
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A focus on detection of polymorphs by dynamic nuclear polarization solid-state nuclear magnetic resonance spectroscopy
Yunhua
Chen†
b,
Jiashan
Mi†
a and
Aaron J.
Rossini
*a
aDepartment of Chemistry, Iowa State University, Ames, IA 50011, USA. E-mail: arossini@iastate.edu
bAnalytical Research & Development, AbbVie, Inc., North Chicago, Illinois 60064, USA
First published on 28th September 2023
Abstract
Solid-state nuclear magnetic resonance (ssNMR) spectroscopy has found increasing application as a method for quantification and structure determination of solid forms (polymorphs) of organic solids and active pharmaceutical ingredients (APIs). However, ssNMR spectroscopy suffers from low sensitivity and resolution, making it challenging to detect dilute solid forms that may be present after recrystallization or reaction with co-formers. Cousin et al. (S. F. Cousin et al., Chem. Sci., 2023, https://doi.org/10.1039/D3SC02063K) have demonstrated that dynamic nuclear polarization (DNP) enhanced 13C cross-polarization (CP) saturation recovery experiments can be used to detect dilute polymorphic forms that are present within a mixture of solid forms. Enhancement of the NMR signal by DNP and differences in signal build-up rates for different polymorphs provide the sensitivity and contrast needed to resolve NMR signals from minor polymorphic forms. This method demonstrated by Cousin et al. should aid the discovery of solid drug forms.
Different solid phases (forms) of active pharmaceutical ingredients (APIs) display varying stability, solubility, and bioavailability and can also be patented.1 Consequently, when solid APIs are being prepared for formulation, crystallization screening experiments are used to search for as many solid phases as possible.1 However, even after extensive solid form screening, new crystal forms can be discovered.2,3 The unexpected emergence of API forms can pose challenges, particularly during late-stage product development or post-launch, as newfound API forms may exhibit undesirable properties, such as reduced solubility.2,3 Diffraction, microscopy, and spectroscopy techniques are used to structurally characterize and detect different solid drug forms.4,5 Solid-state nuclear magnetic resonance (ssNMR) spectroscopy is a powerful technique for polymorph characterization because it can probe the 3D arrangement of atoms and their motions via measurements of chemical shifts, coupling constants, and relaxation times.6–9 However, ssNMR spectroscopy generally suffers from poor sensitivity, meaning it is challenging (and sometimes impossible) to detect minor API phases within mixtures or dilute APIs in drug formulations.
Due to the efforts of Griffin and co-workers, dynamic nuclear polarization (DNP) has emerged as a method to routinely enhance the sensitivity of magic angle spinning (MAS) ssNMR experiments.10 In a DNP experiment the sample is doped with stable free radicals. Microwave irradiation at or near to the electron Larmor frequency is used to facilitate polarization transfer from electron spins to the surrounding nuclear spins, typically resulting in a 1–2 order of magnitude improvement in NMR sensitivity. Relayed DNP experiments have previously been used to enhance the sensitivity of 1D and 2D ssNMR experiments on organic solids, pure APIs, and formulated APIs.11 In a relayed DNP experiment on crystalline APIs, the radicals will be restricted to the surface of the material, resulting in an initial build-up of 1H polarization at the surface of the crystals.12 However, 1H nuclear spin diffusion will spontaneously relay polarization from surface 1H spins to sub-surface or bulk 1H spins.12–14 Polarization from the 1H spins can then be transferred to nearby heteronuclear spins such as 13C, 15N, 17O, etc. using pulsed NMR methods. The efficiency of the 1H nuclear spin diffusion process and DNP enhancements are determined by the particle size distribution, the concentration of 1H spins and 1H T1.12,13
The recent work of Cousin et al. “Exploiting solid-state dynamic nuclear polarization NMR spectroscopy to establish the spatial distribution of polymorphic phases in a solid material” uses the features of relayed DNP experiments to detect minor polymorphic impurity phases present at a concentration of a few weight percent.15 In this paper they studied crystallized samples of meta-aminobenzoic acid (m-ABA) because this is a well-studied model system known to exhibit polymorphism. Most of the m-ABA they studied existed as Form I, however, there was a small amount of Form III m-ABA that was naturally present after recrystallization. DNP-enhanced 1H–13C CPMAS saturation recovery experiments were used to monitor the build-up of 1H polarization in the m-ABA samples as a function of the DNP polarization delay (τ, Fig. 1a). As expected, the 1D 13C ssNMR spectra only show NMR signals from the more concentrated Form I because the Form III NMR signals are much weaker and are obscured by those of Form I (Fig. 1b and c). However, subtraction of the DNP-enhanced 13C ssNMR spectrum recorded with τ of 12 s from the spectrum recorded with τ of 350 s revealed the 13C NMR spectrum of Form III (Fig. 1d). Form III had a much longer 1H DNP build-up time than Form I, allowing the two different drug forms to be resolved in the saturation recovery experiment. The results shown in Fig. 1 are significant as they demonstrate the potential utility of DNP-enhanced 13C ssNMR in the context of solid form screening. The DNP enhancements and signal build-up rates provide the contrast needed to resolve NMR signals from minor polymorphic forms, while the signal enhancement provided by DNP enables the detection of the NMR spectrum of the dilute solid form in the mixture. Furthermore, the authors used numerical simulations of 1H spin diffusion to model DNP build-up curves for different micron-scale spatial distributions of Form I and Form III. The authors’ simulations suggest that spatial segregation of Form I and Form III crystals best models the NMR experiments (Model B, Fig. 2). Overall, the experiments of Cousin et al. illustrate many potential advantages of DNP-enhanced ssNMR spectroscopy for investigating crystallization of APIs.
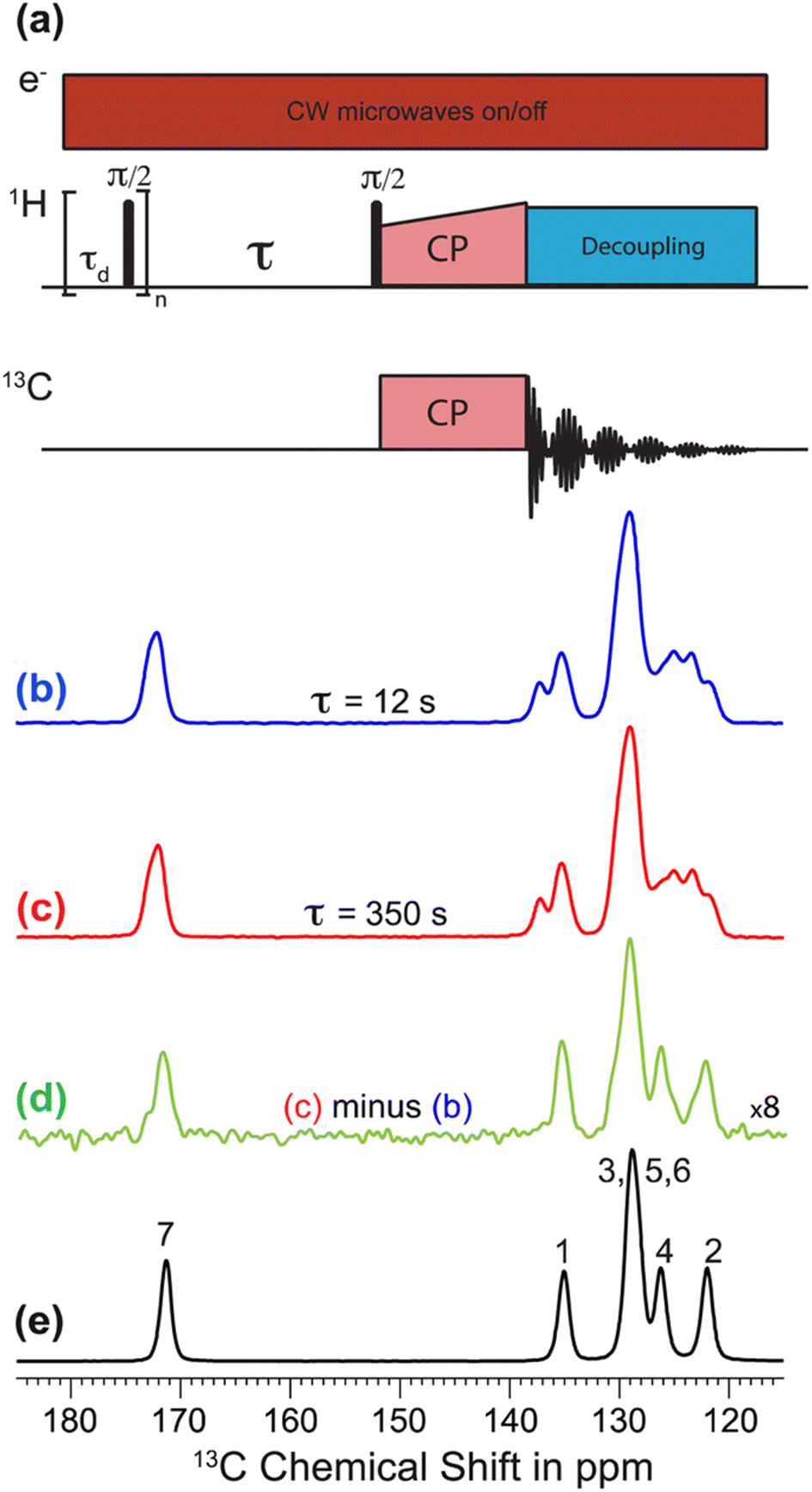 | ||
| Fig. 1 (a) 1H–13C CPMAS saturation–recovery pulse sequence used to record solid-state 13C NMR spectra for different polarization times (τ). (b and c) DNP-enhanced solid-state 13C NMR spectra of powdered m-ABA recorded at 110 K with (b) τ = 12 s and (c) τ = 350 s. (d) Difference spectrum. (e) Solid-state 13C NMR spectrum recorded at 110 K for a sample of pure Form III m-ABA. Reproduced with permission from ref. 15. | ||
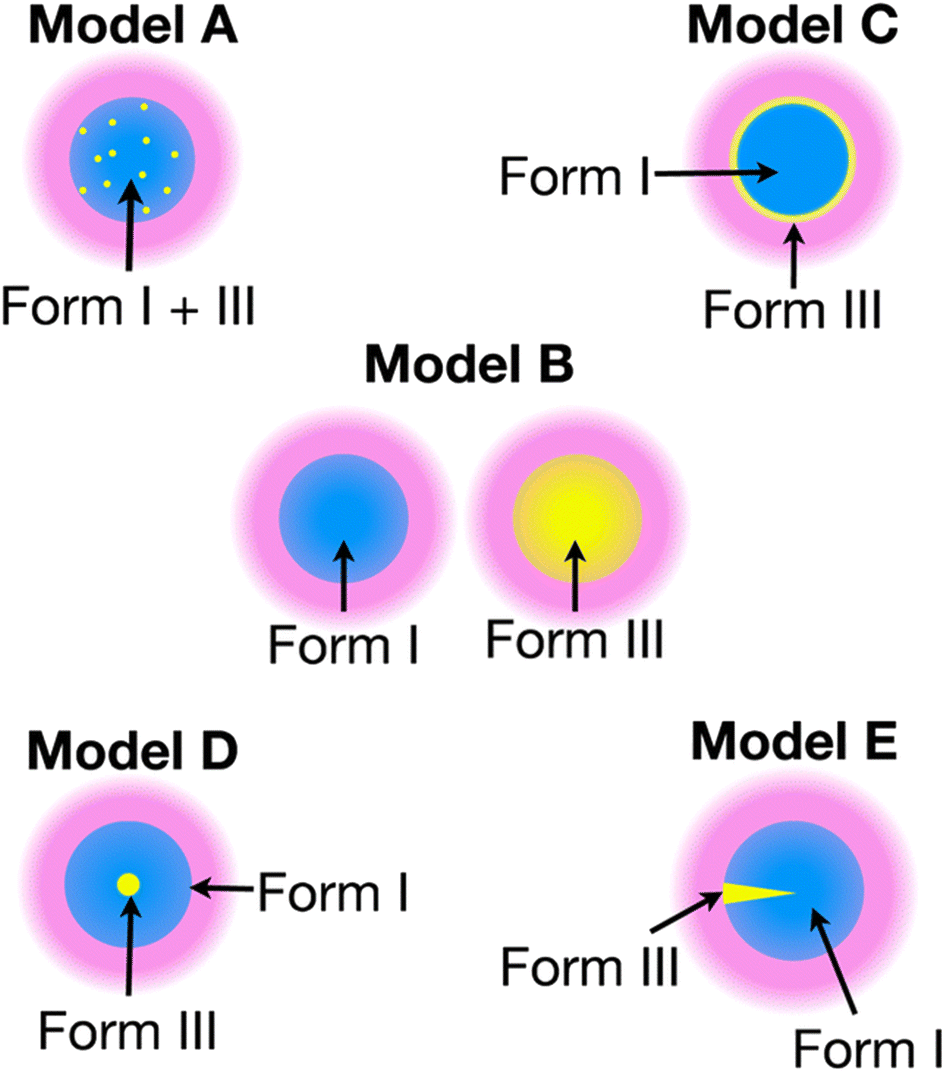 | ||
| Fig. 2 Models A–E show different possible distributions of the two m-ABA polymorphs, Form I and Form III. The solution (pink) containing the polarizing agent is located on the surface of the particles. Numerical simulations of 1H spin diffusion suggested that model B best reproduced the DNP saturation recovery experiments. Reproduced with permission from ref. 15. | ||
There are exciting future directions for these types of NMR experiments, with the most obvious being a better integration with theoretical methods. Computational crystal structure prediction (CSP) has grown increasingly powerful in the past few years, enabling prediction of possible crystal forms and ranking of their lattice energies.16 Relatedly, 1H and 13C chemical shifts can be accurately calculated for different crystal forms using planewave DFT17 or machine-learning based methods.18 The combinations of experimental chemical shift measurement, CSP and calculations of NMR chemical shifts has been dubbed “NMR crystallography” as it enables crystal structure determination from NMR observables.19,20 Therefore, the ability to observe the NMR spectrum of a crystalline organic solid provides the potential means to determine its crystal structure. Using the experiments demonstrated by Cousin et al. and CSP it should be possible to record the NMR signatures of dilute crystal forms present in a mixture, determine their crystal structures and assess if they are worthwhile targets for further development and exploration. Finally, characterization of the spatial distribution of the different crystalline forms by DNP could provide insight into ways to process solid APIs to remove undesired crystal forms or to discover new solid phases.
Author contributions
Y. C., J. M. and A. J. R. equally contributed to writing the article.Conflicts of interest
There are no conflicts to declare.References
- M. Y. Gokhale and R. V. Mantri, in Developing Solid Oral Dosage Forms, ed. Y. Qiu, Y. Chen, G. G. Z. Zhang, L. Yu and R. V. Mantri, Academic Press, Boston, 2nd edn, 2017, pp. 85–112, DOI:10.1016/B978-0-12-802447-8.00004-2.
- J. D. Dunitz and J. Bernstein, Acc. Chem. Res., 1995, 28, 193–200 CrossRef CAS.
- D.-K. Bučar, R. W. Lancaster and J. Bernstein, Angew. Chem., Int. Ed., 2015, 54, 6972–6993 CrossRef PubMed.
- T. L. Threlfall, Analyst, 1995, 120, 2435–2460 RSC.
- S. R. Byrn, R. R. Pfeiffer and J. G. Stowell, Solid-state chemistry of drugs, SSCI, Inc., West Lafayette, Ind., 1999 Search PubMed.
- R. K. Harris, J. Pharm. Pharmacol., 2007, 59, 225–239 CrossRef CAS PubMed.
- M. Li, W. Xu and Y. Su, TrAC, Trends Anal. Chem., 2021, 135, 116152 CrossRef CAS.
- R. T. Berendt, D. M. Sperger, E. J. Munson and P. K. Isbester, TrAC, Trends Anal. Chem., 2006, 25, 977–984 CrossRef CAS.
- M. Geppi, G. Mollica, S. Borsacchi and C. A. Veracini, Appl. Spectrosc. Rev., 2008, 43, 202–302 CrossRef CAS.
- T. Maly, G. T. Debelouchina, V. S. Bajaj, K.-N. Hu, C.-G. Joo, M. L. Mak-Jurkauskas, J. R. Sirigiri, P. C. A. van der Wel, J. Herzfeld, R. J. Temkin and R. G. Griffin, J. Chem. Phys., 2008, 128, 052211 CrossRef PubMed.
- L. Zhao, A. C. Pinon, L. Emsley and A. J. Rossini, Magn. Reson. Chem., 2018, 56, 583–609 CrossRef CAS PubMed.
- A. J. Rossini, A. Zagdoun, F. Hegner, M. Schwarzwälder, D. Gajan, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2012, 134, 16899–16908 CrossRef CAS PubMed.
- A. C. Pinon, J. Schlagnitweit, P. Berruyer, A. J. Rossini, M. Lelli, E. Socie, M. Tang, T. Pham, A. Lesage, S. Schantz and L. Emsley, J. Phys. Chem. C, 2017, 121, 15993–16005 CrossRef CAS.
- P. C. A. van der Wel, K.-N. Hu, J. Lewandowski and R. G. Griffin, J. Am. Chem. Soc., 2006, 128, 10840–10846 CrossRef CAS PubMed.
- S. F. Cousin, C. E. Hughes, F. Ziarelli, S. Viel, G. Mollica, K. D. M. Harris, A. C. Pinon and P. Thureau, Chem. Sci., 2023 10.1039/D3SC02063K.
- A. M. Reilly, R. I. Cooper, C. S. Adjiman, S. Bhattacharya, A. D. Boese, J. G. Brandenburg, P. J. Bygrave, R. Bylsma, J. E. Campbell, R. Car, D. H. Case, R. Chadha, J. C. Cole, K. Cosburn, H. M. Cuppen, F. Curtis, G. M. Day, R. A. DiStasio Jr, A. Dzyabchenko, B. P. van Eijck, D. M. Elking, J. A. van den Ende, J. C. Facelli, M. B. Ferraro, L. Fusti-Molnar, C.-A. Gatsiou, T. S. Gee, R. de Gelder, L. M. Ghiringhelli, H. Goto, S. Grimme, R. Guo, D. W. M. Hofmann, J. Hoja, R. K. Hylton, L. Iuzzolino, W. Jankiewicz, D. T. de Jong, J. Kendrick, N. J. J. de Klerk, H.-Y. Ko, L. N. Kuleshova, X. Li, S. Lohani, F. J. J. Leusen, A. M. Lund, J. Lv, Y. Ma, N. Marom, A. E. Masunov, P. McCabe, D. P. McMahon, H. Meekes, M. P. Metz, A. J. Misquitta, S. Mohamed, B. Monserrat, R. J. Needs, M. A. Neumann, J. Nyman, S. Obata, H. Oberhofer, A. R. Oganov, A. M. Orendt, G. I. Pagola, C. C. Pantelides, C. J. Pickard, R. Podeszwa, L. S. Price, S. L. Price, A. Pulido, M. G. Read, K. Reuter, E. Schneider, C. Schober, G. P. Shields, P. Singh, I. J. Sugden, K. Szalewicz, C. R. Taylor, A. Tkatchenko, M. E. Tuckerman, F. Vacarro, M. Vasileiadis, A. Vazquez-Mayagoitia, L. Vogt, Y. Wang, R. E. Watson, G. A. de Wijs, J. Yang, Q. Zhu and C. R. Groom, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 439–459 CrossRef CAS PubMed.
- C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint, T. Azaïs, S. E. Ashbrook, J. M. Griffin, J. R. Yates, F. Mauri and C. J. Pickard, Chem. Rev., 2012, 112, 5733–5779 CrossRef CAS PubMed.
- F. M. Paruzzo, A. Hofstetter, F. Musil, S. De, M. Ceriotti and L. Emsley, Nat. Commun., 2018, 9, 4501 CrossRef PubMed.
- D. Bryce, IUCrJ, 2017, 4, 350–359 CrossRef CAS PubMed.
- P. Hodgkinson, Prog. Nucl. Magn. Reson. Spectrosc., 2020, 118–119, 10–53 CrossRef CAS PubMed.
Footnote |
| † Y. C. and J. M. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |