
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceO–H bond activation of β,γ-unsaturated oximes via hydrogen atom transfer (HAT) and photoredox dual catalysis†
Liang
Yi
ab,
Chen
Zhu
 a,
Xiangyu
Chen
b,
Huifeng
Yue
a,
Tengfei
Ji
b,
Yiqiao
Ma
b,
Yuanyuan
Cao
b,
Rajesh
Kancherla
a and
Magnus
Rueping
*a
a,
Xiangyu
Chen
b,
Huifeng
Yue
a,
Tengfei
Ji
b,
Yiqiao
Ma
b,
Yuanyuan
Cao
b,
Rajesh
Kancherla
a and
Magnus
Rueping
*a
aKAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: magnus.rueping@kaust.edu.sa
bInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, D-52074 Aachen, Germany
First published on 30th November 2023
Abstract
Hydrogen atom transfer (HAT) and photoredox dual catalysis provides a unique opportunity in organic synthesis, enabling the direct activation of C/Si/S–H bonds. However, the activation of O–H bonds of β,γ-unsaturated oximes poses a challenge due to their relatively high redox potential, which exceeds the oxidizing capacity of most currently developed photocatalysts. We here demonstrate that the combination of HAT and photoredox catalysis allows the activation of O–H bond of β,γ-unsaturated oximes. The strategy effectively addresses the oxime's high redox potential and offers a universal pathway for iminoxyl radical formation. Leveraging the versatility of this approach, a diverse array of valuable heterocycles have been synthesized with the use of different radical acceptors. Mechanistic studies confirm a HAT process for the O–H bond activation.
1 Introduction
In the past few decades, the organic synthesis based on photoredox-catalyzed reactions was boosted by the exploitation of various visible-light-absorbing photocatalysts.1 Hydrogen atom transfer (HAT) is a chemical step consisting of the concerted movement of a proton and an electron occurring between two substrates in a single kinetic step.2 The incorporation of HAT catalysis and photoredox catalysis provides unique opportunities in organic transformations, and has emerged as a powerful tool for C/Si/S–H bond activation.3 In particular, when using quinuclidine as the HAT catalyst, after being oxidized by photocatalyst, the generated quinucildinium radical cation is electrophilic, which could selectively abstract the more hydridic hydrogen atom of C/Si–H bond site according to the polarity-match between them, and regardless of the bond dissociation energies (BDEs) of C/Si–H.4 Oxygen radicals are widely used in both biological processes and organic synthesis.5 The generation of such radicals via direct cleavage of O–H bond is attractive but thermodynamically challenge.6 whereas the hydrogen of the O–H bond is protic, it is inaccessible to activate them through a polarity-match HAT pathway to give the oxygen radicals (Fig. 1a). Nevertheless, in addition to the polarity-match principle, the bond dissociation energies (BDEs) also play an important role during the HAT process. β,γ-Unsaturated oximes are a type of useful building blocks that are used for the construction of isoxazoline molecules.7 The measurement of the redox potential value for β,γ-unsaturated oxime 1a (Ep/2 = 2.2 V vs. SCE in CH3CN) indicates that it is relatively higher than that of most of the currently developed photocatalysts (See ESI Fig. S2†). This makes it challenging for the photocatalysts to directly oxidize the compound.8 As such, we conceived the possibility of activating the O–H bond in oxime 1a through the use of 3-acetoxyquinuclidine as the HAT reagent. This is because the radical cation formed by 3-acetoxyquinuclidine has an H–N+ BDE (quinuclidine) of 100 kcal mol−1,9 which is higher than the O–H BDE of oxime (around 83 kcal mol−1),10 enabling this process thermodynamically favored. If successful, this approach would resolve the challenge posed by the high redox potential of oxime in comparison to that of current photocatalysts, enabling the activation of the O–H bond and expanding the scope of C/Si–H bond activation to include the O–H bond, thus establishing a general pathway for the generation of the iminoxyl radical, as shown in Fig. 1b. Herein, we reported the first visible-light-mediated photoredox-catalyzed O–H bond activation of β,γ-unsaturated oximes by the merge of a thermodynamically favored HAT process, which provides a practical manner towards to a series of potential valuable isoxazoline molecules11 (Fig. 1c).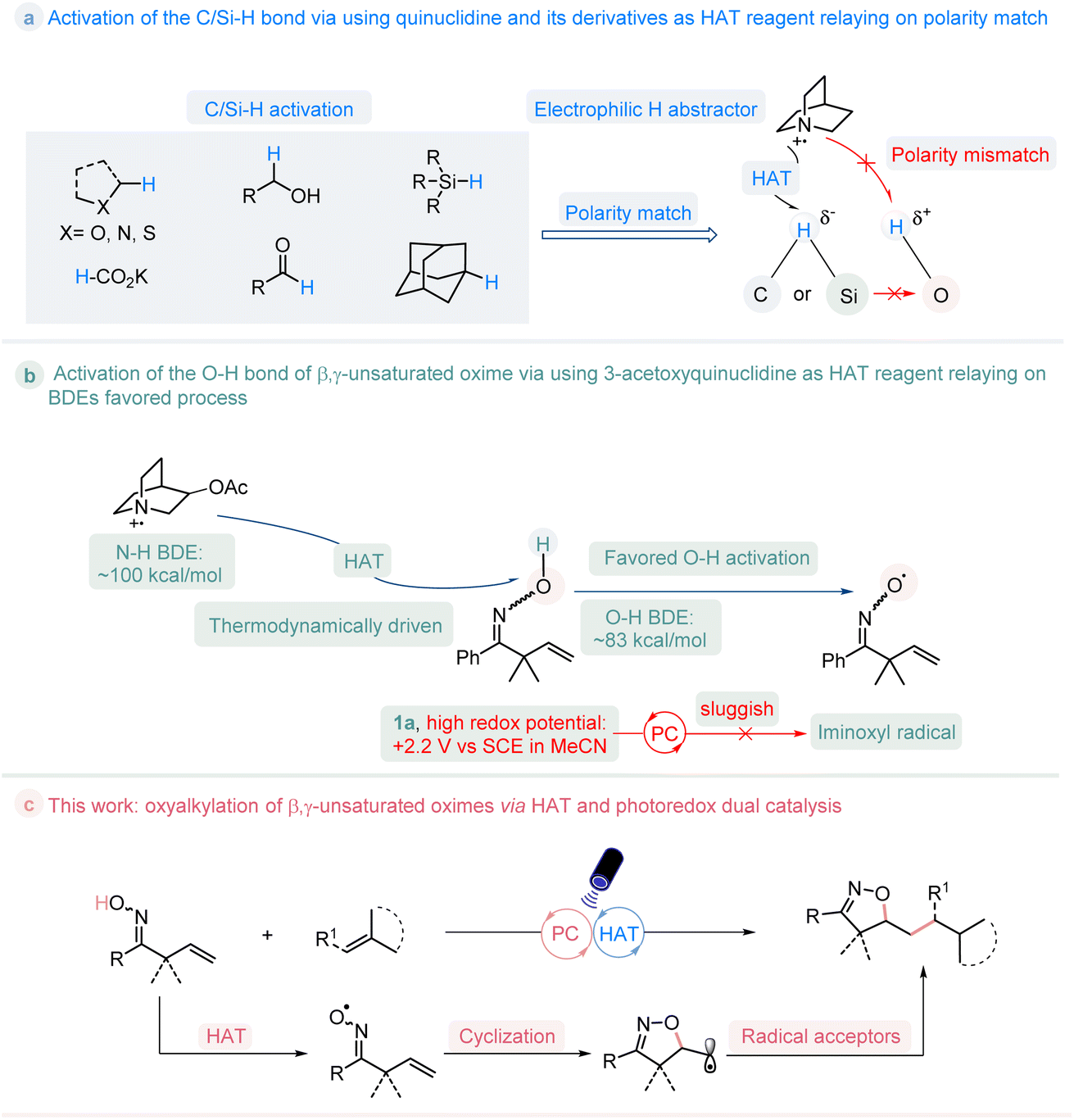 | ||
| Fig. 1 Reaction design: the activation of O–H bond of β,γ-unsaturated oxime by HAT and photoredox dual catalysis. | ||
2 Results and discussion
2.1. Optimization reaction conditions
Our investigation commenced with the visible-light-mediated oxyalkylation of the unactivated alkenes of β,γ-unsaturated oxime 1a with but-3-en-2-one 2a, under 34 W blue LED irradiation. After an exhaustive screening of various photocatalysts, solvents, and bases (see ESI Table S1†), as shown in Table 1, the use of 1.0 mol% Ir[dF(CF3)ppy]2(dtbbpy)PF6 (PC1) and a catalytic amount of 3-acetoxyquinuclidine (20 mol%) in CH3CN provided the desired product in 72% yield (entries 1–6).|
|
||
|---|---|---|
| Entry | Variation from standard conditionsa | Yield%b |
| a Reaction conditions: β,γ-unsaturated oxime 1a (0.4 mmol), but-3-en-2-one 2a (0.2 mmol), photocatalyst (0.002 mmol), 2,6-lutidine (0.4 mmol), 3-acetoxyquinuclidine (0.04 mmol) in CH3CN (2.0 mL) under irradiation of 34 W blue LED for 24 h with fan. b Isolated yield. c 0.04 mmol DABCO in the absence of 3-acetoxyquinuclidine and 2, 6-lutidine. | ||
| 1 | None | 72 |
| 2 | PC2 | 30 |
| 3 | PC3 | Trace |
| 4 | DMF | 7 |
| 5 | DCE | 39 |
| 6 | DABCO | 60 |
| 7 | No base | 64 |
| 8c | DABCO | 51 |
| 9 | No 3-acetoxyquinuclidine, no base | 8 |
| 10 | No 3-acetoxyquinuclidine | 19 |
| 11 | No photocatalyst, no base | 0 |
| 12 | No blue light | 0 |

While the reaction proceeded well without a base, the yield decreased slightly (entry 7), which is reasonable as the optimal base 2,6-lutidine might facilitate the regeneration of the HAT catalyst and the formation of the product via proton transfer. DABCO can in principle also act as a HAT catalyst. However, in the presence of a catalytic amount of DABCO, the product 3a was produced in a lower yield (entry 8).
A significant yield reduction was observed in the absence of 3-acetoxyquinuclidine (entries 9–10). The control experiments demonstrated that light and the photocatalyst are essential for this reaction to occur (entries 11–12).
2.2. Substrate scope investigation
With the optimal conditions in hand, we started to explore the scope of the transformation. As presented in Fig. 2, a series of Michael acceptors, unsaturated ketones, esters or nitriles, reacted smoothly to give the desired products 3a–3g in moderate to good yields. Substituents at the α-position of Michael acceptors (R2 = Me and Ph) were also tolerated, and the corresponding products 3h–3k were obtained with good yields. | ||
| Fig. 2 Scope of different Michael acceptors and β,γ-unsaturated oximes. Reaction conditions: β,γ-unsaturated oxime 1 (0.4 mmol), Michael acceptors 2 (0.2 mmol), Ir[dF(CF3)ppy]2(dtbbpy)PF6 (0.002 mmol), 2,6-lutidine (0.4 mmol), 3-acetoxyquinuclidine (0.04 mmol) in CH3CN (2.0 mL) at room temperature under irradiation of 34 W blue LED for 24 h or 48 h. The diastereomeric ratio of products are determined according to the 1H NMR. [a] 0.2 mmol 1, 0.4 mmol 2. [b] Without 2,6-lutidine. | ||
The reaction of the β-substituted Michael acceptors also proceeded well without an apparent change in the yields 3l–3n. Notably, the cyclic Michael acceptor 2o also exhibited good reactivity. Besides Michael acceptors, styrene 2p proved compatible with this reaction, yielding the desired product 3p in moderate yield, even in the absence of a base. The scope of the reaction with respect to the β,γ-unsaturated oximes were also examined. Both electron-donating (4-MeC6H4, 4-MeOC6H4) and electron-withdrawing groups (4-ClC6H4, 4-BrC6H4 and 4-CNC6H4) were tolerated and gave the desired products 4a–4e in 42–61% yields. Oximes bearing a meta-aryl substituent (3-ClC6H4 and 3-MeC6H4) or a meta/para-disubstitution (3,4-Cl2C6H3 and 3,4-methylenedioxy) also exhibited good reactivity. The reaction of sterically hindered ortho-substituted (2-FC6H4) substrate also proceeded well. Notably, the oxime bearing a non-terminal olefin and 1-naphthyl group was also compatible, giving the corresponding product 4k and 4l in 46% and 52% yield, respectively. This compatibility was extended to heterocyclic substituents, resulting in the products 4m and 4n in 75% and 52% yields, respectively.
The gem-difluoroalkene moiety is viewed as a bioisostere of a carbonyl group and is of considerable interest for drug discovery. This is due to its electronic and steric effects, which resemble those of corresponding aldehydes, ketones, and esters. It has been particularly applied to enhance metabolic stability.12 In our continuing endeavor to expand the utility of this photoredox/HAT dual catalysis strategy, integrating the isoxazoline scaffold with the gem-difluoroalkene moiety presents a compelling avenue for drug discovery. With the same optimized reaction conditions applied, we were delighted to find that a variety of functionally substituted α-CF3 alkenes were effectively applied as radical acceptors in the defluorinative gem-difluorooxylation of β,γ-unsaturated oximes (Fig. 3). Different substitutions involving electron-donating groups (6b–6e, 6h–6j) and weak electron-withdrawing groups (6f and 6g) on the aryl ring of α-CF3 styrenes do not significantly affect the reaction, resulting in desired products with moderate to good yields. It is worth noting that trifluoromethyl styrenes derived from ibuprofen (5k), gemfibrozil (5l) and oxaprozin (5m) reacted smoothly to give the difluorinated products. Moreover, thiophene substituted α-CF3 styrene (6n) also underwent the expected reaction, giving the products with a 54% yield. In additions, we also investigated the scope of oxime moiety, with most oximes proceeding well under the optimized conditions. Interestingly, while exploring the scope of α-CF3 alkenes, we discovered that when α-CF3 styrene bearing an electron-withdrawing group provides the CF3-group containing product 7a–d cyano (7a), acetyl (7b), and methyl ester (7c) with a good yield under standard conditions. For the proposed reaction mechanism, we propose the formation of an anion Int. 7d′, which rationalizes the formation of two distinct products. If Int.7d′ bears an electron-withdrawing group, it could render the carbon anion site more basic, favoring protonation over defluorination. In addition, the oxime moiety was also explored. A series of β,γ-unsaturated oximes participated in the process, giving the product 7e–7i in good yields, regardless of the electronic property and position of the substituent on the aromatic ring. Notably, the thiophene substituted and α-dimethyl unsubstituted oximes also reacted well, providing the final products 7j–7l.
 | ||
| Fig. 3 Scope of different α-trifluoromethyl styrenes and β,γ-unsaturated oximes. Reaction conditions: β,γ-unsaturated oximes 1 (0.4 mmol), α-CF3 styrenes 5 (0.2 mmol), Ir[dF(CF3)ppy]2(dtbbpy)PF6 (0.002 mmol), 2,6-lutidine (0.4 mmol), 3-acetoxyquinuclidine (0.04 mmol) in CH3CN (2.0 mL) under irradiation of 34 W blue LED for 24 h or 48 h with fan. The diastereomeric ratio of products 7 are determined according to the 1H NMR. [a] Without 2,6-lutidine. | ||
2.3. Scale-up reaction and mechanism studies
Given the broad applicability of our newly developed O–H bond activation and application in the synthesis of diverse isoxazolines, we performed a 20-fold scale-up without the use of a base and with a decreased 3-acetoxyquinuclidine loading (10 mol%); still providing an acceptable yield (Fig. 4a). To better understand the reaction mechanism, we performed several mechanistic studies to confirm the involvement of isoxazoline-containing alkyl radical in this reaction. Employing ethene-1,1-diyldibenzene as the radical acceptor under standard conditions and without a base yielded the radical-trapped product 8a in 87% NMR yield (Fig. 4b). Moreover, when 3.0 equivalents of TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) were added under standard conditions, formation of the desired product was completely inhibited. The TEMPO-trapped adduct 8b was obtained instead (Fig. 4c). Two isotope-labeling studies further suggested the presence of carbon anion species in the catalytic cycle (Fig. 4d and e). To ascertain, that the iminoxyl radical was generated via a HAT mechanism rather than direct oxidation or a PT/ET pathway, comprehensive steady-state Stern–Volmer luminescence quenching experiments were performed (Refer to ESI Fig. S3–S17†). Specifically, to confirm the quenching of the excited photocatalyst PC1* by 3-acetoxyquinuclidine, various concentrations of 3-acetoxyquinuclidine were added to PC1*. This yielded a linear fitting of the plots (Fig. 4f and g). The high redox potential value of 1a pointed to a sluggish direct oxidation by PC1*, as confirmed by the lack of observable luminescence quenching (Fig. 4h). To discern that PC1* could be quenched by oxime 1a interacting with lutidine, quenching studies were performed with a mixture of oxime 1a and varying concentrations of lutidine. However, no substantial quenching was observed (Fig. 4i), excluding the PT/ET pathway. Given that 3-acetoxyquinuclidine could possibly act as a base to abstract the proton from oxime or initiate a PCET (proton-coupled electron transfer) process, we conducted a quenching study between PC1* and a mixture of 3-acetoxyquinuclidine with different concentrations of oxime 1a. However, varied concentrations of oxime 1a did not impact the quenching rate, as the quenching rate of the mixture was consistent with that of 3-acetoxyquinuclidine alone (Fig. 4j). Furthermore, to determine any potential interaction of oxime 1a with 3-acetoxyquinuclidine and 2,6-lutidine, we conducted quenching studies using a mixture similar to the standard reaction concentrations. We found no significant quenching with this mixture, inferring that oxime 1a does not interact with the other reaction components (Fig. 4k). In addition, we undertook cyclic voltammetry (CV) experiments involving oxime 1a and 2,6-lutidine (ESI, Fig. S2†). Our findings revealed that the presence of 2,6-lutidine appears to inhibit the oxidation of oxime 1a. This observation prompted us to further explore the impact of 3-acetoxyquinuclidine on the redox potential of 1a. Therefore, we conducted CV measurements on a mixture of oxime 1a and 3-acetoxyquinuclidine. In comparison to the CV diagram of oxime 1a, we noted distinct differences in the shape of the CV diagram for this mixture, particularly within the region corresponding to 1a. This discrepancy arises from the overlap with a portion of the 3-acetoxyquinuclidine region in the diagram. Additionally, we conducted experiments involving oxime 1a in conjunction with both, 3-acetoxyquinuclidine and 2,6-lutidine. Owing to the overlapping regions of these three components, we cannot make direct comparisons. However, despite this complication, 3-acetoxyquinuclidine exhibits the clear propensity for oxidation as the primary species in these mixtures. This can be attributed to the considerably lower redox potential within the system, as detailed in ESI Fig. S2.† These results align with the outcomes of our Stern–Volmer quenching experiments which revealed that, in the presence of 2,6-lutidine and/or oxime 1a, the quenching efficiency of 3-acetoxyquinuclidine towards photocatalyst PC1 remained largely consistent.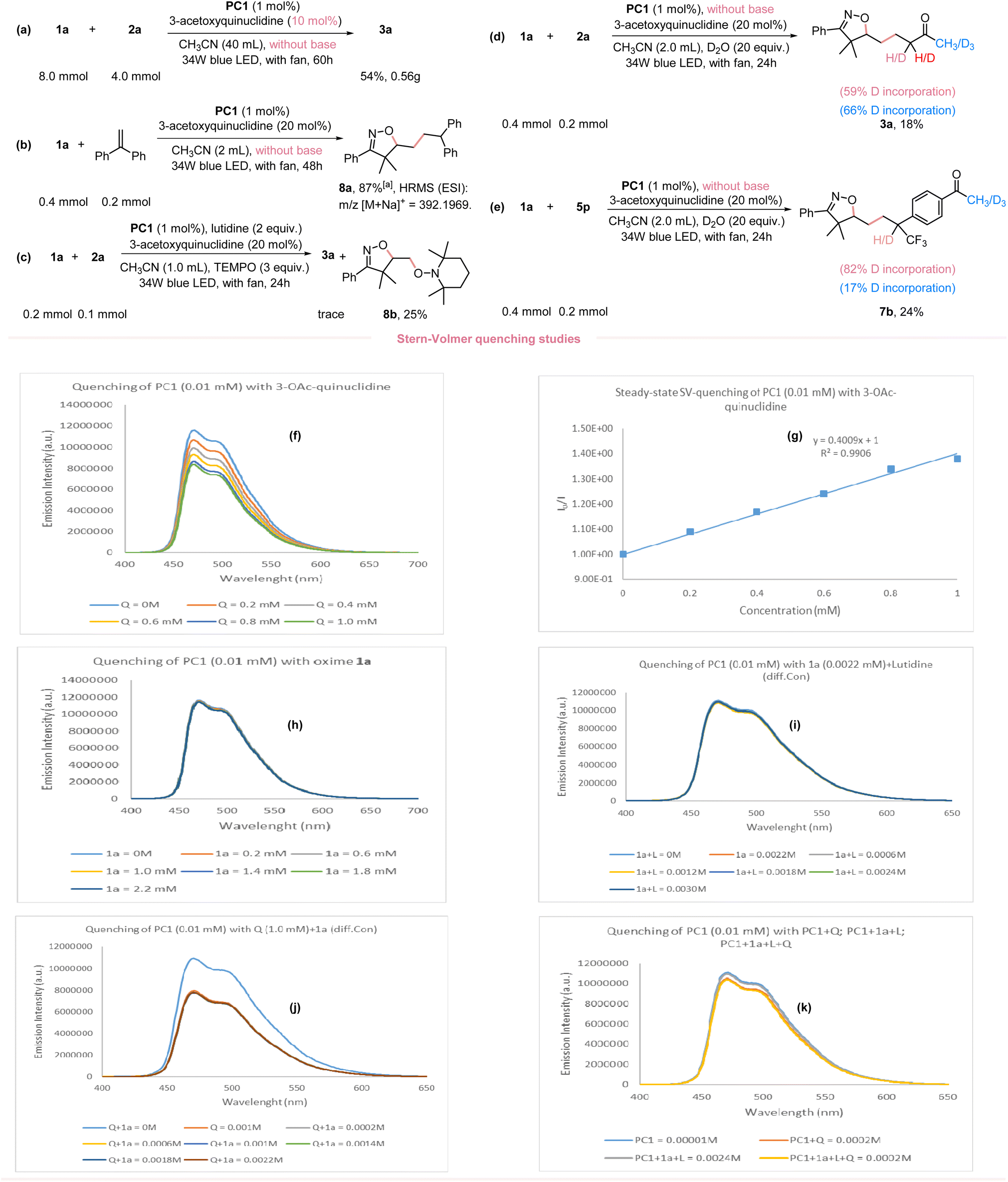 | ||
| Fig. 4 (a) 20-fold scale-up reaction. (b) Isoxazoline-containing alkyl radical capture reaction by styrene. (c) Isoxazoline-containing alkyl radical capture reaction by TEMPO. (d) Isotope-labelling experiment of Michael acceptor (e) isotope-labelling experiment of α-CF3 alkenes. (f–k) Stern–Volmer quenching experiments. [a] NMR yield using CH2Br2 as the internal standard. | ||
2.4. DFT calculation and proposed mechanism
Furthermore, we conducted density functional theory (DFT) calculations (Fig. 5a) to further verify the HAT process. The 3-acetoxylquinuclidinyl radical cation (V), formed through oxidation by the excited *IrIII photocatalyst, can abstract a hydrogen atom from oxime 1a with an energy barrier of 18.9 kcal mol−1. Following the HAT process, the generated iminoxyl radical VI and amine cation X are 17.1 kcal mol−1 more stable. Thus, the HAT process is both kinetically feasible and thermodynamically favored. Subsequently, iminoxyl radical VI undergoes a 5-exo-trig cyclization with an energy barrier of 14.5 kcal mol−1, leading to the formation of the C-centered isoxazoline alkyl radical VII. Based on the experimental and computational mechanistic studies, as well as prior research,3o,7j a plausible mechanism for this photoredox-catalyzed HAT protocol is proposed (Fig. 5b).13 Firstly, the IrIII complex (I) is irradiated by the visible light to give a long-lived triplet excited state *IrIII oxidant (II), which is reduced in the presence of 3-acetoxyquinuclidine (III) to give IrII complex (IV) and amine radical cation (V). Then (V) abstracts the H-atom fromβ,γ-unsaturated oxime 1a to form the iminoxyl radical (VI) which upon 5-exo-trig radical cyclization, yields the C-centered isoxazoline alkyl radical (VII), that subsequently adds to the radical acceptor to form the radical intermediate (VIII). Finally, the IrII complex (IV) is oxidized by (VIII) to regenerate the photocatalyst IrIII complex (I) and the isoxazoline-containing carbon anion (IX). In the case of Michael acceptors, styrene and electron-withdrawing group substituted α-CF3 alkenes (IX), abstracts a proton from amine cation (X) to regenerate HAT catalyst 3-acetoxyquinuclidine (III) to finish the catalytic cycle and to provide the protonated products (3/4/7). Whereas the gem-difluoroalkene products (6) are obtained via defluorination when the α-CF3 styrenes exhibits electron-donating or electron-neutral substituents. The main by-product in this reaction is the is 5-methyl-4,5-dihydroisoxazole which could be formed by HAT of the carbon centered radical (VII) from 1a to generate iminoxyl radical (VI). | ||
| Fig. 5 (a). DFT calculation of hydrogen atom transfer and cyclization steps. (b). Proposed mechanism. | ||
3 Conclusions
In conclusion, we have developed the first visible-light-mediated photoredox and HAT dual catalyzed oxyalkylation of unactivated alkenes of β,γ-unsaturated oximes. This strategy produces a wide range of isoxazoline derivatives through reactions with different Michael acceptors, styrenes, or α-CF3 alkenes. Our approach successfully addresses the high redox potential of the oxime, complements the activation of the O–H bond by quinuclidine, and provides a versatile pathway to generate iminoxyl radicals. Furthermore, this efficient and mild synthetic methodology has the potential to produce a diverse array of valuable isoxazoline compounds, which may contribute to advancements in drug discovery efforts. Further studies may be directed towards the further exploration of photoredox HAT dual catalysis generated iminoxyl radicals in synthesis and reaction development.Data availability
Experiment procedures, characterization of the new compounds, and Cartesian coordinates of DFT calculations are available in the ESI.†Author contributions
L. Y. and M. R. conceived and designed the experiments. L. Y., T. J., Y. M., and Y. C. performed the experiments and analysed the data. C. Z. preformed the DFT calculation. R. K. preformed the Stern–Volmer quenching experiments. M. R. directed the project. All authors discussed the experimental results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication is based upon work supported by the King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under CRG award no. URF/1/5082 and URF/1/4701. L. Y. thanks the China Scholarship Council. C. Z. acknowledges the KAUST Supercomputing Laboratory for providing computational resources from the supercomputer Shaheen II. We thank Dr Jeremy Bau for quantum yield measurement.Notes and references
- (a) M. H. Shaw, J. Twilton and D. W. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (b) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. MacMillan, Nat. Rev. Chem, 2017, 1, 0052 CrossRef CAS; (c) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (d) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef CAS PubMed; (e) Q. Q. Zhou, Y. Q. Zou, L. Q. Lu and W. J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed; (f) C. Zhu, H. Yue, L. Chu and M. Rueping, Chem. Sci., 2020, 11, 4051–4064 RSC; (g) C. Zhu, H. Yue, Ji. Jia and M. Rueping, Angew. Chem., Int. Ed., 2021, 60, 17810–17831 CrossRef CAS PubMed; (h) L. Yi, T. Ji, K.-Q. Chen, X. Yu Chen and M. Rueping, CCS Chem., 2022, 4, 9–30 CrossRef CAS; (i) J. Zhang and M. Rueping, Chem. Soc. Rev., 2023, 52, 4099–4120 RSC.
- J. K. J. T Hynes, H.-H. Limbach and R. L. Schowen, Hydrogen-Transfer Reactions, Wiley-VCH, Weinheim, 2007 Search PubMed.
- Examples: (a) D. Hager and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 16986–16989 CrossRef CAS PubMed; (b) K. Qvortrup, D. A. Rankic and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 626–629 CrossRef CAS PubMed; (c) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (d) Y.-Y. Gui, X.-W. Chen, W.-J. Zhou and D.-G. Yu, Synlett, 2017, 28, 2581–2586 CrossRef CAS; (e) Y.-Y. Gui, L.-L. Liao, L. Sun, Z. Zhang, J.-H. Ye, G. Shen, Z.-P. Lu, W.-J. Zhou and D.-G. Yu, Chem. Commun., 2017, 53, 1192–1195 RSC; (f) R. Zhou, Y. Y. Goh, H. Liu, H. Tao, L. Li and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 16621–16625 CrossRef CAS PubMed; (g) J. Twilton, M. Christensen, D. A. DiRocco, R. T. Ruck, I. W. Davies and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 5369–5373 CrossRef CAS PubMed; (h) J. Ye, I. Kalvet, F. Schoenebeck and T. Rovis, Nat. Chem., 2018, 10, 1037–1041 CrossRef CAS PubMed; (i) V. Dimakos, H. Y. Su, G. E. Garrett and M. S. Taylor, J. Am. Chem. Soc., 2019, 141, 5149–5153 CrossRef CAS PubMed; (j) H.-B. Yang, A. Feceu and D. B. Martin, ACS Catal., 2019, 9, 5708–5715 CrossRef CAS; (k) J. Hou, A. Ee, H. Cao, H. W. Ong, J. H. Xu and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 17220–17224 CrossRef CAS PubMed; (l) M. Jouffroy, C. B. Kelly and G. A. Molander, Org. Lett., 2016, 18, 876–879 CrossRef CAS PubMed; (m) E. L. Tyson, Z. L. Niemeyer and T. P. Yoon, J. Org. Chem., 2014, 79, 1427–1436 CrossRef CAS PubMed; (n) L. Capaldo and D. Ravelli, Eur. J. Org Chem., 2017, 2017, 2056–2071 CrossRef CAS PubMed; (o) M. H. Shaw, V. W. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. MacMillan, Science, 2016, 352, 1304–1308 CrossRef CAS PubMed.
- W. Xiao, X. Wang, R. Liu and J. Wu, Chin. Chem. Lett., 2021, 32, 1847–1856 CrossRef CAS.
- (a) P.-Z. Wang, J.-R. Chen and W.-J. Xiao, J. Am. Chem. Soc., 2023, 145, 17527–17550 CrossRef CAS PubMed; (b) X. Wu, Z. Ma, T. Feng and C. Zhu, Chem. Soc. Rev., 2021, 50, 11577–11613 RSC; (c) A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed; (d) Q. An, Y.-Y. Xing, R. Pu, M. Jia, Y. Chen, A. Hu, S.-Q. Zhang, N. Yu, J. Du and Y. Zhang, J. Am. Chem. Soc., 2022, 145, 359–376 CrossRef PubMed; (e) E. Tsui, H. Wang and R. R. Knowles, Chem. Sci., 2020, 11, 11124–11141 RSC; (f) L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2021, 122, 2429–2486 CrossRef PubMed.
- (a) J. Hartung, Eur. J. Org Chem., 2001, 2001, 619–632 CrossRef; (b) M. H. V. Huynh and T. J. Meyer, Chem. Rev., 2007, 107, 5004–5064 CrossRef CAS PubMed; (c) E. Tsui, A. J. Metrano, Y. Tsuchiya and R. R. Knowles, Angew. Chem., Int. Ed., 2020, 59, 11845–11849 CrossRef CAS PubMed.
- (a) M. D. Mosher, A. L. Norman and K. A. Shurrush, Tetrahedron Lett., 2009, 50, 5647–5648 CrossRef CAS; (b) M.-K. Zhu, J.-F. Zhao and T.-P. Loh, J. Am. Chem. Soc., 2010, 132, 6284–6285 CrossRef CAS PubMed; (c) D. Jiang, J. Peng and Y. Chen, Org. Lett., 2008, 10, 1695–1698 CrossRef CAS PubMed; (d) Y.-T. He, L.-H. Li, Y.-F. Yang, Y.-Q. Wang, J.-Y. Luo, X.-Y. Liu and Y.-M. Liang, Chem. Commun., 2013, 49, 5687–5689 RSC; (e) Q. Wei, J.-R. Chen, X.-Q. Hu, X.-C. Yang, B. Lu and W.-J. Xiao, Org. Lett., 2015, 17, 4464–4467 CrossRef CAS PubMed; (f) F. Chen, F.-F. Zhu, M. Zhang, R.-H. Liu, W. Yu and B. Han, Org. Lett., 2017, 19, 3255–3258 CrossRef CAS PubMed; (g) W. Zhang, Y. Su, K.-H. Wang, L. Wu, B. Chang, Y. Shi, D. Huang and Y. Hu, Org. Lett., 2017, 19, 376–379 CrossRef CAS PubMed; (h) L. Zhu, G. Wang, Q. Guo, Z. Xu, D. Zhang and R. Wang, Org. Lett., 2014, 16, 5390–5393 CrossRef CAS PubMed; (i) R.-H. Liu, D. Wei, B. Han and W. Yu, ACS Catal., 2016, 6, 6525–6530 CrossRef CAS; (j) B. Han, X. L. Yang, R. Fang, W. Yu, C. Wang, X. Y. Duan and S. Liu, Angew. Chem., Int. Ed., 2012, 51, 8816–8820 CrossRef CAS PubMed; (k) J.-Q. Chen, N. Liu, Q. Hu, J. Liu, J. Wu, Q. Cai and J. Wu, Org. Chem. Front., 2021, 8, 5316–5321 RSC; (l) Z. Li, L. Qian, H. Chen and X. Xu, Synlett, 2022, 33, 293–295 CrossRef CAS; (m) X. Q. Hu, J. Chen, J. R. Chen, D. M. Yan and W. J. Xiao, Chem. – Eur. J., 2016, 22, 14141–14146 CrossRef CAS PubMed; (n) X.-L. Yang, Y. Long, F. Chen and B. Han, Org. Chem. Front., 2016, 3, 184–189 RSC; (o) X.-X. Peng, Y.-J. Deng, X.-L. Yang, L. Zhang, W. Yu and B. Han, Org. Lett., 2014, 16, 4650–4653 CrossRef CAS PubMed; (p) X.-L. Yang, F. Chen, N.-N. Zhou, W. Yu and B. Han, Org. Lett., 2014, 16, 6476–6479 CrossRef CAS PubMed.
- X.-Q. Hu, G. Feng, J.-R. Chen, D.-M. Yan, Q.-Q. Zhao, Q. Wei and W.-J. Xiao, Org. Biomol. Chem., 2015, 13, 3457–3461 RSC.
- W.-Z. Liu and F. G. Bordwell, J. Org. Chem., 1996, 61, 4778–4783 CrossRef CAS PubMed.
- (a) D. A. Pratt, J. A. Blake, P. Mulder, J. C. Walton, H.-G. Korth and K. U. Ingold, J. Am. Chem. Soc., 2004, 126, 10667–10675 CrossRef CAS PubMed; (b) S.-S. Chong, Y. Fu, L. Liu and Q.-X. Guo, J. Phys. Chem. A, 2007, 111, 13112–13125 CrossRef CAS PubMed.
- (a) C.-B. Xue, J. Wityak, T. M. Sielecki, D. J. Pinto, D. G. Batt, G. A. Cain, M. Sworin, A. L. Rockwell, J. J. Roderick and S. Wang, J. Med. Chem., 1997, 40, 2064–2084 CrossRef CAS PubMed; (b) S. Castellano, D. Kuck, M. Viviano, J. Yoo, F. López-Vallejo, P. Conti, L. Tamborini, A. Pinto, J. L. Medina-Franco and G. Sbardella, J. Med. Chem., 2011, 54, 7663–7677 CrossRef CAS PubMed; (c) J.-F. Cheng, Y. Huang, R. Penuliar, M. Nishimoto, L. Liu, T. Arrhenius, G. Yang, E. O'leary, M. Barbosa and R. Barr, J. Med. Chem., 2006, 49, 4055–4058 CrossRef CAS PubMed; (d) R. E. Olson, T. M. Sielecki, J. Wityak, D. J. Pinto, D. G. Batt, W. E. Frietze, J. Liu, A. E. Tobin, M. J. Orwat and S. V. Di Meo, J. Med. Chem., 1999, 42, 1178–1192 CrossRef CAS PubMed; (e) P. K. Poutiainen, T. Oravilahti, M. Perakyla, J. J. Palvimo, J. A. Ihalainen, R. Laatikainen and J. T. Pulkkinen, J. Med. Chem., 2012, 55, 6316–6327 CrossRef CAS PubMed; (f) P. K. Poutiainen, T. A. Venäläinen, M. Peräkylä, J. M. Matilainen, S. Väisänen, P. Honkakoski, R. Laatikainen and J. T. Pulkkinen, Bioorg. Med. Chem., 2010, 18, 3437–3447 CrossRef CAS PubMed.
- (a) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed; (b) G. Magueur, B. Crousse, M. Ourévitch, D. Bonnet-Delpon and J.-P. Bégué, J. Fluorine Chem., 2006, 127, 637–642 CrossRef CAS; (c) P. M. Weintraub, A. K. Holland, C. A. Gates, W. R. Moore, R. J. Resvick, P. Bey and N. P. Peet, Bioorg. Med. Chem., 2003, 11, 427–431 CrossRef CAS PubMed; (d) C. Leriche, X. He, C.-w. T. Chang and H.-w. Liu, J. Am. Chem. Soc., 2003, 125, 6348–6349 CrossRef CAS PubMed; (e) Y. Pan, J. Qiu and R. B. Silverman, J. Med. Chem., 2003, 46, 5292–5293 CrossRef CAS PubMed; (f) J.-M. Altenburger, G. Y. Lassalle, M. Matrougui, D. Galtier, J.-C. Jetha, Z. Bocskei, C. N. Berry, C. Lunven, J. Lorrain and J.-P. Herault, Bioorg. Med. Chem., 2004, 12, 1713–1730 CrossRef CAS PubMed.
- Selected articles on hydrogen atom transfer from our group: (a) L. Huang, M. Szewczyk, R. Kancherla, B. Maity, C. Zhu, L. Cavallo and M. Rueping, Nat. Commun., 2023, 14, 548 CrossRef CAS PubMed; (b) C. Zhu, H. Chen, H. Yue and M. Rueping, Nat. Synth., 2023, 2, 1068–1081 CrossRef; (c) R. Kancherla, K. Muralirajan, B. Maity, S. Karuthedath, G. S. Kumar, F. Laquai, L. Cavallo and M. Rueping, Nat. Commun., 2022, 13, 2737 CrossRef CAS PubMed; (d) R. Kancherla, K. Muralirajan, B. Maity, S. Karuthedath, G. S. Kumar, F. Laquai, L. Cavallo and M. Rueping, Nat. Commun., 2022, 13, 2737 CrossRef CAS PubMed; (e) B. Maity, C. Zhu, M. Rueping and L. Cavallo, ACS Catal., 2021, 11, 13973–13982 CrossRef CAS; (f) B. Maity, C. Zhu, H. Yue, L. Huang, M. Harb, Y. Minenkov, M. Rueping and L. Cavallo, J. Am. Chem. Soc., 2020, 142, 16942–16952 CrossRef CAS PubMed; (g) A. Dewanji, P. E. Krach and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 3566–3570 CrossRef CAS PubMed; (h) Y. Cai, Y. Tang, L. Fan, Q. Lefebvre, H. Hou and M. Rueping, ACS Catal., 2018, 8, 9471–9476 CrossRef CAS; (i) L. Huang and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 10333–10337 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04410f |
| This journal is © The Royal Society of Chemistry 2023 |