
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecord release of tetramethylguanidine using a green light activated photocage for rapid synthesis of soft materials†
Kun-You
Chung
,
Ain
Uddin
and
Zachariah A.
Page
 *
*
Department of Chemistry, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: zpage@utexas.edu
First published on 13th September 2023
Abstract
Photocages have enabled spatiotemporally governed organic materials synthesis with applications ranging from tissue engineering to soft robotics. However, the reliance on high energy UV light to drive an often inefficient uncaging process limits their utility. These hurdles are particularly evident for more reactive cargo, such as strong organobases, despite their attractive potential to catalyze a range of chemical transformations. Herein, two metal-free boron dipyrromethene (BODIPY) photocages bearing tetramethylguanidine (TMG) cargo are shown to induce rapid and efficient polymerizations upon exposure to a low intensity green LED. A suite of spectroscopic characterization tools were employed to identify the underlying uncaging and polymerization mechanisms, while also determining reaction quantum efficiencies. The results are directly compared to state-of-the-art TMG-bearing ortho-nitrobenzyl and coumainylmethyl photocages, finding that the present BODIPY derivatives enable step-growth polymerizations that are >10× faster than the next best performing photocage. As a final demonstration, the inherent multifunctionality of the present BODIPY platform in releasing radicals from one half of the molecule and TMG from the other is leveraged to prepare polymers with starkly disparate physical properties. The present findings are anticipated to enable new applications of photocages in both small-molecule photochemistry for medicine and advanced manufacturing of next generation soft materials.
Introduction
Photocages, or chromophores that release molecules (i.e., cargo) or unveil functionality upon exposure to light, have enabled a myriad of applications with precise spatiotemporal control, from drug delivery1,2 to materials fabrication.3–6 Unfortunately, driving this process often requires high energy ultraviolet (UV) light. This limits applicability in biological and materials contexts due to inherent phototoxicity and limited penetration depth. Thus, shifting to longer wavelengths of light as a stimulus to fuel uncaging has received growing attention. However, the scope of visible light reactive photocages, both in composition and function, remains narrow, particularly for the release of reactive cargo. This in-part arises from a low quantum efficiency of uncaging (Φun) and/or extinction coefficient (ε). Overcoming these challenges necessitates a systematic examination of structure–reactivity relationships for a given application, such as advanced manufacturing of polymers as described here.Strong organobases such as 1,1,3,3-tetramethylguanidine (TMG) have played versatile roles in materials science and synthetic chemistry,7,8 from catalysing thiol-Michael addition9–13 and lactone/epoxy ring-opening polymerizations4,14–17 to silyl/acetyl deprotection18 and lanthanide hydroalkoxylation.19 It is thus no surprise that spatiotemporal control over TMG delivery via photouncaging has received considerable attention.17,20 In this manner, photopolymerizations via uncaging of TMG provide an avenue to create soft materials that are inaccessible using traditional radical photoinitiators. For example, unlike contemporary photocurable (i.e., light-induced solidification) acrylic and thiol–ene resins that undergo free radical chain-growth kinetics, base-catalyzed reactions (e.g., thiol-Michael and ring-opening) undergo anionic step-growth kinetics to form polymer networks with fewer defects (i.e., greater uniformity).21 The resultant networks tend to have improved mechanical properties, such as modulus of toughness (i.e., strain-energy density), relative to compositionally analogous materials accessed via a chain-growth mechanism.21 Furthermore, polymers produced in a step-growth manner often contain heteroatoms within each repeat unit along the main-chain, providing opportunities for degradation or dynamic bond exchange towards recyclable and/or stimuli responsive (“smart”) plastics.22,23 Despite these attractive features, the implementation of photocages in photocurable resins remains scarce because of the typical requirement for high photocage concentrations and/or light intensities to achieve curing speeds relevant for coatings, adhesives, and 3D printing (∼seconds to minutes). Thus, efficient visible light driven photouncaging of TMG has immense potential in materials chemistry.
State-of-the-art photocages bearing TMG as the cargo that have been used to drive anionic step-growth polymerizations include those from the classic ortho-nitrobenzyl (oNB)10,13 scaffold and more recently coumarinylmethyl12 (Fig. 1). In the former, oNB-TMG derivatives examined for driving photopolymerizations include those from 6-nitroveratryl chloroformate (NVOC)13 and (3,4-methylenedioxy-6-nitrophenyl)-propyloxycarbonyl (MNPPOC)10 (Fig. 2A). Both undergo a spin-forbidden n–π* transition upon exposure to UV-violet light (∼300–420 nm), followed by heterolytic bond scission and decarboxylation to release TMG. Although these derivates provide high Φun values (∼5–10%), the reliance on UV light coupled with low extinction coefficients (ε < 5000 M−1 cm−1)10,13 reduces their suitability for materials synthesis due to pervasive absorption and scattering in this spectral region, leading to low penetration depth and/or photodamage. Additionally, despite attempts to red-shift absorption of oNB photocages (w/o TMG) via π-extension,24Φun values decrease (≤1%) and extinction coefficients in the visible region (∼450–500 nm) often remain low due to the reliance on non-bonding orbitals to facilitate excited-state uncaging.24,25 Thus, oNB-TMG appears restricted to efficient uncaging in the UV spectral region.
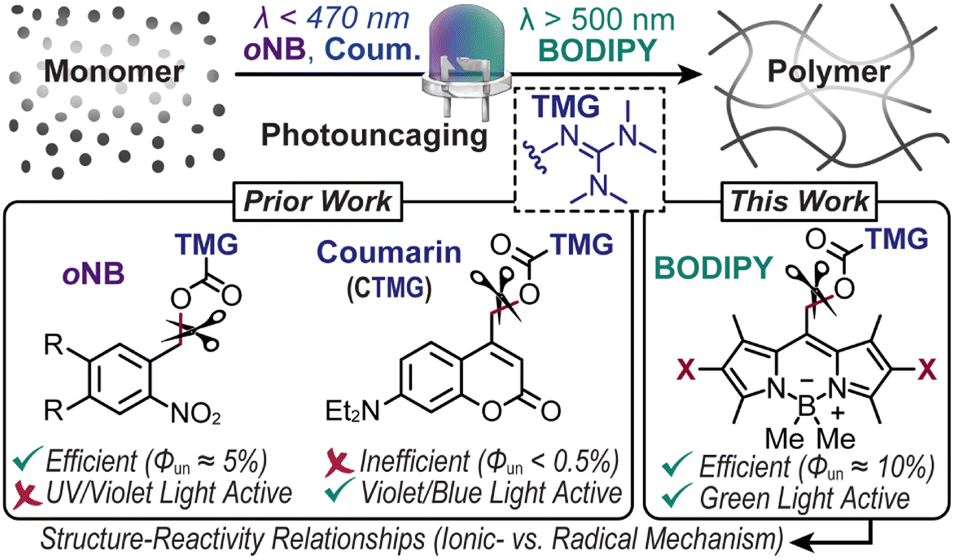 | ||
| Fig. 1 Overview of photocages bearing 1,1,3,3-tetramethylguanidine (TMG) cargo used to drive polymerizations upon exposure to UV or visible light. Prior work with ortho-nitrobenzyl (oNB) and coumarinylmethyl photocages provided either efficient photouncaging (Φun) or activation with visible (violet/blue) light, respectively. Present work with boron dipyrromethene (BODIPY)-TMG photocages increases Φun and extends the spectral activity further into the visible spectrum. | ||
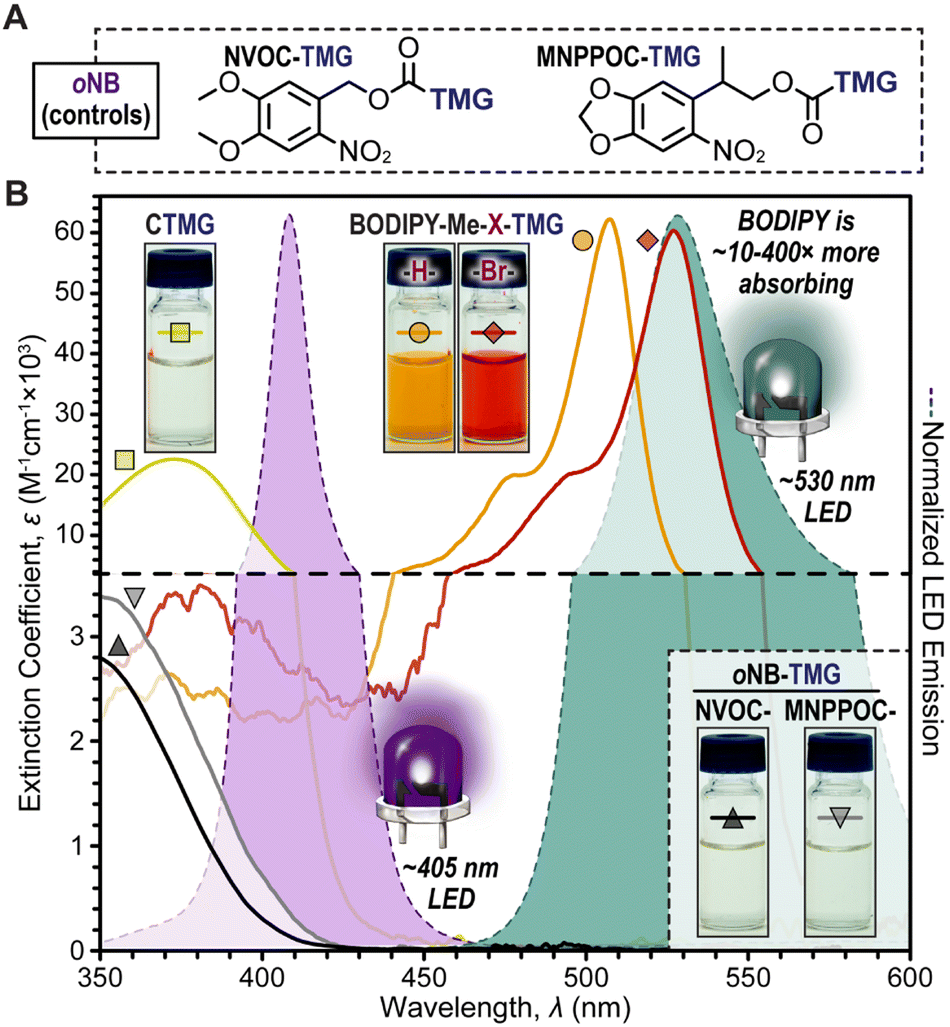 | ||
| Fig. 2 (A) Chemical structures for oNB-TMG photocages used in this study. (B) Spectral profiles for photocage absorption in dilute CH3CN (∼10−5 M) overlaid onto violet (405 nm) and green (530 nm) LED emission profiles. Insets are photographs of photocages in CH3CN (2 mM). | ||
Coumarinylmethyl-TMG (CTMG) represents a more recent alternative used in thiol-Michael addition and epoxy ring-opening polymerizations12,26–30 (Fig. 1). Unlike oNB, these photocages operate by an allowed π–π* transition, resulting in higher extinction coefficients (ε > 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) and the ability to red-shift absorption via π-extension. However, CTMG photocages typically suffer from low Φun (<1%),27,29,31 necessitating high light intensities and/or long exposure times to induce rapid photopolymerizations. For non-TMG cargo, impressive strides have been made to both red-shift absorption and increase Φun from coumarin-based photocages by, for example, introducing halogens (e.g., Br and I) to drive intersystem crossing to long-lived triplet excited states,32,33 and appending vinylic functionality to compete with deactivation pathways via intramolecular cyclization28,30,34 and stabilization of cationic intermediates.35 However, a challenge for many of these derivatives is their complex and thus costly syntheses. Despite this, we recently embarked on applying several of these strategies to the release of TMG to drive thiol-Michael polymerizations.20 Unfortunately, we found that they did not result in high Φun values (∼0.05–0.4%) upon exposure to visible light emitting diodes (LEDs) with peak emission wavelengths (λmax) of ∼405 nm (violet) or 470 nm (blue), and in many cases resulted in radical formation that caused unwanted ene–ene chain-propagation.
000 M−1 cm−1) and the ability to red-shift absorption via π-extension. However, CTMG photocages typically suffer from low Φun (<1%),27,29,31 necessitating high light intensities and/or long exposure times to induce rapid photopolymerizations. For non-TMG cargo, impressive strides have been made to both red-shift absorption and increase Φun from coumarin-based photocages by, for example, introducing halogens (e.g., Br and I) to drive intersystem crossing to long-lived triplet excited states,32,33 and appending vinylic functionality to compete with deactivation pathways via intramolecular cyclization28,30,34 and stabilization of cationic intermediates.35 However, a challenge for many of these derivatives is their complex and thus costly syntheses. Despite this, we recently embarked on applying several of these strategies to the release of TMG to drive thiol-Michael polymerizations.20 Unfortunately, we found that they did not result in high Φun values (∼0.05–0.4%) upon exposure to visible light emitting diodes (LEDs) with peak emission wavelengths (λmax) of ∼405 nm (violet) or 470 nm (blue), and in many cases resulted in radical formation that caused unwanted ene–ene chain-propagation.
Herein, we develop a set of novel boron dipyrromethene (BODIPY)-TMG photocages (Fig. 1) that were inspired by the work of Winter and co-workers,36 among others,5,6 and utilize them to drive anionic step-growth polymerizations with unprecedented speed upon exposure to a low intensity (40 mW cm−2) green LED (λmax = 530 nm). The BODIPY-TMG derivatives required only four to five scalable synthetic steps from inexpensive commercial starting materials, making them accessible and potentially useful in photocurable technologies. Additionally, the rate and mechanism of thiol–ene polymerizations (e.g., radical vs. anionic) were systematically examined and benchmarked against state-of-the-art oNB and CTMG photocages that were exposed to a violet LED (λmax = 405 nm). The result is a record polymerization rate (rp = 0.135 M s−1) that is more than an order of magnitude larger (∼14×) than the best performing benchmark. The unprecedented performance was attributed to a combination of high extinction coefficient (∼60000 M−1 cm−1) and Φun (>10%) and is anticipated to enable both fundamental and applied materials chemistry research.
Results and discussion
Two novel BODIPY photocages bearing TMG cargo were synthesized (Fig. 1) and compared with oNB photocages NVOC and MNPPOC (Fig. 2A) and CTMG (Fig. 1). Both BODIPY derivatives contain methyl (Me) groups off the boron, in-place of the more common fluorine atoms. This was selected to maximize Φun, which has been previously observed for BODIPY photocages bearing alternate cargo (e.g., halides and acetate).37 The nomenclature adopted herein is BODIPY-Z-X-R, where Z is F or Me, X is H or Br, and R is the meso-methyl cargo (e.g., TMG). Synthesis of the two desired BODIPY photocages (BODIPY-Me-X-TMG) followed a straightforward four or five step sequence for X being H or Br, respectively (Scheme S1†). In short, starting from commercially available 2,4-dimethyl-1H-pyrrole, condensation with 2-cholor-2-oxoethyl acetate followed by reaction with boron trifluoride etherate in the same pot provided BODIPY-F-H-OAc. Simultaneous deesterification and fluorine substitution of BODIPY-F-H-OAc was accomplished using methylmagnisium bromide, which provided BODIPY-Me-H-OH. Reacting the –OH functional group with 4-nitrophenyl chloroformate provided a good leaving group that facilitated substitution with TMG to directly provide BODIPY-Me-H-TMG, or BODIPY-Me-Br-TMGvia bromination first with N-bromosuccinimide, followed by TMG substitution.To quantify Φun we first characterized the UV-vis photocage absorption and LED emission spectra (Fig. 2B). Characteristic of halogenation was a red-shift in absorption, providing wavelength maxima at 507 nm (extinction coefficient = 61000 M−1 cm−1) and 527 nm (extinction coefficient = 59200 M−1 cm−1) for BODIPY-Me-H-TMG and -Br-TMG, respectively (Table S1†). We further quantified the relative number of photons absorbed (Φabs) for oNB-TMG and CTMG photocages under irradiation of a violet LED (405 nm) and BODIPY photocages under a green LED (530 nm), using an equivalent intensity (Table S2†). All derivatives were normalized to the strongest absorber being BODIPY-Me-Br-TMG with green light. This provided a Φabs value of 0.37 for BODIPY-Me-H-TMG. In other words, the halogenated photocage absorbed 2.7× more photons from the green LED relative to its non-halogenated counterpart. In comparison, CTMG (Φabs = 0.065), MNPPOC-TMG (Φabs = 0.0059) and NVOC-TMG (Φabs = 0.0025) had considerably less absorption with the violet LED, due to a combination of low extinction coefficients and partial overlap between the spectral profiles for photocage absorption and violet LED emission. Thus, BODIPY photocages absorbed anywhere from ∼6× more photons (BODIPY-Me-H-TMGvs. CTMG) to ∼400× more (BODIPY-Me-Br-TMGvs. NVOC-TMG). This in-turn provides a larger number of opportunities for photouncaging to occur (i.e., excitation events) and release TMG to catalyse thiol-Michael polymerizations.
The photouncaging mechanism for the present BODIPY derivatives was shown to go through a triplet excited-state manifold by using a combination of steady-state and transient absorption and fluorescence spectroscopies. These results are consistent with prior reports on alternate BODIPY photocages,37 and thus our results have been placed in the supporting information for brevity (Fig. S5–S11 and Table S1†). In short, fluorescence spectroscopy revealed low emission quantum yields of 0.37 and 0.092 for BODIPY-Me-H-TMG and -Br-TMG, respectively, while time-correlated single photon counting fluorescence spectroscopy provided short singlet excited state lifetimes of 1.5 and 0.6 ns, respectively. These results were rationalized by efficient intersystem crossing to the triplet excited state manifold, which outcompetes relaxation from the first singlet excited state to the ground state. This was confirmed by nanosecond transient absorption spectroscopy, which provided direct evidence for the formation of long-lived triplet excited states. Specifically, triplet lifetimes of 106 μs and 52 μs were measured for BODIPY-Me-H-TMG and -Br-TMG, respectively. These long excited-state lifetimes were deemed paramount to facilitate TMG photouncaging herein.
The TMG-photocages were next examined for their ability to drive thiol-Michael polymerizations upon exposure to violet (oNB and CTMG) and green (BODIPY) LEDs (Fig. 3). The selected resin comprised inexpensive pentaerythritol tetrakis(3-mercaptopropionate) (PETMP, tetrathiol) and tetra(ethylene glycol) diacrylate (TEGDA) with equimolar thiol (S–H) and ene (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) functionality (Fig. 3A). Initial experiments with BODIPY photocages were performed at a concentration of 0.067 mol%. The complete resin was then placed in a 100 μm gap (d) between glass slides, using shims to set d. Using real-time Fourier transform infrared (FTIR) spectroscopy, the disappearance of CC and S–H functionality were monitored at 3150 cm−1 and 2550 cm−1, respectively, upon irradiation with a low intensity green LED (10 mW cm−2). Integration of these IR bands provided conversion (ρ) values for CC and S–H functionality (Fig. 3B). Both resins containing BODIPY photocages showed excellent temporal control, with no functional group conversion prior to turning the LED ‘on’ (first 15 seconds of measurement), followed by a rapid increase in ρ immediately after irradiation began. To quantify reaction kinetics, the polymerization rates (rp) were determined using the slope shortly after turning the LED ‘on’, along with the time to reach ρ = 0.5 (t50) as a point of comparison (Fig. 3B, dashed line). With this characterization, BODIPY-Me-Br-TMG was found to have an rp that was ∼3× higher than -H-TMG. Specifically, the rp values from CC conversion were 3.10 ± 0.08 M s−1 and 1.03 ± 0.07 M s−1 for halogenated (-Br-) and non-halogenated (-H-) photocages, respectively (Table S3†). These high rates correspond to small times to 50% CC conversion – 0.5 and 2.1 seconds, respectively. However, while both CC and S–H had high rp values, there was an apparent disparity in maximum CC and S–H conversion (Δρ), where CC was ∼quantitative and S–H only reached ∼0.46.
C) functionality (Fig. 3A). Initial experiments with BODIPY photocages were performed at a concentration of 0.067 mol%. The complete resin was then placed in a 100 μm gap (d) between glass slides, using shims to set d. Using real-time Fourier transform infrared (FTIR) spectroscopy, the disappearance of CC and S–H functionality were monitored at 3150 cm−1 and 2550 cm−1, respectively, upon irradiation with a low intensity green LED (10 mW cm−2). Integration of these IR bands provided conversion (ρ) values for CC and S–H functionality (Fig. 3B). Both resins containing BODIPY photocages showed excellent temporal control, with no functional group conversion prior to turning the LED ‘on’ (first 15 seconds of measurement), followed by a rapid increase in ρ immediately after irradiation began. To quantify reaction kinetics, the polymerization rates (rp) were determined using the slope shortly after turning the LED ‘on’, along with the time to reach ρ = 0.5 (t50) as a point of comparison (Fig. 3B, dashed line). With this characterization, BODIPY-Me-Br-TMG was found to have an rp that was ∼3× higher than -H-TMG. Specifically, the rp values from CC conversion were 3.10 ± 0.08 M s−1 and 1.03 ± 0.07 M s−1 for halogenated (-Br-) and non-halogenated (-H-) photocages, respectively (Table S3†). These high rates correspond to small times to 50% CC conversion – 0.5 and 2.1 seconds, respectively. However, while both CC and S–H had high rp values, there was an apparent disparity in maximum CC and S–H conversion (Δρ), where CC was ∼quantitative and S–H only reached ∼0.46.
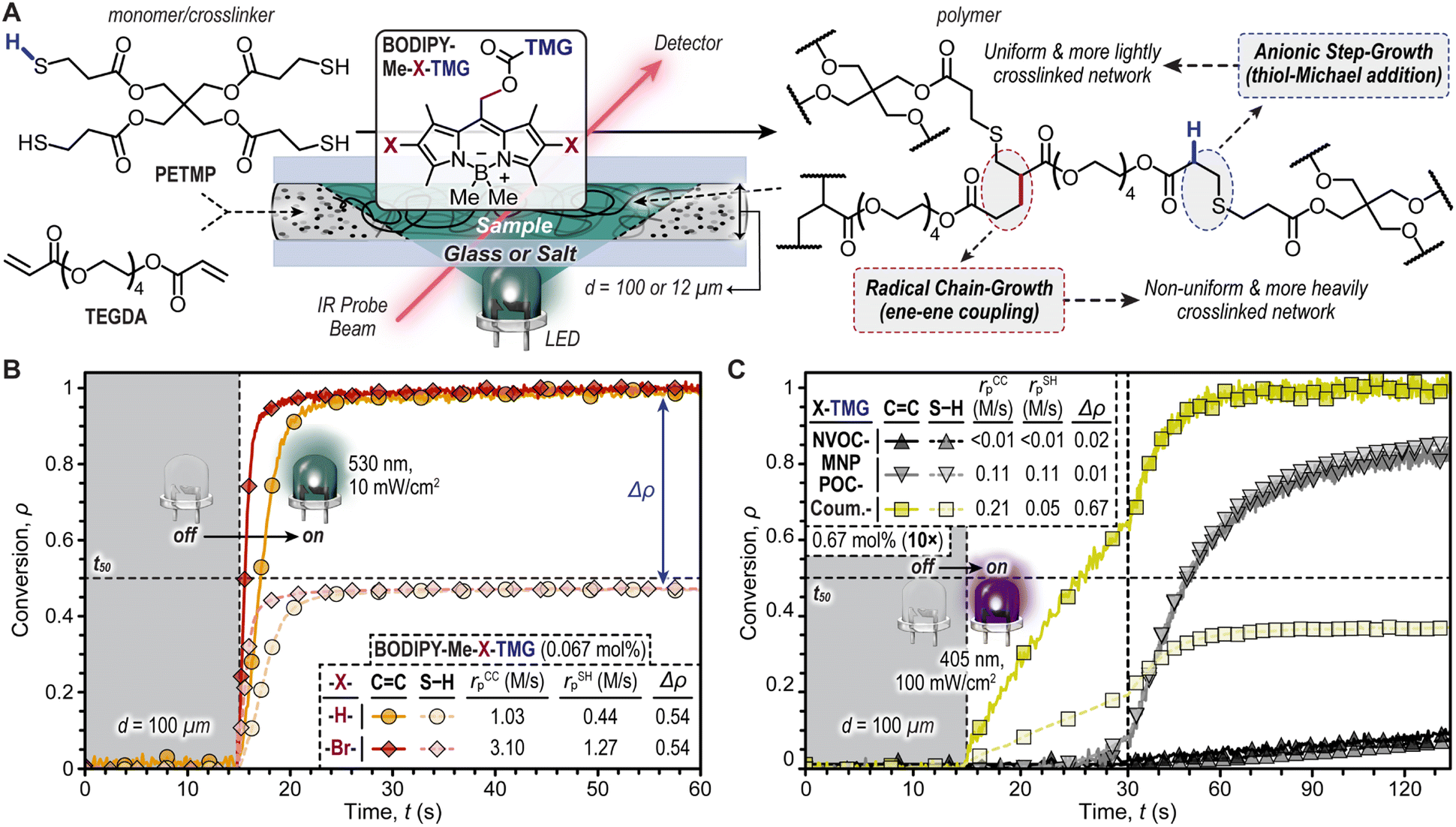 | ||
| Fig. 3 Thiol–ene photopolymerizations monitored using real-time Fourier transform infrared (FTIR) spectroscopy. (A) Resin composition with chemical structures for monomers/crosslinkers PETMP and TEGDA, illustration of transmission FTIR setup, and possible chemical structures for resultant polymer networks based on the reaction mechanism. (B) Photopolymerization kinetics provided as CC and S–H conversion (ρ) vs. time (t) for BODIPY photocages using a green LED (λmax = 530 nm) at an intensity of 10 mW cm−2 and (C) state-of-the-art oNB-TMG and CTMG photocages using a violet LED (λmax = 405 nm) at an intensity of 100 mW cm−2. Symbols: d, sample thickness; t50, time to reach 50% conversion (i.e., ρ = 0.5); rCCp, polymerization rate based on CC conversion; rSHp, polymerization rate based on S–H conversion; Δρ, difference in CC and S–H conversion. | ||
The large Δρ values of ∼0.54 for both BODIPY containing resins indicated the presence of a secondary reaction that consumed CC functionality without simultaneously consuming S–H functionality (Fig. 3B). This secondary reaction was attributed to radical-induced acrylate chain-propagation. The formation of initiating radicals likely arose from homolytic β-scission of the boron–methyl bond, which we recently reported.38 However, at this stage the relative contribution of radical- vs. base-promoted polymerization mechanisms (e.g., step-vs. chain-growth) was not clear, and thus necessitated further investigation.
To better understand the mechanics of photopolymerization when employing BODIPY-TMG photocages, we compared resins comprising them to those with previously reported oNB-TMG and CTMG photocages (Fig. 3C and Table S3†). However, upon attempting photopolymerizations it became evident that the reactivity of resins containing oNB-TMG and CTMG photocages was considerably lower relative to those with BODIPY-TMG. To surmount this issue, a 10× increase in photocage concentration (0.67 mol%) and light intensity (100 mW cm−2, 405 nm LED) was employed. Under these conditions, rp values (based on CC conversion) of 0.21 ± <0.01 M s−1, 0.11 ± <0.01 M s−1, and <0.01 M s−1 for CTMG, MNPPOC-TMG, and NVOC-TMG were respectively measured, with corresponding times to 50% conversion of 10, 34, and 132 seconds. Thus, polymerizations with oNB-TMG and CTMG derivatives were >5× slower than with BODIPY-TMG, despite the considerably higher concentration and light intensity used. However, while CTMG provided a Δρ value of 0.67, similar to BODIPY-TMG, the oNB-TMG photocages had matched CC and S–H functional group conversion (Δρ ≤ 0.02). This suggested that photopolymerization of resins containing oNB-TMG photocages resulted in purely base-promoted thiol-Michael polymerization (i.e., radical-free), which has the potential to provide uniform polymer networks. Nevertheless, the slow polymerization rates and requisite high LED intensities limits utility of oNB-TMG photocages in photocurable technologies.
Purely base-promoted thiol-Michael polymerizations with BODIPY-TMG photocages were achieved by incorporating 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), a radical scavenger, into the resin formulation (Fig. 4A). This aimed to curb the radical contribution and amplify base catalysis. The BODIPY-TMG photocages were directly compared with the fastest oNB-TMG derivative, MNPPOC-TMG, by holding the following factors constant: photocage concentration = 0.25 mol%, LED intensity = 40 mW cm−2, and d = 12 μm. Additionally, two equivalents of TEMPO (0.5 mol%) was found to reduce Δρ to ≤ 0.02 for the BODIPY-TMG photocages, without significantly altering Δρ for MNPPOC-TMG (Fig. S14 and Table S5†). The low Δρ values suggested that radicals had been effectively quenched for the resins containing BODIPY-TMG. Under these same conditions, BODIPY-Me-H-TMG was found to be ∼1.7× faster than the analogous -Br-TMG photocage, and ∼14× faster than MNPPOC-TMG, albeit significantly slower than the corresponding resins without TEMPO (Fig. 4B). Specifically, the rp values (CC conversion) with 0.5 mol% TEMPO were 0.135 ± 0.004 M s−1, 0.078 ± 0.002 M s−1, and 0.010 ± <0.001 M s−1, for resins containing BODIPY-Me-H-TMG and -Br-TMG (green LED) and MNPPOC-TMG (violet LED), respectively. Furthermore, corresponding times to 50% conversion of 25, 50, and 385 seconds were measured (Fig. S14†). The larger rp values for BODIPY-Me-H-TMG relative to -Br-TMG were hypothesized to arise from a higher Φun value (discussed later). Overall, BODIPY-Me-H-TMG in the presence of TEMPO was found to enable thiol-Michael polymerizations at a rate that was more than one order of magnitude larger relative to the state-of-the-art MNPPOC-TMG photocage, and additionally BODIPY uncaging enabled the use of a lower energy visible LED (i.e., green vs. violet).
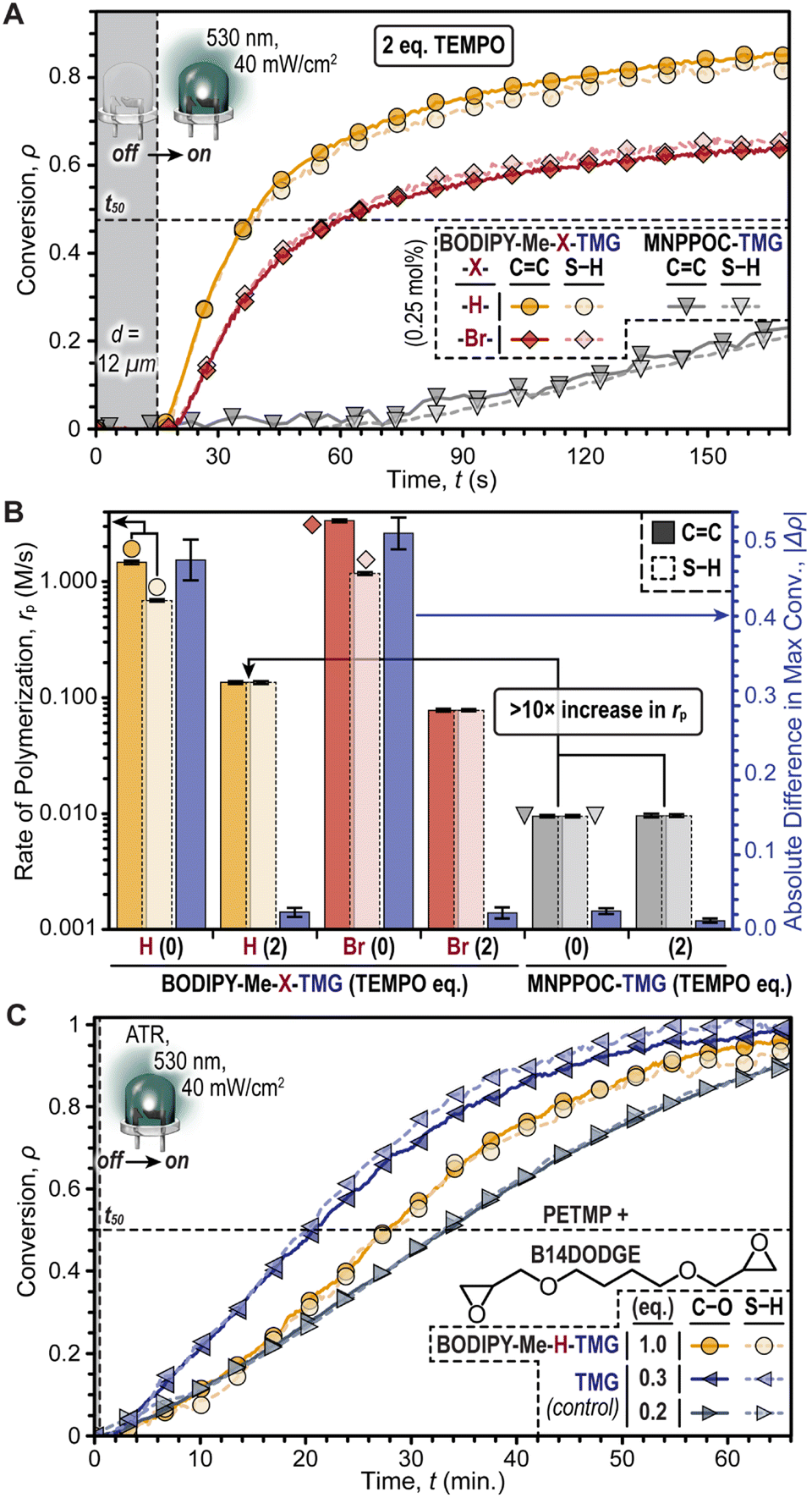 | ||
| Fig. 4 Anionic step-growth photopolymerizations catalysed by TMG. (A) Photopolymerization kinetics for PETMP (tetrathiol) and TEGDA (diacrylate) using BODIPY-Me-X-TMG or MNPPOC-TMG in the presence of two equivalents of TEMPO as a radical scavenger. (B) Bar graph providing the rate of polymerization (log-scale) and absolute difference in max conversion for resins with and without TEMPO. (C) Kinetics of anionic thiol-epoxy ring-opening polymerizations using BODIPY-Me-H-TMG and physical addition of TMG as control experiments. | ||
To further validate that the mechanism was anionic, we next examined the use of BODIPY-Me-H-TMG to induce a thiol-epoxy ring-opening polymerization, which would not proceed through a one-electron (radical) mechanism like the prior thiol–ene reactions (Fig. 4C). The resin comprised 1,4-butanediol diglycidyl ether (B14DODGE) together with the same tetrathiol used previously (PETMP) and the non-halogenated BODIPY photocage. An elevated concentration of BODIPY was used (2.6 mol%) owing to the inherently reduced reactivity of the thiol-epoxy reaction. Irradiation with a low intensity green LED (40 mW cm−2) while monitoring the disappearance of epoxy (∼920 cm−1) and thiol (∼2550 cm−1) functionality with real-time FTIR spectroscopy in an attenuated total reflectance (ATR) configuration provided a rp value of 0.095 ± 0.002 M min−1. To gain insight into the extent of uncaging, polymerizations of the same resin without a photocage were accomplished via direct addition of TMG. Varying the TMG concentration revealed similar polymerization kinetics for ∼0.52 mol% relative to those accomplished using 2.6 mol% BODIPY-Me-H-TMG. Specifically, TMG concentrations of 2.6 mol% (1 eq.), 0.78 mol% (0.3 eq.), 0.52 mol% (0.2 eq.), and 0.26 mol% (0.1 eq.) provided rp values of 0.319 ± 0.008 M s−1, 0.156 ± 0.004 M min−1, 0.082 ± 0.002 M min−1, and 0.008 ± <0.001 M min−1 (Fig. S15 and Table S6†). This result suggested that upon irradiation, BODIPY-Me-H-TMG released ∼0.2 equivalents of TMG, or in other words had a Φun of around ∼0.2.
Quantification of Φun was accomplished using real-time FTIR spectroscopy in an attenuated total reflectance (ATR) configuration and high-performance liquid chromatography coupled with a UV-vis absorption detector (HPLC-Abs) (Fig. 5A). In the former method, the BODIPY photocages (0.5 mol%) were dissolved in 3,6-dioxa-1,8-octanedithiol (DODT) to avoid overlapping IR absorption signals that arise from the previous multifunctional thiol, PETMP, and the disappearance of the C–N stretch from the carbamate on BODIPY was monitored at 1550 cm−1 during a tabulated green LED exposure (Fig. S16, S17 and Table S1†). From this, bond scission quantum yields were calculated and served as a proxy for Φun (i.e., assumes quantitative decarboxylation). As a result, values of 0.16 ± <0.01 and 0.09 ± <0.01 were obtained for BODIPY-Me-H-TMG and -Br-TMG, respectively (Fig. 5B). Notably, the higher Φun value for -H-TMG over -Br-TMG matches the previously discussed photopolymerization performance. The lower Φun value for -Br-TMG was hypothesized to arise from heavy-atom facilitated boron–methyl bond homolysis competing with meso-methyl bond heterolysis (i.e., uncaging). Moreover, bond homolysis preceding TMG uncaging could also generate BODIPY-TMG products that have lower Φun values relative to the parent methylated BODIPY compound. Parallel quantification of Φun was accomplished for MNPPOC-TMG, NVOC-TMG, and CTMG photocages, providing values of 0.09 ± <0.01, 0.06 ± <0.01, and <0.001, respectively (Fig. 5B, S18, and Table S1†). Thus, BODIPY-TMG photocages were found to have similar or better Φun values relative to oNB-TMG derivatives, and a >100× improvement over CTMG.
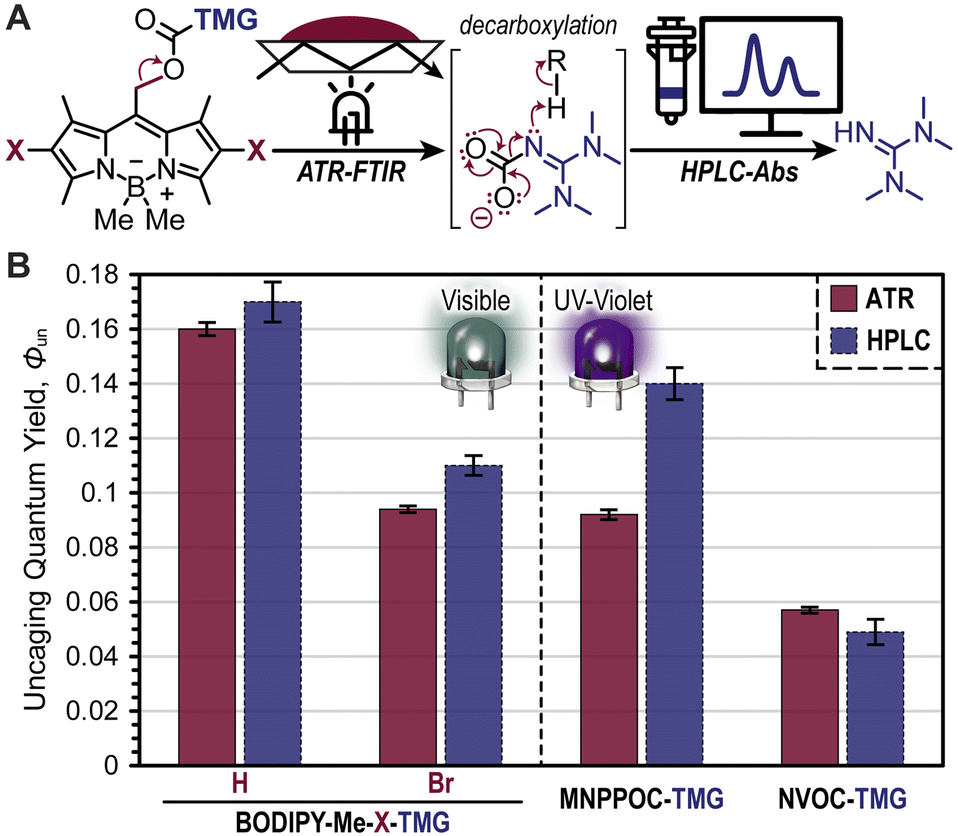 | ||
| Fig. 5 Quantification of TMG photouncaging. (A) Illustration showing the methods used to determine bond scission (ATR-FTIR) and TMG generation (HPLC-Abs). (B) Uncaging quantum yield (Φun) values for BODIPY- and oNB-TMG photocages. | ||
Given the large degree of bond scission for the BODIPY- and oNB-TMG photocages it was possible to directly quantify the amount of TMG released using HPLC with an absorbance detector (Fig. 5B). This was accomplished by dissolving BODIPY photocages in CH3CN:H2O (4:1) to avoid reacting TMG upon uncaging. A BODIPY concentration of 0.34 mM and irradiation pathlength of 10 mm was used to ensure complete photon absorption for ease of calculating Φun (eqn S2†). Upon a predefined dosage of light irradiation, the amount of TMG released was quantified from a TMG absorption calibration curve (Fig. S19†). From this analysis, comparable Φun values to those determined via real-time FTIR were observed. Specifically, Φun values were 0.17 ± <0.01, 0.11 ± <0.01, 0.14 ± <0.01, and 0.05 ± <0.01 for BODIPY-Me-H-TMG, BODIPY-Me-Br-TMG, MNPPOC-TMG, and NVOC-TMG, respectively (Fig. 5B). This coupled with the increased photon absorption provides a good rationale for the observed >10× increase in photopolymerization rates when employing BODIPY-TMG photocages relative to contemporary oNB-TMG derivatives.
To showcase the important implications of radical chain-growth vs. base-promoted step-growth mechanisms on the physical properties of photopolymerized thiol/acrylate resins we performed a rheological study (Fig. 6). The hypothesis was that using only bifunctional monomers, diacrylate (TEGDA) and dithiol (DODT), radical polymerizations (chain-growth) would result in crosslinked networks (i.e., thermoset), while polymerizations that proceed solely via iterative thiol-Michael additions (step-growth) would result in linear polymers (i.e., thermoplastic). This hypothesis was confirmed first qualitatively through a vial inversion test, where irradiation of a resin comprising BODIPY-Me-H-TMG (0.25 mol%) with a green LED (50 seconds, 40 mW cm−2) resulted in a material that did not flow (Fig. 6A). However, performing the same experiment with two equivalents of TEMPO added to the resin resulted in a flowing, viscous fluid. This behaviour was quantified using photorheology (Fig. 6B) by placing the same two resins in a 100 μm gap between parallel plates (oscillating top and transparent bottom plates) and also characterized using real time FTIR (Fig. S21†). The samples were irradiated with the green LED (17 mW cm−2) turned ‘on’ after 15 seconds, monitoring the storage (G′) and loss (G′′) moduli over time. These experiments revealed a stark disparity between resins without and with TEMPO present. Those without TEMPO had a crossover of storage and loss moduli ∼15 seconds after turning the LED ‘on’, which is indicative of gelation. These samples provided self-supporting films with storage moduli of ∼200 kPa. In contrast, irradiating resins without TEMPO ultimately provided a loss modulus in excess of storage modulus (G′′ > G′), which indicates that the material remained fluid (i.e., did not crosslink). Therefore, BODIPY-TMG photocages can be utilized to provide soft materials with a wide-range of physical properties using low intensity green LED illumination.
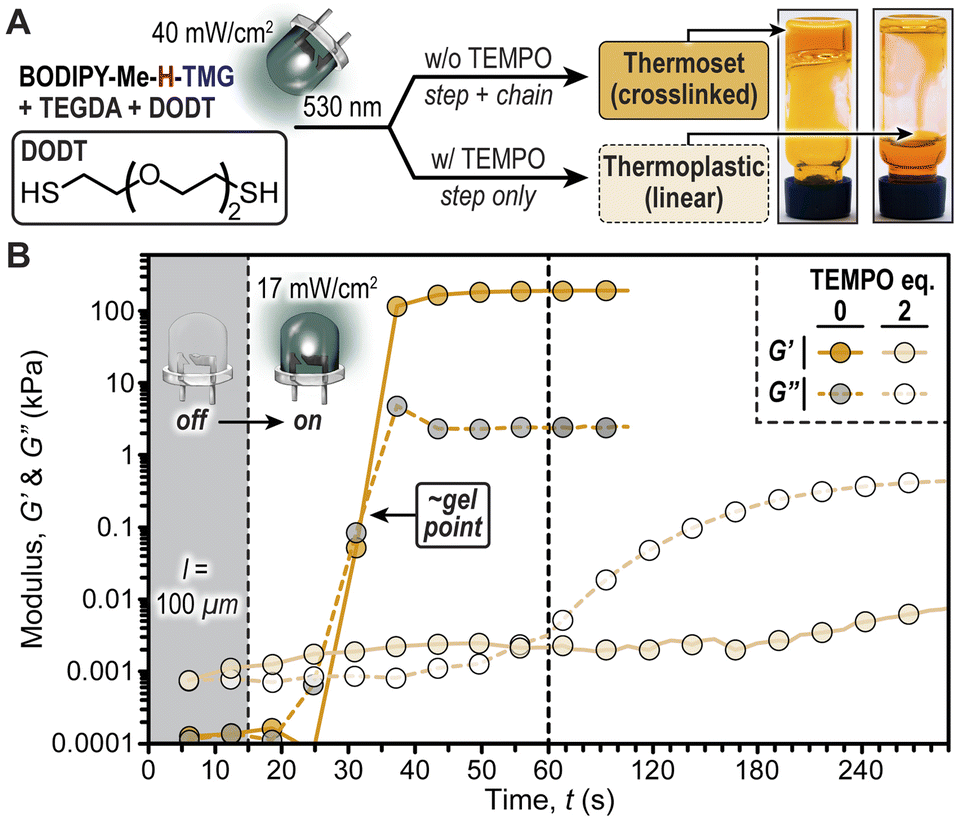 | ||
| Fig. 6 Influence of TEMPO on the physical properties of polymers that result from irradiating bifunctional thiol/acrylate resins containing BODIPY-Me-H-TMG. (A) Chemical structure for DODT, photopolymerization conditions, and images of resultant vial inversion test post-irradiation. (B) Rheological characterization showing how storage (G′) and loss (G′′) moduli change upon irradiation. | ||
Conclusions
In summary, two novel BODIPY photocages bearing TMG cargo and their utility in controlled soft matter synthesis was described. Relative to state-of-the-art TMG photocages (NVOC-TMG, MNPPOC-TMG, and CTMG) the present BODIPY derivatives not only enable the use of low energy green LEDs (λmax = 530 nm) as opposed to UV/violet (λmax ≤ 405 nm) LEDs, but also result in a ≥14× improvement in thiol-Michael photopolymerization rates under equivalent exposure intensities. This was attributed to a beneficial combination of high extinction coefficient (∼60000 M−1 cm−1) and Φun (≥0.1) values measured for the present BODIPY photocages. The incorporation of two equivalents of TEMPO relative to the BODIPY photocages effectively suppressed radical chain-propagation, and revealed that the more synthetically accessible BODIPY-Me-H-TMG compound was found to outperform its halogenated counterpart (BODIPY-Me-Br-TMG). Using the non-halogenated BODIPY photocage, thiol-epoxy anionic ring-opening polymerizations were demonstrated, along with the ability to dramatically alter physical properties of polysulfides from bifunctional thiol/acrylate resins. To the latter point, rapid gelation occurs for resins without TEMPO due to radical-induced crosslinking via acrylate chain-growth, while incorporation of TEMPO enables the formation of linear polymer chains via thiol-Michael step-growth. Looking ahead, further design and optimization of BODIPY-TMG photocages is anticipated to enable the use of even longer wavelengths of light and perform thiol-Michael photopolymerizations in water for biological purposes. Furthermore, the present derivatives have the potential to be used in rapid and efficient advanced manufacturing (e.g., 3D printing) of multifunctional materials that go beyond traditional acrylics, where the non-olefinic polymer backbones (e.g., thioethers) provide a potential handle for breakdown/recyclability. Such materials can be applied in a plethora of manners, including as scaffolds for tissue engineering and core components of soft robotics/electronics.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Conceptualization (KC, AU, ZAP); methodology (KC, AU, ZAP); investigation (KC, AU); visualization (ZAP); funding acquisition (ZAP); project administration (ZAP); supervision (ZAP); writing – original draft (KC, ZAP); writing – review & editing (KC, AU, ZAP).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge primary support by the National Science Foundation under Grant No. MSN-2107877 (KC, AU, ZAP; synthesis and characterization, supervision, and writing). Partial support was provided by the Robert A. Welch Foundation under Grant No. F-2007 and the Camille & Henry Dreyfus Foundation under Award No. TC-23-059 (ZAP; partial materials and supplies support). We acknowledge Prof. Sean Roberts and his research group for the use of a Magnitude Instruments enVISion spectrometer for nanosecond transient absorption measurements.References
- K. Sitkowska, M. F. Hoes, M. Lerch, L. Lameijer, P. van der Meer, W. Szymanski and B. L. Feringa, Chem. Commun., 2020, 56, 5480–5483 RSC.
- G. He, M. He, R. Wang, X. Li, H. Hu, D. Wang, Z. Wang, Y. Lu, N. Xu, J. Du, J. Fan, X. Peng and W. Sun, Angew. Chem., Int. Ed., 2023, e202218768 CAS.
- C. A. DeForest and K. S. Anseth, Angew. Chem., Int. Ed., 2012, 51, 1816–1819 CrossRef CAS PubMed.
- S. P. K. Kuroishi and A. P. Dove, Chem. Commun., 2018, 54, 6264–6267 RSC.
- M. Li, A. P. Dove and V. X. Truong, Angew. Chem., Int. Ed., 2020, 59, 2284–2288 CrossRef CAS PubMed.
- K. Y. Chung, K. N. Halwachs, P. Lu, K. Sun, H. A. Silva, A. M. Rosales and Z. A. Page, Cell Rep. Phys. Sci., 2022, 12, 101185 CrossRef PubMed.
- T. Şucu and M. P. Shaver, Polym. Chem., 2020, 11, 6397–6412 RSC.
- J. Xu, W. Liang, J. Zhang, Z. Dong and C. Lei, Eur. Polym. J., 2022, 179, 111526 CrossRef CAS.
- S. Chatani, T. Gong, B. A. Earle, M. Podgorski and C. N. Bowman, ACS Macro Lett., 2014, 3, 315–318 CrossRef CAS PubMed.
- X. Zhang, W. Xi, G. Gao, X. Wang, J. W. Stansbury and C. N. Bowman, ACS Macro Lett., 2018, 7, 852–857 CrossRef CAS PubMed.
- J. Sinha, M. Podgórski, A. Tomaschke, V. L. Ferguson and C. N. Bowman, Macromolecules, 2020, 53, 6331–6340 CrossRef CAS.
- X. Zhang, W. Xi, C. Wang, M. Podgórski and C. N. Bowman, ACS Macro Lett., 2016, 5, 229–233 CrossRef CAS PubMed.
- W. Xi, H. Peng, A. Aguirre-Soto, C. J. Kloxin, J. W. Stansbury and C. N. Bowman, Macromolecules, 2014, 47, 6159–6165 CrossRef CAS PubMed.
- X. Wang, Y. Liu, Z. Li, H. Wang, H. Gebru, S. Chen, H. Zhu, F. Wei and K. Guo, ACS Macro Lett., 2017, 6, 1331–1336 CrossRef CAS PubMed.
- B. A. Chan, S. Xuan, M. Horton and D. Zhang, Macromolecules, 2016, 49, 2002–2012 CrossRef CAS.
- D. Siefker, B. A. Chan, M. Zhang, J. W. Nho and D. Zhang, Macromolecules, 2022, 55, 2509–2516 CrossRef CAS PubMed.
- K. Li, Z. Li, Y. Shen, X. Fu, C. Chen and Z. Li, Polym. Chem., 2022, 13, 586–591 RSC.
- K. I. Oyama and T. Kondo, Org. Lett., 2003, 5, 209–212 CrossRef CAS PubMed.
- T. E. Janini, R. Rakosi, C. B. Durr, J. A. Bertke and S. D. Bunge, Dalton Trans., 2009, 10601–10608 RSC.
- M. T. Kiker, A. Uddin, L. M. Stevens, K.-Y. Chung, P. Lu and Z. A. Page, Polym. Chem., 2023, 14, 3843–3850 RSC.
- X. Zhao, X. Chen, H. Yuk, S. Lin, X. Liu and G. Parada, Chem. Rev., 2021, 121, 4309–4372 CrossRef CAS PubMed.
- V. X. Truong, J. Bachmann, A. N. Unterreiner, J. P. Blinco and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2022, 61, e202113076 CrossRef CAS PubMed.
- J. L. Pelloth, P. A. Tran, A. Walther, A. S. Goldmann, H. Frisch, V. X. Truong and C. Barner-Kowollik, Adv. Mater., 2021, 33, 2102184 CrossRef CAS PubMed.
- S. Boinapally, B. Huang, M. Abe, C. Katan, J. Noguchi, S. Watanabe, H. Kasai, B. Xue and T. Kobayashi, J. Org. Chem., 2014, 79, 7822–7830 CrossRef CAS PubMed.
- T. K. Bader, F. Xu, M. H. Hodny, D. A. Blank and M. D. Distefano, J. Org. Chem., 2020, 85, 1614–1625 CrossRef CAS PubMed.
- M. Bojtár, A. Kormos, K. Kis-Petik, M. Kellermayer and P. Kele, Org. Lett., 2019, 21, 9410–9414 CrossRef PubMed.
- C. Bao, G. Fan, Q. Lin, B. Li, S. Cheng, Q. Huang and L. Zhu, Org. Lett., 2012, 14, 572–575 CrossRef CAS PubMed.
- Q. Lin, L. Yang, Z. Wang, Y. Hua, D. Zhang, B. Bao, C. Bao, X. Gong and L. Zhu, Angew. Chem., Int. Ed., 2018, 57, 3722–3726 CrossRef CAS PubMed.
- L. Fournier, I. Aujard, T. Le Saux, S. Maurin, S. Beaupierre, J. B. Baudin and L. Jullien, Chem. - Eur. J., 2013, 19, 17494–17507 CrossRef CAS PubMed.
- J. Chaud, C. Morville, F. Bolze, D. Garnier, S. Chassaing, G. Blond and A. Specht, Org. Lett., 2021, 23, 7580–7585 CrossRef CAS PubMed.
- A. Gandioso, M. Palau, A. Nin-Hill, I. Melnyk, C. Rovira, S. Nonell, D. Velasco, J. García-Amorós and V. Marchán, ChemistryOpen, 2017, 6, 375–384 CrossRef CAS PubMed.
- T. Furuta, S. S-h Wang, J. L. Dantzker, T. M. Dore, W. J. Bybee, E. M. Callaway, W. Denk and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1193–1200 CrossRef CAS PubMed.
- X. J. Tang, Y. Wu, R. Zhao, X. Kou, Z. Dong, W. Zhou, Z. Zhang, W. Tan and X. Fang, Angew. Chem., Int. Ed., 2020, 59, 18386–18389 CrossRef CAS PubMed.
- R. Klimek, M. Asido, V. Hermanns, S. Junek, J. Wachtveitl and A. Heckel, Chem. - Eur. J., 2022, 28, e202200647 CrossRef CAS PubMed.
- A. M. Schulte, G. Alachouzos, W. Szymański and B. L. Feringa, J. Am. Chem. Soc., 2022, 144, 12421–12430 CrossRef CAS PubMed.
- T. Slanina, P. Shrestha, E. Palao, D. Kand, J. A. Peterson, A. S. Dutton, N. Rubinstein, R. Weinstain, A. H. Winter and P. Klán, J. Am. Chem. Soc., 2017, 139, 15168–15175 CrossRef CAS PubMed.
- P. Shrestha, D. Kand, R. Weinstain and A. H. Winter, J. Am. Chem. Soc., 2023, 145, 17497–17514 CrossRef CAS PubMed.
- K.-Y. Chung and Z. A. Page, J. Am. Chem. Soc., 2023, 145, 17912–17918 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, instrumentation, synthesis, photophysical and photochemical data, photopolymerization kinetics, uncaging yield characterization, and NMR and mass spectra. See DOI: https://doi.org/10.1039/d3sc04130a |
| This journal is © The Royal Society of Chemistry 2023 |