
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImplications of Ga promotion and metal–oxide interface from tailored PtGa propane dehydrogenation catalysts supported on carbon†
Enzo
Brack
 a,
Milivoj
Plodinec
ab,
Marc-Georg
Willinger‡
b and
Christophe
Copéret
*a
a,
Milivoj
Plodinec
ab,
Marc-Georg
Willinger‡
b and
Christophe
Copéret
*a
aDepartment of Chemistry and Applied Biosciences ETH Zurich, Vladimir Prelog Weg 2/10, CH-8093 Zurich, Switzerland. E-mail: ccoperet@ethz.ch
bScientific Center for Optical and Electron Microscopy (ScopeM) ETH Zurich, Otto-Stern-Weg 3, CH-8093 Zurich, Switzerland
First published on 25th October 2023
Abstract
Propane Dehydrogenation is a key technology, where Pt-based catalysts have widely been investigated in industry and academia, with development exploring the use of promoters (Sn, Zn, Ga, etc.) and additives (Na, K, Ca, Si, etc.) towards improved catalytic performances. Recent studies have focused on the role of Ga promotion: while computations suggest that Ga plays a key role in enhancing catalytic selectivity and stability of PtGa catalysts through Pt-site isolation as well as morphological changes, experimental evidence are lacking because of the use of oxide supports that prevent more detailed investigation. Here, we develop a methodology to generate Pt and PtGa nanoparticles with tailored interfaces on carbon supports by combining surface organometallic chemistry (SOMC) and specific thermolytic molecular precursors containing or not siloxide ligands. This approach enables the preparation of supported nanoparticles, exhibiting or not an oxide interface, suitable for state-of-the art electron microscopy and XANES characterization. We show that the introduction of Ga enables the formation of homogenously alloyed, amorphous PtGa nanoparticles, in sharp contrast to highly crystalline monometallic Pt nanoparticles. Furthermore, the presence of an oxide interface is shown to stabilize the formation of small particles, at the expense of propene selectivity loss (formation of cracking side-products, methane/ethene), explaining the use of additives such as Na, K and Ca in industrial catalysts.
Introduction
The non-oxidative propane dehydrogenation (PDH) reaction has become an intense field of research in both academia and industry, due to a steadily increasing demand for propene and the emergence of shale gas as a major chemical feedstock, resulting in a gap between offer and demand.1 In order to address the supply of propene, industrial processes catalysed by K-CrOx/Al2O3 (Lummus Catofin process, 1986), or Pt-based bimetallic systems such as K–Pt–Sn/Al2O3 (UOP Oleflex process, 1990),2 Pt–Zn–Sn/CaO–Al2O3 (Star process, 1999)3 and K–Pt–Ga/Si–Al2O3 (FDCh process, 2016)3 have been developed for this highly energy intensive process (ΔH298K0 = 124.3 kJ mol−1). These Pt-based systems are rather complex due to the use of various promoters and additives with the possible formation of alloys and interfacial sites, which have been proposed to be driving the catalytic performance. In academic laboratories, numerous supported Pt-based bi- and multi-metallic model systems have been investigated with the goal to understand the effect of promoters, focusing primarily on Sn,2 Zn,4 Ga,4 B,5,6 and In.7 However, most catalysts are quite ill-defined, partially due to the preparation methods, which often rely on (incipient) wetness impregnation – a technique, involving complex dissolution/precipitation processes and leading to the presence of numerous spectator species, rendering molecular-level understanding of promotional effects very challenging even when using advanced spectroscopic techniques or microscopy.In recent years, surface organometallic chemistry (SOMC) has emerged as a versatile tool to prepare silica-supported mono- and bi-metallic catalysts with tailored interfaces and compositions. This approach has been applied to study promoter effects in CO2 hydrogenation and propane dehydrogenation reactions.8–10 In that context, silica-supported PtGa,11 PtZn,12 as well as PtMn13 nanoparticles have been developed and shown to be particularly active and selective in PDH. In these Pt-based systems, the major role of promoters has been linked to the formation of alloyed nanoparticles, which are responsible for improved propene selectivity and increased stability against coking. Computational studies have inferred that the addition of Ga leads to Pt-site isolation and increases surface atom mobility, while the interaction with the oxide interface helps in stabilizing the PtGa nanoparticles against sintering.14 However, assessing these hypotheses and developing structure–activity relationships have been hindered due to experimental limitations, in particular charging effects in electron microscopy. Herein, we thus report the preparation and investigation of carbon-supported Pt and PtGa based catalysts, that unlock the use of state-of-the-art (scanning) transmission electron microscopy ((S)TEM), in order to probe the morphology of the supported nanoparticles and to evaluate the influence of the interface.
Towards this goal, we have developed a SOMC methodology on carbon15 using tailored molecular precursors to control the composition and the interface of the supported nanoparticles (Fig. 1A). Two different classes of organometallic precursors – amidinate/alkyl vs. siloxide – were used as a strategy to prepare a set of Pt and PtGa based catalysts with a tailored interface on carbon. On the one hand, molecular precursors such as [GaMes3]16 and platinum allyl-(N-N′-diisopropyl)acetamidinate ([PtAMD])17 are used for the preparation of carbon-supported Pt and PtGa nanoparticles, while thermolytic molecular siloxide precursors10 [Ga(OSi(OtBu3)3)3(THF)]18 and [Pt(OSi(OtBu3)3)2(COD)]19 are used to prepare the corresponding supported Pt and PtGa nanoparticles with an oxide interface (Fig. 1B). This approach based on molecular precursors containing or not a source of Si thus enables the study of the implications of Ga promotion and metal–oxide interfaces on PtGa PDH catalysts.
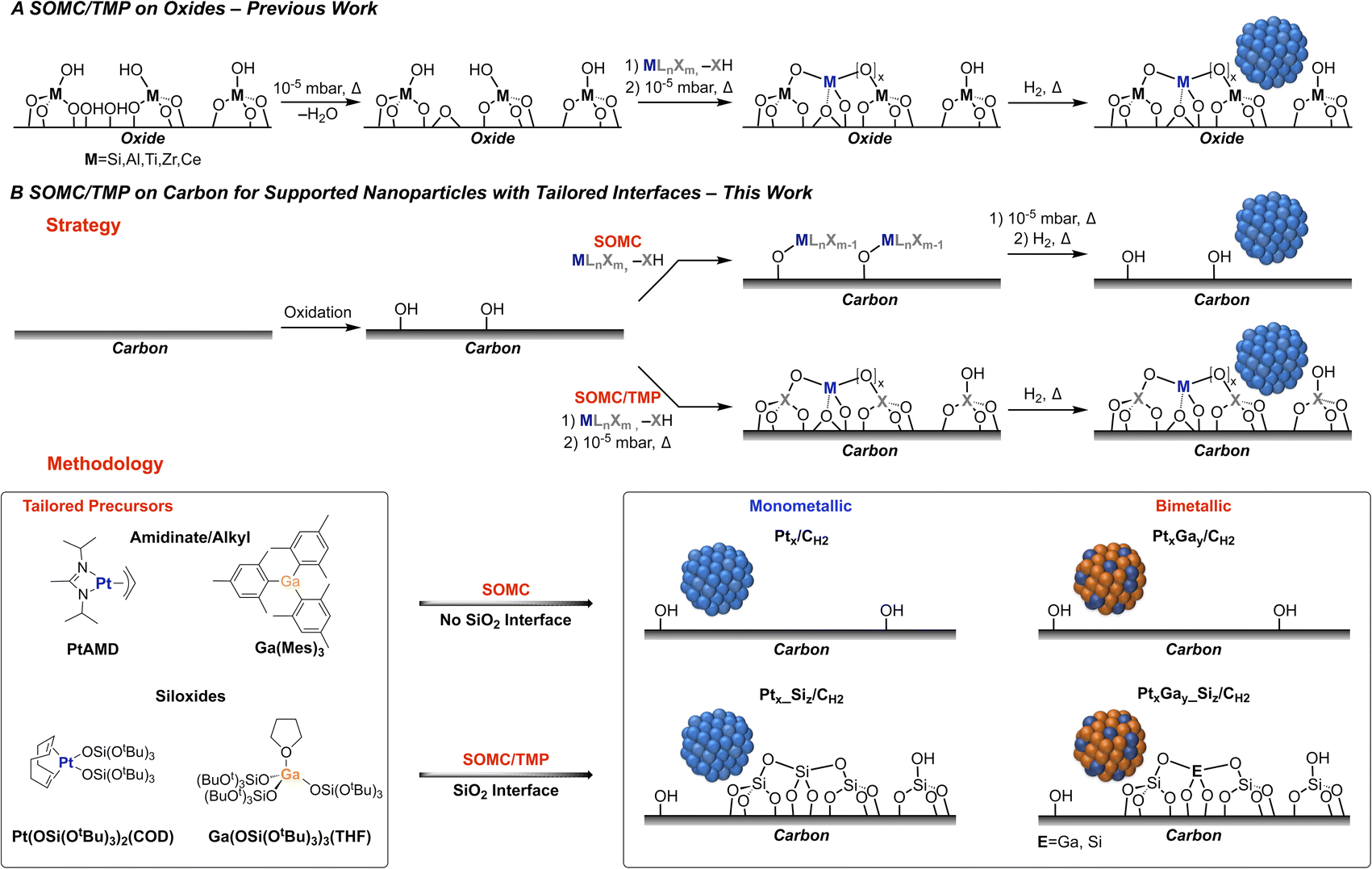 | ||
| Fig. 1 (A) Classical SOMC/TMP approach on oxide supports for the preparation of supported nanoparticles. (B) Strategy for SOMC/TMP on carbon for the generation of carbon-supported nanoparticles with tailored interfaces and methodology for the preparation of carbon-supported (bi)metallic nanoparticles with or without a silica interface, depending on the (non) thermolytic nature of the tailored precursor. | ||
Results and discussion
Surface organometallic chemistry (SOMC) approach on carbon
As a first step to enable grafting, we introduce oxygen-functionalities on carbon via a treatment of a mesoporous carbon support under oxidizing conditions, as previously reported.15,20 The treatment of mesoporous carbon in refluxing nitric acid for 12 hours leads to the incorporation of oxygen functional groups as evidenced from the O 1s XPS spectrum of the material after the treatment, revealing the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–OH functional groups (Fig. S1 and S2†). The amount of protic functional groups (R–OH) is ca. 1.2 OH nm−2 (0.17 mmolOH g−1), as determined by titration with [Mg(Bn)2(THF)2]; a value significantly lower than found for the corresponding carbon black support (0.41 mmol g−1, 3.4 OH nm−2).15 The difference in OH density most likely results from the highly graphitic structure of mesoporous carbon, rendering it more robust against oxidative conditions. Upon nitric acid treatment, the specific surface area of the carbon support slightly increases from 68 to 83 m2 g−1, as evidenced from BET analysis (Fig. S3†), likely as a result of etching.
O, C–OH functional groups (Fig. S1 and S2†). The amount of protic functional groups (R–OH) is ca. 1.2 OH nm−2 (0.17 mmolOH g−1), as determined by titration with [Mg(Bn)2(THF)2]; a value significantly lower than found for the corresponding carbon black support (0.41 mmol g−1, 3.4 OH nm−2).15 The difference in OH density most likely results from the highly graphitic structure of mesoporous carbon, rendering it more robust against oxidative conditions. Upon nitric acid treatment, the specific surface area of the carbon support slightly increases from 68 to 83 m2 g−1, as evidenced from BET analysis (Fig. S3†), likely as a result of etching.
Next, the functionalized carbon is used as a platform for SOMC, using two specific classes of molecular Pt and Ga precursors, with the aim to prepare two sets of carbon-supported Pt and PtGa PDH catalysts (Fig. 1B). First of all, isolated surface metal sites are obtained via grafting of a Ga or Pt precursor, followed by a thermal treatment under reduced pressure (Fig. 1B). Depending on the tailored precursor (siloxide or alkyl/amidinate), two sets (with and without a silica interface) of dispersed metal sites are prepared and denoted Ptx(_Siz)/Cgrafted, where “x” and “z” indicate the measured weight loadings of the corresponding element from elemental analysis (EA) and the subscript “grafted” designates the material after grafting and thermal treatment under reduced pressure. Note, that the formation of isolated metal sites after the grafting step is confirmed via high-resolution high-angle annular dark-field (HAADF)-STEM imaging (Pt0.68_Si0.1/Cgrafted) (Fig. S13†). From these highly dispersed metal sites in Ptx(_Siz)/Cgrafted, supported nanoparticles are formed via exposure to H2 at 600 °C resulting in two monometallic materials: Pt0.68_Si0.1/CH2 and Pt0.60/CH2 (Fig. 1B). The corresponding bimetallic materials are prepared and coined PtxGay(_Siz)/CH2, where “y” additionally denotes the obtained Ga weight loading from EA. In order to prepare the bimetallic PtxGay(_Siz)/CH2 materials, dispersed Ga surface sites are first generated, followed by the introduction of Pt with a matching tailored precursor (siloxide or amidinate/alkyl) and finally a H2 treatment to give Pt0.26Ga0.10/CH2 and Pt0.89Ga1.00_Si1.64/CH2. In order to ensure a more accurate comparison of the catalytic performances of the two sets of materials, an additional low-loading Pt0.15Ga0.10_Si0.31/CH2 material is prepared via sub-stoichiometric grafting. All metal- and silicon-loadings of the catalysts correspond to the EA results of the materials after H2 treatment and are summarized in Table 1.
| Sample | Pt [w%] | Ga [w%] | Si [w%] | Pt [nm−2] | Ga [nm−2] | Si [nm−2] |
|---|---|---|---|---|---|---|
| Pt0.68_Si0.1/CH2 | 0.68 | n.d. | 0.1≤ | 0.25 | n.d. | 0.3≤ |
| Pt0.60/CH2 | 0.60 | n.d. | 0.1≤ | 0.22 | n.d. | 0.3≤ |
| Pt0.89Ga1.00_Si1.64/CH2 | 0.89 | 1.00 | 1.64 | 0.33 | 1.04 | 4.2 |
| Pt0.15Ga0.10_Si0.31/CH2 | 0.15 | 0.10 | 0.31 | 0.06 | 0.10 | 0.8 |
| Pt0.26Ga0.10/CH2 | 0.26 | 0.10 | 0.1≤ | 0.10 | 0.10 | 0.3≤ |
EA shows that the bimetallic PtxGay_Siz/CH2 catalysts prepared using the Ga and Pt siloxide TMPs do indeed contain Si (Table 1), while the other materials do not contain any detectable Si, thus confirming the presence/absence of silicone. For Pt0.68_Si0.1/CH2, the amount of Si is below detection limit (≤0.1 w%), due to the low amount of siloxide precursor decomposed in the preparation (grafting via protonolysis of the ligand, Fig. 1B). For the monometallic Ptx(_Siz)/CH2 catalysts – Pt0.68(_Si0.1)/CH2 and Pt0.60/CH2 – very similar Pt weight loadings of 0.68 w% (0.25 Pt nm−2) and 0.60 w% (0.22 Pt nm−2) are obtained. On the other hand, the Pt and Ga weight loadings of the prepared bimetallic PtxGay(_Siz)/CH2 catalysts vary to a greater extent. The Pt0.89Ga1.00_Si1.64/CH2 and Pt0.26Ga0.10/CH2 catalysts differ significantly in terms of Ga weight loadings, with 1.00 w% Ga (1.04 nm−2) vs. 0.10 w% Ga (0.10 nm−2), depending on the Ga molecular precursor used. The two catalysts also vary in terms of Pt content with 0.89 w% (0.33 Pt nm−2) and 0.26 w% (0.10 Pt nm−2), respectively. The higher Pt loading of Pt0.89Ga1.00_Si1.64/CH2 can be explained by the formation of an in situ formed silicate interface from the thermolysis of the Ga siloxide TMP ([Ga(OSi(OtBu3)3)3(THF)]), whose functional groups can further be exploited for the grafting of the Pt precursor in the second grafting step. This is in line with a higher Pt loading for Pt0.89Ga1.00_Si1.64/CH2 (0.89 w% Pt, 0.33 nm−2) compared to the monometallic Pt0.68_Si0.1/CH2 catalyst (0.68 w% Pt, 0.25 Pt nm−2) (Fig. 1B and Table 1). In the absence of silicon, the Pt content of the bimetallic Pt0.26Ga0.10/CH2 (0.26 w% Pt, 0.10 Pt nm−2) catalyst is lower with respect to the monometallic Pt0.60/CH2 (0.60 w% Pt, Pt 0.22 nm−2), as a result of the partial consumption of functional groups from the grafting of the Ga alkyl precursor ([Ga(Mes)3]) and the absence of oxide formation. In order to assess the effect of the metal–oxide interface on the catalytic performance, a low-loading Pt0.15Ga0.10_Si0.31/CH2 catalyst exhibiting a silicate interface with similar loadings (0.10 Ga nm−2, 0.06 Pt nm−2) compared to the purely carbon-supported Pt0.26Ga0.10/CH2 was prepared via sub-stoichiometric grafting, with respect to the titrated protic R–OH functional groups.
Catalytic propane dehydrogenation (PDH)
We next evaluate the performance of the set of prepared catalysts – Pt0.68_Si0.1/CH2, Pt0.60/CH2, Pt0.89Ga1.00_Si1.64/CH2, Pt0.26Ga0.10/CH2 and Pt0.15Ga0.10_Si0.31/CH2 – in PDH under flow conditions (50 mL min−1, 550 °C, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 C3H8:Ar) as shown in Fig. 2 and summarized in Table 2. As a consequence of their different Pt loading, the catalysts vary in terms of activity. Thus, the amount of catalyst is adjusted for each run, in order to be in a similar conversion range, while still being far from equilibrium conversion levels (∼40% conversion at 550 °C). From the catalytic data, it can be seen, that the bimetallic PtxGay(_Siz)/CH2 catalysts, display relatively good activity (8–3% conversion) and stability over the course of the reaction (>35 hours on stream), with a much-increased stability compared to the monometallic Ptx(_Siz)/CH2 catalysts, as analogously observed for the silica-supported counterparts (Fig. 2).11 After reaching a stable initial state after 1 hour on stream, the deactivation behavior of the bimetallic catalysts (Pt0.89Ga1.00_Si1.64/CH2, kD = 0.013 h−1; Pt0.26Ga0.10/CH2, kD = 0.015 h−1) slightly deviate from one another while both still displaying a slower deactivation compared to the silica-supported counterpart PtGa/SiO2 (kD = 0.04 h−1).11 The low-loading Pt0.15Ga0.10_Si0.31/CH2 showed a deactivation constant of kD = 0.015 h−1, thus lower compared to the one of Pt0.89Ga1.00_Si1.64/CH2.
:4 C3H8:Ar) as shown in Fig. 2 and summarized in Table 2. As a consequence of their different Pt loading, the catalysts vary in terms of activity. Thus, the amount of catalyst is adjusted for each run, in order to be in a similar conversion range, while still being far from equilibrium conversion levels (∼40% conversion at 550 °C). From the catalytic data, it can be seen, that the bimetallic PtxGay(_Siz)/CH2 catalysts, display relatively good activity (8–3% conversion) and stability over the course of the reaction (>35 hours on stream), with a much-increased stability compared to the monometallic Ptx(_Siz)/CH2 catalysts, as analogously observed for the silica-supported counterparts (Fig. 2).11 After reaching a stable initial state after 1 hour on stream, the deactivation behavior of the bimetallic catalysts (Pt0.89Ga1.00_Si1.64/CH2, kD = 0.013 h−1; Pt0.26Ga0.10/CH2, kD = 0.015 h−1) slightly deviate from one another while both still displaying a slower deactivation compared to the silica-supported counterpart PtGa/SiO2 (kD = 0.04 h−1).11 The low-loading Pt0.15Ga0.10_Si0.31/CH2 showed a deactivation constant of kD = 0.015 h−1, thus lower compared to the one of Pt0.89Ga1.00_Si1.64/CH2.
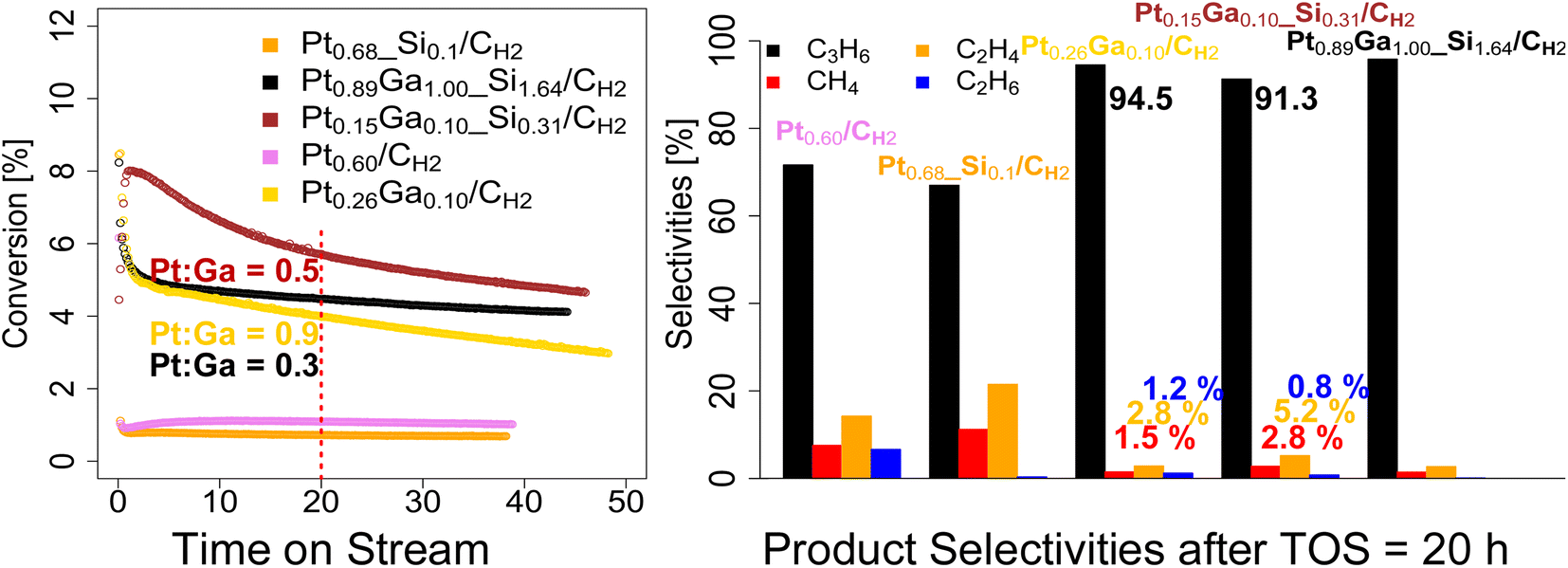 | ||
| Fig. 2 (Left) conversion vs. Time on Stream (TOS) and (right) product selectivities after 20 hours on stream of Pt0.68_Si0.10/CH2, Pt0.60/CH2, Pt0.89Ga1.00_Si1.64/CH2, Pt0.26Ga0.10/CH2 and Pt0.15Ga0.10_Si0.31/CH2 in PDH under flow conditions (50 mL min−1, 1:4 C3H8:Ar, 550 °C). | ||
:4 C3H8:Ar, 550 °C)
| Sample | Time [h] | Conversion [%] | Selectivity [%] | WHSV [gC3H8/(gPt h)] | Initial productivity [gC3H6/(gPt h)] | Carbon Balance | k D [h−1] |
|---|---|---|---|---|---|---|---|
| Pt0.68_Si0.1/CH2 | 0.03 | 8 | 89 | 2536 | 181 | >99% | 0.07 |
| 35 | 1 | 65 | |||||
| Pt0.60/CH2 | 0.03 | 6 | 81 | 1434 | 68 | >99% | 0.05 |
| 35 | 1 | 70 | |||||
| Pt0.89Ga1.00_Si1.64/CH2 | 1 | 8 | 98 | 2315 | 179 | >98% | 0.013 |
| 35 | 4 | 95 | |||||
| Pt0.26Ga0.10/CH2 | 1 | 6 | 99 | 3467 | 486 | >99% | 0.015 |
| 35 | 3 | 94 | |||||
| PtGa/SiO2 (ref. 11) | 0.3 | 32 | >99 | 2243 | 721 | >95% | 0.04 |
| 20 | 17 | >99 | |||||
| Pt0.15Ga0.10_Si0.31/CH2 | 1 | 9 | 95 | 7202 | 903 | >99% | 0.015 |
| 35 | 5 | 90 |
This suggests, that the difference in deactivation behavior between Pt0.89Ga1.00_Si1.64/CH2 and Pt0.26Ga0.10/CH2 could be related to the higher Pt to Ga ratio (0.3 vs. 0.5) of the Pt0.26Ga0.10/CH2 catalyst, rather than an effect caused by the presence of an oxide interface, as recently suggested in PtGa based dehydrogenation of n-heptane.21 Overall, when comparing the initial propene productivity of the silicate decorated Pt0.15Ga0.10_Si0.31/CH2 (904 gC3H6/(gPt h); 4192 molC3H6/(molPt h)) to the purely carbon-supported Pt0.26Ga0.10/CH2 (486 gC3H6/(gPt h); 2254 molC3H6/(molPt h)) catalyst, an enhanced initial propene productivity is observed, likely as a result of the presence of an oxide interface (Table 2 and Fig. S4†). From comparison of the product selectivities after 20 hours on stream, a lower selectivity towards propene along with an increased formation of cracking products (Fig. 2), ethene and methane, is observed for Pt0.26Ga0.10/CH2 compared to the low-loading Pt0.15Ga0.10_Si0.31/CH2 (where an oxide interface is present, Fig. S7†). The shift in selectivity can be attributed to the presence of a metal–oxide interface, since upon increasing the Pt to Ga ratio, the formation of cracking products is enhanced, as evidenced from the comparison of the product selectivities of Pt0.89Ga1.00_Si1.64/CH2 and Pt0.15Ga0.10_Si0.31/CH2 and Pt0.68_Si0.1/CH2 (Fig. 2). Similarly, comparison of the product selectivities of the monometallic Pt0.68_Si0.1/CH2 and Pt0.60/CH2 catalysts, consistently shows a shift in selectivity towards ethene and methane upon presence of an oxide interface (Fig. 2 and S5†). Although typically regarded as an innocent support, these findings demonstrate, that the introduction of a silicate interface enhances the propene productivity, at the expense of promoted cracking activity, as observed from a selectivity shift towards cracking products. This shift in selectivity is likely related to the introduction of acidity from the deposited silicate interface, possibly explaining the use of Na, K and Ca in industrial catalyst formulations,2,3 presumably as a strategy to remove the acidic properties of the oxide support. Additionally, as previously reported, Ga is known to form isolated single-sites on SiO2.18 Therefore, the migration of Ga during reaction conditions from Pt to the silicate interface, might additionally favor a decrease in propene selectivity of Pt0.15Ga0.10_Si0.31/CH2 compared to Pt0.26Ga0.10/CH2.
Electronic structure and morphology of the Ptx(_SiZ)/C and PtxGay(_Siz)/C catalysts
In order to gain further insights on the electronic structure, the morphology, the composition and reasons behind catalytic performances, the set of catalysts is next studied via (in situ) XANES and high-resolution STEM measurements. Ex situ XANES of the silicate decorated materials after grafting (Pt0.68II_Si0.1/Cgrafted and Pt0.89IIGa1.00III_Si1.64/Cgrafted) as well as after treatment under a flow of H2 (Pt0.68II_Si0.1/CH2 and Pt0.89IIGa1.00III_Si1.64/CH2) (Fig. S8–S10†) are found to be consistent with previously reported findings on the silica-supported counterpart PtGa/SiO2.11 Prior to reduction, the Pt L3-Edge (Fig. S8†) and Ga K-edge (Fig. S10†) energies of Pt0.68II_Si0.1/Cgrafted and Pt0.89IIGa1.00III_Si1.64/Cgrafted are identical with their silica-supported counterpart at 11566 eV and 10374 eV, respectively, confirming the preservation of the oxidation states (PtII, GaIII) of the molecular precursors upon grafting and formation of dispersed sites. The same is observed for the catalysts in the absence of an oxide interface. After reduction, the white line intensity decreased along with a shift of the edge energy from 11566 eV to 11565 eV, consistent with a reduction of Pt from Pt+II to Pt0. The decrease in white line intensity and energy shift at the Pt L3-Edge are attributed to an electron transfer from a metallic phase of Ga to the Pt 5d band, leading to a shift in the central transition of the Pt 5d valence band with respect to the Fermi energy level, as observed for the silica-supported systems, thus indicating alloying of the two metals within the nanoparticles.11 The same can be observed from in situ XANES measurements at the Pt L3-Edge of the grafted Pt containing catalysts under a flow of H2 (Fig. 3, S11 and S12†). Notably, the reduction of Pt occurs at lower temperatures in the case of the bimetallic PtGa catalysts compared to the monometallic Pt ones. From in situ XANES measurements at the Ga K-Edge of the bimetallic Pt0.89IIGa1.00III_Si1.64/Cgrafted and Pt0.26IIGa0.10III/Cgrafted catalysts under a flow of H2 (Fig. 3A), the reduction of Ga is observed from a shift of the edge energy to lower energy. Combined with the information gained from in situ XANES measurements at the Pt L3-Edge, it appears that the Pt is reduced at a lower temperature, while Ga being gradually reduced and intercalated into the Pt nanoparticles to form alloyed PtxGay(_Siz)/CH2 nanoparticles, as previously proposed in a computational study on PtGa/SiO2.14 Unfortunately, it is not possible to carry out analysis of the extended X-ray absorption fine structure (EXAFS) due to low weight loadings of the catalysts and consequential poor data quality of the extended fine structure, thus hampering EXAFS fitting. In order to obtain more insights on the alloying of the two metals within the prepared catalysts, they are studied via electron microscopy. The formation of Pt and PtGa nanoparticles in the case of the monometallic Ptx(_Siz)/CH2 (Fig. S16 and S21†) and bimetallic PtxGay(_Siz)/CH2 catalysts (Fig. 3B and S25†) was confirmed from STEM imaging and EDX elemental mapping. Quantitative analysis of the energy dispersive X-ray spectra of Pt0.68_Si0.1/CH2 (Table S1†) and Pt0.89Ga1.00_Si1.64/CH2 catalysts (Table S3†), indicate the presence of Si and confirm the EA results, validating the proposed formation of a silicate interface as a result of the decomposition of the siloxide ligands of the TMPs. The observed silicon most likely deposits in the form of SiO2 domains in close spatial proximity to the nanoparticles, as indicated by the overlap of Si and O signals in the EDX maps (Fig. S16, S25 and S28†). In sharp contrast, no Si was detected (as expected) from the EDX spectra of Pt0.60/CH2 (Tables S2 and S5†) and Pt0.26Ga0.10/CH2 (Fig. 3B). These findings, strongly support the hypothesis that the observed Si domains in the case of Pt0.68_Si0.1/CH2 and Pt0.89Ga1.00_Si1.64/CH2 are a result of the thermolytic decomposition of the siloxide containing ligands present in the TMP used in the grafting step(s). Notably, the absence of a silicate interface in Pt0.26Ga0.10/CH2 allows for the acquisition of a STEM-EDX map of a single nanoparticle. From the spatial superimposition of Ga L and Pt M X-ray peak signals (Fig. 3B) as well as from additional EDX maps (Fig. S25 and S31†), a homogeneously distributed alloy formation of the two metals within the particles is observed. Although, EXAFS fitting is not possible, the spatial correlation of the Ga L and Pt M signals from STEM-EDX mapping, combined with the insights gained from (in situ) XANES measurements are consistent with alloying between Pt and Ga. In order to get further insights on the implications of Ga promotion on the morphology and composition of the prepared catalysts, high-resolution STEM measurements are performed next (Fig. 4).
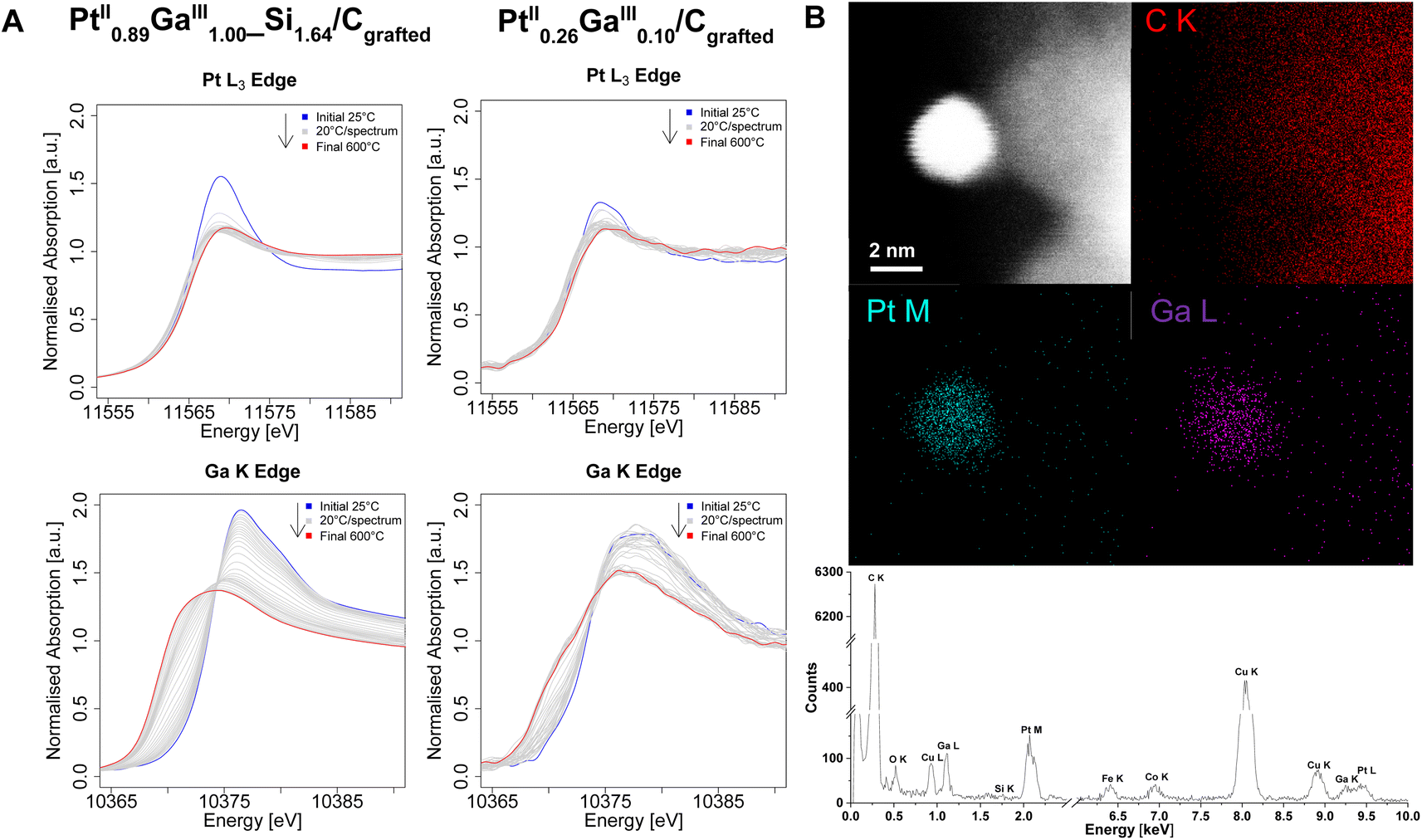 | ||
| Fig. 3 (A) In situ Pt L3 and Ga K-Edge X-ray Absorption Near-Edge Structure (XANES) spectra are shown for Pt0.89IIGa1.00III_Si1.64/Cgrafted (left) and Pt0.26IIGa0.10III/Cgrafted (right), acquired under a flow of 1 barg of H2 (10 mL min−1) over a temperature range of 25 °C to 600 °C. (B) HAADF-STEM image of a single-nanoparticle, corresponding Energy Dispersive X-ray (EDX) maps of Ga L, Pt M, C K and EDX spectrum of Pt0.26IIGa0.10III/Cgrafted. These maps reveal the spatial correlation between Pt and Ga, indicating homogeneous alloying of the two metals within the nanoparticle. The Cu and Fe signals in the EDX spectrum are associated with the polepiece of the microscope, while the Cu signals are related to the TEM grid. | ||
 | ||
| Fig. 4 Particle size distribution of (A) Pt0.68_Si0.1/CH2 and (B) Pt0.89Ga1.00_Si1.64/CH2. Representative high-resolution HAADF-STEM images of (C) Pt0.68_Si0.1/CH2 and (D) Pt0.89Ga1.00_Si1.64/CH2. Enlarged view of a single (E) Pt and (F) PtGa nanoparticle (obtained from red square regions in (C) and (D)). Insets in (E) and (F) show the fast Fourier transforms of a crystalline Pt nanoparticle viewed along the [110] zone-axis and an amorphous, alloyed PtGa nanoparticle, respectively. | ||
From comparison of the particle size distributions of the prepared catalysts, the formation of slightly smaller monometallic Pt nanoparticles compared to the silica-supported counterpart is observed,22 with a mean size of 1.5(6) nm for Pt0.68_Si0.1/CH2 (Fig. 4A) and 1.5(9) nm for Pt0.60/CH2 (Fig. S19†). In contrast, the bimetallic nanoparticles of Pt0.26Ga0.10/CH2 and Pt0.89Ga1.00_Si1.64/CH2 exhibit a mean particle size of 1.4(8) nm (Fig. S29†) and 1.2(5) nm (Fig. 4B) respectively, thus slightly larger than for its SiO2-700-supported counterpart, with a mean of 1.0(2) nm.11 These results confirm the formation of uniformly distributed nanoparticles in analogy to the silica-supported catalysts, while indicating the role of the oxide interface in the stabilisation of the nanoparticles during particle growth, as evidenced by the formation of smaller nanoparticles with a narrower size distribution upon increasing the Si content of the interface. Investigation of the spent catalysts show no apparent sintering effects, remaining similarly uniformly distributed supported nanoparticles after exposure to reaction conditions (Fig. S17, S22, S26 and S32†).
High-resolution HAADF-STEM images confirm the formation of crystalline Pt nanoparticles in Pt0.68_Si0.1/CH2 (Fig. 4C) and Pt0.60/CH2 (Fig. S20†). Furthermore, images of a single Pt nanoparticle (Fig. 4C and E) and its corresponding fast-Fourier transform (FFT) (inset in Fig. 4E) reveal the atomic structure of a fcc structured Pt nanoparticle viewed along the [110] zone-axis. Note, that the graphitic structure of the carbon support is preserved after the nitric acid treatment and SOMC/TMP approach, as evidenced especially from high-resolution annular bright-field STEM (ABF-STEM) images (Fig. S15†), showing the preservation of the graphitic layers. In sharp contrast, bimetallic PtGa/C nanoparticles are amorphous (Fig. 4D and F), according to the high-resolution HAADF-STEM measurements which were validated by 4D STEM. While the Pt catalysts consist of highly crystalline nanoparticles (Fig. S33 and S34†), only convergence-beam electron diffraction (CBED) disks for the graphitic carbon are observed (Fig. S35 and S36†) for the PtGa catalysts. The amorphous nature of the PtGa nanoparticles most likely arises from the intercalation of Ga into Pt nanoparticles during H2 treatment, forming a homogeneous, amorphous, alloy according to in situ XANES and STEM-EDX measurements, consistent with the proposed structure based on our earlier computational studies.14 The homogeneous distribution of Ga and Pt within the PtGa nanoparticles supports the proposed role of Ga promotion via stabilizing the nanoparticles against coke formation through Pt-site isolation.14,23 Notably, the PtGa nanoparticles with high Ga contents are suspected to be in liquid-like state under reaction conditions according to the PtGa phase diagram and as exploited in supported catalytically active liquid metal solution (SCALMS) propane dehydrogenation catalysts.24,25
Conclusion
The transposition of SOMC from classical oxide supports to carbon has enabled the preparation of tailored catalysts suitable for detailed structural analysis, designed to probe the spatial distribution of elements and the morphology of nanoparticles assessed by electron microscopy. STEM-EDX studies confirmed the preparation of a set of catalysts exhibiting or not a siloxide interface in proximity to PtGa nanoparticles depending on the chosen tailored molecular precursors. In particular, a combination of XANES and STEM-EDX confirm a homogenous alloying of Pt and Ga, and high-resolution HAADF-STEM images show the amorphous nature of PtGa nanoparticles, sharply contrasting the formation of highly crystalline monometallic Pt nanoparticles on carbon prepared according to the same approach. Thus, Ga is shown to form amorphous, alloyed PtGa nanoparticles and thereby to enable Pt-site isolation and superior catalytic performance in PDH, as proposed in earlier computational studies on the silica-supported counterpart.14 Furthermore, the presence of a SiO2 interface is shown to enhance the overall propene productivity, but also promotes the formation of cracking products, resulting in loss of propene selectivity. This is consistent with the acidic properties of oxide supports and the use of additional additives such as Na/K/Ca in industrial catalyst formulations.2,3 Additionally, while not necessary for the stabilization of the nanoparticles against sintering, the presence of an oxide interface helps to form smaller nanoparticles. Overall, this strategy based on the development of catalysts, composed of nanoparticles with defined composition and tailored interfaces on carbon, offers a promising platform for the investigation of promotional and support effects in thermal catalysis, and can be readily extended to electrochemical applications of multi-metallic nanoparticles dispersed on conductive supports such as carbon. We are currently exploring these avenues.Data availability
The data supporting the findings of this study is openly accessible on Zenodo at https://doi.org/10.5281/zenodo.8402733.Author contributions
E. B. and C. C. conceptualized the study and were responsible for the methodology. E. B. and M. P. undertook the investigations. C. C. was responsible for the funding acquisition. C. C. and M. G. W. were responsible for supervision. E. B. and C. C. wrote the original draft. All authors edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. C., E. B. and M. P. acknowledge NCCR Catalysis (Grant no. SNF 180544), a National Centre of Competence in Research funded by the Swiss National Science Foundation. The Swiss Light Source is acknowledged for beamtime at the SuperXAS beamline (Proposal no. 20222164). Kazutaka Sakamoto, Domenico Gioffrè and Sumant Phadke are thanked for help with XAS measurements. ScopeM, the Scientific center for optical and electron microscopy of ETH Zurich, is acknowledged for providing access to their instruments for the project no. 2658. The Mougel Group at ETH Zurich is thanked for access to the XPS. Dr Seraphine Zhang is acknowledged for the XPS measurements and corresponding data processing. Dr Lukas Rochlitz is thanked for fruitful discussions and help with XAS data processing.Notes and references
- D. Malakoff, Science, 2014, 344, 1464–1467 CrossRef CAS PubMed.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS.
- S. Chen, X. Chang, G. Sun, T. Zhang, Y. Xu, Y. Wang, C. Pei and J. Gong, Chem. Soc. Rev., 2021, 50, 3315–3354 RSC.
- S. R. de Miguel, E. L. Jablonski, A. A. Castro and O. A. Scelza, J. Chem. Technol. Biotechnol., 2000, 75, 596–600 CrossRef CAS.
- C. Byron, S. Bai, G. Celik, M. S. Ferrandon, C. Liu, C. Ni, A. Mehdad, M. Delferro, R. F. Lobo and A. V. Teplyakov, ACS Catal., 2020, 10, 1500–1510 CrossRef CAS.
- M. Aly, E. L. Fornero, A. R. Leon-Garzon, V. V. Galvita and M. Saeys, ACS Catal., 2020, 10, 5208–5216 CrossRef CAS.
- X. Liu, W.-Z. Lang, L.-L. Long, C.-L. Hu, L.-F. Chu and Y.-J. Guo, Chem. Eng. J., 2014, 247, 183–192 CrossRef CAS.
- S. R. Docherty, L. Rochlitz, P.-A. Payard and C. Copéret, Chem. Soc. Rev., 2021, 50, 5806–5822 RSC.
- S. R. Docherty and C. Copéret, J. Am. Chem. Soc., 2021, 143, 6767–6780 CrossRef CAS PubMed.
- C. Copéret, Acc. Chem. Res., 2019, 52, 1697–1708 CrossRef PubMed.
- K. Searles, K. W. Chan, J. A. Mendes Burak, D. Zemlyanov, O. Safonova and C. Copéret, J. Am. Chem. Soc., 2018, 140, 11674–11679 CrossRef CAS PubMed.
- L. Rochlitz, K. Searles, J. Alfke, D. Zemlyanov, O. V. Safonova and C. Copéret, Chem. Sci., 2020, 11, 1549–1555 RSC.
- L. Rochlitz, Q. Pessemesse, J. W. A. Fischer, D. Klose, A. H. Clark, M. Plodinec, G. Jeschke, P.-A. Payard and C. Copéret, J. Am. Chem. Soc., 2022, 144, 13384–13393 CrossRef CAS.
- P. A. Payard, L. Rochlitz, K. Searles, L. Foppa, B. Leuthold, O. V. Safonova, A. Comas-Vives and C. Copéret, JACS Au, 2021, 1, 1445–1458 CrossRef CAS PubMed.
- C. K. Mavrokefalos, N. Kaeffer, H.-J. Liu, F. Krumeich and C. Copéret, ChemCatChem, 2020, 12, 305–313 CrossRef CAS.
- O. T. Beachley, Jr., M. R. Churchill, J. C. Pazik and J. W. Ziller, Organometallics, 1986, 5, 1814–1817 CrossRef.
- C. Ehinger, X. Zhou, M. Candrian, S. R. Docherty, S. Pollitt and C. Copéret, JACS Au, 2023, 3(8), 2314–2322 CrossRef CAS PubMed.
- K. Searles, G. Siddiqi, O. V. Safonova and C. Copéret, Chem. Sci., 2017, 8, 2661–2666 RSC.
- D. A. Ruddy, J. Jarupatrakorn, R. M. Rioux, J. T. Miller, M. J. McMurdo, J. L. McBee, K. A. Tupper and T. D. Tilley, Chem. Mater., 2008, 20, 6517–6527 CrossRef CAS.
- C. Moreno-Castilla, M. V. López-Ramón and F. Carrasco-Marín, Carbon, 2000, 38, 1995–2001 CrossRef CAS.
- O. Sebastian, A. Al-Shaibani, N. Taccardi, U. Sultan, A. Inayat, N. Vogel, M. Haumann and P. Wasserscheid, Catal. Sci. Technol., 2023, 13, 4435–4450 RSC.
- P. Laurent, L. Veyre, C. Thieuleux, S. Donet and C. Copéret, Dalton Trans., 2013, 42, 238–248 RSC.
- Y. Nakaya, J. Hirayama, S. Yamazoe, K.-i. Shimizu and S. Furukawa, Nat. Commun., 2020, 11, 2838 CrossRef CAS PubMed.
- H. Okamoto, J. Phase Equilib. Diffus., 2007, 28, 494 CrossRef CAS.
- N. Raman, M. Wolf, M. Heller, N. Heene-Würl, N. Taccardi, M. Haumann, P. Felfer and P. Wasserscheid, ACS Catal., 2021, 11, 13423–13433 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04711c |
| ‡ Present address: Department of Chemistry TU Munich, Arcisstraße 21, D-80333 Munich, Germany. |
| This journal is © The Royal Society of Chemistry 2023 |