
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluoroalkylated hypervalent sulfur fluorides: radical addition of arylchlorotetrafluoro-λ6-sulfanes to tetrafluoroethylene†
Eisuke
Yasuo
a,
Kohsuke
Aikawa
 *a,
Kyoko
Nozaki
b and
Takashi
Okazoe
ac
*a,
Kyoko
Nozaki
b and
Takashi
Okazoe
ac
aDepartment of Chemistry and Biotechnology, Graduate School of Engineering, The University of Tokyo, 2-11-16 Yayoi, Bunkyo-ku, Tokyo 113-0032, Japan. E-mail: aikawa@g.ecc.u-tokyo.ac.jp
bDepartment of Chemistry and Biotechnology, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
cYokohama Technical Center, AGC Inc., 1-1 Suehiro-cho, Tsurumi-ku, Yokohama 230-0045, Japan
First published on 19th October 2023
Abstract
Fluorinated groups are essential hydrophobic groups in drug design. Combining a carbon-free tetrafluoro-λ6-sulfanyl (SF4) group with a polyfluoroalkyl group (RF) provides SF4RF groups, exhibiting high hydrophobicity with a short carbon chain. In this study, various aryltetrafluoro(polyfluoroalkyl)-λ6-sulfanes (ArSF4RF) were synthesized through the radical addition of arylchlorotetrafluoro-λ6-sulfanes (ArSF4Cl) to tetrafluoroethylene. In addition, quantification of hydrophobic constants (πPh) indicated that the SF4 group is considerably more hydrophobic than a difluoromethylene (CF2) group. Further transformation reactions revealed the stabilities and reactivities of these novel fluorinated groups. The high hydrophobicity and synthetic utility of the SF4RF group lead to the potential applications of the SF4RF group in the pharmaceutical field.
Introduction
Substituents based on hexacoordinated sulfur fluorides have gained significant attention in medicinal and material chemistry.1 The pentafluoro-λ6-sulfanyl (SF5) group has been regarded as a “super-trifluoromethyl” group owing to its enhanced size, electronegativity, hydrolytic stability, and hydrophobicity (πPh(SF5): 1.23, πPh(CF3): 0.88).2 Therefore, researchers have explored various applications of this moiety and dedicated substantial efforts to developing effective synthetic methods for compounds containing the SF5 group.2–7 Initial direct fluorination methods require strong oxidants, leading to low yields and poor functional group tolerance because of side reactions, such as overfluorination or C–C/C–S fragmentation.3 However, recent advancements in the synthesis of aliphatic compounds with the SF5 group have been achieved through the rapid development of synthetic methods using SF5X reagents, expanding the accessible range.2,4 Arylpentafluoro-λ6-sulfanes (ArSF5) have also gained significant importance in medicinal chemistry attributed to the significant effect of the SF5 group bound to an aromatic ring on hydrophobicity and metabolic stability.8 A notable breakthrough in the synthesis of ArSF5 has been the development of two-step routes via arylchlorotetrafluoro-λ6-sulfanes (ArSF4Cl) (Scheme 1A, left).5,6 These methods have enabled the scalable and inexpensive synthesis of ArSF5 compounds by using less expensive and milder oxidants, such as Cl2 and trichloroisocyanuric acid (TCICA). | ||
| Scheme 1 Synthetic methods for the aromatic compounds with sulfur–fluorine functional groups. (A) Chlorine–fluorine exchange reactions and radical addition reactions of ArSF4Cl compounds. (B) Conventional synthesis of ArSF4CF3 compounds through direct fluorination. (C) This work: radical addition reactions of ArSF4Cl compounds to tetrafluoroethylene. | ||
The more hydrophobic SF4RF (RF = polyfluoroalkyl) group is a promising option for the enhancement of biological activity.9 For instance, the synthesis of aryltetrafluoro(trifluoromethyl)-λ6-sulfanes (ArSF4CF3) and the πPh constant of the SF4CF3 group (2.13) were first reported in 2006, demonstrating the high hydrophobicity of the SF4RF group (Scheme 1B).9c However, direct fluorination is not feasible without deactivating the aromatic ring with a nitro group because of the high reactivity of fluorine gas. Consequently, the synthesis of a wide range of ArSF4CF3 compounds remains challenging thus far.
Based on this background, we envisioned using ArSF4Cl compounds in synthesizing ArSF4RF compounds. Several radical addition reactions of ArSF4Cl compounds have been reported thus far (Scheme 1A, right).5c,10 These synthetic methods are considerably milder than conventional direct fluorination of sulfides and have enabled the preparation of a variety of aromatic compounds with the tetrafluoro-λ6-sulfanyl (SF4) moiety. However, radical addition reactions of ArSF4Cl compounds to fluoroolefins have not been reported. In this study, we discuss radical addition reactions of ArSF4Cl compounds to tetrafluoroethylene (TFE) as a simple synthetic approach for aryltetrafluoro(polyfluoroethyl)-λ6-sulfanes (Scheme 1C).11 The enhanced hydrophobicity of the novel SF4RF groups and their reactivities will also be discussed.
Results and discussion
Radical addition of arylchlorotetrafluoro-λ6-sulfanes to tetrafluoroethylene
We synthesized chlorotetrafluoro(phenyl)-λ6-sulfane 1a using the optimized conditions involving TCICA/KF,5c,d and then the radical addition reaction of 1a to TFE was examined by exploring various radical initiators (Table 1). Although triethylborane effectively catalyzed the addition reaction of ArSF4Cl compounds to terminal alkynes and alkenes,10a–c a low yield was obtained in the reaction with TFE (entry 1). Irradiation with a blue LED, known as the activation method for ArSF4Cl compounds,10b,d afforded the adduct 2a in 49% yield (entry 2). A higher yield was obtained when BDK (2,2-dimethoxy-2-phenylacetophenone) was used as the photoinitiator (entry 3). Subsequently, we examined several thermal initiators, such as AIBN, ADVN, and AMDVN, each requiring different initiation temperatures (entries 4–6). Azo-type initiators exhibited good yields, with ADVN demonstrating the best performance and affording 2a in 88% yield (entry 5). Reducing the reaction time to 20 h resulted in incomplete substrate conversion (entry 7). Although increasing the amount of ADVN or elevating the reaction temperature accelerated the reaction, the yield did not reach the optimal condition attributed to the decomposition of 1a (entries 8–9). Alternation of solvents to acetonitrile, hexane, THF, or 1,2-dichloroethane did not afford satisfactory results (Table S1†), and the formation of a hydrogenated adduct, PhSF4CF2CF2H, was predominant in the case of THF (entry 10). Inspired by the result, we examined the selective formation of 3avia a three-component addition reaction with hydrogenating agents.4f However, several investigations revealed that the hydrogen sources such as (Me3Si)3SiH caused decomposition of 1a rather than the desired reaction (see the ESI†).|
|
|||
|---|---|---|---|
| Entry | Initiator | Temp. | Yielda (%) |
| a Determined by 19F NMR analysis of the crude mixture relative to an internal standard (1,4-bis(trifluoromethyl)benzene). b Irradiated with blue LED. c 20 h. d 30 mol% initiator added. e THF (0.10 M) instead of EtOAc. f Isolated yield. g Yield of 3a. AIBN = 2,2′-azobis(isobutyronitrile), ADVN = 2,2′-azobis(2,4-dimethylvaleronitrile), AMDVN = 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile), BDK = 2,2-dimethoxy-2-phenylacetophenone. | |||
| 1 | BEt3 | rt | 5 |
| 2b | — | rt | 49 |
| 3b | BDK | rt | 68 |
| 4 | AIBN | 60 °C | 80 |
| 5 | ADVN | 40 °C | 88 (77f) |
| 6 | AMDVN | 25 °C | 72 |
| 7c | ADVN | 40 °C | 23 |
| 8c,d | ADVN | 40 °C | 75 |
| 9c | ADVN | 60 °C | 65 |
| 10e | ADVN | 40 °C | 38g |

With the optimal reaction conditions in hand, we explored the substrate scope of this radical addition reaction using various ArSF4Cl compounds. Substrates 1 were synthesized according to the literature method (Scheme 2A).5c,d Notably, the electron-rich diaryl disulfide underwent arene chlorination during the oxidative fluorination step, affording 1j as the major product (Scheme 2B). Bis(pentafluorophenyl) disulfide gave 1n as a mixture of cis- and trans-isomers as previously reported, although the oxidative fluorination generally afforded 1 in trans conformation (Scheme 2C).5a,c,d Initially, the screening of substrates focused on the para-substituents on the benzene ring (Scheme 3). Both electron-withdrawing (2b–e) and electron-donating groups (2f–g) were tolerated, as were protected carboxylic acid and phenol (2h–i). Meta-substituted (2j–l) and ortho-fluorinated substrates (2m–n) afforded the desired products in moderate to high yields. Using a mixture of cis- and trans-ArSF4Cl, only the trans product was obtained (2n). Additionally, this reaction could be applied to pyridines and pyrimidines (2o–q). The structure of 2e was unambiguously determined through X-ray diffraction analysis. Expectedly, the sulfur atom of 2e shows an octahedral geometry characteristic of the hexavalent sulfur species as well as the ArSF4CF3 compound9c (see the ESI†).
 | ||
| Scheme 2 Synthesis of (A) 1 (except 1j and 1n), (B) 1j and (C) 1n. Yields were determined by 19F NMR analysis relative to an internal standard (trifluorotoluene). TCICA = trichloroisocyanuric acid, TFA = trifluoroacetic acid. | ||
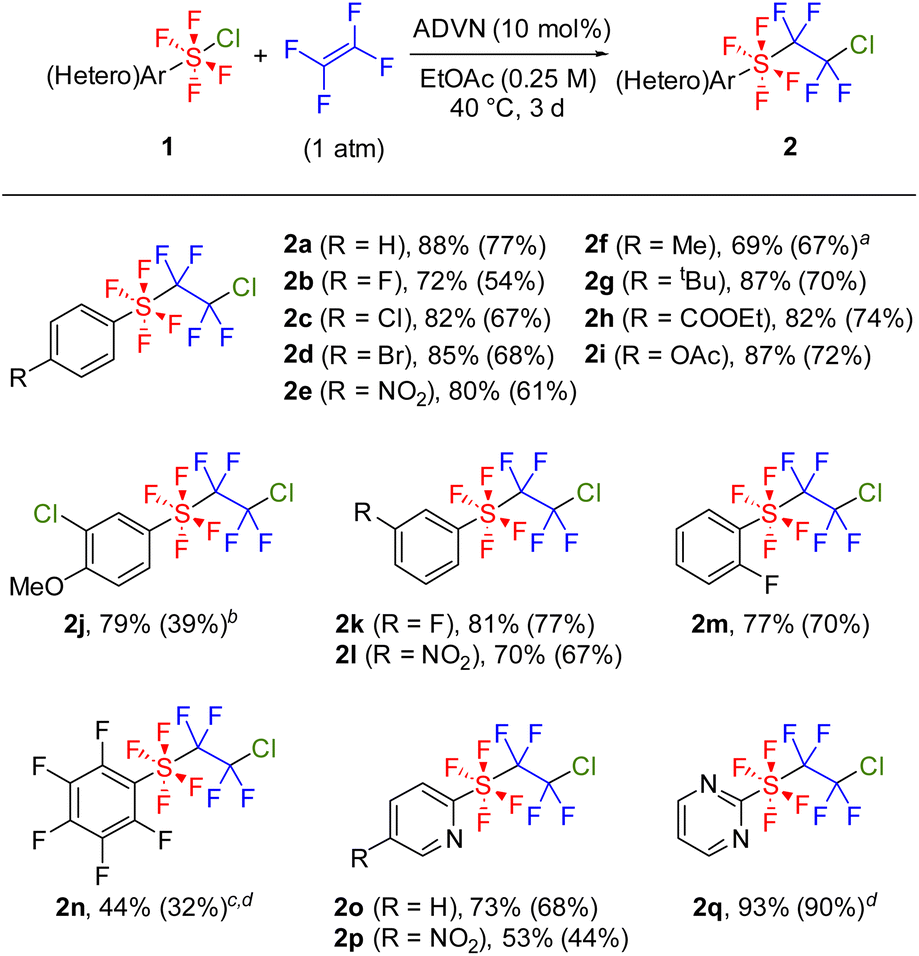 | ||
Scheme 3 Substrate scope. 1 (1.0 equiv.) and ADVN (10 mol%) were stirred in anhydrous EtOAc at 40 °C under TFE (1 atm). Yields were determined by 19F NMR analysis relative to an internal standard (1,4-bis(trifluoromethyl)benzene); isolated yields are shown in parentheses. a AIBN (10 mol%), 60 °C. b ADVN (30 mol%). c Substrate was a mixture of cis- and trans-isomers (39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :61). d EtOAc (0.50 M). :61). d EtOAc (0.50 M). | ||
In several cases where ArSF4Cl compounds were added to TFE, the dimerized products (ArSF4(CF2CF2)2Cl) were also obtained (less than 5%).12 To shed light on the mechanism of the present reactions, a radical addition reaction was performed at higher TFE pressures. Consequently, 2a and the telomer were obtained in 49% and 32% yields, respectively (Scheme 4A). Compared to the standard conditions under atmospheric pressure of TFE, the telomer ratio showed significant improvement. The telomers (2 ≤ n ≤ 7) were detected using GC-MS after treating with silica-gel column chromatography (Fig. S1†).
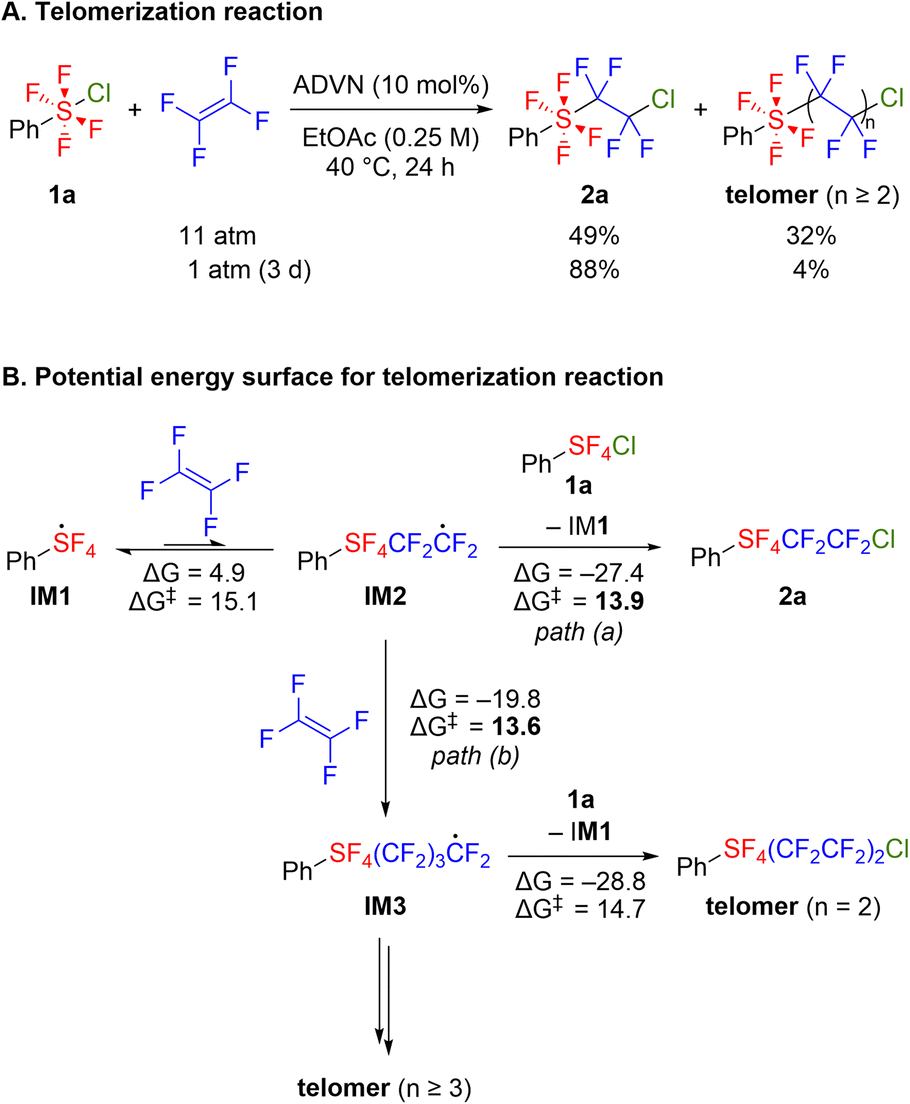 | ||
| Scheme 4 (A) Telomerization reaction. Yields were determined by 19F NMR analysis relative to an internal standard (1,4-bis(trifluoromethyl)benzene). (B) Free energy change and free energy of activation at 25 °C (in kcal mol−1). | ||
DFT calculations
Based on the single addition and telomerization reaction results, our next objective was to elucidate the energy profiles associated with these reactions. In the context of the radical reactions involving hexavalent sulfur fluorides, DFT calculations were performed to study the addition of SF5Cl to alkynes, demonstrating the high stability of the SF5 radical resulting from hyperconjugation.4f According to the literature, DFT calculations were performed at the M06-2X/Def2TZVPP/SMD(EtOAc) level of theory at 25 °C (Scheme 4B, see the ESI† for more details). Firstly, we computed the energy diagram for the formation of 2a (Scheme 4B, path (a)). After abstracting chlorine from 1a, IM1 adds to TFE with a free energy barrier of 15.1 kcal mol−1 to form IM2. Notably, the formation of IM2 from IM1 and TFE was endergonic owing to the high stability of IM1. When the SF5 radical was added, the formation of the SF5CF2CF2 radical was slightly exergonic, indicating apparent resonance stabilization by the benzene ring of IM1 (Scheme S4†). After IM2 is formed, chlorine abstraction from 1a proceeds with a 13.9 kcal mol−1 free energy barrier, regenerating IM1 and yielding 2a. The chlorine transfer is significantly exergonic and is the driving force of a series of reactions. Next, we investigated the propagation reaction (Scheme 4B, path (b)). A dimerized product was obtained from IM2 through a second addition to TFE, followed by chlorine abstraction from 1a. Notably, the formation of 2a through chlorine abstraction was irreversible, and further elongation from 2a could no longer occur. The stability of 2a was also supported by the unsuccessful telomerization from 2a (Scheme S2†). Therefore, the telomer/2a ratio is influenced by the selectivity of chlorine abstraction and propagation from IM2. The calculations showed that these two reactions proceed with comparable activation energies. Considering that the reaction rate depends on the activation energy and the concentrations of reactants, the concentration ratio of TFE and 1a should affect the selectivity. This is consistent with the experimental results (Scheme 4A).Synthetic applications
Compound 2 exhibited stability against silica gel column chromatography and could be stored in air at room temperature for several months. Next, we carried out the transformation of 2 to investigate its stability and reactivity. Several transformations retaining the SF4CF2CF2Cl group were tested initially (Scheme 5A). Bromoarene 2d was converted to 2rvia a Suzuki–Miyaura cross-coupling reaction, facilitating the synthesis of various ArSF4CF2CF2Cl compounds. SF4CF2CF2Cl-substituted aniline 2s and benzoic acid 2t were successfully synthesized from 2e and 2h, respectively. These methods overcome the difficulty in synthesizing 2s and 2t through radical addition because of the low stability of ArSF4Cl with amino and carboxyl groups. In contrast, the SF4CF2CF2Cl group can be reduced to the SF4CF2CF2H group using tris(trimethylsilyl)silane (Scheme 5B). The set of transformation reactions mentioned in Schemes 5A–B enables the synthesis of functional molecules; for instance, an analog of the bioactive molecule Tetrafluron13 was synthesized from 2l in 70% yield over three steps (Scheme 5C). Furthermore, the iodosulfanylation of tetrafluoroethylene followed by fluorination was investigated to prepare a perfluorinated SF4CF2CF3 group (Scheme 5D). After evaluating several external iodine sources, carbon tetraiodide was identified as the most effective iodinating agent in yield and selectivity (Table S4†). Under the optimized reaction conditions, tetrafluoro(tetrafluoroiodoethyl)(phenyl)-λ6-sulfane 4 was synthesized from 1a and TFE in 63% yield. Subsequently, 4 underwent fluorination using AgF2 to form 5. When AgF as a fluorinating agent or 2a (PhSF4CF2CF2Cl) as a substrate was employed, the fluorination did not proceed (Table S5†). We then compared the stability of 5 and PhCF2CF2CF3 to clarify the difference between the SF4 and CF2 linkers (Table 2). Under high temperature conditions, 5 was decomposed by nearly 30%, but PhCF2CF2CF3 was not decomposed at all (entries 1–2). In addition, heating under the basic conditions14 or exposure to large excess of TfOH/C6H6 (ref. 15) resulted in complete degradation of 5, whereas PhCF2CF2CF3 was partially degraded (entries 3–6). These results indicate the higher degradability of the SF4 linker than that of the CF2 linker.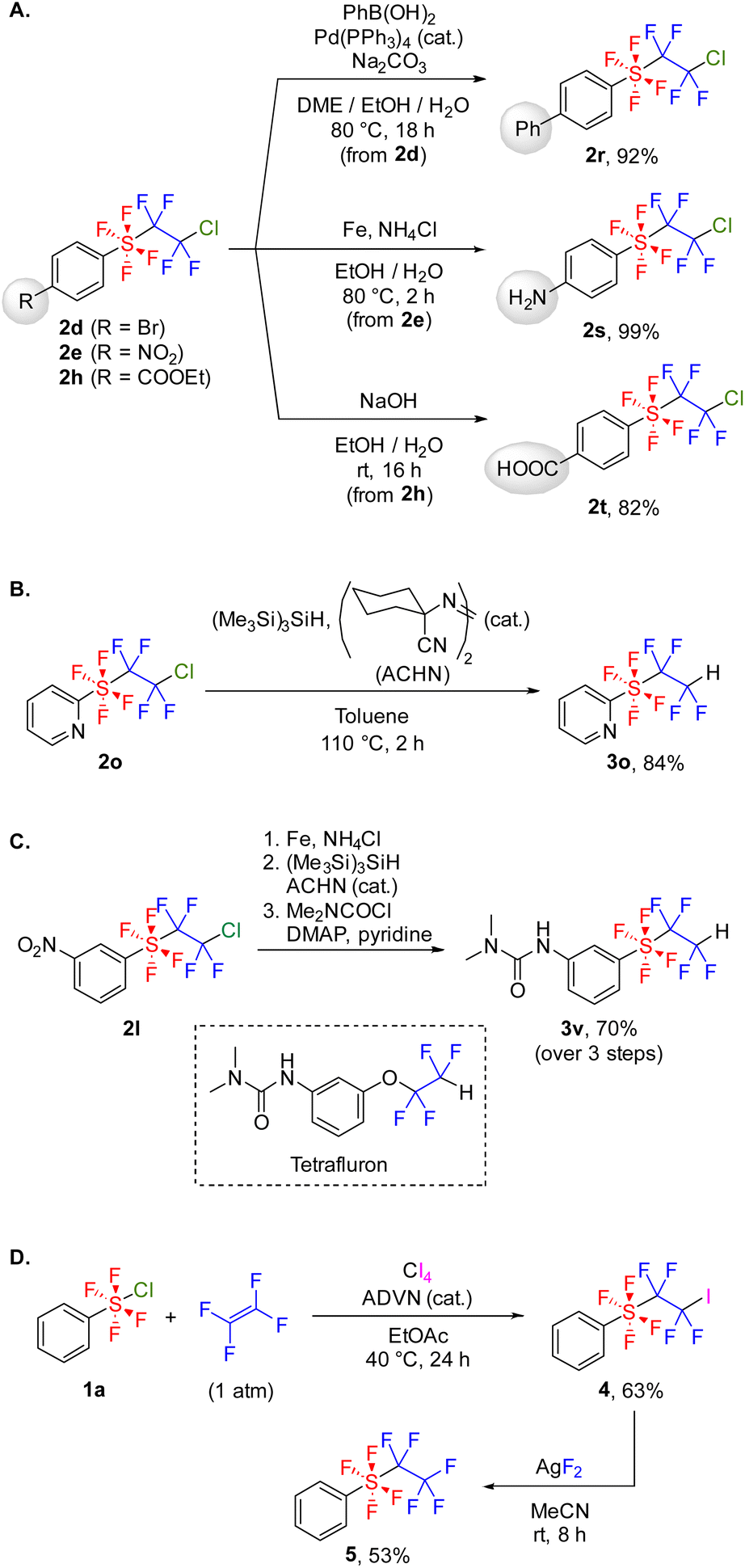 | ||
| Scheme 5 Derivatizations of aryltetrafluoro(polyfluoroethyl)-λ6-sulfanes. Isolated yields are provided. (A) Transformation reactions retaining the SF4CF2CF2Cl group. (B) Reduction of the SF4CF2CF2Cl group. (C) Synthesis of an analog of Tetrafluron. (D) Synthesis of tetrafluoro(perfluoroethyl)(phenyl)-λ6-sulfane 5. | ||
|
|
|||
|---|---|---|---|
| Entry | Substrate | Cond. | Resulta |
| a Conversion of 5 or PhCF2CF2CF3 was determined by 19F NMR analysis of the crude mixture relative to an internal standard (trifluorotoluene or 4,4′-difluorobiphenyl). b 100% recovery of substrate. | |||
| 1 | 5 | 8:1 DMSO:H2O, 120 °C, 24 h |
Decomposition 32% |
| 2 | PhCF2CF2CF3 | 8:1 DMSO:H2O, 120 °C, 24 h |
Decomposition 0%b |
| 3 | 5 | NaOH, 8:1 DMSO:H2O, 120 °C, 24 h |
Decomposition 100% |
| 4 | PhCF2CF2CF3 | NaOH, 8:1 DMSO:H2O, 120 °C, 24 h |
Decomposition 49% |
| 5 | 5 | 1:1 TfOH:C6H6, rt, 4 h |
Decomposition 100% |
| 6 | PhCF2CF2CF3 | 1:1 TfOH:C6H6, rt, 4 h |
Decomposition 77% |

Substituent effects
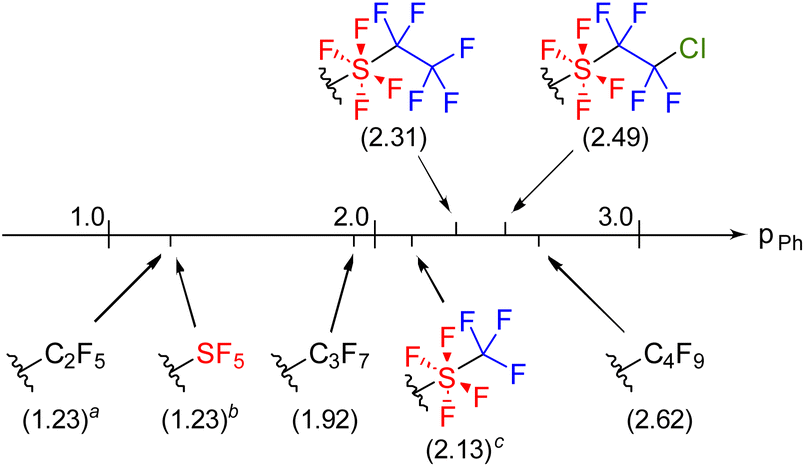
Since aromatic compounds with the SF4CF2CF2Cl and SF4CF2CF3 groups were synthesized for the first time, we investigated their substituent effects such as hydrophobic parameters (πPh)16 and Hammett substituent parameters (σm and σp).17The logP (P being the partition coefficient in an n-octanol/water system) values for 2a and 5 were measured using HPLC.18 The logP(2a) and logP(5) were measured to be 4.62 and 4.44, respectively. Based on these values, πPh(SF4CF2CF2Cl) and πPh(SF4CF2CF3) were estimated to be 2.49 and 2.31, respectively, indicating a slight hydrophobic effect of the terminal chlorine. Subsequently, the hydrophobicity of other fluorinated groups was compared with that of the SF4CF2CF2Cl and SF4CF2CF3 groups (Fig. 1). πPh(SF4CF2CF2Cl) and πPh(SF4CF2CF3) exhibited higher values than πPh(SF5) (1.23) and πPh(SF4CF3) (2.13) owing to their longer carbon chains.2,9c Comparing the SF4CF2CF3 group with the perfluoroalkyl groups, πPh(SF4CF2CF3) was higher than πPh(C3F7) and lower than πPh(C4F9), suggesting that the hydrophobic effect of the SF4 group falls between those of one and two difluoromethylene (CF2) groups.
The Hammett parameters of the SF4CF2CF2Cl group were estimated through the 19F NMR approach developed by Taft (Table 3).19 As a result, the meta- and para-σ values (σm and σp) of the SF4CF2CF2Cl group were estimated to be 0.61 and 0.66, respectively. Among electron-withdrawing functional groups, the values were higher than those of the CF3 group (σm: 0.43, σp: 0.54), comparable to those of the CN (σm: 0.56, σp: 0.66), SF5 (σm: 0.61, σp: 0.68), and SF4CF3 (σp: 0.68) groups, and lower than those of the NO2 group (σm: 0.71, σp: 0.78).9c,17b Although the SF4CF2CF2Cl group has a longer carbon chain than the SF4CF3 group, the electron-withdrawing ability decreases; a similar trend has been reported for linear perfluoroalkyl groups (σp(CF3): 0.54, σp(CF2CF3): 0.52, σp(CF2CF2CF3): 0.48).17b
Conclusions
In summary, we unveiled an unexplored synthetic route for ArSF4RF compounds based on the addition reaction of ArSF4Cl compounds to TFE. It was demonstrated that the inexpensive and scalable radical addition reactions can be applied to the synthesis of an analog of the bioactive molecule and perfluoroalkyl compounds. Additionally, we have estimated the πPh constant for the SF4CF2CF2Cl and SF4CF2CF3 groups, providing preliminary evidence of the significantly higher hydrophobicity of the SF4 group compared to the CF2 group. We believe that the development of the SF4RF groups and the elucidation of their fundamental properties achieved in this study provide promising options for the application of fluorinated functional groups.The ArSF4RF compounds are related to per- and polyfluoroalkyl substances (PFAS), and therefore potential accumulation cannot be ruled out at present.20 Close attention should be paid to this issue for further application.
Data availability
All experimental data and detailed procedures are available in the ESI.†Author contributions
E. Y. conducted the experiments, analysed the data, and wrote the original draft. K. A. and T. O. directed the project. K. N. provided advice and discussed the data. All the authors discussed the results and contributed to the review and editing of the manuscript.Conflicts of interest
An application for a patent (application number: WO2022JP035414) related to this work has been submitted by K. A., E. Y., K. N., and T. O. as co-inventors.Acknowledgements
The computation was performed using Research Center for Computational Science, Okazaki, Japan (Project: 22-IMS-C017). E. Y. would like to thank the Program for Leading Graduate Schools (MERIT-WINGS) for financial support. This work was supported by JSPS KAKENHI, Grant No. 20K05460 (K. A.). AGC Inc. is acknowledged for supporting this study.Notes and references
- (a) D. Lentz and K. Seppelt, in Chemistry of Hypervalent Compounds, ed. K.-Y. Akiba, Wiley-VCH, New York, 1999, pp. 295–325 Search PubMed; (b) P. Kirsch and G.-V. Roschenthaler, in Current Fluoroorganic Chemistry, eds. V. A. Soloshonok, K. Mikami, T. Yamazaki, J. T. Welch and J. F. Honek, American Chemical Society, Washington, DC, 2007, vol. 949, pp. 221–243 Search PubMed; (c) S. Altomonte and M. Zanda, J. Fluorine Chem., 2012, 143, 57–93 CrossRef CAS.
- P. R. Savoie and J. T. Welch, Chem. Rev., 2015, 115, 1130–1190 CrossRef CAS PubMed.
- (a) G. A. Silvey and G. H. Cady, J. Am. Chem. Soc., 1950, 72, 3624–3626 CrossRef CAS; (b) W. A. Sheppard, J. Am. Chem. Soc., 1960, 82, 4751–4752 CrossRef CAS; (c) R. D. Bowden, P. J. Comina, M. P. Greenhall, B. M. Kariuki, A. Loveday and D. Philp, Tetrahedron, 2000, 56, 3399–3408 CrossRef CAS; (d) X. Ou and A. F. Janzen, J. Fluorine Chem., 2000, 101, 279–283 CrossRef CAS.
- For progress in pentafluorosulfanylation reactions using SF5X compounds after ref. 2, see: (a) A. Gilbert, P. Langowski, M. Delgado, L. Chabaud, M. Pucheault and J. F. Paquin, Beilstein J. Org. Chem., 2020, 16, 3069–3077 CrossRef CAS PubMed; (b) F.-F. Feng, J.-A. Ma and D. Cahard, J. Org. Chem., 2021, 86, 13808–13816 CrossRef CAS PubMed; (c) A. Gilbert, M. Birepinte and J.-F. Paquin, J. Fluorine Chem., 2021, 243, 109734 CrossRef CAS; (d) G. Lefebvre, O. Charron, J. Cossy and C. Meyer, Org. Lett., 2021, 23, 5491–5495 CrossRef CAS PubMed; (e) J.-Y. Shou, X. H. Xu and F.-L. Qing, Angew. Chem., Int. Ed., 2021, 60, 15271–15275 CrossRef CAS PubMed; (f) M. Birepinte, P. A. Champagne and J.-F. Paquin, Angew. Chem., Int. Ed., 2022, 61, e202112575 CrossRef CAS PubMed; (g) A. Taponard, T. Jarrosson, L. Khrouz, M. Médebielle, J. Broggi and A. Tlili, Angew. Chem., Int. Ed., 2022, 61, e202204623 CrossRef CAS PubMed; (h) J.-Y. Shou, X.-H. Xu and F.-L. Qing, J. Fluorine Chem., 2022, 261–262, 110018 CrossRef CAS; (i) J.-Y. Shou and F.-L. Qing, Angew. Chem., Int. Ed., 2022, 61, e202208860 CrossRef CAS PubMed; (j) M.-R. Ouellet-Du Berger, M. Boucher and J.-F. Paquin, J. Fluorine Chem., 2023, 268, 110131 CrossRef CAS; (k) R. Kordnezhadian, T. De Bels, K. Su, L. Van Meervelt, E. Ismalaj, J. Demaerel and W. De Borggraeve, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-05387.
- (a) T. Umemoto, L. M. Garrick and N. Saito, Beilstein J. Org. Chem., 2012, 8, 461–471 CrossRef CAS PubMed; (b) O. S. Kanishchev and W. R. Dolbier, Angew. Chem., Int. Ed., 2015, 54, 280–284 CrossRef CAS PubMed; (c) C. R. Pitts, D. Bornemann, P. Liebing, N. Santschi and A. Togni, Angew. Chem., Int. Ed., 2019, 58, 1950–1954 CrossRef CAS PubMed; (d) I. Saidalimu, Y. M. Liang, K. Niina, K. Tanagawa, N. Saito and N. Shibata, Org. Chem. Front., 2019, 6, 1157–1161 RSC.
- (a) K. Lummer, M. V. Ponomarenko, G.-V. Röschenthaler, M. Bremer and P. Beier, J. Fluorine Chem., 2014, 157, 79–83 CrossRef CAS; (b) M. Kosobokov, B. Cui, A. Balia, K. Matsuzaki, E. Tokunaga, N. Saito and N. Shibata, Angew. Chem., Int. Ed., 2016, 55, 10781–10785 CrossRef CAS PubMed; (c) B. Cui, M. Kosobokov, K. Matsuzaki, E. Tokunaga and N. Shibata, Chem. Commun., 2017, 53, 5997–6000 RSC; (d) B. Cui, S. Jia, E. Tokunaga, N. Saito and N. Shibata, Chem. Commun., 2017, 53, 12738–12741 RSC; (e) O. I. Guzyr, V. N. Kozel, E. B. Rusanov, A. B. Rozhenko, V. N. Fetyukhin and Y. G. Shermolovich, J. Fluorine Chem., 2020, 239, 109635 CrossRef CAS; (f) L. Wang and J. Cornella, Angew. Chem., Int. Ed., 2020, 59, 23510–23515 CrossRef CAS PubMed; (g) K. Tanagawa, Z. Zhao, N. Saito and N. Shibata, Bull. Chem. Soc. Jpn., 2021, 94, 1682–1684 CrossRef CAS; (h) L. Wang, S. Ni and J. Cornella, Synthesis, 2021, 53, 4308–4312 CrossRef CAS.
- A milder direct fluorination method with broad functional group tolerance was reported in 2023, please see: T. Gatzenmeier, Y. Liu, M. Akamatsu, T. Okazoe and K. Nozaki, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-jzn11.
- (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- (a) J. I. Darragh, G. Haran and D. W. A. Sharp, J. Chem. Soc., Dalton Trans., 1973, 2289–2293 RSC; (b) K. D. Gupta and J. M. Shreeve, Inorg. Chem., 1985, 24, 1457–1460 CrossRef CAS; (c) P. Kirsch and A. Hahn, Eur. J. Org Chem., 2006, 1125–1131 CrossRef CAS; (d) A. Ikeda, L. Zhong, P. R. Savoie, C. N. von Hahmann, W. Zheng and J. T. Welch, Eur. J. Org Chem., 2018, 772–780 CrossRef CAS; (e) A. Ikeda, A. Capellan and J. T. Welch, Org. Biomol. Chem., 2019, 17, 8079–8082 RSC; (f) G. Chang, L. Chen and Z. Li, Mater. Res. Express, 2020, 7, 075305 CrossRef CAS; (g) K. Liu, L. Chen and Z. Li, J. Appl. Polym. Sci., 2021, 138, 49687 CrossRef CAS; (h) X. Zhao, J.-Y. Shou, J. J. Newton and F.-L. Qing, Org. Lett., 2022, 24, 8412–8416 CrossRef CAS PubMed; (i) M. Deng, M. Wilde and J. T. Welch, J. Org. Chem., 2023, 88, 11363–11366 CrossRef CAS PubMed.
- (a) L. Zhong, P. R. Savoie, A. S. Filatov and J. T. Welch, Angew. Chem., Int. Ed., 2014, 53, 526–529 CrossRef CAS PubMed; (b) L. Zhong, A. S. Filatov and J. T. Welch, J. Fluorine Chem., 2014, 167, 192–197 CrossRef CAS; (c) P. Das, M. Takada, E. Tokunaga, N. Saito and N. Shibata, Org. Chem. Front., 2018, 5, 719–724 RSC; (d) K. Niina, K. Tanagawa, Y. Sumii, N. Saito and N. Shibata, Org. Chem. Front., 2020, 7, 1276–1282 RSC; (e) K. Maruno, K. Niina, O. Nagata and N. Shibata, Org. Lett., 2022, 24, 1722–1726 CrossRef CAS PubMed; (f) K. Iwaki, K. Maruno, O. Nagata and N. Shibata, J. Org. Chem., 2022, 87, 6302–6311 CrossRef CAS PubMed; (g) K. Iwaki, K. Tanagawa, S. Mori, K. Maruno, Y. Sumii, O. Nagata and N. Shibata, Org. Lett., 2022, 24, 3347–3352 CrossRef CAS PubMed; (h) K. Maruno, K. Hada, Y. Sumii, O. Nagata and N. Shibata, Org. Lett., 2022, 24, 3755–3759 CrossRef CAS PubMed; (i) Y. Kraemer, C. Ghiazza, A. N. Ragan, S. Ni, S. Lutz, E. K. Neumann, J. C. Fettinger, N. Nöthling, R. Goddard, J. Cornella and C. R. Pitts, Angew. Chem., Int. Ed., 2022, 61, e202211892 CrossRef CAS PubMed; (j) E. M. Mahmoud, H. Iwasaki, K. Hada, Y. Murata, Y. Sumii and N. Shibata, Bull. Chem. Soc. Jpn., 2023, 96, 110–112 CrossRef CAS; (k) Y. Kraemer, J. A. Buldt, A. M. Stephens, W.-Y. Kong, A. N. Ragan, Z. C. Haidar, A. H. Patel, J. C. Fettinger, D. J. Tantillo and C. R. Pitts, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-dwkhg.
- Radical addition of SF5Cl to TFE was first reported in 1961. However, the milder reaction conditions described in this report offer higher yields and functional group tolerance, please see: J. R. Case, N. H. Ray and H. L. Roberts, J. Chem. Soc., 1961, 2070–2075 RSC.
- (a) R. N. Haszeldine, J. Chem. Soc., 1949, 2856–2861 RSC; (b) R. N. Haszeldine, Nature, 1951, 167, 139–140 CrossRef CAS PubMed.
- (a) D. L. Reasons, L. S. Jeffery and T. C. McCutchen, Weed Sci., 1978, 26, 533–538 CrossRef CAS; (b) H. Pinto and F. T. Corbin, Weed Sci., 1980, 28, 557–565 CrossRef CAS.
- B. Trang, Y. Li, X.-S. Xue, M. Ateia, K. N. Houk and W. R. Dichtel, Science, 2022, 377, 839–845 CrossRef CAS PubMed.
- A. Kethe, A. F. Tracy and D. A. Klumpp, Org. Biomol. Chem., 2011, 9, 4545–4549 RSC.
- (a) C. Hansch, R. M. Muir, T. Fujita, P. P. Maloney, F. Geiger and M. Streich, J. Am. Chem. Soc., 1963, 85, 2817–2824 CrossRef CAS; (b) T. Fujita, J. Iwasa and C. Hansch, J. Am. Chem. Soc., 1964, 86, 5175–5180 CrossRef CAS; (c) C. Hansch and A. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology, Wiley, New York, 1979 Search PubMed.
- (a) C. Hansch, A. Leo, S. H. Unger, K. H. Kim, D. Nikaitani and E. J. Lien, J. Med. Chem., 1973, 16, 1207–1216 CrossRef CAS PubMed; (b) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- OECD, Test No. 117: Partition Coefficient (n-octanol/water), HPLC Method, OECD Publishing, Paris, 2022 Search PubMed.
- (a) R. W. Taft, E. Price, I. R. Fox, I. C. Lewis, K. K. Andersen and G. T. Davis, J. Am. Chem. Soc., 1963, 85, 709–724 CrossRef CAS; (b) R. W. Taft, E. Price, I. R. Fox, I. C. Lewis, K. K. Andersen and G. T. Davis, J. Am. Chem. Soc., 1963, 85, 3146–3156 CrossRef CAS.
- M. G. Evich, M. J. B. Davis, J. P. McCord, B. Acrey, J. A. Awkerman, D. R. U. Knappe, A. B. Lindstrom, T. F. Speth, C. Tebes-Stevens, M. J. Strynar, Z. Wang, E. J. Weber, W. M. Henderson and J. W. Washington, Science, 2022, 375, eabg9065 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2282789. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04837c |
| This journal is © The Royal Society of Chemistry 2023 |