
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical halogen-bonding assisted carbothiophosphorylation reactions of alkenyl and 1,3-dienyl bromides†
Helena F.
Piedra
 ,
Victoria
Gebler
,
Carlos
Valdés
and
Manuel
Plaza
*
,
Victoria
Gebler
,
Carlos
Valdés
and
Manuel
Plaza
*
Departamento de Química Orgánica e Inorgánica, Instituto Universitario de Química Organometálica “Enrique Moles”, Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universidad de Oviedo, Julián Clavería 8, 33006 Oviedo, Spain. E-mail: plazamanuel@uniovi.es
First published on 30th October 2023
Abstract
Herein, we present a synthetic procedure for the facile and general preparation of novel S-alkenyl and dienyl phosphoro(di)thioates for the first time. Extensive mechanistic investigations support that the reactions rely on a photochemical excitation of a halogen-bonding complex, formed with a phosphorothioate salt and an alkenyl or dienyl bromide, which light-induced fragmentation leads to the formation of the desired products through a radical-based pathway. The substrate scope is broad and exhibits a wide functional group tolerance in the formation of the final compounds, including molecules derived from natural products, all with unknown and potentially interesting biological properties. Eventually, a very efficient continuous flow protocol was developed for the upscale of these reactions.
1 Introduction
The development of novel synthetic methodologies for the construction of C–S bonds is an important undertaking in organic chemistry.1 In particular, the design of new organic transformations oriented to the formation of sulfur-containing organophosphorous compounds is particularly attractive. This is due to the interesting properties these compounds feature from both a biological and chemical point of view.2 A prominent class of these organosulfur compounds encompasses phosphorothioate diesters that contain a P–S–C(sp2) structural motif.3 These compounds are reported to be useful as pesticides,4 therapeutic molecules5 and valuable building blocks in organic synthesis.6 The significance of these compounds is underscored by the numerous methodologies that have been documented in recent years centered around their synthesis.7 While both metal-catalyzed8 and metal-free9 procedures have been well established for creating S-arylated phosphorothioates (Scheme 1b), we could only find one example of a synthetic methodology for the preparation of the analogous S-alkenylated compounds.10 However, this protocol contemplates a very limited scope (6 examples), making use of uncommon iodonium salts. When it comes to the S-alkynylation of phosphorothioate diesters, Tang has recently reported an elegant approach through a dual photoredox/copper catalysis under operationally simple conditions.11 However, to the best of our knowledge there are currently still no procedures reported for the S-dienylation of phosphorotioate diesters. Considering the intriguing potential biological attributes inherent in these novel organophosphorous compounds, it is highly desirable to develop general, sustainable and simple procedures for their synthesis.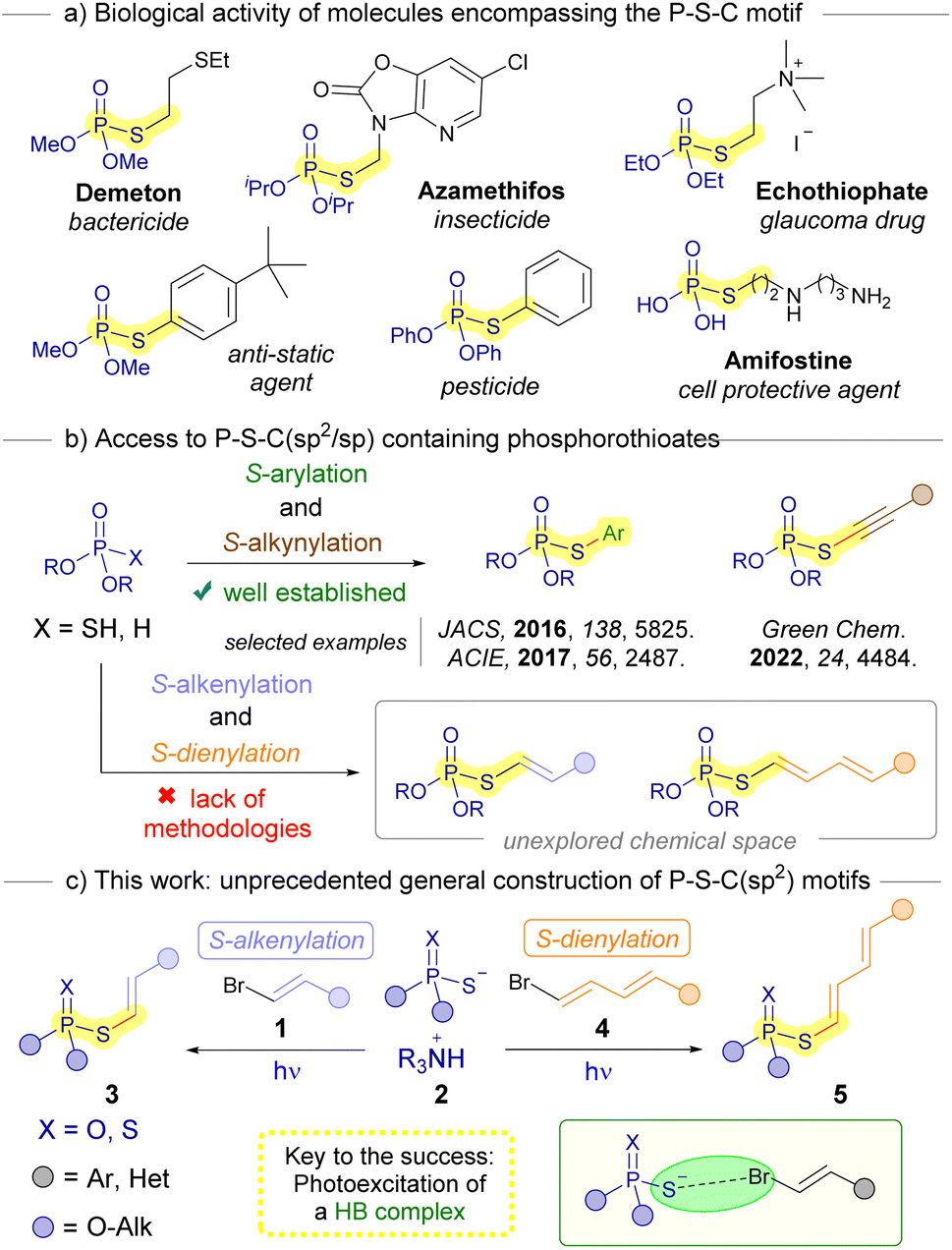 | ||
| Scheme 1 State of the art in the synthesis of P–S–C(sp2/sp) containing molecules. | ||
Our research group has recently developed a procedure for the cross-coupling between alkenyl halides and thiolates, enabled by photochemical excitation of halogen-bonding complexes.12 Notably, this method offers a photocatalyst-free alternative to other contemporary light-driven approaches used for synthesizing similar compounds, such as thiol-yne reactions, which typically involve starting materials like alkynes and thiolates.13
The photochemical activation of halogen-bonding complexes has gained significant attention in recent years for the generation of carbon-centered radicals,14 facilitating novel organic transformations to occur.15 The halogen bond is a type of weak interaction that falls under the category of σ-hole interactions.16 Specifically, it involves a partial n → σ* charge-transfer from a non-bonding orbital of a nucleophilic electron donor (HB acceptor) to an antibonding orbital (σ*) of an electron acceptor, the corresponding organic halide (HB donor).17 This stabilizing interaction is responsible for the formation of halogen-bonding complexes, which are a specific type of EDA (electron donor–acceptor) complexes.18 By inducing a photochemical fragmentation of the HB complex through the reduction of the C–halogen bond, two different radical species can be generated from the synthetic precursors. These radicals can then directly recombine to form the desired cross-coupling product. Based on all the above, we saw potential in the photochemical halogen-bonding-assisted reaction between alkenyl halides 1 and phosphorothioate salts 2 for the construction of S-alkenylated or dienylated phosphorothioates 3 or 5, respectively (Scheme 1c). It should be pointed out that.
2 Results and discussion
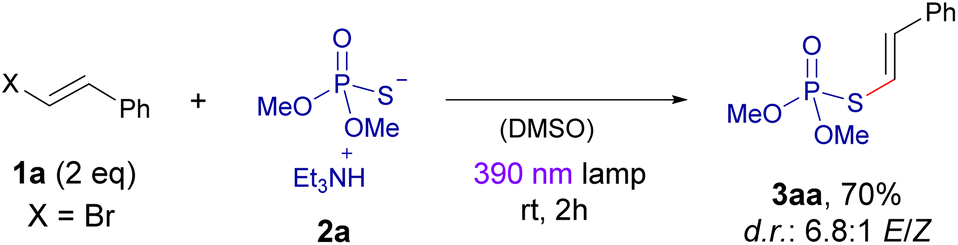
We initiated our experimental studies employing the dimethyl phosphorothioate 2a and β-bromostyrene 1a as the coupling partners in our photochemical reaction. As it will be discussed later in more detail, a preliminary UV-vis analysis of a solution containing both compounds revealed the formation of a charge-transfer (CT) band, which was indicative of the formation of the desired halogen-bonding complex and gave us precious information on which excitation wavelength could potentially be used to promote the coupling reaction (370–400 nm).A summary of the optimization process is presented in the Table 1. Much to our pleasure, our preliminary reaction conditions employing the 390 nm lamp (2 h of irradiation) and DMSO as the solvent, afforded the formation of the desired compound 3aa with a 70% isolated yield. Control experiments revealed that the presence of light was necessary to carry out the transformation (entry 1). It was noticed that longer irradiation times led to significant degradation of the final product, which is not photostable over 16 hours of irradiation with violet light (entry 2). The use of light with either lower (366 nm) or higher (427 nm) wavelength failed to improve the reaction yield (entries 3 and 4). The use of other polar solvents like acetonitrile or DMF was compatible with the reaction, albeit providing lower yields in the obtention of 3aa. A change in the molar ratio of the reaction partners 1a and 2a led to a significant drop in the yield of the transformation (entries 7 and 8). Eventually, the employment of alkenyl chlorides or iodides (entries 9 and 10) provided the coupling product with low and very low yield, respectively; a quite expected result considering our previous experience in halogen-bonding assisted alkenylations.12
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield 3aa |
| 1 | Dark, 24 h | — |
| 2 | 16 h of irradiation | 27% |
| 3 | 366 nm instead of 390 nm lamp | 24% |
| 4 | 427 nm instead of 390 nm lamp | 10% |
| 5 | MeCN instead of DMSO | 45% |
| 6 | DMF instead of DMSO | 58% |
| 7 | 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio 1a/2a :1 molar ratio 1a/2a |
5% |
| 8 | 1:1 molar ratio 1a/2a |
23% |
| 9 | X = Cl | 35% |
| 10 | X = I | 5% |
Having established the optimal conditions for the photochemical alkenylation reaction, we next examined the scope of this novel transformation (Scheme 2a). This methodology demonstrated remarkable versatility by accommodating a diverse array of functional groups on the alkenylated phosphorothioate diesters, validating the robustness of the transformation. For instance, electron-withdrawing moieties could be installed in the final products such as the trifluoromethyl group (3ab), halogens (3ae, 3ai, 3ah), esters (3aj) and nitriles (3al). Neutral aromatic groups such as the phenyl (3aa) or 2-naphtyl (3ad) were also compatible with this methodology. Remarkably, the presence of electron-donating groups at the alkenyl fragment, which theoretically could diminish the strength of the halogen-bond, also enabled the formation of the phosphorothioate diesters. These include for example compounds bearing a methoxy (3ac) or thiomethoxy (3af) functionalities. Importantly, the presence of heterocycles such as benzofuran (3am) or furan (3an) in the alkenyl fragment was compatible with our methodology. Shifting focus to the examination of the phosphorothioate salts 2, we observed tolerance for different alkyl groups such as methyl, ethyl, isopropyl, or homoallyl. This protocol could be extended even to fragments resembling natural products, as demonstrated by the synthesis of vanillin-derived (3ao) and (−)-citronellol-derived (3ap) compounds. However, it should be pointed out that aromatic esters lead to the formation of the S-alkenylated phosphorothioate 3fa in barely traces. Particularly interesting is the synthesis of the compound 3ai, which leaves intact the aromatic bromide at 1i, also prone to form a HB complex with 2a. This result is in line with previous competition experiments carried out in our group in transformations based on photochemical activation of HB complexes.12
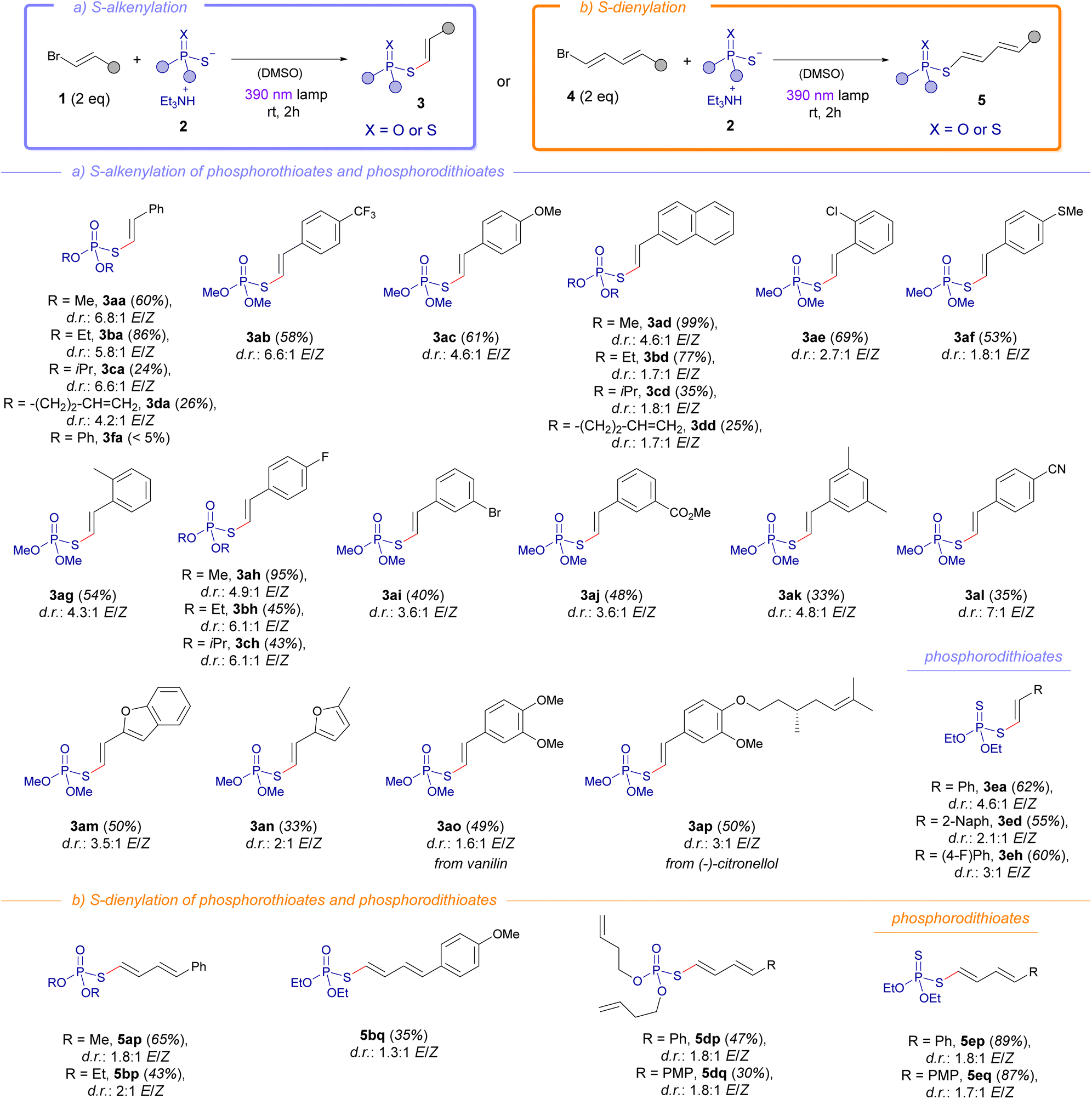 | ||
| Scheme 2 Scope of the S-alkenylation (light purple) and S-dienylation (orange) of phosphorothioate and phosphorodithioate diesters. General reaction conditions: alkenyl bromide 1 (0.3 mmol) and phosphorodithioate salt 2 (0.15 mmol) in 3 mL of DMSO, irradiation with a 390 nm lamp (52 W) over 2 hours. Isolated yields after flash chromatography are presented. PMP = 4-methoxyphenyl. | ||
In an extension of our work, we successfully conducted the alkenylation reaction using phosphorodithioate diesters, yielding the target compounds 3ea, 3ed and 3eh with commendable yields. Given the success of the alkenylation reaction, we wondered if the analogous 1,3-dienylation could take place, which would significantly expand the scope of the photochemical halogen-bonding assisted reaction. Indeed, the reaction of the phosphorothioate salts 2 with the 1,3-dienyl bromides 4 granted the formation of the dienylated phosphorothioates 5 under the same reaction conditions as for the alkenylation reaction (Scheme 2b). In this context, different variations at the alkyl fragment of the phosphorothioate salts 2 were compatible in the reaction with the dienyl bromides, validating the synthetic utility of our S-dienylation reaction for the preparation of the compounds 5ap to 5dp in moderate to good yields. Remarkably, the incorporation of a phosphorodithioate fragment could be accomplished, giving rise to the compounds 5ep and 5eq with very good yields.
A set of mechanistic studies was carried out to support the formation of a halogen-bonding complex during the transformation. First, we recorded the UV-vis profiles of solutions of the individual compounds 1a and 2a. Importantly, when mixing equimolar amounts of both reaction components in DMSO, a distinctive red-shifted charge transfer (CT) band could be observed, a telltale sign of the formation of the halogen-bonding complex 6aa (Scheme 3a).14,19 This observation aligns with our rationale behind selecting the 390 nm lamp, strategically chosen to facilitate the photoexcitation of the HB complex 6aa. Additional evidence for the existence of the halogen-bonding (HB) complex emerged through the implementation of 31P-NMR titration experiments (Scheme 3b).14,19,20 Within this context, we meticulously tracked a striking downfield shift, nearing 1 ppm, of the 31P-NMR signal of 2a as we systematically introduced incremental quantities of 1a while maintaining a consistent molar amount of 2a (see ESI† for details). Eventually, we carried out preliminary DFT computational studies (ω-B97x-D3/Def2TZVPP level) to identify the nature of the halogen-bonding complex 6aa (Scheme 3c). These endeavors led to the identification of a halogen-bonding assembly involving 1a and 2a, validated as a local minimum on the potential energy surface. Notably, the halogen bond interaction between the bromine and sulfur atoms demonstrated a strikingly defined directionality, as evidenced by a dihedral angle of 178.6° within the C(sp2)–Br–S bonds. This linear geometry, characterized by dihedral angles spanning from 160° to 180° within the HB complex, concurs with findings documented in prior investigations.21 Indeed, in the electrostatic potential surface, it is observed that a positively charged bromine σ–hole zone is located at almost the center of the C–Br axis. Furthermore, the distance between the bromine and sulfur atoms within the halogen-bonding complex 6aa (3.35 Å) is distinctly shorter than the cumulative van der Waals radii of these atoms (3.65 Å). This observation bolsters the notion of a substantial non-covalent interaction binding these atoms, most likely attributed to the formation of the halogen bond.
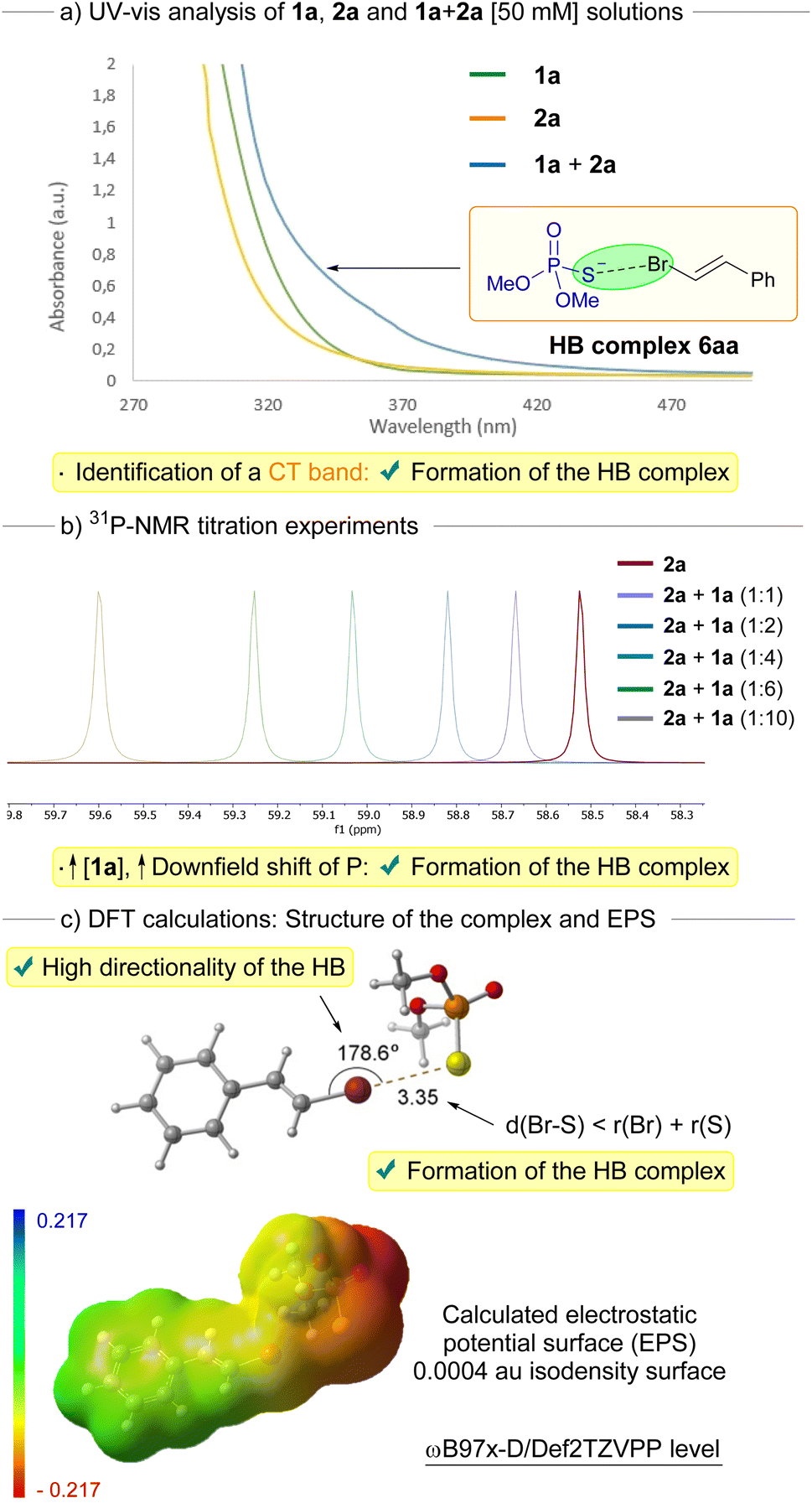 | ||
| Scheme 3 Set of mechanistic studies run towards the identification of the structure and nature of the halogen-bonding complex 6aa. EPS = Electrostatic Potential Surface. | ||
Having initially hypothesized the emergence of alkenyl radicals during our transformation, we undertook a distinct series of mechanistic investigations to probe the radical nature of our reaction. Primarily, we noted that the introduction of radical scavengers, such as TEMPO, caused a complete cessation of the transformation, a compelling endorsement of the radical-mediated pathway (Scheme 4a). Next, the nature of the alkenyl radical formed during the transformation was studied. Since two different structures are normally speculated for the photochemical generation of a vinyl radical (a bent σ-type or a linear π-type),22 we embarked on stereoconvergence experiments to offer deeper insights into the inherent structure of these highly-reactive alkenyl radicals (Scheme 4b).23 To achieve this, we investigated the stereochemical outcomes in the formation of the compound 3aa arising from the reaction between either a cis-configured alkenyl bromide cis-1a or 1a with 2a, respectively. In each of the reactions, a photochemical fragmentation of the HB complex would initially lead to the formation of the sulfur-centered radical 9a and, the angular π-type radicals cis-7a or trans-7a, respectively. At this point, two distinct processes can unfold: (1) a direct diastereoretentive recombination of the radical 9a with either cis-7a or trans-7a, respectively, to forge 3aa; or (2) a kinetic- and/or thermodynamically driven geometric isomerization, transitioning from each of the angular π-type 7a radicals to the linear σ-type radical 8a, which eventually can recombine with 9a to create 3aa, displaying a complete loss of the initial stereogenic information from 1a. Remarkably, the absence of a fully stereoconvergent process in both reactions generating 3aa can be attributed to the coexistence of both these aforementioned processes. This fact effectively accounts for the limited extent of stereoconvergence and the unequal loss of stereochemical information observed across both reactions.22 Interestingly, the group of Kanai recently reported that thiyl radicals emerging from BINOL-derived phosphorothioate diesters can be useful as hydrogen atom transfer catalysts in photochemical processes.24 This is due to the rather long lifetimes these sulfur-centered radicals exhibit (greater than 1000 ns), as demonstrated by transient absorption measurements.
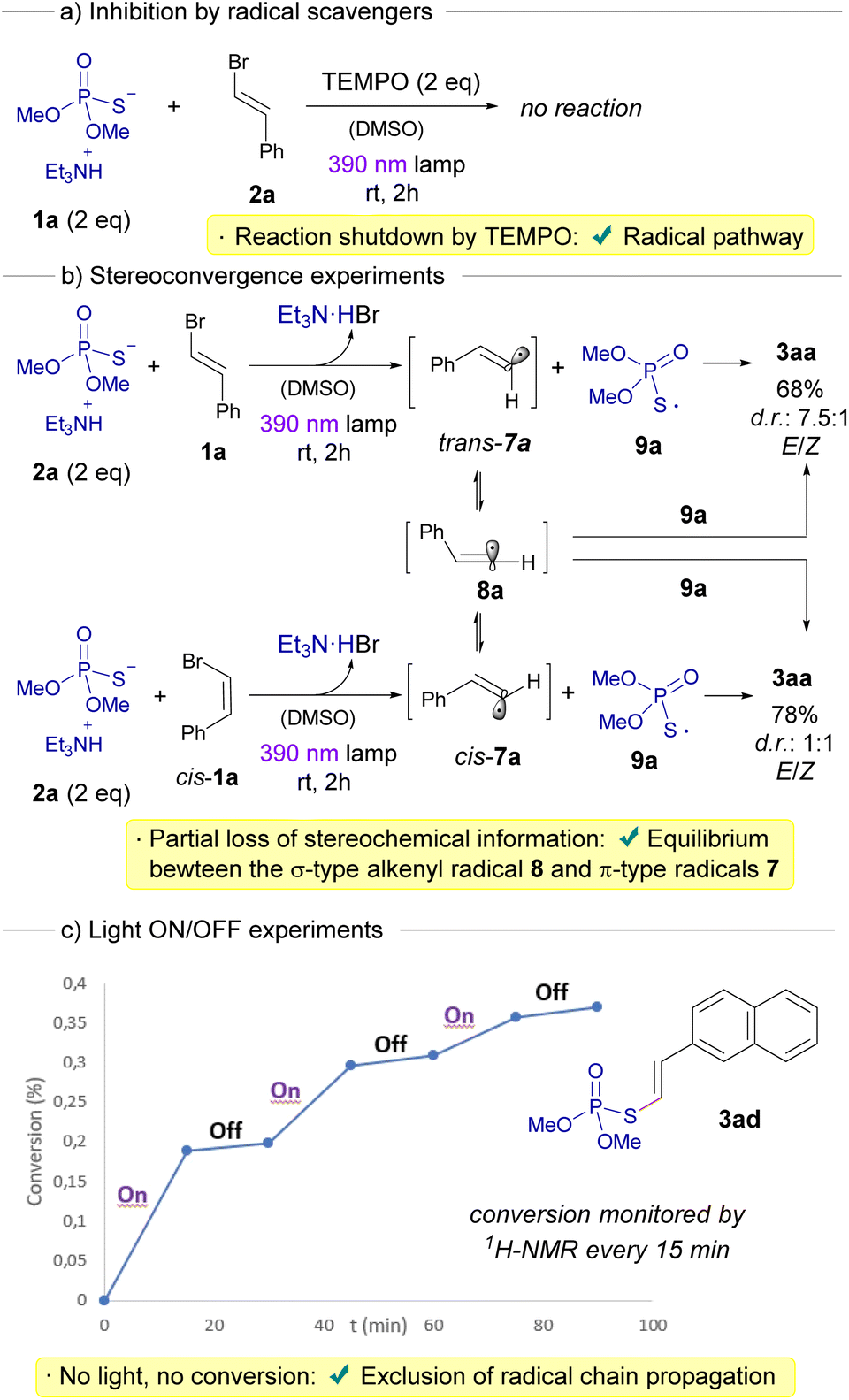 | ||
| Scheme 4 Elucidation of the radical pathway. | ||
Furthermore, we conducted light on/off experiments, strategically designed to both underscore the indispensability of light and ascertain the presence of radical chain propagation events (Scheme 4c).15d,25 The latter were conclusively ruled out, given the absence of conversion in the reaction during long intervals when the light source was deactivated, as gauged by monitoring the reaction progression via1H-NMR.
Based on our mechanistic investigations and the precedent set by other photochemical halogen-bonding assisted reactions,14 the mechanism described in the Scheme 5 is proposed. First, the formation of a halogen-bonding complex 6 between the alkenyl bromide 1 and phosphorothioate salt 2 is proposed to be formed. Photoexcitation and subsequent fragmentation of this aggregate would lead to the creation of the sulfur centered radical 9 and the angular π-type alkenyl radical 7, which is in equilibrium with the corresponding linear σ-type vinyl radical 8. A radical recombination of either 7 or 8 with 9 would eventually forge the final products 3.
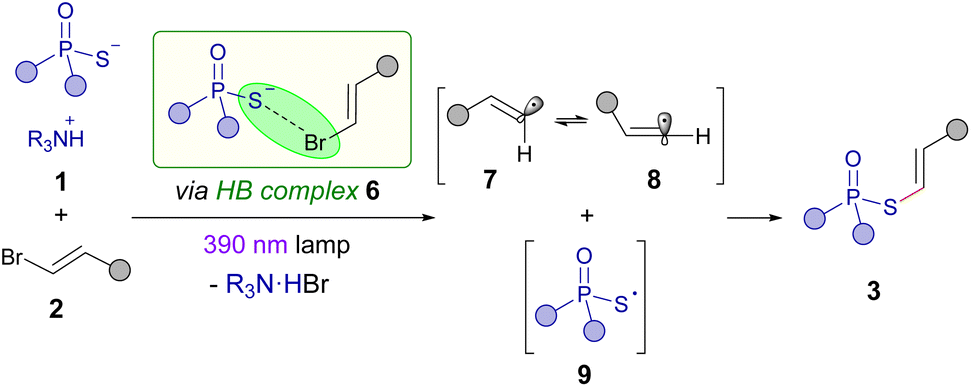 | ||
| Scheme 5 Mechanistic rationale for the photochemical reaction. | ||
The last part of our work was devoted to the development of a continuous flow protocol to enable the scale up of the photochemical transformations. In general, the development of light-driven reactions through continuous flow protocols offers several advantages, which encompass: rapid scalability, enhanced security, better reproducibility and easier control.26 Therefore, we conducted an initial investigation to assess the feasibility of this potential application using a flow reactor constructed from PTFE tubing (see ESI† for details). Our preliminary protocol involved flowing a solution of both reaction components 1 and 2 in DMSO through the flow reactor while subjecting it to 80 min of illumination from a 390 nm lamp. Significantly, thanks to the enhanced efficiency of our process, we have achieved notable improvements in several key aspects compared to the corresponding batch reactions: (a) a remarkable 20–50% general enhancement in reaction yields accross the scope, exemplified, for instance, by the compounds 3ak, 3ca, 5bp, 5bq and 3cd; (b) reduced reaction times (80 min of irradiation versus 120 min in batch); (c) higher diastereomeric ratios in favour of the trans isomer, due to the less exposure time to irradiation of our reaction products (Scheme 6).
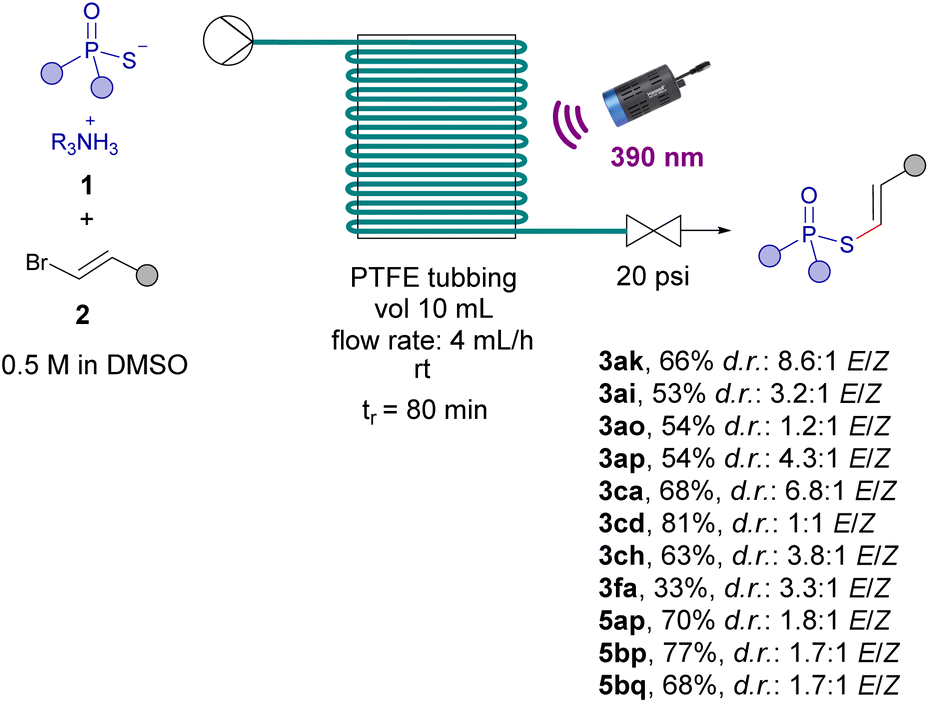 | ||
| Scheme 6 Scale up through a continuous flow protocol. | ||
Remarkably, following our flow protocol, the synthesis of the phenyl phosphorothioate 3fa could be accomplished, which was not possible under batch conditions. All in all, these advancements underscore the value of our flow procedure for facilitating the upscaling and improving reaction efficiencies of the photochemical alkenylations and dienylations.
3 Conclusions
In summary, we have now reported a simple, general and mild procedure for the preparation of novel S-alkenyl and S-dienyl phosphoro(di)thioate esters. The procedure relies on a photochemical excitation with visible-light of a halogen-bonding complex formed between an alkenyl or dienyl bromide with a phosphoro(di)thioate salt. Importantly, there are no other methodologies available for the synthesis of these compounds in a general and straightforward manner. A very efficient flow protocol was developed to upscale the transformations. Overall, we believe the ongoing advancements in the synthesis of novel organosulfur compounds with alkenyl and dienyl substituents within the P–S–C(sp2) structural motif could potentially lead to the discovery of new molecules with improved biological properties and important applications in various fields, such as the agrochemical or drug industries.Data availability
All of the necessary data had been included in the ESI.†Author contributions
H. F. Piedra and V. Gebler conducted all of the reactions, DFT calculations, experimental mechanistic studies and full characterization of the compounds. C. Valdés contributed to the supervision and direction of the project. M. Plaza conceptualized and directed the project and wrote the manuscript. All the authors contributed to scientific discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of this work by a FICYT (Principality of Asturias) “Margarita Salas Joven” postdoctoral grant to M. P. (AYUD/2021/58397) and Ministerio de Ciencia e Innovación of Spain (Agencia Estatal de Investigación: PID2019-107580GB-I00/AEI/10.13039/501100011033). A “Severo Ochoa” predoctoral fellowship to H. F. P. (BP21/050) by FICYT and an Erasmus+ scholarship to V. G. are also gratefully acknowledged.Notes and references
- Selected reviews: (a) C.-F. Lee, R. S. Basha and S. S. Badsara, Top. Curr. Chem., 2018, 376, 25 CrossRef PubMed; (b) P. Annamalai, K. Liu, S. Singh Badsara and C. Lee, Chem. Rec., 2021, 21, 3674–3688 CrossRef CAS PubMed; (c) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2022, 122, 16110–16293 CrossRef CAS PubMed; (d) Z. Wu and D. A. Pratt, Nat. Rev. Chem, 2023, 7, 573–589 CrossRef PubMed; (e) J. Feng, Y. Zhang, X. Wang, J. Liu, V. Benazzi, K. Lu, X. Zhao and S. Protti, Adv. Synth. Catal., 2023, 365, 3413–3431 CrossRef.
- (a) J. Purcell and A. C. Hengge, J. Org. Chem., 2005, 70, 8437–8442 CrossRef CAS PubMed; (b) A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC; (c) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS PubMed; (d) M. Nassir, M. Ociepa, H.-J. Zhang, L. N. Grant, B. J. Simmons, M. S. Oderinde, Y. Kawamata, A. N. Cauley, M. A. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2023, 145, 15088–15093 CrossRef CAS PubMed.
- (a) L. D. Quin, A Guide to Organophosphorus Chemistry, Wiley, New York, 2000 Search PubMed; (b) P. J. Murphy, Organophosphorus Reagents: A Practical Approach in Chemistry, Oxford University Press, New York, 2004 CrossRef.
- (a) L. L. Murdock and T. L. Hopkins, J. Agric. Food Chem., 1968, 16, 954–958 CrossRef CAS; (b) K. D. Wing, A. H. Glickman and J. E. Casida, Pestic. Biochem. Physiol., 1984, 21, 22–30 CrossRef CAS.
- (a) N.-S. Li, J. K. Frederiksen and J. A. Piccirilli, Acc. Chem. Res., 2011, 44, 1257–1269 CrossRef CAS PubMed; (b) J. Duschmalé, H. F. Hansen, M. Duschmalé, E. Koller, N. Albaek, M. R. Møller, K. Jensen, T. Koch, J. Wengel and K. Bleicher, Nucleic Acids Res., 2020, 48, 63–74 CrossRef PubMed; (c) H.-J. Zhang, M. Ociepa, M. Nassir, B. Zheng, S. A. Lewicki, V. Salmaso, H. Baburi, J. Nagel, S. Mirza, B. Bueschbell, H. Al-Hroub, O. Perzanowska, Z. Lin, M. A. Schmidt, M. D. Eastgate, K. A. Jacobson, C. E. Müller, J. Kowalska, J. Jemielity and P. S. Baran, Nat. Chem., DOI:10.1038/s41557-023-01347-2.
- Selected examples: (a) S. Ohuchi, H. Ayukawa and T. Hata, Chem. Lett., 1992, 21, 1501–1504 CrossRef; (b) Y. Nishiyama, Y. Hazama, S. Yoshida and T. Hosoya, Org. Lett., 2017, 19, 3899–3902 CrossRef CAS PubMed.
- Recent reviews: (a) D. J. Jones, E. M. O'Leary and T. P. O'Sullivan, Adv. Synth. Catal., 2020, 362, 2801–2846 CrossRef CAS; (b) X. Liu, L. Zhou, R. Yang, X.-R. Song and Q. Xiao, Adv. Synth. Catal., 2023, 365, 2280–2298 CrossRef CAS.
- Selected examples: (a) J. Bai, X. Cui, H. Wang and Y. Wu, Chem. Commun., 2014, 50, 8860–8863 RSC; (b) Y. Zhu, T. Chen, S. Li, S. Shimada and L.-B. Han, J. Am. Chem. Soc., 2016, 138, 5825–5828 CrossRef CAS PubMed; (c) S. Kovács, B. Bayarmagnai, A. Aillerie and L. J. Gooßen, Adv. Synth. Catal., 2018, 360, 1913–1918 CrossRef.
- Selected examples: (a) S. Song, Y. Zhang, A. Yeerlan, B. Zhu, J. Liu and N. Jiao, Angew. Chem., Int. Ed., 2017, 56, 2487–2491 CrossRef CAS PubMed; (b) J. Shen, Q.-W. Li, X.-Y. Zhang, X. Wang, G.-Z. Li, W.-Z. Li, S.-D. Yang and B. Yang, Org. Lett., 2021, 23, 1541–1547 CrossRef CAS PubMed; (c) P. Ghosh and A. Hajra, Adv. Synth. Catal., 2022, 364, 4157–4165 CrossRef CAS.
- J. Yan and Z.-C. Chen, Synth. Commun., 1999, 29, 3605–3612 CrossRef CAS.
- Z. Zheng, J. He, Q. Ma, Y. Zhang, Y. Liu, G. Tang and Y. Zhao, Green Chem., 2022, 24, 4484–4489 RSC.
- H. F. Piedra and M. Plaza, Chem. Sci., 2023, 14, 650–657 RSC.
- Selected examples on recent photochemical thiol-yne reactions: (a) S. S. Zalesskiy, N. S. Shlapakov and V. P. Ananikov, Chem. Sci., 2016, 7, 6740–6745 RSC; (b) S. Kaur, G. Zhao, E. Busch and T. Wang, Org. Biomol. Chem., 2019, 17, 1955–1961 RSC; (c) J. V. Burykina, N. S. Shlapakov, E. G. Gordeev, B. König and V. P. Ananikov, Chem. Sci., 2020, 11, 10061–10070 RSC.
- For a recent review, see: H. F. Piedra, C. Valdés and M. Plaza, Chem. Sci., 2023, 14, 5545–5568 RSC.
- Selected recent examples: (a) T. Li, K. Liang, J. Tang, Y. Ding, X. Tong and C. Xia, Chem. Sci., 2021, 12, 15655 RSC; (b) Y. Shen, N. Lei, C. Lu, D. Xi, X. Geng, P. Tao, Z. Su and K. Zheng, Chem. Sci., 2021, 12, 15399–15406 RSC; (c) S. Cuadros, C. Rosso, G. Barison, P. Costa, M. Kurbasic, M. Bonchio, M. Prato, G. Filippini and L. Dell'Amico, Org. Lett., 2022, 24, 2961 CrossRef CAS PubMed; (d) N. Kato, T. Nanjo and Y. Takemoto, ACS Catal., 2022, 12, 7843 CrossRef CAS; (e) C. Zhang, H. Zuo, G. Y. Lee, Y. Zou, Q.-D. Dang, K. N. Houk and D. Niu, Nat. Chem., 2022, 14, 686 CrossRef CAS PubMed.
- Reviews and selected publications on the importance of halogen-bonding interactions: (a) T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC; (b) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed; (c) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed; (d) R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS; (e) M. Breugst and J. J. Koenig, Eur. J. Org Chem., 2020, 2020, 5473–5487 CrossRef CAS.
- A. Karpfen, J. Phys. Chem. A, 2000, 104, 6871–6879 CrossRef CAS.
- For some recent reviews covering photochemistry of EDA-complexes, see: (a) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461 CrossRef CAS PubMed; (b) Y. Yuan, S. Majumder, M. Yang and S. Guo, Tetrahedron Lett., 2020, 61, 151506 CrossRef CAS; (c) Y. Sempere, M. Morgenstern, T. Bach and M. Plaza, Photochem. Photobiol. Sci., 2022, 21, 719 CrossRef CAS PubMed.
- M. Erdélyi, Chem. Soc. Rev., 2012, 41, 3547 RSC.
- P. Metrangolo, W. Panzeri, F. Recupero and G. Resnati, J. Fluorine Chem., 2002, 114, 27–33 CrossRef CAS.
- (a) M. G. Sarwar, B. Dragisic, L. J. Salsberg, C. Gouliaras and M. S. Taylor, J. Am. Chem. Soc., 2010, 132, 1646–1653 CrossRef CAS PubMed; (b) L. Maugeri, E. M. G. Jamieson, D. B. Cordes, A. M. Z. Slawin and D. Philp, Chem. Sci., 2017, 8, 938–945 RSC.
- (a) Free Radicals in Organic Chemistry, ed. J. Fossey, D. Lefort and J. Sorba, Wiley and Masson, 1995 Search PubMed; (b) T. P. M. Goumans, K. Van Alem and G. Lodder, Eur. J. Org Chem., 2008, 2008, 435–443 CrossRef.
- S. K. Pagire, T. Föll and O. Reiser, Acc. Chem. Res., 2020, 53, 782 CrossRef CAS PubMed.
- H. Fuse, Y. Irie, M. Fuki, Y. Kobori, K. Kato, A. Yamakata, M. Higashi, H. Mitsunuma and M. Kanai, J. Am. Chem. Soc., 2022, 144, 6566–6574 CrossRef CAS PubMed.
- A. Nandy, I. Kazi, S. Guha and G. Sekar, J. Org. Chem., 2021, 86, 2570–2581 CrossRef CAS PubMed.
- For some reviews on continuous flow photochemistry: (a) D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed; (b) L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05263j |
| This journal is © The Royal Society of Chemistry 2023 |