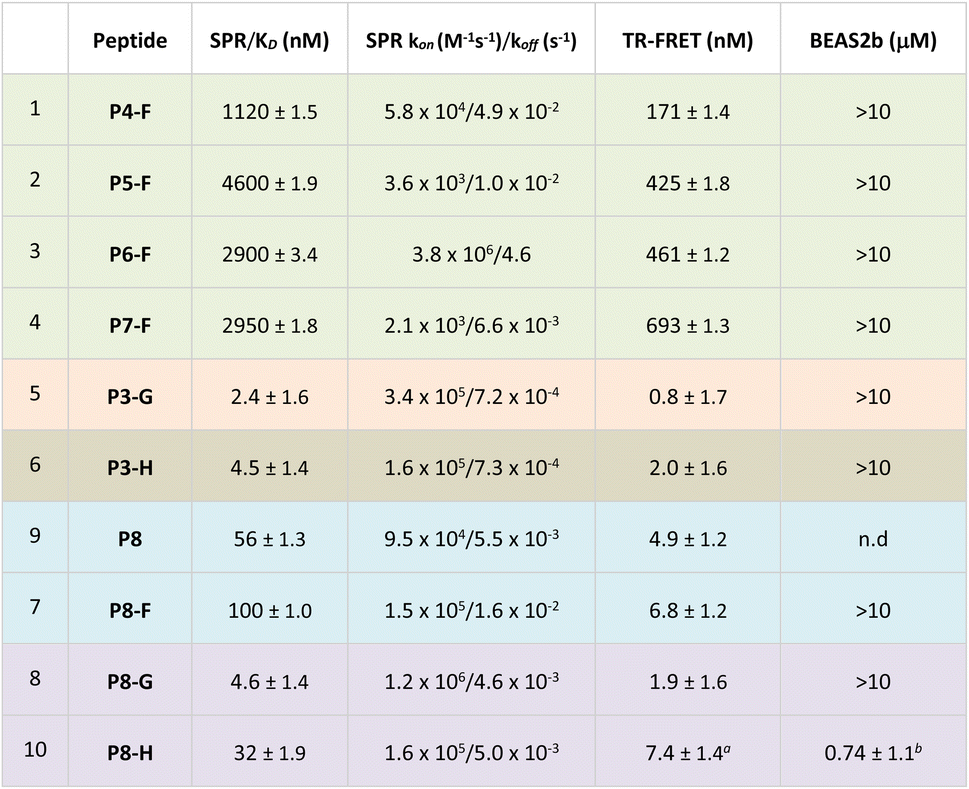
Disappointingly, no Nrf2-dependent gene expression was observed in the cell assay, indicating either lack of internalisation or lack of intracellular methyl ester hydrolysis.
Incorporation of GWWG-polyarginine peptide onto the staple
Another strategy to enhance the cell permeability of peptides is the addition of a tri-(L)Arg motif onto the staple coupled with GWWG sequence. Tri-(L)Arg tags were previously reported to be able to enhance the cellular uptake of stapled peptides into cells, while the GWWG motif is a hydrophobic sequence known to enhance the endosomal escape.33,34 Therefore, synthesis of peptides P3-G and P7-G was attempted (Fig. 2). Solubility issues hampered the synthesis of P7-G whereas peptide P3-G was successfully synthesised. Despite showing low nM activity in the biophysics and enzymatic assays (Table 2), no activity was detected in the cellular experiments.Peptide shortening
Guided by the X-ray structure of P3-F in complex with Keap1, we envisaged that the removal of amino acids not essential for Keap1 binding would result in reduction of the number of hydrogen-bond donors that might limit membrane permeability. Therefore, it was decided to make use of the shorter sequence obtainable by removing the two N-terminal residues of P2-D, leading to P8 and sequentially cyclise the peptide using staples F and G (Fig. 2 and 4). Peptide P8-F showed a KD of 100 nM in the SPR assay, in line with the values seen for the P2 analogues. We believe the higher affinity observed for P8-G to be due to interaction of the tag with the SPR surface. Both peptides showed low nM inhibition in the TR-FRET assay.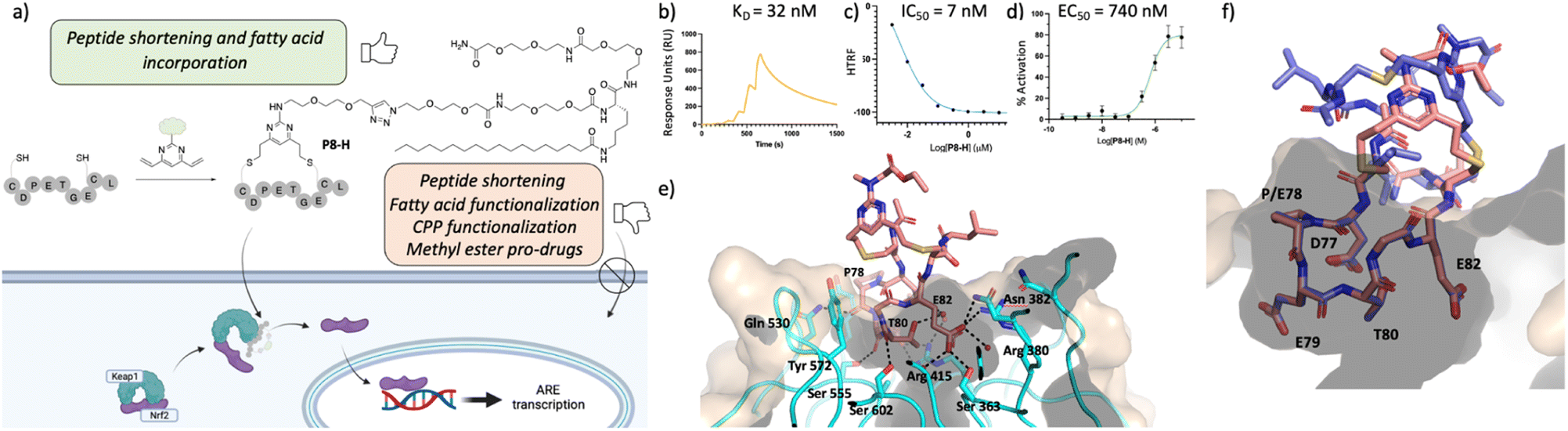 | ||
| Fig. 4 Profiling of P8-H. (a) structure of the lead peptide obtained vie DVP peptide stapling and it's proposed mechanism of action; (b) SPR data of the peptide showing a KD of 92 nM in two independent repeats; (c) HRFT data of the peptide shown as a geometric mean of 3 independent repeats; (d) qPCR results in BEAS-2B cells shown as the SEM of 5 independent repeats; (e) crystal structure of peptide P8-F (pink, PDB: 8Q1R) in complex with Keap1. (f) Overlay of P8-F (pink) with P3-F (purple). While P8-F is shorter, as designed, all the key interactions with Keap1 are maintained. | ||
Pleasingly, an X-ray structure of P8-F revealed identical binding of the ETGE motif to Keap1 as compared with P3-F, confirming the design hypothesis (Fig. 4e and f). Different from the larger P3-F macrocycle, the staple in P8-F is in closer proximity to Keap1, and a hydrogen bond between the thioether and the phenol oxygen of Tyr334 of 3.4 Å can be observed (Fig. SI_1†). However, shortening of the peptide on its own (P8-F) or combining peptide shortening with the cell penetrant staple G (P8-G) did not lead to any activity in the qPCR assay. Shortening of the methyl ester containing peptides was not possible due to solubility issues encountered during their synthesis.
Incorporation of C18 motif onto the staple
We decided to modify the staple by incorporating a C18 motif (H, Fig. 2 and 4a). Fatty acid tags have been reported to effectively transport small molecules and peptides into cells,35 and therefore, this was regarded as a reasonable strategy to transport the Nrf2 peptides into cells (Fig. 4). The C18 motif to attach to the stapled peptides P3-H and P8-H was designed in such a way that it could be easily made on by solid phase peptide synthesis and would overcome solubility issues, often encountered in the synthesis of the C18-bearing stapled peptides.¶P3-H and P8-H showed nM activity in the SPR and HTRF assays with KD's of 4 and 32 nM and IC50's of 2 and 7 nM respectively. Comparable kON and kOFF rates were observed for both peptides (Table 2). Whilst the more potent, but larger macrocycle did not induce Nrf2-dependent gene expression, we were pleased to observe that the shorter peptide P8-H, was active in BEAS-2B cells with an EC50 of 740 nM. In addition, P8-H showed no cytotoxicity when tested in a CellTiter Glo assay up to 30 μM (Fig. SI_2†). These results suggest that the peptide disrupts the Nrf2/Keap1 PPI and acts as a chemical probe since it engages Keap1 in both the enzymatic and cellular settings and structural information is available.Conclusions
In this work, we showcased the potential of the DVP-2C peptide stapling to access a variety of functionalised Nrf2-like peptides. Peptide P3-F, featuring a simple functionalisation on the staple, yielded an X-ray structure in complex with Keap1 of a high resolution (1.4 Å). The structure revealed that the peptide retains the hotspot of the native epitope whilst the staple and functionality sit outside the binding site. Furthermore, the high resolution allowed us to accurately observe the position of each atom and water network, serving as an excellent starting point for the design of peptidomimetic inhibitors. The DVP-2C stapling methodology was also well-suited in the further functionalisation of the cyclic peptides. Among the strategies explored to aid cell penetration, the combination of peptide shortening and fatty acid tag was the most promising. In addition to exhibiting low nM activity in the enzymatic assays, stapled peptide P8-H, functionalised with a stearic fatty acid, was able to induce the expression of the Nrf2-dependent gene NQO1 in cells at submicromolar concentration – the most promising cellular activity reported to date for a peptide. The ease of functionalisation and exploration of different macrocycle sizes coupled with the high-resolution X-ray structure and the submicromolar cellular activity make the peptides presented in this work an excellent starting point to develop further peptide Nrf2/Keap1 inhibitors.Data availability
The coordinates and structure factors have been deposited to the Protein Data Bank (PDB) with accession codes, 8Q1Q and 8Q1R.Author contributions
JI: project conception, performed experiments, wrote the manuscript; SK: performed experiments, approved the manuscript; AG: performed experiments, approved the manuscript Lisa Wissler – performed experiments, approved the manuscript; HK: performed experiments, approved the manuscript; AL: performed experiments, approved the manuscript; ST: performed experiments, approved the manuscript; FN: project conception, project supervision, wrote the manuscript; DRS: project conception, project supervision, approved the manuscript.Conflicts of interest
A. G., L. W., H. K., A. L., S. T. and F. N. are AstraZeneca full-time employees and may own stock or stock options.Acknowledgements
The Spring group would like to thank UKRI and AstraZeneca for funding. JI acknowledges funding from the EPSRC and Wellcome Trust. SK is grateful to the Experientia Foundation (https://www.experientia.cz/), Czech Science Foundation (GA CR 22-07138O) and Cambridge Isaac Newton Trust (Grant Ref No.: 22.39(l)) for their financial support. Authors are grateful to Dr Raoul Walther to provide several milligrams of staple F. We acknowledge MAX IV Laboratory for time on Beamline BioMAX under Proposal 20220052. Research conducted at MAX IV, a Swedish national user facility, is supported by the Swedish Research council under contract 2018-07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019-02496.Notes and references
- K. Itoh, T. Chiba, S. Takahashi, T. Ishii, K. Igarashi, Y. Katoh, T. Oyake, N. Hayashi, K. Satoh, I. Hatayama, M. Yamamoto and Y.-i. Nabeshima, BBRC, 1997, 236, 313–322 CrossRef CAS PubMed.
- A. Cuadrado, A. I. Rojo, G. Wells, J. D. Hayes, S. P. Cousin, W. L. Rumsey, O. C. Attucks, S. Franklin, A.-L. Levonen, T. W. Kensler and A. T. Dinkova-Kostova, Nat. Rev. Drug Discovery, 2019, 18, 295–317 CrossRef CAS PubMed.
- E. Crisman, P. Duarte, E. Dauden, A. Cuadrado, M. I. Rodríguez-Franco, M. G. López and R. León, Med. Res. Rev., 2023, 43, 237–287 CrossRef CAS PubMed.
- L. Torrente and G. M. DeNicola, Annu. Rev. Pharmacol. Toxicol., 2022, 62, 279–300 CrossRef PubMed.
- P. Canning, F. J. Sorrell and A. N. Bullock, Free Radical Biol. Med., 2015, 88, 101–107 CrossRef CAS PubMed.
- S.-C. Lo, X. Li, M. T. Henzl, L. J. Beamer and M. Hannink, EMBO J., 2006, 25, 3605–3617 CrossRef CAS PubMed.
- N. D. Georgakopoulos, S. K. Talapatrea, J. Gatliff, F. Kosielzki and G. Wells, ChemBioChem, 2018, 19, 1810–1816 CrossRef CAS PubMed.
- M.-C. Lu, Z.-Y. Chen, Y.-L. Wang, Y.-L. Jiang, Z.-W. Yuan, Q.-D. You and Z.-Y. Jiang, RSC Adv., 2015, 5, 85983–85987 RSC.
- M.-C. Lu, Q. Jiao, T. Liu, S.-J. Tan, H.-S. Zhou, Q.-D. You and Z.-Y. Jiang, Eur. J. Med. Chem., 2018, 143, 1578–1589 CrossRef CAS PubMed.
- K. Chen, L. Huang and B. Shen, Biophys. Chem., 2019, 244, 22–28 CrossRef CAS PubMed.
- M.-C. Lu, X. Zhang, F. Wu, S.-J. Tan, J. Zhao, Q.-D. You and Z.-Y. Jiang, J. Med. Chem., 2019, 62, 6796–6813 CrossRef CAS PubMed.
- R. J. Steel, M. A. O'Connell and M. Searcey, Bioorg. Med. Chem. Lett., 2018, 28, 2728–2731 CrossRef CAS PubMed.
- R. Hancock, M. Shaap, H. Pfister and G. Wells, Org. Biomol. Chem., 2013, 11, 3553–3557 RSC.
- R. Steel, J. Cowan, E. Payerne, M. A. O'Connel and M. Searcey, ACS Med. Chem. Lett., 2012, 3, 407–410 CrossRef CAS PubMed.
- J. Ontoria, I. Biancofiore, P. Fezzardi, F. Ferrigno, E. Torrente, S. Colarusso, E. Bianchi, M. Andreini, A. Patsilinakos and G. Kempf, et al. , ACS Med. Chem. Lett., 2020, 11, 740–746 CrossRef CAS PubMed.
- S. Colarusso, D. De Simone, T. Frattarelli, M. Andreini, M. Cerretani, A. Missineo, A. Moretti, S. Tambone, G. Kempf and M. Augustin, et al. , Bioorg. Med. Chem., 2020, 28, 115738 CrossRef CAS PubMed.
- H. Salim, J. Song, A. Sahni and D. Pei, J. Org. Chem., 2020, 85, 1416–1424 CrossRef CAS PubMed.
- H. Yin, Y.-H. Huang, S. A. Best, K. D. Sutherland, D. J. Craik and C. K. Wanf, ACS Chem. Biol., 2021, 16, 1276–1287 CrossRef CAS PubMed.
- P. C. Ortet, S. N. Muellers, L. A. Viarengo-Baker, K. Streu, B. R. Szymczyna, A. B. Beeler, K. N. Allen and A. Whitty, J. Am. Chem. Soc., 2021, 143, 3779–3793 CrossRef CAS PubMed.
- R. Hancock, H. Bertrand, T. T. Sujita, S. Naz, A. El-Bakry, J. Laoruchupong, J. D. Hayes and G. Wells, Free Radical Biol. Med., 2012, 52, 444–451 CrossRef CAS PubMed.
- G. Wells, Biochem. Soc. Trans., 2015, 43, 674–679 CrossRef CAS PubMed.
- A. E. Owens, J. A. Iannuzzelli, Y. Gu and R. Fasan, ACS Cent. Sci., 2020, 6, 368–381 CrossRef CAS PubMed.
- Y. Wu, S. Fan, M. Dong, J. Li, C. Kong, J. Zhuang, X. Meng, S. Lu, Y. Zhao and C. Wu, Chem. Sci., 2022, 13, 7780 RSC.
- C. Zhuang, Z. Wu, C. Xing and Z. Niao, MedChemComm, 2017, 8, 286 RSC.
- T. G. Davies, W. E. Wixted, J. E. Coyle, C. Griffiths-Jones, K. Hearn, R. McMenamin, D. Norton, S. J. Rich, C. Richardson and G. Saxty, et al , J. Med. Chem., 2016, 59, 3991–4006 CrossRef CAS PubMed.
- D. Norton, W. G. Bonnette, J. F. Callahan, M. G. Carr, C. M. Griffiths-Jones, T. D. Heightman, J. K. Kerns, H. Nie, S. J. Rich, C. Richardson, W. Rumsey, Y. Sanchez, M. L. Verdonk, H. M. G. Willems, W. E. Wixted, L. Wolfe, A. J. A. Woolford, Z. Wu and T. G. Davies, J. Med. Chem., 2021, 64, 15949–15972 CrossRef CAS PubMed.
- N. Georgakopoulos, S. Talapatra, D. Dikovskaya, S. Dayalan Naidu, M. Higgins, J. Gatliff, A. Ayhan, R. Nikoloudaki, M. Schaap, K. Valko, F. Javid, A. T. Dinkova-Kostova, F. Kozielski and G. Wells, J. Med. Chem., 2022, 65, 7380–7398 CrossRef CAS PubMed.
- F. Begnini, S. Geschwindner, P. Johansson, L. Wissler, R. J. Lewis, E. Danelius, A. Luttens, P. Matricon, J. Carlsson, S. Lenders, B. König, A. Friedel, P. Sjö, S. Schiesser and J. Kihlberg, J. Med. Chem., 2022, 65, 3473–3517 CrossRef CAS PubMed.
- D. Narayanan, K. T. Tran, J. S. Pallesen, S. M. Ø. Solbak, Y. Qin, E. Mukminova, M. Luchini, K. O. Vasilyeva, D. González Chichón, G. Goutsiou, C. Poulsen, N. Haapanen, G. M. Popowicz, M. Sattler, D. Olagnier, M. Gajhede and A. Bach, J. Med. Chem., 2022, 65, 14481–14526 CrossRef CAS PubMed.
- K. Otake, M. Ubukata, N. Nagahashi, N. Ogawa, Y. Hantani, R. Hantani, T. Adachi, A. Nomura, K. Yamaguchi, M. Maekawa, H. Mamada, T. Motomura, M. Sato and K. Harada, ACS Med. Chem. Lett., 2023, 14, 658–665 CrossRef CAS PubMed.
- S. J. Walsh, S. Omarjee, W. R. J. D. Galloway, T. T.-L. Kwan, H. F. Sore, J. S. Parker, M. Hyvonen, J. S. Carroll and D. R. Spring, Chem. Sci., 2019, 10, 694–700 RSC.
- H. Maag, Prodrugs of Carboxylic Acids, Springer, pp. 703–729, DOI:10.1007/978-0-387-49785-3_20.
- J. Iegre, P. Brear, D. J. Baker, Y. S. Tan, E. L. Atkinson, H. Sore, D. H. O'Donovan, C. Verma, M. Hyvonen and D. R. Spring, Chem. Sci., 2019, 10, 5056–5063 RSC.
- P. Lönn, A. D. Kacsinta, X.-S. Cui, A. S. Hamil, M. Kaulich, K. Gohoi and S. F. Dowdy, Sci. Rep., 2016, 6, 32301 CrossRef PubMed.
- S. A. Ibrahim and S. K. Li, Pharm. Res., 2010, 27, 115–125 CrossRef CAS PubMed.
- G. Li Petri, S. Di Martino and M. De Rosa, J. Med. Chem., 2022, 65, 7438–7475 CrossRef CAS PubMed.
- S. Yoshida, S. Uehara, N. Kondo, Y. Takahashi, S. Yamamoto, A. Kameda, S. Kawagoe, N. Inoue, M. Yamada, N. Yoshimura and Y. Tachibana, J. Med. Chem., 2022, 65, 10655–10673 CrossRef CAS PubMed.
- M. Zhong, A. Lynch, S. N. Muellers, S. Jehle, L. Luo, D. R. Hall, R. Iwase, J. P. Carolan, M. Egbert and A. Wakefield, Biochem, 2020, 59, 563–581 CrossRef CAS PubMed.
- S. R. Trevino, S. Schaefer, J. M. Scholtz and C. N. Pace, J. Mol. Biol., 2007, 373, 211–218 CrossRef CAS PubMed.
- D. Ross and D. Siegel, Front. Physiol., 2017, 8, 595 CrossRef PubMed.
- D. Wang, L. Zou, Q. Jin, J. Hou, G. Ge and L. Yang, Acta Pharm. Sin. B, 2018, 8, 699–712 CrossRef PubMed.
- J. M. Veranth, N. S. Cutler, E. G. Kaser, C. A. Reilly and G. S. Yost, Toxicol. in Vitro, 2008, 22, 498–509 CrossRef CAS PubMed.
- F. Zhao and W. T. Klimecki, J. Appl. Toxicol., 2014, 35, 945–951 CrossRef PubMed.
- J. F. Lechner, A. Haugen, I. A. McClendon and A. M. Shamsuddin, Differentiation, 1984, 25, 1–3 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04083f |
| ‡ These authors contributed equally. |
| § In the context of this study we chose BEAS-2B cells as a representative model for epithelial cells. The presence of serum has been reported to affect BEAS-2B cell phenotype. Specifically, exposure of BEAS-2B to FBS has been associated with squamous differentiation, alteration in cytokine secretion and response to toxicants.42–44 Therefore, we did not use FBS in the cell assay media. |
| ¶ Synthesis of the ester bearing peptides and corresponding shorter analogues was not possible due to solubility issues. |
| This journal is © The Royal Society of Chemistry 2023 |