
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA chiral pentanidium and pyridinyl-sulphonamide ion pair as an enantioselective organocatalyst for Steglich rearrangement†
Ziqi
Yang
a,
Chaoran
Xu
a,
Xianxian
Zhou
a,
Choon Boon
Cheong
 *ab,
Choon Wee
Kee
*b and
Choon-Hong
Tan
*a
*ab,
Choon Wee
Kee
*b and
Choon-Hong
Tan
*a
aSchool of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, 21 Nanyang Link, Singapore 637371, Republic of Singapore. E-mail: choonhong@ntu.edu.sg
bInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology and Research (A*STAR), 1 Pesek Road, Jurong Island, Singapore 627833, Republic of Singapore. E-mail: cheong_choon_boon@isce2.a-star.edu.sg; kee_choon_wee@isce2.a-star.edu.sg
First published on 9th November 2023
Abstract
Enantioselective ion pair catalysis has gained significant attention due to its ability to exert selectivity control in various reactions. Achiral counterions have been found to play crucial roles in modulating reactivity and selectivity. The modular nature of an ion pair catalyst allows rapid alterations of the achiral counterion to achieve optimal outcomes, without the need to modify the more onerous chiral component. In this study, we report the successful development of a stable chiral pentanidium pyridinyl-sulphonamide ion pair as a nucleophilic organocatalyst for asymmetric Steglich rearrangement. The ion pair catalyst demonstrated excellent performance, leading to enantioenriched products with up to 99% ee through simple alterations of the achiral anions. We conducted extensive ROESY experiments and concluded that the reactivity and enantioselectivity were correlated to the formation of a tight ion pair in solution. Further computational analyses provided greater clarity to the structure of the ion pair catalyst in solution. Our findings reveal the critical roles of NMR experiments and computational analyses in the design and optimisation of ion pair catalysts.
Introduction
Asymmetric ion pair catalysis is a powerful strategy to achieve an enantioselective outcome in a reaction.1,2 The chiral cation or anion can transmit stereochemical information to the reaction centres on their respective counterions, thus enabling enantioselectivity to be achieved. The use of chiral cations such as quaternary ammonium salts in phase transfer catalysis (PTC) has been extensively reported in the literature.2–5 Some prominent strategies that evolved from chiral cation PTC include asymmetric counteranion-directed catalysis,6 chiral anion PTC,7 and cation- or anion-binding catalysis.8,9The use of a chiral cation ion pair catalyst with transition metal binding to a ligand on the chiral cation (Ooi10 and Peters11) or a ligand on the achiral anion (Phipps12,13) has been previously reported (Fig. 1A). An alternative strategy will be chiral cation-directed transition metal catalysis, where the active metal centre is achiral and chiral information from the cation is transmitted by virtue of proximity through tight ion pairing. In this aspect, we have contributed to the use of chiral guanidinium cations such as bisguanidinium (BG) and pentanidium (PN) for various enantioselective oxidation reactions.14–17 Recently, the group of Jacobsen has reported an asymmetric anion-binding transition metal catalysis via hydrogen bond based organocatalysts.18
 | ||
| Fig. 1 (A) Representative chiral cation catalysts. (B) Stable pyridinyl amide ion pairs. (C) This work: chiral pentanidium pyridinyl-sulphonamide ion pair. | ||
Ion pair catalysis allows modular design of an asymmetric catalytic system. For instance, a privileged chiral scaffold can be deployed as the cation or anion for optimal stereochemical induction, while the counterion can be an achiral catalyst that is responsible for activation of the reactants. This modularity enables a variety of cooperative catalytic concepts to be possible. Chiral cation-directed transition metal catalysis19 and asymmetric photocatalysis via ion-pairing20 are two examples of such concepts.
Building on our prior research on chiral guanidinium-directed transition metal catalysis,14–17,19,21 we now introduce the utilization of an anionic nucleophilic catalyst in chiral cation-directed nucleophilic organocatalysis. The DMAP scaffold, known for its versatility in catalysing an array of reactions including esterification, acylation, and halogenation, has been extensively investigated as a nucleophilic organocatalyst.22 Over the past few years, chiral DMAP catalysts have also been developed. For example, Ruble and Fu have designed ferrocene-based planar DMAP derivatives,23,24 while Suga and his colleagues have developed BINOL-based chiral DMAP derivatives.25,26 The research groups of Vedejs,27,28 Conon,29 Sibi,30 and Spivey31 have each prepared various chiral DMAP derivatives, adding different chiral handles to the C-2 or C-3 positions of the pyridine ring. In 2020, Zipse32 and co-workers introduced stable pyridinyl amide ion pairs with achiral cations such as ammonium, phosphonium, and benzimidazolium as counterions (Fig. 1B).
These ion pairs served as a Lewis basic catalyst in urethane synthesis and aza-Morita–Baylis–Hillman reactions.32 This work points to the potential of ion pairs operating as chiral Lewis basic catalysts using a chiral cation and a nucleophilic anion.
The enantioselective Steglich rearrangement of oxindole derivatives is an acyl transfer reaction and has been reported to be catalysed by chiral DMAP derivatives, chiral DMAP-N-oxides and Cinchona alkaloid-derived zwitterions.24–26,33–36 In 2011, Seidel and co-workers reported an enantioselective Steglich rearrangement on O-acylazlactones using the strategy that combines a DMAP-based Lewis base together with a chiral organocatalyst via hydrogen bonding.37 In this work, we built on our previous foundation and developed a chiral ion pair organocatalyst, which composed of a chiral PN cation38 and an achiral anionic pyridinyl-sulphonamide (Fig. 1C). By successful application of this ion pair, we hope to enlarge the scope of chiral cation ion pair catalysis by expanding the range of suitable achiral anions.
Results and discussion
We embarked on our investigation by exploring the pyridinyl-sulphonamide due to the synthetic ease of accessing its anion (see the ESI†). Theoretical calculations suggest a highly diminished catalytic capability of the protonated pyridinyl sulphonamide of 2a (H-2a) relative to DMAP (calc. TOFDMAP = 2.8 h−1vs. calc. TOFH-2a = 6.9 × 10−10 h−1), which is validated experimentally (Table S1,† entry 2). Besides the low catalytic activity of H-2a, its low solubility in toluene further contributes unfavourably to achieve the required transformation. Therefore, accessing its anionic derivative via deprotonation of the sulphonamide proton is critical to achieve reactivity (Table S1,† entry 3). However, a mixture of chiral pentanidium chloride 1a-Cl and the sodium pyridinyl-sulphonamide Na-2a exhibited low yield and enantioselectivity (Table 1, entry 2). In sharp contrast, when utilizing a pre-formed ion pair 1a–2a, the reactivity was significantly boosted, with yields reaching 99% (Table 1, entry 3). When the reaction temperature was lowered to −20 °C, this ion pair 1a–2a afforded the product in 87% ee and 74% yield (Table 1, entry 4). Further optimization of the reaction was performed by screening different combinations of chiral pentanidiums and pyridinyl-sulphonamides (Table 1). The chiral ion pair 1a–2a demonstrated the best control over enantioselectivity in toluene (74% yield and 87% ee), whereas the chiral ion pair 1b–2a gave very high yield but poorer ee (entries 4–5). Therefore, pentanidium 1a was employed to pair with anionic pyridinyl-sulphonamide containing different substituents (2b–2g). Different pyridinyl-sulphonamides did indeed affect the yield and enantioselectivity of products (entries 4 and 7–12). Solvent (entries 4 and 13–17), reaction temperature (entries 17–18) and catalyst loading (entries 17 and 19) were then explored with the chiral ion pair catalyst 1a–2a. A strong solvent effect was observed using solvents that have a higher dielectric constant than toluene or mesitylene39 (such as acetonitrile or dichloromethane), giving products with severely degraded ee values and varyingly worsened yields, whereas the products afforded in less polar solvents had higher ee values (entries 16–17). Meanwhile, increasing the temperature or decreasing the catalyst loading both decreased the yields and ee values of products (entries 17–19). In summary, the product 4a can be afforded with the best yield and ee value using 5 mol% chiral ion pair catalyst 1a–2a in mesitylene at −20 °C.|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Yieldb | eec |
| a Reaction conditions: 3a (0.04 mmol, 1.0 equiv.), catalyst (5 mol%), and solvent (0.4 mL) at −20 °C for 24 h. b Isolated yield. c Determined by chiral HPLC analysis. d Reaction was carried out at room temperature. e Reaction was carried out at 0 °C. f The catalyst loading was 3 mol%. | ||||
| 1d | 1a-Cl, DMAP | Toluene | 39% | 0% |
| 2d | 1a-Cl, Na-2a | Toluene | 8% | 7% |
| 3d | 1a–2a | Toluene | 99% | 0% |
| 4 | 1a–2a | Toluene | 74% | 87% |
| 5 | 1b–2a | Toluene | 82% | 73% |
| 6 | 1c–2a | Toluene | 63% | 55% |
| 7 | 1a–2b | Toluene | 90% | 70% |
| 8 | 1a–2c | Toluene | 68% | 84% |
| 9 | 1a–2d | Toluene | — | — |
| 10 | 1a–2e | Toluene | 66% | 77% |
| 11 | 1a–2f | Toluene | 97% | 79% |
| 12 | 1a–2g | Toluene | 63% | 83% |
| 13 | 1a–2a | MeCN | <5% | 20% |
| 14 | 1a–2a | DCM | 63% | 25% |
| 15 | 1a–2a | THF | 65% | 30% |
| 16 | 1a–2a | Et2O | 94% | 75% |
| 17 | 1a–2a | Mesitylene | 99% | 92% |
| 18e | 1a–2a | Mesitylene | 81% | 86% |
| 19f | 1a–2a | Mesitylene | 91% | 86% |
To gain a better understanding on the nature of our ion pair catalyst, an investigation into the substrate scope was carried out (Fig. 2). We first probed the migrating acyl group on the substrates. Alkyl and benzyl substituents on the migrating acyl group (3b–3e) showed good compatibility with the ion pair catalyst 1a–2a to achieve moderate to good ee values, albeit with poorer yield for the ethyl ester derivative (4c). It is partially due to the purification process as the substrate 3c and the product 4c share similar polarity. The substitution groups on the oxindole scaffold were also examined. For C-3 substitution, excellent ee values of the corresponding products were achieved with the catalyst 1a–2a when using a larger alkyl (3f–3g) or a 2-thienyl substituent (3i). The bromine substitution on the C-6 position had a minimal effect on the enantioselectivity but had a negative impact on the yield (3h). The ion pair catalyst 1a–2f was also able to affect the rearrangement for the substrates bearing C-3 benzyl or para-methoxyphenyl groups (3j–3k) in excellent yields and good to excellent ee values. Meanwhile, sensitive substituents such as the acidic cyanomethyl group could be tolerated (3l). The acyloxyethyl pendant was also particularly noteworthy as it demonstrated insensitivity to β-elimination or loss of the acyloxy group even after 48 h (3m). Both 4l and 4m were obtained in excellent yields and ee values. The absolute configuration of the major enantiomer of 4e was determined to be S by comparing the HPLC trace of a known compound in the literature.25
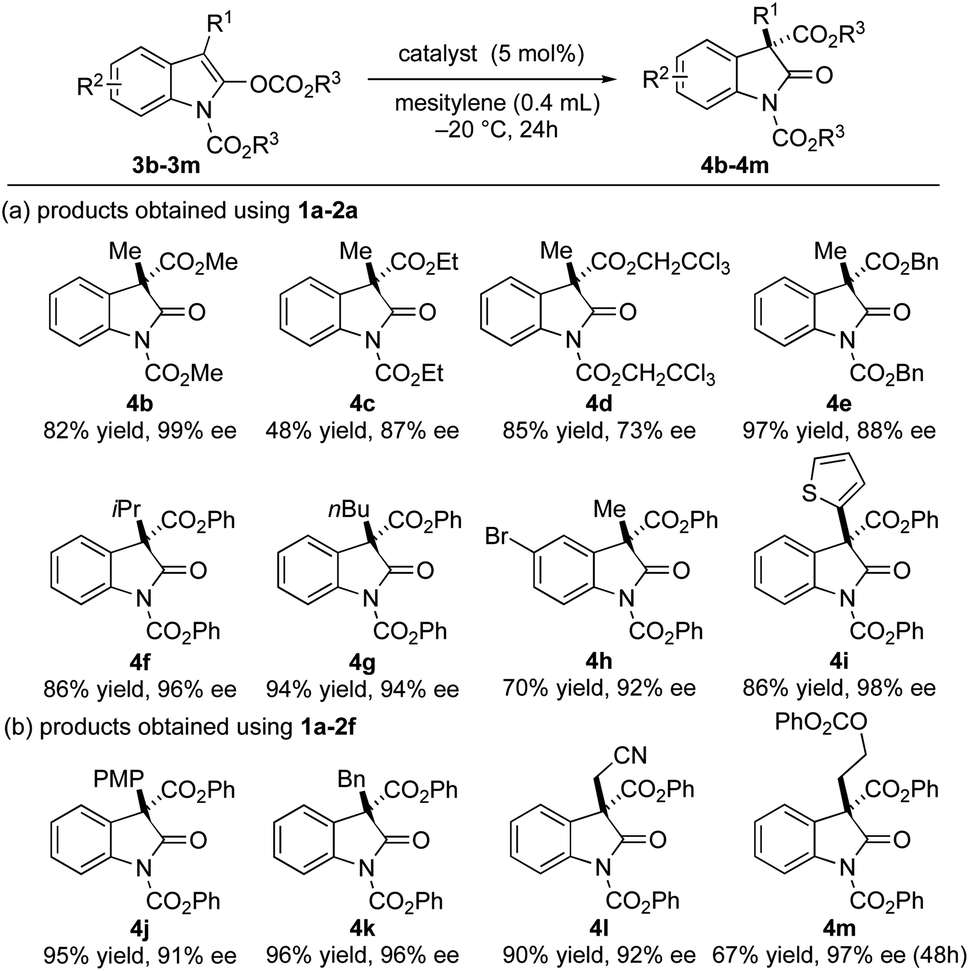 | ||
| Fig. 2 Substrate scope of enantioselective Steglich rearrangement of oxindole derivatives. | ||
The influence of temperature and the properties of organic solvent on the viability of ion pair formation and the strength of the association between ions is well-documented.41,42 NMR spectroscopy serves as a powerful method to examine the behaviour of the chiral ion pair catalyst in the solution state. This information can then be correlated to the results observed in our optimization process (Table 1).
In our NMR investigation, we varied both temperatures and solvents using the ion pair catalyst 1a–2f as a model. The catalyst 1a–2f was chosen for the study as it demonstrated slightly better and more consistent solubility across all the temperatures during NMR studies.
Two-dimensional ROESY (Rotating-frame Overhauser Effect Spectroscopy) experiments can reveal spatial correlations between two groups. A negative off-diagonal cross-peak, represented by a blue signal that maps to two different signals, suggests that the associated protons are generally less than 5 Å apart. ROESY experiments were conducted on the ion pair catalyst 1a–2f in two solvents: chloroform-d and toluene-d8. The experiments were performed at two different temperatures: 25 °C and −20 °C. At 25 °C, the reaction produced a racemic product. However, at −20 °C, a high degree of enantioselectivity was observed (Table 1, entries 3 and 4). No intermolecular spatial correlations were found in the 2D ROESY spectrum of 1a–2f in chloroform-d at either temperature (refer to Fig. S1–S4†). Instead, only intramolecular contacts were present. The 2D ROESY spectra for 1a–2f in toluene-d8, a less polar solvent, showed distinct spatial correlations between two of the aryl protons in 2f and the “arms” of the 1a cation, specifically the tert-butyl protons. These cross-peaks were absent at 25 °C (see Fig. 3A) but appeared at −20 °C (refer to Fig. 3B).
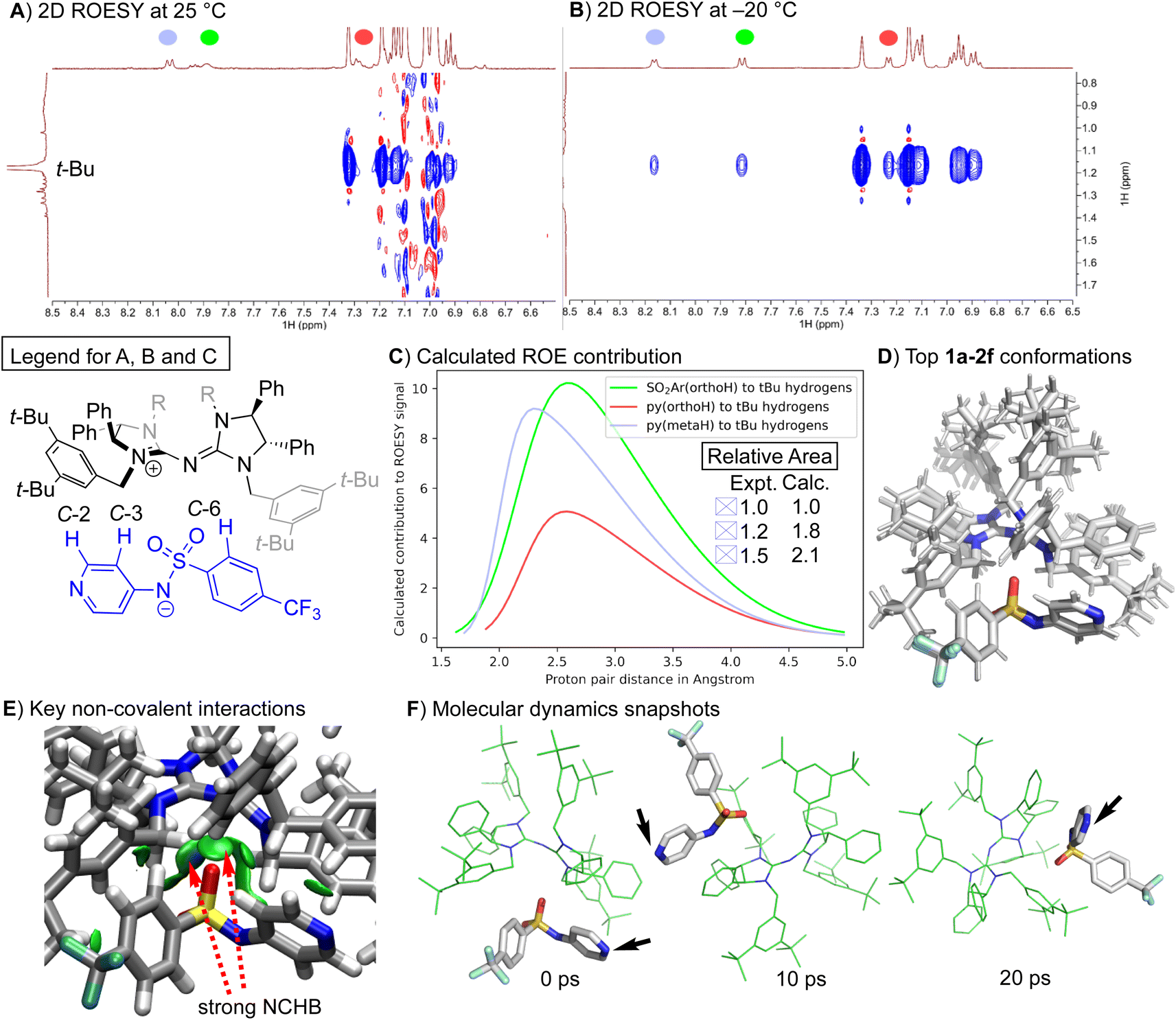 | ||
| Fig. 3 Assignment of peaks to the structure is indicated by the colour code in the legend. (A) Selected region of the 2D ROESY spectrum of 1a–2f in toluene-d8 at 25 °C. (B) Selected region of the 2D ROESY spectrum of 1a–2f in toluene-d8 at −20 °C. (C) 1D ROESY contribution estimated from molecular dynamics simulation (refer to the ESI† for details) and experimental integrals from 1D ROESY of selected peaks. (D) Overlay of the top seven conformations for 1a–2f optimized at ALPB(toluene)/GFN2-XTB:r2SCAN-3c. (E) NCHB: Non-classical hydrogen bond. Independent Gradient Model (IGM) isosurface (isovalue at 0.01 a.u.) visualized by VMD40 to reveal key intermolecular interactions between 1a and 2f. (F) MD snapshot from a 20 ps trajectory of the most stable conformation at the same level of theory. Black arrow indicates the position of the nucleophilic nitrogen of the anion 2f. | ||
Molecular Dynamics (MD) simulations of 1a–2f using a multiscale approach43 with ORCA44–46 provided a distribution of distances that contribute to the observed ROE signals (as seen in Fig. 3C). The relative areas derived from the MD simulations align qualitatively with the experimental 1D ROESY results (calculation details can be found in the ESI†). The pyridyl C-3 protons (depicted in red in Fig. 3) and the aryl protons on C-6 (depicted in green in Fig. 3) are, on average, in closer proximity to the tert-butyl protons of the cation 1a than the pyridyl C-2 protons (depicted in blue in Fig. 3). These findings suggest that aromatic solvents with low polarity (such as toluene) coupled with lower temperatures result in a tighter association of the ion pair. This increased association enhances the ability of the chiral cation to exert its enantio-discriminating influence on substrates activated by the nucleophilic anion.
The solution-state structure of the ion pair catalyst 1a–2f was determined via theoretical calculations. The top 26 conformations located47 account for 95% of the total Boltzmann population. The top seven conformations, which represent 70% of the population, show 1a binding to 2f in a similar orientation. The primary differences between these conformations involve the tert-butyl groups' arrangement (Fig. 3D). Electron density-based independent gradient model (IGM) analysis48,49 indicates that the main intermolecular interaction consists of non-classical hydrogen bonds (NCHBs) between the benzylic C–H or aryl C–H and the sulphonamide group's oxygens (Fig. 3E). The intermolecular interaction between 1a and 2f is estimated to be −36 kcal mol−1 from promolecular density with IGM analysis. The nitrogen atom on the pyridine ring faces outward, positioned ideally for substrate activation during the reaction. This orientation is maintained despite the facile anion's movement around the cation (black arrows in Fig. 3F). This can be attributed to the kinetically stable NCHB between 1a and 2f (Fig. 3E).
The chiral ion pair catalysed Steglich rearrangement50 is calculated to proceed via a stepwise mechanism (Fig. 4). The pyridinyl-sulphonamide anion 1f activates the reactant 3aviaTS1 (nucleophilic activation in Fig. 4). The dissociation of the intermediate to the 1a-enolate and py-carboxylate allows the subsequent product formation step to occur. The nucleophilic C-attack on the carbonyl group of the py-carboxylate furnishes the oxindole product 4a, together with regeneration of the catalyst. The turnover frequency is calculated to be 6.8 h−1 with TS2 being the turnover-determining TS.51,52
 | ||
| Fig. 4 A) Free energy profile of the 1a–2f catalyzed enantioselective Steglich reaction. Values of free energy profiles are calculated at SMD(toluene)/M06-2X54/def2-TZVPP//ALPB(toluene)/GFN2-XTB:r2SCAN-3c. (B) Activation strain analysis on the best TS structures from each set of TS: negative values favor TS2-R-15, while positive values favor TS2-S-96 (leads to the experimentally observed enantiomer). IGM isosurface (0.01 a.u.) based on promolecular density to highlight the origin of the substrate strain. | ||
The calculated ee value of 93% from the ensemble of 69 R- and S- carbonyl substitution transition state (TS) structures is higher than the experimental ee value of 79% (Table 1, entry 11). The % ee calculated from ΔΔH‡ is 71%, which is in good agreement with the experimental value. The discrepancy arises mainly from the entropy contribution that is likely to have significant numerical noise due to the numerical nature of the hessian. Full DFT on the selected optimized TS reduces the positive contribution of entropy to the calculated level of enantioselectivity (see the ESI†). Given the considerable reduction in computational effort by using a multiscale method, the agreement is reasonable.
An activation strain analysis53 of the two most stable conformations from the ensemble of TS structures reveals that the key contributions to enantioselectivity arise from the more stable substrate geometry in the TS and solvation. As revealed by the IGM analysis, the substrates in TS2-S-96 have essentially one additional NCHB between the SO2 of 1f and aryl C–H, which leads to a more favourable substrate geometry and thus less strain. From the less favourable binding energy of TS2-S-96 relative to TS2-R-15, it can be concluded that the geometry in TS2-S-96 was adopted to maximize substrate stability and solvent interaction.
Conclusions
In conclusion, we have extended the capability of chiral cation-directed catalysis by pairing with anionic nucleophiles. In this work, the nucleophilic pyridinyl-sulphonamide works synergistically with pentanidium to form an ion pair capable of inducing a high level of enantioselectivity. This chiral ion pair catalyst has been successfully utilized in the enantioselective Steglich rearrangement of oxindole derivatives, achieving up to 99% ee. Through ROE integrals obtained under experimentally relevant conditions, we conclude that a tightly bound ion pair existence is solvent and temperature dependent. Its existence is important for the high level of enantioselectivities observed in the Steglich rearrangement. Molecular dynamics simulation provided microscopic details on the structural preferences of the ion pair catalyst. A stepwise mechanism and the origin of enantioselectivity were postulated via theoretical calculations. This study demonstrates the potential for pairing chiral cations with achiral anionic organocatalysts through hydrogen bonding. We anticipate that this concept could also be extended to organocatalysts that are activated via halogen bonding or carbene formation.Data availability
The data that support the findings of this study are available in the ESI† of this article. Data for this paper, including computational results, total energies, thermochemical corrections, and cartesian coordinates of all optimized structures are available at FigShare at https://doi.org/10.6084/m9.figshare.23294474.Author contributions
Z. Y. and C.-H. T conceived the project and contributed to the methodology development. Z. Y. performed the experiments (catalysts synthesis, reaction testing, and characterization). C. X. and X. Z. contributed to part of experimental data. Z. Y., C. W. K. and C.-H. T. wrote the manuscript. C. W. K. performed the theoretical calculations. C. B. C. supervised the NMR experiments performed by Z. Y. All authors contributed to revising the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledge Nanyang Technological University for Tier 1 grants RG2/20 and RG61/21 and the Singapore National Research Foundation Competitive Research Program (NRF-CRP22-2019-0002). This work was supported by the A*STAR Computational Resource Centre through its high-performance computing facilities. The computational work for this article was partially performed on resources of the National Supercomputing Centre, Singapore (https://www.nscc.sg).Notes and references
- K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534 CrossRef CAS PubMed.
- E. Miller, P. J. Moon and F. D. Toste, in Catalytic Asymmetric Synthesis, John Wiley & Sons, Inc., Hoboken, 2022, pp. 117–156 Search PubMed.
- S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312 CrossRef CAS PubMed.
- N. Patel, R. Sood and P. V. Bharatam, Chem. Rev., 2018, 118, 8770 CrossRef CAS PubMed.
- D. Qian and J. Sun, Eur. J. Chem., 2019, 25, 3740 CrossRef CAS PubMed.
- M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518 CrossRef CAS PubMed.
- R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603 CrossRef CAS PubMed.
- A. P. Jadhav, S. Y. Park, J.-W. Lee, H. Yan and C. E. Song, Acc. Chem. Res., 2021, 54, 4319 CrossRef CAS PubMed.
- G. Pupo and V. Gouverneur, J. Am. Chem. Soc., 2022, 144, 5200 CrossRef CAS PubMed.
- K. Ohmatsu, S. Kawai, N. Imagawa and T. Ooi, ACS Catal., 2014, 4, 4304 CrossRef CAS.
- M. Mechler and R. Peters, Angew. Chem., Int. Ed., 2015, 54, 10303 CrossRef CAS PubMed.
- G. R. Genov, J. L. Douthwaite, A. S. Lahdenperä, D. C. Gibson and R. J. Phipps, Science, 2020, 367, 1246 CrossRef CAS PubMed.
- A. Fanourakis, B. D. Williams, K. J. Paterson and R. J. Phipps, J. Am. Chem. Soc., 2021, 143, 10070 CrossRef CAS PubMed.
- C. Wang, L. Zong and C.-H. Tan, J. Am. Chem. Soc., 2015, 137, 10677 CrossRef CAS PubMed.
- L. Zong, C. Wang, A. M. P. Moeljadi, X. Ye, R. Ganguly, Y. Li, H. Hirao and C.-H. Tan, Nat. Commun., 2016, 7, 13455 CrossRef PubMed.
- X. Ye, A. M. P. Moeljadi, K. F. Chin, H. Hirao, L. Zong and C.-H. Tan, Angew. Chem., Int. Ed., 2016, 55, 7101 CrossRef CAS PubMed.
- K. F. Chin, X. Ye, Y. Li, R. Lee, A. M. Kabylda, D. Leow, X. Zhang, E. C. X. Ang and C.-H. Tan, ACS Catal., 2020, 10, 2684 CrossRef CAS.
- J. M. Ovian, P. Vojáčková and E. N. Jacobsen, Nature, 2023, 616, 84 CrossRef CAS PubMed.
- X. Ye and C.-H. Tan, Chem. Sci., 2021, 12, 533 RSC.
- T. E. Schirmer and B. König, J. Am. Chem. Soc., 2022, 144, 19207 CrossRef CAS PubMed.
- L. Zong and C.-H. Tan, Acc. Chem. Res., 2017, 50, 842 CrossRef CAS PubMed.
- R. P. Wurz, Chem. Rev., 2007, 107, 5570 CrossRef CAS PubMed.
- J. C. Ruble and G. C. Fu, J. Org. Chem., 1996, 61, 7230 CrossRef CAS PubMed.
- J. C. Ruble and G. C. Fu, J. Am. Chem. Soc., 1998, 120, 11532 CrossRef CAS.
- H. Mandai, K. Fujii, H. Yasuhara, K. Abe, K. Mitsudo, T. Korenaga and S. Suga, Nat. Commun., 2016, 7, 11297 CrossRef PubMed.
- H. Mandai, T. Fujiwara, K. Noda, K. Fujii, K. Mitsudo, T. Korenaga and S. Suga, Org. Lett., 2015, 17, 4436 CrossRef CAS PubMed.
- E. Vedejs and X. Chen, J. Am. Chem. Soc., 1996, 118, 1809 CrossRef CAS.
- S. A. Shaw, P. Aleman and E. Vedejs, J. Am. Chem. Soc., 2003, 125, 13368 CrossRef CAS PubMed.
- C. Ó. Dálaigh, S. J. Hynes, J. E. O'Brien, T. McCabe, D. J. Maher, G. W. Watson and S. J. Connon, Org. Biomol. Chem., 2006, 4, 2785 RSC.
- G. Ma, J. Deng and M. P. Sibi, Angew. Chem., Int. Ed., 2014, 126, 12012 CrossRef.
- A. C. Spivey, T. Fekner, S. E. Spey and H. Adams, J. Org. Chem., 1999, 64, 9430 CrossRef CAS.
- J. Helberg, T. Ampßler and H. Zipse, J. Org. Chem., 2020, 85, 5390 CrossRef CAS PubMed.
- I. D. Hills and G. C. Fu, Angew. Chem., Int. Ed., 2003, 42, 3921 CrossRef CAS PubMed.
- S. A. Shaw, P. Aleman, J. Christy, J. W. Kampf, P. Va and E. Vedejs, J. Am. Chem. Soc., 2006, 128, 925 CrossRef CAS PubMed.
- M.-S. Xie, Y.-F. Zhang, M. Shan, X.-X. Wu, G.-R. Qu and H.-M. Guo, Angew. Chem., Int. Ed., 2019, 58, 2839 CrossRef CAS PubMed.
- Y.-P. Lam, J. Huang, X. Jiang and Y.-Y. Yeung, ChemCatChem, 2022, 14, e202200136 CrossRef CAS.
- C. K. De, N. Mittal and D. Seidel, J. Am. Chem. Soc., 2011, 133, 16802 CrossRef CAS PubMed.
- X. Zhang, J. Ren, S. M. Tan, D. Tan, R. Lee and C.-H. Tan, Science, 2019, 363, 400 CrossRef CAS PubMed.
- M. Chastrette, M. Rajzmann, M. Chanon and K. F. Purcell, J. Am. Chem. Soc., 1985, 107, 1 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS PubMed.
- H. Sadek and R. M. Fuoss, J. Am. Chem. Soc., 1954, 76, 5897 CrossRef CAS.
- S. Winstein, E. Clippinger, A. H. Fainberg and G. C. Robinson, J. Am. Chem. Soc., 1954, 76, 2597 CrossRef CAS.
- L. W. Chung, W. M. C. Sameera, R. Ramozzi, A. J. Page, M. Hatanaka, G. P. Petrova, T. V. Harris, X. Li, Z. Ke, F. Liu, H.-B. Li, L. Ding and K. Morokuma, Chem. Rev., 2015, 115, 5678 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652 CrossRef CAS PubMed.
- S. Grimme, A. Hansen, S. Ehlert and J.-M. Mewes, J. Chem. Phys., 2021, 154, 064103 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169 RSC.
- C. Lefebvre, G. Rubez, H. Khartabil, J.-C. Boisson, J. Contreras-García and E. Hénon, Phys. Chem. Chem. Phys., 2017, 19, 17928 RSC.
- C. Lefebvre, J. Klein, H. Khartabil, J.-C. Boisson and E. Hénon, J. Comput. Chem., 2023, 44, 1750 CrossRef CAS PubMed.
- P. P. D. Castro, J. A. D. Santos, M. M. D. Siqueira, G. M. F. Batista, H. F. D. Santos and G. W. Amarante, J. Org. Chem., 2019, 84, 12573 CrossRef PubMed.
- S. Kozuch and S. Shaik, J. Am. Chem. Soc., 2006, 128, 3355 CrossRef CAS PubMed.
- S. Kozuch, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 795 CAS.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070 CrossRef CAS PubMed.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and full characterization for all compounds, spectroscopic data, copies of NMR spectra, MS data and computational details. See DOI: https://doi.org/10.1039/d3sc04397e |
| This journal is © The Royal Society of Chemistry 2023 |