
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Steering competitive N2 and CO adsorption toward efficient urea production with a confined dual site†
Zhe
Chen
 ab,
Yonghua
Liu
b and
Tao
Wang
*bcd
ab,
Yonghua
Liu
b and
Tao
Wang
*bcd
aDepartment of Chemistry, Zhejiang University, 38 Zheda Road, Hangzhou 310027, Zhejiang Province, China
bCenter of Artificial Photosynthesis for Solar Fuels and Department of Chemistry, School of Science and Research Center for Industries of the Future, Westlake University, 600 Dunyu Road, Hangzhou 310030, Zhejiang Province, China. E-mail: twang@westlake.edu.cn
cInstitute of Natural Sciences, Westlake Institute for Advanced Study, 18 Shilongshan Road, Hangzhou 310024, Zhejiang Province, China
dDivision of Solar Energy Conversion and Catalysis at Westlake University, Zhejiang Baima Lake Laboratory Co., Ltd, Hangzhou 310000, Zhejiang, China
First published on 23rd October 2023
Abstract
Electrocatalytic urea synthesis under mild conditions via the nitrogen (N2) and carbon monoxide (CO) coupling represents an ideal and green alternative to the energy-intensive traditional synthetic protocol. However, this process is challenging due to the more favorable CO adsorption than N2 at the catalytic site, making the formation of the key urea precursor (*NCON) extremely difficult. Herein, we theoretically construct a spatially isolated dual-site (DS) catalyst with the confinement effect to manipulate the competitive CO and N2 adsorption, which successfully guarantees the dominant horizontal N2 adsorption and subsequent efficient *NCON formation via C–N coupling and achieves efficient urea synthesis. Among all the computationally evaluated candidates, the catalyst with dual V sites anchored on 4N-doped graphene (DS-VN4) stands out and shows a moderate energy barrier for C–N coupling and a low theoretical limiting potential of −0.50 V for urea production, which simultaneously suppresses the ammonia production and hydrogen evolution. The confined dual-site introduced in this computational work has the potential to not only properly address part of the challenges toward efficient urea electrosynthesis from CO and N2 but also provide an elegant theoretical strategy for fine-tuning the strength of chemical bonds to achieve a rational catalyst design.
1. Introduction
As a crucial artificial fertilizer, urea (NH2CONH2) has laid a solid foundation for the development of agriculture to produce food and avoid the mass starvation of human beings.1–3 Meanwhile, it is an essential feedstock for manufacturing high-value-added chemicals such as plastics, adhesives, potassium cyanate, and urea nitrate. In industry, large-scale urea synthesis was achieved at a high temperature of 150–200 °C and high pressure of 150–250 bar using ammonia (NH3) and carbon dioxide (CO2) as reactants. Another key issue of this route lies in its dependence on NH3 as the nitrogen source, which is industrially produced via the energy- and capital-intensive Haber–Bosch process under harsh conditions.4 It consumes ∼2% of the global fossil energy and emits ∼300 million tons of CO2 annually during the inert N2 fixation to NH3 due to the usage of gray hydrogen, and ultimately impacts the sustainability of the whole urea production industry.4 Therefore, developing a low-carbon and alternative sustainable urea synthesis protocol is significant for achieving and maintaining the sustainable development of human society.Recently, the direct coupling of carbon-based feedstocks (CO2 and CO2-derived CO) with NH3-free nitrogen sources (N2, NO, nitrite, and nitrate) has been proposed as a promising and efficient one-step method to produce urea via electrochemical reduction, which has attracted increasing attention from academia due to the sustainability and affordability.5–20 For example, Chen et al. experimentally prepared an electrocatalyst by anchoring PdCu nanoparticles on TiO2 nanosheets for aqueous N2 and CO2 coupling to synthesize urea, which achieved a formation rate of 3.36 mmol g−1 h−1 and a faradaic efficiency of 8.92% at −0.4 V versus RHE.5 From electrochemical CO2 and N2 coupling, Zhang and co-workers reported the urea yield rates/faradaic efficiency of 5.91 mmol g−1 h−1/12.55% and 9.70 mmol g−1 h−1/20.36% on the Mott–Schottky Bi–BiVO4 heterostructures and flower-like nickel borate [Ni3(BO3)2], respectively.6,7 Zhu et al. theoretically predicted Mo2B2, Ti2B2, and Cr2B2 as potential efficient electrocatalysts for urea production via N2 and CO2 coupling under ambient conditions based on the data from mechanistic calculations with the computational hydrogen electrode (CHE) model.8 Moreover, compared with the chemically inert N2 molecule, higher urea yields can be generally obtained by using more activated nitrite and nitrate, but have trouble in large-scale applications due to the limited availabilities of those nitrogen sources.13–20
Despite the impressive recent progress in electrochemical urea synthesis, the sluggish reaction kinetics and limited yield still seriously impede its industrial integration. In principle, the formation of key *NCON species is a prerequisite for urea synthesis,5–7,21–23 while the direct coupling of N2 with CO represents an ideal approach to generating this crucial intermediate. Unfortunately, this route faces several formidable challenges due to the intrinsic difference in electronic structures of N2 and CO molecules, as well as their similar bonding modes with active sites. As shown in Fig. 1A, the foremost challenge is the competitive adsorption of N2 and CO. Mechanistically, the binding of the polarized CO molecule with the metallic active site is usually more favorable than the non-polarized N2 molecule, forming a σ bond via the C atom due to the significant character of the highest occupied molecular orbital (HOMO) of C 2s (Fig. 1B).24 Meanwhile, the HOMO of the CO molecule is energetically closer to the metal d-orbital than that of the N2 molecule, resulting in a more stable metal-CO σ bond. As a result, N2 is usually a poorer π-acceptor ligand than CO,25 while achieving horizontal N2 adsorption is even more challenging. In principle, the horizontal N2 adsorption should be energetically ensured (Fig. 1A) to achieve the efficient formation of the *NCON intermediate via N2 coupling with CO, but unfortunately is much less competitive than CO adsorption. Clearly, this dilemma greatly restricts the feasibility of *NCON formation, leading to either CO poisoning or CO reduction to hydrocarbons during the electrochemical process. Therefore, manipulating the strong competitive CO adsorption with N2 at the catalytic site thereby guaranteeing the dominant N2 horizontal adsorption represents one of the key challenges for efficient urea electrosynthesis. Besides, as shown in Fig. 1A, even if the dominance of horizontal N2 adsorption is guaranteed, the competition between C–N coupling and N2 direct electroreduction to *N2H is the second grand challenge to obtain a preferential formation of the *NCON intermediate. Then, the production of urea via the protonation of the *NCON intermediate should be achieved at an affordable energy cost, thereby requiring high reactivity of the catalyst, which clearly denotes the third challenge. Noteworthily, the hydrogen evolution reaction (HER) is inevitable in almost all electrocatalytic processes, true also for urea electrosynthesis, which represents the fourth formidable challenge.
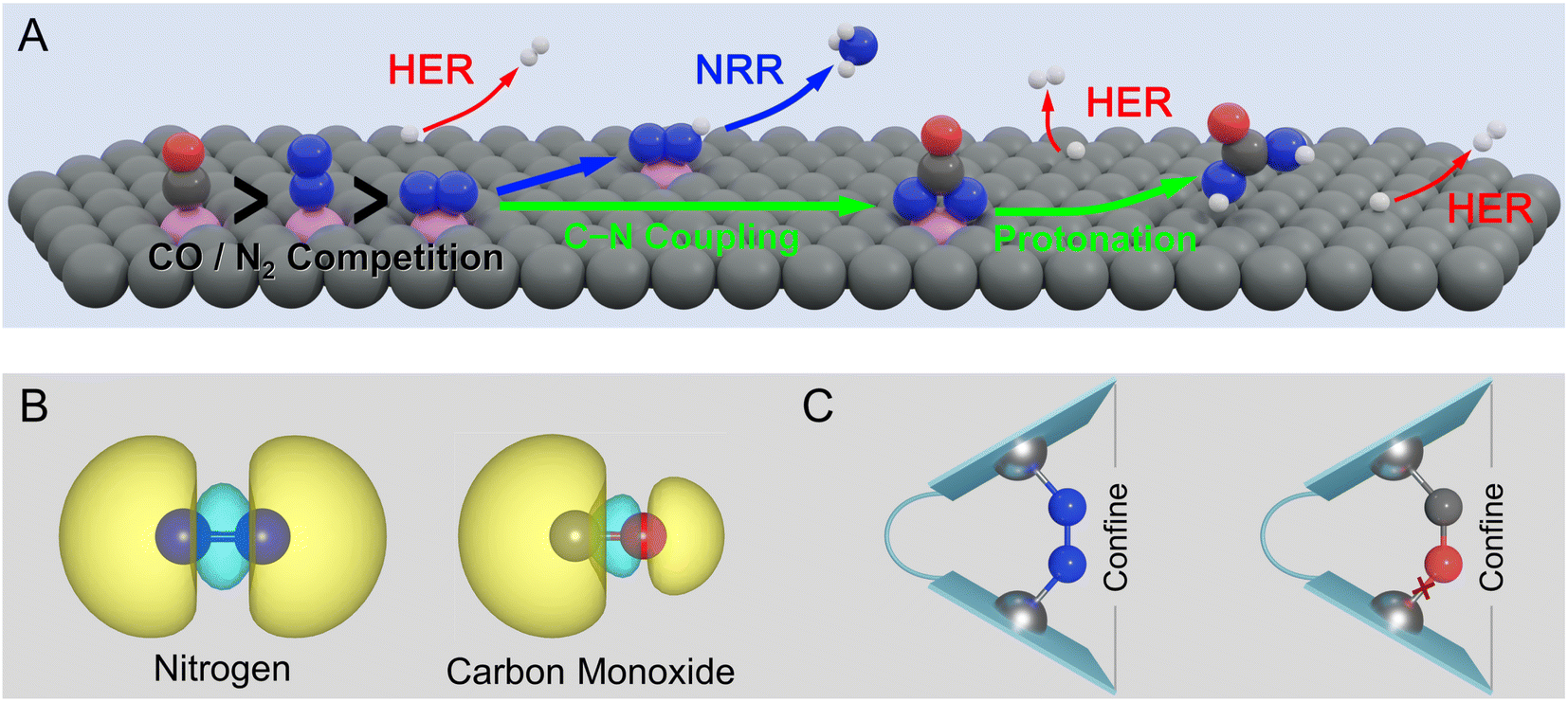 | ||
| Fig. 1 (A) Typical challenges to urea electrosynthesis by N2 and CO coupling from a view of the reaction mechanism. (B) HOMO electron density of N2 and CO. (C) Schematic illustration of N2/CO adsorption steering with the confined dual-site strategy. | ||
In this work, we proposed a strategy to properly address the aforementioned challenges by steering the competitive N2 and CO adsorption with a confined dual active site, which provided an elegant proposal to achieve efficient urea production from N2 and CO. As presented in Fig. 1C, the confined dual active site could stabilize non-polar N2 adsorption by forming one additional stable TM–N bond, whereas the polar CO molecule binding was hardly affected or even destabilized, thereby making horizontal N2 adsorption very competitive and eventually promoting the formation of the *NCON intermediate. A group of metals, mainly first-row transition metals, was chosen as active centers and anchored on nitrogen-doped graphene to construct our confined dual-site catalysts shown in Fig. 1C. Our simulations successfully predict the dual-site VN4 (DS-VN4) catalyst to be a very promising candidate to drive the electrochemical production of urea via the *NCON protonation with a low limiting potential of −0.50 V. Meanwhile, this DS-VN4 catalyst shows tremendous suppression of undesirable ammonia formation and hydrogen evolution, enabling the high reaction selectivity toward urea products. This work not only identified the spatially isolated dual site with the confinement effect for steering the competitive N2 and CO adsorption but also rationalized this theoretical strategy to fine-tune the strengths of chemical bonds, thus paving a promising route to obtain green urea electrosynthesis via rational catalyst design.
2. Results and discussion
2.1. Competitive N2 and CO adsorption on single and dual sites
To achieve an efficient urea electrochemical synthesis via N2 and CO coupling, the first priority is to ensure preferential N2 adsorption with horizontal configuration. However, as mentioned in Fig. 1, the polar CO molecule is more likely to cover the catalytic site than the non-polar N2 molecule based on the analysis from molecular orbital theory. To this end, we first chose ten transition metals (TM) as active sites (Ti, V, Cr, Mn, Fe, Co, Ni, Mo, Ru, and Rh) and embedded them in graphene with different coordination environments (i.e., TMN4, TMN3C1, TMN2C2, and TMN1C3) shown in Fig. S1.† Then, we calculated and compared their binding strengths with N2 and CO. As presented in Fig. 2 and the detailed energetic data in Table S1,† the N2 molecule energetically favors the one-sided vertical binding mode to the single active site, and the binding strength of CO is much stronger than that of the N2 molecule. The two exceptions, NiN4 and NiN3C1, with similar CO and N2 binding strengths are due to their physical adsorptions toward adsorbates. In general, our calculated binding strengths of CO and N2 on the single active site follow the vertical CO > vertical N2 > horizontal N2, which is consistent with the theoretical expectation shown in Fig. 1A. Therefore, the CO adsorption and subsequent reduction will be the dominant reaction route on the single-site catalyst if co-feeding CO with N2, thereby eliminating the N2 attacking and blocking the C–N coupling for urea synthesis.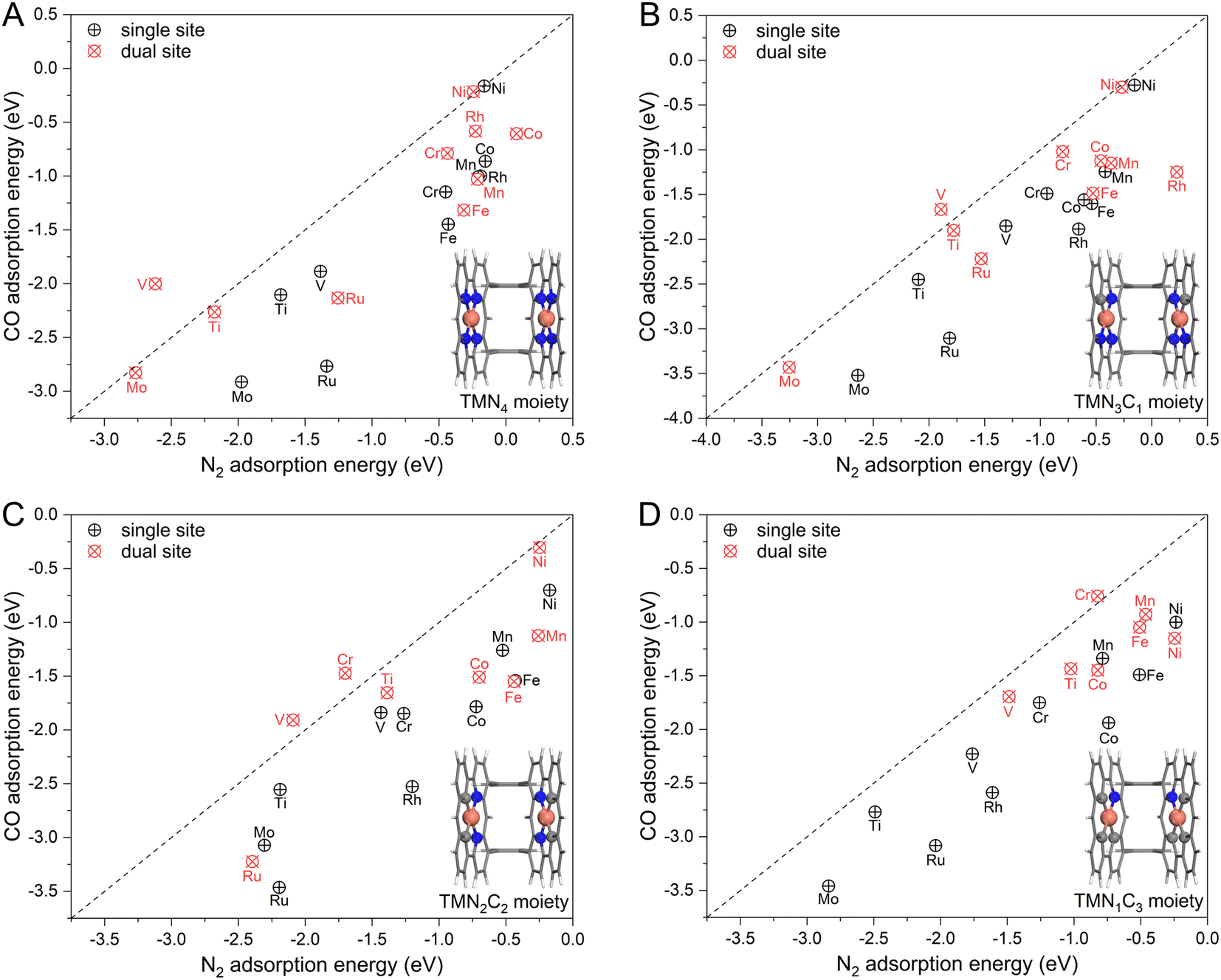 | ||
| Fig. 2 The calculated adsorption energy of N2 and CO molecules on the (A) TMN4 moiety, (B) TMN3C1 moiety, (C) TMN2C2 moiety, and (D) TMN1C3 moiety with single site and spatially isolated dual sites. The black dashed line represents the equivalent adsorption, and above the line is the N2-dominated adsorption region. | ||
Based on the proposed strategy in Fig. 1C, we spatially introduced a second active site with weak O binding strength but strong binding with N, namely the dual active site catalyst, to steer the competitive N2 and CO adsorption. Noteworthily, our recent work has successfully validated this strategy to enhance N2 adsorption and activation with the dual-site catalyst, making near-ambient conditions of ammonia synthesis possible.26 In our model catalyst shown in Fig. S2,† we sterically linked two localized single TM site catalysts by using two benzene rings. Noteworthily, this designed catalyst is very similar to the experimentally reported Pacman dinuclear porphyrins27–30 for O2 electroreduction27 and CO2 electroreduction.28 Thus, the adsorption of N2 and CO molecules was systematically examined on the designed dual-site catalysts. As presented in Fig. 2 and Table S2,† different from the cases of the single-site catalyst dominated by CO adsorption, a group of dual-site catalysts form the N2-dominated adsorption such as DS-VN4, DS-VN3C1, DS-VN2C2, DS-CrN2C2, and DS-CrN1C3. The para-configuration of the TMN2C2 moiety was also considered (Fig. S3†), which showed similar functionality and activity to the ortho-configuration in Fig. 2. Furthermore, we applied a Boltzmann function to evaluate the distribution of N2 and CO on the active sites. Our results in Fig. S4, Tables S3, and S4† clearly indicate the dominant population of N2 on the DS-VN4, DS-VN3C1, DS-VN2C2, DS-CrN2C2, and DS-CrN1C3 catalysts, which effectively suppresses the CO adsorption. The aforementioned results clearly reveal the capability of the designed dual-site catalyst in regulating the competitive N2 and CO adsorption, which lays a good basis for efficient C–N coupling.
2.2. Functionality of the confined dual site
Before moving forward to urea synthesis mechanism calculations on different confined dual-site catalysts, we further explored the origins behind these fine-tuned N2 and CO adsorptions. To this end, the spin-charge density maps were plotted to reveal the interactions of the adsorbates with the active sites, where the DS-VN4 catalyst was chosen as an example. As shown in Fig. 3A, the dual V site has an obviously large spin charge but gradually decreases as the adsorbates approach, indicating the involvement of spin electrons in V sites during the adsorption process. In principle, a more significant change in spin-charge represents a stronger bonding interaction. For the case of N2 adsorption, the V sites on both sides retain small and equal spin-charge density. For CO adsorption, the spin charge of the V site on the C atom side is negligible, while that on the O atom side is still obvious. Thus, based on the changes in the spin-charge density of the V site before and after adsorption, the bonding strength will follow the order of V–C > V–N > V–O. A similar phenomenon could be observed on the DS-VN3C1, DS-VN2C2, and DS-CrN1C3 catalysts as shown in Fig. S5.†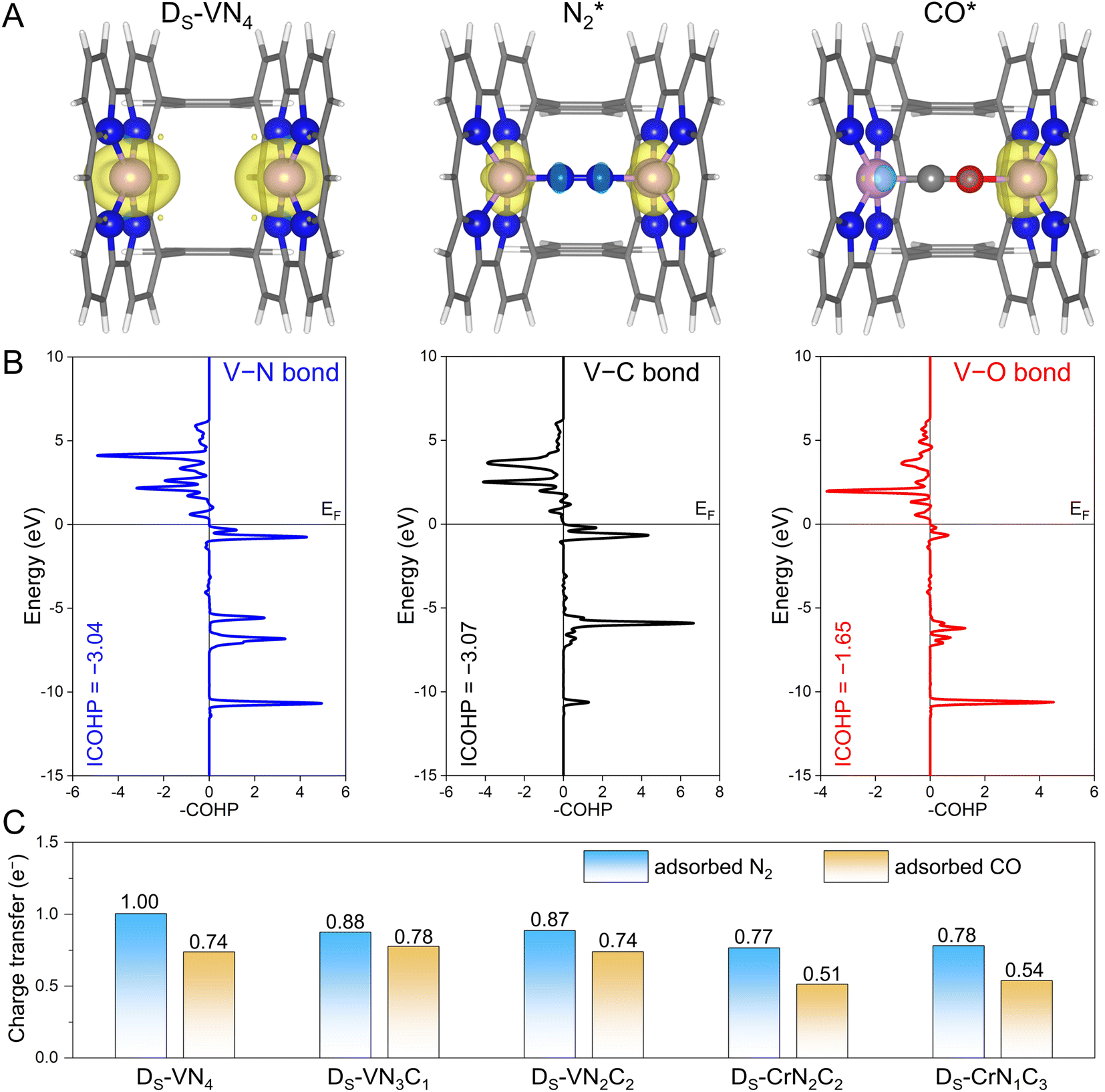 | ||
| Fig. 3 (A) The spin-charge density of pure Ds-VN4, N2 adsorption, and CO adsorption, where the isosurface value was set to be 0.009 e Å−3. (B) The calculated COHP between DS-VN4 and the N2 molecule as well as the CO molecule. The Fermi level (EF) was set to zero, and the bonding and antibonding orbitals were presented on the right and left, respectively. (C) The calculated charge transfer to adsorbed N2 and CO molecules. | ||
To further quantitatively evaluate the strength of chemical bonds, we performed the crystal orbital Hamilton population (COHP) analysis31 on the DS-VN4 catalyst. As shown in Fig. 3B, fewer bonding orbitals of V–O can be observed compared with V–N and V–C, showing a weak V–O interaction. Furthermore, the integrated COHP (ICOHP) values of V–N, V–C, and V–O bonds were calculated to be −3.04, −3.07, and −1.65, respectively. Typically, a more positive ICOHP value represents a weaker chemical bond. Thus, the strength of the chemical bonds at the DS-VN4 sites follows the order of V–C > V–N > V–O. The corresponding ICOHP values for the other four catalysts with N2-dominant adsorption are summarized in Table S5.† A general trend of bonding strength of TM–N > TM–O can be obtained on the designed dual-site candidates. Indeed, this trend is in line with our proposal in Fig. 1C that the sterically introduced second site can better enhance the binding strength of the N atom compared to the O atom, and eventually makes N2 adsorption more competitive than CO.
Meanwhile, the confined dual-site catalysts will transfer more electrons to the adsorbed N2 molecule via the additionally constructed stable TM–N bond shown in Fig. 3C. By contrast, no significant difference was found in the amount of charge transfer for the adsorbed N2 and CO molecules on the single-site catalysts shown in Fig. S6.† Clearly, the enhanced N2 adsorption and its dominance on the active site by the dual-site strategy are again supported by the electron transfer analysis. Note that the implementation of this strategy is based on the active site with strong binding to the N atom, i.e., guaranteeing a strong TM–N bond. We further examined it at the single TM site by calculating the binding energy of a single N atom using the N2 molecule as the energy reference. As listed in Table S6,† the core active sites of the five candidates with dominant N2 distribution all exhibit strong N binding, implying the effectiveness of the dual-site strategy for modulating competitive adsorption. As a summary, the aforementioned spin-charge density, COHP, and charge transfer analysis provide the rationale for the fine-tuning of N2 and CO adsorption using our proposed confined dual-site catalysts.
2.3. Feasibility of confined dual-sites for urea electrosynthesis
Since our proposed dual-site strategy was already able to properly address the challenge from the N2 and CO competitive adsorption, we further evaluated the capability of this strategy in tackling the competition between CO–N2 coupling and N2 electrochemical hydrogenation, where the five candidates with dominant N2 adsorption were chosen as model systems. At first, we examined the free energy changes of the coupling process via the Eley–Rideal mechanism (*N2 + CO(g) → *NCON) and the N2 electroreduction process (*N2 + H+ + e− → *N2H) for convenient comparison. As shown in Fig. 4A and S7,† negative reaction free energies can be observed for the coupling process (−0.49 eV for DS-VN4, −0.34 eV for DS-VN3C1, −0.43 eV for DS-VN2C2, −0.01 eV for DS-CrN2C2, and −0.15 eV for DS-CrN1C3) on the five catalysts, indicating the thermodynamic feasibility. In contrast, the protonation of the adsorbed N2 molecule to the *N2H intermediate needs to overcome the positive free energy. Thus, at 0 V potential, the C–N coupling is thermodynamically more favorable than N2 electroreduction on the five dual-site catalysts.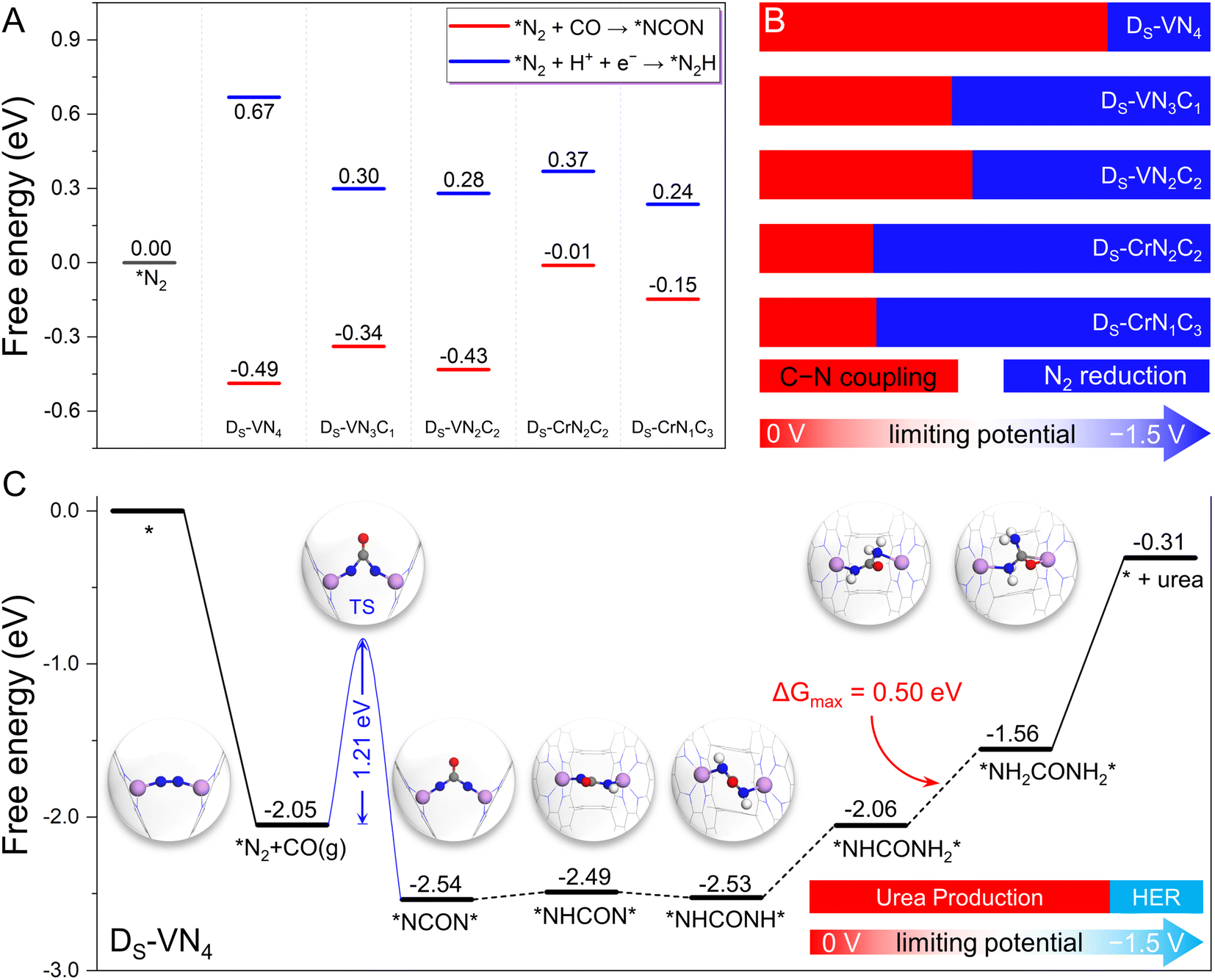 | ||
| Fig. 4 (A) The calculated free energy change of C–N coupling and N2 electroreduction at 0 V vs. RHE. (B) Reaction competition between C–N coupling and N2 electroreduction under the applied potential. (C) The calculated free energy diagram of urea synthesis by C–N coupling and reduction at 0 V vs. RHE on the designed DS-VN4 catalyst, as well as the reaction competition between urea production and H2 evolution under different applied potentials. | ||
However, the formation of the *N2H intermediate via N2 reduction would be electrochemically promoted by increasing the applied potential (U), while that of *NCON was less affected due to the noninvolvement of the proton-electron coupling step during the CO–N2 coupling. In other words, the free energy change of N2 protonation will become more negative at the high U, making N2 reduction more favorable than the C–N coupling at a certain U. Thus, the effect of U was further considered to explore this competitive process. As shown in Fig. 4A, there is a free energy gap of 1.16 eV between *N2H and *NCON intermediates on the DS-VN4 catalyst, which indicates that at least a U of −1.16 V is needed to make the formation of *N2H more competitive than *NCON. In principle, the DS-VN4 catalyst will preferentially form the *NCON intermediate at U < −1.16 V (Fig. 4B), which is named the potential tolerance limitation (PTL) in this work. Similarly, the PTL of DS-VN3C1, DS-VN2C2, DS-CrN2C2, and DS-CrN1C3 is calculated to be −0.64, −0.71, −0.38, and −0.39 eV, respectively. Therefore, the CO–N2 coupling process through the Eley–Rideal mechanism will be preferentially triggered unless the applied limiting potential exceeds the PTL (Fig. 4B). Besides, the possibility of adsorbed N2 dissociation into two isolated N atoms was also considered over these five candidates shown in Fig. S8.† Due to the significant energy requirements, N2 dissociation is thermodynamically very unfavorable to occur under electrochemical conditions on these model catalysts.
The above results clearly show that the five designed dual-site catalysts have the potential to achieve a high reaction selectivity for C–N coupling to form the *NCON intermediate, which could be converted into valuable urea by the subsequent electrochemical reduction process. Clearly, the DS-VN4 catalyst with the largest potential tolerance range in Fig. 4B is the most interesting candidate in our framework. Notably, the successful synthesis of a single VN4 site was recently reported experimentally,32 laying a good basis for the synthesis of the DS-VN4 catalyst.27,28 Therefore, we focus on the DS-VN4 catalyst to specifically explore the subsequent reaction pathway and catalytic activity for urea synthesis in the following section.
As presented in Fig. 4C, we plotted the free energy diagram of the most favorable reaction pathway for urea electrochemical production over the DS-VN4 catalyst, while the atomic structures of important intermediates were also provided. As mentioned above, the N2 molecule forms a horizontal adsorption mode on the dual site with two TM–N bonds, which is exothermic by 2.05 eV. Then, the formation of the *NCON intermediate from the C–N coupling occurs via the Eley–Rideal mechanism with a free energy change of −0.49 eV. Notably, a moderate reaction energy barrier of 1.21 eV is observed for this coupling process (Fig. S9†), which is lower than that of reported in-plane dual site catalysts (e.g., 1.62 eV for Co2@N6G and 1.35 eV for FeNi@N6G),21 indicating the kinetic feasibility of generating the *NCON intermediate. Afterward, the *NCON intermediate undergoes consecutive electrochemical protonation to produce the *NHCON, *NHCONH, *NHCONH2, *NH2CONH2 intermediates. The corresponding free energy changes are +0.05, −0.04, +0.47, and +0.50 eV, respectively. In this case, the final protonation process (*NHCONH2 + H+ + e− → *NH2CONH2) shows the largest uphill free energy of +0.50 eV, and is therefore the theoretical potential-determining step. Finally, the desorption of the *NH2CONH2 intermediate is endothermic by 1.25 eV. Theoretically, only a low limiting potential of −0.50 V is required to drive this reduction process toward urea synthesis, indicating the high activity of the designed DS-VN4 catalyst.
Due to the inevitable occurrence of the hydrogen evolution reaction (HER) in any electrochemical reactions, we further took it into consideration. Our calculated binding free energies of two H atoms on the Ds-VN4 catalyst (Fig. S10†) are only +0.22 eV, and much weaker than the chemisorption of the N2 molecule (−2.05 eV), making the binding of H atoms much less competitive than N2 molecules. Despite the electrochemical binding of H with the catalyst being enhanced with increasing applied potential, N2 adsorption will remain dominant on the DS-VN4 catalyst at UL < −1.135 V and properly suppress the HER due to the lack of active sites to bind H. Based on the above energetic analysis of the whole reaction pathways for urea synthesis, the DS-VN4 catalyst could effectively and selectively produce urea in the limiting potential range of −0.50–−1.135 V, which are able to suppress both the N2 reduction and the HER. We anticipate that our proposed strategy based on confined dual-site catalysts will shed light on the experimental catalyst design with high activity and selectivity toward urea production via CO and N2 coupling.
3. Conclusion
In summary, we proposed a theoretically feasible strategy to steer the competitive adsorption of reactants by constructing a catalyst with a spatially isolated dual-site. With the help of the spatial formation of a more stable TM–N bond than TM–O, the interaction of the N2 molecule with the catalytic dual-site can be enhanced, which guarantees the effective coupling of the pre-adsorbed N2 with CO and facilitates the formation of the *NCON intermediate as the key precursor for urea electrosynthesis. Based on our systematic calculations on a group of transition metal-based dual-site catalysts, DS-VN4, DS-VN3C1, DS-VN2C2, DS-CrN2C2, and DS-CrN1C3 were computationally found to exhibit dominant N2 distribution in a horizontal mode, effectively blocking CO adsorption. Furthermore, the formation of the *NCON intermediate on these screened catalysts is thermodynamically more favorable than the *N2H formation from electrochemical N2 hydrogenation. Among all the theoretically predicted promising candidates, DS-VN4 stands out as an efficient electrocatalyst for urea synthesis with high activity and selectivity. This study demonstrates the feasibility and functionality of the confined dual-site strategy in regulating the competitive adsorption of reactants to achieve efficient C–N coupling and urea production, which can bring important theoretical guidance for the design of experimental catalysts for practical and sustainable urea synthesis.4. Computational details
All the density functional theory (DFT) calculations with spin-polarization were performed using the Vienna Ab Initio Simulation Package (VASP) code.33 The revised Perdew–Burke–Ernzerhof (RPBE) functional was employed to describe the exchange–correlation interactions within the generalized gradient approximation.34,35 The electron–ion interactions were represented by the projector augmented wave (PAW) method.36 The kinetic energy cutoff of the plane wave was set to be 500 eV and the convergence criterion for the residual forces and total energies were set to be 0.03 eV Å−1 and 10−5 eV, respectively. The empirical correction in Grimme's method (DFT + D3) was adopted to describe the van der Waals interaction.37 The transition state with only one imaginary frequency was identified using the climbing image nudged elastic band (CI-NEB) method.38 Bader charge calculation was performed to analyze the charge population and charge transfer.39 Other computational details can be found in the ESI.†Data availability
Computational data supporting the findings can be found in the article and ESI,† and are available from the authors upon reasonable request.Author contributions
T. W. led and supervised this project; Z. C. performed all the DFT computations and data analysis; Y. H. L. contributed to the plot of Fig. 1; all authors discussed the results, and contributed to the writing of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFA0911900) and the National Natural Science Foundation of China (22273076); T. W. is thankful for the start-up packages from Westlake University and the Research Center for Industries of the Future (RCIF) at Westlake University for supporting this work. We thank Westlake University HPC Center for computation support. This work is dedicated to Prof. Haijun Jiao on the occasion of his 60th birthday.References
- J. Li, Y. Zhang, K. Kuruvinashetti and N. Kornienko, Nat. Rev. Chem., 2022, 6, 303–319 CrossRef CAS PubMed.
- C. Chen, N. He and S. Wang, Small Sci., 2021, 1, 2100070 CrossRef CAS.
- Z. Mei, Y. Zhou, W. Lv, S. Tong, X. Yang, L. Chen and N. Zhang, ACS Sustainable Chem. Eng., 2022, 10, 12477–12496 CrossRef CAS.
- Z. Chen, C. Liu, L. Sun and T. Wang, ACS Catal., 2022, 12, 8936–8975 CrossRef CAS.
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 CrossRef CAS PubMed.
- M. Yuan, J. Chen, Y. Xu, R. Liu, T. Zhao, J. Zhang, Z. Ren, Z. Liu, C. Streb, H. He, C. Yang, S. Zhang and G. Zhang, Energy Environ. Sci., 2021, 14, 6605–6615 RSC.
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Wang, S. Li, H. He and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 10910–10918 CrossRef CAS PubMed.
- X. Zhu, X. Zhou, Y. Jing and Y. Li, Nat. Commun., 2021, 12, 4080 CrossRef CAS PubMed.
- X. Liu, Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2022, 13, 5471 CrossRef CAS PubMed.
- L. Kong, D. Jiao, Z. Wang, Y. Liu, Y. Shang, L. Yin, Q. Cai and J. Zhao, Chem. Eng. J., 2023, 451, 138885 CrossRef CAS.
- J. Mukherjee, S. Paul, A. Adalder, S. Kapse, R. Thapa, S. Mandal, B. Ghorai, S. Sarkar and U. K. Ghorai, Adv. Funct. Mater., 2022, 32, 2200882 CrossRef CAS.
- M. Wang, L. Y. Chu, Z. Y. Li, A. M. Messinis, Y. Q. Ding, L. Hu and J. B. Ma, J. Phys. Chem. Lett., 2021, 12, 3490–3496 CrossRef CAS PubMed.
- C. Lv, L. Zhong, H. Liu, Z. Fang, C. Yan, M. Chen, Y. Kong, C. Lee, D. Liu, S. Li, J. Liu, L. Song, G. Chen, Q. Yan and G. Yu, Nat. Sustainable, 2021, 4, 868–876 CrossRef.
- D. Zhang, Y. Xue, X. Zheng, C. Zhang and Y. Li, Multi-heterointerfaces for selective and efficient urea production, Natl. Sci. Rev., 2023, 10, nwac209 CrossRef PubMed.
- Y. Feng, H. Yang, Y. Zhang, X. Huang, L. Li, T. Cheng and Q. Shao, Nano Lett., 2020, 20, 8282–8289 CrossRef CAS PubMed.
- C. Lv, C. Lee, L. Zhong, H. Liu, J. Liu, L. Yang, C. Yan, W. Yu, H. H. Hng, Z. Qi, L. Song, S. Li, K. P. Loh, Q. Yan and G. Yu, ACS Nano, 2022, 16, 8213–8222 CrossRef CAS PubMed.
- N. Meng, Y. Huang, Y. Liu, Y. Yu and B. Zhang, Cell Rep. Phys. Sci., 2021, 2, 100378 CrossRef CAS.
- H. Wang, Y. Jiang, S. Li, F. Gou, X. Liu, Y. Jiang, W. Luo, W. Shen, R. He and M. Li, Appl. Catal., B, 2022, 318, 121819 CrossRef CAS.
- X. Wei, X. Wen, Y. Liu, C. Chen, C. Xie, D. Wang, M. Qiu, N. He, P. Zhou, W. Chen, J. Cheng, H. Lin, J. Jia, X. Z. Fu and S. Wang, J. Am. Chem. Soc., 2022, 144, 11530–11535 CrossRef CAS PubMed.
- X. Zhang, X. Zhu, S. Bo, C. Chen, M. Qiu, X. Wei, N. He, C. Xie, W. Chen, J. Zheng, P. Chen, S. P. Jiang, Y. Li, Q. Liu and S. Wang, Nat. Commun., 2022, 13, 5337 CrossRef CAS PubMed.
- C. Zhu, M. Wang, C. Wen, M. Zhang, Y. Geng, G. Zhu and Z. Su, Adv. Sci., 2022, 9, e2105697 CrossRef PubMed.
- Y. Xiao, C. Shen, Z. Xiong, J. Li and W. Zhang, Mater. Today Phys., 2022, 26, 100726 CrossRef CAS.
- S. Dutta and S. K. Pati, ChemPhysChem, 2023, 24, e202200453 CrossRef CAS PubMed.
- M. Kaushik, A. Singh and M. Kumar, Eur. J. Chem., 2012, 3, 367–394 CrossRef CAS.
- E. Lu, B. E. Atkinson, A. J. Wooles, J. T. Boronski, L. R. Doyle, F. Tuna, J. D. Cryer, P. J. Cobb, I. J. Vitorica-Yrezabal and G. F. Whitehead, Nat. Chem., 2019, 11, 806–811 CrossRef CAS.
- T. Wang and F. Abild-Pedersen, Proc. Natl. Acad. Sci. U.S.A., 2021, 118, e2106527118 CrossRef CAS PubMed.
- Y. Liu, G. Zhou, Z. Zhang, H. Lei, Z. Yao, J. Li, J. Lin and R. Cao, Chem. Sci., 2020, 11, 87–96 RSC.
- A. Mohamed, Z. N. Zahran and Y. Naruta, Chem. Commun., 2015, 51, 16900–16903 RSC.
- Y. Shimazaki, T. Nagano, H. Takesue, B. H. Ye, F. Tani and Y. Naruta, Angew. Chem., Int. Ed., 2004, 43, 98–100 CrossRef PubMed.
- P. Lang and M. Schwalbe, Chem. – Eur. J., 2017, 23, 17398–17412 CrossRef CAS PubMed.
- R. Dronskowski and P. E. Blöchl, J. Phys. Chem., 1993, 97, 8617–8624 CrossRef CAS.
- L. Han, H. Cheng, W. Liu, H. Li, P. Ou, R. Lin, H.-T. Wang, C.-W. Pao, A. R. Head and C.-H. Wang, Nat. Mater., 2022, 21, 681–688 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04688e |
| This journal is © The Royal Society of Chemistry 2023 |