
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure-guided optimisation of N-hydroxythiazole-derived inhibitors of factor inhibiting hypoxia-inducible factor-α†
Thomas P.
Corner
 a,
Ryan Z. R.
Teo
a,
Yue
Wu
b,
Eidarus
Salah
a,
Yu
Nakashima
c,
Giorgia
Fiorini
a,
Anthony
Tumber
a,
Amelia
Brasnett
a,
James P.
Holt-Martyn
a,
William D.
Figg
Jr.
a,
Xiaojin
Zhang
*b,
Lennart
Brewitz
*a and
Christopher J.
Schofield
*a
a,
Ryan Z. R.
Teo
a,
Yue
Wu
b,
Eidarus
Salah
a,
Yu
Nakashima
c,
Giorgia
Fiorini
a,
Anthony
Tumber
a,
Amelia
Brasnett
a,
James P.
Holt-Martyn
a,
William D.
Figg
Jr.
a,
Xiaojin
Zhang
*b,
Lennart
Brewitz
*a and
Christopher J.
Schofield
*a
aChemistry Research Laboratory, Department of Chemistry and the Ineos Oxford Institute for Antimicrobial Research, University of Oxford, 12 Mansfield Road, OX1 3TA, Oxford, United Kingdom. E-mail: lennart.brewitz@chem.ox.ac.uk; christopher.schofield@chem.ox.ac.uk
bState Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of Drug Design and Optimization and Department of Chemistry, China Pharmaceutical University, Nanjing 211198, China. E-mail: zxj@cpu.edu.cn
cInstitute of Natural Medicine, University of Toyama, 2630-Sugitani, 930-0194, Toyama, Japan
First published on 27th October 2023
Abstract
The human 2-oxoglutarate (2OG)- and Fe(II)-dependent oxygenases factor inhibiting hypoxia-inducible factor-α (FIH) and HIF-α prolyl residue hydroxylases 1–3 (PHD1–3) regulate the response to hypoxia in humans via catalysing hydroxylation of the α-subunits of the hypoxia-inducible factors (HIFs). Small-molecule PHD inhibitors are used for anaemia treatment; by contrast, few selective inhibitors of FIH have been reported, despite their potential to regulate the hypoxic response, either alone or in combination with PHD inhibition. We report molecular, biophysical, and cellular evidence that the N-hydroxythiazole scaffold, reported to inhibit PHD2, is a useful broad spectrum 2OG oxygenase inhibitor scaffold, the inhibition potential of which can be tuned to achieve selective FIH inhibition. Structure-guided optimisation resulted in the discovery of N-hydroxythiazole derivatives that manifest substantially improved selectivity for FIH inhibition over PHD2 and other 2OG oxygenases, including Jumonji-C domain-containing protein 5 (∼25-fold), aspartate/asparagine-β-hydroxylase (>100-fold) and histone Nε-lysine demethylase 4A (>300-fold). The optimised N-hydroxythiazole-based FIH inhibitors modulate the expression of FIH-dependent HIF target genes and, consistent with reports that FIH regulates cellular metabolism, suppressed lipid accumulation in adipocytes. Crystallographic studies reveal that the N-hydroxythiazole derivatives compete with both 2OG and the substrate for binding to the FIH active site. Derivatisation of the N-hydroxythiazole scaffold has the potential to afford selective inhibitors for 2OG oxygenases other than FIH.
Introduction
In order to maintain an adequate supply of dioxygen (O2) to tissues and cells, animals have evolved mechanisms to sense and respond to limiting O2 availability (hypoxia).1–3 One such mechanism involves the hypoxia-inducible factor (HIF) system, which regulates genes that work to ameliorate the effects of hypoxia and restore normal O2 supply.4–6 HIF is an α/β-heterodimeric transcription factor that plays important roles in both normal physiology and diseases, including cancer, in particular renal cell carcinoma.7,8 The abundance and transcriptional activity of HIF are controlled in an O2-dependent manner through post-translational modifications of HIF-α isoforms that are catalysed by the Fe(II)- and 2-oxoglutarate (2OG)-dependent oxygenases prolyl residue hydroxylase domain-containing proteins 1–3 (PHD1–3) and factor inhibiting hypoxia-inducible factor-α (FIH) (Fig. 1A–C).5,6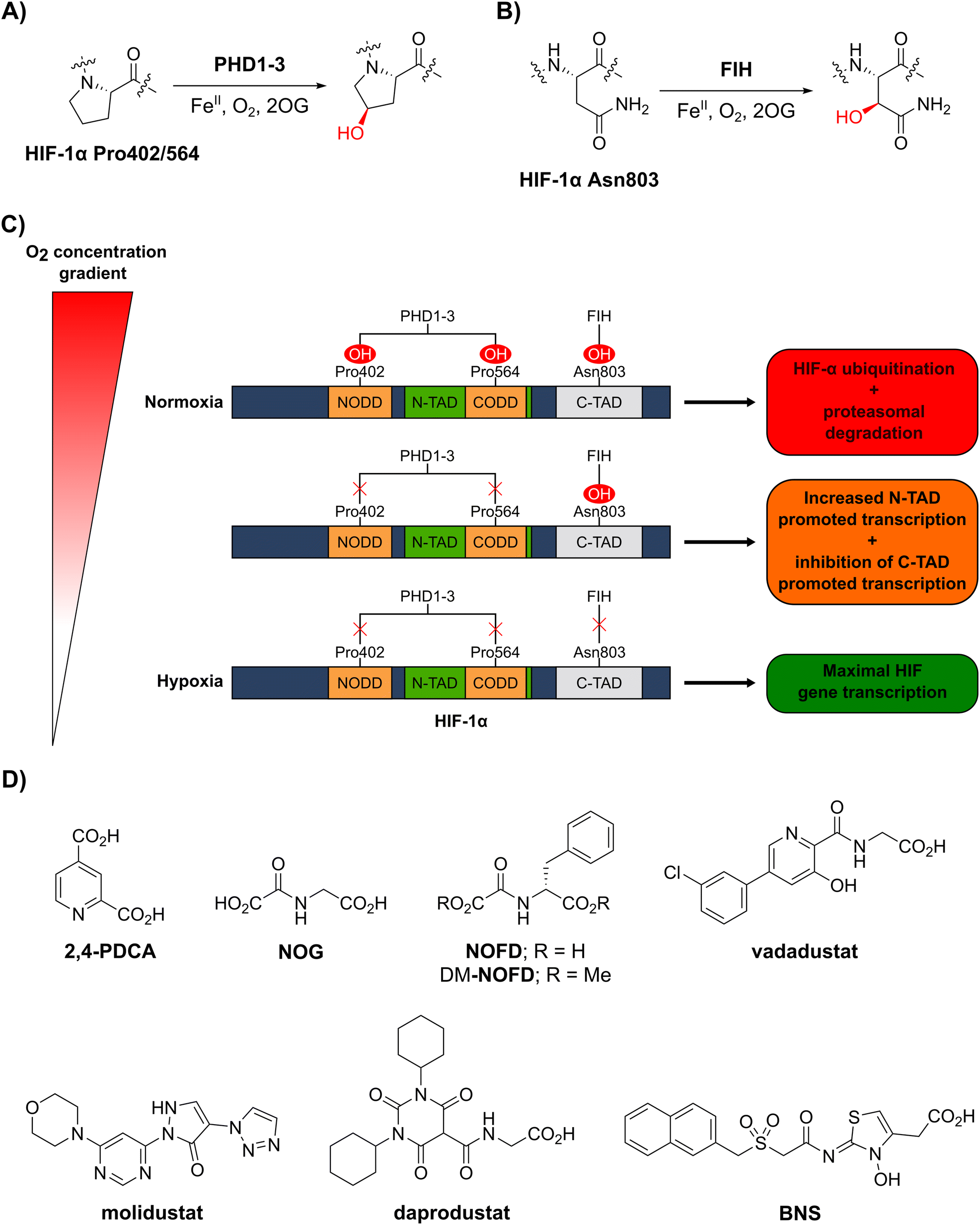 | ||
| Fig. 1 The human 2-oxoglutarate and Fe(II)-dependent oxygenases PHD1–3 and FIH regulate the transcriptional activity of hypoxia-inducible factor (HIF) isoforms in an O2 availability-dependent manner by catalysing hydroxylation of HIF-α residues. (A) PHD1–3 catalyse the C4-prolyl-residue hydroxylation of HIF-α (Pro402 and Pro564 in HIF-1α), a modification that promotes HIF-α degradation via the ubiquitin-proteasome pathway.9–12 (B) FIH catalyses the C3-asparaginyl-residue hydroxylation of HIF-α (Asn803 in HIF-1α) resulting in context-dependent suppression of HIF C-terminal activation domain (C-TAD) transcription activity.14–17 (C) Schematic representation of the role of the PHDs and FIH in the HIF O2/hypoxia sensing system. (D) Reported FIH inhibitors: the broad-spectrum 2OG oxygenase inhibitors pyridine-2,4-dicarboxylic acid27 (2,4-PDCA) and N-oxalylglycine28 (NOG), the FIH-selective inhibitor N-oxalyl-D-phenylalanine (NOFD)29 and its dimethyl ester prodrug form dimethyl N-oxalyl-D-phenylalanine (DM-NOFD). Reported PHD2 inhibitors: vadadustat,30 molidustat,31 daprodustat32 and BNS.17,33 | ||
PHD1–3 catalyse the C4-hydroxylation of two prolyl residues (Pro402 and Pro564 in human HIF-1α) in the N- and C-terminal oxygen-dependent degradation domains (ODDs) of HIF-α isoforms (Fig. 1A).9 The von Hippel-Lindau (pVHL) E3 ubiquitin ligase complex recognizes HIF-α prolyl hydroxylation, leading to polyubiquitination of HIF-α and its proteasomal degradation.10–12 PHD inhibitors increase HIF-α isoform levels and are used for the treatment of anaemia in chronic kidney disease.13 By contrast with the PHDs, FIH, which is part of a different structural subfamily of 2OG oxygenases,14 catalyses the C3-hydroxylation of a HIF-α asparagine residue (Gln803 in human HIF-1α) within the C-terminal activation domains (C-TADs) of HIF-1α and HIF-2α (Fig. 1B).14,15 HIF-α asparagine residue hydroxylation inhibits the interaction between the HIF-α C-TADs and the histone acetyl transferases and transcriptional coactivators p300/CBP resulting in suppression of C-TAD-mediated promotion of the transcriptional activity of α,β-HIF.16,17
O2 is a cosubstrate of both PHD1–3 and FIH, with the PHDs being more sensitive to O2 availability than FIH, as evidenced by kinetic analyses (PHD Km(O2) = 230–250 μM;18 FIH Km(O2) = 90 μM19).20–22 As O2 levels are depleted, PHD activity decreases, resulting in increased levels of HIF-α. In combination with HIF-β, HIF-α promotes the cellular hypoxic response by increasing the context-dependent expression of genes, including those encoding for erythropoietin (EPO)23 and vascular endothelial growth factor (VEGF).24 Under severe hypoxia,25 the activities of both the PHDs and FIH in cells are inhibited, and C-TAD-mediated promotion of HIF-α,β transcription is increased (Fig. 1C).1,4,6,26
Cellular studies have provided evidence that the sets of α,β-HIF target genes that are upregulated in hypoxia vary dependent on the context, including the cell type used.17,34,35 Exploiting the context-dependent nature of α,β-HIF gene expression is important from a therapeutic perspective, e.g. if anaemia treatment via α,β-HIF-mediated upregulation of the EPO gene is being targeted, concomitant upregulation of VEGF may be undesirable, as the latter has the potential to promote cancer.36 Interestingly, studies comparing the effects of hypoxia with those of broad spectrum 2OG oxygenase inhibitors or (at least) partially selective PHD inhibitors indicate that broad-spectrum inhibitors might better mimic hypoxia, at least in cells under laboratory conditions.17
Factors other than FIH, including other 2OG oxygenase related mechanisms, clearly have potential to regulate α,β-HIF target gene expression, e.g. the 2OG dependent JmjC histone Nε-lysine demethylase 4A (KDM4A) is reported to regulate HIF-1α abundance.37 The modulation of FIH activity is, however, of special interest with respect to controlling the set of α,β-HIF target genes upregulated, because, like the PHDs, it directly modifies HIF-α isoforms.14,15 There is good evidence that the role of FIH in the HIF system is context dependent.17 Thus, there is potential for the use of combinations of PHD and FIH inhibitors to ‘tune’ the set of HIF target genes upregulated, or, preferably, to have a single compound that manifests PHD and FIH inhibition activity, in a manner achieving the desired upregulation of a specific set of α,β-HIF target genes.
The rational control of FIH activity in a manner that leads to predicted physiological outcomes is, however, challenging, in part because of the general complexity of eukaryotic transcriptional regulation and the dynamic O2 availability-dependent nature of HIF-α levels. Further, by contrast with PHD1–3, for which substrates other than HIF-α have not yet been fully validated,38 there is biochemical and cellular evidence that FIH catalyses the C3-asparaginyl hydroxylation of multiple non-HIF substrates, including ankyrin repeat domain (ARD)-containing proteins NF-κB,39,40 apoptosis-stimulating p53-binding protein 2 (ASPP2)41,42 and Notch.43 FIH is also reported to catalyse the C3 hydroxylation of residues other than asparagine, including histidine-,44 tryptophan-45,46 and aspartate-residues.47
Cellular studies employing genetic methods have found that FIH also regulates small-molecule metabolism in addition to its role within the HIF system,48 potentially reflecting its apparent promiscuity with respect to protein substrates. Interestingly, deletion of the FIH gene in mice results in stimulation of both oxidative metabolism and glycolysis, resulting in an increase in cellular energy consumption.48 Furthermore, cellular peroxide inhibits hydroxylation of multiple FIH substrates at relatively low concentrations (<0.5 μM), suggesting that FIH may function as a sensor of oxidative stress.49 The underlying molecular mechanism of how FIH affects small-molecule metabolism and the pathophysiological relevance of the apparent pleiotropic cellular functions of FIH remain unclear. Selective small-molecule FIH inhibitors will therefore be of use to inform on the physiological functions of FIH.50
Whilst extensive work has focused on the development of PHD2 inhibitors,51 resulting e.g. in the clinical approval of daprodustat,32 vadadustat30 and FG-4592 (roxadustat),52 few small-molecule FIH inhibitors have been reported (Fig. 1D), and none have been approved for therapeutic use.53 The broad-spectrum 2OG oxygenase inhibitors pyridine-2,4-dicarboxylic acid (2,4-PDCA, FIH IC50: 5.0 μM)54 and N-oxalylglycine (NOG, FIH IC50: 0.36 μM), which are structural mimetics of 2OG, inhibit FIH via competitive displacement of 2OG at the active site.27,28 In addition, the reported PHD2 inhibitors vadadustat,30 molidustat,31 daprodustat32 and IOX455 inhibit isolated recombinant FIH with weak/moderate potency (IC50s: 29 μM, 66 μM, 21 μM and 31 μM, respectively).56
A derivative of NOG has been identified with improved selectivity for FIH inhibition, i.e. N-oxalyl-D-phenylalanine (NOFD; FIH IC50: 0.24 μM).29NOFD manifests excellent selectivity for FIH inhibition over PHD2, and its dimethyl ester prodrug (DM-NOFD) has been used to inhibit FIH-catalysed HIF-1α Asn803 hydroxylation in cells.17,57 However, due to the limited options to chemically modify NOFD for further potency and selectivity optimization, and evidence that N-oxalyl amino acid derivatives can inhibit other 2OG oxygenases, including aspartate/asparagine-β-hydroxylase (AspH)58 and the JmjC KDMs,59 alternative lead scaffolds for FIH inhibitor development, suitable for fine tuning with respect to FIH and PHD inhibition, are required.
Here, we report mass spectrometric and crystallographic evidence that the reported N-hydroxythiazole-based PHD inhibitor BNS17,33 (Fig. 1D) is a broad-spectrum inhibitor of 2OG oxygenases, including FIH. Structure-guided SAR studies on the N-hydroxythiazole scaffold resulted in the discovery of potent N-hydroxythiazole-based small-molecule FIH inhibitors with similar potency to NOFD.29 Mass spectrometry (MS)-based inhibition assays reveal the potential of the N-hydroxythiazole scaffold to achieve high levels of selectivity with respect to inhibiting PHD2, and other 2OG oxygenases, i.e. Jumoni-C domain-containing protein 5 (JMJD5), AspH and KDM4A. Crystallographic studies inform on the mechanism of FIH inhibition and on the mode of metal binding at the active site. In cell-based studies, optimised N-hydroxythiazole-derived FIH inhibitors were found to upregulate the expression of the FIH-dependent HIF target gene EGLN3 and to reduce adipocyte lipid accumulation, an observation which supports a proposed role of FIH in metabolic regulation.48 Importantly, our results imply that modifying the N-hydroxythiazole scaffold has potential for the design of selective inhibitors of 2OG oxygenases other than FIH.
Results and discussion
BNS is a broad-spectrum 2OG oxygenase inhibitor and a suitable lead scaffold for FIH-targeted inhibitor development
Considering that the PHD-selective inhibitors vadadustat30 and molidustat31 and daprodustat32 have been shown to moderately inhibit FIH in vitro,56 we initially investigated the effect of other reported PHD2 inhibitors on isolated recombinant human FIH (ESI Table S1†). IC50 values were determined using a solid-phase extraction coupled to mass spectrometry (SPE-MS)-based FIH inhibition assay, which monitors the mass change (i.e. +16 Da) associated with the FIH-catalysed hydroxylation of a HIF-1α-derived peptide, i.e. HIF-1α788–822.60,61 We observed that the reported PHD2 inhibitor BNS17,33 (Fig. 1D) inhibits FIH with similar potency (IC50 = 0.30 μM; Table 1, entry iii) as the FIH inhibitor NOFD (IC50 = 0.24 μM; Table 1, entry iv).29| Entry | Cmpd | IC50 [μM]a | ||||
|---|---|---|---|---|---|---|
| FIHb | PHD2c | AspHd | KDM4Ae | JMJD5f | ||
| a Mean average ± standard deviation (SD) of two independent experiments (each composed of technical duplicates). b Using 0.15 μM FIH, 10.0 μM 2OG and 5.0 μM of a HIF-1α C-terminal transactivation domain fragment (HIF-1α C-TAD788–822).61 c Using 0.15 μM PHD2181–426, 10.0 μM 2OG and 5.0 μM of a HIF-1α C-terminal oxygen-dependent degradation domain fragment (HIF-1α CODD556–574).61 d Using 0.05 μM His6-AspH315–758, 3.0 μM 2OG and 1.0 μM of a cyclic peptide based on human Factor X (hFX-CP101–119).58 e Using 0.15 μM KDM4A, 10.0 μM 2OG and 10.0 μM of a variant of histone 3 (H31–15K9me31–15).69 f Using 0.15 μM JMJD5, 2.0 μM 2OG and 2.0 μM of a 40S ribosomal protein S6 fragment (RSP6128–148).70 Inhibition assays were performed using SPE-MS as described in the ESI. | ||||||
| i | 2,4-PDCA | 5.0 ± 2.1 (ref. 54) | 5.3 ± 3.4 (ref. 54) | 0.03 ± 0.01 (ref. 54) | 0.10 ± 0.00 | 0.33 ± 0.07 (ref. 70) |
| ii | NOG | 0.36 ± 0.03 | 12.3 ± 4.4 | 1.1 ± 0.3 (ref. 58) | 22.1 ± 1.1 | 0.15 ± 0.02 (ref. 70) |
| iii | BNS | 0.30 ± 0.07 | 0.11 ± 0.00 | 3.4 ± 0.1 | 67.4 ± 39.8 | 0.25 ± 0.01 |
| iv | NOFD | 0.24 ± 0.02 | >100 | 15.5 ± 1.2 (ref. 60) | 14.1 ± 0.0 | >100 (ref. 70) |
Most reported 2OG oxygenase inhibitors, including the above mentioned PHD inhibitors, coordinate to the active site Fe(II) and compete with 2OG for binding, as pioneered in studies on inhibition of collagen prolyl hydroxylase and plant 2OG oxygenases.62,63 However, it is unclear how BNS binds to the active site of the PHDs; pioneering modelling and kinetic studies with N-hydroxythiazole-derived PHD inhibitors suggested two possible 2OG-competitive PHD2 binding modes.33,64 In each of these, the terminal carboxylate of BNS was proposed to bind in a similar manner to the 2OG C5 carboxylate, i.e. forming hydrogen bonds with the side chains of Tyr329 and Arg383. The mode of Fe(II) coordination was less clear, with two potential bidentate binding modes being considered – one involving coordination via the N-hydroxyl group and the exocyclic nitrogen atom of the N-hydroxythiazole unit (i.e. a 5-membered chelate ring) and the other via the N-hydroxyl group and the acetamide oxygen atom (i.e. a 7-membered chelate ring; Fig. 2A).
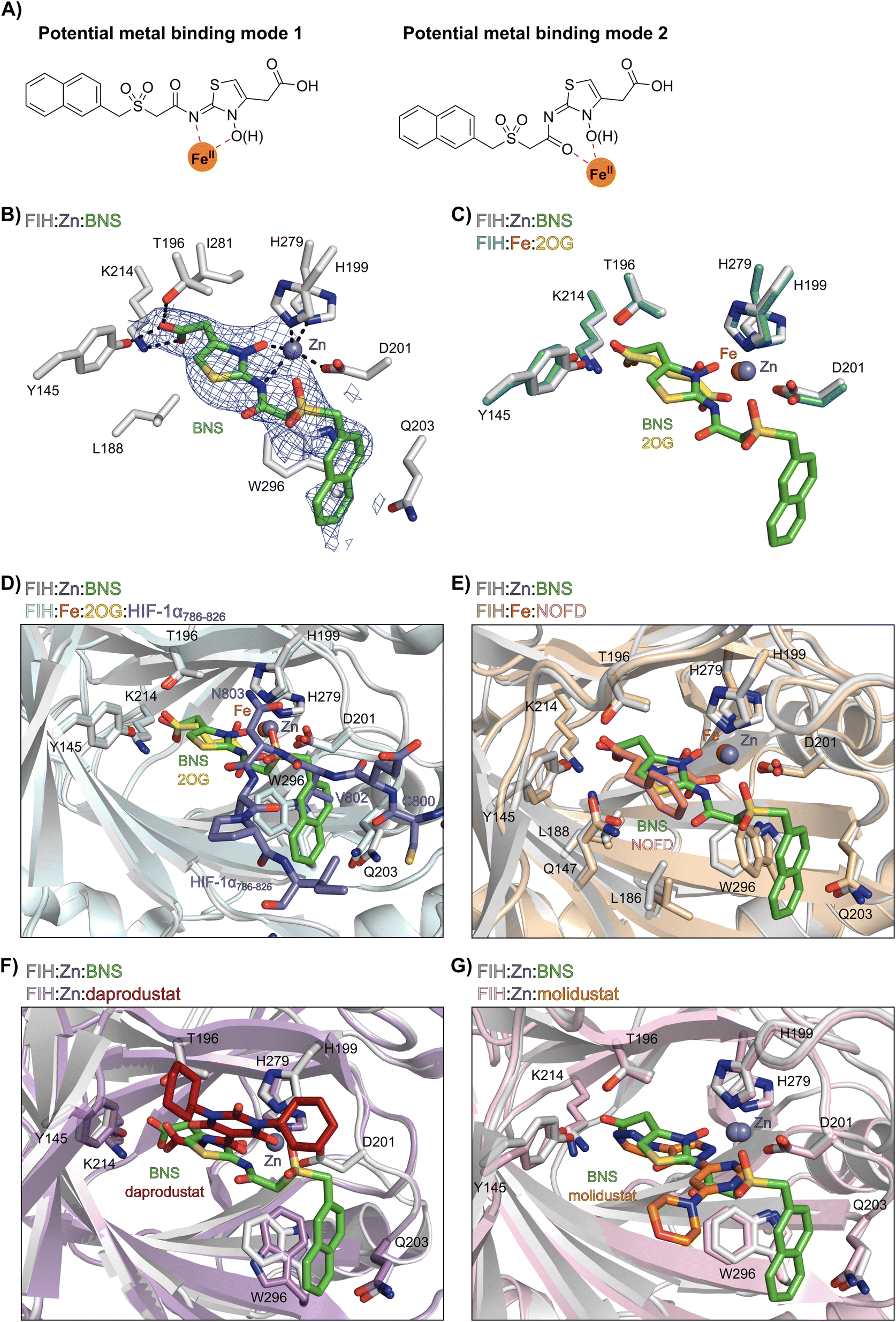 | ||
| Fig. 2 Crystallographic studies indicate that BNS binds to the FIH active site in a 2OG- and HIF-1α-competitive manner. Colour code: light grey: FIH; green: carbon-backbone of BNS; yellow: carbon-backbone of 2OG; dark grey: zinc; orange: iron; red: oxygen; blue: nitrogen and yellow: sulphur. (A) Two alternative modes have been proposed for how BNS binds to Fe(II) in the PHD2 active site.33 (B) Active site view from the FIH:Zn:BNS complex structure (PDB ID: 8K71) showing the OMIT electron density map (mFo-DFc) contoured to 2.1 σ around BNS. (C–F) Superimposition of active site views from the FIH:Zn:BNS complex structure and reported (C) FIH:Fe:2OG (PDB ID: 1MZF;66 teal: FIH), (D) FIH:Fe:2OG:HIF-1α786–826 (PDB ID: 1H2L;28 light blue: FIH; purple: carbon-backbone of HIF-1α), (E) FIH:Fe:NOFD (PDB ID: 1YCI;29 ochre: FIH; pink: carbon-backbone of NOFD), (F) FIH:Zn:daprodustat (PDB ID: 5OP6;56 light purple: FIH; deep red: carbon-backbone of daprodustat) and (G) FIH:Zn:molidustat (PDB ID: 5OP8;56 light pink: FIH; orange: carbon-backbone of molidustat) complex structures. | ||
To investigate its binding mode to the active site of a 2OG oxygenase, BNS was co-crystallised with FIH in the presence of Zn(II), which was used as a catalytically inert surrogate for Fe(II). The structure was solved by molecular replacement (MR) using a reported FIH structure (PDB ID: 4B7K65) as a search model. The FIH:Zn:BNS complex structure (PDB ID: 8K71; space group: P41212, resolution: 2.23 Å) reveals that BNS binds at the active site and coordinates Zn(II) in a bidentate manner via the N-hydroxyl group (O–Zn distance: 2.1 Å) and the exo-nitrogen atom (N–Zn distance: 2.7 Å) of its N-hydroxythiazole unit (Fig. 2B). Thus, the structural analysis implies that BNS binds to FIH via the proposed metal binding mode 1 (Fig. 2A); note, that BNS might bind to the PHDs via different modes; however, our efforts to crystallize BNS with PHD2 were unsuccessful.
Superimposition of the FIH:Zn:BNS complex structure with reported FIH:Fe:2OG (PDB ID: 1MZF;66 Cα RMSD = 0.30 Å) FIH:Fe:2OG:HIF-1α786–826 (PDB ID: 1H2L;28 Cα RMSD = 0.28 Å) complex structures indicates that BNS competes with both 2OG and HIF-1α for binding FIH (Fig. 2C and D). Notably, the orientation of the metal coordination mode of BNS is different to that observed crystallographically for 2OG (Fig. 2C) and NOFD (PDB ID: 1YCI;29Fig. 2E), as previously observed in FIH structures in complex with daprodustat (PDB ID: 5OP6;56Fig. 2F), molidustat (PDB ID: 5OP8;56Fig. 2G) and vadadustat (PDB ID: 5OPC;56 ESI Fig. S5†).56,67 The hydroxyl group of BNS binds Zn(II) in the same manner as the 2OG ketone carbonyl (i.e. trans to Asp201); however, the interaction between Zn(II) and the N-hydroxythiazole exo-nitrogen, which occupies the coordination site trans to His279, is perpendicular to that observed for the C1 carboxylate of 2OG, which binds trans to His199. The carboxylate of BNS is positioned to interact with the side chains of FIH residues Tyr145 (2.2 Å), Thr196 (3.0 Å), and Lys214 (2.5 Å), mimicking the interactions of the 2OG C5 carboxylate with FIH (Fig. 2C). In complex with FIH, daprodustat has also been found to engage in hydrogen bonding with Tyr145, Thr196 and Lys214 through its carboxylate group (Fig. 2F).56 By contrast, in the FIH:Zn:vadadustat complex structure, the carboxylate of vadadustat orients away from the side chains of Tyr145 and Thr196, and only interacts with Lys214 (ESI Fig. S5†).56 The triazole ring of molidustat is observed crystallographically to engage in hydrogen bonds with Tyr145 and Lys214; however, not with Thr196 (Fig. 2G).56
The FIH structure in complex with BNS reveals that the naphthalene group of BNS extends into the HIF-1α substrate binding pocket and is positioned to engage in π–π and amide-π stacking interactions with the side chains of Trp296 (face-to-face) and Gln203 (edge-to-face), respectively (Fig. 2D). As a result, the binding of BNS to FIH likely disrupts the hydrophobic interaction between the indole group of Trp296 and the conserved hydrophobic substrate residue (e.g. Val802 in HIF-1α) that is adjacent to the asparagine residue undergoing hydroxylation, which biochemical studies have shown is essential for efficient FIH catalysis.68 Analysis of a reported FIH:Zn:2OG:HIF-1α786–826 complex structure (PDB ID: 1H2L;28Fig. 2D) indicates that the primary amide side chain of Gln203 is involved in substrate recognition, by forming a hydrogen bond with the backbone amide oxygen atom of HIF-1α Cys800. Interestingly, neither daprodustat or molidustat appear to interact with Gln203 and/or Trp296 in complex with FIH (Fig. 2F and G). Given the importance of Gln203 and, in particular, Trp296 for substrate binding,68 the hydrophobic interactions between the naphthalene unit of BNS, Gln203 and Trp296 are potentially contributing factors behind the observed increased in FIH inhibitory activity of BNS compared with other PHD inhibitors (>50-fold, as judged by IC50 comparison, ESI Table S1†),56 in addition to the three hydrogen bonds made by the BNS carboxylate with FIH, unlike molidustat and vadadustat (ESI Fig. S5†).
To investigate the potential of BNS for selective FIH inhibition, its selectivity was investigated with respect to other 2OG-dependent protein oxidising enzymes, that is PHD2, Jumonji-C domain-containing protein 5 (JMJD5), aspartate/asparagine-β-hydroxylase (AspH) and KDM4A, using reported SPE-MS inhibition assays.58,61,69,70 In addition to PHD2 and KDM4A, we included AspH and JMJD5 in our selectivity studies, because AspH, like FIH, catalyses protein aspartate- and asparagine-residue hydroxylation (although in epidermal growth factor-like domains),71,72 and because JMJD5 is structurally closely related to FIH.73,74 The results reveal that, in addition to being a potent PHD2 (IC50 = 0.11 μM; Table 1, entry iii) and FIH inhibitor, BNS inhibits JMJD5 (IC50 = 0.25 μM) and AspH (IC50 = 3.36 μM), whereas inhibition of KDM4A was not observed (IC50 > 100 μM).
The combined results thus indicate that BNS is in fact a relatively broad-spectrum inhibitor of 2OG oxygenases, similar to 2,4-PDCA and NOG (Table 1, entries i and ii). Importantly, however, the results also reveal that the selectivity profile of BNS for 2OG oxygenase inhibition differs from that of both 2,4-PDCA and NOG. For example, 2,4-PDCA inhibits JMJD5, AspH, and KDM4A substantially more efficiently that PHD2 and FIH, whereas NOG manifests more potent inhibition of FIH, JMJD5 and AspH than PHD2 and KDM4A. This observation is of interest as it indicates that the use of particular broad-spectrum 2OG oxygenase inhibitors could result in different biological outcomes due to the inhibition of a different subset of 2OG oxygenases.
BNS likely does not inhibit KDM4A because of the different geometry of the KDM4A active site compared to those of FIH, PHD2, JMJD5 and AspH.75 Thus, superimposition of the FIH:Zn:BNS complex structure and a reported KDM4A:Ni:2OG complex structure (PDB ID: 6H8P76) indicates that the naphthalene group of BNS likely clashes with KDM4A residues Thr289 and Asn290, which form part of β-strand VIII of the rigid β-barrel core fold of the KDM4A active site, therefore preventing efficient binding of BNS to KDM4A (ESI Fig. S6†). By contrast, modelling studies predict that BNS can likely adopt binding modes within the PHD2, JMJD5 and AspH active sites that are analogous to the FIH:Zn:BNS complex structure (Fig. 3A–D).
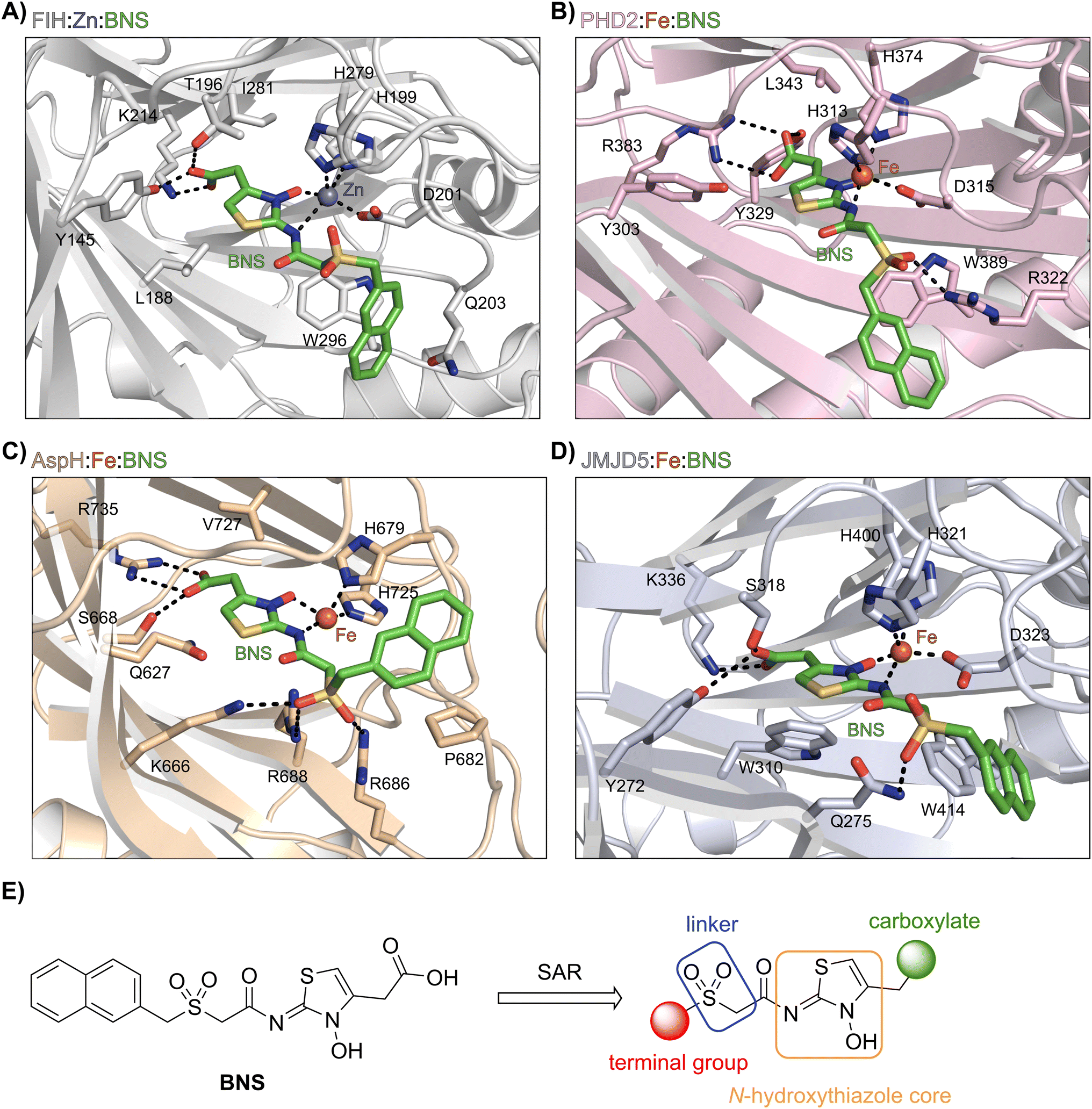 | ||
| Fig. 3 Docking studies predict that BNS may bind to the active sites of PHD2, AspH and JMJD5 with similar binding modes to that observed in the FIH:Zn:BNS complex structure. Colour code: green: carbon-backbone of BNS; dark grey: zinc; orange: iron; red: oxygen; blue: nitrogen and yellow: sulphur. (A) Active site view from the FIH:Zn:BNS complex structure (PDB ID: 8K71; light grey: FIH). (B–D) Active site views from: (B) the PHD2:Fe:BNS docking prediction (pink: PHD2), (C) the AspH:Fe:BNS docking prediction (ochre: AspH) and (D) the JMJD5:Fe:BNS docking prediction (light blue: JMJD5). (E) Outline of the strategies used for selectivity optimisation of the N-hydroxythiazole scaffold. | ||
The N-hydroxythiazole core of BNS is predicted to bind within the 2OG binding pockets of PHD2, AspH and JMJD5 and to coordinate to the metal in a bidentate manner, consistent with the observation that N-hydroxythiazoles inhibit recombinant PHD2 via a 2OG-competitive mechanism.64 The BNS carboxylate group is predicted to form hydrogen bond interactions with residues that interact with the 2OG C5 carboxylate (Tyr329 and Arg383 in PHD2;77 Ser668 and Arg735 in AspH;78 Tyr272, Ser318 and Lys336 in JMJD5;74Fig. 3B–D), as observed in the FIH:Zn:BNS complex structure, and the naphthalene unit likely extends into the respective peptide substrate binding pockets.
The docking results also indicate that the sulfone moiety of BNS will be positioned to form hydrogen bonds with the side chains of active site residues in PHD2, AspH and JMJD5 (Arg322 in PHD2; Lys666, Arg686 and Arg688 in AspH; Gln275 in JMJD5), an observation that contrasts with the FIH:Zn:BNS complex structure, in which the oxygen atoms of the sulfone group are solvent exposed. The residues that are predicted to interact with the sulfone of BNS in, at least, AspH78 and PHD2,79 have been observed by crystallography to be involved in substrate recognition, suggesting that the sulfone of BNS may be important to prevent productive substrate binding to AspH and PHD2.
Notably, the BNS modelling study implied that the orientation of the naphthalene unit of BNS may be different for BNS in complex with PHD2, AspH and JMJD5, as compared with that observed in the FIH:Zn:BNS complex structure (Fig. 3A–D). The difference in naphthalene binding may be due to the flexibility of the sulfone and methylene units that connect the N-hydroxythiazole core and naphthalene ring of BNS, and likely contributes to BNS being a relatively broad spectrum 2OG oxygenase inhibitor compared to more structurally rigid 2OG-competitive inhibitors such as daprodustat.
Overall, the above described results indicate that the N-hydroxythiazole scaffold of BNS is attractive for the development of selective FIH inhibitors, because (i) the FIH:Zn:BNS complex structure can be used to guide inhibitor design, (ii) the structure of BNS is modular and can be chemically modified, (iii) the BNSN-hydroxythiazole scaffold has potential for fine tuning inhibition of FIH and the PHDs, and (iv) BNS is active in cell-based studies.17,80 We therefore carried out SAR studies directed at optimising the selectivity of BNS for FIH inhibition. As part of the SAR study, the four main structural features of BNS, i.e. the N-hydroxythiazole core, the carboxylate group, the linker region and the terminal naphthalene moiety, were systematically varied (Fig. 3E).
Structure–activity relationship studies towards optimised N-hydroxythiazole-based FIH inhibitors
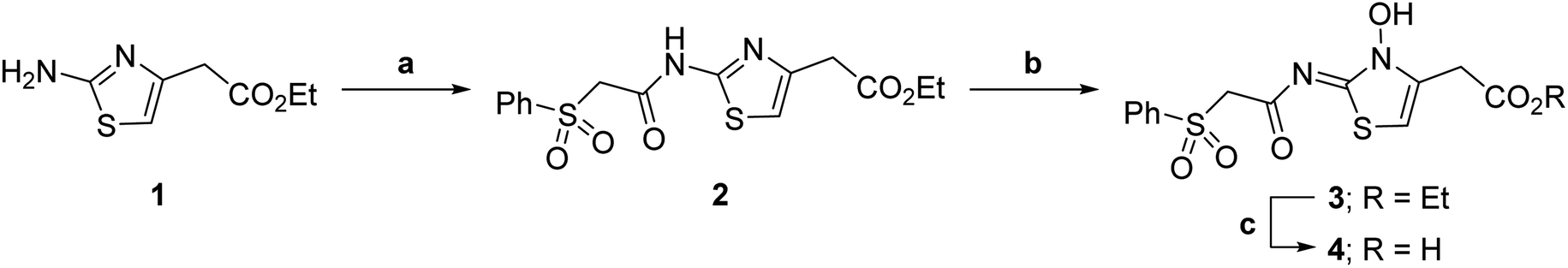 | ||
| Scheme 1 Synthesis of N-hydroxythiazole derivative 4.a aReagents and conditions: (a) 2-(phenylsulfonyl)acetic acid, T3P,82iPr2NEt, DMF, 0 °C to rt, 82%; (b) mCPBA, CHCl3, rt, 71%; (c) LiOH, MeOH/H2O, 0 °C to rt, 43%. | ||
Importantly, the naphthyl to phenyl group substitution of BNS did not affect the potency of FIH inhibition; however, it appeared that 4 inhibited PHD2, AspH and JMJD5 less efficiently than BNS, thus supporting our proposal that changes in the BNS structure can alter its selectivity profile (Table 2, entries i and ii). To investigate the effect of the N-hydroxyl group of 4 in FIH inhibition, we synthesized derivative 5 which lacks the N-hydroxyl group (ESI Scheme S1†). Notably, 5 did not inhibit the tested 2OG oxygenases (Table 2, entry iii), highlighting the importance of metal chelation for efficient inhibition by N-hydroxythiazoles, as indicated by the FIH:Zn:BNS complex structure (Fig. 2B).
| Entry | Cmpda |
|
IC50 [μM]b | ||||||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | FIHc | PHD2d | AspHe | KDM4Af | JMJD5g | ||
| a All chiral N-hydroxythiazole derivatives were prepared as racemic mixtures. b Mean average ± SD of two independent experiments (each composed of technical duplicates). c Using 0.15 μM FIH, 10.0 μM 2OG and 5.0 μM HIF-1α C-TAD788–822.61 d Using 0.15 μM PHD2181–426, 10.0 μM 2OG and 5.0 μM HIF-1α CODD556–574.61 e Using 0.05 μM His6-AspH315–758, 3.0 μM 2OG and 1.0 μM hFX-CP101–119.58 f Using 0.15 μM KDM4A, 10.0 μM 2OG and 10.0 μM H31–15K9me31–15.69 g Using 0.15 μM JMJD5, 2.0 μM 2OG and 2.0 μM RSP6128–148.70 h BNS has a 2-naphthylmethyl sulfonyl group rather than a phenyl sulfonyl group. Inhibition assays were performed using SPE-MS as described in the ESI. | |||||||||
| i | BNS | OH |
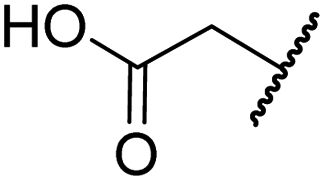
|
H | 0.30 ± 0.07 | 0.11 ± 0.00 | 3.4 ± 0.1 | 67.4 ± 39.8 | 0.25 ± 0.01 |
| ii | 4 | OH |
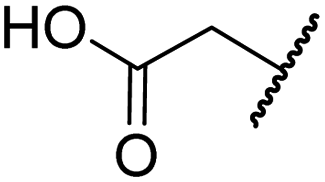
|
H | 0.28 ± 0.00 | 0.50 ± 0.05 | 9.6 ± 1.3 | >100 | 0.48 ± 0.01 |
| iii | 5 | H |
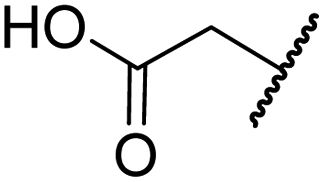
|
H | >100 | >100 | >100 | >100 | >100 |
| iv | 6 | OH |
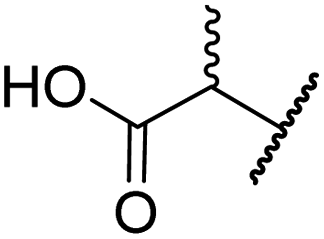
|
H | 61.0 ± 3.1 | 5.2 ± 2.6 | 25.6 ± 12.7 | >100 | 60.5 ± 1.0 |
| v | 7 | OH |
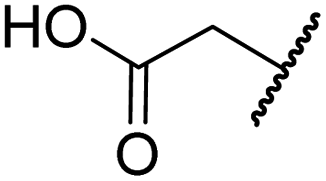
|
Me | 36.7 ± 4.9 | 3.1 ± 1.4 | 24.3 ± 12.3 | >100 | >100 |
| vi | 8 | OH | H | H | >100 | >100 | >100 | >100 | >100 |
| vii | 9 | OH |
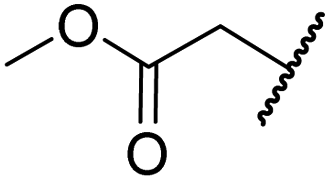
|
H | >100 | >100 | 96.7 ± 3.2 | >100 | >100 |
| viii | 3 | OH |
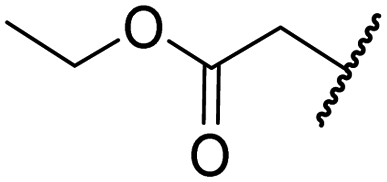
|
H | >100 | >100 | 68.0 ± 26.8 | >100 | >100 |
| ix | 10 | OH |
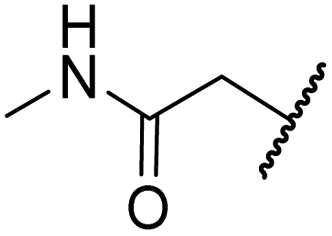
|
H | >100 | >100 | 43.6 ± 3.7 | >100 | 77.5 ± 23.8 |
| x | 11 | OH |
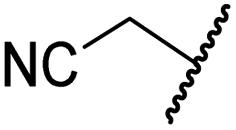
|
H | >100 | >100 | >100 | >100 | >100 |
| xi | 12 | OH |
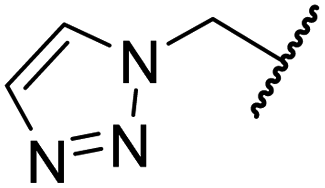
|
H | >100 | >100 | 83.3 ± 6.7 | >100 | >100 |
| xii | 13 | OH |
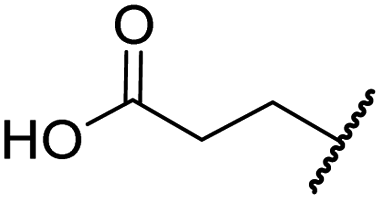
|
H | 2.2 ± 0.6 | 3.0 ± 1.3 | 47.3 ± 4.7 | >100 | >100 |
In the FIH:Zn:BNS complex structure, the C5 position of the N-hydroxythiazole ring of BNS and the methylene unit that connects the N-hydroxythiazole ring with the terminal carboxylate are positioned adjacent to the hydrophobic side chains of Leu188 and Ile281, respectively (Fig. 2B). To investigate whether substituents at these positions affect inhibitor potency and selectivity by enhancing hydrophobic interactions with Leu188 and/or Ile281, the corresponding methyl-substituted derivatives of 4 were synthesised, i.e.6 and 7 (ESI Scheme S2†). 6 and 7 showed substantially reduced levels of FIH inhibition compared to 4 (>100-fold; Table 2, entries iv and v), indicating a potential steric clash with the side chains of Leu188 and/or Ile281. Hence, further substitutions at these positions were not explored. Note that, while 7 inhibited PHD2 ∼ 30-fold less efficiently than 4, its selectivity for PHD2 inhibition over FIH inhibition increased by ∼20-fold compared to that of 4 (12-fold for 7vs. not PHD2 selective for 4). 7 also showed no inhibition of JMJD5 and KDM4A (IC50 > 100 μM), an observation which may be relevant for the future development of optimised N-hydroxythiazole-based PHD2 inhibitors, including with an improved selectivity profile.
The removal of the N-hydroxythiazole carboxylate side chain (as in 8), as well as the substitution of the carboxylate with bioisosteres such as amide (as in 10), nitrile (as in 11) and triazole (as in 12) groups resulted in the complete loss of FIH inhibition (Table 2, entries vi–xi), highlighting the importance of the hydrogen bonding interactions between the carboxylate and the FIH active site residues. Increasing the distance between the carboxylate and N-hydroxythiazole ring, as e.g. in compound 13 (ESI Scheme S3f†), led to a ∼10-fold reduction in both PHD2 and FIH inhibition relative to 4 (FIH IC50: 2.2 μM; Table 2, entry xii). Hence, further modifications of the BNS carboxylate group or thiazole C4 substituent were not investigated.
Whilst the truncated N-acetylated BNS derivative 14 was >200-fold less efficient in inhibiting FIH than 4 (IC50: 52.4 μM; Table 3, entry ii), compounds 15–23 were all relatively efficient FIH inhibitors (IC50 < 10 μM; Table 3, entries iii–xi). This observation indicates that the terminal phenyl substituent is important for FIH inhibition. Sulfones 4 (IC50: 0.28 μM) and 15 (IC50: 0.30 μM), and sulfonamide 21 (IC50: 0.36 μM) were the most potent FIH inhibitors within this series (Table 3, entries i, iii and ix), an observation which may reflect their increased ability to engage in hydrophobic contacts with the side chains of Gln203 and Trp296, as observed in the FIH:Zn:BNS complex structure (Fig. 2B). Both 4 and 21 were also efficient PHD2 inhibitors (IC50s: 0.50 μM and 0.57 μM, respectively). By contrast, 15, in which an ethylene group connects the N-hydroxythiazole core and the sulfone linker, showed decreased inhibition of PHD2 (IC50: 2.7 μM), JMJD5 (IC50: 3.3 μM) and AspH (IC50: 33.0 μM), relative to 4.
| Entry | Cmpd |
|
IC50 [μM]a | ||||
|---|---|---|---|---|---|---|---|
| R | FIHb | PHD2c | AspHd | KDM4Ae | JMJD5f | ||
| a Mean average ± SD of two independent experiments (each composed of technical duplicates). b Using 0.15 μM FIH, 10.0 μM 2OG and 5.0 μM HIF-1α C-TAD788–822.61 c Using 0.15 μM PHD2181–426 and 5.0 μM HIF-1α CODD556–574.61 d Using 0.05 μM His6-AspH315–758, 3.0 μM 2OG and 1.0 μM hFX-CP101–119. e Using 0.15 μM KDM4A, 10.0 μM 2OG and 10.0 μM H31–15K9me31–15.69 f Using 0.15 μM JMJD5, 2.0 μM 2OG and 2.0 μM RSP6128–148.70 Inhibition assays were performed using SPE-MS as described in the ESI. | |||||||
| i | 4 |
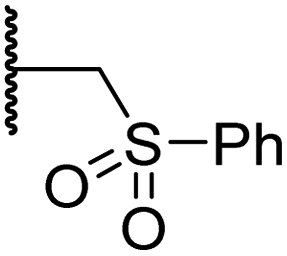
|
0.28 ± 0.00 | 0.50 ± 0.05 | 9.6 ± 1.3 | >100 | 0.48 ± 0.01 |
| ii | 14 | Me | 52.4 ± 30.4 | >100 | >100 | >100 | 3.2 ± 0.7 |
| iii | 15 |
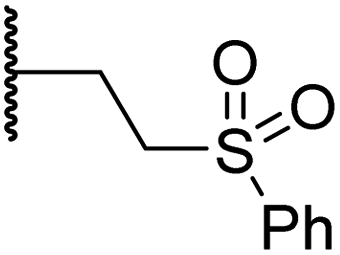
|
0.30 ± 0.05 | 2.7 ± 0.2 | 33.0 ± 12.1 | >100 | 3.3 ± 1.5 |
| iv | 16 |
|
1.2 ± 0.2 | 1.2 ± 0.2 | 2.4 ± 1.1 | >100 | 1.7 ± 0.3 |
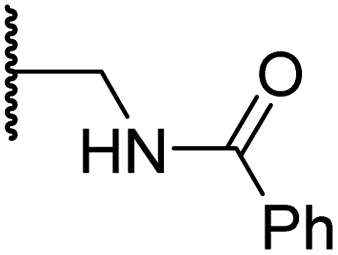
| v | 17 |
|
4.6 ± 0.7 | 3.6 ± 0.4 | 28.3 ± 6.5 | >100 | 4.6 ± 1.7 |
| vi | 18 |
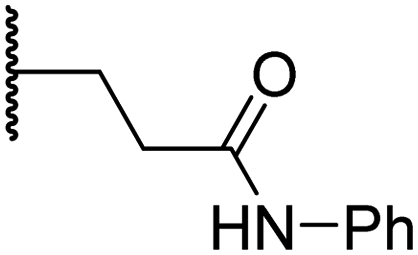
|
6.0 ± 1.2 | 2.3 ± 0.1 | 3.7 ± 1.5 | >100 | 8.4 ± 2.7 |
| vii | 19 |
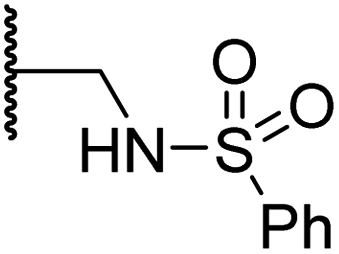
|
3.7 ± 1.5 | 15.5 ± 4.6 | 3.1 ± 0.4 | 17.4 ± 0.2 | 12.0 ± 0.6 |
| viii | 20 |
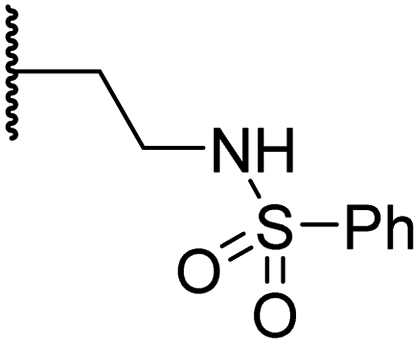
|
1.0 ± 0.1 | 6.5 ± 1.8 | 23.3 ± 10.1 | >100 | 13.4 ± 6.5 |
| ix | 21 |
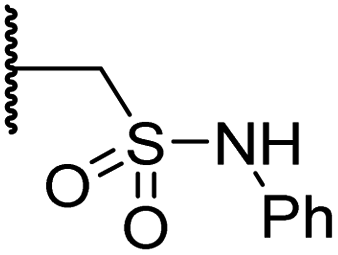
|
0.36 ± 0.21 | 0.57 ± 0.18 | 0.76 ± 0.10 | >100 | 2.8 ± 0.9 |
| x | 22 |
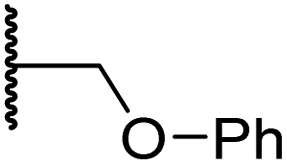
|
0.75 ± 0.23 | 2.6 ± 0.2 | 20.2 ± 6.6 | >100 | 1.2 ± 0.2 |
| xi | 23 |
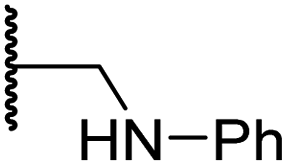
|
3.2 ± 1.1 | 10.3 ± 2.9 | 11.1 ± 0.4 | 71.6 ± 11.8 | 4.7 ± 1.1 |
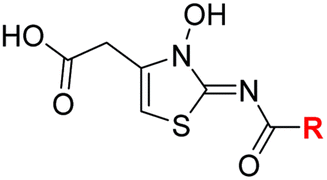
Sulfonamide 20, which, like sulfone 15, contains an ethylene chain that bridges the N-hydroxythiazole core and sulfonamide linker, and ether 22 also retained potent FIH inhibition (IC50s: 1.0 μM and 0.75 μM, respectively; Table 3, entries viii and x). Furthermore, 20 and 22 did not substantially inhibit KDM4A or AspH, and 20 displayed ∼6-fold selectivity for FIH inhibition over PHD2 inhibition and ∼13-fold selectivity over JMJD5 inhibition, which is ∼3-fold and ∼8-fold greater than that of 4, respectively. This observation indicates that extending the alkylene unit connecting the N-hydroxythiazole unit and linker group may be a viable approach to increase selectivity for FIH inhibition, potentially resulting from disruption of the predicted hydrogen bonding interactions by the BNS sulfone in the PHD2, JMJD5 and AspH substrate binding sites (Fig. 3B–D). Replacement of the sulfonamide of 20 with an amide (17; IC50: 4.6 μM) and truncation of the ethylene chain (19; IC50: 3.7 μM) led to a ∼5-fold and ∼4-fold loss in FIH inhibition, respectively (Table 3, entries v and vii).
Interestingly, sulfonamide 21 was the most active inhibitor of AspH among the tested N-hydroxythiazole derivatives (IC50: 0.76 μM; Table 3, entry ix), being ∼5-fold more potent than 4. Docking studies indicate that the sulfonamide moiety of 21 may be appropriately positioned to interact with the polar side chains of Arg686, Arg688 and Glu617 (ESI Fig. S10†). AspH is a proposed medicinal chemistry target54,88 associated with the pathologies of various cancers,89,90 including hepatocellular carcinoma88,91 and pancreatic cancer,93 hence, derivatisation of 21 to develop potent AspH inhibitors warrants further investigation.92
Analysis of the FIH:Zn:20 complex structure revealed that 20 adopts an FIH binding pose similar to that of BNS (Fig. 4A and C). Notably, the phenyl ring of 20 did not engage in a π-stacking interaction with the side chain of Trp296, as observed for the naphthalene unit of BNS in complex with FIH, but instead forms a face-to-face amide-π interaction with the primary amide group of Gln203. The reduced potency of FIH inhibitor 20 compared with 4 and BNS (i.e. ∼3-fold), may be a result of the lack of interaction with Trp296. Importantly, however, there appeared to be sufficient space adjacent to the ethylene unit of 20 in the FIH:Zn:20 complex structure to potentially allow for its substitution with a carbocycle, which may also interact productively with the indole side chain of Trp296. Thus, N-hydroxythiazole derivatives 24–30, in which carbocycles substitute for the ethylene chain of 20, were synthesised to test this proposal (ESI Scheme S5†) and their inhibition of FIH, PHD2, JMJD5, AspH and KDM4A was determined (Table 4).
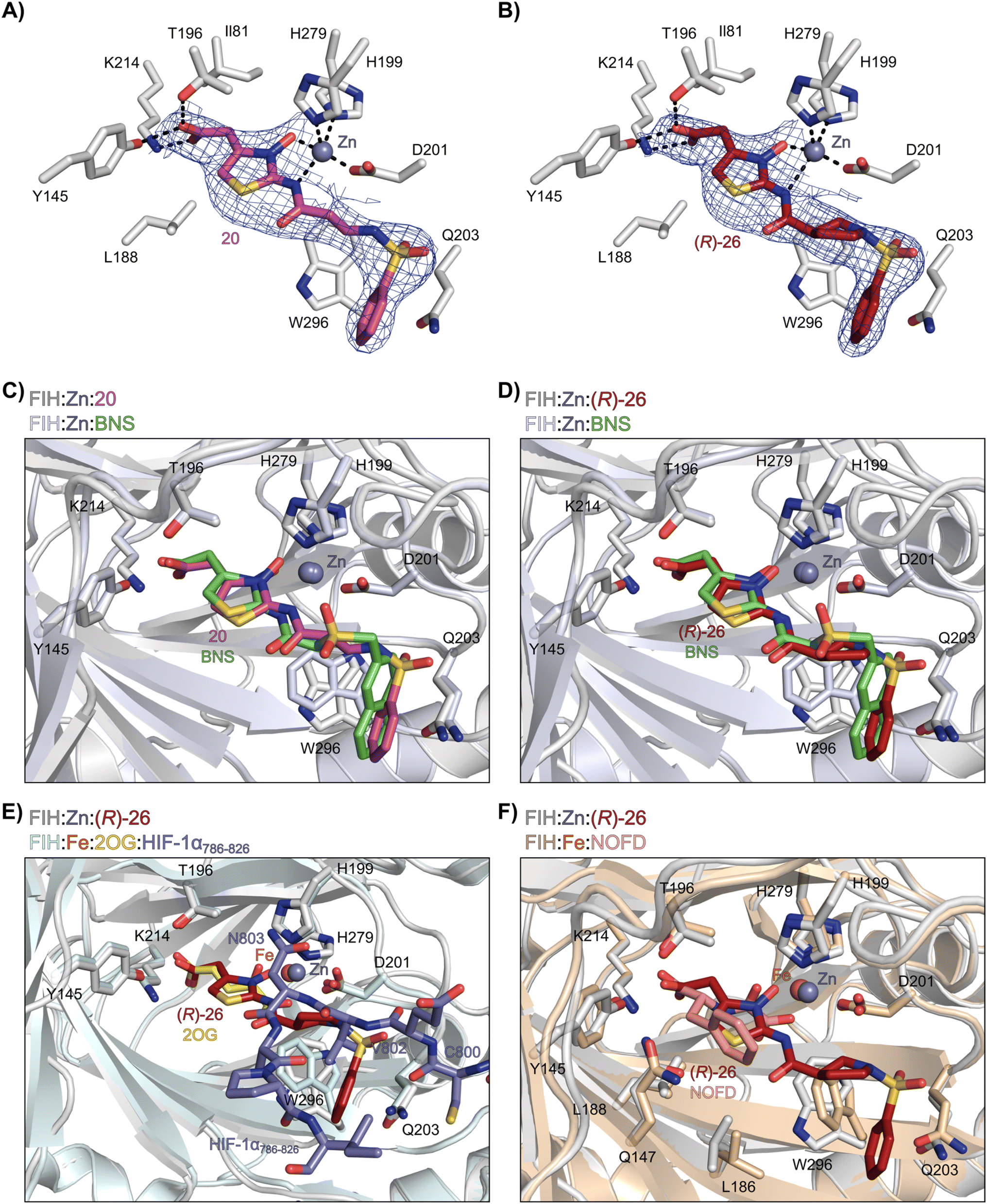 | ||
| Fig. 4 N-Hydroxythiazole derivatives 20 and 26 bind to the FIH active site in a similar manner to BNS. Colour code: light grey: FIH; purple: carbon-backbone of 20; ruby: carbon-backbone of (R)-26; green: carbon-backbone of BNS; dark grey: zinc; orange: iron; red: oxygen; blue: nitrogen and yellow: sulphur. (A) View from the FIH:Zn:20 complex structure (PDB ID: 8K72) showing the OMIT electron density map (mFo-DFc) contoured to 2.1 σ around 20. (B) View from the FIH:Zn:26 complex structure (PDB ID: 8K73) showing the OMIT electron density map (mFo-DFc) contoured to 2.1 σ around 26. (C) Superimposition of active site views from the FIH:Zn:20 and the FIH:Zn:BNS (PDB ID: 8K71, FIH: light blue) complex structures. (D–F) Superimposition of active site views from the FIH:Zn:26 complex structure and (D) the FIH:Zn:BNS complex structures (PDB ID: 8K71, FIH: light blue), (E) a reported FIH:Fe:2OG:HIF-1α786–826 complex structure (PDB ID: 1H2L;28 light blue: FIH; yellow: carbon-backbone of 2OG; purple: carbon-backbone of HIF-1α786–826), and (F) a reported FIH:Fe:NOFD complex structure (PDB ID: 1YCI;29 ochre: FIH; pink: carbon-backbone of NOFD). | ||
| Entry | Cmpda |
|
IC50 [μM]b | ||||
|---|---|---|---|---|---|---|---|
| R | FIHc | AspHd | PHD2e | KDM4Af | JMJD5g | ||
| a All chiral N-hydroxythiazole derivatives were prepared as racemic mixtures. b Mean average ± SD of two independent experiments (each composed of technical duplicates). c Using 0.15 μM FIH, 10.0 μM 2OG and 5.0 μM HIF-1α C-TAD788–822.61 d Using 0.05 μM His6-AspH315–758, 3.0 μM 2OG and 1.0 μM hFX-CP101–119.58 e Using 0.15 μM PHD2181–426, 10.0 μM 2OG and 5.0 μM HIF-1α CODD556–574.61 f Using 0.15 μM KDM4A, 10.0 μM 2OG and 10.0 μM H31–15K9me31–15.69 g Using 0.15 μM JMJD5, 2.0 μM 2OG and 2.0 μM RSP6128–148.70 Inhibition assays were performed using SPE-MS as described in the ESI. | |||||||
| i | 20 |
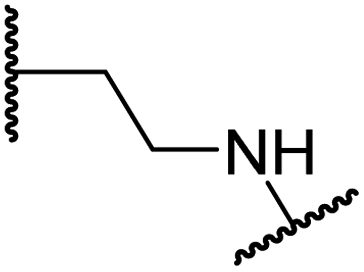
|
1.0 ± 0.0 | 23.3 ± 10.1 | 6.5 ± 1.8 | >100 | 13.4 ± 6.5 |
| ii | 24 |
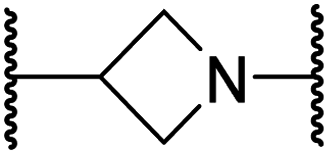
|
0.45 ± 0.17 | 60.9 ± 24.1 | 5.2 ± 0.7 | >100 | 29.1 ± 4.4 |
| iii | 25 |
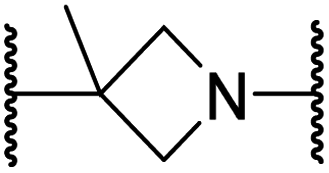
|
1.2 ± 0.2 | >100 | 33.5 ± 1.0 | >100 | 32.6 ± 6.7 |
| iv | 26 |
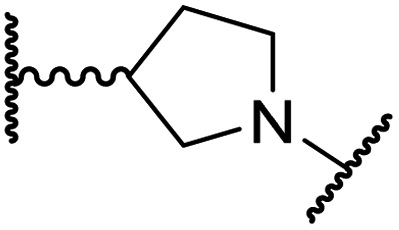
|
0.50 ± 0.02 | 37.8 ± 11.3 | 12.4 ± 0.8 | >100 | 12.5 ± 1.0 |
| v | 27 |
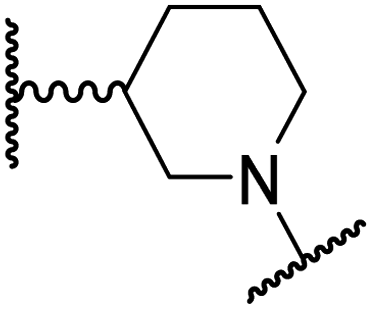
|
0.87 ± 0.04 | 49.1 ± 1.7 | 11.0 ± 1.6 | >100 | 13.4 ± 1.0 |
| vi | 28 |
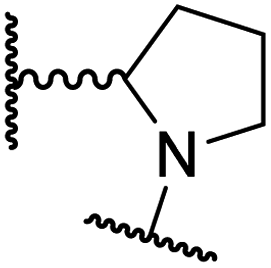
|
2.1 ± 1.5 | 24.9 ± 12.0 | 12.7 ± 1.8 | >100 | 2.6 ± 0.2 |
| vii | 29 |
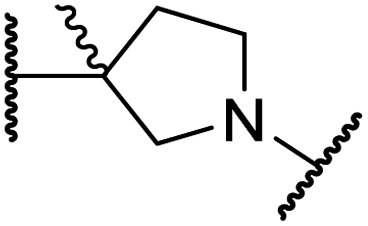
|
>100 | >100 | >100 | >100 | >100 |
| viii | 30 |
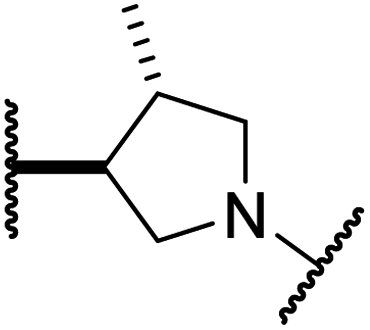
|
1.7 ± 0.4 | >100 | 71.7 ± 26.6 | >100 | 25.6 ± 0.6 |
The use of a β-pyrrolidine linker (26; Table 4, entry iv) increased FIH inhibition potency by ∼2-fold relative to 20 (IC50: 0.50 μM) and enhanced selectivity for FIH inhibition over PHD2 (∼25-fold), JMJD5 (∼25-fold) and AspH (∼75-fold) inhibition; 26 did not inhibit KDM4A (IC50 > 100 μM). The increased rigidity of the pyrrolidine ring of 26, compared with the ethylene unit of 20, is likely responsible for the increased selectivity of 26, by limiting the potential binding modes of the terminal phenyl ring. Decreasing the linker ring size from a β-pyrrolidine to an azetidine (24; Table 4, entry ii) had no effect on FIH inhibition (IC50: 0.45 μM); however, the selectivity of compound 24 for FIH over PHD2 inhibition (12-fold) was reduced by ∼2-fold compared to 26. 3-Methylazetidine (25; Table 4, entry iii) and β-piperidine (27; Table 3, entry v) analogues showed decreased FIH inhibition (IC50s: 1.2 and 0.87 μM, respectively) relative to 26.
Analysis of a crystal structure of FIH complexed with β-pyrrolidine derivative 26 (FIH:Zn:26, PDB ID: 8K73; space group: P41212, resolution: 2.25 Å) reveals that 26 binds to the FIH active site in a similar manner to ethylamine derivative 20, with its phenyl sulfonamide moiety adopting an almost identical conformation for both compounds (Fig. 4A and B), that is engaging in a face-to-face amide-π stacking interaction with the side chain of Gln203. Notably, the β-pyrrolidine ring of 26 is positioned to form hydrophobic interactions with Trp296, as observed with the naphthalene ring of BNS (Fig. 2B), which likely contributes to the increased FIH inhibition by 26, compared with 20, in addition to the reduced entropic cost of binding. Analysis of the electron density maps for the FIH:Zn:26 structure indicates that only (R)-26 binds to FIH, despite 26 being present as a racemic mixture, suggesting that the inhibitory activity of the two 26 enantiomers likely differs. Thus, the structural analyses support our proposal that the observed increase in selectivity for FIH inhibition may be a result of stabilising the conformation that binds to the FIH active site.
A substantial decrease in inhibition potency was observed for the α-pyrrolidine analogue of 26 (i.e.28; IC50: 2.1 μM, Table 4, entry vi), potentially reflecting a steric clash with the FIH active site, in particular with Trp296, and/or loss of the favourable amide-π stacking interaction between the phenyl ring and Gln203, as observed in the FIH:Zn:26 complex (Fig. 4B), due to the geometry of the α-pyrrolidine ring. These results indicate that a β-pyrrolidine ring, as in 26, is preferred for achieving both FIH inhibition and selectivity over PHD2, JMJD5, AspH and KDM4A. The addition of a methyl substituent at the C3 position of the β-pyrrolidine ring (29; Table 4, entry vii) results in a complete loss of FIH inhibition (IC50 > 100 μM), while the trans-4-methylpyrrolidine derivative (30; Table 4, entry viii) shows decreased levels of FIH inhibition (IC50: 1.7 μM), but improved selectivity for FIH inhibition over PHD2 and AspH relative to 26.
 | ||
| Scheme 2 Synthesis of N-hydroxythiazole derivatives 33–43.a,b a(a) N-Boc-pyrrolidine-3-carboxylic acid, T3P,82iPr2NEt, DMF, 0 °C to rt, 99%; (b) HCl/dioxane, 0 °C to rt, 83%; (c) PhSO2Cl, Et3N, CH2Cl2, 0 °C to rt, 78–95%; (d) mCPBA, CHCl3, rt, 24–67%; (e) LiOH, MeOH/H2O, 0 °C to rt, 28–82%. bChemical structures of R groups are shown in Table 5. | ||
The introduction of a chlorine substituent at the ortho- (33; IC50: 0.39 μM), meta- (34; IC50: 0.44 μM) or para-position (35; IC50: 0.46 μM) of the phenyl ring of 26 had negligible effects on inhibitor potency (Table 5, entries ii–iv). Similarly, FIH inhibition was not affected by electron donating (36; IC50: 0.46 μM) or electron withdrawing (37; IC50: 0.47 μM) substituents at the para-position of the phenyl ring (Table 5, entries v and vi), although selectivity over JMJD5 was enhanced ∼5-fold for trifluoromethyl-substituted 37, relative to 26. Cyclopentyl derivative 43 manifested reduced levels of FIH inhibition (IC50: 0.81 μM, Table 5; entry xii), which might relate to the loss of the amide-π stacking interaction between the phenyl ring of 26 and Gln203, as observed in the FIH:Zn:26 complex structure.
| Entry | Cmpda |
|
IC50 [μM]b | ||||
|---|---|---|---|---|---|---|---|
| R | FIHc | PHD2d | AspHe | KDM4Af | JMJD5g | ||
| a All chiral N-hydroxythiazole derivatives were prepared as racemic mixtures. b Mean average ± SD of two independent experiments (each composed of technical duplicates). c Using 0.15 μM FIH, 10.0 μM 2OG and 5.0 μM HIF-1α C-TAD788–822.61 d Using 0.15 μM PHD2181–426, 10.0 μM 2OG and 5.0 μM HIF-1α CODD556–574.61 e Using 0.05 μM His6-AspH315–758, 3.0 μM 2OG and 1.0 μM hFX-CP101–119.58 f Using 0.15 μM KDM4A, 10.0 μM 2OG and 10.0 μM H31–15K9me31–15.69 g Using 0.15 μM JMJD5, 2.0 μM 2OG and 2.0 μM RSP6128–148.70 Inhibition assays were performed using SPE-MS as described in the ESI. | |||||||
| i | 26 |
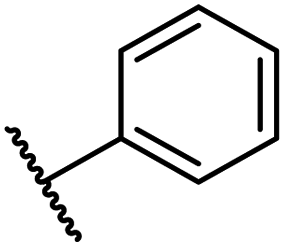
|
0.45 ± 0.17 | 12.4 ± 0.8 | 37.8 ± 11.3 | >100 | 12.5 ± 1.0 |
| ii | 33 |
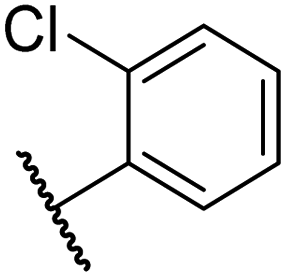
|
0.39 ± 0.04 | 8.0 ± 0.2 | 30.3 ± 12.6 | >100 | 11.9 ± 1.8 |
| iii | 34 |
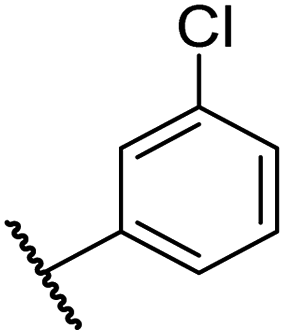
|
0.44 ± 0.03 | 14.6 ± 3.2 | 35.0 ± 0.0 | >100 | 18.5 ± 1.0 |
| iv | 35 |
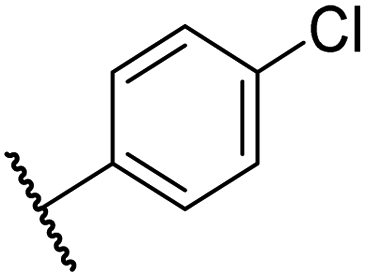
|
0.46 ± 0.01 | 9.2 ± 0.4 | 28.8 ± 8.9 | >100 | 37.7 ± 0.9 |
| v | 36 |
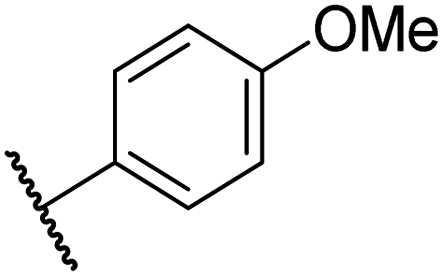
|
0.46 ± 0.01 | 9.8 ± 0.5 | 36.6 ± 2.8 | >100 | 16.0 ± 6.8 |
| vi | 37 |
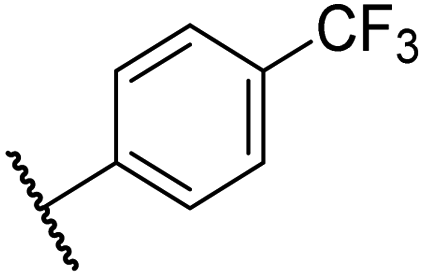
|
0.47 ± 0.07 | 15.2 ± 3.6 | 36.0 ± 0.7 | >100 | 63.2 ± 6.1 |
| vii | 38 |
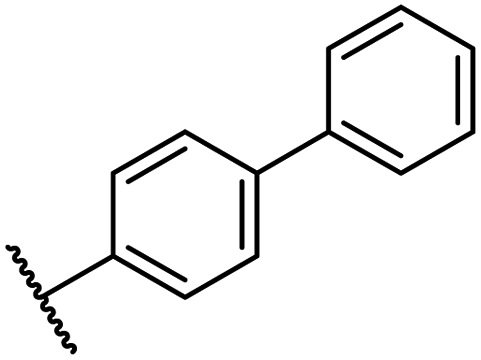
|
0.26 ± 0.01 | 3.1 ± 0.1 | 30.2 ± 10.9 | >100 | 31.9 ± 2.0 |
| viii | 39 |
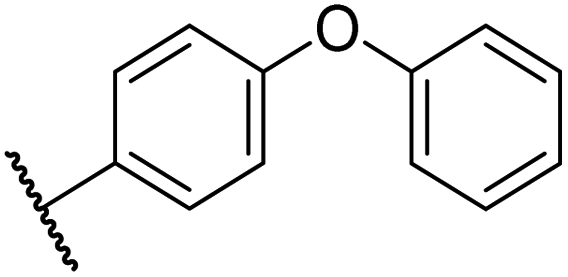
|
0.29 ± 0.03 | 3.1 ± 0.2 | 29.8 ± 4.3 | >100 | 8.6 ± 0.2 |
| ix | 40 |
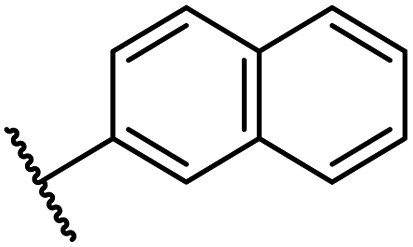
|
0.45 ± 0.08 | 5.2 ± 1.1 | 40.1 ± 2.2 | >100 | 62.8 ± 0.4 |
| x | 41 |
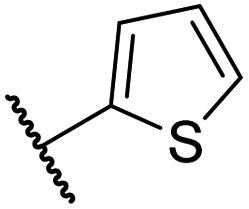
|
0.47 ± 0.07 | 11.2 ± 2.3 | 30.3 ± 8.0 | >100 | 15.4 ± 7.1 |
| xi | 42 |
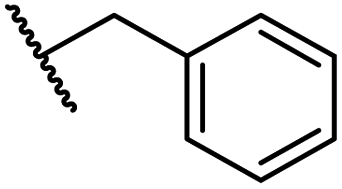
|
0.28 ± 0.02 | 6.9 ± 0.5 | 43.1 ± 18.7 | >100 | 7.2 ± 2.8 |
| xii | 43 |
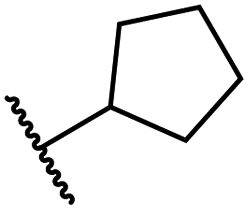
|
0.81 ± 0.18 | 9.9 ± 0.5 | 35.5 ± 3.2 | >100 | 17.4 ± 6.0 |
The most potent FIH inhibitors identified from this series were biphenyl sulfonamide derivative 38 (IC50: 0.26 μM), ether 39 (IC50: 0.29 μM), and the benzyl derivative 42 (IC50: 0.28 μM) (Table 5; entries vii, viii and xi). These compounds displayed comparable levels of FIH inhibition to the (partially) selective FIH inhibitor NOFD (IC50: 0.24 μM) and the broad-spectrum 2OG oxygenase inhibitor NOG (IC50: 0.36 μM), and were ∼10-fold more potent than 2,4-PDCA (IC50: 5.0 μM)54 (Table 6, entries i–iii). Excellent selectivity for FIH inhibition over AspH and KDM4A inhibition was also observed (>100-fold and >300-fold, respectively); 42 manifested ∼25-fold selectivity for FIH inhibition over PHD2 and JMJD5 inhibition.
| Entry | Cmpd | IC50 [μM]a | ||||
|---|---|---|---|---|---|---|
| FIHb | PHD2c | AspHd | KDM4Ae | JMJD5f | ||
| a Mean average ± SD of two independent experiments (each composed of technical duplicates). b Using 0.15 μM FIH, 10.0 μM 2OG and 5.0 μM HIF-1α C-TAD788–822.61 c Using 0.15 μM PHD2181–426, 10.0 μM 2OG and 5.0 μM HIF-1α CODD556–574.61 d Using 0.05 μM His6-AspH315–758, 3.0 μM 2OG and 1.0 μM hFX-CP101–119.58 e Using 0.15 μM KDM4A, 10.0 μM 2OG and 10.0 μM H31–15K9me31–15.69 f Using 0.15 μM JMJD5, 2.0 μM 2OG and 2.0 μM RSP6128–148.70 Inhibition assays were performed using SPE-MS as described in the ESI. | ||||||
| i | 2,4-PDCA | 5.0 ± 2.1 (ref. 54) | 5.3 ± 3.4 (ref. 54) | 0.03 ± 0.01 (ref. 54) | 0.10 ± 0.00 | 0.33 ± 0.07 (ref. 70) |
| ii | NOG | 0.36 ± 0.03 | 12.3 ± 4.4 | 1.1 ± 0.3 (ref. 58) | 22.1 ± 1.1 | 0.15 ± 0.02 (ref. 70) |
| iii | NOFD | 0.24 ± 0.02 | >100 | 15.5 ± 1.2 (ref. 60) | 14.1 ± 0.0 | >100 (ref. 70) |
| iv | BNS | 0.30 ± 0.07 | 0.11 ± 0.00 | 3.4 ± 0.1 | 67.4 ± 39.8 | 0.25 ± 0.01 |
| v | 26 | 0.45 ± 0.17 | 12.4 ± 0.8 | 37.8 ± 11.3 | >100 | 12.5 ± 1.0 |
| vi | 38 | 0.26 ± 0.01 | 3.1 ± 0.1 | 30.2 ± 10.9 | >100 | 31.9 ± 2.0 |
| vii | 39 | 0.29 ± 0.03 | 3.1 ± 0.2 | 29.8 ± 4.3 | >100 | 8.6 ± 0.2 |
| viii | 42 | 0.28 ± 0.02 | 6.9 ± 0.5 | 43.1 ± 18.7 | >100 | 7.2 ± 2.8 |
Interestingly, the results indicate that 42 exhibits a different selectivity profile compared to NOFD with respect to the other 2OG oxygenases tested. NOFD displays greater selectivity for inhibition of FIH over PHD2 and JMJD5 (NOFD: >300-fold selective; 42: ∼25-fold selective, as judged by IC50 values; Table 6). Whereas, 42 manifests improved selectivity for AspH (NOFD: ∼65-fold selective; 42: >100-fold selective) and KDM4A (NOFD: ∼60-fold selective; 42: >300-fold selective). The difference in 2OG oxygenase selectivity likely reflects the different binding modes of NOFD compared with the N-hydroxythiazole-derived FIH inhibitors, as indicated by superimposition of the FIH:Fe:NOFD and FIH:Zn:26 and complex structures (Fig. 4F).
Crystallographic studies have shown that the benzyl side chain of NOFD, which occupies a hydrophobic pocket in the FIH active site (formed by Tyr102, Tyr145, Gln147 and Leu186; ESI Fig. S11†), which has not yet been observed in the active sites of some other 2OG oxygenases, including PHD2, is responsible for the selectivity of NOFD for FIH inhibition over PHD2. In the reported PHD2:Mn:NOG complex structure (PDB ID: 5L9R),79 the pro-R methylene hydrogen atom of NOG is orientated towards the side chain of Leu343, which would likely clash with the benzyl substituent of NOFD, so preventing efficient binding of NOFD to PHD2. By contrast, we propose that the FIH selectivity of 26 and 42 arises, at least in part, due to the rigidity of the β-pyrrolidine ring and the formation of productive interactions with the side chains of residues involved in FIH substrate recognition, i.e. Gln203 and Trp296.
Cellular studies
Quantitative real time PCR (qRT-PCR) analyses revealed a dose-dependent increase in EGLN3 mRNA levels following the treatment of human hepatocyte carcinoma-derived Hep3B cells with 38, 39, 42, or 42b (Fig. 5A). At 50 μM, carboxylic acids 38, 39 and 42 increased EGLN3 expression by ∼2.5-fold relative to the negative inhibition control (i.e. DMSO). An apparently greater increase (∼3.4-fold) was observed for the ethyl ester 42b at 50 μM. By comparison, use of 50 μM DM-NOFD increased EGLN3 levels by ∼1.8-fold, indicating the N-hydroxythiazole derivatives manifest similar, if not greater, cellular efficacy compared to DM-NOFD (Fig. 5A). Since they also display weak PHD2 inhibition (Table 6), it cannot be ruled out that 38, 39, 42 or 42b cause weak HIF-α upregulation, that, along with FIH inhibition, contributes to the upregulation of EGLN3 levels observed in their presence. Note, however, that NOFD upregulates EGLN3 levels and has little, if any, PHD2 inhibition activity (Table 6).
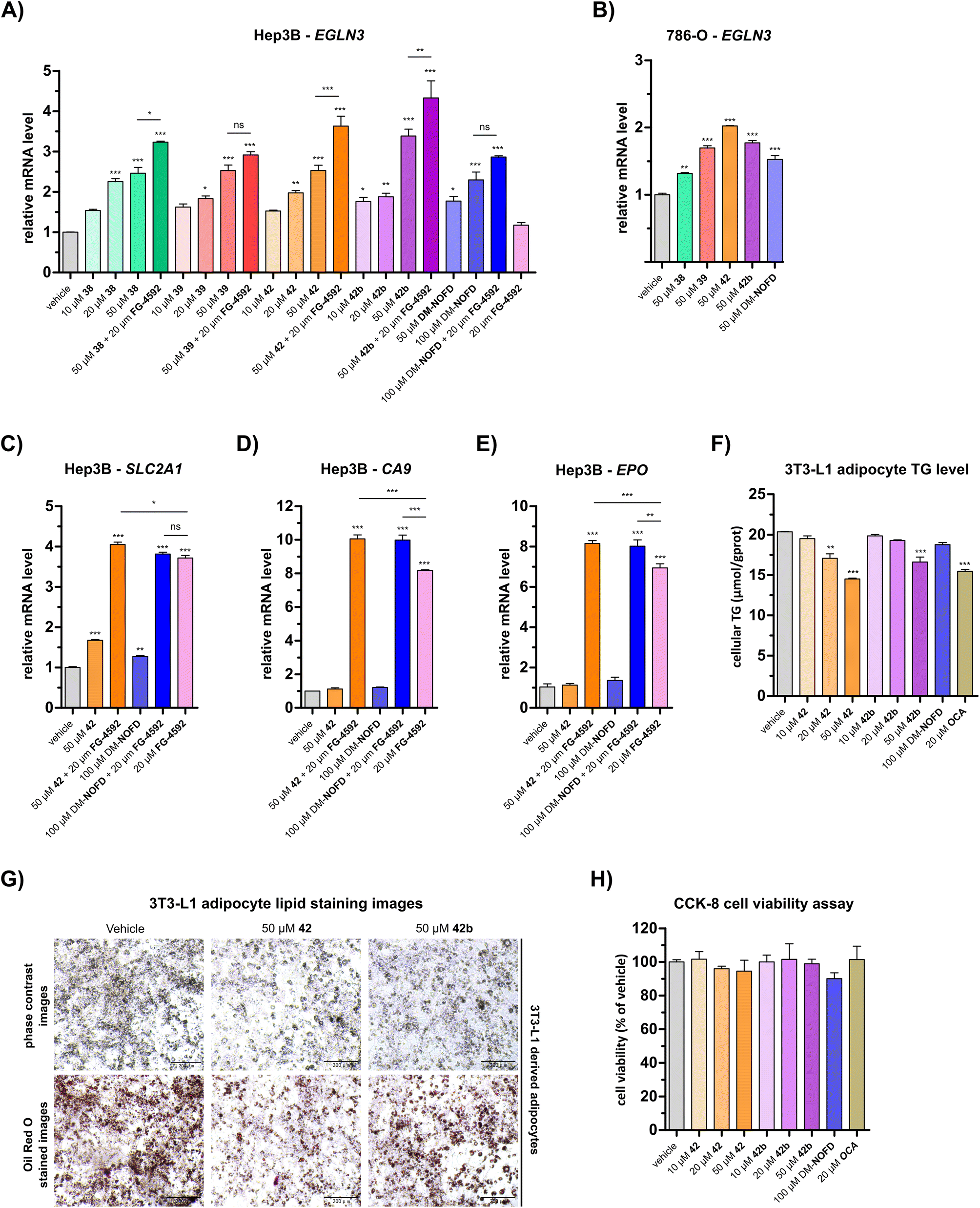 | ||
| Fig. 5 N-Hydroxythiazole-based FIH inhibitors modulate HIF target gene expression in cells and reduce adipocyte lipid accumulation. (A and B) Quantitative Real Time PCR (qRT-PCR) analyses showing the effects of N-hydroxythiazole-based FIH inhibitors 38, 39, 42 and the ethyl ester prodrug form of 42 (i.e.42b), dimethyl N-oxalyl-D-phenylalanine (DM-NOFD) and FG-4592 on prolyl hydroxylase domain-containing protein 3 (EGLN3) expression levels in (A) Hep3B and (B) 786-O cells relative to a negative inhibition control (DMSO). (C–E) qRT-PCR analyses showing the effects of 42, DM-NOFD, and FG-4592 on (C) solute carrier family 2 member 1 (SLC2A1), (D) carbonic anhydrase 9 (CA9) and (E) erythropoietin (EPO) expression levels in Hep3B cells relative to negative inhibition control (DMSO). (F) The cellular triglyceride (TG) levels of 3T3-L1 derived adipocytes following treatment with 42, 42b, DM-NOFD, the reported farnesoid X receptor (FXR) agonist obeticholic acid (OCA) and a negative inhibition control (DMSO), as determined by a triglyceride colorimetric assay. (G) Representative images of 3T3-L1 derived adipocytes treated with 42 and 42b, after staining with Oil Red O. Scale bar, 200 μm. (H) The effects of 42, 42b, DM-NOFD and OCA on 3T3-L1 derived adipocyte cell viability, as measured using the CCK-8 cell viability assay.95 Values shown are percentages (%) relative to the negative inhibition control (DMSO). Mean average ± SD of three independent experiments are shown. Results were analysed with one-way ANOVA followed by Tukey's multiple comparison test (ns, p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001). Cell-based studies were performed as described in the ESI.† | ||
We investigated the effect of 38, 39, 42 and 42b in the presence of the PHD inhibitor FG-4592 (FG-4592, roxadustat),52 which stimulates an increase in HIF-α levels, to explore whether the effect on EGLN3 expression would alter. When co-administered with 38, 42 or 42b, FG-4592 caused an greater increase in EGLN3 upregulation relative to the effect of 38, 42 or 42b alone (Fig. 5A); FG-4592 alone had no effect on Hep3B EGLN3 levels. These results support the proposal that FIH catalysis negatively regulates expression of the HIF target gene EGLN3 through a HIF-dependent mechanism. Interestingly, no additional effect on EGLN3 levels was observed for FG-4592 with either 39 or DM-NOFD, even at 100 μM DM-NOFD, compared to use of 39 or DM-NOFD alone, an observation requiring further investigation, and one which illustrates the complexity of the biochemistry of HIF transcriptional regulation. We also investigated the effect of the N-hydroxythiazole-derived FIH inhibitors 38, 39, 42 and 42b, and DM-NOFD on EGLN3 gene expression in VHL-deficient renal cell carcinoma-derived 786-O cells. Due to the absence of VHL, PHD-mediated HIF degradation does not operate in 786-O cells leading to elevated HIF levels. As with Hep3B cells, we observed upregulation of EGLN3 with all compounds tested, including DM-NOFD (Fig. 5B).
The effect of 42 and DM-NOFD on the expression of three additional proposed HIF target genes in Hep3B cells,17i.e. solute carrier family 2 member 1 (SLC2A1; Fig. 5C), carbonic anhydrase 9 (CA9; Fig. 5D) and erythropoietin (EPO; Fig. 5E), with and without FG-4592, was subsequently investigated by qPCR. SLC2A1 levels were increased significantly with FG-4592 (∼4-fold), and, to a lesser extent with 42 (∼1.5-fold) and DM-NOFD (∼1.3-fold). EPO and CA9 were also strongly upregulated by FG-4592 (∼7-fold and ∼8-fold, respectively) and showed no response to treatment with either 42 or DM-NOFD. By contrast, an increase in CA9 and, to a lesser extent, EPO levels was observed when 42 and DM-NOFD were administered in combination with FG-4592, compared with when FG-4592 was used alone. By contrast, no synergistic effect was observed for 42 and DM-NOFD in combination with FG-4592 on SLC2A1 gene expression.
Overall, the cellular results are in agreement with previous work that implying dual inhibition of both FIH and PHDs may be necessary to induce substantial expression of some, but not necessarily all, HIF target genes. Note, it cannot be ruled out that other mechanisms, including inhibition of the PHDs or other 2OG oxygenases potentially involved in HIF target gene expression by 42 or DM-NOFD, or (e.g.) inhibitor mediated effects on the location or the availability of HIF-α isoforms, may contribute towards the observed effects.
Conclusions
The physiological roles of FIH are incompletely understood, including how its substrate and, potentially, its 2-oxo acid cosubstrate60 promiscuity relate to its role in hypoxia sensing. Potent and selective small-molecule FIH inhibitors will be useful for in vivo functional assignment studies. Reported studies on the cellular roles of FIH have employed a prodrug diester form of the small-molecule NOFD,17,29 which is a derivative of the broad-spectrum 2OG oxygenase inhibitor NOG. However, we have previously shown that NOFD inhibits other 2OG oxygenases than FIH, including AspH,58 albeit substantially less efficiently. Nonetheless, this observation indicates that NOFD may not be perfectly selective for FIH inhibition considering that ∼60–70 2OG oxygenases are present in humans.96 Thus, we were interested in identifying alternative scaffolds suitable for optimisation as in vivo active FIH inhibitors.We investigated reported PHD inhibitors for FIH inhibition on the basis that PHD inhibitors such as daprodustat are reported to inhibit both PHDs and FIH,56 to identify novel lead structures for the development of potent and selective FIH inhibitors (ESI Table S1†). Interestingly, we observed that the reported N-hydroxythiazole-based PHD inhibitor BNS33 inhibits other 2OG oxygenases than the PHDs, including FIH, and can thus be considered as a relatively broad-spectrum 2OG oxygenase inhibitor (Table 1, entry iii). This observation suggests that previous cellular studies performed with BNS(-derivatives) should be interpreted with care,17,80 including with respect to potential toxicity.
N-Hydroxythiazoles are chemically interesting metalloenzyme inhibitors because of the heavily functionalised and polar nature of the core heterocycle. Thus, BNS was an attractive lead structure for the development of selective FIH inhibitors, inter alia because its scaffold can be modified at multiple positions. Rigidification of the BNS structure and replacement of its sulfone and naphthalene groups afforded N-hydroxythiazoles that display potent FIH inhibition (IC50 < 0.3 μM) and a substantially improved selectivity for FIH inhibition over PHD2 compared with BNS (∼25-fold for 42vs. ∼0.4-fold for BNS). High levels of selectivity were also achieved over JMJD5 (∼25-fold), KDM4A (>300-fold) and AspH (∼100-fold), the latter being of interest as AspH catalyses, like FIH, the β-hydroxylation of asparaginyl residues.71,72
Importantly, MS studies with isolated recombinant human 2OG oxygenases reveal that the N-hydroxythiazole-based FIH inhibitors show a different selectivity profile for inhibiting 2OG oxygenases than does NOFD (Table 6),77 an observation that reflects the differences in their FIH binding modes, as observed in co-crystal structures with FIH (Fig. 2 and 4). Analysis of the FIH:Zn:26 complex structure suggests that the FIH inhibition selectivity of 26, and by implication 42, is as a result, at least in part, of interactions formed between 26 and residues involved in FIH substrate recognition, i.e. Gln203 and Tyr296. By contrast, NOFD achieves selectivity for FIH through its benzyl side chain, which occupies a hydrophobic pocket formed by FIH residues Tyr102, Tyr145, Gln147 and Leu186 (ESI Fig. S11†).29 This observation implies there are, at least, two complementary strategies that can be employed to achieve selectivity for FIH inhibition over other 2OG oxygenases.
The N-hydroxythiazole-based FIH inhibitors have potential to help identify the physiologically relevant phenotypes of FIH inhibition, together with other inhibitors such as DM-NOFD. We showed that the FIH inhibitors 38, 39 and 42, and the ethyl ester prodrug form of 42 (i.e.42b) induce a dose-dependent increase in EGLN3 expression, a reported FIH-dependent HIF target gene,17,56 which was comparable to that induced by DM-NOFD, the dimethyl ester prodrug of NOFD (Fig. 5A and B). It also appeared that both 42 and 42b reduced lipid accumulation in 3T3-L1 derived adipocytes (Fig. 5F and G), an observation which is consistent with the proposed function of FIH as a regulator of cellular metabolism.48 The combined results thus indicate that the N-hydroxythiazole-based FIH inhibitors have potential for enabling cell-based and in vivo studies directed at investigating the potential therapeutic benefit of FIH inhibition. However, given the complexity of HIF and FIH biochemistry in cells, it cannot be ruled out that other mechanisms contribute to the biologically observed effects.
Crystallographic analyses in combination with docking studies guided compound design and will be valuable for future FIH inhibitor development programs. The FIH:Zn:N-hydroxythiazole complex structures indicate that the N-hydroxythiazole-based inhibitors bind at the FIH active site via competitive displacement of 2OG and its HIF-α substrate. The limited inhibitory activity of truncated N-hydroxythiazole derivative 14 suggests that the formation of productive interactions with the HIF-1α substrate binding site, for example with the side chains of Gln203 and Trp296, as observed in the FIH:Zn:26 structure, is important for efficient FIH inhibition (Fig. 4B). Efficient binding to residues that engage the HIF-1α substrate may be responsible for the increased FIH potency of N-hydroxythiazoles compared with other PHD inhibitors, such as daprodustat, that appear to bind FIH in a similar manner.56
Given the modular structure and broad-spectrum 2OG oxygenase inhibitory activity of BNS, it is likely that the N-hydroxythiazole scaffold can be modified to generate selective inhibitors of other 2OG oxygenases including PHD2, JMJD5 or, at least some, JmjC KDMs. The latter is of interest because of potential roles for JmjC KDMs in the hypoxic response.97 Note that the addition of substituents to the broad-spectrum 2OG oxygenase inhibitors NOG and 2,4-PDCA has resulted in development of inhibitors of FIH29 and JMJD5,98 respectively, which display an improved selectivity profile with respect to the parent compounds.
The FIH:Zn:N-hydroxythiazole complex structures also showed that N-hydroxythiazoles can bind to the active site metal of FIH in a bidentate manner through the exocyclic nitrogen atom and nucleophilic hydroxyl group of the N-hydroxythiazole unit. Although previous studies have demonstrated the promising biological activity of N-hydroxythiazole-derived compounds, in addition to PHD2 inhibition,33 for example antibacterial properties99 and inhibition of metallo-β-lactamases (MBLs),81 their ability to bind metals in this manner had not yet been observed in crystallographic studies. It is likely that this metal coordination motif may be useful in the development of inhibitors for other metal-containing enzymes, including for 2OG oxygenases and beyond.
Abbreviations
| FIH | Factor inhibiting hypoxia-inducible factor-α |
| HIF | Hypoxia-inducible factor |
| 2OG | 2-Oxoglutarate |
| PHD | Prolyl hydroxylase domain-containing protein |
| AspH | Aspartate/asparagine-β-hydroxylase |
| KDM | N ε-Lysine demethylase |
| C-TAD | C-Terminal activation domain |
| NOG | N-Oxalylglycine |
| EPO | Erythropoietin |
| PDCA | Pyridine-2,4-dicarboxylic acid |
| (DM)-NOFD | (Dimethyl) N-oxalyl-D-phenylalanine |
Data availability
The crystal structure data for the FIH:Zn:BNS, FIH:Zn:20 and FIH:Zn:26 complex structures have been deposited in the protein data bank under PDB accession codes: 8K71 (FIH:Zn:BNS), 8K72 (FIH:Zn:20), and 8K73 (FIH:Zn:26).Author contributions
T. P. C., and R. Z. R. T. synthesised the inhibitors, with assistance from J. H. M. T. P. C. carried out modelling studies/analysis of crystal structures. T. P. C., A. T., and L. B. performed the SPE-MS assays; T. P. C., R. Z. R. T., and W. F. performed the FIH:N-hydroxythiazole co-crystallizations. Y. N. solved and refined the FIH:N-hydroxythiazole complex structures. E. S, L. B., G. F., and A. B. produced and purified recombinant proteins. Y. W. performed the cell-based experiments. L. B., X. Z., and C. J. S. supervised the research. T. P. C. (original draft), L. B., and C. J. S. wrote the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank Dr Michael A. McDonough and Dr Rashed Chowdhury for initial studies on crystallography of N-hydroxythiazole complexes. This research was funded in part by the Wellcome Trust (106244/Z/14/Z). We thank Cancer Research UK (C8717/A18245) and the Biotechnology and Biological Sciences Research Council (BB/J003018/1 and BB/R000344/1) for funding. T. P. C. thanks the Centre for Doctoral Training in Synthesis for Biology and Medicine for a studentship, generously supported by GlaxoSmithKline, MSD, Syngenta, and Vertex. T. P. C. thanks the Royal Commission for the Exhibition 1851 for an industrial fellowship. X. Z. thanks the National Natural Science Foundation of China (grants 81973173, 82273769, and 82322062) and the Jiangsu Provincial Funds for Distinguished Young Scientists (grant BK20211527) for funding. We thank the Diamond Light Source and staff for allocation of beam time and support. We thank the MS and NMR facilities from the Chemistry Research Laboratory, University of Oxford, for excellent support.References
- W. G. Kaelin Jr. and P. J. Ratcliffe, Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway, Mol. Cell, 2008, 30, 393–402 CrossRef PubMed.
- J. López-Barneo, R. Pardal and P. Ortega-Sáenz, Cellular mechanism of oxygen sensing, Annu. Rev. Physiol., 2001, 63, 259–287 CrossRef PubMed.
- R. H. Wenger, Mammalian oxygen sensing, signalling and gene regulation, J. Exp. Biol., 2000, 203, 1253–1263 CrossRef CAS PubMed.
- R. K. Bruick, Oxygen sensing in the hypoxic response pathway: regulation of the hypoxia-inducible transcription factor, Genes Dev., 2003, 17, 2614–2623 CrossRef CAS PubMed.
- M. Safran and W. G. Kaelin Jr., HIF hydroxylation and the mammalian oxygen-sensing pathway, J. Clin. Invest., 2003, 111, 779–783 CrossRef CAS PubMed.
- C. J. Schofield and P. J. Ratcliffe, Oxygen sensing by HIF hydroxylases, Nat. Rev. Mol. Cell Biol., 2004, 5, 343–354 CrossRef CAS PubMed.
- M. Nangaku and K.-U. Eckardt, Hypoxia and the HIF system in kidney disease, J. Mol. Med., 2007, 85, 1325–1330 CrossRef PubMed.
- O. Martínez-Sáez, P. G. Borau, T. Alonso-Gordoa, J. Molina-Cerrillo and E. Grande, Targeting HIF-2 α in clear cell renal cell carcinoma: a promising therapeutic strategy, Crit. Rev. Oncol. Hematol., 2017, 111, 117–123 CrossRef PubMed.
- A. C. R. Epstein, J. M. Gleadle, L. A. McNeill, K. S. Hewitson, J. O'Rourke, D. R. Mole, M. Mukherji, E. Metzen, M. I. Wilson, A. Dhanda, Y.-M. Tian, N. Masson, D. L. Hamilton, P. Jaakkola, R. Barstead, J. Hodgkin, P. H. Maxwell, C. W. Pugh, C. J. Schofield and P. J. Ratcliffe, C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation, Cell, 2001, 107, 43–54 CrossRef CAS PubMed.
- W.-C. Hon, M. I. Wilson, K. Harlos, T. D. W. Claridge, C. J. Schofield, C. W. Pugh, P. H. Maxwell, P. J. Ratcliffe, D. I. Stuart and E. Y. Jones, Structural basis for the recognition of hydroxyproline in HIF-1α by pVHL, Nature, 2002, 417, 975–978 CrossRef CAS PubMed.
- J.-H. Min, H. Yang, M. Ivan, F. Gertler, W. G. Kaelin Jr. and N. P. Pavletich, Structure of an HIF-1α-pVHL complex: hydroxyproline recognition in signaling, Science, 2002, 296, 1886–1889 CrossRef CAS PubMed.
- K. Tanimoto, Y. Makino, T. Pereira and L. Poellinger, Mechanism of regulation of the hypoxia-inducible factor-1α by the von Hippel-Lindau tumor suppressor protein, EMBO J., 2000, 19, 4298–4309 CrossRef CAS PubMed.
- A. A. Joharapurkar, V. B. Pandya, V. J. Patel, R. C. Desai and M. R. Jain, Prolyl hydroxylase inhibitors: a breakthrough in the therapy of anemia associated with chronic diseases, J. Med. Chem., 2018, 61, 6964–6982 CrossRef CAS PubMed.
- K. S. Hewitson, L. A. McNeill, M. V. Riordan, Y.-M. Tian, A. N. Bullock, R. W. Welford, J. M. Elkins, N. J. Oldham, S. Bhattacharya, J. M. Gleadle, P. J. Ratcliffe, C. W. Pugh and C. J. Schofield, Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family, J. Biol. Chem., 2002, 277, 26351–26355 CrossRef CAS PubMed.
- D. Lando, D. J. Peet, J. J. Gorman, D. A. Whelan, M. L. Whitelaw and R. K. Bruick, FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor, Genes Dev., 2002, 16, 1466–1471 CrossRef CAS PubMed.
- P. C. Mahon, K. Hirota and G. L. Semenza, FIH-1: a novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity, Genes Dev., 2001, 15, 2675–2686 CrossRef CAS PubMed.
- M. C. Chan, N. E. Ilott, J. Schödel, D. Sims, A. Tumber, K. Lippl, D. R. Mole, C. W. Pugh, P. J. Ratcliffe, C. P. Ponting and C. J. Schofield, Tuning the transcriptional response to hypoxia by inhibiting hypoxia-inducible factor (HIF) prolyl and asparaginyl hydroxylases, J. Biol. Chem., 2016, 291, 20661–20673 CrossRef CAS PubMed.
- M. Hirsilä, P. Koivunen, V. Günzler, K. I. Kivirikko and J. Myllyharju, Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor, J. Biol. Chem., 2003, 278, 30772–30780 CrossRef PubMed.
- P. Koivunen, M. Hirsilä, V. Günzler, K. I. Kivirikko and J. Myllyharju, Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases, J. Biol. Chem., 2004, 279, 9899–9904 CrossRef CAS PubMed.
- F. Dayan, D. Roux, M. C. Brahimi-Horn, J. Pouyssegur and N. M. Mazure, The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1α, Cancer Res., 2006, 66, 3688–3698 CrossRef CAS PubMed.
- H. Tarhonskaya, R. Chowdhury, I. K. H. Leung, N. D. Loik, J. S. O. McCullagh, T. D. W. Claridge, C. J. Schofield and E. Flashman, Investigating the contribution of the active site environment to the slow reaction of hypoxia-inducible factor prolyl hydroxylase domain 2 with oxygen, Biochem. J., 2014, 463, 363–372 CrossRef CAS PubMed.
- E. Flashman, L. M. Hoffart, R. B. Hamed, J. M. Bollinger Jr., C. Krebs and C. J. Schofield, Evidence for the slow reaction of hypoxia-inducible factor prolyl hydroxylase 2 with oxygen, FEBS J., 2010, 277, 4089–4099 CrossRef CAS PubMed.
- G. L. Wang and G. L. Semenza, Molecular basis of hypoxia-induced erythropoietin expression, Curr. Opin. Hematol., 1996, 3, 156–162 CrossRef CAS PubMed.
- Y. Liu, S. R. Cox, T. Morita and S. Kourembanas, Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells: identification of a 5’ enhancer, Circ. Res., 1995, 77, 638–643 CrossRef CAS PubMed.
- H. Tarhonskaya, A. P. Hardy, E. A. Howe, N. D. Loik, H. B. Kramer, J. S. O. McCullagh, C. J. Schofield and E. Flashman, Kinetic investigations of the role of factor inhibiting hypoxia-inducible factor (FIH) as an oxygen sensor, J. Biol. Chem., 2015, 290, 19726–19742 CrossRef CAS PubMed.
- E. Berra, E. Benizri, A. Ginouvès, V. Volmat, D. Roux and J. Pouysségur, HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia, EMBO J., 2003, 22, 4082–4090 CrossRef CAS PubMed.
- A. Conejo-Garcia, M. A. McDonough, C. Loenarz, L. A. McNeill, K. S. Hewitson, W. Ge, B. M. Liénard, C. J. Schofield and I. J. Clifton, Structural basis for binding of cyclic 2-oxoglutarate analogues to factor-inhibiting hypoxia-inducible factor, Bioorg. Med. Chem. Lett., 2010, 20, 6125–6128 CrossRef CAS PubMed.
- J. M. Elkins, K. S. Hewitson, L. A. McNeill, J. F. Seibel, I. Schlemminger, C. W. Pugh, P. J. Ratcliffe and C. J. Schofield, Structure of factor-inhibiting hypoxia-inducible factor (HIF) reveals mechanism of oxidative modification of HIF-1α, J. Biol. Chem., 2003, 278, 1802–1806 CrossRef CAS PubMed.
- M. A. McDonough, L. A. McNeill, M. Tilliet, C. A. Papamicaël, Q.-Y. Chen, B. Banerji, K. S. Hewitson and C. J. Schofield, Selective inhibition of factor inhibiting hypoxia-inducible factor, J. Am. Chem. Soc., 2005, 127, 7680–7681 CrossRef CAS PubMed.
- P. E. Pergola, B. S. Spinowitz, C. S. Hartman, B. J. Maroni and V. H. Haase, Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease, Kidney Int., 2016, 90, 1115–1122 CrossRef CAS PubMed.
- H. Beck, M. Jeske, K. Thede, F. Stoll, I. Flamme, M. Akbaba, J.-K. Ergüden, G. Karig, J. Keldenich, F. Oehme, H.-C. Militzer, I. V. Hartung and U. Thuss, Discovery of molidustat (BAY 85-3934): a small-molecule oral HIF-prolyl hydroxylase (HIF-PH) inhibitor for the treatment of renal anemia, ChemMedChem, 2018, 13, 988–1003 CrossRef CAS PubMed.
- R. A. Brigandi, B. Johnson, C. Oei, M. Westerman, G. Olbina, J. de Zoysa, S. D. Roger, M. Sahay, N. Cross, L. McMahon, V. Guptha, E. A. Smolyarchuk, N. Singh, S. F. Russ, S. Kumar, A. V. Borsukov, V. V. Marasaev, G. Prasad, G. Y. Timokhovskaya, E. V. Kolmakova, V. A. Dobronravov, E. V. Zakharova, G. Abraham, D. Packham, D. A. Zateyshchikov, G. P. Arutyunov, G. V. Volgina, K. S. Lipatov, D. V. Perlin, B. Cooper, T. Kumar Saha, O. A. Zagrebelnaya, K. S. Mehta, N. A. Koziolova, R. Fassett, N. P. Alexeeva and L. V. Lysenko, A novel hypoxia-inducible factor− prolyl hydroxylase inhibitor (GSK1278863) for anemia in CKD: a 28-day, phase 2A randomized trial, Am. J. Kidney Dis., 2016, 67, 861–871 CrossRef CAS PubMed.
- C. M. Tegley, V. N. Viswanadhan, K. Biswas, M. J. Frohn, T. A. N. Peterkin, C. Chang, R. W. Bürli, J. H. Dao, H. Veith, N. Rogers, S. C. Yoder, G. Biddlecome, P. Tagari, J. R. Allen and R. W. Hungate, Discovery of novel hydroxy-thiazoles as HIF-α prolyl hydroxylase inhibitors: SAR, synthesis, and modeling evaluation, Bioorg. Med. Chem. Lett., 2008, 18, 3925–3928 CrossRef CAS PubMed.
- V. L. Dengler, M. D. Galbraith and J. M. Espinosa, Transcriptional regulation by hypoxia inducible factors, Crit. Rev. Biochem. Mol. Biol., 2014, 49, 1 CrossRef CAS PubMed.
- A. Ortiz-Barahona, D. Villar, N. Pescador, J. Amigo and L. del Peso, Genome-wide identification of hypoxia-inducible factor binding sites and target genes by a probabilistic model integrating transcription-profiling data and in silico binding site prediction, Nucleic Acids Res., 2010, 38, 2332–2345 CrossRef CAS PubMed.
- P. Carmeliet, VEGF as a key mediator of angiogenesis in cancer, Oncology, 2005, 69, 4–10 CrossRef CAS PubMed.
- G. Dobrynin, T. E. McAllister, K. B. Leszczynska, S. Ramachandran, A. J. Krieg, A. Kawamura and E. M. Hammond, KDM4A regulates HIF-1 levels through H3K9me3, Sci. Rep., 2017, 7, 11094 CrossRef PubMed.
- M. E. Cockman, K. Lippl, Y.-M. Tian, H. B. Pegg, W. D. Figg Jr., M. I. Abboud, R. Heilig, R. Fischer, J. Myllyharju, C. J. Schofield and P. J. Ratcliffe, Lack of activity of recombinant HIF prolyl hydroxylases (PHDs) on reported non-HIF substrates, Elife, 2019, 8, e46490 CrossRef PubMed.
- M. E. Cockman, D. E. Lancaster, I. P. Stolze, K. S. Hewitson, M. A. McDonough, M. L. Coleman, C. H. Coles, X. Yu, R. T. Hay, S. C. Ley, C. W. Pugh, N. J. Oldham, N. Masson, C. J. Schofield and P. J. Ratcliffe, Posttranslational hydroxylation of ankyrin repeats in IκB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH), Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 14767–14772 CrossRef CAS PubMed.
- M. E. Cockman, J. D. Webb and P. J. Ratcliffe, FIH-dependent asparaginyl hydroxylation of ankyrin repeat domain-containing proteins, Ann. N. Y. Acad. Sci., 2009, 1177, 9–18 CrossRef CAS PubMed.
- K. Janke, U. Brockmeier, K. Kuhlmann, M. Eisenacher, J. Nolde, H. E. Meyer, H. Mairbäurl and E. Metzen, Factor inhibiting HIF-1 (FIH-1) modulates protein interactions of apoptosis-stimulating p53 binding protein 2 (ASPP2), J. Cell Sci., 2013, 126, 2629–2640 CAS.
- T. M. Leissing, A. P. Hardy, H. Chan, Y. Wang, A. Tumber, R. Chowdhury, T. Feng, M. L. Coleman, M. E. Cockman, H. B. Kramer, G. Berridge, R. Fischer, B. M. Kessler, P. J. Ratcliffe, X. Lu and C. J. Schofield, Factor inhibiting HIF can catalyze two asparaginyl hydroxylations in VNVN motifs of ankyrin fold proteins, J. Biol. Chem., 2022, 298, 102020 CrossRef CAS PubMed.
- M. L. Coleman, M. A. McDonough, K. S. Hewitson, C. Coles, J. Mecinovicí, M. Edelmann, K. M. Cook, M. E. Cockman, D. E. Lancaster, B. M. Kessler, N. J. Oldham, P. J. Ratcliffe and C. J. Schofield, Asparaginyl hydroxylation of the Notch ankyrin repeat domain by factor inhibiting hypoxia-inducible factor, J. Biol. Chem., 2007, 282, 24027–24038 CrossRef CAS PubMed.
- M. Yang, R. Chowdhury, W. Ge, R. B. Hamed, M. A. McDonough, T. D. W. Claridge, B. M. Kessler, M. E. Cockman, P. J. Ratcliffe and C. J. Schofield, Factor-inhibiting hypoxia-inducible factor (FIH) catalyses the post-translational hydroxylation of histidinyl residues within ankyrin repeat domains, FEBS J., 2011, 278, 1086–1097 CrossRef CAS PubMed.
- J. Kang, Y.-S. Chun, J. Huh and J.-W. Park, FIH permits NAA10 to catalyze the oxygen-dependent lysyl-acetylation of HIF-1α, Redox Biol., 2018, 19, 364–374 CrossRef CAS PubMed.
- M. Mantri, Z. Zhang, M. A. McDonough and C. J. Schofield, Autocatalysed oxidative modifications to 2-oxoglutarate dependent oxygenases, FEBS J., 2012, 279, 1563–1575 CrossRef CAS PubMed.
- M. Yang, W. Ge, R. Chowdhury, T. D. W. Claridge, H. B. Kramer, B. Schmierer, M. A. McDonough, L. Gong, B. M. Kessler, P. J. Ratcliffe, M. L. Coleman and C. J. Schofield, Asparagine and aspartate hydroxylation of the cytoskeletal ankyrin family is catalyzed by factor-inhibiting hypoxia-inducible factor, J. Biol. Chem., 2011, 286, 7648–7660 CrossRef CAS PubMed.
- J. Sim, A. S. Cowburn, A. Palazon, B. Madhu, P. A. Tyrakis, D. Macías, D. M. Bargiela, S. Pietsch, M. Gralla, C. E. Evans, T. Kittipassorn, Y. C. J. Chey, C. M. Branco, H. Rundqvist, D. J. Peet and R. S. Johnson, The factor inhibiting HIF asparaginyl hydroxylase regulates oxidative metabolism and accelerates metabolic adaptation to hypoxia, Cell Metab., 2018, 27, 898–913 CrossRef CAS PubMed.
- N. Masson, R. S. Singleton, R. Sekirnik, D. C. Trudgian, L. J. Ambrose, M. X. Miranda, Y.-M. Tian, B. M. Kessler, C. J. Schofield and P. J. Ratcliffe, The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity, EMBO Rep., 2012, 13, 251–257 CrossRef CAS PubMed.
- Y. L. Volkova, C. Pickel, A. E. Jucht, R. H. Wenger and C. C. Scholz, The asparagine hydroxylase FIH: a unique oxygen sensor, Antioxid. Redox Signaling, 2022, 37, 913–935 CrossRef CAS PubMed.
- P. H. Maxwell and K.-U. Eckardt, HIF prolyl hydroxylase inhibitors for the treatment of renal anaemia and beyond, Nat. Rev. Nephrol., 2016, 12, 157–168 CrossRef CAS PubMed.
- K. Wu, K. Zhou, Y. Wang, Y. Zhou, N. Tian, Y. Wu, D. Chen, D. Zhang, X. Wang, H. Xu and X. Zhang, Stabilization of HIF-1α by FG-4592 promotes functional recovery and neural protection in experimental spinal cord injury, Brain Res., 2016, 1632, 19–26 CrossRef CAS PubMed.
- Y. Wu, Z. Li, M. A. McDonough, C. J. Schofield and X. Zhang, Inhibition of the oxygen-sensing asparaginyl hydroxylase factor inhibiting hypoxia-inducible factor: a potential hypoxia response modulating strategy, J. Med. Chem., 2021, 64, 7189–7209 CrossRef CAS PubMed.
- L. Brewitz, A. Tumber, A. Thalhammer, E. Salah, K. E. Christensen and C. J. Schofield, Synthesis of novel pyridine-carboxylates as small-molecule inhibitors of human Aspartate/asparagine-β-hydroxylase, ChemMedChem, 2020, 15, 1139–1149 CrossRef CAS PubMed.
- M. C. Chan, O. Atasoylu, E. Hodson, A. Tumber, I. K. H. Leung, R. Chowdhury, V. Gómez-Pérez, M. Demetriades, A. M. Rydzik, J. Holt-Martyn, Y.-M. Tian, T. Bishop, T. D. W. Claridge, A. Kawamura, C. W. Pugh, P. J. Ratcliffe and C. J. Schofield, Potent and selective triazole-based inhibitors of the hypoxia-inducible factor prolyl-hydroxylases with activity in the murine brain, PLoS One, 2015, 10, e0132004 CrossRef PubMed.
- T.-L. Yeh, T. M. Leissing, M. I. Abboud, C. C. Thinnes, O. Atasoylu, J. P. Holt-Martyn, D. Zhang, A. Tumber, K. Lippl, C. T. Lohans, I. K. H. Leung, H. Morcrette, I. J. Clifton, T. D. W. Claridge, A. Kawamura, E. Flashman, X. Lu, P. J. Ratcliffe, R. Chowdhury, C. W. Pugh and C. J. Schofield, Molecular and cellular mechanisms of HIF prolyl hydroxylase inhibitors in clinical trials, Chem. Sci., 2017, 8, 7651–7668 RSC.
- C. J. Brereton, L. Yao, E. R. Davies, Y. Zhou, M. Vukmirovic, J. A. Bell, S. Wang, R. A. Ridley, L. S. N. Dean, O. G. Andriotis, F. Conforti, L. Brewitz, S. Mohammed, T. Wallis, A. Tavassoli, R. M. Ewing, A. Alzetani, B. G. Marshall, S. V. Fletcher, P. J. Thurner, A. Fabre, N. Kaminski, L. Richeldi, A. Bhaskar, C. J. Schofield, M. Loxham, D. E. Davies, Y. Wang and M. G. Jones, Pseudohypoxic HIF pathway activation dysregulates collagen structure-function in human lung fibrosis, Elife, 2022, 11, e69348 CrossRef CAS PubMed.
- L. Brewitz, A. Tumber, I. Pfeffer, M. A. McDonough and C. J. Schofield, Aspartate/asparagine-β-hydroxylase: a high-throughput mass spectrometric assay for discovery of small molecule inhibitors, Sci. Rep., 2020, 10, 8650 CrossRef CAS PubMed.
- N. R. Rose, E. C. Y. Woon, G. L. Kingham, O. N. F. King, J. Mecinović, I. J. Clifton, S. S. Ng, J. Talib-Hardy, U. Oppermann, M. A. McDonough and C. J. Schofield, Selective inhibitors of the JMJD2 histone demethylases: combined nondenaturing mass spectrometric screening and crystallographic approaches, J. Med. Chem., 2010, 53, 1810–1818 CrossRef CAS PubMed.
- Y. Nakashima, L. Brewitz, A. Tumber, E. Salah and C. J. Schofield, 2-Oxoglutarate derivatives can selectively enhance or inhibit the activity of human oxygenases, Nat. Commun., 2021, 12, 6478 CrossRef CAS PubMed.
- J. P. Holt-Martyn, R. Chowdhury, A. Tumber, T.-L. Yeh, M. I. Abboud, K. Lippl, C. T. Lohans, G. W. Langley, W. Figg Jr., M. A. McDonough, C. W. Pugh, P. J. Ratcliffe and C. J. Schofield, Structure-activity relationship and crystallographic studies on 4-hydroxypyrimidine HIF prolyl hydroxylase domain inhibitors, ChemMedChem, 2020, 15, 270–273 CrossRef CAS PubMed.
- N. R. Rose, M. A. McDonough, O. N. F. King, A. Kawamura and C. J. Schofield, Inhibition of 2-oxoglutarate dependent oxygenases, Chem. Soc. Rev., 2011, 40, 4364–4397 RSC.
- R. I. Dowell and E. M. Hadley, Novel inhibitors of prolyl 4-hydroxylase, J. Med. Chem., 1992, 35, 800–804 CrossRef CAS PubMed.
- J. H. Dao, R. J. M. Kurzeja, J. M. Morachis, H. Veith, J. Lewis, V. Yu, C. M. Tegley and P. Tagari, Kinetic characterization and identification of a novel inhibitor of hypoxia-inducible factor prolyl hydroxylase 2 using a time-resolved fluorescence resonance energy transfer-based assay technology, Anal. Biochem., 2009, 384, 213–223 CrossRef CAS PubMed.
- M. Yang, A. P. Hardy, R. Chowdhury, N. D. Loik, J. S. Scotti, J. S. O. McCullagh, T. D. W. Claridge, M. A. McDonough, W. Ge and C. J. Schofield, Substrate selectivity analyses of Factor Inhibiting Hypoxia-Inducible Factor, Angew. Chem., 2013, 125, 1744–1748 CrossRef.
- C. E. Dann III, R. K. Bruick and J. Deisenhofer, Structure of factor-inhibiting hypoxia-inducible factor 1: an asparaginyl hydroxylase involved in the hypoxic response pathway, Proc. Natl. Acad. Sci. U.S.A., 2002, 99, 15351–15356 CrossRef PubMed.
- W. D. Figg Jr., M. A. McDonough, R. Chowdhury, Y. Nakashima, Z. Zhang, J. P. Holt-Martyn, A. Krajnc and C. J. Schofield, Structural basis of prolyl hydroxylase domain inhibition by molidustat, ChemMedChem, 2021, 16, 2082–2088 CrossRef PubMed.
- S. Linke, C. Stojkoski, R. J. Kewley, G. W. Booker, M. L. Whitelaw and D. J. Peet, Substrate requirements of the oxygen-sensing asparaginyl hydroxylase factor-inhibiting hypoxia-inducible factor, J. Biol. Chem., 2004, 279, 14391–14397 CrossRef CAS PubMed.
- S. E. Hutchinson, M. V. Leveridge, M. L. Heathcote, P. Francis, L. Williams, M. Gee, J. Munoz-Muriedas, B. Leavens, A. Shillings, E. Jones, P. Homes, S. Baddeley, C.-w. Chung, A. Bridges and A. Argyrou, Enabling lead discovery for histone lysine demethylases by high-throughput RapidFire mass spectrometry, J. Biomol. Screening, 2012, 17, 39–48 CrossRef CAS PubMed.
- A. Tumber, E. Saleh, L. Brewitz, T. P. Corner and C. J. Schofield, Kinetic and inhibition studies on human Jumonji-C (JmjC) domain-containing protein 5, RSC Chem. Biol., 2023, 4, 399–413 RSC.
- J. Stenflo, E. Holme, S. Lindstedt, N. Chandramouli, L.-H. Huang, J. P. Tam and R. B. Merrifield, Hydroxylation of aspartic acid in domains homologous to the epidermal growth factor precursor is catalyzed by a 2-oxoglutarate-dependent dioxygenase, Proc. Natl. Acad. Sci. U.S.A., 1989, 86, 444–447 CrossRef CAS PubMed.
- L. Brewitz, B. C. Onisko and C. J. Schofield, Combined proteomic and biochemical analyses redefine the consensus sequence requirement for epidermal growth factor-like domain hydroxylation, J. Biol. Chem., 2022, 298, 102129 CrossRef CAS PubMed.
- H. Wang, X. Zhou, M. Wu, C. Wang, X. Zhang, Y. Tao, N. Chen and J. Zang, Structure of the JmjC-domain-containing protein JMJD5, Acta Crystallogr. D, 2013, 69, 1911–1920 CrossRef CAS PubMed.
- P. A. Del Rizzo, S. Krishnan and R. C. Trievel, Crystal structure and functional analysis of JMJD5 indicate an alternate specificity and function, Mol. Cell. Biol., 2012, 32, 4044–4052 CrossRef CAS PubMed.
- Z. Chen, J. Zang, J. Whetstine, X. Hong, F. Davrazou, T. G. Kutateladze, M. Simpson, Q. Mao, C.-H. Pan, S. Dai, J. Hagman, K. Hansen, Y. Shi and G. Zhang, Structural insights into histone demethylation by JMJD2 family members, Cell, 2006, 125, 691–702 CrossRef CAS PubMed.
- L. J. Walport, R. J. Hopkinson, R. Chowdhury, Y. Zhang, J. Bonnici, R. Schiller, A. Kawamura and C. J. Schofield, Mechanistic and structural studies of KDM-catalysed demethylation of histone 1 isotype 4 at lysine 26, FEBS Lett., 2018, 592, 3264–3273 CrossRef CAS PubMed.
- R. Chowdhury, J. I. Candela-Lena, M. C. Chan, D. J. Greenald, K. K. Yeoh, Y.-M. Tian, M. A. McDonough, A. Tumber, N. R. Rose, A. Conejo-Garcia, M. Demetriades, S. Mathavan, A. Kawamura, M. K. Lee, F. van Eeden, C. W. Pugh, P. J. Ratcliffe and C. J. Schofield, Selective small molecule probes for the hypoxia inducible factor (HIF) prolyl hydroxylases, ACS Chem. Biol., 2013, 8, 1488–1496 CrossRef CAS PubMed.
- I. Pfeffer, L. Brewitz, T. Krojer, S. A. Jensen, G. T. Kochan, N. J. Kershaw, K. S. Hewitson, L. A. McNeill, H. Kramer, M. Münzel, R. J. Hopkinson, U. Oppermann, P. A. Handford, M. A. McDonough and C. J. Schofield, Aspartate/asparagine-β-hydroxylase crystal structures reveal an unexpected epidermal growth factor-like domain substrate disulfide pattern, Nat. Commun., 2019, 10, 4910 CrossRef PubMed.
- R. Chowdhury, I. K. H. Leung, Y.-M. Tian, M. I. Abboud, W. Ge, C. Domene, F.-X. Cantrelle, I. Landrieu, A. P. Hardy, C. W. Pugh, P. J. Ratcliffe, T. D. W. Claridge and C. J. Schofield, Structural basis for oxygen degradation domain selectivity of the HIF prolyl hydroxylases, Nat. Commun., 2016, 7, 12673 CrossRef CAS PubMed.
- K. Thirstrup, S. Christensen, H. A. Møller, A. Ritzén, A.-L. Bergström, T. N. Sager and H. S. Jensen, Endogenous 2-oxoglutarate levels impact potencies of competitive HIF prolyl hydroxylase inhibitors, Pharmacol. Res., 2011, 64, 268–273 CrossRef CAS PubMed.
- A. Makena, S. S. van Berkel, C. Lejeune, R. J. Owens, A. Verma, R. Salimraj, J. Spencer, J. Brem and C. J. Schofield, Chromophore-linked substrate (CLS405): probing metallo-β-lactamase activity and inhibition, ChemMedChem, 2013, 8, 1923–1929 CrossRef CAS PubMed.
- H. Wissmann and H.-J. Kleiner, New peptide synthesis, Angew. Chem., Int. Ed., 1980, 19, 133–134 CrossRef.
- S. B. Hatch, C. Yapp, R. C. Montenegro, P. Savitsky, V. Gamble, A. Tumber, G. F. Ruda, V. Bavetsias, O. Fedorov, B. Atrash, F. Raynaud, R. Lanigan, L. Carmichael, K. Tomlin, R. Burke, S. M. Westaway, J. A. Brown, R. K. Prinjha, E. D. Martinez, U. Oppermann, C. J. Schofield, C. Bountra, A. Kawamura, J. Blagg, P. E. Brennan, O. Rossanese and S. Müller, Assessing histone demethylase inhibitors in cells: lessons learned, Epigenet. Chromatin, 2017, 10, 9 CrossRef PubMed.
- G. Joberty, M. Boesche, J. A. Brown, D. Eberhard, N. S. Garton, P. G. Humphreys, T. Mathieson, M. Muelbaier, N. G. Ramsden, V. Reader, A. Rueger, R. J. Sheppard, S. M. Westaway, M. Bantscheff, K. Lee, D. M. Wilson, R. K. Prinjha and G. Drewes, Interrogating the druggability of the 2-oxoglutarate-dependent dioxygenase target class by chemical proteomics, ACS Chem. Biol., 2016, 11, 2002–2010 CrossRef CAS PubMed.
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Järvinen and J. Savolainen, Prodrugs: design and clinical applications, Nat. Rev. Drug Discovery, 2008, 7, 255–270 CrossRef CAS PubMed.
- T. Wang, R. Zhang, Y. Liu, Z. Fang, H. Zhang, Y. Fan, S. Yang and R. Xiang, Discovery of a new class of JMJD6 inhibitors and structure–activity relationship study, Bioorg. Med. Chem. Lett., 2021, 44, 128109 CrossRef CAS PubMed.
- S. M. Westaway, A. G. S. Preston, M. D. Barker, F. Brown, J. A. Brown, M. Campbell, C.-w. Chung, G. Drewes, R. Eagle, N. Garton, L. Gordon, C. Haslam, T. G. Hayhow, P. G. Humphreys, G. Joberty, R. Katso, L. Kruidenier, M. Leveridge, M. Pemberton, I. Rioja, G. A. Seal, T. Shipley, O. Singh, C. J. Suckling, J. Taylor, P. Thomas, D. M. Wilson, K. Lee and R. K. Prinjha, Cell penetrant inhibitors of the KDM4 and KDM5 families of histone lysine demethylases. 2. Pyrido[3,4-d]pyrimidin-4(3H)-one derivatives, J. Med. Chem., 2016, 59, 1370–1387 CrossRef CAS PubMed.
- A. Aihara, C. K. Huang, M. J. Olsen, Q. Lin, W. Chung, Q. Tang, X. Dong and J. R. Wands, A cell-surface β-hydroxylase is a biomarker and therapeutic target for hepatocellular carcinoma, Hepatol, 2014, 60, 1302–1313 CrossRef CAS PubMed.
- W. Zheng, X. Wang, J. Hu, B. Bai and H. Zhu, Diverse molecular functions of aspartate β-hydroxylase in cancer (Review), Oncol. Rep., 2020, 44, 2364–2372 CrossRef CAS PubMed.
- M. Kanwal, M. Smahel, M. Olsen, J. Smahelova and R. Tachezy, Aspartate β-hydroxylase as a target for cancer therapy, J. Exp. Clin. Cancer Res., 2020, 39, 163 CrossRef CAS PubMed.
- L. Lavaissiere, S. Jia, M. Nishiyama, S. de la Monte, A. M. Stern, J. R. Wands and P. A. Friedman, Overexpression of human aspartyl(asparaginyl)beta-hydroxylase in hepatocellular carcinoma and cholangiocarcinoma, J. Clin. Invest., 1996, 98, 1313–1323 CrossRef CAS PubMed.
- L. Brewitz, Y. Nakashima, A. Tumber, E. Salah and C. J. Schofield, Fluorinated derivatives of pyridine-2,4-dicarboxylate are potent inhibitors of human 2-oxoglutarate dependent oxygenases, J. Fluorine Chem., 2021, 247, 109804 CrossRef CAS PubMed.
- X. Dong, Q. Lin, A. Aihara, Y. Li, C.-K. Huang, W. Chung, Q. Tang, X. Chen, R. Carlson, C. Nadolny, G. Gabriel, M. Olsen and J. R. Wands, Aspartate β-hydroxylase expression promotes a malignant pancreatic cellular phenotype, Oncotarget, 2015, 6, 1231–1248 CrossRef PubMed.
- R. Pellicciari, S. Fiorucci, E. Camaioni, C. Clerici, G. Costantino, P. R. Maloney, A. Morelli, D. J. Parks and T. M. Willson, 6α-Ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity, J. Med. Chem., 2002, 45, 3569–3572 CrossRef CAS PubMed.
- M. Ishiyama, H. Tominaga, M. Shiga, K. Sasamoto, Y. Ohkura and K. Ueno, A combined assay of cell viability and in vitro cytotoxicity with a highly water-soluble tetrazolium salt, neutral red and crystal violet, Biol. Pharm. Bull., 1996, 19, 1518–1520 CrossRef CAS PubMed.
- M. S. Islam, T. M. Leissing, R. Chowdhury, R. J. Hopkinson and C. J. Schofield, 2-Oxoglutarate-dependent oxygenases, Annu. Rev. Biochem., 2018, 87, 585–620 CrossRef CAS PubMed.
- R. L. Hancock, N. Masson, K. Dunne, E. Flashman and A. Kawamura, The activity of JmjC histone lysine demethylase KDM4A is highly sensitive to oxygen concentrations, ACS Chem. Biol., 2017, 12, 1011–1019 CrossRef CAS PubMed.
- L. Brewitz, Y. Nakashima, S. K. Piasecka, E. Salah, S. C. Fletcher, A. Tumber, T. P. Corner, T. J. Kennedy, G. Fiorini, A. Thalhammer, K. E. Christensen, M. L. Coleman and C. J. Schofield, 5-Substituted pyridine-2,4-dicarboxylate derivatives Have potential for selective inhibition of human Jumonji-C Domain-Containing Protein 5, J. Med. Chem., 2023, 66, 10849–10865 CrossRef CAS PubMed.
- E. Perrone, M. Alpegiani, F. Giudici, F. Buzzetti, G. Nannini, G. Meinardi, S. Grasso, A. Bianchi and I. de Carneri, Cephalosporins. VII. Synthesis and antibacterial activity of new cephalosporins bearing a 2-imino-3-hydroxythiazoline (2-aminothiazole N-oxide) in the C-7 acylamino side chain, J. Antibiot., 1984, 37, 1423–1440 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04253g |
| This journal is © The Royal Society of Chemistry 2023 |