
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-throughput and high-purity separation of malignant tumor cells in pleural and peritoneal effusions using interfacial elasto-inertial microfluidics†
Nan
Xiang
 *a,
Zhonghua
Ni
a and
Dan
Wu
*b
*a,
Zhonghua
Ni
a and
Dan
Wu
*b
aSchool of Mechanical Engineering and Jiangsu Key Laboratory for Design and Manufacture of Micro-Nano Biomedical Instruments, Southeast University, Nanjing, 211189, China. E-mail: nan.xiang@seu.edu.cn
bDepartment of Oncology, Jiangyin People's Hospital, Jiangyin, 214400, China. E-mail: wudan96121@163.com
First published on 13th June 2023
Abstract
Efficient separation of exfoliated malignant tumor cells (MTCs) from pleural and peritoneal effusions is critical for improving the detection sensitivity and reducing the time consumed for cytological assessment. Herein, an interfacial elasto-inertial microfluidic device was developed for the high-throughput and high-purity separation of MTCs from malignant pleural and peritoneal effusions. Through using hyaluronic acid as an elastic enhancer, a high sample throughput of 120 μL min−1 could be achieved. We first explored the effects of the flow-rate ratio and flow rate on particle separation. The results indicated that stable particle separation could be achieved over wide flow rates. We next characterized the device separation performance using blood samples spiked with various tumor cells (A549, MCF-7, and MDA-MB-231 cells) of different concentrations. A removal ratio of blood cells of over 99.8%, an average recovery ratio of tumor cells of 94%, and an average purity of tumor cells as high as 40% could be successfully achieved in a high-throughput and label-free manner. Finally, we successfully applied our device for the separation of MTCs in clinical pleural and peritoneal effusions from patients of different cancers. Our interfacial elasto-inertial microfluidic device may provide a potential sample preparation tool for cytological diagnosis of pleural and peritoneal effusions.
Introduction
Malignant pleural and peritoneal effusions are defined as the existence of exfoliated malignant tumor cells (MTCs) in abnormal pleural and peritoneal exudates.1,2 Metastatic malignancy is the main etiology for generating malignant pleural or peritoneal effusions, which is associated with poor prognosis and short survival time.3 Therefore, determining the malignancy of pleural and peritoneal effusions is critical for rapid and accurate diagnosis.Cytological diagnosis is still the widely used and recognized method for determining the malignancy of pleural and peritoneal effusions by manually enumerating MTCs in a cell smear under a microscope.4 However, the rarity of MTCs in large-volume fluids and the interference of a large number of background cells result in low detection sensitivity and time-consuming assessment. Therefore, it is essential to separate and purify the MTCs before smearing. In addition, the separated MTCs are important liquid biopsy sources for studying cancer metastasis and precision oncology.
Recently, microfluidics has been recognized as a powerful tool for various biomedical applications by engineering cells or fluids at a scale that is comparable to the cell diameter.5,6 To date, various novel microfluidic separators have been reported and some of which are successfully commercialized.7,8 According to the employed working principles, the reported microfluidic separators fall into two categories: active and passive microfluidic separators. The active microfluidic separators commonly employ external fields (e.g., magnetic,9 electric,10 optical,11 and acoustic12) to exert an external force on cells. Separation could be achieved by moving the different cells towards differential positions or trapping target cells at specific positions. As the external forces exerted on cells could be carefully tuned, a high manipulation accuracy could be realized by the active microfluidic separators. However, the processing throughput of these active separators is commonly very low due to the delayed time of external forces, which prevents the application of these active separators for processing large-volume effusions.
Meanwhile, the passive microfluidic separators achieve the cell separation based on the hydrodynamic forces induced by the fluidic effects or specific microstructures.13 Representative techniques in this category include inertial microfluidics,14,15 viscoelastic microfluidics,16,17 deterministic lateral displacement,18 hydrodynamic filtration,19 pinched flow fractionation,20 and so on. As a relatively high throughput could be offered by the passive microfluidic separators, they have been widely employed for the separation of rare cells (e.g., MTCs,21 circulating tumor cells (CTCs),22 and fetal nucleated red blood cells23) from complex cell populations. Specifically, spiral inertial microfluidics has been successfully used to separate MTCs from pleural and peritoneal effusions.21,24 Although a high throughput could be provided, the separation accuracy is still limited, which results in the low purity of the separated MTCs. Among these passive microfluidic separators, viscoelastic microfluidics has attracted increasing interest over recent years because a high manipulation accuracy could be offered by this technique.16,17 Through adding high molecular polymers or some biological macromolecules (e.g., DNA) into the carrying fluid, a viscoelasticity effect could be induced in viscoelastic microfluidics for achieving cell focusing.25 Further coupling the viscoelasticity effect with an inertial effect enables three-dimensional elasto-inertial focusing of cells at specific positions.26,27 According to the differential focusing statuses (central single-line focusing or symmetrical two-line focusing), the separation of differently-sized cells could be realized.28 However, the close distance between the focusing positions of differently-sized cells limits the separation performance. As compared with single-phase viscoelastic microfluidics, the interfacial viscoelastic microfluidics offers an improved performance for cell separation by using a co-flowing system.29 Currently, the interfacial viscoelastic microfluidics is successfully applied to separate Escherichia coli clusters from their singlets,30 circulating tumor cells from background blood cells,31,32 and large extracellular vesicles from small exosomes.33,34 As can be seen from the above progress, great successes have been achieved in the field of viscoelastic microfluidics. However, high-throughput and high-purity separation of MTCs from malignant pleural and peritoneal effusions has not been reported.
In this work, we developed interfacial elasto-inertial microfluidics for the high-throughput and high-purity separation of MTCs from malignant pleural and peritoneal effusions. We first explored the effects of critical operational parameters (i.e., flow-rate ratio and flow rate) on the particle separation performance. It was found that stable particle separation could be realized over wide flow rates using our interfacial elasto-inertial microfluidic device. We next characterized the device separation performance using blood samples spiked with tumor cells (A549, MCF-7, and MDA-MB-231 cells) of different concentrations. A removal ratio of blood cells of over 99.8%, an average recovery ratio of tumor cells of 94%, and an average purity of tumor cells as high as 40% could be successfully achieved at a high sample throughput of 120 μL min−1 in a totally passive manner. Finally, we successfully applied our device for the separation of MTCs in clinical pleural and peritoneal effusions from patients of five different cancers. Our work may provide a potential sample preparation tool for the cytological diagnosis of pleural and peritoneal effusions.
Materials and methods
Device design and working principle
Fig. 1A shows the schematic diagram of our interfacial elasto-inertial microfluidic device which is composed of three inlets (i.e., two sample inlets and one sheath inlet), a straight main channel, and three outlets (i.e., two waste outlets and one target outlet). The sample fluids containing differently-sized cells are pumped into the device via the two side sample inlets, while the viscoelastic fluid is employed as the sheath fluid and injected into the device via the middle sheath inlet. Through controlling the flow rates of the sample and sheath fluids, the three fluids simultaneously co-flow through the main channel, generating a stable fluidic interface between each other. Fig. 1B shows the migration process of cells and force competition on cells along the channel. As the cells are initially confined within the sample flow near the channel wall, the inertial lift force (FL) caused by the wall effect will laterally move the cells towards the middle sheath fluid (I in Fig. 1B). When the cells approach the fluidic interface, the interfacial elastic lift force (FE) induced by the asymmetry of compressive elastic stresses across the cell surface will force the cells towards the channel wall (II in Fig. 1B). Both the FL and FE are heavily dependent on the cell size, and thus the large cells will suffer from strong forces and are able to traverse the fluidic interface into the middle sheath fluid. When entering the viscoelastic fluid (middle sheath fluid), the direction of FE gets reversed due to the offset effects of the compressive elastic stresses acting from the near-wall side (III in Fig. 1B). When approaching the channel centerline, the direction of FL gets reversed due to the dominance of the shear gradient effect. Finally, the dominant FE (FE > FL) will force the large cells towards the channel centerline (IV in Fig. 1B). Therefore, the large cells could be focused on the channel centerline. Meanwhile, the small cells are still intercepted by the fluidic interface and remain in the original sample fluids. Using this principle, the large MTCs are focused on the channel centerline and could be collected via the middle target outlet, while the small blood cells (WBCs and RBCs) are removed via the two side waste outlets, enabling the high-purity size-dependent separation of MTCs from background blood cells in a high-throughput manner. In the current design, the channel height was designed to be 50 μm, while the width and length of the main channel were determined to be 100 μm and 40 mm, respectively. At the end of the main channel, an expansion channel was used to facilitate the cell separation. The CAD drawing and detailed dimensions of our device can be found in Fig. S1.† | ||
| Fig. 1 (A) Schematic diagram of our interfacial elasto-inertial microfluidic device for the high-throughput and high-purity separation of MTCs from background blood cells (WBCs and RBCs). (B) Migration process of cells and force competition on cells along the channel. Due to the symmetry, only half of the cross-section is illustrated. (C) Demonstration of the device separation principle using a particle mixture of 5 μm, 10 μm, and 15 μm. The particle distributions near the inlet and outlet are illustrated. The red bar indicates the distribution of 15 μm particles, while the blue bars indicate the distributions of 5 μm and 10 μm particles. | ||
Fig. 1C shows the demonstration of the device separation principle using a particle mixture of 5 μm, 10 μm, and 15 μm. At the outlet, the 15 μm particles were found to be focused on the channel centerline and well separated from the small 10 μm and 5 μm particles, which validated the effectiveness of our device. The whole separation process is totally passive and the device could be operated by simply controlling the flow rates of the sample and sheath fluids without pre-labeling the cells.
Device fabrication
Our device was made of three layers, including one upper cover layer #1, one middle channel layer #2, and one bottom cover layer #3 (see Fig. 2A). The upper cover layer #1 consisted of through holes as inlets and outlets, while the middle channel layer #2 contained the channel pattern. A polysiloxane film material (Dudao United Chemical Company) with a thickness of 50 μm was used as the middle channel layer #2, while polyethylene terephthalate (PET) films were employed as the two cover layers # 1 and #3. As shown in Fig. 2B, the patterns in these three layers were designed using the AutoCAD software (Autodesk) and cut using a UV laser cutting machine (10 W, 355 nm laser source, TH-UV200A, Tianhong). After cutting, the materials in the channel and holes were removed to form the through grooves in the films. Then, the three layers were treated with an oxygen plasma cleaner (PDC-002, Harrick Plasma) and bonded together to create the polymer-film chip. The alignment of these three layers was completed with the assistance of locating holes at the corners of each layer and locating pins in the custom fixture. Fig. 2C shows the photograph of the fabricated device with a total thickness of less than 300 μm. More importantly, the material cost of our device is very low and the whole fabrication process could be finished within 10 minutes. The low-cost and rapid fabrication enables the mass production and disposable use of our device in future. Fig. 2D shows the microscopy image of the fabricated channel. After fabrication, the device was clamped between two PMMA plates, as presented in Fig. 2E. The plates served as the world-to-chip interface for sample introduction and exportation. | ||
| Fig. 2 (A) CAD designs of each layer. (B) Schematic diagram illustrating the fabrication process of our device using laser cutting and bonding. (C) Photograph of the fabricated device. The channel was fully filled with red ink for clear visualization. (D) Microscopy image illustrating the fabricated channel. (E) Photograph of the assembled device. The PMMA plates served as the world-to-chip interface for sample introduction and exportation. | ||
Experimental setup
The assembled device was fixed on the platform of an inverted fluorescence microscope (IX 71, Olympus) to visualize the separation performance. The particle and cell motions in the channels were captured using a high-speed CCD camera (Exi Blue, QImaging) attached to the microscope and stored as image frames. The sample and sheath fluids were driven using two separate precise syringe pumps (Legato 270, KD Scientific) at specific flow rates, respectively. The tubing was used to connect the device with the syringe or collecting tubes for fluid introduction and exportation. To obtain the statistical distributions of particles/cells, the captured image frames were stacked vertically to create composite images using the ImageJ software (NIH), as shown in Fig. S2.† The fluorescence intensity profile across the channel width was also measured using this software. To characterize the separation performance, the cell concentrations in the initial sample and the collected liquids were measured using an automated cell counter (Countess II FL, Thermo Fisher Scientific).Sample preparation
Fluorescent polystyrene particles with diameters of 5 μm, 10 μm, and 15 μm (1% solid content, Thermo Fisher Scientific) were employed to mimic RBCs (∼6 μm), WBCs (∼10 μm), and MTCs (15–25 μm), respectively. The particle suspensions were prepared by re-suspending the particles in phosphate-buffered saline (PBS, Sigma-Aldrich) at concentrations of 105 particles per mL. Human lung cancer cells (A549) and breast cancer cells (MCF-7 and MDA-MB-231) obtained from Zhongda Hospital were also used to evaluate the separation performance of our device. These tumor cells were cultured using high-glucose Dulbecco's modified Eagle's medium (DMEM, Thermo Fisher Scientific) with 10% fetal bovine serum (Thermo Fisher Scientific) and 1% penicillin–streptomycin (Thermo Fisher Scientific) at 5% CO2 atmosphere. Before experiments, the tumor cells were dissociated by adding 0.25% Trypsin–EDTA solution (Thermo Fisher Scientific) and re-suspended in PBS. The sheath fluid was viscoelastic fluid and was prepared by adding 50 ppm hyaluronic acid (HA, Shanghai Future Industrial) with a molecular weight of 1650 kDa into PBS.For cell separation experiments, the blood samples were collected from healthy consenting volunteers and diluted with PBS 100 fold to mimic pleural and peritoneal effusions. Then, the three tumor cells were, respectively, spiked into the diluted blood samples at specific concentrations. To identify the tumor cells from background blood cells, the tumor cells were stained with calcein AM (Thermo Fisher Scientific) dye before spiking.
For experiments using clinical samples, the pleural and peritoneal effusions were collected from patients who were diagnosed with advanced metastatic pancreatic, ovarian, breast, liver, and lung cancers. All the blood and pleural and peritoneal effusion samples were obtained from Zhongda Hospital. The collection and use of blood samples and pleural and peritoneal effusions were approved by the institutional committee of the Institutional Ethical Committee for Clinical Research of Zhongda Hospital (2020ZDSYLL043) and informed consent was obtained from patient donors or healthy volunteers. All experiments were performed in compliance with Chinese laws and followed institutional guidelines. To prevent channel blocking caused by large impurities, the pleural and peritoneal effusions were first filtered using a cell strainer (BD Biosciences). After separation, the immunofluorescence staining technique was used to distinguish target MTCs from residual WBCs. The detailed procedure concerning the immunofluorescence staining can be found in our previous work.21,35 After smearing, the strained cells on the glass slides were counted using a fluorescence microscope (ECLIPSE 80i, Nikon) equipped with a camera (DS-Ri1, Nikon). The target MTCs and WBCs were enumerated according to the established criteria, respectively.
Results and discussion
Stable particle separation over wide flow rates
We first explored the effect of the flow-rate ratio on particle separation. The 5 μm, 10 μm, and 15 μm particle suspensions were pumped into the device at a total flow rate of 160 μL min−1, respectively. The flow-rate ratios of sheath to sample were controlled to be 0.25, 0.5, 1, and 2, respectively. Fig. 3A shows the distributions of 5 μm, 10 μm, and 15 μm particles before the outlet system at different flow-rate ratios.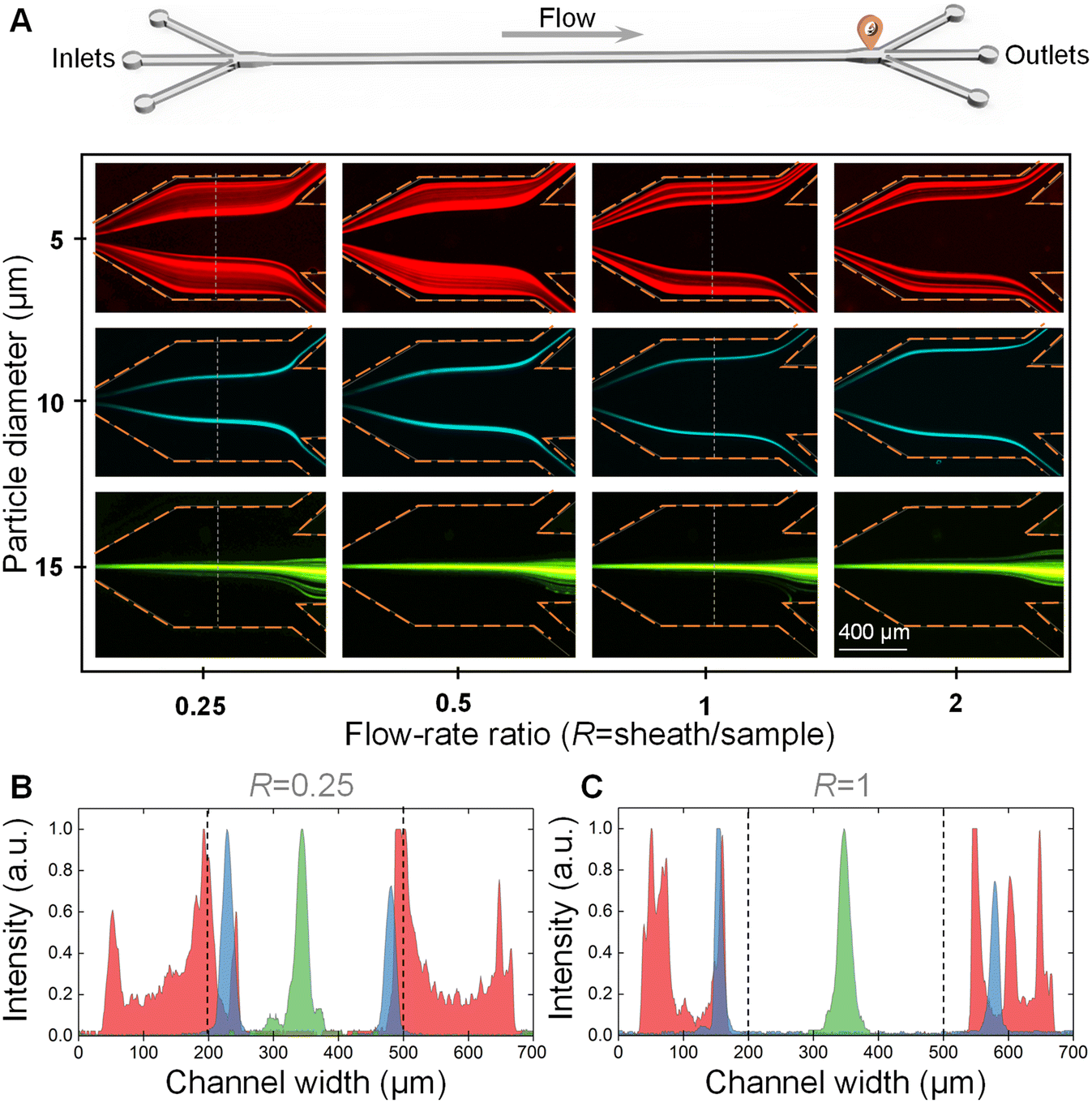 | ||
| Fig. 3 Effect of the flow-rate ratio on particle separation. (A) Images illustrating the distributions of 5 μm, 10 μm, and 15 μm particles before the outlet system at different flow-rate ratios of 0.25, 0.5, 1, and 2. The yellow dotted lines indicate the channel walls. The 5 μm, 10 μm, and 15 μm particles were pseudo-colored with red, blue, and green, respectively. (B and C) Fluorescence intensities across the channel width at the flow-rate ratios (R) of 0.25 (B) and 1 (C). The fluorescence intensities were measured at the positions of white dotted lines in subfigure A. The intensities were normalized. The red, blue, and green represent the 5 μm, 10 μm, and 15 μm particles. | ||
From these images, it was clearly observed that only the 15 μm large particles could traverse the fluidic interface into the middle sheath fluid and be focused into a stream at the channel centerline at all the tested flow-rate ratios. Meanwhile, the 5 μm and 10 μm small particles were still confined within the original sample fluids near the two channel side walls due to the relatively weak inertial lift force. The differential particle distribution positions facilitated the successful separation of 15 μm large particles from 5 μm and 10 μm small particles. Different from the band focusing of 5 μm particles, the 10 μm particles were able to form two focusing streams in the original sample fluids.
We also plotted the fluorescence intensities across the channel width at the flow-rate ratios (R) of 0.25 and 1 (see Fig. 3B and C). From these two figures, we found that the two focusing bands/streams of 5 μm and 10 μm small particles were pushed towards the channel side walls with the increase of the flow-rate ratio, which was beneficial for the complete removal of 5 μm and 10 μm small particles. Therefore, it would be better to select an R of larger than 1 for the highly efficient separation of cells with polydisperse sizes. However, the increase of R would decrease the actual processing throughput of the sample fluids. Therefore, the R was determined to be 1 for the downstream experiments.
We next explored the effect of the total flow rate on particle separation. The three particle suspensions were pumped into the device at different total flow rates of 120–240 μL min−1 with an interval of 40 μL min−1, while the flow-rate ratio was controlled to be 1. The experimental results illustrating the distributions of 5 μm, 10 μm, and 15 μm particles before the outlet system at different total flow rates are presented in Fig. S3A.† Fig. S3B and C† show the fluorescence intensities across the channel width at the selected flow rates of 160 μL min−1 and 240 μL min−1, respectively. It was found that the total flow rate did not obviously affect the particle distribution over the tested flow rate range, which indicated that our device could be operated over a wide flow rate range. To increase the processing throughput, the total flow rate for downstream cell separation was determined to be 240 μL min−1. Further increasing the flow rate would cause the failure of our device due to the leakage under high pressures.
Characterization of cell separation performance
We next characterized the separation performance of our device using blood samples spiked with different tumor cells. The blood samples were diluted 100 fold and three types of tumor cells (A549, MCF-7, and MDA-MB-231 cells) were, respectively, spiked into the diluted blood at a concentration of 104 cells per mL. The prepared blood samples were injected into the device at the total flow rate of 240 μL min−1 and the flow-rate ratio of 1. Fig. 4A shows the bright and fluorescence images illustrating the cell distributions near the inlet, at the main channel, and near the outlet. It was clearly observed that the co-flowing of the sample–sheath–sample was formed at the inlet. When migrating along the channel, the large tumor cells migrated into the middle sheath fluid, while the small blood cells were still confined within the original sample fluids. Through using a three-branch outlet system, the tumor cells could be separated from the blood cells and collected by the middle target outlet. Fig. S4† shows the sampled microscopy images of the fluids collected from the waste and target outlets. From the microscopy images, it could be seen that the blood cells are well removed via the waste outlets and the high-purity tumor cells could be observed in the liquid from the target outlet.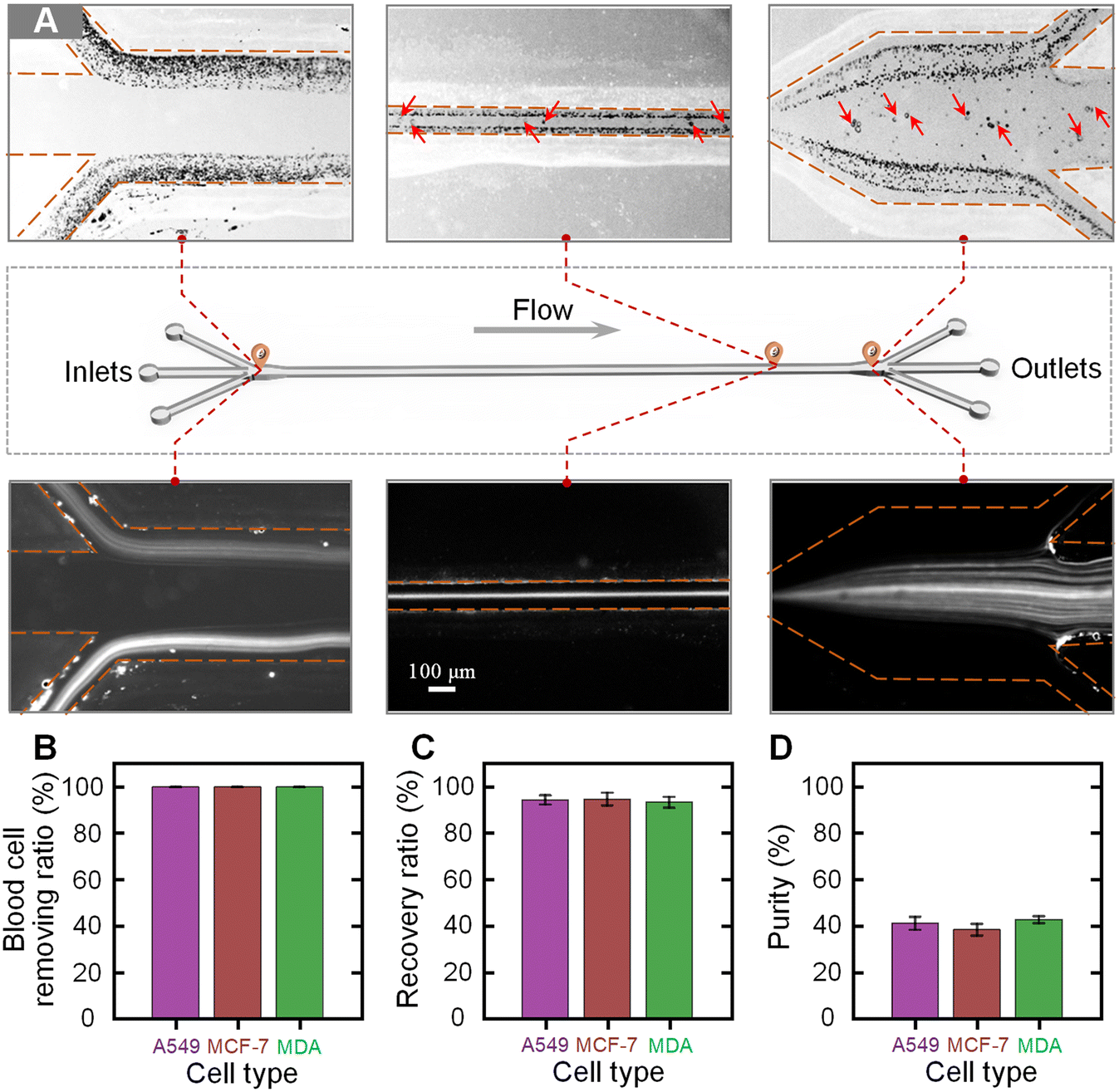 | ||
| Fig. 4 Characterization of the cell separation performance. (A) Bright and fluorescence images illustrating the cell distributions near the inlet, at the main channel, and near the outlet. The yellow dotted lines indicate the channel walls. The tumor cells were stained before being spiked into the diluted blood samples. The fluorescent streams in the fluorescence images indicate the trajectories of the stained tumor cells. (B–D) Calculated blood cell removal ratio (B), recovery ratio (C), and purity (D) for the samples spiked with three types of tumor cells (A549, MCF-7, and MDA-MB-231 cells). | ||
To quantitatively evaluate the separation performance of our device, the liquids collected from the target and waste outlets were analyzed. Three parameters, the removal ratio of blood cells, the recovery ratio of tumor cells, and the purity of tumor cells, were used to evaluate the separation performance. The blood cell removal ratio is defined by dividing the number of blood cells in the waste by the total number of blood cells in the initial sample. The recovery ratio of tumor cells is calculated by dividing the number of tumor cells in the liquid collected from the target outlet by the number of tumor cells in the initial sample. The purity of the separated tumor cells is defined by dividing the number of target tumor cells by the total number of cells in the liquid collected from the target outlet. Fig. 4B–D show the calculated blood cell removal ratio, recovery ratio, and purity for samples spiked with three types of tumor cells. The removal ratios of blood cells for the samples spiked with A549, MCF-7, and MDA-MB-231 cells were 99.85% ± 0.03%, 99.83% ± 0.02%, and 99.86% ± 0.03%, respectively. The recovery ratios of A549, MCF-7, and MDA-MB-231 cells were 94.31% ± 2.03%, 94.65% ± 2.75%, and 93.28% ± 2.35%, respectively. In addition, the purities of the separated A549, MCF-7, and MDA-MB-231 cells were 40.99% ± 2.78%, 38.20% ± 2.53%, and 42.55% ± 1.48%, respectively. It was found that a high blood cell removal ratio of over 99.8% could be stably achieved using our device, while the average recovery ratio and purity of tumor cells for these three samples were as high as 94% and 40%, respectively.
We next evaluated the separation performance of our device using samples spiked with low-concentration tumor cells (∼1000 cells per mL). The experimental results are shown in Fig. S5 and S6.† From the experimental results, it was found that excellent separation performance was also successfully achieved for these three low-concentration samples. The quantitative data indicated that only the separation parameter of purity decreased due to the decrease of spiked tumor cell concentrations. The purities of A549, MCF-7, and MDA-MB-231 cells decreased to 12.99% ± 2.76%, 13.18% ± 3.47%, and 14.12% ± 1.81%, respectively. As the number of residual blood cells in the target outlet would not change significantly, the purities of recovered tumor cells decreased with decreasing number of the spiked tumor cells.
The above experimental results well validated the excellent separation performance of our device. The whole separation process is totally label-free and does not affect the activity of cells, which enables various downstream applications (e.g., single cell analysis and next-generation sequencing) of the separated cells to be possible. As compared with single-phase inertial microfluidics, precise high-purity cell separation could be achieved using our current interfacial elasto-inertial microfluidics as the small blood cells are unable to traverse the fluidic interface of co-flowing fluids. In our device, the viscoelastic fluid was used as the middle sheath fluid, while the sample fluid was directly pumped into the device from the two side inlets without adding an elastic enhancer, which reduces the complexity of sample pretreatment. To increase the processing throughput, low-viscosity HA solution was used as the viscoelastic sheath fluid. Therefore, a sample throughput as high as 120 μL min−1 was reported in our device, which was much higher than that of previous work using PEO as the elastic enhancer.29–32 The good biocompatibility of HA solution was well validated in many previous studies.36
Separation of MTCs from clinical pleural and peritoneal effusions
To examine the practical application of our device for clinical samples, we applied our device to separate MTCs from clinical pleural and peritoneal effusions. The pleural and peritoneal effusions were collected from patients with five different cancers (i.e., pancreatic cancer, ovarian cancer, breast cancer, liver cancer, and lung cancer). The detailed clinical information on the involved patients can be found in Table S1.† After filtering with a strainer to remove the large impurities, the prepared samples were pumped into the device at the total flow rate of 240 μL min−1 and the flow-rate ratio of 1.Fig. 5A shows the microscopy images illustrating the cell distribution at the outlet. It was clearly observed that the large cells migrated into the middle sheath fluid and could be collected via the middle target outlet (II), while most small cells were removed by the two side waste outlets (I and III). As it is difficult to directly identify the MTCs in Fig. 5A, we collected the liquids from the three outlets (I, II, and III). Fig. 5B shows the photograph of the collected liquids. It was observed that the liquids from the two side waste outlets appeared as the original color of pleural and peritoneal effusions, while the liquid from the middle target outlet showed a lighter color. We further analyzed the liquid collected from the middle target outlet (II). The immunofluorescence staining technique was used to distinguish the target MTCs from residual WBCs. The cells satisfying the criterion of CK+, CD45−, and DAPI+ were identified as MTCs, while the cells satisfying the criterion of CK−, CD45+, and DAPI+ were recognized as WBCs. Fig. 5C shows the sampled microscopy image of the liquid collected from outlet II in the bright-field mode. Fig. 5D shows the fluorescence images after immunofluorescence staining. The rare MTCs could be clearly observed in the sampled window. However, there still existed some large-sized WBCs which could also traverse the fluidic interface into the middle sheath fluid. The incapacity for separating MTCs from similar-sized WBCs is the inherent limitation of size-dependent separation techniques. To further address this limitation, our device could be coupled with other size-independent separation techniques (e.g., immunomagnetic separation). More interestingly, we found that the intact cluster of MTCs was successfully recovered using our device. Previous studies proved that tumor cell clusters had a potentially high capacity of metastasis, and thus the separation and single-cell analysis of tumor cell clusters offer new insights into tumor metastasis and could facilitate precision cancer diagnosis.37,38
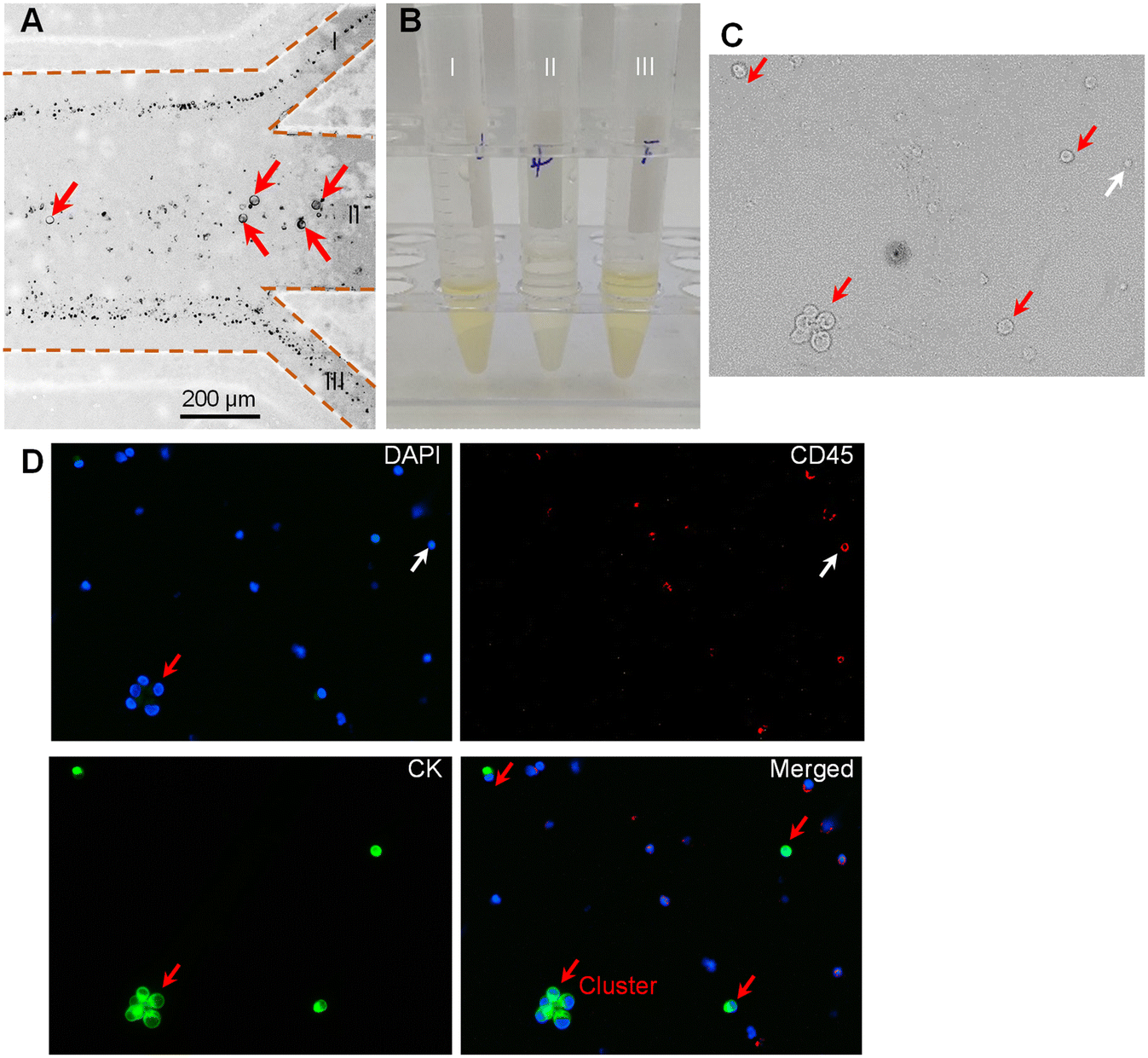 | ||
| Fig. 5 Separation of MTCs from clinical pleural and peritoneal effusions. (A) Microscopy image illustrating the cell distribution at the outlet. (B) Photograph of liquids collected from the three outlets (I, II, and III). (C) Microscopy image of the liquid from outlet II in the bright-field mode. (D) Fluorescence images after immunofluorescence staining. The cells satisfying the criterion of CK+, CD45−, and DAPI+ were identified as MTCs, while the cells satisfying the criterion of CK−, CD45+, and DAPI+ were recognized as WBCs. The red arrows indicate the MTCs, while the white arrows indicate the WBCs. | ||
We further identified and counted the cells in the liquids collected from the target outlet. The number of MTCs in the pleural and peritoneal effusions from patients of pancreatic cancer, ovarian cancer, breast cancer, liver cancer, and lung cancer was 154 cells per mL, 66 cells per mL, 243 cells per mL, 492 cells per mL, and 271 cells per mL, respectively. The number of MTCs is highly related to the cancer type and disease progression, and may vary before and after treatment. As compared with CTCs in peripheral blood (typically 0–50 cells per mL), the number of MTCs in pleural and peritoneal effusions was much larger. As our device separated the MTCs in a label-free manner, the live MTCs could be further used for various downstream biological applications (e.g., drug resistance test and next-generation sequencing). The purity of MTCs was, respectively, calculated to be 6.75%, 5.84%, 10.32%, 6.46%, and 3.89% for effusions of pancreatic cancer, ovarian cancer, breast cancer, liver cancer, and lung cancer. The purities were slightly lower than those of experiments using cultured cells possibly due to the polydisperse size distribution of cells in real clinical samples.
Conclusion
In this work, we developed interfacial elasto-inertial microfluidics for the high-throughput and high-purity separation of MTCs from malignant pleural and peritoneal effusions. The device was fabricated from low-cost polymer films using a rapid process of laser cutting and bonding. Through using hyaluronic acid as an elastic enhancer, a throughput much higher than previous interfacial elasto-inertial microfluidics could be achieved. We first explored the effects of critical operational parameters (i.e., flow-rate ratio and flow rate) on particle separation. It was interesting to find that stable particle separation over wide flow rates could be realized using our device. We next characterized the device separation performance using blood samples spiked with tumor cells (A549, MCF-7, and MDA-MB-231 cells) of different concentrations. A removal ratio of blood cells of over 99.8%, an average recovery ratio of tumor cells of 94%, and an average purity of tumor cells as high as 40% could be successfully achieved at a high sample throughput of 120 μL min−1. Finally, we successfully applied our device for the separation of MTCs in clinical pleural and peritoneal effusions from patients of different cancers. Our work may provide a potential sample preparation tool for cytological diagnosis of pleural and peritoneal effusions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work is supported by the National Key Research and Development Program of China (2021YFC2103300), the National Natural Science Foundation of China (51875103 and 81727801), the Project of Jiangyin Health Committee (M202204), and the Natural Science Foundation of Jiangsu Province (BK20190064).References
- J. K. T. Dermawan and M. L. Policarpio-Nicolas, Arch. Pathol. Lab. Med., 2020, 144, 1086–1091 CrossRef PubMed
.
- D. Feller-Kopman and R. Light, N. Engl. J. Med., 2018, 378, 740–751 CrossRef CAS PubMed
- A. M. Egan, D. McPhillips, S. Sarkar and D. P. Breen, QJM, 2014, 107, 179–184 CrossRef CAS PubMed
- J. M. Porcel, Curr. Opin. Pulm. Med., 2019, 25(4), 362–368 CrossRef CAS PubMed
- E. K. Sackmann, A. L. Fulton and D. J. Beebe, Nature, 2014, 507, 181–189 CrossRef CAS PubMed
- S. Battat, D. A. Weitz and G. M. Whitesides, Lab Chip, 2022, 22, 530–536 RSC
- M. A. Witek, I. M. Freed and S. A. Soper, Anal. Chem., 2020, 92, 105–131 CrossRef CAS PubMed
- S. Zhu, F. Jiang, Y. Han, N. Xiang and Z. Ni, Analyst, 2020, 145, 7103–7124 RSC
- N. Pamme, Lab Chip, 2006, 6, 24–38 RSC
- X. Xuan, Electrophoresis, 2019, 40, 2484–2513 CAS
- P. Paiè, T. Zandrini, R. M. Vázquez, R. Osellame and F. Bragheri, Micromachines, 2018, 9, 200 CrossRef PubMed
- P. Zhang, H. Bachman, A. Ozcelik and T. J. Huang, Annu. Rev. Anal. Chem., 2020, 13, 17–43 CrossRef CAS PubMed
- Y. Song, D. Li and X. Xuan, Electrophoresis, 2023, 44(11–12), 910–937 CrossRef PubMed
- N. Xiang and Z. Ni, Lab Chip, 2022, 22, 4792–4804 RSC
- J. Zhang, S. Yan, D. Yuan, G. Alici, N.-T. Nguyen, M. E. Warkiani and W. Li, Lab Chip, 2016, 16, 10–34 RSC
- D. Yuan, Q. Zhao, S. Yan, S.-Y. Tang, G. Alici, J. Zhang and W. Li, Lab Chip, 2018, 18, 551–567 RSC
- J. Zhou and I. Papautsky, Microsyst. Nanoeng., 2020, 6, 113 CrossRef CAS PubMed
- A. Hochstetter, R. Vernekar, R. H. Austin, H. Becker, J. P. Beech, D. A. Fedosov, G. Gompper, S.-C. Kim, J. T. Smith, G. Stolovitzky, J. O. Tegenfeldt, B. H. Wunsch, K. K. Zeming, T. Krüger and D. W. Inglis, ACS Nano, 2020, 14, 10784–10795 CrossRef CAS PubMed
- N. Xiang, Q. Li and Z. Ni, Anal. Chem., 2020, 92, 6770–6776 CrossRef CAS PubMed
- X. Lu and X. Xuan, Anal. Chem., 2015, 87, 4560–4565 CrossRef CAS PubMed
- Z. Zhu, S. Li, D. Wu, H. Ren, C. Ni, C. Wang, N. Xiang and Z. Ni, Lab Chip, 2022, 22, 2097–2106 RSC
- H. Pei, L. Li, Z. Han, Y. Wang and B. Tang, Lab Chip, 2020, 20, 3854–3875 RSC
- X. Wei, K. Chen, S. Guo, W. Liu and X.-Z. Zhao, ACS Appl. Bio Mater., 2021, 4, 1140–1155 CrossRef CAS PubMed
- P.-H. Tsou, P.-H. Chiang, Z.-T. Lin, H.-C. Yang, H.-L. Song and B.-R. Li, Lab Chip, 2020, 20, 4007–4015 RSC
- M. D. Graham, Annu. Rev. Fluid Mech., 2011, 43, 273–298 CrossRef
- S. Yang, J. Y. Kim, S. J. Lee, S. S. Lee and J. M. Kim, Lab Chip, 2011, 11, 266–273 RSC
- N. Xiang, X. Zhang, Q. Dai, J. Cheng, K. Chen and Z. Ni, Lab Chip, 2016, 16, 2626–2635 RSC
- C. Liu, C. Xue, X. Chen, L. Shan, Y. Tian and G. Hu, Anal. Chem., 2015, 87, 6041–6048 CrossRef CAS PubMed
- F. Tian, W. Zhang, L. Cai, S. Li, G. Hu, Y. Cong, C. Liu, T. Li and J. Sun, Lab Chip, 2017, 17, 3078–3085 RSC
- T. Zhang, A. K. Cain, L. Semenec, L. Liu, Y. Hosokawa, D. W. Inglis, Y. Yalikun and M. Li, Anal. Chem., 2023, 95, 2561–2569 CrossRef CAS PubMed
- F. Tian, L. Cai, J. Chang, S. Li, C. Liu, T. Li and J. Sun, Lab Chip, 2018, 18, 3436–3445 RSC
- Y. Cheng, S. Zhang, L. Qin, J. Zhao, H. Song, Y. Yuan, J. Sun, F. Tian and C. Liu, Anal. Chem., 2023, 95, 3468–3475 CrossRef CAS PubMed
- Y. Zhou, Z. Ma, M. Tayebi and Y. Ai, Anal. Chem., 2019, 91, 4577–4584 CrossRef CAS PubMed
- C. Liu, J. Guo, F. Tian, N. Yang, F. Yan, Y. Ding, J. Wei, G. Hu, G. Nie and J. Sun, ACS Nano, 2017, 11, 6968–6976 CrossRef CAS PubMed
- N. Xiang and Z. Ni, Lab Chip, 2022, 22, 757–767 RSC
- E. J. Lim, T. J. Ober, J. F. Edd, S. P. Desai, D. Neal, K. W. Bong, P. S. Doyle, G. H. McKinley and M. Toner, Nat. Commun., 2014, 5, 4120 CrossRef CAS PubMed
- E. Schuster, R. Taftaf, C. Reduzzi, M. K. Albert, I. Romero-Calvo and H. Liu, Trends Cancer, 2021, 7, 1020–1032 CrossRef CAS PubMed
- N. Aceto, A. Bardia, D. T. Miyamoto, M. C. Donaldson, B. S. Wittner, J. A. Spencer, M. Yu, A. Pely, A. Engstrom, H. Zhu, B. W. Brannigan, R. Kapur, S. L. Stott, T. Shioda, S. Ramaswamy, D. T. Ting, C. P. Lin, M. Toner, D. A. Haber and S. Maheswaran, Cell, 2014, 158, 1110–1122 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sd00107e |
| This journal is © The Royal Society of Chemistry 2023 |