
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnprecedented sesterterpenoids, orientanoids A–C: discovery, bioinspired total synthesis and antitumor immunity†
Cheng-Yu
Zheng‡
ac,
Jin-Xin
Zhao‡
 a,
Chang-Hao
Yuan‡
acd,
Xia
Peng
a,
Meiyu
Geng
acd,
Jing
Ai
*ac,
Yao-Yue
Fan
*ac and
Jian-Min
Yue
*abc
a,
Chang-Hao
Yuan‡
acd,
Xia
Peng
a,
Meiyu
Geng
acd,
Jing
Ai
*ac,
Yao-Yue
Fan
*ac and
Jian-Min
Yue
*abc
aState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. E-mail: jai@simm.ac.cn; s040500290@126.com; jmyue@simm.ac.cn
bResearch Units of Discovery of New Drug Lead Molecules, Chinese Academy of Medical Sciences, Shanghai 201203, China
cUniversity of Chinese Academy of Science, No. 19A Yuquan Road, Beijing 100049, China
dSchool of Pharmaceutical Science and Technology, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, China
First published on 6th November 2023
Abstract
Sesterterpenoids are a very rare class of important natural products. Three new skeletal spiro sesterterpenoids, named orientanoids A–C (1–3), were isolated from Hedyosmum orientale. Their structures were determined by a combination of spectroscopic data, X-ray crystallography, and total synthesis. To obtain adequate materials for biological research, the bioinspired total syntheses of 1–3 were effectively achieved in 7–8 steps in overall yields of 2.3–6.4% from the commercially available santonin without using any protecting groups. In addition, this work also revised the stereochemistry of hedyosumins B (6) and C (10) as 11R-configuration. Tumor-associated macrophages (TAMs) have emerged as important therapeutic targets in cancer therapy. The in-depth biological evaluation revealed that these sesterterpenoids antagonized the protumoral and immunosuppressive functional phenotype of macrophages in vitro. Among them, the most potent and major compound 1 inhibited protumoral M2-like macrophages and activated cytotoxic CD8+ T cells, and consequently inhibited tumor growth in vivo.
Introduction
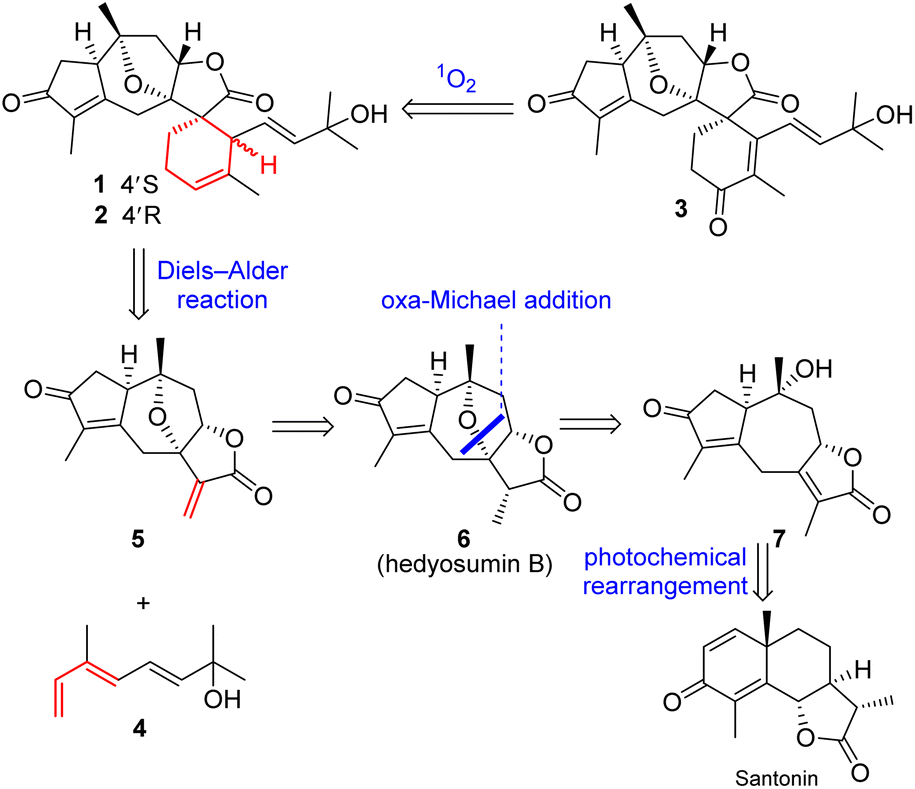
Natural products (NPs) are a valuable source for the development of drugs that are of particularly high importance to modulate inflammation and immunity,1,2 and some of the bioactive NPs often serve as handy tools for chemical biology to probe unknown biological processes and/or to identify drug targets.3,4 Terpenoids (C5n skeletons) are the largest compound class of NPs, and account for over one-third (ca. 110![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) of the total number of NPs identified hitherto.5–7 Meanwhile, sesterterpenoids (C25) are the smallest group (ca. 1000) among the terpenoid family, of which the vast majority have marine origins, and only very limited numbers have been reported from terrestrial plants.8–13 The fascinating structures and significant bioactivities of sesterterpenoids have been attracting broad interest from many associated scientific communities.14–19 Our preliminary studies on Hedyosmum orientale Merr. et Chun led to the isolation of three guaianolides, a guaiane-type sesquiterpenoid dimer, and a eudesmane–guaiane heterodimeric sesquiterpenoid.20–22 As part of our continuing efforts for the discovery of structurally unique and biologically potent natural products from medicinal plants,23–25 three sesterterpenoids, orientanoids A–C (1–3), were further identified from H. orientale. Compounds 1–3 possessed an unprecedented spiro carbon skeleton by incorporating the main components of a decahydro-4,7-epoxyazulene and a contiguous 2-oxaspiro[4.5]decane. Biosynthetically, compounds 1–3 could be derived from a monoterpenoid diene 4 and the coexisting guaiane sesquiterpenoid, hedyosumin A (5) through an intermolecular Diels–Alder reaction. To mimic the biosynthetic proposal and acquire sufficient amounts of samples for further biological study,26,27 the bioinspired total syntheses of compounds 1–3 were thus achieved in a very concise and elegant manner without using any protecting groups.
000) of the total number of NPs identified hitherto.5–7 Meanwhile, sesterterpenoids (C25) are the smallest group (ca. 1000) among the terpenoid family, of which the vast majority have marine origins, and only very limited numbers have been reported from terrestrial plants.8–13 The fascinating structures and significant bioactivities of sesterterpenoids have been attracting broad interest from many associated scientific communities.14–19 Our preliminary studies on Hedyosmum orientale Merr. et Chun led to the isolation of three guaianolides, a guaiane-type sesquiterpenoid dimer, and a eudesmane–guaiane heterodimeric sesquiterpenoid.20–22 As part of our continuing efforts for the discovery of structurally unique and biologically potent natural products from medicinal plants,23–25 three sesterterpenoids, orientanoids A–C (1–3), were further identified from H. orientale. Compounds 1–3 possessed an unprecedented spiro carbon skeleton by incorporating the main components of a decahydro-4,7-epoxyazulene and a contiguous 2-oxaspiro[4.5]decane. Biosynthetically, compounds 1–3 could be derived from a monoterpenoid diene 4 and the coexisting guaiane sesquiterpenoid, hedyosumin A (5) through an intermolecular Diels–Alder reaction. To mimic the biosynthetic proposal and acquire sufficient amounts of samples for further biological study,26,27 the bioinspired total syntheses of compounds 1–3 were thus achieved in a very concise and elegant manner without using any protecting groups.
Guaiane sesquiterpenoids, such as englerins with nanomolar anticancer activity, are an important class of biological natural products and have attracted broad interest from both chemistry and biology communities.28,29 As the pivotal Diels–Alder precursor, compound 5 has been effectively synthesized in 13 or 14 steps.30,31 However, a more concise synthesis of 5 remains a challenge. In the present study, a bioinspired synthesis of 5 was achieved in 6 steps from santonin involving a photochemical rearrangement and a subsequent intramolecular oxa-Michael reaction as the key steps. During the synthesis course, its two biosynthetic upstream guaiane sesquiterpenoids, hedyosumins B (6) and C (10),20 were also obtained and structurally revised as their C-11 epimers.
TAMs have been demonstrated to suppress the recruitment and function of cytotoxic T cells, promote angiogenesis, and affect multiple other aspects of tumor immunity, which together generate an immunosuppressive tumor microenvironment (TME) that finally promotes tumor growth and metastasis as well as mediating resistance to anticancer therapy.32 Generally, in contrast to the anti-tumor M1-like macrophages, the pro-tumor macrophages appear as M2-like polarized ones.33 It was observed that the T cell exhaustion was associated with the M2-like macrophages in the immune-rich TME.34 Thus, TAM-centric therapy has emerged as an important strategy for cancer treatment clinically.35,36 In a TAM-centric anticancer drug screening project, natural compounds 1–3 and their synthetic analogs showed inhibition toward the protumoral and immunosuppressive phenotype of macrophages in vitro. Furthermore, in the TAM-rich immunocompetent murine tumor models, the most potent and major compound 1 exhibited significant activities to remodel the TME by inhibiting protumoral M2-like macrophage infiltration and increasing activated CD8+ T cell infiltration, and consequently inhibited tumor growth in vivo.
Herein, we present a holistic study of compounds 1–3 and/or their synthetic analogs that spans the isolation, structure elucidation, total syntheses, and biological evaluation.
Results and discussion
Isolation, structure elucidation and plausible biosynthetic consideration of 1–3
An EtOH extract prepared from the powder of the twigs and leaves of H. orientale was suspended in water and partitioned with EtOAc. Sequential separations of the EtOAc extract by column chromatography and semi-preparative HPLC purification afforded compounds 1–3.Orientanoid A (1), colorless crystals, has a molecular formula of C25H32O5 with 10 double-bond equivalents (DBEs) as deduced from the HRESIMS ion at m/z 435.2145 [M + Na]+ (calcd for C25H32O5Na, 435.2147) and its 13C NMR data (Table S1, ESI†). The IR spectrum displayed absorption bands for hydroxy (3470 cm−1), carbonyl (1768 cm−1), and olefin (1655 cm−1) functionalities. The 1H NMR data (Table S1, ESI†) showed typical proton resonances for five methyls and three olefinic methines. With the aid of DEPT and HSQC experiments, 25 carbon resonances observed in the 13C NMR spectrum were distinguished as two carbonyls, three double bonds, five methyls, five sp3 methylenes, three sp3 methines, and four sp3 non-proton-bearing carbons. The two carbonyls and three double bonds accounted for 5 DBEs, and the remaining ones thus required compound 1 being pentacyclic.
The 2D structure of 1 was mainly delineated by 2D NMR interpretation (Fig. S1, ESI†). Four proton-bearing fragments as drawn in bold bonds were defined by the 1H–1H COSY spectrum, and were then assembled with the non-proton-bearing carbons and oxygen atoms to furnish the scaffold of 1 by the HMBC spectrum. Specifically, the HMBC correlation networks of H-2 and H3-14/C-3, C-4, and C-5; H-6/C-1, C-4, C-8, and C-7; H-8/C-12; H-13/C-11 and C-12; and H3-15/C-1, C-9, and C-10, as well as the 1H–1H COSY correlations of H-1/H-2 and H-8/H-9, enabled the establishment of a guaiane-type sesquiterpenoid framework. An oxo-bridge between C-7 (δC 91.3) and C-10 (δC 86.7) was recognized by their chemical shifts to form a unique 8-oxabicyclo[3.2.1]octane moiety. The HMBC cross-peaks of H-13/C-4′; H-4′/C-7; and H-10′/C-2′, C-3′, and C-4′, along with the 1H–1H COSY cross-peaks of H-13/H-1′/H-2′ forged the six-membered E ring, which fused with the D-ring via the C-11 to shape a novel oxaspiro[4.5]decane system. The HMBC correlations of H3-8′ (9′)/C-6′ and C-7′ (δC 71.1), as well as the 1H–1H COSY correlations of H-4′/H-5′/H-6′, constructed the C-4′ appendage of a 2-methylbut-3-en-2-ol moiety. Thus, compound 1 was elucidated to be a unique 5/6/5/5/6-pentacyclic spiro sesterterpenoid.
The relative configuration of 1 was primarily assigned using the NOESY spectrum (Fig. S1, ESI†). The NOESY correlations of H3-15 with H-1 and H-9α revealed that these protons were cofacial, and were randomly assigned to be α-oriented. Subsequently, the NOESY correlations of H-6β with H-8 and H3-14, as well as H-8 with H-9β indicated that they were β-oriented. The vital NOESY cross-peaks of H-6β with H-5′, and H-4′ with H-13, H-6′, and H3-10′ fixed the orientation of the spiro E ring, as well as an E-geometry for the Δ5′ double bond that was consistent with the large coupling constant (J5′,6′ = 15.1 Hz). The absolute configuration of 1 was finally determined to be 1R, 7S, 8S, 10R, 11S, 4′S, 5′E [absolute structure parameter: 0.10(7); CCDC 2002619]37 by X-ray crystallography study with Cu Kα radiation (Fig. 1).
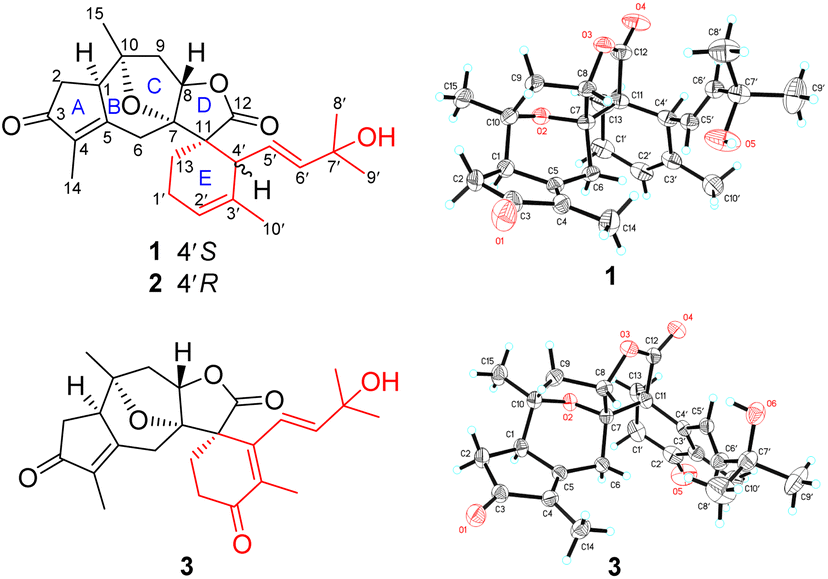 | ||
| Fig. 1 The structures of compounds 1–3. | ||
Orientanoid B (2), an amorphous white powder, shared the same molecular formula (C25H32O5) as 1 based on the HRESIMS ion at m/z 435.2157 [M + Na]+ (calcd 435.2147) and its 13C NMR data (Table S1, ESI†), suggestive of isomeric analogs. The 1D NMR data of compounds 1 and 2 (Table S1, ESI†) showed high similarity with the major differences evident in the chemical shifts of C-13, C-4′, C-5′, and C-6′. Further analysis of the 1H–1H COSY and HMBC spectra of 2 (Fig. S2a, ESI†) indicated that its 2D structure was identical to that of 1. Examination of the NOESY spectrum (Fig. S2b, ESI†) and the key coupling constants revealed that the stereochemistry of A–D rings and the E-geometry of the Δ5′ double bond (J5′,6′ = 15.4 Hz) in 2 were retained as those of 1, and the only variation was likely the C-4′ configuration. This was validated by the key NOESY correlations of H-4′ with H-8 and H-6β, as well as H-13 with H-5′. Compared with compound 1, the chemical shift of C-13 in 2 was severely resonated upfield (ΔδC −6.4 ppm) due to the γ-gauche effects from the Δ5′ double bond (Fig. 2a). The absolute configuration of 2 was then assigned as 1R, 7S, 8S, 10R, 11S, 4′R, 5′E from the roughly matched experimental and calculated ECD curves (Fig. 2b).
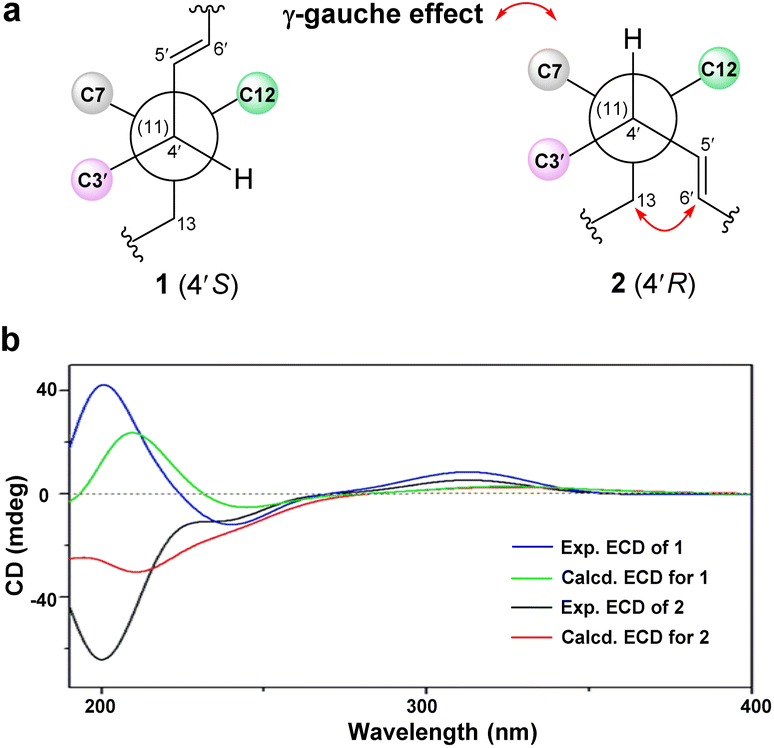 | ||
| Fig. 2 (a) Newman projections along the C4′/C11 bond for 1 and 2. (b) Experimental and calculated ECD spectra of compounds 1 and 2. | ||
Orientanoid C (3), colorless crystals, has a molecular formula of C25H30O6 with 11 DBEs based on the HRESIMS ion at m/z 471.2018 [M + HCO2]− (calcd 471.2019) and its 13C NMR data (Table S1, ESI†), which is one oxygen atom more and two hydrogen atoms less than that of 1, suggesting it is likely an oxidative analog of 1. This was supported by their similar NMR data, and in particular, the 13C NMR spectrum of 3 showed characteristic resonances for three carbonyls (one ester and two ketones) and three double bonds (one disubstituted and two persubstituted). Specifically, the additional ketone carbonyl (δC 199.5) was assigned to C-2′ by the vital HMBC correlations (Fig. S3a, ESI†) from H2-13 and H3-10′ to C-2′. A Δ3′ double bond was further located by the HMBC correlations from H3-10′ to C-3′ (δC 136.7) and C-4′ (δC 148.2), as well as from H2-13 to C-4′. Comprehensive 2D NMR data analysis (Fig. S3a, ESI†) showed that the A–D rings and the C-4′ appendage of 3 remained the same as those of 1 and 2, and the only discrepancy was that the E ring of 3 was modified to a cyclohex-2-en-1-one. The NOESY correlations of H-1 with H-6α and H3-15; H-6β with H-8 and H3-14; H-8 with H-9β and H-5′; H3-14 with H-10′; and H3-15 with H-2α and H-9α (Fig. S3b, ESI†), as well as the key coupling constant (J5′,6′ = 16.1 Hz) allowed the establishment of its relative configuration as depicted. The absolute configuration of 3 was finally determined to be 1R, 7S, 8S, 10R, 11S, 5′E [absolute structure parameter: −0.08(9); CCDC 2002618] by X-ray crystallographic study with Cu Kα radiation (Fig. 1).
Biosynthetically, the origins of sesterterpenoids 1–3 could be readily tracked back to the assembly of a coexisting major guaiane-type sesquiterpenoid, hedyosumin A (5), with a monoterpenoid (4) via an intermolecular Diels–Alder reaction (Scheme 1). The biosynthesis of guaiane-type sesquiterpenoids, e.g. hedyosumin A, was proposed to originate from the farnesyl cation via a pivotal monocyclic sesquiterpenoid intermediate i. The intermediate ivia the path A of an acidic catalyzed annulation produced the guaiyl cation, which then underwent a cascade of oxidative modifications to yield hedyosumin A. Meanwhile, the intermediate ivia an alternative path B of cyclization could generate the eudesmyl cation, which would finally produce the famous molecule of NPs, santonin.38 This indicated that guaiane-type and eudesmane-type sesquiterpenoids have inherent connections biosynthetically, suggesting that they might be mutually transformed conditionally. This was supported by a successfully photochemical rearrangement from santonin to the guaiane-type sesquiterpenoid framework.39–41
 | ||
| Scheme 1 Plausible biosynthetic consideration of 1–3. | ||
Chemical synthesis of 1–3
The scarce sample amounts (1.9–3.5 mg) of compounds 1–3 have unfortunately hampered deeper investigations on these unique spirocyclic scaffolds. In order to obtain sufficient materials for further bioactivity studies, the chemical syntheses of 1–3 were implemented.Inspired by the proposed biosynthetic pathway, the retrosynthetic stream of thought toward 1–3 (Scheme 2) materialized as follows: (1) an intermolecular Diels–Alder reaction between hedyosumin A (5) and a monoterpenoid (4) was intended to directly construct compounds 1 and 2; (2) compound 3 was envisioned to be descended from 1 and/or 2via a sequential singlet oxygen ene reaction and dehydration; (3) a thought-provoking transformation from the commercially available material santonin to the crucial intermediate hedyosumin A (5) was deliberated through several steps, in which compound 5 could be formed by a challenging direct α,β-dehydrogenation of 6 that could be forged by a intramolecular oxa-Michael addition from 7.
 | ||
| Scheme 2 Retrosynthetic analysis of 1–3. | ||
The total synthesis commenced with the construction of hedyosumin A (5), and a six-step efficient approach to 5 in an enantioselective manner was thus developed (Scheme 3). First, the known compound 8 was made from santonin in one step according to the reported procedure.42 Next, the allylic hydroxylation of enone 8 proved challenging. After screening various reaction conditions (Table S15, ESI†), enone 8 was successfully hydroxylated to provide 9 in 30% yield by employing the enone oxidation method developed by us quite recently.43 The structure of 9 was confirmed by single crystal X-ray diffraction [absolute structure parameter: −0.07(9); CCDC 2216319]. Having secured compound 9, the photochemical rearrangement/lactonization/alkene migration cascade reaction40 was achieved to give the desired 7 in 90% yield by irradiating a solution of 9 in AcOH/H2O,44 with 365 nm LEDs (50 W). Then our effort was focused on the installation of the 7,10-epoxy bridge to produce hedyosumin B (6) via the intramolecular oxa-Michael addition. After a brief survey of the reaction conditions (Table S17, ESI†), we were delighted to find that upon treatment with NaHCO3 in dioxane/H2O at room temperature, compound 7 smoothly underwent oxa-Michael addition to afford product 6 as a single diastereomer in 72% yield. The stereospecificity is attributable to the strong stereochemical bias of the substrate. The absolute structure of 6 was determined as depicted by single crystal X-ray diffraction [absolute structure parameter: −0.05(6); CCDC 2002637], which is actually the C-11 epimer of hedyosumin B. However, its spectroscopic data and physical constants well matched those of the latter, and allowed the revision of the structure of hedyosumin B as 6. With the key intermediate 6 in hand, we carried out direct oxidative dehydrogenation of 6 under various conditions with different oxidants, such as DDQ and IBX, aiming to generate the exocyclic double bond of 5, which was proved to be unsuccessful and the starting material was kept unreacted in all the cases. As an alternative, the reduction of 6 with NaBH4 in MeOH provided an epimeric mixture of compound 10 and its C-3 epimer 11 (10:1) in 95% combined yield. The absolute structures of 10 and 11 were confirmed by single crystal X-ray diffraction [10: absolute structure parameter: −0.08(11); CCDC 2022064. 11: absolute structure parameter: 0.03(4); CCDC 2022065]. Similarly, the structure of hedyosumin C was revised to be 10 by comparing the spectroscopic data of the NP and the synthetic counterpart, as well as the correlation of this chemical transformation. To our delight, a very effective dehydrogenation of 10 and its C-3 epimer was exploited to yield dienophile 5 in a one-pot reaction and protecting-group-free manner. Briefly, the mixture of 10 and its C-3 epimer was treated with LDA in THF, and the resultant enolates were then reacted with PhSeBr to give the corresponding mixture of selenylated lactones, which were finally subjected to a Dess–Martin oxidation and subsequent m-CPBA-mediated oxidative elimination45 to give compound 5 in 83% yield in one pot. Compound 5 was verified to be hedyosumin A by X-ray diffraction analysis [absolute structure parameter: 0.09(8); CCDC 2002638].
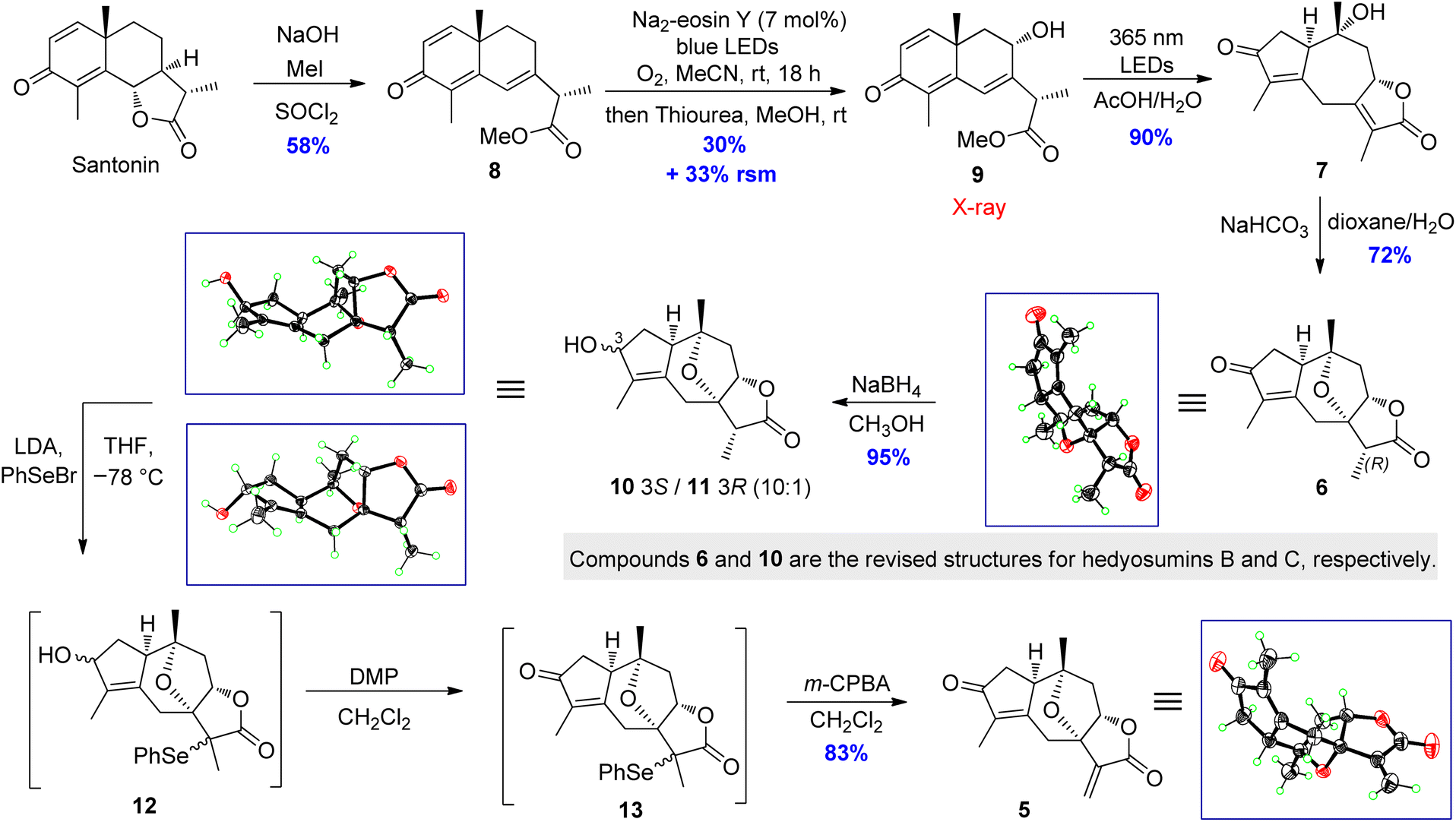 | ||
| Scheme 3 Synthesis of key intermediate hedyosumin A (5). LED = light-emitting diode; RSM = recovered starting material; LDA = lithium diisopropylamide; Ph = phenyl; DMP = Dess–Martin periodinane; m-CPBA = meta-chloroperoxybenzoic acid. | ||
The monoterpenoid, octa-3(E),5(E)-trien-2-ol (4), was synthesized from the commercially available ethyl (E)-3-methyl-4-oxo-2-butenoate in 6 steps according to the literature procedures.46,47 With both diene 4 and dienophile 5 in hand, we next explored the intermolecular Diels–Alder reaction (Scheme 4). After several attempts, the desired products of orientanoids A (1, Si-exo) and B (2, Si-endo), as well as two undesired minor compounds 14 (Re-exo) and 15 (Re-endo) were obtained in 81% combined yield by heating the mixture of compounds 4 and 5 in a neat form at 80 °C for 18 h without any catalysts. In this operation, the neat mixture of 4 and 5 was well managed in thin film and foiled to the reaction flask. This is a regio-specific Diels–Alder reaction, in which four expected products as mentioned above were produced and no other positional isomers were detected. The structures of the synthetic orientanoid A and 14 were confirmed by X-ray diffraction analysis [1: absolute structure parameter: −0.03(10); CCDC 2002639. 14: absolute structure parameter: 0.08(7); CCDC 2181591], and the structures with absolute configurations of the synthetic compounds 2 and 15 were assigned by extensive spectroscopic analysis and ECD calculation (Fig. S5 and S6, ESI†). With synthetic minor isomers 14 and 15 in hand, we performed LC-MS analysis of crude extracts of the title plant. Based on the LC-MS analysis reports (pages 106–111 in the ESI†), we propose that compounds 14 and 15 may also be natural products, which were likely lost in the processes of isolation and/or purification due to their much lower concentrations.
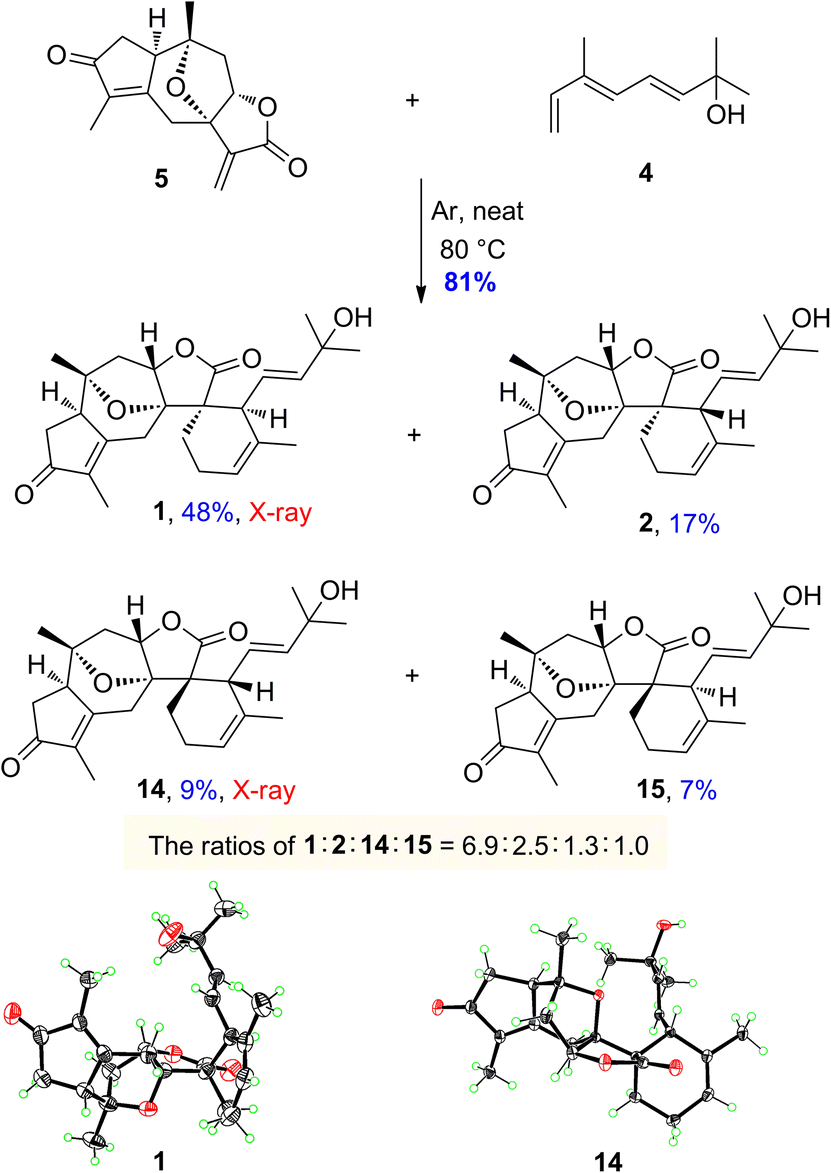 | ||
| Scheme 4 Syntheses of orientanoids A (1) and B (2). | ||
The endo products of Diels–Alder addition are normally preferred when the electron-withdrawing groups, such as carbonyl, are conjugated with the dienophile owing to the following secondary orbital interactions. However, in the current study, despite a carbonyl group being conjugated to the dienophile, the exo products were abnormally predominant in the resultant reaction. The unexpected ratios of endo and exo products attracted our great interest in seeking the immanent mechanism. Four plausible transition states a–d for compounds 1, 2, 14 and 15 in the Diels–Alder reaction were proposed as depicted in Fig. 3, respectively. The products (1 and 2) yielded by the diene reaching from the Si-face (referring to the C-12 carbonyl group) of dienophile 5 were much more abundant than those (14 and 15) formed via the Re-face addition due largely to the presence of steric hindrances from the 7,10-epoxy bridge in the transition states c and d. Meanwhile, the yields of exo adducts (1 and 14) were unusually higher than those of endo ones (2 and 15) owing to the balanced net dipole moments between the dienophile enone system and diene in the exo cases being smaller than those in the endo ones, resulting in the relatively lower free energy for the exo transition states.48–50 The total stability for each of the transition states a–d was regulated by both the steric effects and the net balanced dipole moments, which finally determined the yields of the products. In recent years, various bimolecular stereoselective Diels–Alderases in nature have been discovered,51,52 so it is also possible that 1 and 2 are selectively formed due to enzyme-dependent [4 + 2] cycloaddition in the biogenetic pathway, which needs further verification.
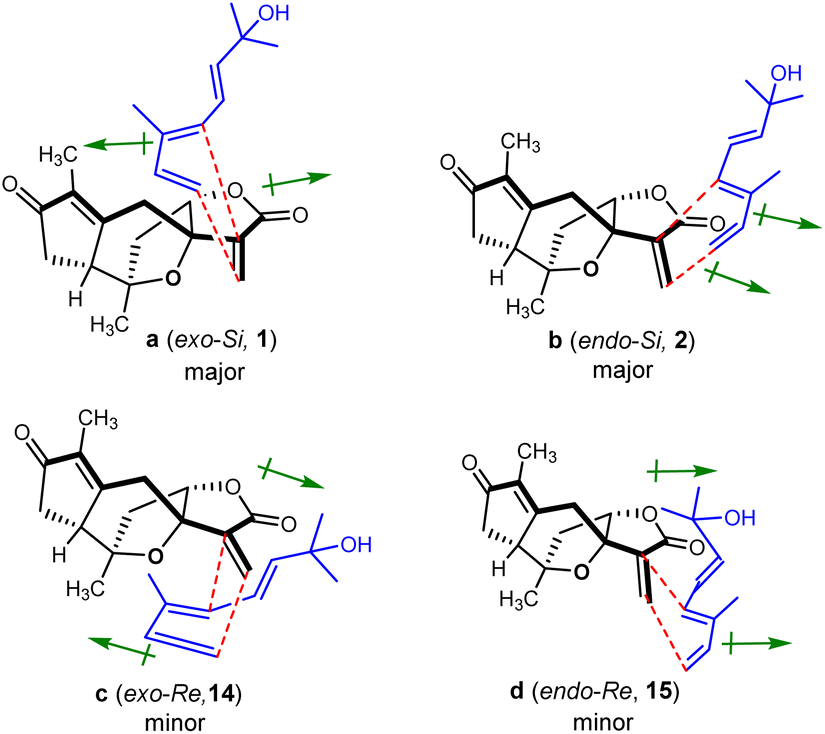 | ||
| Fig. 3 Transition states proposed for the regio-specific intermolecular Diels–Alder reactions of 4 and 5 (↦: denotes dipole moments of the dienophile enone and diene systems). | ||
According to the biosynthetic proposal (Scheme 1), we believed that orientanoid C is the more downstream metabolite that might be derived from orientanoids A and/or B via oxidative modifications. Therefore, after completing the syntheses of orientanoids A and B, we directed our focus to make orientanoid C from the synthetic formers via the initiative of oxidative modifications.
After enormous efforts, we finally focused on the singlet oxygen ene reaction aiming to furnish the enone functionality in orientanoid C (3). After extensive optimization, we found that irradiating an oxygen-bubbled solution of compound 1 and methylene blue in MeCN with a 23 W fluorescent lamp at room temperature smoothly produced the expected key intermediate hydroperoxide, which was then immediately exposed to 2-iodoxybenzoic acid (IBX)53 to afford the desired compound 3 in 72% yield in a one-pot manner (Scheme 5a).
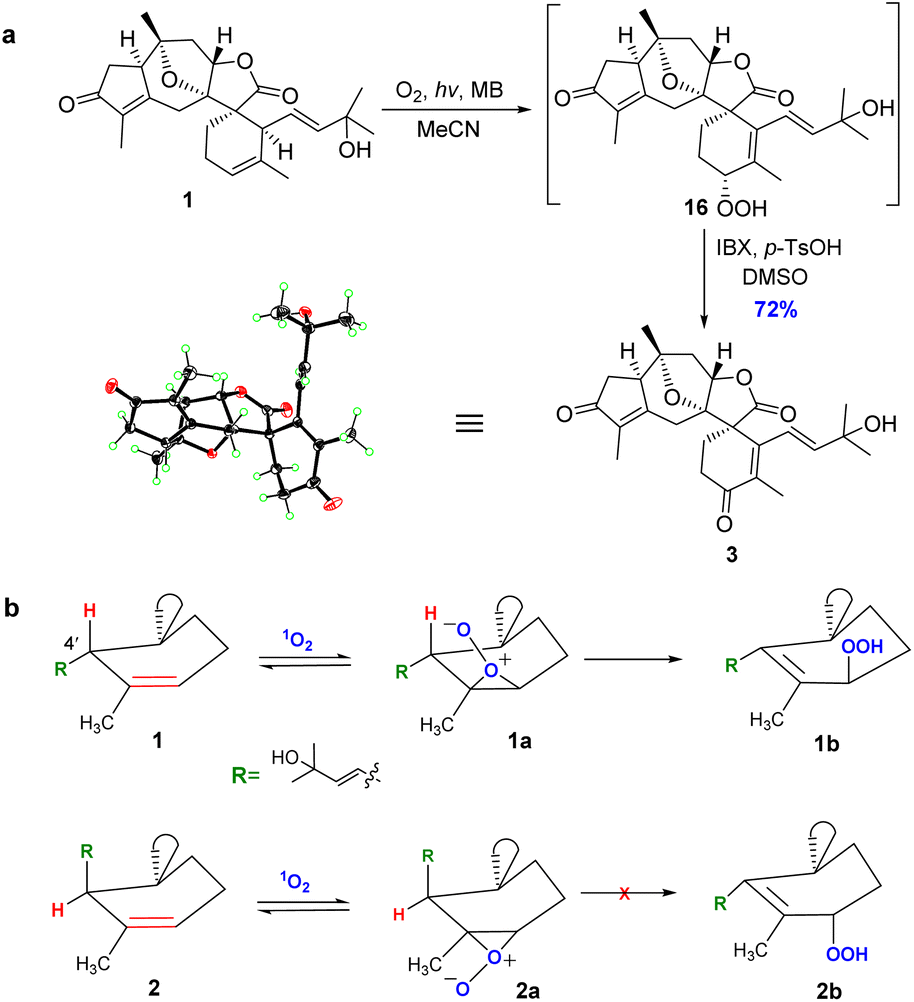 | ||
| Scheme 5 (a) Synthesis of orientanoid C (3). MB = methylene blue; IBX = 2-iodoxybenzoic acid; p-TsOH = p-toluenesulfonic acid; DMSO = dimethylsulfoxide. (b) Stereoselectivity for the singlet oxygen ene reaction of compounds 1 and 2. | ||
The structure of the synthetic orientanoid C was verified by X-ray diffraction study [absolute structure parameter: −0.02(4); CCDC 2013505]. Meanwhile, orientanoid B (2) could not be transformed into orientanoid C (3) under the same conditions. The cause of this stereo specific transformation was likely the conformational difference between two key reaction intermediates of 1 and 2.54,55 As depicted in Scheme 5b, the allylic hydrogen H-4′ of 1 is perfectly perpendicular to the Δ2′ double bond (see the X-ray structure), which is spatially conducive to form the key intermediate 1a required for the singlet oxygen ene reaction. Meanwhile, the H-4′ of 2 is almost parallel to the Δ2′ double bond, which resulted in the facts that the corresponding key intermediate 2a doesn't meet the conformational requirements for the singlet oxygen ene reaction, and is also likely unstable.
Antitumor immunity by antagonizing TAMs
With adequate quantity of compounds 1–3 and synthetic analogs in hand, we explored the effects of these compounds on the protumor phenotype of macrophages. In this test, the classic mouse monocyte-macrophages leukemia cell line RAW 264.7 was stimulated with IL-4/IL-13 to obtain M2-like protumoral phenotype macrophages, in which the mRNA expression levels of two molecules, namely MRC1 (CD206),56 a typical M2-type marker, and ARG1,56 a key functional effector to mediate T cells suppression, were observed to be significantly increased. The addition of compounds 1–3, 5, and 10 effectively inhibited this up-regulation (Fig. 4a and b) without obvious influence on cell viability (Fig. S7a, ESI†). Among them, compound 1 was found to be the most potent one and was thus selected for further evaluation both in vitro and in vivo.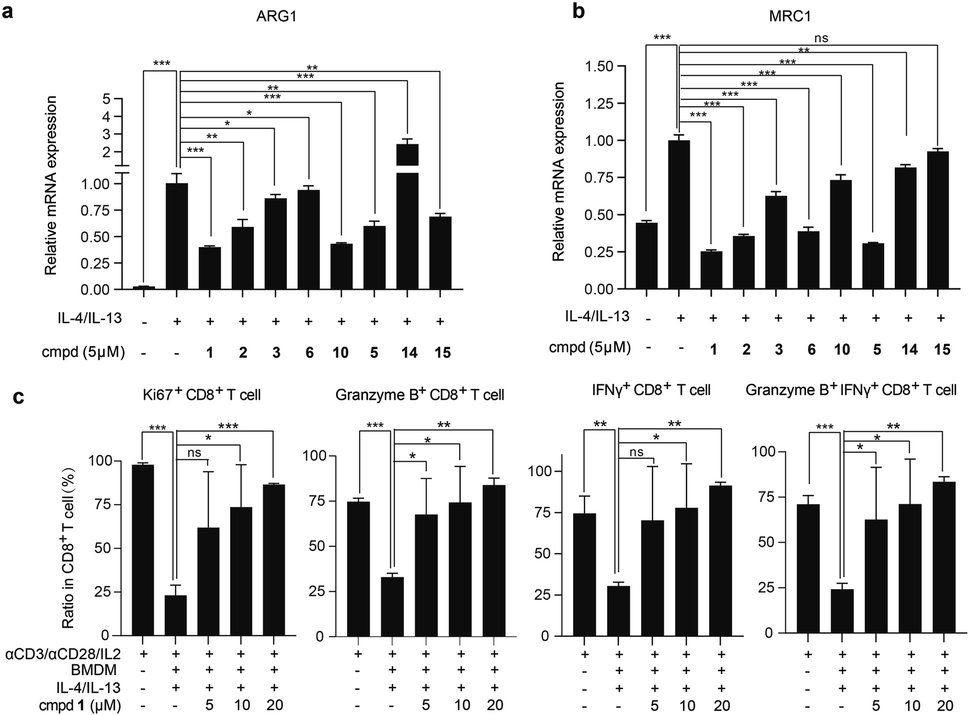 | ||
| Fig. 4 The effects of compounds on the protumor phenotype and immunosuppressive function of macrophages in vitro. (a and b) Quantitative RT-PCR analysis for mRNA levels of ARG1 (a) and MRC1 (b) in RAW 264.7 cells treated with IL-4/IL-13 alone or combined with indicated compounds for 12 h. (c) Compound 1 reverses macrophage-mediated CD8+ T suppression. BMDMs were treated with IL-4/IL-13 alone or combined with compound 1 for 48 h, followed by coculturing with spleen cells that were stimulated with anti-CD3/CD28 beads and IL-2 for 72 h. The ratio of the indicated CD8+ T cell subpopulation was assessed by flow cytometry. Data represent mean ± SD from triplicates. *P < 0.05, **P < 0.01, ***P < 0.001, ns, P > 0.05, determined by Student's t-test. | ||
We subsequently tested the inhibitory effects of compound 1 at multiple concentrations on the mRNA expression level of ARG1, MRC1, and another classic M2-marker CD163 (ref. 57) in IL-4/IL-13-induced RAW 264.7 cells and primary murine macrophages derived from bone marrow (BMDMs), respectively. Compound 1 dose-dependently inhibited the mRNA expression levels of ARG1, MRC1, and CD163 in both cells (Fig. S7c and d, ESI†), with no significant influence on cell viability (Fig. S7a and b, ESI†), further confirming that compound 1 suppressed the immunosuppressive and M2-polarized phenotype of macrophages. Protumoral M2-like TAMs have been demonstrated to directly suppress the proliferation and activation of CD8+ T cells.35,58 When co-cultured with autologous spleen cells, as expected, the IL-4/IL-13-induced BMDMs suppressed the proliferation and activation of CD8+ T cells. Interestingly, the suppression was reversed by the pretreatment of BMDMs with compound 1, as evidenced by a significant increase in the proportion of Ki67+, granzyme B+, and IFNγ+, as well as granzyme B+ IFNγ+ CD8+ T cells in total CD8+ T cells (Fig. 4c). The above data indicated that compound 1 reversed the suppression of T cell proliferation and activation induced by protumoral macrophages.
Finally, we investigated the in vivo antitumor efficacy of compound 1 in immune-competent and nude mice, respectively (Fig. 5a and b and S8a, ESI†). In subcutaneous E0771 (ref. 59) and Hepa1-6 (ref. 60) immunocompetent murine tumor models with abundant macrophages, compound 1 consistently and significantly delayed tumor growth (Fig. 5a and S8a, ESI†) at well-tolerated doses (Fig. S8b and c, ESI†). Concurrently, the infiltration of macrophages and CD8+ T cells in tumor tissue of the representative murine E0771 model was analyzed, which indicated that compound 1 notably reduced the infiltration of protumoral M2-like CD206+ macrophages and increased the infiltration of IFNγ+ and TNFα+ CD8+ T cells (Fig. 5c) without significant impact on the infiltration of total immune cells, macrophages and CD8+ T cells (Fig. S8e, ESI†). We also established the Hepa1-6 subcutaneous nude mouse tumor model, which lacks T cells, the key target cell suppressed by TAMs. By contrast, the antitumor effect of compound 1 was apparently greatly attenuated in nude mice (Fig. 5b). Additionally, compound 1 exhibited no significant inhibition on the cell viability of Hepa1-6 and E0771 cells in vitro (Fig. S9, ESI†). The above evidence suggested that compound 1 exerted antitumor effects in vivo mainly by suppressing protumoral macrophages and activating antitumoral CD8+ T cells, rather than through a direct cytotoxic effect on tumor cells.
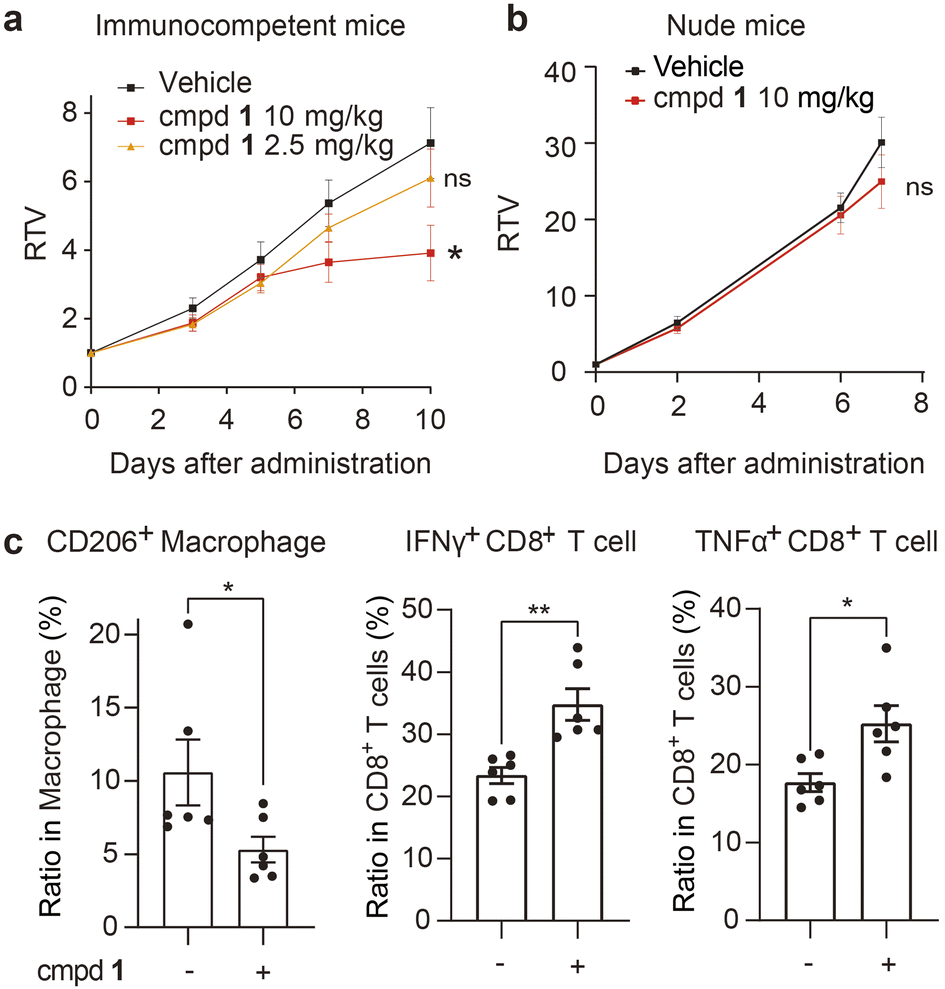 | ||
| Fig. 5 Compound 1 reshapes the tumor immune microenvironment and inhibits tumor growth in vivo. (a and b) Immune-competent mice bearing Hepa1-6 xenograft (n = 11 per group) (a) or nude mice bearing Hepa1-6 xenograft (n = 10 per group) (b) were intratumorally administered with compound 1 or the vehicle daily for the indicated period. The relative tumor volume (RTV) is shown as the mean ± SEM. (c) Flow cytometric analysis of tumor infiltrated immune cell subsets in the E0771 tumor model treated with compound 1 (5 mg kg−1) (n = 6 per group). See details in the ESI.† Data are shown as the mean ± SEM. *P < 0.05, **P < 0.01, ns P > 0.05 vs. the vehicle group, determined by Student's t-test. | ||
Conclusions
In summary, three unprecedented sesterterpenoids (1–3) were isolated and structurally characterized from H. orientale. Further to this, the concise and asymmetric syntheses of compounds 1–3 were accomplished in 7–8 steps with overall yields of 2.3–6.4% starting from the commercially available santonin without using any protecting groups, which not only corroborated the structural assignments but also imitated the biosynthetic proposal for these sesterterpenoids. Chemically, it is notable that a concise access to the guaiane-type sesquiterpenoids, such as hedyosumins A–C, was exploited involving a photochemical rearrangement of a santonin derivative as the key step, and a highly robust and catalyst-free intermolecular Diels–Alder reaction was accomplished to afford the desired products under very mild conditions by managing two reaction substrates in a well-mixed film of neat form. Biologically, this sesterterpenoid class, e.g. compound 1, inhibited tumor growth by reshaping the TME via antagonizing TAMs and activating antitumor immunity. The unprecedented structures and the unique anticancer mechanism of these rare sesterterpenoids provided a new class of drug leads for TAM-centric cancer therapy.Data availability
General information, detailed experimental procedures, and characterization data for all new compounds can be found in the ESI.†Author contributions
Y. Y. F. performed the isolation and structural elucidation of natural compounds. C. Y. Z. and J. X. Z. performed the biomimetic syntheses. C. H. Y., X. P., M. G., and J. A. contributed to the bioactive evaluation. J. M. Y. designed and supervised the project, and edited the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation (22237007, 81821005, and 82122062) of the People's Republic of China, the CAMS Innovation Fund for Medical Sciences (2019-I2M-5-080), the CAS Biological Resources Program (KFJ-BRP-008), the Collaborative Innovation Cluster Project of Shanghai Municipal Commission of Health and Family Planning (No. 2020CXJQ02), and the Shanghai Municipal Science and Technology Major Project. We also thank Prof. S.-M. Huang of Hainan University for the identification of the plant material.Notes and references
- A. F. Majdalawieh and M. W. Fayyad, Int. Immunopharmacol., 2015, 28, 295–304 CrossRef CAS PubMed.
- M. Yin, Y. Zhang and H. Li, Front. Immunol., 2019, 10, 145 CrossRef CAS PubMed.
- B. J. Huffman and R. A. Shenvi, J. Am. Chem. Soc., 2019, 141, 3332–3346 CrossRef CAS PubMed.
- J. W. H. Li and J. C. Vederas, Science, 2009, 325, 161–165 CrossRef PubMed.
- T. Zeng, Z. Liu, J. Zhuang, Y. Jiang, W. He, H. Diao, N. Lv, Y. Jian, D. Liang, Y. Qiu, R. Zhang, F. Zhang, X. Tang and R. Wu, J. Chem. Inf. Model., 2020, 60, 2082–2090 CrossRef CAS PubMed.
- D. W. Christianson, Chem. Rev., 2017, 117, 11570–11648 CrossRef CAS PubMed.
- D. Tholl, Adv. Biochem. Eng./Biotechnol., 2015, 148, 63–106 CrossRef CAS PubMed.
- K. Li and K. R. Gustafson, Nat. Prod. Rep., 2021, 38, 1251–1281 RSC.
- K. Guo, Y. Liu and S. H. Li, Nat. Prod. Rep., 2021, 38, 2293–2314 RSC.
- P. Máximo and A. Lourenço, Curr. Org. Chem., 2018, 22, 2381–2393 CrossRef.
- L. Wang, B. Yang, X.-P. Lin, X.-F. Zhou and Y. Liu, Nat. Prod. Rep., 2013, 30, 455–473 RSC.
- Y. Liu, L. Wang, J. H. Jung and S. Zhang, Nat. Prod. Rep., 2007, 24, 1401–1429 RSC.
- J. R. Hanson, Nat. Prod. Rep., 1996, 13, 529–535 RSC.
- M. Liu, W. Sun, L. Shen, Y. He, J. Liu, J. Wang, Z. Hu and Y. Zhang, Angew. Chem., Int. Ed., 2019, 58, 12091–12095 CrossRef CAS PubMed.
- C. Yuan, B. Du, L. Yang and B. Liu, J. Am. Chem. Soc., 2013, 135, 9291–9294 CrossRef CAS PubMed.
- D. Q. Thach, Z. G. Brill, H. K. Grover, K. V. Esguerra, J. K. Thompson and T. J. Maimone, Angew. Chem., Int. Ed., 2020, 59, 1532–1536 CrossRef CAS PubMed.
- N. Zhao, S. Yin, S. Xie, H. Yan, P. Ren, G. Chen, F. Chen and J. Xu, Angew. Chem., Int. Ed., 2018, 57, 3386–3390 CrossRef CAS PubMed.
- Z. G. Brill, H. K. Grover and T. J. Maimone, Science, 2016, 352, 1078–1082 CrossRef CAS PubMed.
- C. L. Hugelshofer and T. Magauer, J. Am. Chem. Soc., 2015, 137, 3807–3810 CrossRef CAS PubMed.
- Z.-S. Su, S. Yin, Z.-W. Zhou, Y. Wu, J. Ding and J.-M. Yue, J. Nat. Prod., 2008, 71, 1410–1413 CrossRef CAS PubMed.
- Y.-Y. Fan, Y.-L. Sun, B. Zhou, J.-X. Zhao, L. Sheng, J.-Y. Li and J.-M. Yue, Org. Lett., 2018, 20, 5435–5438 CrossRef CAS PubMed.
- Y.-Y. Fan, L.-S. Gan, S.-X. Chen, Q. Gong, H.-Y. Zhang and J.-M. Yue, J. Org. Chem., 2021, 86, 11277–11283 CrossRef CAS PubMed.
- C.-P. Liu, C.-Y. Xie, J.-X. Zhao, K.-L. Ji, X.-X. Lei, H. Sun, L.-G. Lou and J.-M. Yue, J. Am. Chem. Soc., 2019, 141, 6812–6816 CrossRef CAS PubMed.
- Y.-Z. Ge, B. Zhou, R.-X. Xiao, X.-J. Yuan, H. Zhou, Y.-C. Xu, M. A. Wainberg, Y.-S. Han and J.-M. Yue, Sci. China: Chem., 2018, 61, 1430–1439 CrossRef CAS.
- Y.-Y. Fan, H. Zhang, Y. Zhou, H.-B. Liu, W. Tang, B. Zhou, J.-P. Zuo and J.-M. Yue, J. Am. Chem. Soc., 2015, 137, 138–141 CrossRef CAS PubMed.
- J.-X. Zhao, Y.-Y. Yu, S.-S. Wang, S.-L. Huang, Y. Shen, X.-H. Gao, L. Sheng, J.-Y. Li, Y. Leng, J. Li and J.-M. Yue, J. Am. Chem. Soc., 2018, 140, 2485–2492 CrossRef CAS PubMed.
- B. Zhang, Y. Wang, S.-P. Yang, Y. Zhou, W.-B. Wu, W. Tang, J.-P. Zuo, Y. Li and J.-M. Yue, J. Am. Chem. Soc., 2012, 134, 20605–20608 CrossRef CAS PubMed.
- R. A. Fernandes, S. Moharana and G. N. Khatun, Org. Biomol. Chem., 2023, 21, 6652–6670 RSC.
- Z. Wu, S. Zhao, D. M. Fash, Z. Li, W. J. Chain and J. A. Beutler, J. Nat. Prod., 2017, 80, 771–781 CrossRef CAS PubMed.
- W.-B. Sun, X. Wang, B.-F. Sun, J.-P. Zou and G.-Q. Lin, Org. Lett., 2016, 18, 1219–1221 CrossRef CAS PubMed.
- K.-Y. Wang, Y. Li, S.-L. Zhang, J.-H. Chen and Z. Yang, Tetrahedron Lett., 2022, 102, 153946 CrossRef CAS.
- D. DeNardo and B. Ruffell, Nat. Rev. Immunol., 2019, 19, 369–382 CrossRef CAS PubMed.
- A. Mantovani, F. Marchesi, A. Malesci, L. Laghi and P. Allavena, Nat. Rev. Clin. Oncol., 2017, 14, 399–416 CrossRef CAS PubMed.
- A. Combes, B. Samad, J. Tsui, N. Chew, P. Yan, G. Reeder, D. Kushnoor, A. Shen, B. Davidson, A. Barczak, M. Adkisson, A. Edwards, M. Naser, K. Barry, T. Courau, T. Hammoudi, R. Argüello, A. Rao, A. Olshen, C. Cai, J. Zhan, K. Davis, R. Kelley, J. Chapman, C. Atreya, A. Patel, A. Daud, P. Ha, A. Diaz, J. Kratz, E. Collisson, G. Fragiadakis, D. Erle, A. Boissonnas, S. Asthana, V. Chan and M. Krummel, Cell, 2022, 185, 184–203 CrossRef CAS PubMed.
- A. Mantovani, P. Allavena, F. Marchesi and C. Garlanda, Nat. Rev. Drug Discovery, 2022, 21, 799–820 CrossRef CAS PubMed.
- M. Pittet, O. Michielin and D. Migliorini, Nat. Rev. Clin. Oncol., 2022, 19, 402–421 CrossRef PubMed.
- H. D. Flack and G. Bernardinelli, Chirality, 2008, 20, 681–690 CrossRef CAS PubMed.
- P. M. Dewick, Medicinal Natural Products: A Biosynthetic Approach, John Wiley & Sons, Ltd, Chichester, 3rd edn, 2009, p. 212 Search PubMed.
- D. H. R. Barton, P. de Mayo and M. Shafiq, J. Chem. Soc., 1957, 929–935 RSC.
- G. Blay, V. Bargues, L. Cardona, B. García and J. R. Pedro, J. Org. Chem., 2000, 65, 6703–6707 CrossRef CAS PubMed.
- L. Zhang, X. Dai, L. Tao, C. Xie, M. Zhang and M. Wang, Chin. J. Chem., 2017, 35, 1284–1288 CrossRef CAS.
- G. Blay, M. Luz Cardona, B. Garcia and J. R. Pedro, J. Org. Chem., 1991, 56, 6172–6175 CrossRef CAS.
- C.-Y. Zheng and J.-M. Yue, Nat. Commun., 2023, 14, 2399 CrossRef CAS PubMed.
- F. A. Macías, A. Santana, A. Yamahata, R. M. Varela, F. R. Fronczek and J. M. G. Molinillo, J. Nat. Prod., 2012, 75, 1967–1973 CrossRef PubMed.
- J. Li, W. Zhang, F. Zhang, Y. Chen and A. Li, J. Am. Chem. Soc., 2017, 139, 14893–14896 CrossRef CAS PubMed.
- S. Yildizhan and S. Schulz, Synlett, 2011, 2011, 2831–2833 CrossRef.
- G. Majetich, Y. Wang, Y. Li, J. K. Vohs and G. H. Robinson, Org. Lett., 2003, 5, 3847–3850 CrossRef CAS PubMed.
- F. Fotiadu, F. Michel and G. Buono, Tetrahedron Lett., 1990, 31, 4863–4866 CrossRef CAS.
- K. Takeda, I. Imaoka and E. Yoshii, Tetrahedron, 1994, 50, 10839–10848 CrossRef CAS.
- W. J. Lording, T. Fallon, M. S. Sherburn and M. N. Paddon-Row, Chem. Sci., 2020, 11, 11915–11926 RSC.
- L. Gao, C. Su, X. Du, R. Wang, S. Chen, Y. Zhou, C. Liu, X. Liu, R. Tian, L. Zhang, K. Xie, S. Chen, Q. Guo, L. Guo, Y. Hano, M. Shimazaki, A. Minami, H. Oikawa, N. Huang, K. N. Houk, L. Huang, J. Dai and X. Lei, Nat. Chem., 2020, 12, 620–628 CrossRef CAS PubMed.
- L. Gao, Y. Zou, X. Liu, J. Yang, X. Du, J. Wang, X. Yu, J. Fan, M. Jiang, Y. Li, K. N. Houk and X. Lei, Nat. Catal., 2021, 4, 1059–1069 CrossRef CAS.
- T. Kuga, Y. Sasano and Y. Iwabuchi, Chem. Commun., 2018, 54, 798–801 RSC.
- M. Orfanopoulos, I. Smonou and C. S. Foote, J. Am. Chem. Soc., 1990, 112, 3607–3614 CrossRef CAS.
- E. L. Clennan, in Synthetic Organic Photochemistry, ed. A. G. Griesbeck and J. Mattay, Marcel Dekker, New York, 2005, pp. 365–390 Search PubMed.
- X. Li, R. Liu, X. Su, Y. Pan, X. Han, C. Shao and Y. Shi, Mol. Cancer, 2019, 18, 177 CrossRef PubMed.
- Y. Wang, Y. Lin, S. Qiao, J. Wang and H. Wang, Adv. Biosyst., 2019, 3, e1800232 CrossRef PubMed.
- E. Peranzoni, J. Lemoine, L. Vimeux, V. Feuillet, S. Barrin, C. Kantari-Mimoun, N. Bercovici, M. Guérin, J. Biton, H. Ouakrim, F. Régnier, A. Lupo, M. Alifano, D. Damotte and E. Donnadieu, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, E4041–E4050 CrossRef CAS PubMed.
- T. Muliaditan, J. Caron, M. Okesola, J. Opzoomer, P. Kosti, M. Georgouli, P. Gordon, S. Lall, D. Kuzeva, L. Pedro, J. Shields, C. Gillett, S. Diebold, V. Sanz-Moreno, T. Ng, E. Hoste and J. Arnold, Nat. Commun., 2018, 9, 2951 CrossRef PubMed.
- T. Kimura, Y. Kato, Y. Ozawa, K. Kodama, J. Ito, K. Ichikawa, K. Yamada, Y. Hori, K. Tabata, K. Takase, J. Matsui, Y. Funahashi and K. Nomoto, Cancer Sci., 2018, 109, 3993–4002 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2002618, 2002619, 2002637–2002639, 2013505, 2022064, 2022065, 2181591 and 2216319. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04238c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |