
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing the effect of LiOH on forming a SEI using a Co magnetic “probe”†
Zhiqiang
Zhao‡
a,
Wanneng
Ye‡
 a,
Fengling
Zhang‡
a,
Yuanyuan
Pan
a,
Zengqing
Zhuo
b,
Feihu
Zou
a,
Xixiang
Xu
a,
Xiancheng
Sang
a,
Weiqi
Song
a,
Yue
Zhao
a,
Hongsen
Li
a,
Kuikui
Wang
a,
Chunfu
Lin
a,
Han
Hu
*c,
Qinghao
Li
*a,
Wanli
Yang
b and
Qiang
Li
*a
a,
Fengling
Zhang‡
a,
Yuanyuan
Pan
a,
Zengqing
Zhuo
b,
Feihu
Zou
a,
Xixiang
Xu
a,
Xiancheng
Sang
a,
Weiqi
Song
a,
Yue
Zhao
a,
Hongsen
Li
a,
Kuikui
Wang
a,
Chunfu
Lin
a,
Han
Hu
*c,
Qinghao
Li
*a,
Wanli
Yang
b and
Qiang
Li
*a
aCollege of Physics, Weihai Innovation Research Institute, Institute of Materials for Energy and Environment, Qingdao University, Qingdao 266071, P. R. China. E-mail: qhli@qdu.edu.cn; liqiang@qdu.edu.cn
bAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
cCollege of Chemical Engineering, China University of Petroleum (East China), Qingdao 266580, P. R. China. E-mail: hhu@upc.edu.cn
First published on 12th October 2023
Abstract
The solid-electrolyte-interphase (SEI) plays a critical role in lithium-ion batteries (LIBs) because of its important influence on electrochemical performance, such as cycle stability, coulombic efficiency, etc. Although LiOH has been recognized as a key component of the SEI, its influence on the SEI and electrochemical performance has not been well clarified due to the difficulty in precisely controlling the LiOH content and characterize the detailed interface reactions. Here, a gradual change of LiOH content is realized by different reduction schemes among Co(OH)2, CoOOH and CoO. With reduced Co nanoparticles as magnetic “probes”, SEI characterization is achieved by operando magnetometry. By combining comprehensive characterization and theoretical calculations, it is verified that LiOH leads to a composition transformation from lithium ethylene di-carbonate (LEDC) to lithium ethylene mono-carbonate (LEMC) in the SEI and ultimately results in capacity decay. This work unfolds the detailed SEI reaction scenario involving LiOH, provides new insights into the influence of SEI composition, and has value for the co-development between the electrode materials and electrolyte.
Introduction
Lithium-ion batteries (LIBs) have greatly facilitated the development of portable electronic devices, and more recently, enabled the realization of electric vehicles.1–4 Because of its important role in cycle stability, coulombic efficiency etc., the solid-electrolyte-interphase (SEI) has attracted extensive attention.5,6 The SEI consists of multiple components, and the influence of each individual component has been a central topic in SEI studies. It has been verified that the main components of the SEI include LiF, Li2CO3, Li2O, LiOH and some organic compounds.7,8 These components have different roles in the SEI. For example, LiF can act as an effective passivation layer to prevent further electrolyte decomposition,9 while Li2CO3 mainly acts as an ionic conductor to transport lithium ions, thus facilitating charge transfer.10–12 Although extensive investigations have been performed,13 many questions remain open owing to the complexity and instability of SEI components.The influence of LiOH, a key component in the SEI, on the SEI has been widely considered.14–22 In previous research, LiOH in the SEI was generally believed to come from water contamination.15,17 While others found that LiOH can naturally form during the SEI formation.14,16 Recently, some viewpoints about the influence of LiOH on the SEI were proposed.21–23 Hu et al. found that the conversion of LiOH to Li2O and LiH may exist in the SEI,23 which can provide extra storage capacity. However Wang et al.22 and Xie et al.21 believed that LiOH can influence the SEI components and result in interface instability, because LiOH can lead to LEMC production, instead of the LEDC as commonly thought.24,25
Despite these efforts and the proposed reaction mechanism, the detailed influence of LiOH on the SEI has not been studied specifically, and the change of electrochemical performance from this influence has not been explained. The challenge on this topic lies in the difficulty of meticulously controlling the LiOH content in the SEI, so the changes of the SEI and electrochemical performance under LiOH influence are too weak to monitor. Interestingly, the transition metal hydroxide reduction process will produce LiOH26–28 and the conversion reactions and SEI formation voltages do not overlap. This provides the opportunity of controlling the content of LiOH when the SEI is formed in a low voltage range. Moreover, transition metal nanoparticles can significantly promote the SEI formation,29–31 which further facilitates SEI characterization. Technically, operando magnetometry is highly sensitive to magnetic changes caused by the SEI,32,33 which provides a new and intuitive characterization technique for studying the influence of LiOH on the SEI.
In this work, the control of different LiOH content levels was achieved through the reduction reactions with Co(OH)2, CoOOH and CoO, as shown below.
| Co(OH)2 + 2Li+ + 2e− → Co0 + 2LiOH | (1) |
| CoOOH + 3Li+ + 3e− → Co0 + 1LiOH + Li2O | (2) |
| CoO + 2Li+ + 2e− → Co0 + 0LiOH + Li2O | (3) |
Other than the control of the LiOH content, the product Co0 can be used as a “probe” to detect the magnetic change by operando magnetometry. Fig. 1 shows the diagram of this experimental design. Combined with X-ray absorption spectroscopy (XAS) and other characterization techniques, our results show that although LiOH provides some additional capacity, it can result in component variation from LEDC to LEMC in the SEI, along with the formation of some Li2CO3 aggregates. These reaction effects directly result in SEI instability, eventually leading to capacity decay. We further verified that the LiOH influence is mainly related to EC decomposition by adjusting electrolyte composition. These findings are important for understanding the effects of various components in the SEI and the synergistic development between electrodes and electrolyte materials.34,35
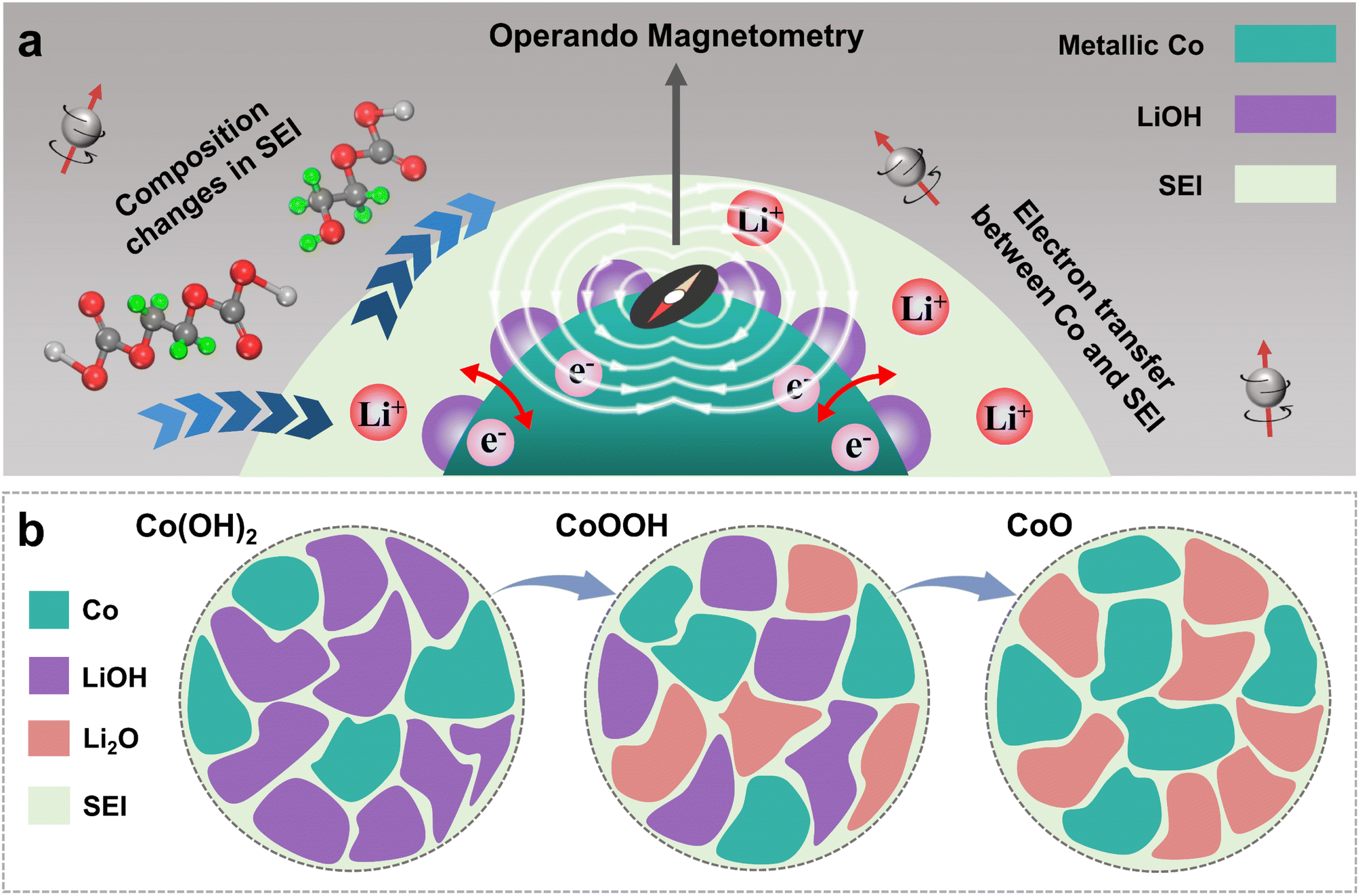 | ||
| Fig. 1 Diagram of experimental design. (a) Diagram of a Co magnetic “probe”. (b) Diagram of the LiOH content of Co(OH)2, CoOOH and CoO (reduced product). | ||
Results and discussion
Co(OH)2, CoOOH and CoO were selected as research objects because the obtained Co nanoparticles can catalyze SEI formation and LiOH presents different concentrations (Fig. 1b), and both of them are produced in the reduction process.29,30 Three materials were designed with the same morphology to ensure that morphology would not lead to differences in electrochemical performance. Pure nano-disk-like Co(OH)2 particles (β-Co(OH)2) were prepared using a simple wet chemical precipitation method, while CoO and CoOOH were obtained by thermal decomposition and oxidation reactions from Co(OH)2, respectively (Fig. S1 ESI†). As shown in Fig. S2a,† XRD patterns of the three materials clearly reveal that all characteristic peaks are in perfect agreement with the standard card (JCPDS card no. 74-1057, 73-1213, and 75-0418) without any impurities. Magnetic hysteresis curves in Fig. S1b† confirm that all materials show paramagnetic behavior. The high-resolution Co 2p spectrum (Fig. S2c and d†) shows the Co valence states of Co(OH)2, CoOOH and CoO, respectively. In Fig. S2c and e,† satellite peaks center at 785.9 eV and 802.7 eV, and two peaks located at 780.2 eV and 795.9 eV correspond to Co2+. The characteristic peaks of Co3+ are located at 779.8 and 781.2 eV,36 as seen in Fig. S2d.† The Co–OH content difference of the three materials can be proved in the high-resolution O 1s spectrum of Fig. S3a–c†. The ratio of Co–OH and Co–O bonds shows the materials change from Co(OH)2 to CoO.37 The Co–OH bond in CoO can be attributed to unavoidable surface water contamination.38 As shown in Fig. S2f–h,† although CoOOH and CoO experience structural changes during oxidation and annealing reactions, they maintain nano-disk-like morphology with a size of about 5 μm. Fig. S4a–i† show the high-resolution TEM (HRTEM) images of the three materials, and all of them show nano-disk-like morphology with high crystallinity. EDS mapping images (Fig. S5a–c†) provide clear evidence of the high homogeneity of Co and O elements in the three materials.The electrochemical cycling performance of Co(OH)2, CoOOH and CoO was investigated with Li metal as the anode. In Fig. 2a, the initial capacity of Co(OH)2 and CoOOH reaches 2001.9 and 2183.5 mA h g−1 under 100 mA g−1, which are far beyond their theoretical capacity based on conversion reactions (577 and 856 mA h g−1, respectively). According to previous studies, this kind of extra capacity in transition metal compounds may be related to space charge39 and hydride formation reaction.23,27 However, these two aspects are not sufficient to support such a high capacity. And what is strange is that the capacities of Co(OH)2 and CoOOH drop dramatically to 1179.8 and 1326.2 mA h g−1 over the next ten cycles, while CoO does not exhibit this phenomenon. As shown in Fig. 1a, CoO LIBs yield an initial capacity of 1129.4 mA h g−1 under 100 mA g−1, but demonstrate a capacity increase in the subsequent ten cycles to 1399.6 mA h g−1. This difference in electrochemical performance can be seen more clearly in Fig. 2b. Capacity increases can be attributed to surface layer activation or morphological changes,40 while capacity decay during the initial cycles is believed to be related to SEI instability.41–43 As shown in Fig. 2c, the capacity decay of Co(OH)2 and CoOOH mostly comes from the voltage range from about 0.8 to 0.01 V, which is the main range of SEI formation. Note that voltage plateau changes of the three materials here can be attributed to amorphous transformation and particle crushing.44 These results indicate that the capacity decay of Co(OH)2 and CoOOH may be related to SEI instability. Actually, in transition metal hydroxide materials, this inevitable capacity decay in the initial cycles is very widespread (Table S1†),26–28,45–47 and the intrinsic reason should be related to the reduction product LiOH. We conducted electrochemical impedance spectroscopy (EIS) on Co(OH)2, CoOOH and CoO (initial and discharged to 0.01 V), as shown in Fig. S6† and 2d. It can be seen that initial impedances of the three materials are small and close to each other (Fig. S6†). However, after discharging to 0.01 V, all of them show obvious SEI characteristics,48 and clear semicircle curves appear in a high frequency range (Fig. 2d). And the SEI impedance decreases with the decrease of LiOH content, which agrees with their different cycle performance. Obviously, SEI difference leads to these different impedance results. All of these electrochemical characterization results indicate that the SEI instability directly causes capacity decay during the initial cycles, and can be attributed to the influence of LiOH.
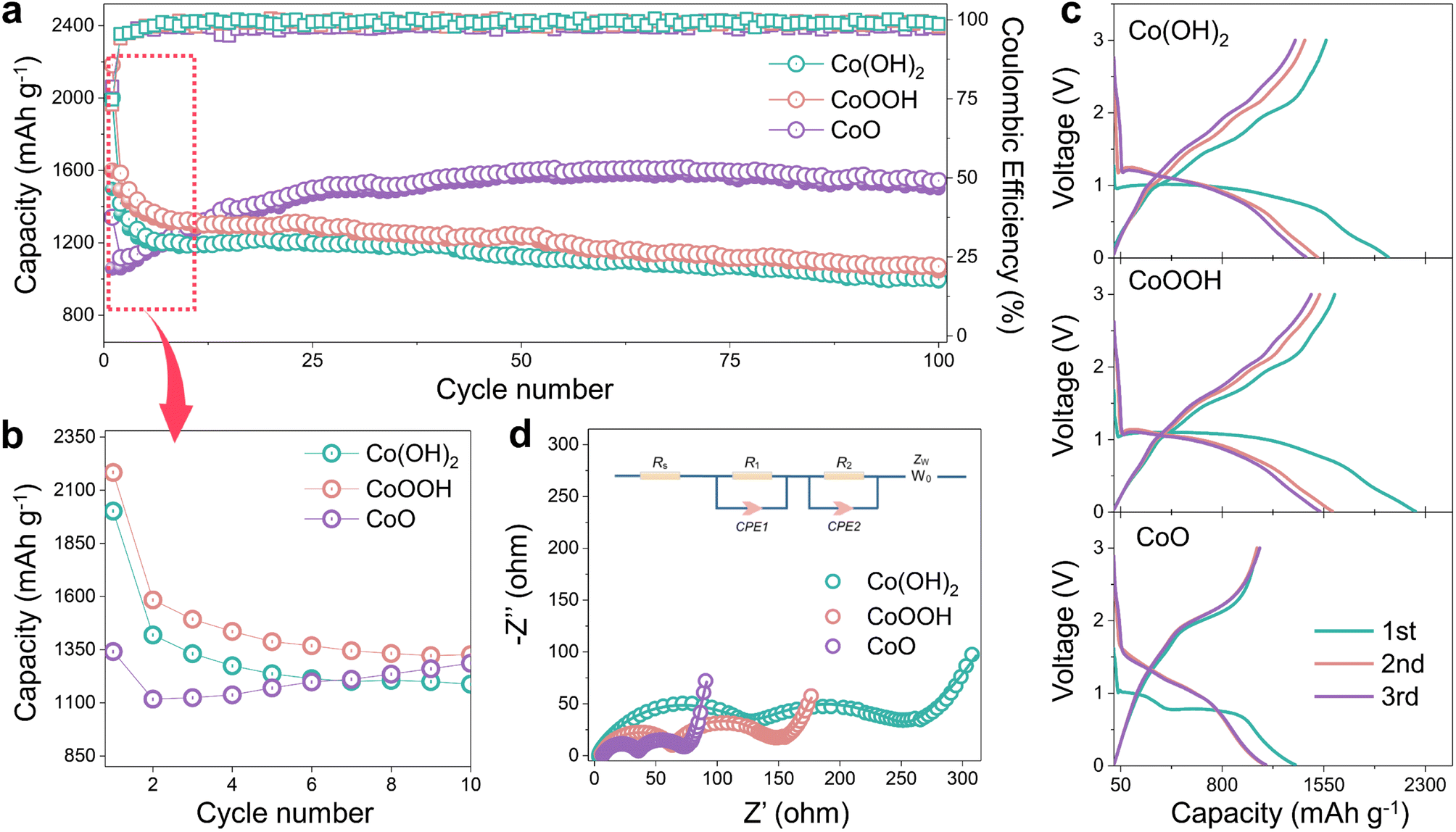 | ||
| Fig. 2 Electrochemical characterization of the three materials. (a) Cycling performance of Co(OH)2, CoOOH and CoO half cells at a current density of 100 mA g−1. (b) The initial ten cycles of the three materials. (c) Galvanostatic charge–discharge curves of Co(OH)2, CoOOH and CoO at a current density of 100 mA g−1. (d) The Nyquist curves of the three products after discharged to 0.01 V. | ||
TEM was carried out to analyze the SEI difference between Co(OH)2, CoOOH and CoO. TEM images are exhibited in Fig. 3a–c, corresponding to the SEI of Co(OH)2, CoOOH, and CoO (discharged to 0.01 V), respectively. TEM images show that the SEIs of Co(OH)2, CoOOH and CoO are significantly different. The SEIs of Co(OH)2 and CoOOH are very uneven. For Co(OH)2, the SEI thickness varies from 10.3 to 19.1 nm, and for CoOOH from 16.7 to 23.7 nm. However, the SEI of CoO is well maintained at 13.2 nm. Moreover, we found some agglomerating inorganic substances in the SEI of Co(OH)2 and CoOOH (Fig. S7a and b†). In sharp contrast, the SEI of CoO maintains a good amorphous state and no agglomerated particles can be found (Fig. S7c†). SEM tests showed similar results. As shown in Fig. S8,† the SEI formed on the surface of Co(OH)2 and CoOOH electrodes is thick, while the SEI on the CoO electrode surface is very thin. This contrast indicates that compared to CoO, the SEIs of Co(OH)2 and CoOOH are unstable. Fig. S9† shows the uniform distribution of Co and LiOH (or Li2O) nanoparticles after the discharging process. Interestingly, LiOH, Co and Li2O can be found on Co(OH)2 and CoOOH electrode surfaces (Fig. 3d–f). Co and LiOH come from the conversion reaction, while Li2O should come from the lithiation reaction of LiOH (LiOH + Li+ + e− → Li2O + LiH).23 These TEM results agree well with previous reports,23,27,45 in which similar results were verified by nuclear magnetic resonance (NMR), TEM, and theoretical calculation, indicating that LiOH lithiation indeed exists. To sum up, TEM results indicate that although the LiOH lithiation reaction can bring some extra capacity, it can also lead to SEI instability.
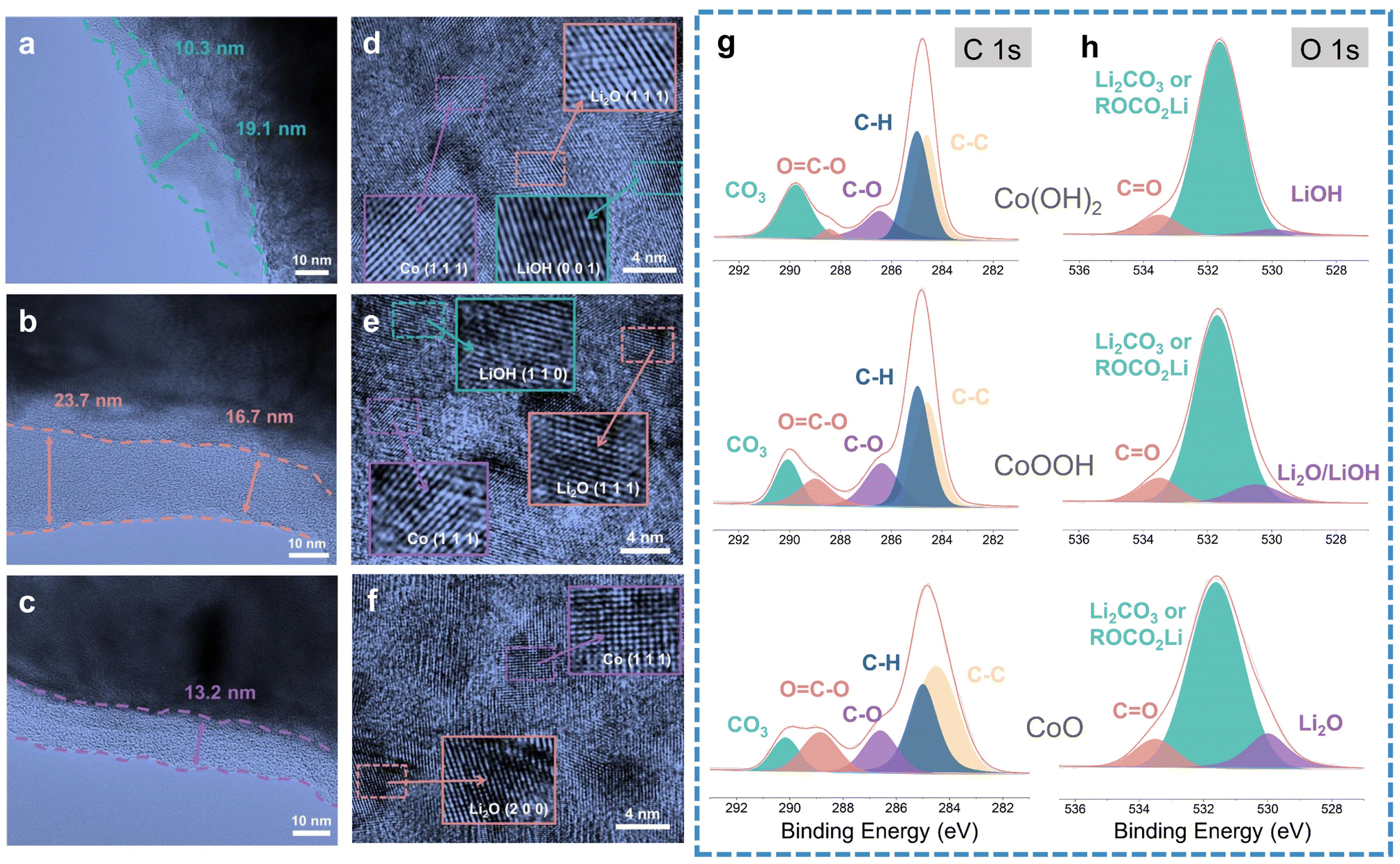 | ||
| Fig. 3 TEM and XPS characterization of the three materials. TEM images of (a) Co(OH)2, (b) CoOOH and (c) CoO electrodes (discharged to 0.01 V). HRTEM images of (d) Co(OH)2, (e) CoOOH and (f) CoO electrodes (discharged to 0.01 V). XPS high-resolution (g) C 1s and (h) O 1s spectra for the three materials (discharged to 0.01 V). | ||
In order to analyze the LiOH influence on SEI components, we performed XPS characterization on Co(OH)2, CoOOH and CoO (discharge to 0.01 V), with high-resolution C 1s, O 1s, F 1s and Li 1s XPS spectrum analysis, respectively. The XPS analysis results of F 1s and Li 1s spectrum are overall consistent among the three materials (Fig. S10a–f†), while the XPS analysis results of C 1s and O 1s spectra have obvious difference. As seen in Fig. 3g, the components observed at about 290, 288.5, 286.4, 285, and 284.4 eV can be attributed to CO3, O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O, C–O, C–H and C–C bonds, respectively.49–51 With the decrease of LiOH content (from Co(OH)2 to CoOOH and CoO), the relative content of C–O and OC–O in the XPS C 2p spectrum gradually increases, while CO3 gradually reduces. This phenomenon is also reflected in the O 1s spectrum. With the decrease of LiOH, the relative content of CO increases. Due to the higher main peak of Li2CO3 or ROCO2Li, the difference of CO is not as obvious as that in high-resolution C 1s. Note that the Li2O bond in the CoO O 1s spectrum should come from the conversion reaction of CoO.52,53 The SEI of CoO is thinner, allowing XPS to detect the electrode material. In contrast to one OC–O functional group in each LEMC, LEDC has a longer chain structure, with two such functional groups in each molecule (Fig. S11†). Combined with such structural difference and our characterization results, the composition change from LEDC to LEMC in the SEI under LiOH influence can be confirmed, which is consistent with the theoretical speculations of Wang et al.22 and Xie et al.21 Considering the change of CO3 content in high-resolution C 1s spectrum analysis, the inorganic substances gathered in the SEI can be attributed to Li2CO3. In addition, the XPS etching technique shows that with the increase of etching depth, the CO bonds in Co(OH)2 and CoOOH disappear along with the appearance of Li2O/LiOH peaks (Fig. S12a and b†). In contrast, the Li2O peak appears when CoO is etched at only 5 nm, and it has little change with the increase of etching depth (Fig. S12c†). These results were similar to TEM and SEM results, indicating a thinner thickness of the CoO SEI.
C–O, C–O, C–H and C–C bonds, respectively.49–51 With the decrease of LiOH content (from Co(OH)2 to CoOOH and CoO), the relative content of C–O and OC–O in the XPS C 2p spectrum gradually increases, while CO3 gradually reduces. This phenomenon is also reflected in the O 1s spectrum. With the decrease of LiOH, the relative content of CO increases. Due to the higher main peak of Li2CO3 or ROCO2Li, the difference of CO is not as obvious as that in high-resolution C 1s. Note that the Li2O bond in the CoO O 1s spectrum should come from the conversion reaction of CoO.52,53 The SEI of CoO is thinner, allowing XPS to detect the electrode material. In contrast to one OC–O functional group in each LEMC, LEDC has a longer chain structure, with two such functional groups in each molecule (Fig. S11†). Combined with such structural difference and our characterization results, the composition change from LEDC to LEMC in the SEI under LiOH influence can be confirmed, which is consistent with the theoretical speculations of Wang et al.22 and Xie et al.21 Considering the change of CO3 content in high-resolution C 1s spectrum analysis, the inorganic substances gathered in the SEI can be attributed to Li2CO3. In addition, the XPS etching technique shows that with the increase of etching depth, the CO bonds in Co(OH)2 and CoOOH disappear along with the appearance of Li2O/LiOH peaks (Fig. S12a and b†). In contrast, the Li2O peak appears when CoO is etched at only 5 nm, and it has little change with the increase of etching depth (Fig. S12c†). These results were similar to TEM and SEM results, indicating a thinner thickness of the CoO SEI.
During the discharging process, Co0 nanoparticles can generate in all three materials, which can promote SEI formation.29,30,32,33 There is electron exchange between Co and the SEI during SEI formation and decomposition.32,33 This makes Co nanoparticles suitable magnetic “probes” to indirectly detect the SEI differences among the three materials (Fig. 1a).32,33 Considering this effect and the sensitivity of operando magnetometry to this process,54–56 cyclic voltammetry (CV) along with operando magnetometry in real time was carried out for Co(OH)2, CoOOH and CoO (Fig. S13a–f†). As shown in Fig. 4a–c, we selected the magnetic response of the second cycle to better compare the magnetization change. Note that magnetic background has been removed, and magnetic signals of other components during the cycle can be ignored (ESI, Section S4†). Therefore, magnetization was calculated per gram of Co. The real-time magnetization response has multiple peaks and valleys at different voltages, which can be indexed as V1 to V5.
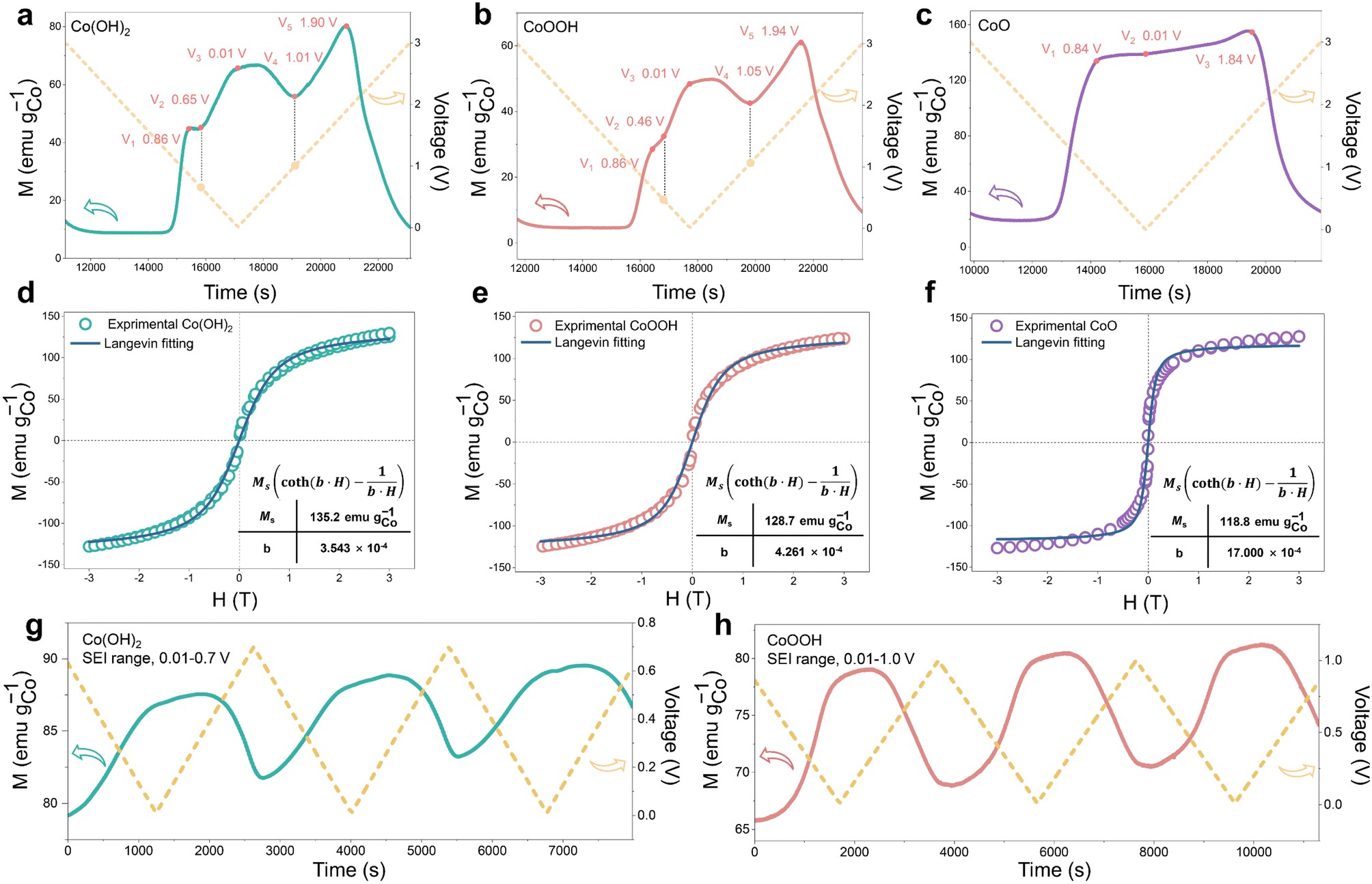 | ||
| Fig. 4 Magnetometry of the three materials. Operando magnetometry (second cycle) curves of (a) Co(OH)2, (b) CoOOH and (c) CoO. Ex situ M–H curves of (d) Co(OH)2, (e) CoOOH and (f) CoO (discharge to 0.01 V), accompanied by a Langevin fit. Operando magnetometry (SEI range) curves of (g) Co(OH)2 and (h) CoOOH. | ||
Due to the different structures and reaction kinetics, reaction potentials (V1 to V5) of the three materials have a little difference. Ex situ M–H measurements accompanied by a Langevin fit were carried out to analyze the reaction degree of the three materials.57 Electrons filled in Co0 nanoparticles based on space charge can be released in an ex situ environment,32,33,39,57,58 so the magnetization of Co0 can be fully reflected. As we can see in Fig. 4d–f, all of them show superparamagnetism and can be fitted by the Langevin equation59 (ESI, Section S5†). The fitting results (Fig. 4d–f) show that the magnetization of Co(OH)2, CoOOH and CoO reaches 135.2, 128.7 and 118.75 emu gCo−1, respectively. The saturation magnetization almost reaches the general magnetization of Co0 nanoparticles,60,61 indicating that the conversion reaction has overall completed. In addition, ex situ MH measurement and Langevin fitting were performed on the three materials with different cycle numbers to analyze the comminution situation of the materials. As shown in Fig. S14,† particle sizes of all three materials did not change dramatically during the first ten cycles, and the Co particle size basically remained at about 3 nm. This result excludes the possibility that the difference of capacity decay is caused by a different particle comminution situation.
The magnetic response can be divided into two ranges, the conversion reaction range (before V1 and after V5) and the interfacial reaction range (V1–V5),40 the latter is the focus of our research. From the magnetic response of interfacial reaction, it can be clearly found that compared to CoO, the magnetizations of Co(OH)2 and CoOOH have a totally different variation tendency in a low voltage range (from about 0.8 to 0.01 V), and the magnetization increases and decreases reversibly between V2 and V4. However, the magnetization of CoO has no obvious change in the range of V1 to V3. In order to determine the reaction mechanism of the low voltage range, CV measurement and operando magnetometry were conducted. As shown in Fig. S15, S16†, 3g and h, the results of Co(OH)2 and CoOOH in the range from V4 to V5 show space charge characteristics, and the trend of magnetization is consistent with voltage. While the range from V3 to V4 shows SEI characteristics, the trend of magnetization is opposite to voltage (Fig. 3g and h).32,33,39 These results verify the contribution of space charge in extra capacity. More importantly, these results indicate that the different magnetic response trends of Co(OH)2 and CoOOH are related to SEI changes influenced by LiOH. In combination with the theory perspectives of Co promoting SEI formation,29,30 it can be safely inferred that the SEI has a more drastic reaction process under the influence of LiOH, resulting in more electron transfer between Co and the SEI (ESI, Section S6†). This drastic reaction should be related to the transition from LEDC to LEMC mentioned above. Due to this transition, the SEI becomes unstable, which makes continuous electrode and electrolyte contact and incessant SEI formation. As a result, the initial capacity of Co(OH)2 and CoOOH is unusually high and decline rapidly in subsequent cycles.41–43
Besides, we noticed that the operando magnetization of Co(OH)2 and CoOOH at 0.01 V is much lower than that of CoO, which may be due to the influence of space charge caused by the complex interface. It was proposed that further lithiation reaction would occur at the interface between Co nanoparticles and LiOH, to form CoxHy in the discharging process.27 Also, previous research has shown that adsorption of H ions on the Co surface can cause orbital reconstruction, thus leading to magnetic changes.62 Considering the possibility of these two influences, the magnetization of Co adsorption of H and CoxHy formation were calculated theoretically, respectively (ESI, Section S7.1†). The calculated results show that both reactions can inevitably lead to Co magnetization reduction, and Co magnetization decreases along with the increase of H atoms, which is consistent with our characterization results. This indicates that the overall low magnetization of Co(OH)2 and CoOOH is attributed to the influence of space charge and/or H atom adsorption. Obviously, results of magnetometry and theoretical calculation indicate that CoxHy formation is also an important origin of extra capacity. We found that the oxidation potential (V5) of CoOOH was very close to that of Co(OH)2 (V5). The reaction potential of oxidation to Co3+ should be significantly different from that of oxidation to Co2+. The difference of 0.04 V here is obviously unreasonable. Based on this, Co K-edge XAS spectra were recorded to analyze the oxidation process of CoOOH. As shown in Fig. S17,† only Co2+ can be found in the electrode material (fully charged to 3 V). This indicates that the electrochemically driven oxidation process cannot oxidize Co0 to Co3+, which is similar to the oxidation process of Fe.39,58,63 This result debunks many previous studies on the Co3+ material mechanism.64–67
It has been verified that SEI instability dominates the rapid capacity decay in the initial cycles. In order to further analyze the composition change in the SEI, soft XAS spectra analysis with a total electron yield (TEY) model was carried out. The three materials were discharged to 0.01 V and charged back to 1.7 V, respectively.68,69 Note that the conversion reaction of LiOH cannot be triggered in this voltage range, so the spectral contrast here is mostly attributed to the SEI reactions. In Fig. 5a–c, the features of O K-edge soft XAS spectra located at 532.4 eV and 533.7 eV correspond to LiOH and Li2CO3, respectively.70,71 The data (Fig. 5a–c) show clearly that Li2CO3 forms in the SEI of the three materials at the discharge state. In the SEI of Co(OH)2 and CoOOH, the content of LiOH demonstrates an obvious increase during the charging process to 1.7 V. While the content of LiOH in the SEI of CoO is almost unchanged. This indicates that LiOH is actively involved in the SEI related reaction. SEI becomes unstable under LiOH influence during the charging process, and partial decomposition or dissolution occurs, leading to a thinner SEI. As a result, XAS results showed more signals of LiOH, which is on the electrode material surface. Note that the LiOH in CoO stems from surface water contamination, which is consistent with the XPS results (Fig. S3c†). The comparison of Li2CO3 content among the three materials can be found in Fig. S18.† The Li2CO3 level of Co(OH)2 and CoOOH samples is higher than CoO at both 0.01 V and 1.7 V. In C K-edge soft XAS spectra (Fig. 5d–f), the 288.6 eV feature originates from the C–H, while the feature at 290.4 eV originates from the CO functional group.70,71 The intensity contrast between the peaks of C–H and CO changes significantly among the samples, and the intensity of the CO peak, relative to the C–H peak, gradually increases from Co(OH)2 and CoOOH to CoO. This can be attributed to the influence of LiOH, under which LEDC transformed into LEMC.22 These results are consistent with the XPS results discussed above. SEI differences among the three materials can also be reflected in Co L-edge soft XAS spectra. As shown in Fig. S19,† signals of Co0 in Co(OH)2 and CoOOH are negligible, but detectable in CoO. XAS has a limited probe depth of the electron yield, which indicates that the SEIs of Co(OH)2 and CoOOH are thicker than that of CoO, which is related to the continuous generation of the SEI under the influence of LiOH. When the monitoring depth was increased, Co particles were successfully detected in all three materials (Fig. S20†). Based on all soft XAS characterization results and theory in related studies,21 a transformation path from LEDC to LEMC under the influence of LiOH can be proposed, which can be formulated as:
| LEDC + LiOH → LEMC + Li2CO3 | (4) |
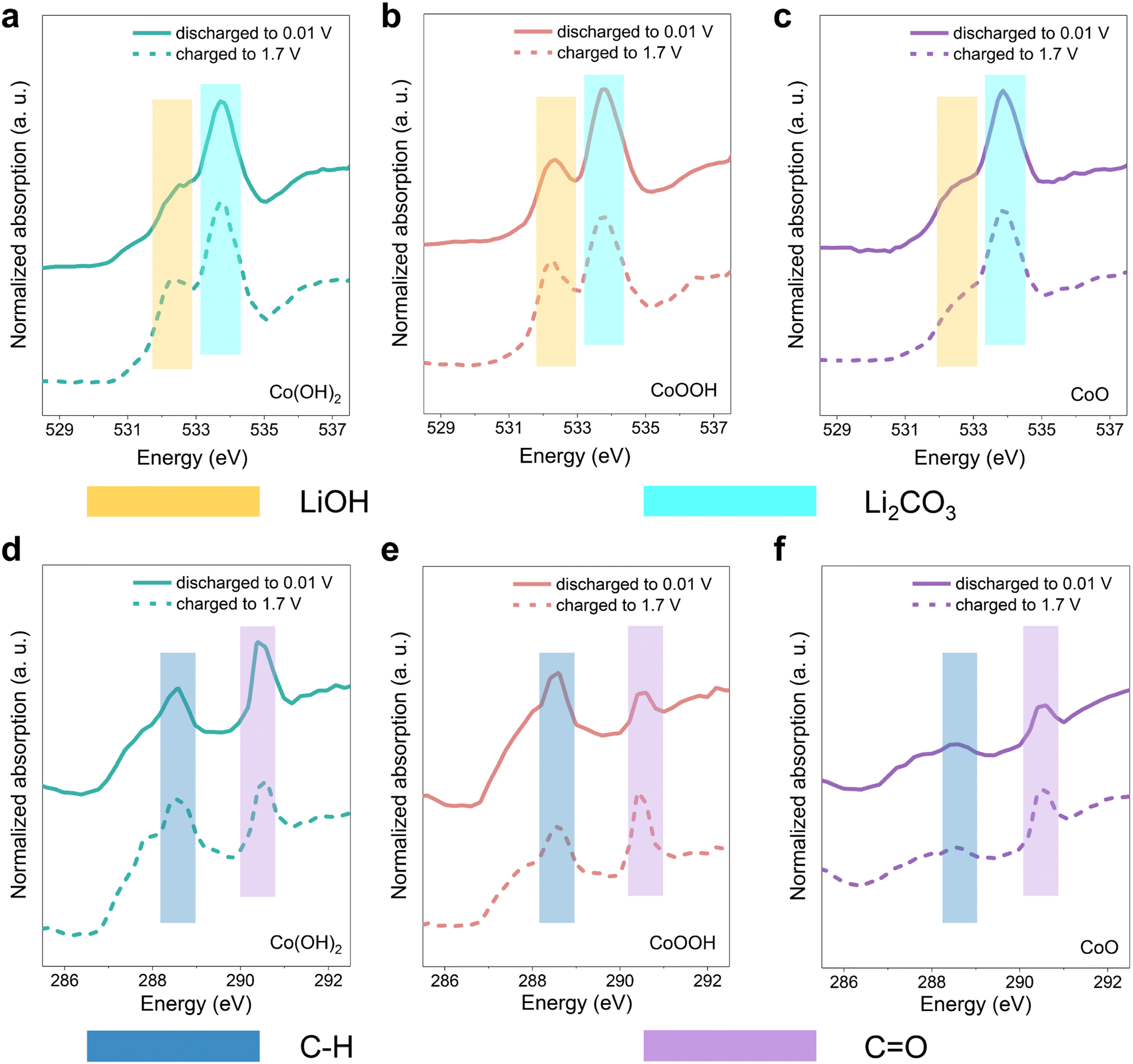 | ||
| Fig. 5 Soft XAS characterization (TEY model) of the three materials, probe depth is about 5–10 nm. O K-edge soft XAS spectra of (a) Co(OH)2, (b) CoOOH and (c) CoO. C K-edge soft XAS spectra of (d) Co(OH)2, (e) CoOOH and (f) CoO. | ||
Gibbs free energy calculation was used to determine the possibility of this reaction path. Theoretical calculation (ESI, Section S7.2†) was carried out in the presence and absence of Co, respectively. Theoretical results show that ΔG of this reaction is −0.56 eV. When the reaction is performed on the Co surface, ΔG is −1.48 eV (Fig. 6a). This suggests that this reaction can take place spontaneously, which is similar to the results of Xie et al.21 However, it is worth noting that water cannot directly react with LEDC, and it is LiOH that converted from water that directly reacts with LEDC. This reaction is easier to take place after adsorption on the Co surface, which indicates that Co plays an obvious catalytic role. This is consistent with the results of operando magnetometry. As shown in Fig. 4a and b magnetization of Co(OH)2 and CoOOH shows a significant change between V2 and V4, which can be attributed to the electron exchange between the SEI and Co during Co the catalysis process.29,30
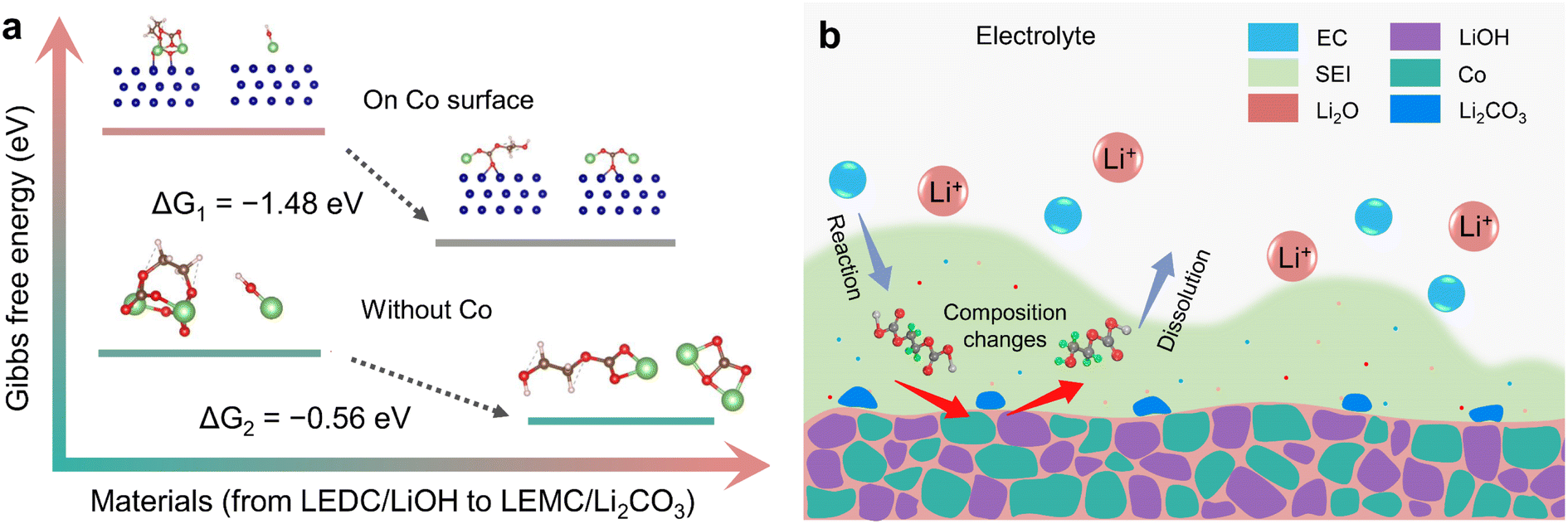 | ||
| Fig. 6 Diagram of theoretical calculation and SEI composition changes. (a) Theoretical calculation of the transformation from LEDC to LEMC, the calculation was carried out in the absence of Co and on the surface of Co particles, respectively. (b) Schematic diagram of the mechanism of SEI composition change. | ||
To further unfold the detailed reaction procedure, the electrolyte composition was adjusted to analyze the reaction path of the transformation from LEDC to LEMC. Considering that EC is the most reactive component in the electrolyte, which will form LEDC through the reaction path: EC + 2e− + 2Li+ → LEDC + C2H4,7,72,73 we replaced the EC component in the electrolyte with more chemically inert FEC.22,74,75 As we can see in Fig. S21,† the initial capacity decay of Co(OH)2 and CoOOH is significantly improved through this electrolyte adjustment. At the same time, the electrolyte adjustment inevitably leads to a different degree of capacity decline among all three materials. This suggests that the influence of LiOH is directly related to EC decomposition. The composition adjustment of the electrolyte reduces LEDC formation and brings about more LiF,76,77 which isolates the contact between LiOH and the SEI, thus inhibiting the influence of LiOH. Obviously, the cycle performance of transition metal hydroxides can be improved by adjusting the composition of the electrolyte. Note that LiOH is the main impurity caused by water contamination in typical electrode materials,15,17,21,23 and adjusting electrolyte composition, especially stabilizing EC decomposition, can be a promising strategy to suppress the influence of water on cycle performance. Considering the uncertainty of the transformation path from LEDC to LEMC under water influence,78 these results provide significant experimental evidence for this issue.
Based on the above discussion, a complete mechanism diagram of the influence of LiOH on the SEI was presented as follows:22,24,25
| EC + 2e− + 2Li+ → LEDC + CH2 CH2 | (5) |
| LEDC + LiOH → LEMC + Li2CO3 | (6) |
| LEMC + DMC → LMC + EC + CH3OH | (7) |
Three equations give the complex variation of SEI composition. In the process of discharging to low voltage, the SEI begins to form (eqn (5)) along with further lithiation of LiOH and CoxHy formation. These reactions occur almost simultaneously, along with space charge, resulting in unusually high capacity. With the formation of the SEI, LiOH nanoparticles at the interface between the SEI and electrode material gradually affect the components of the SEI (eqn (6) and (7)), resulting in SEI fragmentation and uneven distribution (Fig. 6b). This process may be related to the side reaction of LEMC (eqn (7)) and its high solubility in the electrolyte.18 SEI instability results in continuous contact between the electrolyte and electrode material, continuously generating a thicker SEI and bringing unusually high capacity. Li2CO3 gradually accumulates in the SEI and forms obvious particles during this process. As a result, the rapid capacity decay of Co(OH)2 and CoOOH in the initial cycles is due to SEI instability from LiOH. As the cycle progresses, this kind of LiOH effect stabilizes after about ten cycles, and some extra capacity is retained.
Conclusions
In summary, using Co as a magnetic “probe”, the effect of LiOH on the SEI was systematically analyzed by operando magnetometry, and the relationship between the effects and electrochemical performance was clarified for the first time. We found that LiOH can lead to the transformation of SEI components from LEDC to LEMC, during which part of the Li2CO3 will accumulate in the SEI. These effects lead to an inevitable capacity decay in the first few cycles. Adjusting the electrolyte composition can effectively eliminate this detrimental reaction, which is found to be directly related to the EC component in the electrolyte. In addition, it is proved that origins of extra capacity of Co(OH)2 and CoOOH are space charge storage, hydride formation reaction and continuous SEI formation/dissolution. This work is of vital significance for promoting the understanding of the SEI and the synergistic effect of electrode materials and electrolyte. Practically, the findings are also valuable for the development of electrode materials, which often involve the LiOH formation from humid air exposure.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
H. H, Q.-H. Li and Q. Li designed this study. Z. Zhao and F. Zhang wrote the manuscript and carried out most of the experiments with the exception at the following. Z. Zhuo and W. Yang carried out XAS experiments. F. Zou, W. Song and Y. Zhao carried out theoretical analysis. W. Ye performed experiments with TEM. X. Xu and X. Sang performed experiments with magnetometry. Y. Pan supervised DFT calculation. K. Wang, and C. Lin supervised XRD and STEM experiments. H. Li contributed to editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China 22179066, 11504192 and Natural Science Foundation of Shandong, China ZR2021QE061. Thanks for the Advanced Light Source support by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Thanks for the Advanced Light Source support by Beamline 02B02 at Shanghai Synchrotron Radiation Facility (SSRF).Notes and references
- J. M. Tarascon and M. Armand, Issues and challenges facing rechargeable lithium batteries, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- J. W. Choi and D. Aurbach, Promise and reality of post-lithium-ion batteries with high energy densities, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- J. B. Goodenough and K. S. Park, The Li-ion rechargeable battery: a perspective, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Challenges in the development of advanced Li-ion batteries: a review, Energy Environ. Sci., 2011, 4, 3243–3262 RSC.
- N. Takenaka, A. Bouibes, Y. Yamada, M. Nagaoka and A. Yamada, Frontiers in Theoretical Analysis of Solid Electrolyte Interphase Formation Mechanism, Adv. Mater., 2021, 33, e2100574 CrossRef PubMed.
- K. Xu, Electrolytes and interphases in Li-ion batteries and beyond, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- S. K. Heiskanen, J. Kim and B. L. Lucht, Generation and Evolution of the Solid Electrolyte Interphase of Lithium-Ion Batteries, Joule, 2019, 3, 2322–2333 CrossRef CAS.
- P. Verma, P. Maire and P. Novák, A review of the features and analyses of the solid electrolyte interphase in Li-ion batteries, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS.
- H. Wu, H. Jia, C. Wang, J. G. Zhang and W. Xu, Recent Progress in Understanding Solid Electrolyte Interphase on Lithium Metal Anodes, Adv. Energy Mater., 2020, 11, 2003092 CrossRef.
- N. Qin, L. Jin, Y. Lu, Q. Wu, J. Zheng, C. Zhang, Z. Chen and J. P. Zheng, Over-Potential Tailored Thin and Dense Lithium Carbonate Growth in Solid Electrolyte Interphase for Advanced Lithium Ion Batteries, Adv. Energy Mater., 2022, 12, 2103402 CrossRef CAS.
- Q. Zhang, J. Pan, P. Lu, Z. Liu, M. W. Verbrugge, B. W. Sheldon, Y. T. Cheng, Y. Qi and X. Xiao, Synergetic Effects of Inorganic Components in Solid Electrolyte Interphase on High Cycle Efficiency of Lithium Ion Batteries, Nano Lett., 2016, 16, 2011–2016 CrossRef CAS PubMed.
- S. Shi, Y. Qi, H. Li and L. G. Hector, Defect Thermodynamics and Diffusion Mechanisms in Li2CO3 and Implications for the Solid Electrolyte Interphase in Li-Ion Batteries, J. Phys. Chem. C, 2013, 117, 8579–8593 CrossRef CAS.
- Y. Chu, Y. Shen, F. Guo, X. Zhao, Q. Dong, Q. Zhang, W. Li, H. Chen, Z. Luo and L. Chen, Advanced Characterizations of Solid Electrolyte Interphases in Lithium-Ion Batteries, Electrochem. Energy Rev., 2019, 3, 187–219 CrossRef.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood, The state of understanding of the lithium-ion-battery graphite solid electrolyte interphase (SEI) and its relationship to formation cycling, Carbon, 2016, 105, 52–76 CrossRef CAS.
- T. Fujieda, Y. Xia, S. Koike, M. Shikano and T. Sakai, Effect of acid on passivation of a copper electrode in LiCF3SO3/propylene carbonate in underpotential region, J. Power Sources, 1999, 83, 186–192 CrossRef CAS.
- K.-I. Morigaki and A. Ohta, Analysis of the surface of lithium in organic electrolyte by atomic force microscopy, Fourier transform infrared spectroscopy and scanning auger electron microscopy, J. Power Sources, 1998, 76, 159–166 CrossRef CAS.
- K. Kanamura, H. Tamura and Z.-I. Takehara, XPS analysis of a lithium surface immersed in propylene carbonate solution containing various salts, J. Electroanal. Chem., 1992, 333, 127–142 CrossRef CAS.
- K. Tasaki and S. J. Harris, Computational Study on the Solubility of Lithium Salts Formed on Lithium Ion Battery Negative Electrode in Organic Solvents, J. Phys. Chem. C, 2010, 114, 8076–8083 CrossRef CAS.
- A. Kominato, E. Yasukawa, N. Sato, T. Ijuuin, H. Asahina and S. Mori, Analysis of surface films on lithium in various organic electrolytes, J. Power Sources, 1997, 68, 471–475 CrossRef CAS.
- M. Odziemkowski and D. E. Irish, An Electrochemical Study of the Reactivity at the Lithium Electrolyte/Bare Lithium Metal Interface: II. Unpurified Solvents, J. Electrochem. Soc., 1993, 140, 1546–1555 CrossRef CAS.
- X. Xie, E. W. Clark Spotte-Smith, M. Wen, H. D. Patel, S. M. Blau and K. A. Persson, Data-Driven Prediction of Formation Mechanisms of Lithium Ethylene Monocarbonate with an Automated Reaction Network, J. Am. Chem. Soc., 2021, 143, 13245–13258 CrossRef CAS PubMed.
- L. Wang, A. Menakath, F. Han, Y. Wang, P. Y. Zavalij, K. J. Gaskell, O. Borodin, D. Iuga, S. P. Brown, C. Wang, K. Xu and B. W. Eichhorn, Identifying the components of the solid-electrolyte interphase in Li-ion batteries, Nat. Chem., 2019, 11, 789–796 CrossRef CAS PubMed.
- Y. Y. Hu, Z. Liu, K. W. Nam, O. J. Borkiewicz, J. Cheng, X. Hua, M. T. Dunstan, X. Yu, K. M. Wiaderek, L. S. Du, K. W. Chapman, P. J. Chupas, X. Q. Yang and C. P. Grey, Origin of additional capacities in metal oxide lithium-ion battery electrodes, Nat. Mater., 2013, 12, 1130–1136 CrossRef CAS PubMed.
- F. Shi, P. N. Ross, H. Zhao, G. Liu, G. A. Somorjai and K. Komvopoulos, A catalytic path for electrolyte reduction in lithium-ion cells revealed by in situ attenuated total reflection-Fourier transform infrared spectroscopy, J. Am. Chem. Soc., 2015, 137, 3181–3184 CrossRef CAS PubMed.
- A. L. Michan, M. Leskes and C. P. Grey, Voltage Dependent Solid Electrolyte Interphase Formation in Silicon Electrodes: Monitoring the Formation of Organic Decomposition Products, Chem. Mater., 2015, 28, 385–398 CrossRef.
- T. Li and X. Nie, One-Step Fast-Synthesized Foamlike Amorphous Co(OH)2 Flexible Film on Ti Foil by Plasma-Assisted Electrolytic Deposition as a Binder-Free Anode of a High-Capacity Lithium-Ion Battery, ACS Appl. Mater. Interfaces, 2018, 10, 16943–16946 CrossRef CAS PubMed.
- H. Kim, W. I. Choi, Y. Jang, M. Balasubramanian, W. Lee, G. O. Park, S. B. Park, J. Yoo, J. S. Hong, Y. S. Choi, H. S. Lee, I. T. Bae, J. M. Kim and W. S. Yoon, Exceptional Lithium Storage in a Co(OH)2 Anode: Hydride Formation, ACS Nano, 2018, 12, 2909–2921 CrossRef CAS PubMed.
- X.-L. Huang, J. Chai, T. Jiang, Y.-J. Wei, G. Chen, W.-Q. Liu, D. Han, L. Niu, L. Wang and X.-B. Zhang, Self-assembled large-area Co(OH)2 nanosheets/ionic liquid modified graphene heterostructures toward enhanced energy storage, J. Mater. Chem., 2012, 22, 3404–3410 RSC.
- S. Laruelle, S. Grugeon, P. Poizot, M. Dollé, L. Dupont and J.-M. Tarascon, On the Origin of the Extra Electrochemical Capacity Displayed by MO/Li Cells at Low Potential, J. Electrochem. Soc., 2002, 149, A627–A634 CrossRef CAS.
- S. Grugeon, S. Laruelle, L. Dupont and J. M. Tarascon, An update on the reactivity of nanoparticles Co-based compounds towards Li, Solid State Sci., 2003, 5, 895–904 CrossRef CAS.
- L. Su, J. Hei, X. Wu, L. Wang and Z. Zhou, Ultrathin Layered Hydroxide Cobalt Acetate Nanoplates Face-to-Face Anchored to Graphene Nanosheets for High-Efficiency Lithium Storage, Adv. Funct. Mater., 2017, 27, 1605544 CrossRef.
- H. Li, Z. Hu, Q. Xia, H. Zhang, Z. Li, H. Wang, X. Li, F. Zuo, F. Zhang, X. Wang, W. Ye, Q. Li, Y. Long, Q. Li, S. Yan, X. Liu, X. Zhang, G. Yu and G. X. Miao, Operando Magnetometry Probing the Charge Storage Mechanism of CoO Lithium-Ion Batteries, Adv. Mater., 2021, 33, e2006629 CrossRef PubMed.
- X. Li, Z. Li, Y. Liu, H. Liu, Z. Zhao, Y. Zheng, L. Chen, W. Ye, H. Li and Q. Li, Transition metal catalysis in lithium-ion batteries studied by operando magnetometry, Chin. J. Catal., 2022, 43, 158–166 CrossRef.
- X. Wang, M. Salari, D.-E. Jiang, J. Chapman Varela, B. Anasori, D. J. Wesolowski, S. Dai, M. W. Grinstaff and Y. Gogotsi, Electrode material–ionic liquid coupling for electrochemical energy storage, Nat. Rev. Mater., 2020, 5, 787–808 CrossRef CAS.
- J. Chen, X. Fan, Q. Li, H. Yang, M. R. Khoshi, Y. Xu, S. Hwang, L. Chen, X. Ji, C. Yang, H. He, C. Wang, E. Garfunkel, D. Su, O. Borodin and C. Wang, Electrolyte design for LiF-rich solid–electrolyte interfaces to enable high-performance microsized alloy anodes for batteries, Nat. Energy, 2020, 5, 386–397 CrossRef CAS.
- J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu, Y. Jiang, Z. Pan and S. Wei, CoOOH Nanosheets with High Mass Activity for Water Oxidation, Angew. Chem., Int. Ed., 2015, 54, 8722–8727 CrossRef CAS PubMed.
- Z. Y. Wang, L. Wang, S. Liu, G. R. Li and X. P. Gao, Conductive CoOOH as Carbon-Free Sulfur Immobilizer to Fabricate Sulfur-Based Composite for Lithium–Sulfur Battery, Adv. Funct. Mater., 2019, 29, 1901051 CrossRef.
- S. C. Petitto, E. M. Marsh, G. A. Carson and M. A. Langell, Cobalt oxide surface chemistry: the interaction of CoO(100), Co3O4(110) and Co3O4(111) with oxygen and water, J. Mol. Catal. A: Chem., 2008, 281, 49–58 CrossRef CAS.
- Q. Li, H. Li, Q. Xia, Z. Hu, Y. Zhu, S. Yan, C. Ge, Q. Zhang, X. Wang, X. Shang, S. Fan, Y. Long, L. Gu, G. X. Miao, G. Yu and J. S. Moodera, Extra storage capacity in transition metal oxide lithium-ion batteries revealed by in situ magnetometry, Nat. Mater., 2021, 20, 76–83 CrossRef CAS PubMed.
- H. Kim, W. Choi, J. Yoon, J. H. Um, W. Lee, J. Kim, J. Cabana and W. S. Yoon, Exploring Anomalous Charge Storage in Anode Materials for Next-Generation Li Rechargeable Batteries, Chem. Rev., 2020, 120, 6934–6976 CrossRef CAS PubMed.
- S. Kim, J. Choi, S. M. Bak, L. Sang, Q. Li, A. Patra and P. V. Braun, Reversible Conversion Reactions and Small First Cycle Irreversible Capacity Loss in Metal Sulfide-Based Electrodes Enabled by Solid Electrolytes, Adv. Funct. Mater., 2019, 29, 1901719 CrossRef.
- C. Liao, Y. Wen, B. Shan, T. Zhai and H. Li, Probing the capacity loss of Li3VO4 anode upon Li insertion and extraction, J. Power Sources, 2017, 348, 48–56 CrossRef CAS.
- A. L. Michan, G. Divitini, A. J. Pell, M. Leskes, C. Ducati and C. P. Grey, Solid Electrolyte Interphase Growth and Capacity Loss in Silicon Electrodes, J. Am. Chem. Soc., 2016, 138, 7918–7931 CrossRef CAS PubMed.
- S. H. Yu, S. H. Lee, D. J. Lee, Y. E. Sung and T. Hyeon, Conversion Reaction-Based Oxide Nanomaterials for Lithium Ion Battery Anodes, Small, 2016, 12, 2146–2172 CrossRef CAS PubMed.
- S. Zhao, Z. Wang, Y. He, H. Jiang, Y. W. Harn, X. Liu, C. Su, H. Jin, Y. Li, S. Wang, Q. Shen and Z. Lin, A Robust Route to Co2(OH)2CO3 Ultrathin Nanosheets with Superior Lithium Storage Capability Templated by Aspartic Acid-Functionalized Graphene Oxide, Adv. Energy Mater., 2019, 9, 1901093 CrossRef.
- Z. Liu, J. Huang, B. Liu, D. Fang, T. Wang, Q. Yang, L. Dong, G.-H. Hu and C. Xiong, Constructing enhanced pseudocapacitive Li+ intercalation via multiple ionically bonded interfaces toward advanced lithium storage, Energy Storage Mater., 2020, 24, 138–146 CrossRef.
- K.-Y. Niu, F. Lin, L. Fang, D. Nordlund, R. Tao, T.-C. Weng, M. M. Doeff and H. Zheng, Structural and Chemical Evolution of Amorphous Nickel Iron Complex Hydroxide upon Lithiation/Delithiation, Chem. Mater., 2015, 27, 1583–1589 CrossRef CAS.
- S. S. Zhang, K. Xu and T. R. Jow, EIS study on the formation of solid electrolyte interface in Li-ion battery, Electrochim. Acta, 2006, 51, 1636–1640 CrossRef CAS.
- X. Han, G. Xu, Z. Zhang, X. Du, P. Han, X. Zhou, G. Cui and L. Chen, An In Situ Interface Reinforcement Strategy Achieving Long Cycle Performance of Dual-Ion Batteries, Adv. Energy Mater., 2019, 9, 1804022 CrossRef.
- R. I. R. Blyth, H. Buqa, F. P. Netzer, M. G. Ramsey, J. O. Besenhard, P. Golob and M. Winter, XPS studies of graphite electrode materials for lithium ion batteries, Appl. Surf. Sci., 2000, 167, 99–106 CrossRef CAS.
- R. Dedryvère, S. Laruelle, S. Grugeon, P. Poizot, D. Gonbeau and J. M. Tarascon, Contribution of X-ray Photoelectron Spectroscopy to the Study of the Electrochemical Reactivity of CoO toward Lithium, Chem. Mater., 2004, 16, 1056–1061 CrossRef.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nano-sized transition-metaloxides as negative-electrode materials for lithium-ion batteries, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- R. Dedryvere, S. Laruelle, S. Grugeon, P. Poizot, D. Gonbeau and J. M. Tarascon, Contribution of X-ray photoelectron spectroscopy to the study of the electrochemical reactivity of CoO toward lithium, Chem. Mater., 2004, 16, 1056–1061 CrossRef CAS.
- G. Gershinsky, E. Bar, L. Monconduit and D. Zitoun, Operando electron magnetic measurements of Li-ion batteries, Energy Environ. Sci., 2014, 7, 2012–2016 RSC.
- G. Klinser, M. Stückler, H. Kren, S. Koller, W. Goessler, H. Krenn and R. Würschum, Charging processes in the cathode LiNi0.6Mn0.2Co0.2O2 as revealed by operando magnetometry, J. Power Sources, 2018, 396, 791–795 CrossRef CAS.
- T. Yamada, In situ seamless magnetic measurements for solid-state electrochemical processes in Prussian blue analogues, Angew. Chem., Int. Ed., 2013, 125, 6358–6361 CrossRef.
- Z. Li, Y. Zhang, X. Li, F. Gu, L. Zhang, H. Liu, Q. Xia, Q. Li, W. Ye, C. Ge, H. Li, H. Hu, S. Li, Y. Z. Long, S. Yan, G. X. Miao and Q. Li, Reacquainting the Electrochemical Conversion Mechanism of FeS2 Sodium-Ion Batteries by Operando Magnetometry, J. Am. Chem. Soc., 2021, 143, 12800–12808 CrossRef CAS PubMed.
- F. Zhang, Z. Li, Q. Xia, Q. Zhang, C. Ge, Y. Chen, X. Li, L. Zhang, K. Wang, H. Li, L. Gu, S. Yan, G.-X. Miao and Q. Li, Li-ionic control of magnetism through spin capacitance and conversion, Matter, 2021, 4, 3605–3620 CrossRef CAS.
- M. Gossler, M. Albu, G. Klinser, E. M. Steyskal, H. Krenn and R. Wurschum, Magneto-Ionic Switching of Superparamagnetism, Small, 2019, 15, 1904523 CrossRef PubMed.
- M. Respaud, J. M. Broto, H. Rakoto and A. R. Fert, Surface effects on the magnetic properties of ultrafine cobalt particles, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 64, 012408 Search PubMed.
- D. M. Clifford, C. E. Castano and J. V. Rojas, Highly magnetic Co nanoparticles fabricated by X-ray radiolysis, Radiat. Phys. Chem., 2018, 144, 111–115 CrossRef CAS.
- K. Klyukin, G. Beach and B. Yildiz, Hydrogen tunes magnetic anisotropy by affecting local hybridization at the interface of a ferromagnet with nonmagnetic metals, Phys. Rev. Mater., 2020, 4, 104416 CrossRef CAS.
- X. Hua, P. K. Allan, C. Gong, P. A. Chater, E. M. Schmidt, H. S. Geddes, A. W. Robertson, P. G. Bruce and A. L. Goodwin, Non-equilibrium metal oxides via reconversion chemistry in lithium-ion batteries, Nat. Commun., 2021, 12, 561 CrossRef CAS PubMed.
- Z.-S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H.-M. Cheng, Graphene Anchored with Co3O4 Nanoparticles as Anode ofLithium Ion Batteries with Enhanced Reversible Capacity and Cyclic Performance, ACS Nano, 2010, 4(6), 3187–3194 CrossRef CAS PubMed.
- Y. Fu, L. Li, S. Ye, P. Yang, P. Liao, X. Ren, C. He, Q. Zhang and J. Liu, Construction of cobalt oxyhydroxide nanosheets with rich oxygen vacancies as high-performance lithium-ion battery anodes, J. Mater. Chem. A, 2021, 9, 453–462 RSC.
- H. Sun, G. Xin, T. Hu, M. Yu, D. Shao, X. Sun and J. Lian, High-rate lithiation-induced reactivation of mesoporous hollow spheres for long-lived lithium-ion batteries, Nat. Commun., 2014, 5, 4526 CrossRef CAS PubMed.
- Y. Wang, H. J. Zhang, J. Wei, C. C. Wong, J. Lin and A. Borgna, Crystal-match guided formation of single-crystal tricobalt tetraoxygen nanomesh as superior anode for electrochemical energy storage, Energy Environ. Sci., 2011, 4, 1845–1854 RSC.
- R. Qiao, Q. Li, Z. Zhuo, S. Sallis, O. Fuchs, M. Blum, L. Weinhardt, C. Heske, J. Pepper, M. Jones, A. Brown, A. Spucces, K. Chow, B. Smith, P. A. Glans, Y. Chen, S. Yan, F. Pan, L. F. Piper, J. Denlinger, J. Guo, Z. Hussain, Y. D. Chuang and W. Yang, High-efficiency in situ resonant inelastic x-ray scattering (iRIXS) endstation at the Advanced Light Source, Rev. Sci. Instrum., 2017, 88, 033106 CrossRef PubMed.
- R. Qiao and W. Yang, Interactions at the electrode-electrolyte interfaces in batteries studied by quasi-in-situ soft x-ray absorption spectroscopy, J. Electron Spectrosc. Relat. Phenom., 2017, 221, 58–64 CrossRef CAS.
- Z. Zhuo, P. Lu, C. Delacourt, R. Qiao, K. Xu, F. Pan, S. J. Harris and W. Yang, Breathing and oscillating growth of solid-electrolyte-interphase upon electrochemical cycling, Chem. Commun., 2018, 54, 814–817 RSC.
- M. Schellenberger, R. Golnak, W. G. Quevedo Garzon, S. Risse and R. Seidel, Accessing the solid electrolyte interphase on silicon anodes for lithium-ion batteries in-situ through transmission soft X-ray absorption spectroscopy, Mater. Today Adv., 2022, 14, 100215 CrossRef CAS.
- W. Huang, J. Wang, M. R. Braun, Z. Zhang, Y. Li, D. T. Boyle, P. C. McIntyre and Y. Cui, Dynamic Structure and Chemistry of the Silicon Solid-Electrolyte Interphase Visualized by Cryogenic Electron Microscopy, Matter, 2019, 1, 1232–1245 CrossRef.
- K. Ushirogata, K. Sodeyama, Y. Okuno and Y. Tateyama, Additive effect on reductive decomposition and binding of carbonate-based solvent toward solid electrolyte interphase formation in lithium-ion battery, J. Am. Chem. Soc., 2013, 135, 11967–11974 CrossRef CAS PubMed.
- J. J. Xu, J. X. Zhang, T. P. Pollard, Q. D. Li, S. Tan, S. Y. Hou, H. L. Wan, F. Chen, H. X. He, E. Y. Hu, K. Xu, X. Q. Yang, O. Borodin and C. S. Wang, Electrolyte design for Li-ion batteries under extreme operating conditions, Nature, 2023, 614, 694–700 CrossRef CAS PubMed.
- H. Wang, J. Liu, J. He, S. Qi, M. Wu, F. Li, J. Huang, Y. Huang and J. Ma, Pseudo-concentrated electrolytes for lithium metal batteries, eScience, 2022, 2, 557–565 CrossRef.
- J. Tan, J. Matz, P. Dong, J. Shen and M. Ye, A Growing Appreciation for the Role of LiF in the Solid Electrolyte Interphase, Adv. Energy Mater., 2021, 11, 2100046 CrossRef CAS.
- L. Dong, Y. Liu, K. Wen, D. Chen, D. Rao, J. Liu, B. Yuan, Y. Dong, Z. Wu, Y. Liang, M. Yang, J. Ma, C. Yang, C. Xia, B. Xia, J. Han, G. Wang, Z. Guo and W. He, High-Polarity Fluoroalkyl Ether Electrolyte Enables Solvation-Free Li+ Transfer for High-Rate Lithium Metal Batteries, Adv. Sci., 2021, 9, 2104699 CrossRef PubMed.
- E. J. McShane, H. K. Bergstrom, P. J. Weddle, D. E. Brown, A. M. Colclasure and B. D. McCloskey, Quantifying Graphite Solid-Electrolyte Interphase Chemistry and its Impact on Fast Charging, ACS Energy Lett., 2022, 7, 2734–2744 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04377k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |