
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon dots from alcohol molecules: principles and the reaction mechanism†
Hanyu
Tu
,
Huaxin
Liu
,
Laiqiang
Xu
,
Zheng
Luo
,
Lin
Li
,
Ye
Tian
,
Wentao
Deng
,
Guoqiang
Zou
 ,
Hongshuai
Hou
* and
Xiaobo
Ji
*
,
Hongshuai
Hou
* and
Xiaobo
Ji
*
State Key Laboratory of Powder Metallurgy, College of Chemistry and Chemical Engineering, Central South University, Changsha, 410083, China. E-mail: hs-hou@csu.edu.cn; xji@csu.edu.cn
First published on 9th October 2023
Abstract
Carbon dots (CDs) have attracted significant attention in the energy, environment, and biology fields due to their exceptional physicochemical properties. However, owing to the multifarious precursors and complex reaction mechanisms, the production of carbon dots from organic molecules is still a mysterious process. Inspired by the color change of sodium hydroxide ethanol solution after standing for some time, in this work, we thoroughly investigated the reaction mechanism from alcohol molecules to carbon dots through a lot of experiments and theoretical calculations, and it was found that the rate-controlling reaction is the formation of aldehydes, and it is also confirmed that there is a self-catalysis reaction, which can accelerate the conversion from alcohol to aldehyde, further facilitating the final formation of CDs. After the rate-controlling reaction of alcohol to aldehyde, under strongly alkaline conditions, an aldol reaction occurs to form unsaturated aldehydes, followed by further condensation and polymerization reactions to form long carbon chains, which are cross-linked and dehydrated to form carbon dots with a carbon core and surface functional groups. Additionally, it is found that the reaction can be largely accelerated with the assistance of electricity, which indicates the great prospect of industrial production. Furthermore, the obtained CDs with rich functional groups can be utilized as electrolyte additives to optimize the deposition behavior of Na metal, manifesting great potential towards safe and stable Na metal batteries.
Introduction
Carbon dots (CDs) have become a subject of great interest due to their distinct characteristics as zero-dimensional carbon nanomaterials as well as owing to their small size, good solubility in different solvents, the abundance of surface functional groups, remarkable photoluminescence, electrochemical properties and so forth. These appealing and unique properties of CDs present a wide range of potential applications, including their utilization in energy conversion/storage devices, biomedical imaging, and catalysis.1–5 Typically, they are nanostructured materials with a diameter of less than 10 nm in all three dimensions, consisting of two parts: the carbon core and surface functional layer.6Carbon dots can be prepared by two main categories of methods: top-down and bottom-up approaches.7 Top-down methods apply to the etching and exfoliating of large-size carbon targets such as graphite, carbon nanotubes and graphene into smaller pieces.8–11 Nevertheless, the practical application of top-down methods is limited because of their tedious operation steps and the requirement for harsh experimental conditions.12 Bottom-up methods seem to be the most widely administered route in terms of the simple equipment and cost-effectiveness principles, containing combustion, hydrothermal, solvothermal, microwave, and pyrolysis to carbonize small organic molecules such as glucose, citric acid, carbohydrate, biomass precursors.13–16 Current research in the strategies towards green, low-cost raw materials and large-scale production greatly boosts the application of carbon dots.17,18 However, the low yields and the difficulty in achieving stable regulation of their properties are the main causes hindering the further development of CDs. Bottom-up methods exhibit the complex formation process of CDs, and precursors contribute significantly to the synthesis process. Hence, revealing the reaction mechanism of the formation process of CDs is of great theoretical significance to improve the yield of CDs and to realize the precise design of the structures with specific functional groups on the edges, which is an urgent challenge to be solved.12
Ten years ago, we observed that the solution would turn yellow after leaving the ethanol solution of sodium hydroxide for several days. We then configured a 1.5 M ethanolic solution of NaOH and confirmed the observed color change (Fig. S1†). The initial solution was colorless (day 1) and then changed to light yellow (day 7), dark yellow (day 8), reddish brown (day 10), and brown (day 11). The microstructure of this product was investigated, and it was found to be carbon dots with the sodium element on the surface (Fig. S2†). Interestingly, 10 mL of the solution on day 7 was added to the solution on day 1, and the solution turned brown after 7 days, which is consistent with the solution that was left for 11 days. We speculated that the presence of a self-catalyst in this reaction would accelerate the reaction. Also, it was found that the color change rate of the resting solution was influenced by the amount of sodium hydroxide (1 mol L−1 to 3 mol L−1, Fig. S1†). Subsequently, we also noticed that a similar phenomenon was reported with a faster reaction rate during the electrolysis of ethanol solutions.19 However, the chemical reaction mechanism explaining why ethanol can generate carbon dots in alkaline solutions and how electrolysis enhances the reaction rate remains unknown.
We spent years attempting to unravel the mystery behind this interesting phenomenon, but it was not until recently that we were successful and obtained a satisfactory answer. Under alkaline conditions, ethanol is oxidized to aldehydes, which generate carbon chains and tiny clusters through their condensation and substitution reactions, and then carbon cores are formed by agglomerating and carbonizing carbon chains and clusters, finally, carbon dots with various short chains containing abundant functional groups, cross-linked, and anchored on the surface of the carbon nucleus were obtained.
Due to their unique features, carbon dots have been shown to be excellent electrolyte additives for metal batteries. It is worth noting that metal batteries, including lithium metal batteries, sodium metal batteries, and zinc metal batteries, are the most promising high specific energy battery systems. Nonetheless, they all confront serious challenges, such as dendrite growth and electrolyte side effects on the metal anode side, leading to poor electrochemical performance and potential safety hazards.20 We previously found that nitrogen and sulfur co-doped carbon dots with good solubility in electrolytes can function as electrolyte additives for inducing uniform deposition of Li+, which effectively inhibits the growth of Li dendrites.21 Additionally, we applied graphene quantum dots to the electrolyte solution of zinc ion batteries to get a robust Zn anode by lowering the nuclear barrier of zinc formation and reducing the reactivity of water.22 Taking inspiration from this observation and considering the presence of sodium on the surface of carbon dots prepared from alcohol and NaOH, we predicted that these types of carbon dots could be used as electrolyte additives for sodium metal batteries to modulate the transport behavior of sodium ions at the electrolyte and interface, to inhibit the growth of sodium dendrites; meanwhile, it can also supplement sodium ions in the electrolyte.
Herein, we comprehensively investigated the mechanism from alcohols to carbon dots. We systematically studied the effect of various types of alcohol (ethanol, ethylene glycol, glycerol, and n-butanol), the water content in the solution, and other relevant factors on carbon dots. It is shown that the Na content of CDs and the process of synthesizing CDs are strongly associated with the water ratio in electrolytes. Further, we applied CDs containing the highest Na content as electrolyte additives to sodium metal batteries to get a robust Na anode by lowering the nuclear barrier of sodium formation and boosting the Na+ ion transference number. After introducing Na-doped CDs, the Na metal anode exhibited high CE (99.8%) and long-term cycling stability (1200 h). The SEM and XPS confirmed the uniform post-cycling morphology and F-containing SEI protection layer.
Results and discussion
The electrolytic method was employed to prepare carbon dots (Fig. S3†). Ethanol, ethylene glycol, glycerol and n-butanol were used as electrolytes to investigate the effect of carbon chain length and functional groups of the carbon source on the synthesis process of carbon dots (abbreviated as EA-CDs, EG-CDs, GL-CDs, and NBA-CDs, respectively). Meanwhile, we also investigated the effect of water content in electrolytes on carbon dot generation (abbreviated as 100EA-CDs, 90EA-CDs, 80EA-CDs and so on, indicating the percentage of ethanol content in the ethanol-based electrolyte).Compared with the generation process of 100EA-CDs, 90EA-CDs, and 80EA-CDs (Fig. S4–S13†), the increasing amount of water content in the electrolyte leads to a faster reaction, which we assume is due to the fact that sodium hydroxide dissociates OH− and Na+ more easily in water than in ethanol. Additionally, the existence of a self-catalyst in the middle of the reaction further accelerated the color change, particularly in the later stages of the reaction. Furthermore, the initial reaction of EG-CDs, GL-CDs, and NBA-CDs was faster than that of EA-CDs. Initially, a large number of bubbles are generated due to the formation of H2 and O2 during the electrolysis of aqueous sodium hydroxide solution at 20 V, indicating that the by-product is high value-added H2, presenting industrial prospects. The shorter initial discoloration time of the solution indicated an intense reaction. However, as the reaction progresses towards its endpoint, the reaction end time increases. This leads us to speculate whether the decreasing solubility of NaOH in EA, EG, GL, and NBA sequentially contributes to the lengthening of the reaction time, which will be further investigated and verified in the subsequent experiments.
Various characterization experiments are performed to characterize the structure of the reaction products. Transmission electron microscope (TEM) images of 100EA-CDs (Fig. 1) demonstrate the good dispersibility and uniformity of CDs with average sizes of about 2–3 nm. The high-resolution (HRTEM) image of an individual 100EA-CD, as shown in Fig. S14,† reveals a high crystallinity with a lattice fringe of 0.21 nm.23 X-ray diffraction (XRD) patterns of ten kinds of CDs are shown in Fig. S15(a) and (b),† which display similar broad diffraction peaks at 23–26° attributed to highly cross-linked carbon skeletons.24 UV/Vis absorbance spectra of ten kinds of CDs are presented in Fig. S15(c) and (d).† The absorbance in the 200–250 nm range is attributed to the n–σ* transition of C–OH.25 The peaks in the range from 250 nm to 300 nm indicate the strong π–π* transition of the conjugated double bond in the CDs.23,25
 | ||
| Fig. 1 TEM images of prepared CDs. (a) 100EA-CDs. (b) 90EA-CDs. (c) 80EA-CDs. (d) 60EA-CDs. (e) 50EA-CDs. (f) 100EG-CDs. (g) 80EG-CDs. (h) 100GL-CDs. (i) 80GL-CDs. (j) 100NBA-CDs. | ||
For chemical composition, Fourier transform infrared spectrometer (FTIR, Fig. 2(a) and (b)) reveal the characteristic absorption peaks for CDs, which are assigned to O–H (3435 cm−1), C–H (2970 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1627 cm−1), O–CO (1700 cm−1), –CH3 (1425 cm−1), and C–O (1079 cm−1) stretching vibrations, respectively.17,26–28 The element content and surface functional groups of CDs can be further proved by X-ray photoelectron spectroscopy (XPS, Fig. 2(c) and (d)). CDs mainly contain C, O, and Na elements, and their corresponding binding energies are around 285.3, 532.5, and 1072.3 eV, respectively. Like the results of previous measurements, ten kinds of CDs show similar results for surface groups. High-resolution C 1s spectra (Fig. S16(a1)†) of CDs show a C 1s band containing four main peaks near 284.8, 285.4, 286.8, and 288.4 eV, attributed to C–C/CC, C–O, CO, and O–CO groups, respectively.21,28–30 Fig. S16(a2)† shows four fitting peaks for the high-resolution O 1s spectrum: CO (531.4 eV), O–H (532.4 eV), C–O (533.4 eV), and the Na KLL Auger peak appears at approximately 536.4 eV.31–33 However, no obvious Na KLL Auger peak for other kinds of carbon dots was observed, owing to the very low Na element content in CDs and the different binding forms of sodium to functional groups on the surface of CDs. The Na 1s spectra (Fig. S16(a3)†) exhibit one peak near 1071.7 eV for the C–O–Na bond.34 Both XPS and FTIR results indicate that the surface of carbon dots is rich in oxygen-containing functional groups. XPS spectrum analysis of the products revealed that with the increasing water content in the electrolyte, the Na content of five kinds of EA-CDs decreased to some extent (Fig. S16 and S17†). The reduced Na content can be associated with the diluted alcohol solution of sodium hydroxide after introducing water. Furthermore, the aqueous solution of sodium hydroxide is electrolyzed during the electrolysis of the alcohol solution of sodium hydroxide to produce hydrogen and other by-products, owing to the fewer hydroxyl radicals involved in the formation process of CDs. At the same time, after introducing water, the binding form of sodium element and organic functional group on the surface of CDs changed, while the ionic form of sodium can be easily removed in the dialysis purification process, thus an increase of water content in electrolyte leading to a decrease of sodium content in CDs. By comparison, n-butanol contains a lower ratio of elemental oxygen to carbon than ethanol, decreasing the proportion of oxygen-containing functional groups. Moreover, 100NBA-CDs have a longer carbon dot polymerization time, which is consistent with the phenomenon in Fig. S13;† thus, more NaOH is consumed during the side reactions, leading to the reduction of sodium content incorporated with oxygen-containing functional groups.
O (1627 cm−1), O–CO (1700 cm−1), –CH3 (1425 cm−1), and C–O (1079 cm−1) stretching vibrations, respectively.17,26–28 The element content and surface functional groups of CDs can be further proved by X-ray photoelectron spectroscopy (XPS, Fig. 2(c) and (d)). CDs mainly contain C, O, and Na elements, and their corresponding binding energies are around 285.3, 532.5, and 1072.3 eV, respectively. Like the results of previous measurements, ten kinds of CDs show similar results for surface groups. High-resolution C 1s spectra (Fig. S16(a1)†) of CDs show a C 1s band containing four main peaks near 284.8, 285.4, 286.8, and 288.4 eV, attributed to C–C/CC, C–O, CO, and O–CO groups, respectively.21,28–30 Fig. S16(a2)† shows four fitting peaks for the high-resolution O 1s spectrum: CO (531.4 eV), O–H (532.4 eV), C–O (533.4 eV), and the Na KLL Auger peak appears at approximately 536.4 eV.31–33 However, no obvious Na KLL Auger peak for other kinds of carbon dots was observed, owing to the very low Na element content in CDs and the different binding forms of sodium to functional groups on the surface of CDs. The Na 1s spectra (Fig. S16(a3)†) exhibit one peak near 1071.7 eV for the C–O–Na bond.34 Both XPS and FTIR results indicate that the surface of carbon dots is rich in oxygen-containing functional groups. XPS spectrum analysis of the products revealed that with the increasing water content in the electrolyte, the Na content of five kinds of EA-CDs decreased to some extent (Fig. S16 and S17†). The reduced Na content can be associated with the diluted alcohol solution of sodium hydroxide after introducing water. Furthermore, the aqueous solution of sodium hydroxide is electrolyzed during the electrolysis of the alcohol solution of sodium hydroxide to produce hydrogen and other by-products, owing to the fewer hydroxyl radicals involved in the formation process of CDs. At the same time, after introducing water, the binding form of sodium element and organic functional group on the surface of CDs changed, while the ionic form of sodium can be easily removed in the dialysis purification process, thus an increase of water content in electrolyte leading to a decrease of sodium content in CDs. By comparison, n-butanol contains a lower ratio of elemental oxygen to carbon than ethanol, decreasing the proportion of oxygen-containing functional groups. Moreover, 100NBA-CDs have a longer carbon dot polymerization time, which is consistent with the phenomenon in Fig. S13;† thus, more NaOH is consumed during the side reactions, leading to the reduction of sodium content incorporated with oxygen-containing functional groups.
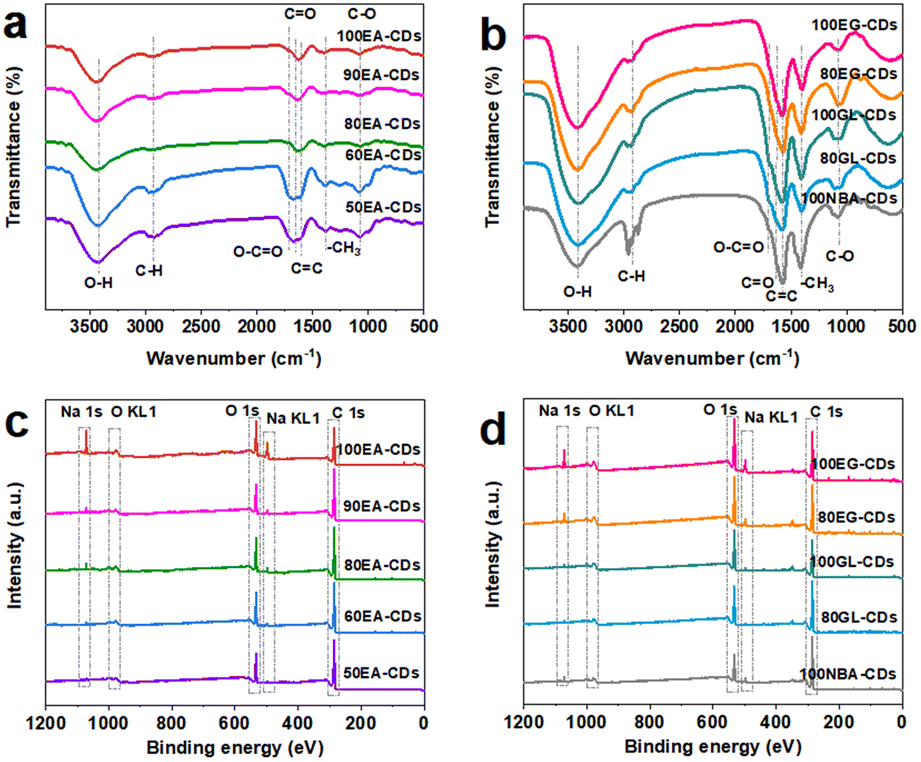 | ||
| Fig. 2 (a and b) FTIR spectrum of prepared CDs. (c and d) XPS survey spectrum of prepared CDs. | ||
Similarly, under 100% alcohol conditions, the Na content of 100EG-CDs is higher than that of 100GL-CDs, owing to the lower number of hydroxyl groups and carbon chains in ethylene glycol, which takes less time to form CDs in the late stage of electrolysis than in the case of GL; thus, the amount of sodium hydroxide involved in the side reaction is lower. In the case of the same type of alcohol, such as EG and GL, an increase in the water content in the electrolyte leads to a decrease in the Na content.
Gas chromatography-mass spectrometry (GC-MS) was performed to detect the intermediate products of the reaction. For the purpose of reducing the rate of electrolysis and facilitating the desalination process later, we reduced the amount of sodium hydroxide to 1 g and selected the electrolyte when the color of the solution turned from yellow to brown. We chose the C18 HyperSep column to remove the sodium hydroxide until the solution reaches neutrality (Fig. 3(a)). To eliminate the influence of the activator, the extraction column was activated using different activators (ethyl acetate, acetonitrile and tetrahydrofuran). This step was taken to ensure that any observed effects could be attributed entirely to the electrochemical process rather than the specific activator used. Acetic acid, ethanol, ethyl acetate, cyclohexane, and acetal were generated in the process of reaction (Fig. S19†). The formation of ethyl acetate is a typical Tishchenko reaction.35 CH3CH2OH first turns into CH3CH2O−, which can be converted to a hemiacetal structure intermediate with the participation of CH3CHO; the final ion migration occurs to obtain the product of ethyl acetate. Acetal is the product of the reaction of ethanol and acetaldehyde.36 The composition of cyclohexane is more complex, including the hydrolysis of esters, oxidation of alcohols, hydroxylation of aldehydes, hydrogenation of aldehydes, dehydration, etc. It can be seen that acetaldehyde, which is invisible in multiple tests, plays an essential role during the reaction and exhibits instability when reacting with other intermediate products.
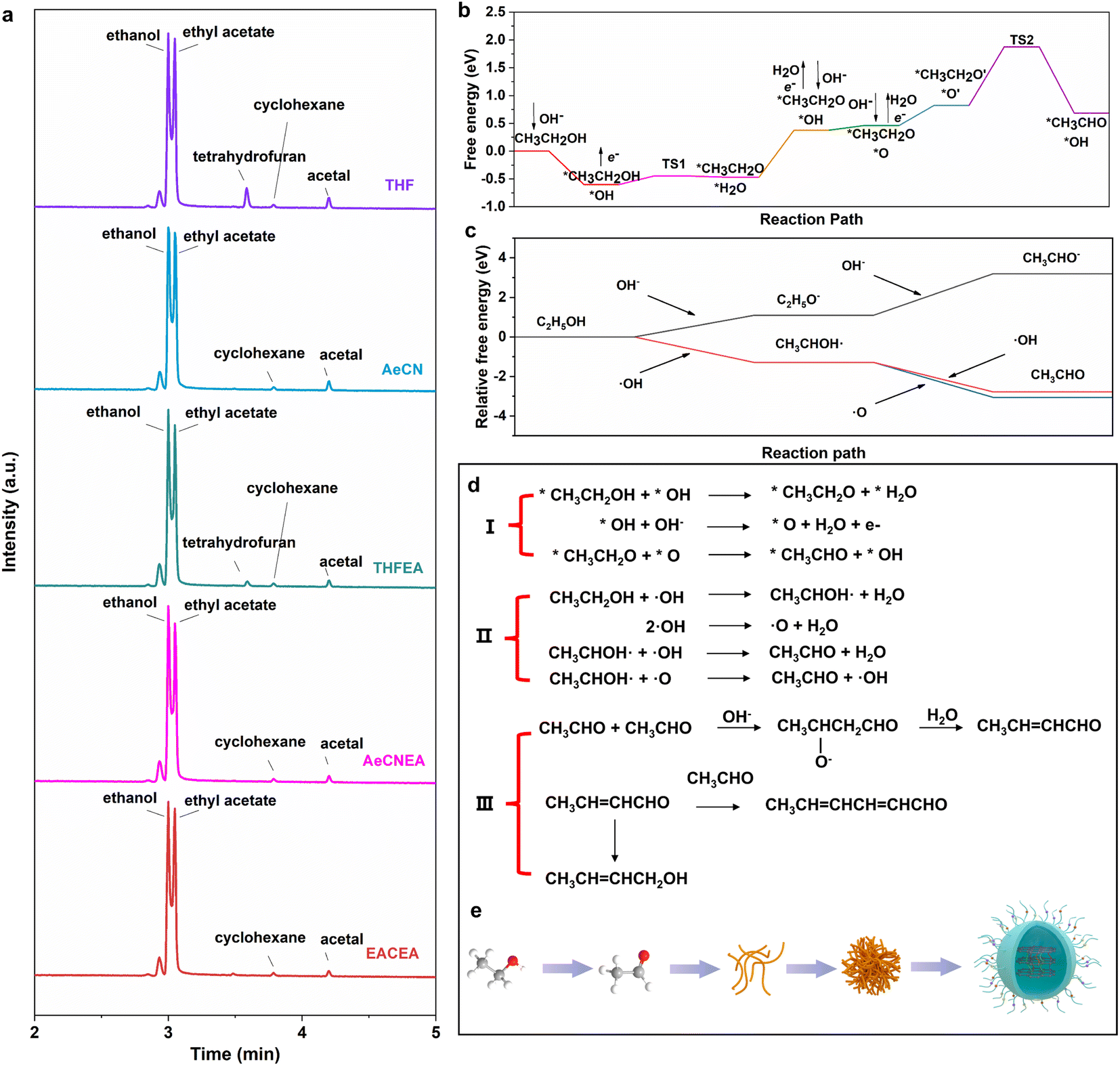 | ||
| Fig. 3 (a) GC-MS spectrum of 100EA-CDs. (b) DFT calculated reaction procedure on the electrode surface. (c) DFT calculated reaction procedure of ethanol solution of NaOH. (d) The chemical reactions that may occur during the formation of carbon dots from ethanol. (e) Scheme of the synthesis of carbon dots. | ||
Density functional theory (DFT) calculations are further applied to understand the reaction procedure. We explored the possible mechanism for the alcohol oxidation reactions under alkaline conditions based on the previous phenomenon (Fig. 3(d)). As schematically illustrated in Fig. 3(d), the involved adsorption energies (Ea) of all intermediates on the electrode surface were meticulously calculated. CH3CH2OH and OH− are the initial reactants; they are adsorbed on the electrode surface to become the adsorbed states *CH3CH2OH and adsorbed radicals *OH at the beginning of the reaction (* + CH3CH2OH → *CH3CH2OH, * + OH− → *OH + e−). * represents the adsorbed substrate. Then *CH3CH2OH is oxidized to remove the hydroxyl H to form *CH3CH2O, while the adsorbed state *H2O is generated (R1: *CH3CH2OH + *OH → *CH3CH2O + *H2O). The configurations of the initial state (IS1), transition state (TS1) and final state (FS1) of these steps are illustrated in Fig. S20.† The reaction energy based on this step is 0.154 eV. The following step is the desorption of water and the adsorption of ·OH (*H2O → H2O + *, * + OH− → *OH + e−). In addition, we calculated the ease of production of oxidation-like substances (·OH, ·O, and ·OOH) in the electrode surface by the OER four-electron transfer process. It proves that H2O is easily cleaved to *O, indicating that it is *O that participates in the oxidation reaction (Fig. S21†). The production of OH− is advantageous to ensure the pH of the system and subsequently promote the aldol condensation of acetaldehyde. After the dehydrogenation of *OH to produce *O and water (*OH + OH− → *O + H2O + e−), *CH3CH2O and *O underwent a co-adsorption conformational transition to get the most stable configuration for the reaction (*CH3CH2O → *CH3CH2O′, *O→ *O′). Then, *CH3CH2O removes the first H from the carbon to produce *CH3CHO, and the reaction energy barrier of this step is 1.051 eV (R2: *CH3CH2O′ + *O′ → *CH3CHO + *OH). It can be inferred that this reaction of producing CH3CHO cannot take place automatically without additional energy. Based on this, we introduce an electric energy to overcome the reaction energy barrier and enable the reaction to proceed. During the electrochemical process, *OH accumulates on the electrode surface and can be desorbed to the solution as ·OH. Then, we calculated the reaction procedure in an ethanol solution of NaOH. As shown in Fig. 3(c), CH3CH2OH can easily react with ·OH, and CH3CHO can be generated quickly with the assistance of ·OH and ·O (Fig. S22†). Besides, it can be seen that it's such a difficult process if OH− acts as an ion to dominate the reaction. CH3CH2OH transfers H to OH− through the hydrogen bonding network, but the resulting CH3CH2O− is unstable, which will revert back to CH3CH2OH quickly (Fig. S23†). The energy barriers are too high to carry out the dehydrogenation reaction of CH3CH2O−. Therefore, the reaction in solution proceeds via free radicals. After that, the intermediate acetaldehyde is easily transformed into carbon dots by the aldol condensation reaction.17 The α-H addition of an aldehyde to another carbonyl carbon produces β-hydroxy aldehyde, which is easily dehydrated to produce α,β-unsaturated aldehyde. The unsaturated aldehyde can then undergo a series of substitution and condensation reactions with acetaldehyde or intermediates to produce plenty of carbon chains and tiny clusters.
Additionally, a series of side reactions occurred, leading to the crosslink and dehydration of carbon chains, resulting in the formation of a carbon core. Meanwhile, short chains containing abundant functional groups cross-linked and anchored on the surface of the carbon nucleus to form carbon dots.
The reaction mechanism verifies the previous observation that an increase in water content gradually accelerates the reaction when ethanol is used as the electrolyte solvent. Since the ionization of sodium hydroxide in water is relatively easy, along with the generation of ·OH from the electrolysis of water, it indicates that the addition of water can enhance the concentration of reactants and, in turn, accelerate the generation of the transition state reactive oxygen radical to catalyze the following reaction. Additionally, oxygen is produced by the electrolysis of water, promoting the oxidation of alcohols. In the absence of water addition, the reaction rate of EG and GL is accelerated due to the fact that the increasing number of hydroxyl groups boosts the chance of exposure to *OH; consequently, the reaction process is facilitated, leading to an overall acceleration of the reaction. Under the condition of water addition, the initial reaction is indeed accelerated; however, the production of CDs decreased due to the reduced amount of alcohol.
Furthermore, in the absence of electricity, the process of ethanol solution of NaOH is the same as in Fig. 3(c). However, in the absence of electricity, ·OH can only be produced by dissolved oxygen, which is extremely difficult. Hence, it takes longer for the color change compared to the case of using electricity (Fig. S1 and S4†). Once ·OH is formed, the following process is spontaneous with the involvement of radicals (Fig. S24†), corresponding to the color change phenomenon in Fig. S1.†
DFT calculation was performed to dissect the interaction between Na ions and CDs (Fig. S25†). For convenience, one sodium ion is positioned on graphite containing three different functional groups (carbonyl, hydroxyl, and carboxyl), respectively. We first examine that Na+ tends to be absorbed on carbon dots owing to the strong coordination ability. Besides, carbonyl, hydroxyl, and carboxyl exhibit higher binding energy with Na+ of −0.13 Ha, −0.13 Ha, and −0.29 Ha, respectively, indicating CDs assembled with abundant oxygen-containing groups have the capability of absorbing Na+ ions. Combined with the situation that introducing CDs into the electrolyte can boost the dissociation of NaPF6 and increase the Na+ ion transference number, it provides faster mass transfer kinetics and induces a uniform deposition of sodium ions. Additionally, Na+ ions can be dissociated from Na-CDs under low potential conditions and deposited on the Na metal anode; to some extent, it replenishes the sodium source. A series of battery tests and characterization studies are presented below to further explore and demonstrate the role of CDs in the electrolyte.
According to the above-mentioned XPS results, 100EA-CDs, containing the most sodium content, were employed as electrolyte additives (represented by Na-CDs). Various concentrations of Na-CDs (from 0.1 mg mL−1 to 1.0 mg mL−1) were dissolved in the blank electrolyte (1.0 M NaPF6 in diglyme) to obtain a series of electrolyte solutions. Fig. S26† displays good dispersibility and stability of Na-CDs under sunlight and ultraviolet lamps. Linear sweep voltammetry (LSV) was performed on Na‖stainless steel cells to approximately evaluate the electrochemical stability windows of electrolytes with various concentrations of Na-CDs. In comparison, the optimal Na-CD concentration displaying higher oxidation potential is 0.2 mg mL−1 and 0.5 mg mL−1 (0.2Na-CDs and 0.5Na-CDs). The transference number of sodium ions (tNa+) was measured through impedance measurements in symmetric Na‖Na cells by a combined DC polarization and AC impedance method. The tNa+ of the electrolyte with Na-CDs is 0.80 and 0.83 for 0.2 mg mL−1 and 0.5 mg mL−1, respectively, which are higher than 0.77 for blank electrolyte (Fig. S27†). The increased migration number indicates a higher concentration of freely moving sodium ions in solution and a faster mass transfer kinetics of the batteries. In this case, the steady state of ion concentrations at the sodium metal electrode during the charging–discharging process achieves a homogeneous deposition of sodium ions, verifying the above-mentioned stronger binding capacity between Na-CDs containing polar-rich functional groups and sodium ions, facilitating the dissociation of NaPF6.
Meanwhile, the solution structure in electrolyte with dissociated anion and more free solvent molecules is performed by Raman spectroscopy (Fig. S28†). As previously reported, the obtained spectrum of pure diglyme exhibits three overlapping peaks at 805, 826, and 850 cm−1.37 The ion-dipole attraction between the dissociated Na+ and the carbonyl oxygen of the solvent molecule leads to two additional peaks of solvent molecules at 842 and 865 cm−1. Comparing Fig. S29(b) and (c),† it is observed that the vibrating bands exhibit a blue shift. The bonding formation between the anion and Na+ through fluorine atoms is displayed by the peak at 740 cm−1, corresponding to the P–F stretching of PF6−, while pure diglyme indicates no cation–anion aggregates. According to the previous studies,37 there are two feasible 6-fold coordination in the diglyme solution between the sodium ion and the solvent molecules and PF6− ions: the solvent-separated ion pair (SSIP) and contact ion pair (CIP). After adding 0.2Na-CDs and 0.5Na-CDs, the total integral intensity of the coordination peaks increases, indicating a reducing number of free solvent molecules, which eventually enhances the oxidation potential of the electrolyte and results in the PF6− anion preferential reduction at the anode, contributing to the formation of F-containing SEI.38
Then, the electrochemical performance of Na plating/stripping with and without Na-CDs was studied in a Na‖Cu half-cell at a current density of 0.5 and 1.0 mA cm2 with a capacity of 1.0 mA h cm2 (Fig. S29(c) and (e)†). Under 0.5 mA cm2 to 1.0 mA h cm2 conditions, 130 cycles of Na plating/stripping with an average coulombic efficiency (CE) of 99.8% on a Cu electrode were achieved, while the cell without additives failed after 80 cycles. The addition of carbon dots increases the utilization of Na plating/stripping. Also, Fig. S29† f exhibits the unstable cycling condition and high nucleation overpotential at 1.0 mA cm2 to 1.0 mA h cm2 in the blank electrolyte. However, 0.5Na-CDs may aggregate at low current in the first few cycles, leading to different cycling performances at 0.5 and 1.0 mA cm2. Large polarization (40 mV) was observed in blank electrolytes (Fig. S29(d)†), while the electrolyte with the Na-CD additive showed a low value of 19 mV. Indeed, lower nucleation overpotentials indicate a lower energy barrier for the nucleation and growth to overcome for the first nucleation of Na+ ions, which implies that it becomes easier for Na+ ions to nucleate on the copper foil and subsequently easier to obtain a dense deposit layer. The scanning electron microscopy (SEM) image of Cu foil at 5.0 mA cm−2 to 1.0 mA h cm−2 was recorded to further understand the surface state after cycling. Fig. 4(e)–(g) show a smooth surface of the Cu side after stripping in Na-CD electrolyte, while the one in blank electrolyte had a mass of loose needle-like irregular dendrites with apparent holes. The irregular dendrite morphology can lead to unstable cycling performance and cause the accumulation of dead Na, which accounts for the increased CE instability of the Na‖Cu half-cell without adding CDs. Furthermore, symmetrical Na‖Na cells were assembled to investigate the role of the Na-CD additive in suppressing dendrite proliferation. For the case with the 0.2Na-CD additive, the symmetrical Na‖Na cells displayed a stable cycling response over 1200 h at a current density of 1.0 mA cm2 with a capacity of 1.0 mA h cm2 (Fig. 4(a) and (b)), suggesting the more stable plating/stripping behavior of Na. The deposition and dissolution overpotential were maintained at around 25 mV. In sharp contrast, the cell with blank electrolyte disordered after 300 h with a large overpotential. Furthermore, rate performance tests were conducted on symmetrical Na‖Na cells at increased current density. Notably, the symmetrical Na‖Na in 0.2Na-CD electrolyte always exhibits the lowest voltage at subsequent increased current density (Fig. 4(c) and (d)).
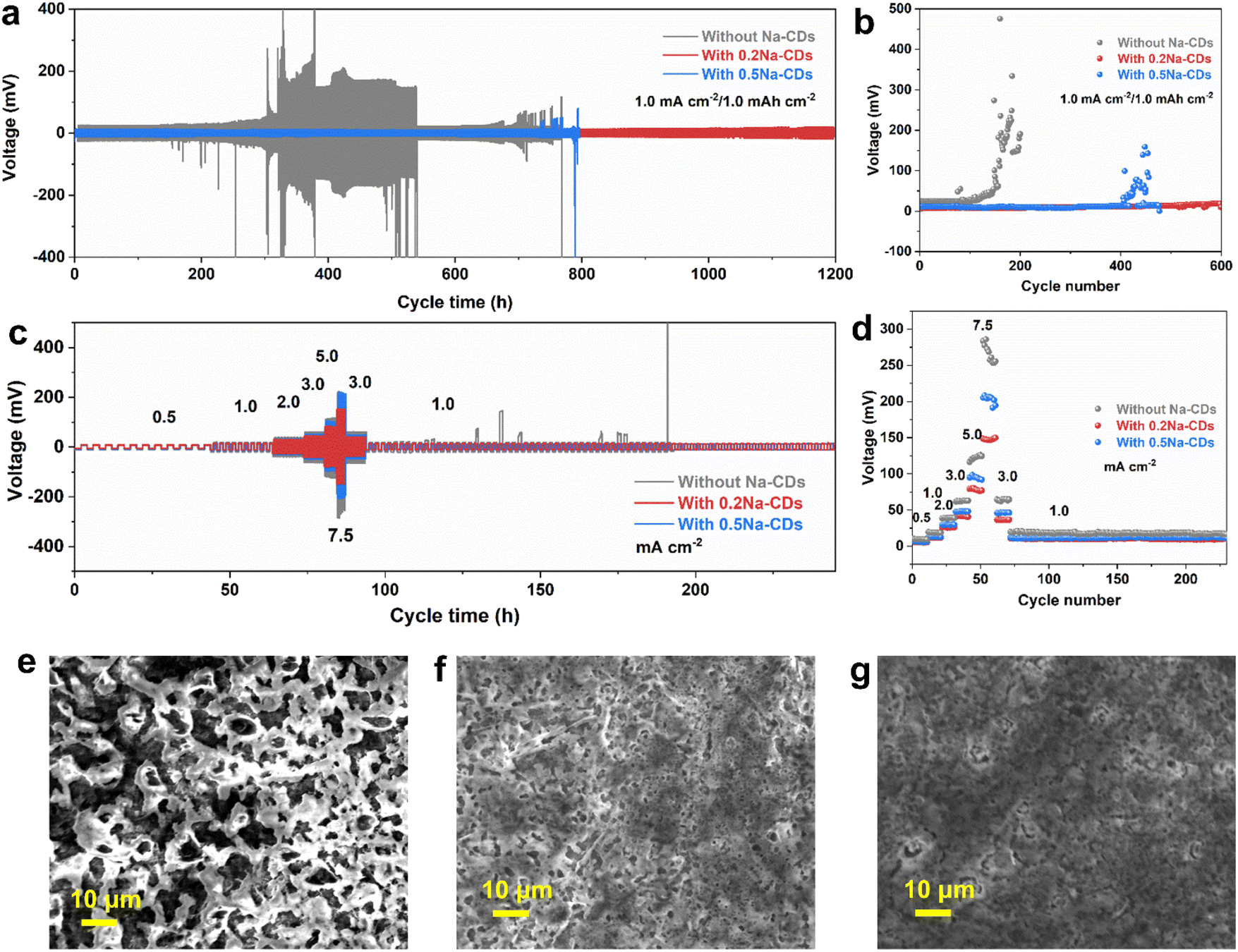 | ||
| Fig. 4 (a and b) Charge–discharge voltage profiles of Na‖Na symmetric cells with and without the Na-CD additive at 1.0 mA cm−2 to 1.0 mA h cm−2. (c and d) Rate performance of Na‖Na symmetric cells with and without the Na-CD additive. (e–g) SEM images of the sodium ion deposition on the copper foil in the blank electrolyte, in electrolyte with 0.2Na-CDs, and 0.5Na-CDs at 5.0 mA cm−2 to 1.0 mA h cm−2. | ||
Electrochemical impedance spectroscopy (EIS) of the symmetric Na‖Na cells after 10 and 30 cycles at 1.0 mA cm2 to 1.0 mA h cm2 was performed to reveal the resistance of the surface film resistance (Rs) and charge transfer resistance (Rct). Rs and Rct both decreased, exhibiting lower mass transfer impedance and easier diffusion behavior, which demonstrates that the interfacial layer effectively suppresses side reactions at the electrode–electrolyte interface, resulting in a highly reversible sodium metal deposition and stripping process after adding the 0.2Na-CD additive (Fig. S30†). Additionally, the surface morphology of cycled Na metal was investigated with and without the 0.2Na-CD additive after 10 and 30 cycles under 1.0 mA cm2 to 1.0 mA h cm2. As displayed in Fig. 5(a3), (a4), (b3) and (b4), a loose and porous structure with cracks and a mass of needle-like dendrites is exhibited on the surface of Na metal with different cycles in the blank electrolyte. In contrast, the electrode in the 0.2Na-CD electrolyte shows a smooth and compact surface with no obvious dendrite growth.39 The SEI components formed on the cycled sodium metal anode were analyzed in detail using depth XPS. The normalized XPS fitting results of high-resolution C 1s and F 1s spectra at different etching depths after 10 cycles are shown in Fig. 5(a1), (a2), (b1) and (b2), respectively. The C 1s spectrum (no etching) can be fitted using 4 peaks with binding energies of 284.8 eV (C–C/C–H), 286.3 eV (C–O), 288.4 eV (O–CO), and 289.7 eV (CO32−), while 3 peaks at 284.8 eV (C–C/C–H), 286.7 eV (C–O), and 289.1 eV (CO32−) were exhibited after 1 min and 3 min of sputtering to remove the top surface in the blank electrolyte.22,40,41 For the case with the 0.2Na-CD additive, the three peaks at 284.8 eV, 286.5 eV, and 289.0 eV could be assigned to C–C/C–H, C–O, and CO32− after 0 min, 1 min, and 3 min of sputtering, respectively. As for O 1s, the peaks at 530.1 eV, 531.3 eV, and 535.7 eV correspond to Na–O, C–O, and Na KLL, respectively.40,42,43 The C–H/C–C, C–O, O–CO peaks could be from RCH2ONa, a major organic reduction product of diglyme. In the F 1s and P 2p spectra, the inorganic NaxPFy and NaxPFyOz are clearly observed, which hold a higher relative ratio, indicating the severe side reaction of NaPF6.22,44 In addition, the inorganic component of NaF (683.5 eV) is found, and its ratio is significantly higher than that in the blank electrolyte (Fig. S31†).44 Fewer organics and more inorganics (Na–O, CO32−, NaF) in the inner-SEI can contribute to stable mechanical strength and high ionic conductivity, which is consistent with the lower interfacial resistance. Additionally, the NaF-rich SEI can be observed on the cycled anode in 0.2Na-CD electrolyte after 30 cycles (Fig. 5(c1), (c2), (d1), (d2) and S32†), providing more convincing evidence for the superior mechanical and chemical stability of the SEI, which inhibits the side reactions between the sodium metal and the electrolyte and effectively mitigates the volume changes caused by Na metal deposition and stripping, finally realizing the improvement of electrochemical performance. Na-CDs with small size and large specific surface area can easily absorb Na+ ions. Na-CDs with absorbed Na+ ions are transported to the surface of the anode under ion diffusion and electric field force. The Na+ ions dissociated from Na-CDs under low potential conditions might contribute to the increased Na content in the SEI and, to some extent, replenish the sodium source. After that, Na-CDs and Na+ ions can strip off to the electrolyte. The electrochemical performance of Na-CDs as electrolyte additives is further assessed in a full cell with Na3V2(PO4)3 (NVP) as a cathode (∼2.5 mg cm−2). Na‖NVP batteries were tested with a charging voltage of 3.8 V. The full cells were operated at a high rate of 5C, which is equal to a current density of 0.59 A g−1. By comparison, the full cell with the Na-CD additive sustains a longer cycle lifespan, delivering a stable discharge capacity for over 1000 cycles with a capacity retention of 95% (Fig. S33a†). After the addition of the Na-CD additive, the full cell also shows excellent rate performance, delivering a discharge capacity of 101 mA h g−1 at 1C and a discharge capacity of 25 mA h g−1 at a high current density of 20C, superior to the one adopting blank electrolyte (Fig. S33c†). Cyclic voltammetry (CV) at various scanning rates from 0.1 to 1.0 mV s−1 in the potential range of 2.5–3.8 V (vs. Na+/Na) further verifies the availability of the Na-CD additive (Fig. S34†). The electrolyte with the Na-CD additive exhibits better compatibility with the NVP cathode and lower polarization voltage, manifesting fast kinetics for sodium intercalation and de-intercalation.
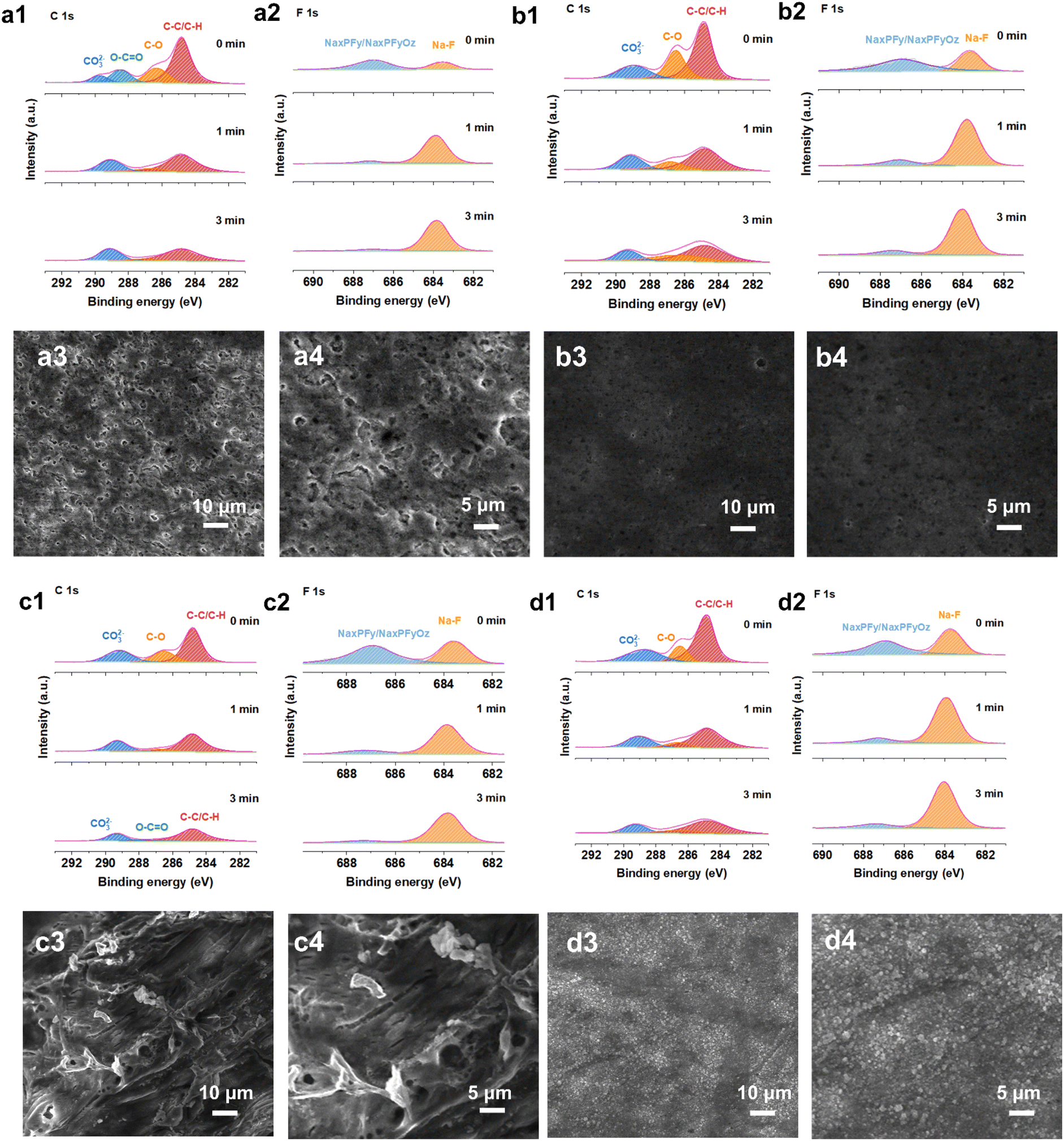 | ||
| Fig. 5 XPS depth profiles of C 1s and F 1s for the Na anode after 10 cycles in (a1 and a2) blank and (b1 and b2) Na-CD electrolytes. SEM images of the sodium anode surface in (a3 and a4) blank and (b3 and b4) Na-CD electrolytes after 10 cycles. XPS depth profiles of C 1s and F 1s for the Na anode after 30 cycles in (c1 and c2) blank and (d1 and d2) Na-CD electrolytes. SEM images of the sodium anode surface in (c3 and c4) blank and (d3 and d4) Na-CD electrolytes after 30 cycles. | ||
Conclusions
In summary, inspired by the color change of sodium hydroxide ethanol solution after standing for some time, we found that the alcohol can be transformed into carbon dots, and the reaction mechanism from alcohols to carbon dots was investigated in-depth. Under alkaline conditions, alcohol was oxidized to aldehyde, which was the key molecule for the formation of CDs, and this process was proved to be the rate-controlling step. It was confirmed that there is a self-catalysis reaction during the reaction process of alcohol to aldehyde, and it is also found that the reaction from alcohol to aldehyde can be largely accelerated with the assistance of electricity. Under strongly alkaline conditions, the newly formed aldehyde molecules can soon be susceptible to producing carbon chain materials and clusters through the aldol condensation reaction. Meanwhile, different carbon chains crosslinked and dehydrated to form the carbon core, which is covered by short chains rich in functional groups on the surface, eventually forming carbon dots. As the water content of the electrolyte increases, the production of *O accelerates, which catalyzes subsequent reactions. In the case of choosing other alcohols as electrolytes, the oxidation reaction of alcohols was accelerated at the beginning with the increase of hydroxyl groups and the increase of reaction active sites. The obtained Na-doped carbon dots contained abundant functional groups, displaying good compatibility with the electrolyte of SMBs. It was proved through calculations that carbon dots having rich polar functional groups exhibit an adsorption effect on sodium ions; hence, higher sodium migration numbers and faster transport kinetics could be achieved due to the free Na+ dissociated by NaPF6. Besides, more PF6− engaged in pairing with free solvent molecules to exhibit lower reducibility. Moreover, a lower nucleation overpotential in Na‖Cu half cells and long-term Na plating/stripping over 1200 h with 25 mV voltage in Na‖Na symmetrical cells were achieved by the Na-CD additive, which demonstrates that Na+ ions are more likely to nucleate on the anode and exhibit more stable plating/stripping behavior, eventually reaching a dense deposit layer and excellent stable cycling performance. More prominently, due to their facile synthesis using low-cost and non-toxic electrolytes, electrochemical methods containing high-value by-products such as hydrogen hold great promise in providing CDs with various physical and chemical properties, which opens up vast opportunities for their application in other emerging fields.Data availability
All the data supporting this article have been uploaded as part of the ESI.†Author contributions
The concept of the work was designed by H. T., H. H., and X. J.; H. T., L. X., H. L., Z. L, and G. Z. obtained research data; H. T., L. X., Z. L., L. L., and Y. T. analysed data; H. T., H. L., Z. L., and W. D. performed the experiments with assistance from L. X., and Y. T.; G. Z. performed the computational strategy. H. T., H. L., and L. X. wrote the manuscript under the supervision of H. H., and X. J. All authors contributed to the revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52074359, 22379165, 52261135632, and U21A20284) and the Science and Technology Innovation Program of Hunan Province (2021RC3014).Notes and references
- Q. Zhang, R. Wang, B. Feng, X. Zhong and K. Ostrikov, Photoluminescence mechanism of carbon dots: triggering high-color-purity red fluorescence emission through edge amino protonation, Nat. Commun., 2021, 12, 6856 CrossRef CAS PubMed.
- B. Zhao and Z. a. Tan, Fluorescent Carbon Dots: Fantastic Electroluminescent Materials for Light-Emitting Diodes, Advanced Science, 2021, 8, 2001977 CrossRef CAS PubMed.
- S. Tao, C. Zhou, C. Kang, S. Zhu, T. Feng, S.-T. Zhang, Z. Ding, C. Zheng, C. Xia and B. Yang, Confined-domain crosslink-enhanced emission effect in carbonized polymer dots, Light: Sci. Appl., 2022, 11, 56 CrossRef CAS PubMed.
- D. Li, C. Liang, E. V. Ushakova, M. Sun, X. Huang, X. Zhang, P. Jing, S. J. Yoo, J.-G. Kim, E. Liu, W. Zhang, L. Jing, G. Xing, W. Zheng, Z. Tang, S. Qu and A. L. Rogach, Thermally Activated Upconversion Near-Infrared Photoluminescence from Carbon Dots Synthesized via Microwave Assisted Exfoliation, Small, 2019, 15, 1905050 CrossRef CAS PubMed.
- Y. Liu, N. Chen, W. Li, M. Sun, T. Wu, B. Huang, X. Yong, Q. Zhang, L. Gu, H. Song, R. Bauer, J. S. Tse, S.-Q. Zang, B. Yang and S. Lu, Engineering the synergistic effect of carbon dots-stabilized atomic and subnanometric ruthenium as highly efficient electrocatalysts for robust hydrogen evolution, SmartMat, 2022, 3, 249–259 CrossRef CAS.
- L. Ai, Y. Yang, B. Wang, J. Chang, Z. Tang, B. Yang and S. Lu, Insights into photoluminescence mechanisms of carbon dots: advances and perspectives, Sci. Bull., 2021, 66, 839–856 CrossRef CAS PubMed.
- S. Li, L. Li, H. Tu, H. Zhang, D. S. Silvester, C. E. Banks, G. Zou, H. Hou and X. Ji, The development of carbon dots: from the perspective of materials chemistry, Mater. Today, 2021, 51, 188–207 CrossRef CAS.
- L. Bao, Z.-L. Zhang, Z.-Q. Tian, L. Zhang, C. Liu, Y. Lin, B. Qi and D.-W. Pang, Electrochemical Tuning of Luminescent Carbon Nanodots: From Preparation to Luminescence Mechanism, Adv. Mater., 2011, 23, 5801–5806 CrossRef CAS PubMed.
- H. Ming, Z. Ma, Y. Liu, K. Pan, H. Yu, F. Wang and Z. Kang, Large scale electrochemical synthesis of high quality carbon nanodots and their photocatalytic property, Dalton Trans., 2012, 41, 9526–9531 RSC.
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S.-Y. Xie, Quantum-sized carbon dots for bright and colorful photoluminescence, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed.
- X. Y. Xu, R. Ray, Y. L. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, Electrophoretic analysis and purification of fluorescent single-walled carbon nanotube fragments, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef CAS PubMed.
- A. Khayal, V. Dawane, M. A. Amin, V. Tirth, V. K. Yadav, A. Algahtani, S. H. Khan, S. Islam, K. K. Yadav and B.-H. Jeon, Advances in the Methods for the Synthesis of Carbon Dots and Their Emerging Applications, Polymers, 2021, 13, 3190 CrossRef CAS PubMed.
- S. Lu, L. Sui, J. Liu, S. Zhu, A. Chen, M. Jin and B. Yang, Near-Infrared Photoluminescent Polymer-Carbon Nanodots with Two-Photon Fluorescence, Adv. Mater., 2017, 29, 1603443 CrossRef PubMed.
- X. Yang, X. Yang, Z. Li, S. Li, Y. Han, Y. Chen, X. Bu, C. Su, H. Xu, Y. Jiang and Q. Lin, Photoluminescent carbon dots synthesized by microwave treatment for selective image of cancer cells, J. Colloid Interface Sci., 2015, 456, 1–6 CrossRef CAS PubMed.
- H. Zhu, X. Wang, Y. Li, Z. Wang, F. Yang and X. Yang, Microwave synthesis of fluorescent carbon nanoparticles with electrochemiluminescence properties, Chem. Commun., 2009, 5118–5120, 10.1039/b907612c.
- C. Kang, Y. Huang, H. Yang, X. F. Yan and Z. P. Chen, A Review of Carbon Dots Produced from Biomass Wastes, Nanomaterials, 2020, 10, 2316 CrossRef CAS PubMed.
- L. Li, Y. Li, Y. Ye, R. Guo, A. Wang, G. Zou, H. Hou and X. Ji, Kilogram-Scale Synthesis and Functionalization of Carbon Dots for Superior Electrochemical Potassium Storage, ACS Nano, 2021, 15, 6872–6885 CrossRef CAS PubMed.
- B. Bartolomei, J. Dosso and M. Prato, New trends in nonconventional carbon dot synthesis, Trends Chem., 2021, 3, 943–953 CrossRef CAS.
- J. Zhu, M. Zhu, R. Zhang, Z. He, L. Xiong and L. Guo, Corrosion inhibition behavior of electrochemically synthesized carbon dots on Q235 carbon steel, J. Adhes. Sci. Technol., 2023, 37, 1997–2009 CrossRef CAS.
- K. Ding, Y. Ye, J. Hu, L. Zhao, W. Jin, J. Luo, S. Cai, B. Weng, G. Zou, H. Hou and X. Ji, Aerophilic Triphase Interface Tuned by Carbon Dots Driving Durable and Flexible Rechargeable Zn-Air Batteries, Nano-Micro Lett., 2023, 15, 28 CrossRef CAS PubMed.
- S. Li, Z. Luo, H. Tu, H. Zhang, W. Deng, G. Zou, H. Hou and X. Ji, N,S-codoped carbon dots as deposition regulating electrolyte additive for stable lithium metal anode, Energy Storage Mater., 2021, 42, 679–686 CrossRef.
- Z. Luo, S. Tao, Y. Tian, L. Xu, Y. Wang, X. Cao, Y. Wang, W. Deng, G. Zou, H. Liu, H. Hou and X. Ji, Robust artificial interlayer for columnar sodium metal anode, Nano Energy, 2022, 97, 107203 CrossRef CAS.
- X. Wang, B. Wang, H. Wang, T. Zhang, H. Qi, Z. Wu, Y. Ma, H. Huang, M. Shao, Y. Liu, Y. Li and Z. Kang, Carbon-Dot-Based White-Light-Emitting Diodes with Adjustable Correlated Color Temperature Guided by Machine Learning, Angew. Chem., Int. Ed., 2021, 60, 12585–12590 CrossRef CAS PubMed.
- S. Tao, S. Zhu, T. Feng, C. Xia, Y. Song and B. Yang, The polymeric characteristics and photoluminescence mechanism in polymer carbon dots: a review, Mater. Today Chem., 2017, 6, 13–25 CrossRef.
- Y. Xu, C. Wang, T. Wu, G. Ran and Q. Song, Template-Free Synthesis of Porous Fluorescent Carbon Nanomaterials with Gluten for Intracellular Imaging and Drug Delivery, ACS Appl. Mater. Interfaces, 2022, 14, 21310–21318 CrossRef CAS PubMed.
- Y. Xiang, L. Xu, L. Yang, Y. Ye, Z. Ge, J. Wu, W. Deng, G. Zou, H. Hou and X. Ji, Natural Stibnite for Lithium-/Sodium-Ion Batteries: Carbon Dots Evoked High Initial Coulombic Efficiency, Nano-Micro Lett., 2022, 14, 136 CrossRef CAS PubMed.
- H. Zhang, R. Guo, S. Li, C. Liu, H. Li, G. Zou, J. Hu, H. Hou and X. Ji, Graphene quantum dots enable dendrite-free zinc ion battery, Nano Energy, 2022, 92, 106752 CrossRef CAS.
- Q.-y. Li, X. Yu, X. Li, L.-n. Bao, Y. Zhang, S.-l. Wang, M. Jiang, K. Huang and L. Xu, Congo Red-Derived Carbon Dots: Simultaneously as Fluorescence Probe for Protein Aggregates, Inhibitor for Protein Aggregation, and Scavenger of Free Radicals, Small, 2023, 19, 2205634 CrossRef CAS PubMed.
- J. Li, J. Kong, S. Ma, J. Li, M. Mao, K. Chen, Z. Chen, J. Zhang, Y. Chang, H. Yuan, T. Liu, Z. Zhang and G. Xing, Exosome-Coated 10B Carbon Dots for Precise Boron Neutron Capture Therapy in a Mouse Model of Glioma In Situ, Adv. Funct. Mater., 2021, 31, 2100969 CrossRef CAS.
- M. Abdelhameed, M. Elbeh, N. S. Baban, L. Pereira, J. Matula, Y.-A. Song and K. B. Ramadi, High-yield, one-pot upcycling of polyethylene and polypropylene waste into blue-emissive carbon dots, Green Chem., 2023, 25, 1925–1937 RSC.
- L. Xu, X. Xiao, H. Tu, F. Zhu, J. Wang, H. Liu, W. Huang, W. Deng, H. Hou, T. Liu, X. Ji, K. Amine and G. Zou, Engineering Functionalized 2D Metal-Organic Frameworks Nanosheets with Fast Li+ Conduction for Advanced Solid Li Batteries, Adv. Mater., 2023, 2303193 CrossRef CAS PubMed.
- X. Yang, L. Ai, J. Yu, G. I. N. Waterhouse, L. Sui, J. Ding, B. Zhang, X. Yong and S. Lu, Photoluminescence mechanisms of red-emissive carbon dots derived from non-conjugated molecules, Sci. Bull., 2022, 67, 1450–1457 CrossRef CAS PubMed.
- L. Wang, M. Peng, J. Chen, T. Hu, K. Yuan and Y. Chen, Eliminating the Micropore Confinement Effect of Carbonaceous Electrodes for Promoting Zn-Ion Storage Capability, Adv. Mater., 2022, 34, 2203744 CrossRef CAS PubMed.
- J.-L. Xia, D. Yan, L.-P. Guo, X.-L. Dong, W.-C. Li and A.-H. Lu, Hard Carbon Nanosheets with Uniform Ultramicropores and Accessible Functional Groups Showing High Realistic Capacity and Superior Rate Performance for Sodium-Ion Storage, Adv. Mater., 2020, 32, 2000447 CrossRef CAS PubMed.
- J. Mlynarski, Direct asymmetric Aldol-Tishchenko reaction, Eur. J. Org Chem., 2006, 2006, 4779–4786 CrossRef.
- C. Liu, Y. Shen, Z. Xiao, H. Yang, X. Han, K. Yuan and Y. Ding, Direct anodic (thio)acetalization of aldehydes with alcohols (thiols) under neutral conditions, and computational insight into the electrochemical formation of the acetals, Green Chem., 2019, 21, 4030–4034 RSC.
- N. R. Dhumal and S. P. Gejji, Theoretical studies on blue versus red shifts in diglyme-M+-X- (M = Li, Na, and K and X = CF3SO3, PF6, and (CF3SO2)(2)N), J. Phys. Chem. A, 2006, 110, 219–227 CrossRef CAS PubMed.
- N. S. Katorova, S. S. Fedotov, D. P. Rupasov, N. D. Luchinin, B. Delattre, Y.-M. Chiang, A. M. Abakumov and K. J. Stevenson, Effect of Concentrated Diglyme-Based Electrolytes on the Electrochemical Performance of Potassium-Ion Batteries, ACS Appl. Energy Mater., 2019, 2, 6051–6059 CrossRef CAS.
- H.-B. Sun, H.-Y. Fu, Y.-Y. Huang, X.-Y. Zheng, Y. Huang, R. Lin and W. Luo, Design of a 3D mixed conducting scaffold toward stable metallic sodium anodes, Rare Met., 2022, 41, 3336–3342 CrossRef CAS.
- D. Wu, C. Zhu, M. Wu, H. Wang, J. Huang, D. Tang and J. Ma, Highly Oxidation-Resistant Electrolyte for 4.7 V Sodium Metal Batteries Enabled by Anion/Cation Solvation Engineering, Angew. Chem., Int. Ed., 2022, 61, e202214198 CrossRef CAS PubMed.
- H. M. Law, J. Yu, S. C. T. Kwok, G. Zhou, M. J. Robson, J. Wu and F. Ciucci, A hybrid dual-salt polymer electrolyte for sodium metal batteries with stable room temperature cycling performance, Energy Storage Mater., 2022, 46, 182–191 CrossRef.
- Y.-Q. Zheng, M.-Y. Sun, F.-D. Yu, L. Deng, Y. Xia, Y.-S. Jiang, L.-F. Que, L. Zhao and Z.-B. Wang, Utilizing weakly-solvated diglyme-based electrolyte to achieve a 10,000-cycles durable Na3V2(PO4)2F3 cathode endured at – 20 °C, Nano Energy, 2022, 102, 107693 CrossRef CAS.
- X. Zheng, Z. Gu, X. Liu, Z. Wang, J. Wen, X. Wu, W. Luo and Y. Huang, Bridging the immiscibility of an all-fluoride fire extinguishant with highly-fluorinated electrolytes toward safe sodium metal batteries, Energy Environ. Sci., 2020, 13, 1788–1798 RSC.
- Z. W. Seh, J. Sun, Y. Sun and Y. Cui, A Highly Reversible Room-Temperature Sodium Metal Anode, ACS Cent. Sci., 2015, 1, 449–455 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04606k |
| This journal is © The Royal Society of Chemistry 2023 |