
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDivergent 1,2-carboallylation of terminal alkynes enabled by metallaphotoredox catalysis with switchable triplet energy transfer†
Jian
Qin
,
Zhuzhu
Zhang
,
Yi
Lu
,
Shengqing
Zhu
 and
Lingling
Chu
*
and
Lingling
Chu
*
State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Chemistry and Chemical Engineering, Center for Advanced Low-Dimension Materials, Donghua University, Shanghai 201620, China. E-mail: lingling.chu1@dhu.edu.cn
First published on 13th October 2023
Abstract
We report a metallaphotoredox strategy for stereodivergent three-component carboallylation of terminal alkynes with allylic carbonates and alkyl trifluoroborates. This redox-neutral dual catalytic protocol utilizes commercially available organic photocatalyst 4CzIPN and nickel catalysts to trigger a radical addition/alkenyl–allyl coupling sequence, enabling straightforward access to functionalized 1,4-dienes in a highly chemo-, regio-selective, and stereodivergent fashion. This reaction features a broad substrate generality and a tunable triplet energy transfer control with pyrene as a simple triplet energy modulator, offering a facile synthesis of complex trans- and cis-selective skipped dienes with the same set of readily available substrates.
Introduction
The 1,4-diene motifs, also known as skipped dienes, are widely present in different classes of natural products and biologically active compounds and serve as valuable synthetic synthons in chemical synthesis due to their versatile reactivity.1 Therefore, the development of efficient methods for constructing these skipped dienes has received increasing attention. Among various synthetic methods developed,2–5 transition metal-catalyzed hydroallylation of alkynes represents one of the most straightforward and reliable approaches to accessing skipped dienes.5 Since the seminal report by Trost in 1998,5a a number of elegant examples of catalytic hydroallylation of alkynes, enabled by stoichiometric reductants or hydrides in the presence of nickel, copper, or cobalt catalysts, have later been developed by several other groups, providing efficient and regioselective methods for the synthesis of skipped alkenes.5 Despite these advancements, selective assembly of highly substituted 1,4-dienes remains challenging.Transition metal-catalyzed 1,2-carboallylation of alkynes, in which an allyl moiety and another carbon-centered functionality are spontaneously incorporated in one pot, represents a practical tactic for synthesizing substituted 1,4-dienes.6,7 Despite attractive, only a few examples of 1,2-carboallylation of alkynes, which generally proceed through a two-electron redox process, i.e., carbo-metalation of alkynes followed by cross-couplings, have been successfully developed via nickel catalysis (Fig. 1a). Typically, highly active organometallic agents such as Me3Al, Me2Zn, or alkynyltins are employed for requisite couplings.6 To our knowledge, catalytic three-component carboallylation of alkynes enabled by transition metal-catalyzed photoinduced electron transfer strategies,8 an emerging area that has attracted increasing attention in chemical synthesis, remains unknown. Recently, elegant progress has been made in geometric isomerization of alkenes facilitated by triplet–triplet energy transfer (EnT) catalysis, providing a facile approach to access thermodynamically less stable yet synthetically challenging Z-alkenes.9–11 More importantly, a metallaphotoredox system combining EnT process with single-electron transfer (SET) events allows for the stereo-selective and even -divergent construction of structurally diverse alkenes from simple starting materials.11 Inspired by these elegant examples, we envisage that metallaphotoredox-enabled carboallylation of alkynes would not only enable a precise assembly of substituted 1,4-dienes but also allow for a stereoselective modulation for the resulting dienes. Herein, we describe a redox-neutral, stereo-divergent 1,2-carboallylation of alkynes with alkyl trifluoroborates and allylic electrophiles via dual photoredox and nickel catalysis (Fig. 1b). This reaction takes advantage of the versatile reactivity of open-shell alkenyl radicals,12in situ generated via radical addition to alkynes, to ensure the facile assembly of substituted skipped dienes with complementary reactivity and selectivity. More importantly, this synergistic protocol allows for a divergent regulation of the stereochemistry of 1,4-diene products with pyrene as a simple EnT modulator, providing a complementary strategy to the previously developed photoinduced divergent synthesis of substituted alkenes that rely on using different photocatalysts or metal complexes.11d–g,11i–k
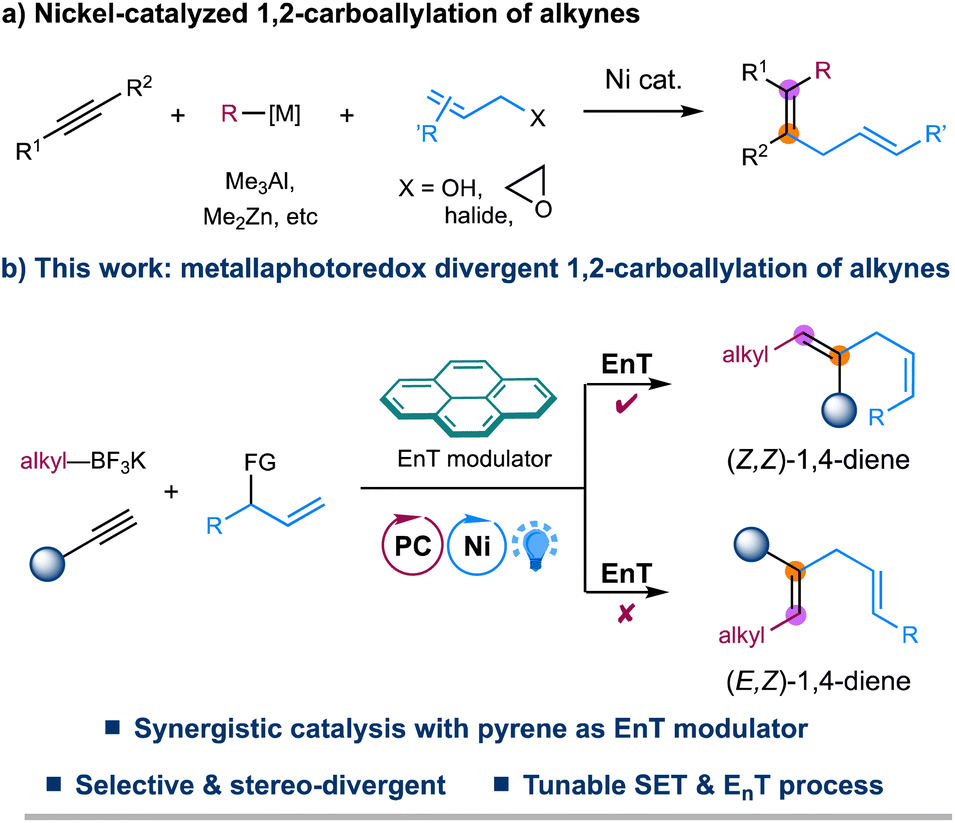 | ||
| Fig. 1 Divergent three-component carboallylation of alkynes via metallaphotoredox catalysis. | ||
Results and discussion
We began our investigations by employing terminal alkyne 1a as the template substrate, allylic carbonate 2a as the electrophilic coupling partner, and tert-butyl trifluoroborate 3a as the alkyl radical precursor. As shown in Table 1, the optimal results were obtained with a catalytic combination of Ni(phen)Cl2 as a pre-catalyst, 4,4′-di-tert-butyl-2,2′-bipyridine (dtbbpy) as a ligand, 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) as a photocatalyst,13 and pyrene as an additive. The reaction is performed in a DMAc/MeCN solvent with an irradiation of blue LED (λmax = 467 nm) at 35 °C, delivering the desired (E,Z)-1,4-diene product 4a in 84% yield and high stereoselectivity (E/Z = 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8) (entry 1). The use of Ir[dF(CF3)ppy]2(dtbbpy)PF6
:8) (entry 1). The use of Ir[dF(CF3)ppy]2(dtbbpy)PF6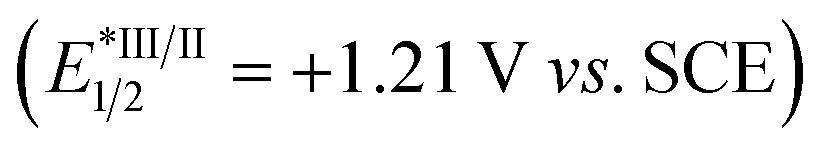 ,14 a commonly employed photocatalyst in synergistic nickel catalysis, results in a significant decrease in the yield of product 4a/5a with an inversed E/Z selectivity of double bond 1, probably due to its higher triplet energy (ET = 60.1 kcal mol−1)9c (entry 2). Switching it to Ir(ppy)3, a more reducing photocatalyst, leads to a trace amount of product (entry 3). Control experiments indicate that both nickel catalysts and visible light are essential to the success of this transformation, and the addition of exogenous nitrogen ligands is beneficial to improving the coupling efficiency (entries 6–8). Interestingly, product 4a (E,Z/Z,Z = 97:3) was still observed in 23% yield in the absence of 4CzIPN, while only a trace amount of products 4a/5a was detected in the absence of 4CzIPN and pyrene (entries 4 and 5). These results suggest that pyrene might also function as a photocatalyst to gear the nickel cycle yet cannot promote the E to Z isomerization of double bond 1. Intriguingly, performing the reaction in the absence of pyrene, the stereoisomer (Z,Z)-1,4-diene 5a was obtained in 79% yield with opposite stereoselectivity (E/Z = 16:84) (entry 9). Switching the solvent to DME improved the Z-selectivity of 5a to 94:6 with comparable yields (entry 10). This protocol represents an operationally simple approach to accessing both Z/E-alkenes by adding or removing pyrene additive. It should be noted that no stereoisomers regarding double bond 4 of 1,4-dienes were observed in these cases, probably due to the augmented A1,3-strains in the 4Z isomers (Table 1).9b,c
,14 a commonly employed photocatalyst in synergistic nickel catalysis, results in a significant decrease in the yield of product 4a/5a with an inversed E/Z selectivity of double bond 1, probably due to its higher triplet energy (ET = 60.1 kcal mol−1)9c (entry 2). Switching it to Ir(ppy)3, a more reducing photocatalyst, leads to a trace amount of product (entry 3). Control experiments indicate that both nickel catalysts and visible light are essential to the success of this transformation, and the addition of exogenous nitrogen ligands is beneficial to improving the coupling efficiency (entries 6–8). Interestingly, product 4a (E,Z/Z,Z = 97:3) was still observed in 23% yield in the absence of 4CzIPN, while only a trace amount of products 4a/5a was detected in the absence of 4CzIPN and pyrene (entries 4 and 5). These results suggest that pyrene might also function as a photocatalyst to gear the nickel cycle yet cannot promote the E to Z isomerization of double bond 1. Intriguingly, performing the reaction in the absence of pyrene, the stereoisomer (Z,Z)-1,4-diene 5a was obtained in 79% yield with opposite stereoselectivity (E/Z = 16:84) (entry 9). Switching the solvent to DME improved the Z-selectivity of 5a to 94:6 with comparable yields (entry 10). This protocol represents an operationally simple approach to accessing both Z/E-alkenes by adding or removing pyrene additive. It should be noted that no stereoisomers regarding double bond 4 of 1,4-dienes were observed in these cases, probably due to the augmented A1,3-strains in the 4Z isomers (Table 1).9b,c
|
|
|||
|---|---|---|---|
| Entry | Variations from “standard” condition | Yield of 4a/5a | E/Z |
|
a Reaction conditions: alkyne 1a (0.1 mmol), allylic ester 2a (1.5 equiv.), tert-Bu trifluoroborates 3a (1.5 equiv.), 4CzIPN (1 mol%), Ni(phen)Cl2 (5 mol%), dtbbpy (5 mol%), pyrene (0.5 equiv.), DMAc/CH3CN [0.05 M] (v/v 3:1), blue LED (90 W, λmax = 467 nm), ∼35 °C, 18 h. Yields and E/Z ratios were determined by GC analysis of the reaction mixture with dodecane as an internal standard. Isolated yields are given in parentheses.
b With DME as solvent.
|
|||
| 1 | None | 87% (84%) | 92/8 |
| 2 | Ir[dF(CF3)ppy]2(dtbbpy)PF6, instead of 4CzIPN | 37% | 17/83 |
| 3 | Ir(ppy)3, instead of 4CzIPN | 8% | 63:37 |
| 4 | No 4CzIPN | 23% | 97/3 |
| 5 | No pyrene & 4CzIPN | 8% | — |
| 6 | No Ni catalyst | 0 | — |
| 7 | No ligand | 64% | 87/13 |
| 8 | No light | 0 | — |
| 9 | No pyrene | 79% | 16/84 |
| 10b | No pyrene | 75% (71%) | 6/94 |

|
|||
With the optimal conditions in hand, we turned our attention to investigating the generality of this protocol. As shown in Scheme 1, a wide range of aryl-substituted branched allylic carbonates couple efficiently with aryl alkynes and tBuBF3K under the standard E,Z-selective conditions (with pyrene) (Table 1, entry 1), affording the corresponding tri-substituted 1,4-dienes with high yield and excellent stereoselectivity (4a–4j, E/Z up to 98:2). Installation of electron-donating or electron-poor substituents at the para-, meta-, or ortho-positions of the aromatic rings has no significant effect on the reaction efficiency or stereoselectivity (4a–4j). Alkyl-substituted allylic carbonates are applicable coupling partners, affording the desired products in moderate yields, yet as mixtures of trans/cis isomers (E/Z = 3:1 for product S4an in ESI†).
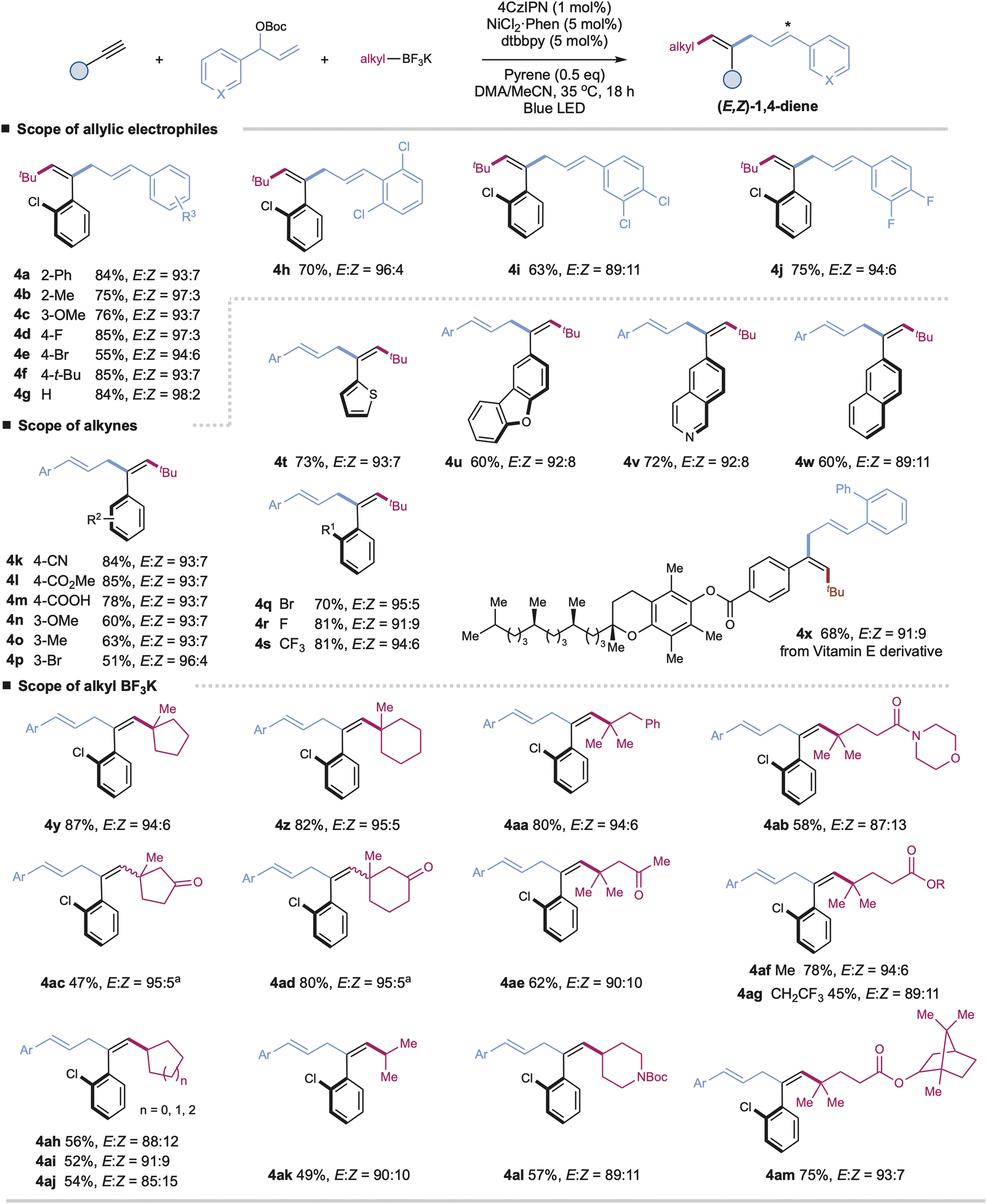 | ||
| Scheme 1 Substrate scope for (1E,4Z)-1,4-dienes. Reaction conditions: alkyne (0.2 mmol), allylic carbonate (1.5 equiv.), alkyl trifluoroborate (1.5 equiv.), 4CzIPN (1 mol%), Ni(phen)Cl2 (5 mol%), dtbbpy (5 mol%), pyrene (0.5 equiv.), DMAc/CH3CN [0.05 M] (v/v 3:1), blue LED (90 W, λmax = 467 nm), ∼35 °C, 18 h. Isolated yields. E/Z ratios were determined by 1H NMR. aE/Z ratio of double bond 4 is around 1:1. | ||
A wide range of terminal aryl alkynes with various electron-donating or withdrawing functional groups are applicable with high efficiency and excellent stereoselectivity in this catalytic system (4k–4x, E/Z up to 96:4). The electronic property or steric hindrance of the aromatic rings of alkynes has no significant effect on the stereoselectivity of resulting 1,4-dienes. The mild conditions are tolerated with many important functional groups, including cyanides, esters, carboxylic acids, and halides (4k–4m, 4p, 4q–4r). Moreover, heteroaryl-, such as thiophenyl-, dibenzo[b,d]furanyl-, quinolinyl-, and naphthalenyl-substituted alkynes, are suitable substrates with good yields and stereoselectivity under the optimal conditions (4t–4w). The reactions with complex terminal alkynes, exemplified by the one derived from vitamin E, proceed smoothly to afford corresponding functionalized 1,4-dienes under mild conditions (4x). Nevertheless, internal alkynes are unsuitable coupling partners due to the competitive cross-couplings between allyl carbonates and alkyl trifluoroborates and self-couplings of allyl carbonates.
Next, we further evaluate the generality regarding alkyl precursors (4y–4am, E/Z up to 95:5). Both linear and cyclic tertiary alkyl trifluoroborates, including those derived from biologically active molecules, show high reactivity, allowing for the facile installation of alkyl units at the end of 1,4-dienes (4y–4ag, 4am). Besides tertiary alkyl precursors, cyclic and acyclic secondary alkyl trifluoroborates are applicable with excellent chemo- and regioselectivity, albeit with slightly decreased yields and stereoselectivity (4ah–4al). The tethered ketones, amides, and esters are well-compatible (4ac–4ae, 4ab, 4al, 4af–4ag, and 4am). Generally, this chemistry affords excellent control for the configuration of double bond 4 of 1,4-dienes, probably due to the less sterically hindered E-alkenyl radicals combining faster with nickel complexes to form trans-adduct alkenylnickel species.15 Nevertheless, in the cases of cyclic ketone-derived alkyl trifluoroborates, we observed a significant loss in the stereoselectivity of double bond 4 (4ac and 4ad). We reason that the potential chelation between the cyclic ketone and nickel species16 could affect the stereoselective radical capture process, leading to the formation of trans/cis mixtures of alkenylnickel species and, finally, the Z/E isomers of double bond 4 (see Table S8†).
Then, we turned our attention to exploring the scope of these components under the Z-selective conditions (without pyrene) (Table 1, entry 10). As shown in Scheme 2, this protocol is applicable to a wide array of terminal aryl alkynes, allylic carbonates, tertiary and secondary alkyl trifluoroborates, affording the corresponding Z,Z-skipped dienes with moderate to good yields and high selectivity (5a–5s). Pleasingly, performing the reaction on a gram scale still led to the formation of product 5a with excellent selectivity, albeit in a decreased yield. The presence of an ortho-substituent on the aromatic ring of allylic carbonates is vital to maintain the excellent Z-selectivity of double bond 4 of 1,4-dienes due to the augmented A1,3-strain.9b,c
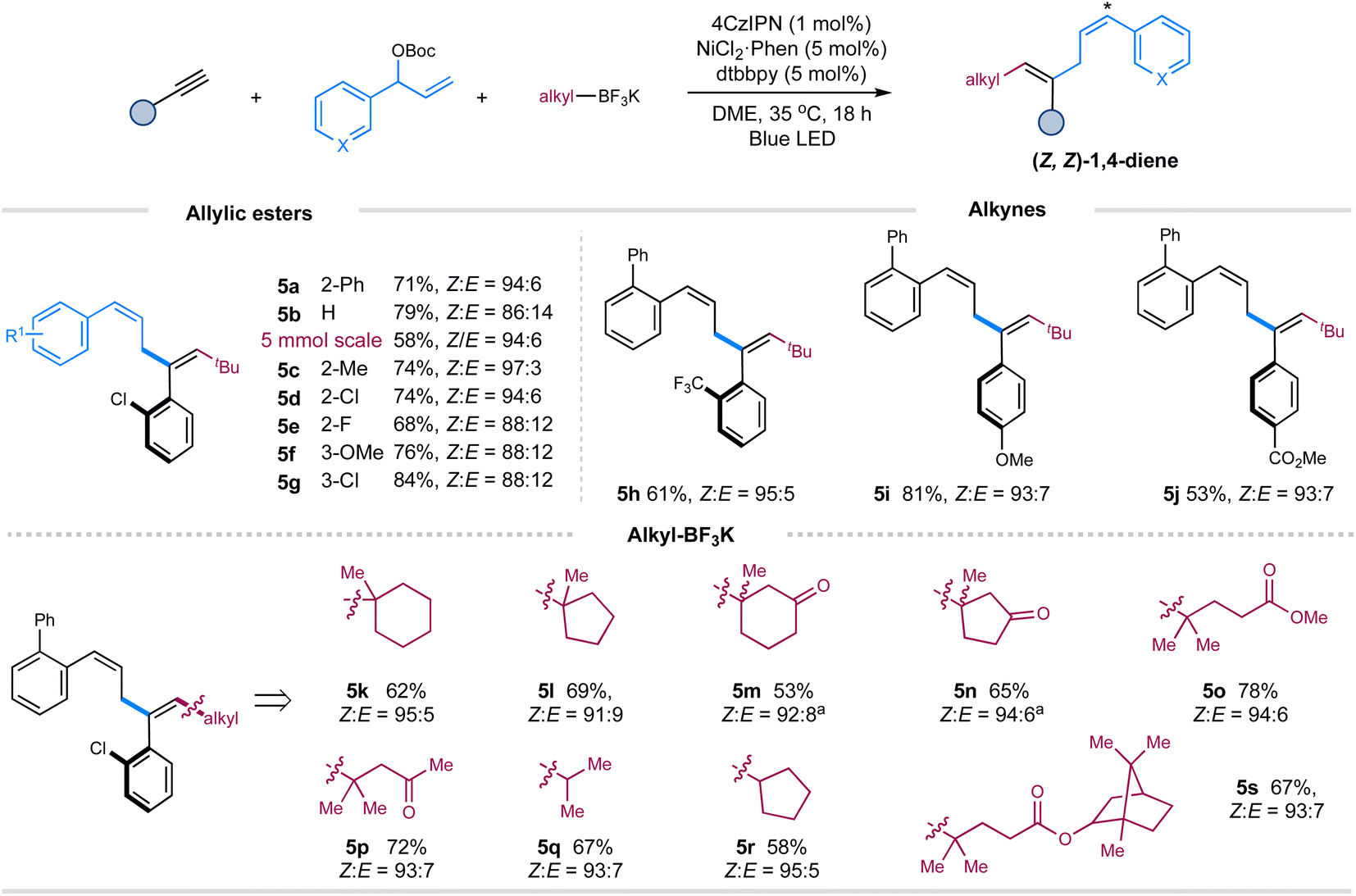 | ||
| Scheme 2 Substrate scope of (Z,Z)-1,4-dienes. Reaction conditions: alkynes (0.2 mmol), allylic esters (1.5 equiv.), alkyl trifluoroborates (1.5 equiv.), 4CzIPN (1 mol%), Ni(phen)Cl2 (5 mol%), dtbbpy (5 mol%), DME [0.05 M] (v/v 3:1), blue LED (90 W, λmax = 467 nm), ∼35 °C, 18 h. Isolated yields. E/Z ratios were determined by 1H NMR. aE/Z ratio of double bond 4 is around 1:1. | ||
We further performed several preliminary experiments to gain insight into the mechanism (Scheme 3). Light-on/off experiments demonstrated that continuous light irradiation is required for product formation and stereoselectivity in these reactions (see Fig. S12–S15 in ESI†). Moreover, Stern–Volmer fluorescence quenching experiments revealed that the photoexcited state of 4CzIPN was quenched by alkyl trifluoroborates instead of alkynes and allylic esters (see Fig. S7–S10 in ESI†). Furthermore, the radical inhibition experiment with 2.0 equivalents of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) led to no formation of the coupling product 4a (Scheme 3a). To probe the nature of radical species, (1-cyclopropylvinyl) benzene was used as a radical probe to react with allylic ester 2a and t-BuBF3K under the standard condition (Scheme 3b). 20% yield of product 6, probably generated via a radical addition/radical ring-opening/cyclization sequence, was observed, suggesting the involvement of t-Bu˙ radical. On the other hand, the reaction of allylic ester 2a with alkyl trifluoroborate 3p in the absence of alkyne 1a gave 42% of allyl-alkyl coupling product 7/7′ (branch & linear),17 together with 14% of allyl–allyl homo-coupling product 8/8′ (branch & linear) and 18% of reductive product 9/9′ (Scheme 3c). In contrast, the standard 3-component reaction of 2a afforded the desired product 5p in 72% yield, with only an 11% yield of by-product 7/7′. These results imply that the addition rate of t-Bu˙ to alkyne might be faster than its coupling rate with allylic carbonate, thus leading to the desired 3-component carboallylation reaction.18 The observation of the allyl-derived side products also implies the potential involvement of allylnickel species. To verify this hypothesis, we prepared π-allyl nickel complex I. The reaction of Ni(COD)2, 1,10-phen, and allylic carbonate in THF gave a 22% yield of nickel complex I as a dark purple powder (Scheme 3d).19 The stoichiometric reaction of allylnickel complex I with alkyne 1a and tBuBF3K in the presence of catalytic 4CzIPN and pyrene with blue LED irradiation gave 26% yield of 1,4-diene product 4g (E/Z = 94:6); and the reaction of 1a, allylic carbonate, and tBuBF3K with catalytic complex I under otherwise same condition gave product 4g in 63% yield (E/Z = 89:11). These results suggest that allylnickel species could be involved in this carboallylation reaction.
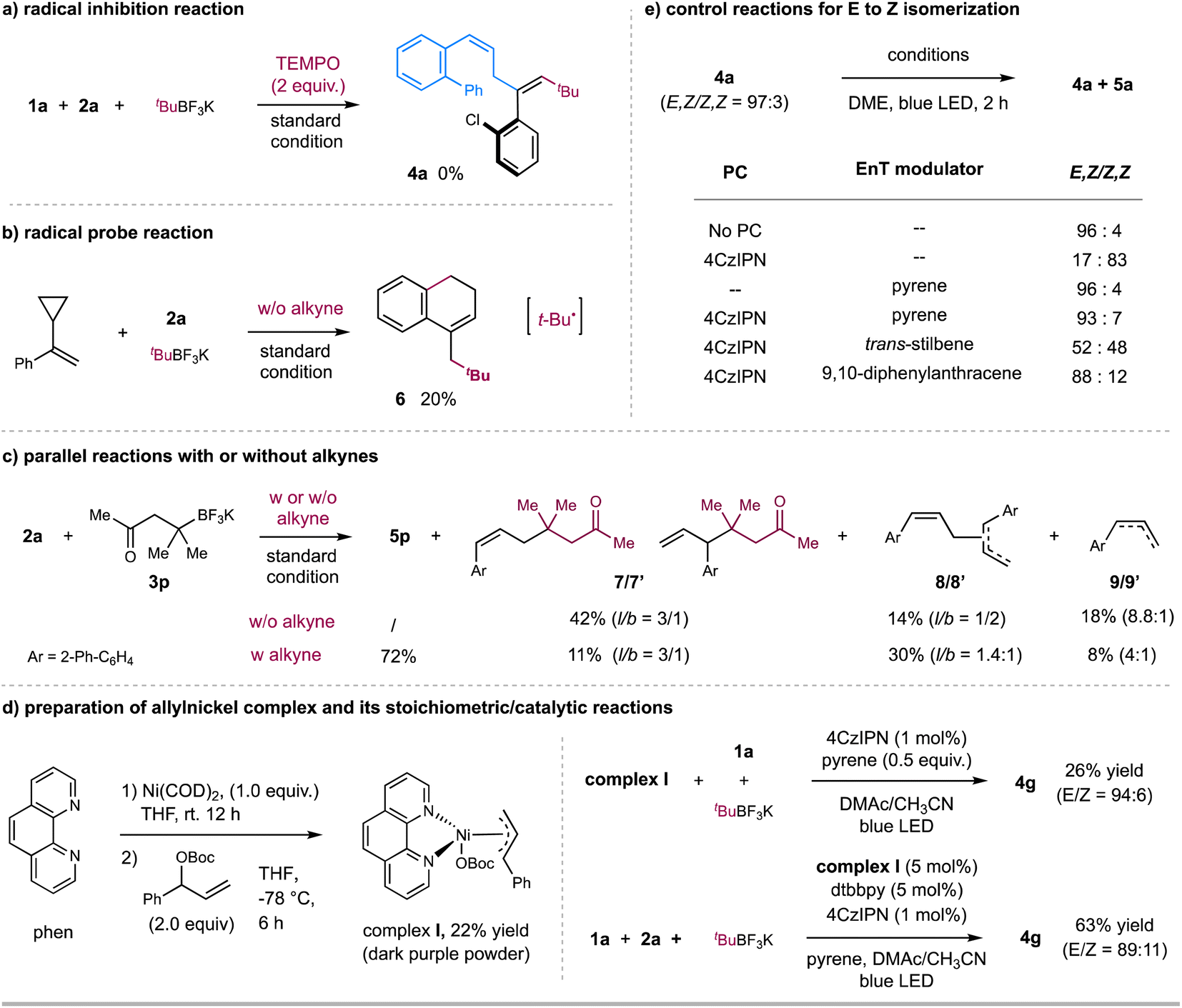 | ||
| Scheme 3 Mechanistic studies. (a) Radical inhibition reaction; (b) radical probe reaction; (c) parallel reactions with or without alkyne; (d) stoichiometric reaction with allylnickel complex; (e) control reactions regarding E to Z isomerization. | ||
Next, we carried out several control reactions to investigate the stereoselectivity. As shown in Scheme 3e, (E,Z)-1,4-diene 4a (E/Z = 97:3) was efficiently converted to corresponding (Z,Z)-isomer 5a (E/Z = 17:83) in the presence of catalytic 4CzIPN and blue LED, while with no significant isomerization in the presence of catalytic pyrene (E/Z = 96:4). Based on these results, we reason that (E,Z)-1,4-dienes could undergo a selective EnT process with photoexcited [4CzIPN]* (ET = 56.4 kcal mol−1),9c,20 to afford (Z,Z)-1,4-dienes; while the addition of pyrene, likely acting as a triplet energy modulator (ET = 2.10 eV),21 suppresses the EnT process to leave (E,Z)-1,4-dienes as they are. This assumption is also consistent with the fluorescence quenching experimental results, as pyrene showed a slightly more significant quenching to photoexcited [4CzIPN]* than (E,Z)-1,4-diene 4a, which could proceed via a selective EnT21 or triplet–triplet annihilation22 process (Fig. S11 and S18–S21†).23 We also tested several potential triplet energy modulators, such as trans-stilbene24 and 9,10-diphenylanthracene (Scheme 3e). 9,10-Diphenylanthracene, the structure of which is similar to pyrene, gave a comparable effect to pyrene (Scheme 3e); at the same time, the addition of trans-stilbene led to the formation of cis/trans-isomers (Scheme 3e), echoed with its no quenching effect to [4CzIPN]* (Fig. S11†).
We propose a potential catalytic cycle for this metallaphotoredox-catalyzed carboallylation of alkynes, as depicted in Fig. 2. A single-electron oxidation of alkyl trifluoroborate (Ered1/2 = +1.26 V vs. SCE)25 by the photoexcited [4CzIPN]* (E1/2 = +1.35 V vs. SCE)13 generates alkyl radical I, which then adds to an alkyne to form alkenyl radical II. The nickel cycle starts from Ni(0) species. Oxidative addition of allylic ester IV to Ni(0) III affords allylnickel intermediate V,19,26 which then capture alkenyl radical II to form trans-(alkenyl)(allyl)Ni(III) VI. Subsequent reductive elimination of Ni(III) VI would deliver the (E,Z)-skipped diene product and Ni(I) VII. The final SET event between the reducing photocatalyst and Ni(I) will regenerate the ground-state photocatalyst and Ni(0) to close the two catalytic cycles. While we are unable to rigorously rule out the possibility of alkenylnickel intermediate, generated via direct radical capture of alkenyl radical by Ni(0). Concerning the E/Z selectivity of products, we assume that (E,Z)-skipped diene remains intact in the presence of pyrene, while undergoing a photoinduced *4CzIPN-enabled E to Z isomerization in the absence of pyrene, thus securing divergent and modular access to both trans- and cis-1,4-dienes.
 | ||
| Fig. 2 Proposed mechanism. | ||
Conclusions
In summary, we report a metallaphotoredox-catalyzed stereo-divergent carboallylation strategy for the straightforward synthesis of highly functionalized skipped dienes with readily available terminal alkynes, allylic carbonates, and alkyl trifluoroborates. Utilizing 4CzIPN as an organic photocatalyst and Ni(phen)Cl2 as a nickel catalyst, this dual protocol enables stereoselective access to a wide range of E,Z-1,4-dienes with pyrene as a triplet energy modulator. Furthermore, this protocol allows for modular synthesis of corresponding Z,Z-isomers with the same substrates and catalysts by removing pyrene additive. These transformations show a broad substrate scope with excellent chemo-, regio-, and stereo-selective controls. Mechanistic investigations shed some light on the reaction pathways and stereoselectivity.Author contributions
J. Q., Z. Z., Y. L. conducted the investigation under L. C.'s supervision. L. C. wrote the manuscript and J. Q., S. Z. prepared the ESI.† All authors contributed to the conceptualization of the project and the editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Zheng Tang at Donghua University for the helpful discussion on triplet–triplet annihilation and Prof. Jin Wen at Donghua University for the calculation of the triplet energy of (E,Z)-1,4-dienes. We are grateful for the financial support provided by the National Natural Science Foundation of China (21971036) and the National Science Fund for Excellent Young Scholars (22122101).Notes and references
- (a) J. Pospíšil and I. E. Markó, Total Synthesis of Jerangolid D, J. Am. Chem. Soc., 2007, 129, 3516–3517 CrossRef PubMed; (b) F. Surup, H. Shojaei, P. von Zezschwitz, B. Kunze and S. Grond, Iromycins from Streptomyces sp. and from synthesis: New inhibitors of the mitochondrial electron transport chain, Bioorg. Med. Chem., 2008, 16, 1738–1746 CrossRef PubMed; (c) T. Sato, T. Suto, Y. Nagashima, S. Mukai and N. Chida, Total Synthesis of Skipped Diene Natural Products, Asian J. Org. Chem., 2021, 10, 2486–2502 CrossRef.
- (a) A. Zhang and T. V. RajanBabu, Hydrovinylation of 1,3-Dienes: A New Protocol, an Asymmetric Variation, and a Potential Solution to the Exocyclic Side Chain Stereochemistry Problem, J. Am. Chem. Soc., 2006, 128, 54–55 CrossRef PubMed; (b) V. A. Schmidt, C. R. Kennedy, M. J. Bezdek and P. J. Chirik, Selective [1,4]-Hydrovinylation of 1,3-Dienes with Unactivated Olefins Enabled by Iron Diimine Catalysts, J. Am. Chem. Soc., 2018, 140, 3443–3453 CrossRef PubMed; (c) X. Pang, Z.-Z. Zhao, X.-X. Wei, L. Qi, G.-L. Xu, J. Duan, X.-Y. Liu and X.-Z. Shu, Regiocontrolled Reductive Vinylation of Aliphatic 1,3-Dienes with Vinyl Triflates by Nickel Catalysis, J. Am. Chem. Soc., 2021, 143, 4536–4542 CrossRef PubMed.
- (a) F. Kolundzic and G. C. Micalizio, Synthesis of Substituted 1,4-Dienes by Direct Alkylation of Allylic Alcohols, J. Am. Chem. Soc., 2007, 129, 15112–15113 CrossRef PubMed; (b) Y. Lee, K. Akiyama, D. G. Gillingham, M. K. Brown and A. H. Hoveyda, Highly Site- and Enantioselective Cu-Catalyzed Allylic Alkylation Reactions with Easily Accessible Vinylaluminum Reagents, J. Am. Chem. Soc., 2008, 130, 446–447 CrossRef PubMed; (c) K. Akiyama, F. Gao and A. H. Hoveyda, Stereoisomerically pure trisubstituted vinylaluminum reagents and their utility in copper-catalyzed enantioselective synthesis of 1,4-dienes containing Z or E alkenes, Angew. Chem., Int. Ed., 2010, 49, 419–423 CrossRef PubMed; (d) S. M. Sarkar, Y. Uozumi and Y. M. Yamada, A highly active and reusable self-assembled poly(imidazole/palladium) catalyst: allylic arylation/alkenylation, Angew. Chem., Int. Ed., 2011, 50, 9437–9441 CrossRef; (e) M. S. McCammant, L. Liao and M. S. Sigman, Palladium-Catalyzed 1,4-Difunctionalization of Butadiene To Form Skipped Polyenes, J. Am. Chem. Soc., 2013, 135, 4167–4170 CrossRef PubMed; (f) J. Y. Hamilton, D. Sarlah and E. M. Carreira, Iridium-catalyzed enantioselective allylic vinylation, J. Am. Chem. Soc., 2013, 135, 994–997 CrossRef PubMed; (g) C. P. McGeough, A. E. Strom and T. F. Jamison, Ni-Catalyzed Cross-Electrophile Coupling for the Synthesis of Skipped Polyenes, Org. Lett., 2019, 21, 3606–3609 CrossRef PubMed.
- O. Bernardo, K. Yamamoto, I. Fernández and L. A. López, Reactivity of Stabilized Vinyldiazo Compounds toward Alkenyl- and Alkynylsilanes under Gold Catalysis: Regio- and Stereoselective Synthesis of Skipped Dienes and Enynes, Org. Lett., 2021, 23, 4452–4456 CrossRef PubMed.
- (a) B. M. Trost, G. D. Probst and A. Schoop, Ruthenium-Catalyzed Alder Ene Type Reactions. A Formal Synthesis of Alternaric Acid, J. Am. Chem. Soc., 1998, 120, 9228–9236 CrossRef; (b) G. Hilt and J. Treutwein, Cobalt-Catalyzed Alder–Ene Reaction, Angew. Chem., Int. Ed., 2007, 46, 8500–8502 CrossRef PubMed; (c) D. P. Todd, B. B. Thompson, A. J. Nett and J. Montgomery, Deoxygenative C–C Bond-Forming Processes via a Net Four-Electron Reductive Coupling, J. Am. Chem. Soc., 2015, 137, 12788–12791 CrossRef PubMed; (d) G. Xu, H. Zhao, B. Fu, A. Cang, G. Zhang, Q. Zhang, T. Xiong and Q. Zhang, Ligand-Controlled Regiodivergent and Enantioselective Copper-Catalyzed Hydroallylation of Alkynes, Angew. Chem., Int. Ed., 2017, 56, 13130–13134 CrossRef PubMed; (e) M. Mailig, A. Hazra, M. K. Armstrong and G. Lalic, Catalytic Anti-Markovnikov Hydroallylation of Terminal and Functionalized Internal Alkynes: Synthesis of Skipped Dienes and Trisubstituted Alkenes, J. Am. Chem. Soc., 2017, 139, 6969–6977 CrossRef PubMed; (f) D.-W. Ji, Y.-C. Hu, H. Zheng, C.-Y. Zhao, Q.-A. Chen and V. M. Dong, A regioselectivity switch in Pd-catalyzed hydroallylation of alkynes, Chem. Sci., 2019, 10, 6311–6315 RSC; (g) J. Chen, J. Ying and Z. Lu, Cobalt-catalyzed branched selective hydroallylation of terminal alkynes, Nat. Commun., 2022, 13, 4518 CrossRef PubMed.
- (a) S.-i. Ikeda, D.-M. Cui and Y. Sato, Regio- and Stereoselective Synthesis of 3,6-Dien-1-ynes by Nickel-Catalyzed Coupling Reaction of Allyl Chlorides, 1-Alkynes, and Alkynyltins, J. Org. Chem., 1994, 59, 6877–6878 CrossRef; (b) S.-i. Ikeda, H. Miyashita and Y. Sato, Tandem Coupling of Allyl Electrophiles, Alkynes, and Me3Al or Me2Zn in the Presence of a Nickel Catalyst, Organometallics, 1998, 17, 4316–4318 CrossRef; (c) T. Mori, T. Nakamura and M. Kimura, Stereoselective Coupling Reaction of Dimethylzinc and Alkyne toward Nickelacycles, Org. Lett., 2011, 13, 2266–2269 CrossRef PubMed; (d) W. Li, S. Yu, J. Li and Y. Zhao, Nickel-Catalyzed Allylmethylation of Alkynes with Allylic Alcohols and AlMe3: Facile Access to Skipped Dienes and Trienes, Angew. Chem., Int. Ed., 2020, 59, 14404–14408 CrossRef PubMed.
- (a) J. Mateos, E. Rivera-Chao and M. Fañanás-Mastral, Synergistic Copper/Palladium Catalysis for the Regio- and Stereoselective Synthesis of Borylated Skipped Dienes, ACS Catal., 2017, 7, 5340–5344 CrossRef; (b) A. M. Y. Suliman, E.-A. M. A. Ahmed, T.-J. Gong and Y. Fu, Cu/Pd-Catalyzed cis-Borylfluoroallylation of Alkynes for the Synthesis of Boryl-Substituted Monofluoroalkenes, Org. Lett., 2021, 23, 3259–3263 CrossRef PubMed; (c) N. Vázquez-Galiñanes, I. Velo-Heleno and M. Fañanás-Mastral, Bifunctional Skipped Dienes through Cu/Pd-Catalyzed Allylboration of Alkynes with B2pin2 and Vinyl Epoxides, Org. Lett., 2022, 24, 8244–8248 CrossRef PubMed.
- (a) M. D. Levin, S. Kim and F. D. Toste, Photoredox Catalysis Unlocks Single-Electron Elementary Steps in Transition Metal Catalyzed Cross-Coupling, ACS Cent. Sci., 2016, 2, 293–301 CrossRef PubMed; (b) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Merging Visible Light Photoredox and Gold Catalysis, Acc. Chem. Res., 2016, 49, 2261–2272 CrossRef PubMed; (c) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, The merger of transition metal and photocatalysis, Nat. Rev. Chem, 2017, 1, 0052 CrossRef; (d) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Alkyl Carbon–Carbon Bond Formation by Nickel/Photoredox Cross-Coupling, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef PubMed; (e) A. Hossain, A. Bhattacharyya and O. Reiser, Copper's rapid ascent in visible-light photoredox catalysis, Science, 2019, 364, eaav9713 CrossRef PubMed; (f) P. Chuentragool, D. Kurandina and V. Gevorgyan, Catalysis with Palladium Complexes Photoexcited by Visible Light, Angew. Chem., Int. Ed., 2019, 58, 11586–11598 CrossRef PubMed; (g) M. Kojima and S. Matsunaga, The Merger of Photoredox and Cobalt Catalysis, Trends Chem., 2020, 2, 410–426 CrossRef; (h) H.-H. Zhang, H. Chen, C. Zhu and S. Yu, A review of enantioselective dual transition metal/photoredox catalysis, Sci. China: Chem., 2020, 63, 637–647 CrossRef; (i) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis, Chem. Rev., 2022, 122, 1485–1542 CrossRef PubMed.
- (a) J. J. Molloy, T. Morack and R. Gilmour, Positional and Geometrical Isomerisation of Alkenes: The Pinnacle of Atom Economy, Angew. Chem., Int. Ed., 2019, 58, 13654–13664 CrossRef PubMed; (b) T. Neveselý, M. Wienhold, J. J. Molloy and R. Gilmour, Advances in the E → Z Isomerization of Alkenes Using Small Molecule Photocatalysts, Chem. Rev., 2022, 122, 2650–2694 CrossRef PubMed; (c) J. Corpas, P. Mauleón, R. Gómez Arrayás and J. C. Carretero, E/Z Photoisomerization of Olefins as an Emergent Strategy for the Control of Stereodivergence in Catalysis, Adv. Synth. Catal., 2022, 364, 1348–1370 CrossRef; (d) Y. Sakakibara and K. Murakami, Switchable Divergent Synthesis Using Photocatalysis, ACS Catal., 2022, 12, 1857–1878 CrossRef.
- (a) K. Singh, S. J. Staig and J. D. Weaver, Facile Synthesis of Z-Alkenes via Uphill Catalysis, J. Am. Chem. Soc., 2014, 136, 5275–5278 CrossRef PubMed; (b) J. B. Metternich and R. Gilmour, A Bio-Inspired, Catalytic E → Z Isomerization of Activated Olefins, J. Am. Chem. Soc., 2015, 137, 11254–11257 CrossRef PubMed; (c) J. J. Molloy, J. B. Metternich, C. G. Daniliuc, A. J. B. Watson and R. Gilmour, Contra-Thermodynamic, Photocatalytic E→Z Isomerization of Styrenyl Boron Species: Vectors to Facilitate Exploration of Two-Dimensional Chemical Space, Angew. Chem., Int. Ed., 2018, 57, 3168–3172 CrossRef PubMed; (d) J. J. Molloy, M. Schäfer, M. Wienhold, T. Morack, C. G. Daniliuc and R. Gilmour, Boron-enabled geometric isomerization of alkenes via selective energy-transfer catalysis, Science, 2020, 369, 302–306 CrossRef PubMed; (e) T. Neveselý, J. J. Molloy, C. McLaughlin, L. Brüss, C. G. Daniliuc and R. Gilmour, Leveraging the n→π* Interaction in Alkene Isomerization by Selective Energy Transfer Catalysis, Angew. Chem., Int. Ed., 2022, 61, e202113600 CrossRef PubMed; (f) T. J. B. Zähringer, M. Wienhold, R. Gilmour and C. Kerzig, Direct Observation of Triplet States in the Isomerization of Alkenylboronates by Energy Transfer Catalysis, J. Am. Chem. Soc., 2023, 145, 21576–21586 CrossRef PubMed.
- (a) J. B. Metternich and R. Gilmour, One Photocatalyst, n Activation Modes Strategy for Cascade Catalysis: Emulating Coumarin Biosynthesis with (−)-Riboflavin, J. Am. Chem. Soc., 2016, 138, 1040–1045 CrossRef PubMed; (b) H.-H. Zhang, J.-J. Zhao and S. Yu, Enantioselective Allylic Alkylation with 4-Alkyl-1,4-dihydro-pyridines Enabled by Photoredox/Palladium Cocatalysis, J. Am. Chem. Soc., 2018, 140, 16914–16919 CrossRef PubMed; (c) L. Guo, F. Song, S. Zhu, H. Li and L. Chu, syn-Selective alkylarylation of terminal alkynes via the combination of photoredox and nickel catalysis, Nat. Commun., 2018, 9, 4543 CrossRef PubMed; (d) C. Zhu, H. Yue, B. Maity, I. Atodiresei, L. Cavallo and M. Rueping, A multicomponent synthesis of stereodefined olefins via nickel catalysis and single electron/triplet energy transfer, Nat. Catal., 2019, 2, 678–687 CrossRef; (e) J. Xu, Z. Li, Y. Xu, X. Shu and H. Huo, Stereodivergent Synthesis of Both Z- and E-Alkenes by Photoinduced, Ni-Catalyzed Enantioselective C(sp3)–H Alkenylation, ACS Catal., 2021, 11, 13567–13574 CrossRef; (f) F. Song, F. Wang, L. Guo, X. Feng, Y. Zhang and L. Chu, Visible-Light-Enabled Stereodivergent Synthesis of E- and Z-Configured 1,4-Dienes by Photoredox/Nickel Dual Catalysis, Angew. Chem., Int. Ed., 2020, 59, 177–181 CrossRef PubMed; (g) X. Shen, C. Huang, X.-A. Yuan and S. Yu, Diastereoselective and Stereodivergent Synthesis of 2-Cinnamylpyrrolines Enabled by Photoredox-Catalyzed Iminoalkenylation of Alkenes, Angew. Chem., Int. Ed., 2021, 60, 9672–9679 CrossRef PubMed; (h) H. Zhang, C. Huang, X.-A. Yuan and S. Yu, Photoexcited Chiral Copper Complex-Mediated Alkene E → Z Isomerization Enables Kinetic Resolution, J. Am. Chem. Soc., 2022, 144, 10958–10967 CrossRef PubMed; (i) T. Long, C. Zhu, L. Li, L. Shao, S. Zhu, M. Rueping and L. Chu, Ligand-controlled stereodivergent alkenylation of alkynes to access functionalized trans- and cis-1,3-dienes, Nat. Commun., 2023, 14, 55 CrossRef PubMed; (j) L. Huang, M. Szewczyk, R. Kancherla, B. Maity, C. Zhu, L. Cavallo and M. Rueping, Modulating stereoselectivity in allylic C(sp3)–H bond arylations via nickel and photoredox catalysis, Nat. Commun., 2023, 14, 548 CrossRef PubMed; (k) H. Zhang, X. He, X.-A. Yuan and S. Yu, Kinetic Resolution of 2-Cinnamylpyrrolines Enabled by Photoexcited Chiral Copper Complex-Mediated Alkene E → Z Isomerization, ACS Catal., 2023, 13, 2857–2866 CrossRef.
- (a) U. Wille, Radical Cascades Initiated by Intermolecular Radical Addition to Alkynes and Related Triple Bond Systems, Chem. Rev., 2013, 113, 813–853 CrossRef PubMed; (b) W. Liu and W. Kong, Ni-Catalyzed stereoselective difunctionalization of alkynes, Org. Chem. Front., 2020, 7, 3941–3955 RSC; (c) S. Zhu, X. Zhao, H. Li and L. Chu, Catalytic three-component dicarbofunctionalization reactions involving radical capture by nickel, Chem. Soc. Rev., 2021, 50, 10836–10856 RSC.
- T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Recent advances of 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) in photocatalytic transformations, Chem. Commun., 2019, 55, 5408–5419 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113, 5322–5363 CrossRef PubMed.
- Y. Dai, F. Wang, S. Zhu and L. Chu, Selective Ni-catalyzed cross-electrophile coupling of alkynes, fluoroalkyl halides, and vinyl halides, Chin. Chem. Lett., 2022, 33, 4074–4078 CrossRef.
- F. Wang, S. Pan, S. Zhu and L. Chu, Selective Three-Component Reductive Alkylalkenylation of Unbiased Alkenes via Carbonyl-Directed Nickel Catalysis, ACS Catal., 2022, 12, 9779–9789 CrossRef.
- H. Chen, X. Jia, Y. Yu, Q. Qian and H. Gong, Nickel-Catalyzed Reductive Allylation of Tertiary Alkyl Halides with Allylic Carbonates, Angew. Chem., Int. Ed., 2017, 56, 13103–13106 CrossRef PubMed.
- L. Guo, M. Yuan, Y. Zhang, F. Wang, S. Zhu, O. Gutierrez and L. Chu, General Method for Enantioselective Three-Component Carboarylation of Alkenes Enabled by Visible-Light Dual Photoredox/Nickel Catalysis, J. Am. Chem. Soc., 2020, 142, 20390–20399 CrossRef PubMed.
- K. Liu, Z. Wang, A. N. Kunzel, M. Layh and A. Studer, Regioselective Formal beta-Allylation of Carbonyl Compounds Enabled by Cooperative Nickel and Photoredox Catalysis, Angew. Chem., Int. Ed., 2023, 62, e202303473 CrossRef PubMed.
- (a) J. Lu, B. Pattengale, Q. Liu, S. Yang, W. Shi, S. Li, J. Huang and J. Zhang, Donor–Acceptor Fluorophores for Energy-Transfer-Mediated Photocatalysis, J. Am. Chem. Soc., 2018, 140, 13719–13725 CrossRef PubMed; (b) S. E. Chérif, A. Ghosh, S. Chelli, I. M. Dixon, J. Kraiem and S. Lakhdar, Merging Grubbs second-generation catalyst with photocatalysis enables Z-selective metathesis of olefins: scope, limitations, and mechanism, Chem. Sci., 2022, 13, 12065–12070 RSC.
- (a) C. Mongin, S. Garakyaraghi, N. Razgoniaeva, M. Zamkov and F. N. Castellano, Direct observation of triplet energy transfer from semiconductor nanocrystals, Science, 2016, 351, 369–372 CrossRef PubMed; (b) P. Herr, C. Kerzig, C. B. Larsen, D. Häussinger and O. S. Wenger, Manganese(I) complexes with metal-to-ligand charge transfer luminescence and photoreactivity, Nat. Chem., 2021, 13, 956–962 CrossRef PubMed.
- (a) D. V. Kozlov and F. N. Castellano, Anti-Stokes delayed fluorescence from metal–organic bichromophores, Chem. Commun., 2004, 2860–2861 RSC; (b) W. Zhao and F. N. Castellano, Upconverted Emission from Pyrene and Di-tert-butylpyrene Using Ir(ppy)3 as Triplet Sensitizer, J. Phys. Chem. A, 2006, 110, 11440–11445 CrossRef PubMed; (c) M. Marchini, G. Bergamini, P. G. Cozzi, P. Ceroni and V. Balzani, Photoredox Catalysis: The Need to Elucidate the Photochemical Mechanism, Angew. Chem., Int. Ed., 2017, 56, 12820–12821 CrossRef PubMed.
- Note: Due to the compliance of this photoinduced system, currently, we cannot distinguish which pathway (EnT or TTA) is involved in the quenching of *4CzIPN by pyrene; please see ESI† for related discussions.
- X. Cheng, T. Li, Y. Liu and Z. Lu, Stereo- and Enantioselective Benzylic C–H Alkenylation via Photoredox/Nickel Dual Catalysis, ACS Catal., 2021, 11, 11059–11065 CrossRef.
- D. N. Primer and G. A. Molander, Enabling the Cross-Coupling of Tertiary Organoboron Nucleophiles through Radical-Mediated Alkyl Transfer, J. Am. Chem. Soc., 2017, 139, 9847–9850 CrossRef PubMed.
- Y. Dai, F. Wu, Z. Zang, H. You and H. Gong, Ni-Catalyzed Reductive Allylation of Unactivated Alkyl Halides with Allylic Carbonates, Chem.–Eur. J., 2012, 18, 808–812 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on experimental procedures and characterization data for all new compounds. See DOI: https://doi.org/10.1039/d3sc04645a |
| This journal is © The Royal Society of Chemistry 2023 |