
Tuning electrocatalytic water oxidation by MnOx through the incorporation of abundant metal cations†
Jens
Melder‡
 *a,
Stefan
Mebs
*b,
Florian
Lessing
a,
Holger
Dau
b and
Philipp
Kurz
a
*a,
Stefan
Mebs
*b,
Florian
Lessing
a,
Holger
Dau
b and
Philipp
Kurz
a
aInstitut für Anorganische und Analytische Chemie and Freiburger Materialforschungszentrum (FMF), Albert-Ludwigs-Universität Freiburg, Albertstraße 21, 79104 Freiburg, Germany. E-mail: jens.melder@dlr.de
bFachbereich Physik, Freie Universität Berlin, Arnimallee 14, 14195 Berlin, Germany. E-mail: stebs@chemie.fu-berlin.de
First published on 17th November 2022
Abstract
Layered manganese oxides which were deposited on carbon fibre paper (MnOx/CFP) via a redox-deposition process show promising activities and stabilities for the OER over a very wide pH range. In the study presented here, the influence of the incorporation of metal cations (Mn+, M = Ca, Co, Ni, Fe) into MnOx layers on the OER performance at different pH values (2.5/7.0/14.0) is explored. Spectroscopic analyses of MyMnOx/CFP reveal that doping with other metal cations does not alter the basic birnessite-type MnOx structure. However, the average manganese oxidation state can be tuned and is found to depend on the incorporated metal cations. Furthermore, a clear dependence of the catalytic activity and stability on the incorporated metal ion is observed and especially Ni- and Co-doping lead to a significant performance boost under alkaline (Ni and Co) and neutral conditions (Co). A comparison to other state-of-the-art metal oxide catalysts (also deposited on CFP for comparison) shows that the metal-doped MyMnOx/CFP anodes are able to compete with the most active materials known so far.
1 Introduction
The need for a sustainable energy supply that is not based on fossil fuels like crude oil or natural gas is urgent not only because their resources are finite but also because their combustion produces CO2. The conversion of solar energy, the most ubiquitous energy source, into storable fuels like H2 by catalytic processes has recently attracted much attention. For this, the splitting of water to H2 and O2 might play a central technological role.1–3The energy (ΔG = 237 kJ mol(H2)−1)4 required to power this highly endothermic reaction can be provided by several renewable sources, one of which is electricity from wind or solar energy (e.g. by photovoltaics or wind turbines). The electrochemical splitting of water can be divided into two half-cell reactions, the hydrogen evolving reaction (HER) and the oxygen evolving reaction (OER).
From these two reactions, the OER is kinetically more demanding due to the combined release of four electrons and four protons from two water molecules during the production of a single oxygen molecule. In an ideal system, O2 and H2 can be evolved when the potential difference between the cathode and the anode exceeds 1.23 V vs. the reversible hydrogen electrode (RHE).5 However, often high overpotentials η need to be applied to achieve reasonable current densities j. Earth-abundant, cheap, scalable, non-toxic, stable and first and foremost efficient catalysts are urgently needed in order to make the production of hydrogen from water splitting energetically more efficient and at the same time industrially feasible.
Great efforts have been made over the last few years in finding such earth abundant catalysts for the OER. The most promising candidates are based on Co,6 Ni,7,8 Fe9,10 or Mn,11–15 which are able to reach benchmark current densities of j = 10 mA cm−2 at overpotentials as low as η < 300 mV in alkaline or η ∼ 550 mV in neutral media.15–21 Unfortunately, most of the before mentioned catalysts suffer from corrosion when used under more acidic pH conditions22–25 and thus, acid-stable, well-performing catalysts from non-critical sources are still scarce. Currently, the only convincing materials for OER catalysis at pH < 7 are based on Ir and Ru, two of the rarest metals on earth, with a global Ir production of less than 9 tons per year.26,27
On the other hand, OER catalysis in the acidic pH regime is of great interest due to its possible application in proton exchange membrane (PEM) electrolyzers, photoelectrochemical (PEC) cells or other electrochemical devices, which have to be operated at pH < 7 (e.g. some microbial electrosynthesis cells).28–31 PEM electrolyzers can be operated at high current densities, are able to respond quickly to the power input due to the fast proton conduction across the membrane and show a compact cell design.4 Hence, they are especially applicable for highly dynamic operations, one of the main requirements if power from renewable sources is to be used. Furthermore, PEM electrolyzers are widely seen as the most promising water electrolysis systems.32,33
Manganese oxides (MnOx) have extensively been investigated as OER catalysts. With the oxygen evolving complex (OEC), the Mn4CaO5(H2O)x cluster of photosystem II, biology provides a template for a catalyst able to efficiently accelerate the OER under near-neutral conditions.34–36 Inspired by this, the water oxidation catalysis (WOC) activities of many MnOx polymorphs have been investigated. In comparative studies, amorphous layered or tunneled phases often showed the best performance13–15,37–44 with birnessites and phyllomanganates consisting of edge-sharing [MnO6] octahedra with layer-to-layer distances of ∼7 Å, as prominent examples. The average oxidation states of the manganese ions in these oxides are very flexible ranging typically from +3.3 to +3.9.38,45
We recently developed a method to deposit birnessite-type MnOx on high surface area graphitic carbon fiber papers by an easily scalable in situ redox deposition approach and obtained promising WOC electrodes.13 In this process, the carbon itself acts as a reducing agent to permanganate ions and (unlike many other electrode preparation routes) no further binders (e.g. polyethylene oxide, PEO) or additional polymers (e.g. Nafion®) are needed to attach MnOx to the support material. In a consecutive study, we showed that MnOx/CFP is a nanostructured volume catalyst exhibiting good WOC activities and stabilities over nearly the entire pH range from 1–14.14 For all pH values of this range, current densities > 1 mA cm−2 could be achieved at overpotentials η of 350–500 mV, which are very good values for electrodes containing only earth-abundant elements (Mn, O, K and C). Especially the very substantial performance of MnOx/CFP in the mildly acidic pH regime (pH 2–6) has to be highlighted.14 The good stability of manganese oxide catalysts under acidic pH conditions was also found by other groups. For example, Li et al. showed that γ-MnO2 is stable for over 8000 h at 10 mA cm−2 and pH 2.46
Birnessites can be modified by incorporating a wide range of different cations into their layered structure. The physical and chemical properties of layered MnOx are greatly influenced by the type, amount and location of the dopants.47–51 This has been used for the selective removal of toxic metals from wastewater, the optimization of MnOx-based capacitors, and also for MnOx catalysis e.g. amine–imine transformations,51 alcohol oxidation,52 alkene oxidation,53 and water oxidation.48–50,54–57 Co doped MnOx are particularly noteworthy in the latter case. It has already been shown that the incorporation of Co into the MnOx framework can have a positive effect on its OER activity.55–57 Moreover, Nakamura and co-workers demonstrated that the OER stability under acidic pH of cobalt spinel oxide can be significantly enhanced by the selective incorporation of manganese.58 However, no study comparing the effect on the electrocatalytic activity and stability in a wide pH range of the incorporation of different OER active transition metal cations into birnessite electrocatalysts has been reported.
Here, we present the results of an extensive investigation of the influence of the incorporation of abundant metal ions (Ni, Fe, Co and Ca) into MnOx/CFP electrodes by means of ion exchange. The choice fell on these metals, either because their oxides are themselves among the most active OER catalysts (in the cases of Ni, Fe and Co) or because they are part of the biological OEC (Ca). The prime motivation was to find out whether the already good performance of MnOx/CFP can be further enhanced by metal doping. Therefore, the electrochemical activities and stabilities of ion-exchanged MyMnOx/CFP electrodes were tested at three different pH values either in potassium phosphate buffer (pH 2.5/7.0) or in a potassium hydroxide electrolyte (pH 14.0). The influence of the incorporation of these foreign ions on the oxide's structure, composition and morphology both before and after electrocatalysis was also probed by means of infra-red spectroscopy (IR), powder X-ray diffraction (XRD), scanning electron microscopy (SEM) and X-ray absorption spectroscopy (XAS). The elemental composition was determined by flame atomic absorption spectroscopy (fAAS) and energy dispersive X-ray (EDX) spectroscopy. Finally, the activities of the MyMnOx/CFP anodes were compared to those of common state-of-the-art OER electrocatalysts.
2 Results and discussion
2.1 Electrode preparation, ion-exchange properties and characterization
MnOx/CFP electrodes were prepared by the direct redox reaction of potassium permanganate solutions with carbon fiber papers.13 This process leads to carbon fibers uniformly covered by a birnessite-type MnOx coating with an average Mn oxidation state of +3.7.14 The manganese oxide coating additionally contains potassium ions with an atomic K![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn ratio of 0.1:1.0 as well as water. Starting from the K0.1MnOx/CFP electrodes, the metal doped electrodes were obtained by an ion-exchange reaction from solutions of M2+/3+(NO3)2/3 (M = Ni2+, Fe3+, Co2+, Ca2+) (Fig. 1). Electrodes with different molar M2+/3+:Mn ratios were synthesized by varying the time for which the electrodes were exposed to the metal nitrate solutions. After the ion-exchange reactions, the electrodes were thoroughly washed with water and afterwards sintered at 400 °C for 1 h in air, which was known from former studies to be essential for activation.14 To determine the elemental composition of the electrodes, the oxide layers were dissolved in acid and the resulting solutions were analyzed by fAAS (see Fig. S1 in the ESI†).
:Mn ratio of 0.1:1.0 as well as water. Starting from the K0.1MnOx/CFP electrodes, the metal doped electrodes were obtained by an ion-exchange reaction from solutions of M2+/3+(NO3)2/3 (M = Ni2+, Fe3+, Co2+, Ca2+) (Fig. 1). Electrodes with different molar M2+/3+:Mn ratios were synthesized by varying the time for which the electrodes were exposed to the metal nitrate solutions. After the ion-exchange reactions, the electrodes were thoroughly washed with water and afterwards sintered at 400 °C for 1 h in air, which was known from former studies to be essential for activation.14 To determine the elemental composition of the electrodes, the oxide layers were dissolved in acid and the resulting solutions were analyzed by fAAS (see Fig. S1 in the ESI†).
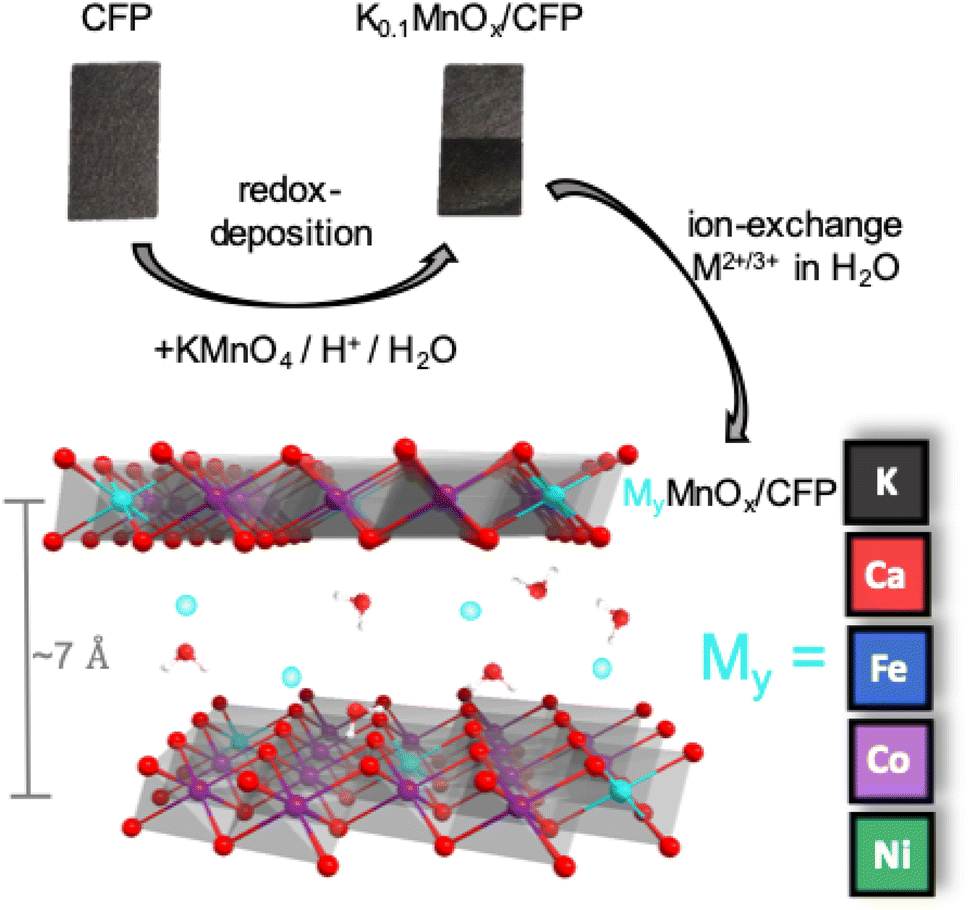 | ||
| Fig. 1 Schematic representation of the preparation of MyMnOx/CFP by ion exchange from K0.1MnOx/CFP. | ||
The ion-exchange reactions with Ni2+/Fe3+/Ca2+ were found to be slow reaching a steady state only after ∼8 h. A good correlation between the decrease of K+ ions and the increase of Ni2+/Fe3+/Ca2+ as a function of time can be observed. Nevertheless, even after 24 h only maximum molar ratios of M2+/3+:Mn ∼ 0.1:1 were found suggesting that other species, most likely H+, must also be involved in the ion-exchange reaction to keep the overall charge neutral. A dissolution of Mn ions during the ion-exchange process can be excluded as no loss of Mn from the electrodes was found by fAAS.
For Co, the ion-exchange reaction is much faster and could therefore be carried out using a 0.1 M Co2+ nitrate solution. Furthermore, many more Co ions can be incorporated into the as-prepared K0.1MnOx/CFP electrode leading to maximum molar ratios of Co:Mn ∼ 0.3:1 already after 4 h. A faster and higher uptake of Co-ions in comparison to Ni- and Fe-ions by birnessites was several times observed before.59–61 The ion-exchange reactions were confirmed by EDX analysis (Fig. S2 in the ESI†), which showed similar M:Mn ratios.
In order to determine morphological and structural changes induced by the incorporation of the metal ions during the ion exchange and the subsequent annealing steps, a number of analytical measurements were carried out.
First, possible changes of the morphology were analyzed by SEM (see Fig. 2 and S3†). The exposure of the as-prepared electrodes to the metal nitrate solution has no impact on the stability and the uniformity of the coverage of the carbon fibers by MnOx. No cracks or loss of metal oxide could be observed and all metal oxide layers show very similar, sponge-like structures typical of birnessites.11,13,14,37 Minor differences are visible for the Ca0.1MnOx/CFP electrode where the porosity of the overall structure appears less pronounced.
Interestingly, larger morphological deviations can be found for the Co0.3MnOx/CFP electrode. Here, the disordered structure is still present but the threads from which the basic structure is built are wider and more structured on the surface. Despite the different morphology, CoOx or MnOx phase separations can be excluded as EDX mapping shows a uniform distribution of both metals within the structure (see Fig. S4†).
The findings from SEM/EDX could be supported by powder XRD measurements, where no additional reflexes are visible in all MyMnOx/CFP patterns in comparison to K0.1MnOx/CFP (see Fig. 2B), indicating that no additional crystalline phases/islands, e.g. Co/Ni/Ca/Fe (oxy)hydroxides or oxides, are formed.
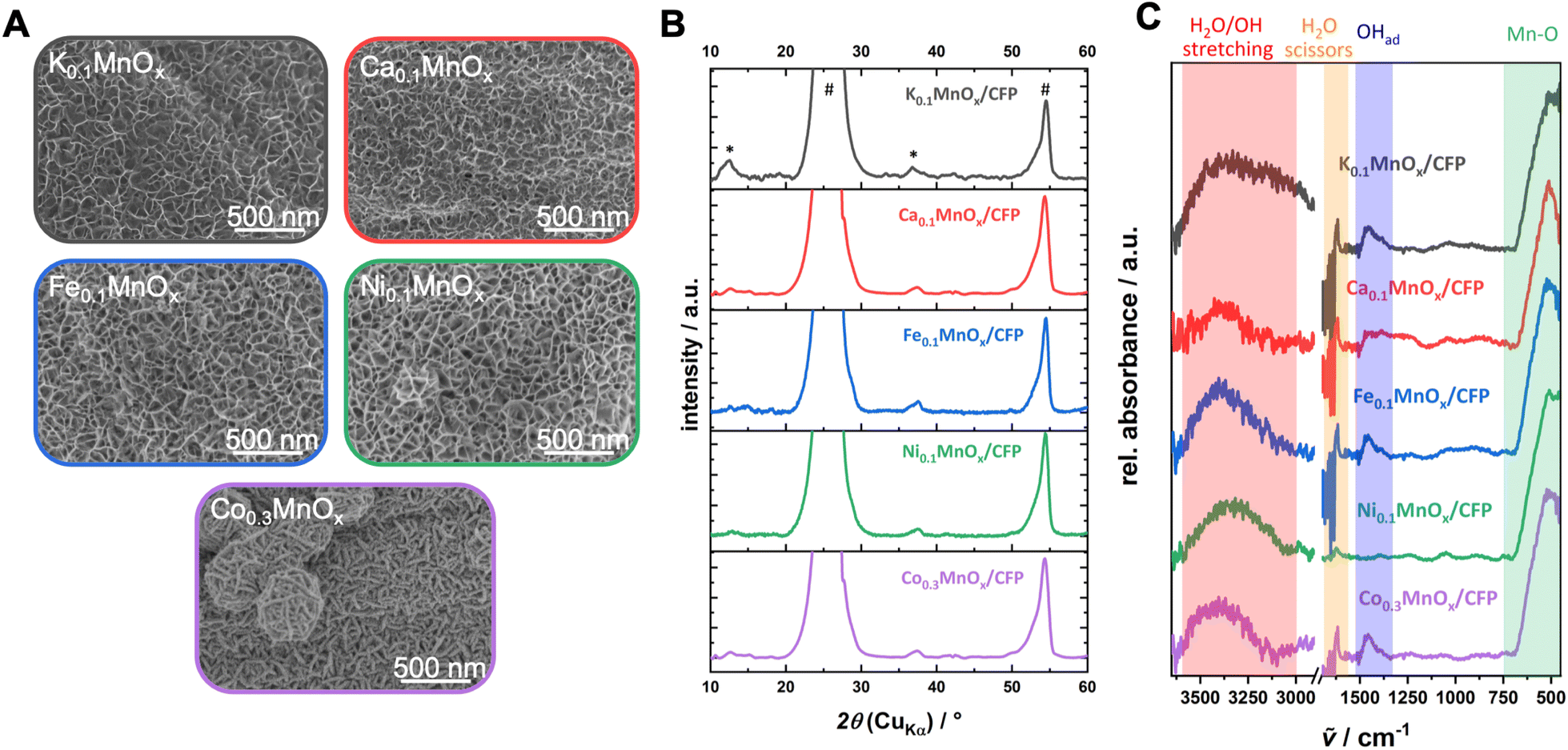 | ||
| Fig. 2 (A) Scanning electron microscopy images, (B) powder XRD patterns (reflexes of the graphitic carbon support and the birnessite-like catalyst are labeled with * and #, respectively) and (C) ATR-FTIR spectra of a K0.1MnOx/CFP electrode and electrodes after the ion-exchange reaction before operation as anodes for water oxidation catalysis. Additional SEM images can be found in Fig. S3 in the ESI.† | ||
The intense reflections visible at 2θ ∼ 26° and ∼54° in all XRD patterns stem from the graphitic carbon support and can be attributed to the (002) and (004) planes of graphite.62 Much less intense, very broad reflections at 2θ ∼ 12° (001) and ∼37° (100) are characteristic of highly disordered birnessite. The reflection at ∼12° which is indicative for an ordered stacking of the octahedral sheets is only well pronounced for the K0.1MnOx/CFP electrode. Its disappearance is indicative of a further amorphization introduced by the ion-exchange process by increasing the stacking disorder of the [MnO6]-octahedral sheets, most likely due to the incorporation of additional ions into the interlayer space. Such a loss of crystallinity has already been previously observed for Ca-, Ni- and Co-doped birnessites.63–65 The reflection at ∼37° which arises from regular distances within the layers of edge-sharing [MnO6] octahedra remains visible for all electrodes. Compared to the reflex for the parent K0.1MnOx/CFP electrode, which can be found at 2θ = 36.8°, a slight shift to higher values can be observed for the ion-exchanged electrodes (between 2θ = 37.4° and 37.7°). This is an indication that the incorporated ions also intercalate into the layers of the birnessite backbone.
The ATR-FTIR spectra of all electrodes are also typical of birnessites and after doping does not show new bands (Fig. 2). The observed signals at 750–500 cm−1 are indicative of Mn–O vibrations from the [MnO6] octahedra,13,37,66 while the absorptions between 3600 and 3000 cm−1 and 1700–1600 cm−1 belong to incorporated water molecules.67 For all MyMnOx/CFP electrodes (except for Ca0.1MnOx/CFP and Ni0.1MnOx/CFP electrodes), an absorption at ∼1460 cm−1 is visible, which can be attributed to the vibrations of adsorbed hydroxides.67
Thus, the analytical methods used so far indicate that ion exchange results in interlayer as well as intralayer incorporation. The formation of larger crystalline domains consisting of Ni/Fe/Co oxide can be excluded and a uniform statistical distribution of the ions can be assumed. To obtain complementary structural and electronic information, further characterization was performed by X-ray absorption spectroscopy (XAS) experiments. XAS is an element-specific method that can be applied to almost any material under nearly any environmental conditions and is not restricted to crystalline structures (like XRD) or specific electronic states (like NMR and EPR). This spectroscopic method has been successfully used for the analyses of birnessites by our groups before.11,12,38,43,68 Accordingly, X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS, Fig. 4) spectra at the Mn, Co, Ni and Fe K-edges were recorded for the as-prepared MyMnOx/CFP electrodes in order to determine the average oxidation states and local coordination environments of the different metal ions. The XANES and EXAFS spectra of all metals can be found in Fig. S19–S26 in the ESI.† In order to determine the average oxidation states of Mn, Co, Ni and Fe, the X-ray energy values at a normalized absorption of 0.5 of common reference compounds were used (see ESI Fig. S19–S25†). The mean oxidation states of the metals are visualized in Fig. 3, where it can be seen that the mean manganese oxidation states differ in a rather narrow range from +3.2 to +3.6.
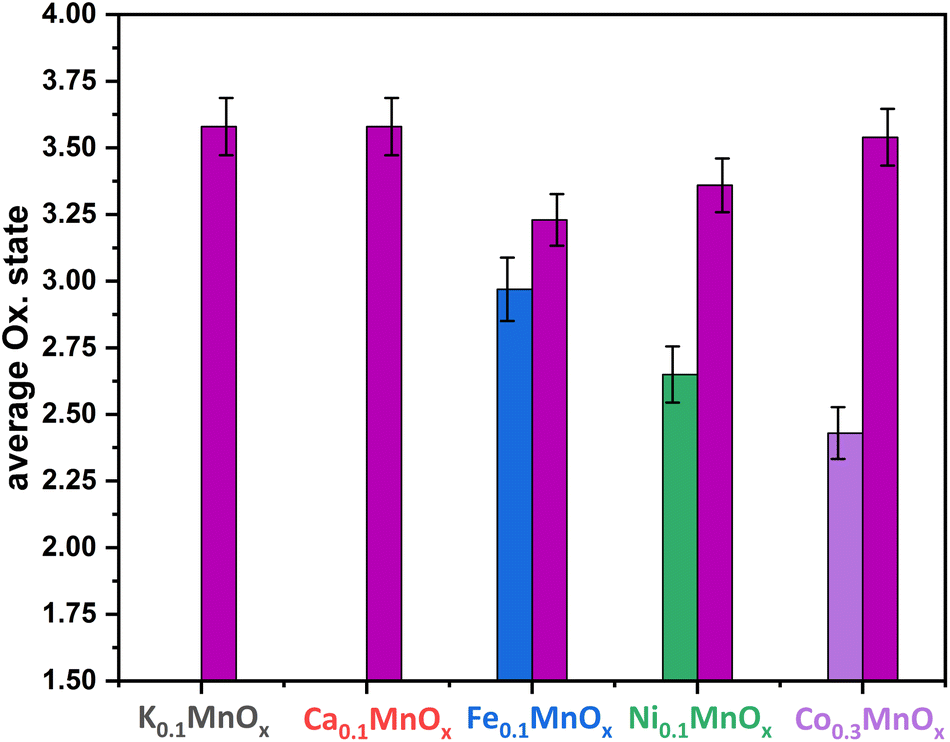 | ||
| Fig. 3 Average oxidation states of Mn (plum), Fe (blue), Ni (green) and Co (purple) for MyMnOx/CFP electrodes. The oxidation states are derived from a linear fit of the data extracted at a normalized X-ray absorption intensity of 0.5 for reference compounds. See Fig. S19–S26 in the ESI† for more information. The error bars represent one standard deviation. | ||
Especially, the incorporation of Fe and Ni leads to a reduction of the Mn ions in comparison to K0.1MnOx/CFP, while the Mn oxidation state of the Ca and Co doped samples is very similar. On the other hand, an oxidation of Co and Ni occurs relative to the M2+ ions resulting in relative average oxidation states of +2.6 and +2.4, respectively, whereas Fe is always present in the form of Fe3+. Thus, the number of Mn3+ ions in the birnessite structure is increased by Ni, Co and Fe-doping. This is an important finding, as Mn3+ sites are believed to be an essential active site feature in OER catalysis by MnOx.69–72
Layered manganese oxides exhibit two different ion-exchange sites: metals can be incorporated into the interlayer space as well as into the layers themselves, while Co and Ni ions are able to fill in-layer vacancies within the [MnO6] octahedra of the sheets.60,61,73,74 The latter, i.e. the incorporation into the oxide layer, was confirmed by Ni and Co EXAFS, as both spectra show Co–Mn/Ni–Mn scattering at ∼2.9 Å. This distance is close to the Mn–Mn distance (app. 2.85 Å) in the MnOx layer and indicates that some Ni/Co ions are incorporated into the layers of [MnO6] octahedra. Such a distance can also be found for Fe, indicating that Fe ions can also become a part of the [MnO6] layers.75 However, the doping of Ca2+ into the octahedral layers is unlikely as the ion has a much larger radius (∼114 pm) than Fe/Co/Ni/Mn cations (∼65–75 pm).76
Notably, a specific structural motif is obtained for Mn in the Fe doped samples. For the Ca, Co, and Ni doped electrodes, the local environment of the Mn ions is similar to two Mn–O distances at 1.84 and 1.95 Å and two Mn–M distances at roughly 2.9 and 3.4 Å, whereas three Mn–O shells at 1.84, 1.98 and 2.24 Å and two Mn–M shells at roughly 2.8 and 3.0 Å are present in the case of Fe (Fig. 4, S23, S24 and Table S4†). Moreover, the Mn–O or Mn–M shells sum up to populations (coordination number as determined by EXAFS simulations, Table S2†) of 4.8–5.1 (Mn–O) or 2.7–3.4 (Mn–M) for Ca, Co, and Ni, but for Fe they increase to 6.0 (Mn–O) and decrease to 2.4 (Mn–Fe). This suggests a lower degree of connectivity of the MnO6 octahedra in the case of Fe and is most likely an effect of the lower Mn oxidation state.
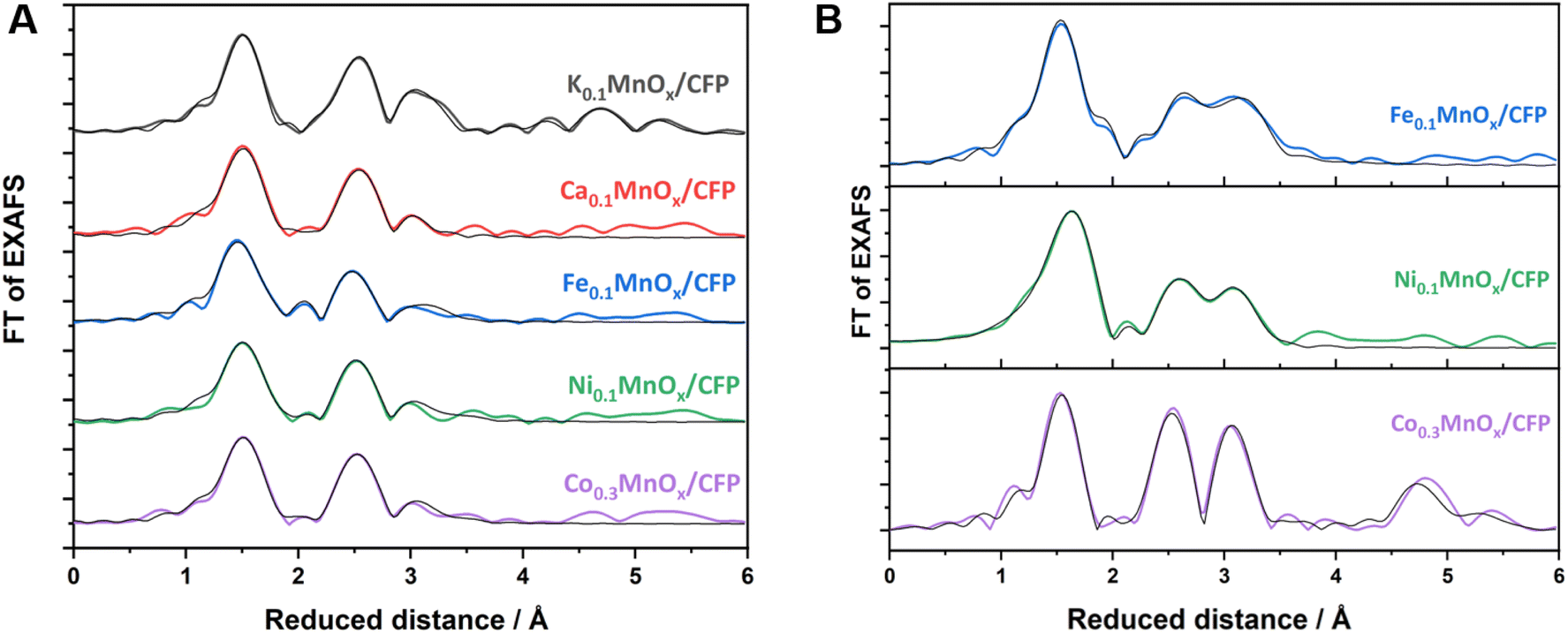 | ||
| Fig. 4 FT of Mn (A) and Fe, Co, Ni (B) K-edge EXAFS spectra for MyMnOx/CFP electrodes. EXAFS simulations (parameters are given in Tables S2–S4 in the ESI†) are shown as black lines. | ||
The local environment of the doping ions, however, is different for all of them. Fe and Ni both exhibit M–O shells at circa 1.93 and 2.07 Å, but with quite different populations of 4.2/1.8 (Fe) or 2.1/3.4 (Ni). In contrast, Co has only one Co–O shell at 1.91 Å and the smallest overall first shell population of 4.6. The first M–M shell distance increases in the order Co (2.85 Å) < Ni (2.90 Å) < Fe (2.95 Å), whereas the second M–M shell is very similar for all three (3.37–3.38 Å). The populations of all M–M shells, however, vary considerably between 1.3 and 4.6. By far the highest degree of ordering is obtained for Co, which, in addition to the highest M–M populations in the 3 Å regime, show pronounced Co–M shells at 4.99 and 5.46 Å. Accordingly, there is a tendency for higher ordering in the local Mn environment of the Co doped samples compared to Ca, Fe, and Ni. This higher order, which is also seen in the FT of the Co K-edge EXAFS spectrum, is most likely due to statistical reasons, as the number of [CoO6] octahedra is increased due to stoichiometry. The local formation of (CoOx)n or CoOxHy building blocks next to incorporated CoMnOxHy clusters or even local phases of pure CoOx, however, can also not be totally excluded by the EXAFS data. In sum, the characterization methods used here indicate with a very high probability that there is a uniform, statistical distribution of Co ions in the birnessite backbone.
In conclusion, Ca/Fe/Co/Ni-doped MnOx/CFP electrodes can be obtained via simple ion-exchange reactions, which is much faster in the case of Co2+. All MyMnOx/CFP electrodes exhibit a layered, birnessite-type MnOx structure. The doping of Fe/Ni cations leads to a reduction of the Mn ions, while Co and Ni are oxidized beyond the M2+ level. EXAFS revealed an incorporation of Fe/Co/Ni ions into the [MnO6] layers, whereas Ca2+ only occupies the interlayer space. Interestingly, the incorporation of Fe leads to a higher disorder within the MnOx backbone, whereas for Co the opposite can be said.
2.2 Electrochemical characterization of MyMnOx/CFP electrodes
Next, we studied water oxidation electrocatalysis by MyMnOx/CFP in order to evaluate the influence of metal doping on the catalytic activities and stabilities. Three different pHs were chosen for this study: 1 M potassium phosphate buffer (pH 2.5 and 7.0, roughly corresponding to the first two pKa values of phosphoric acid) or 1 M KOH (pH ∼14.0).All electrochemical measurements were manually corrected for ohmic losses at 85%, for which the electrolyte resistance Ru was determined by electrochemical impedance spectroscopy. Potentials were converted to the reversible hydrogen electrode (RHE) in order to obtain comparable values independent of the solvent pH.
Fig. 5 shows cyclic voltammograms in the anodic potential range from +1.0 to +1.8 V for MyMnOx/CFP electrodes operated at the three different pHs and show two distinct regions, which are typical of transition metal oxide electrodes immersed in aqueous electrolytes:
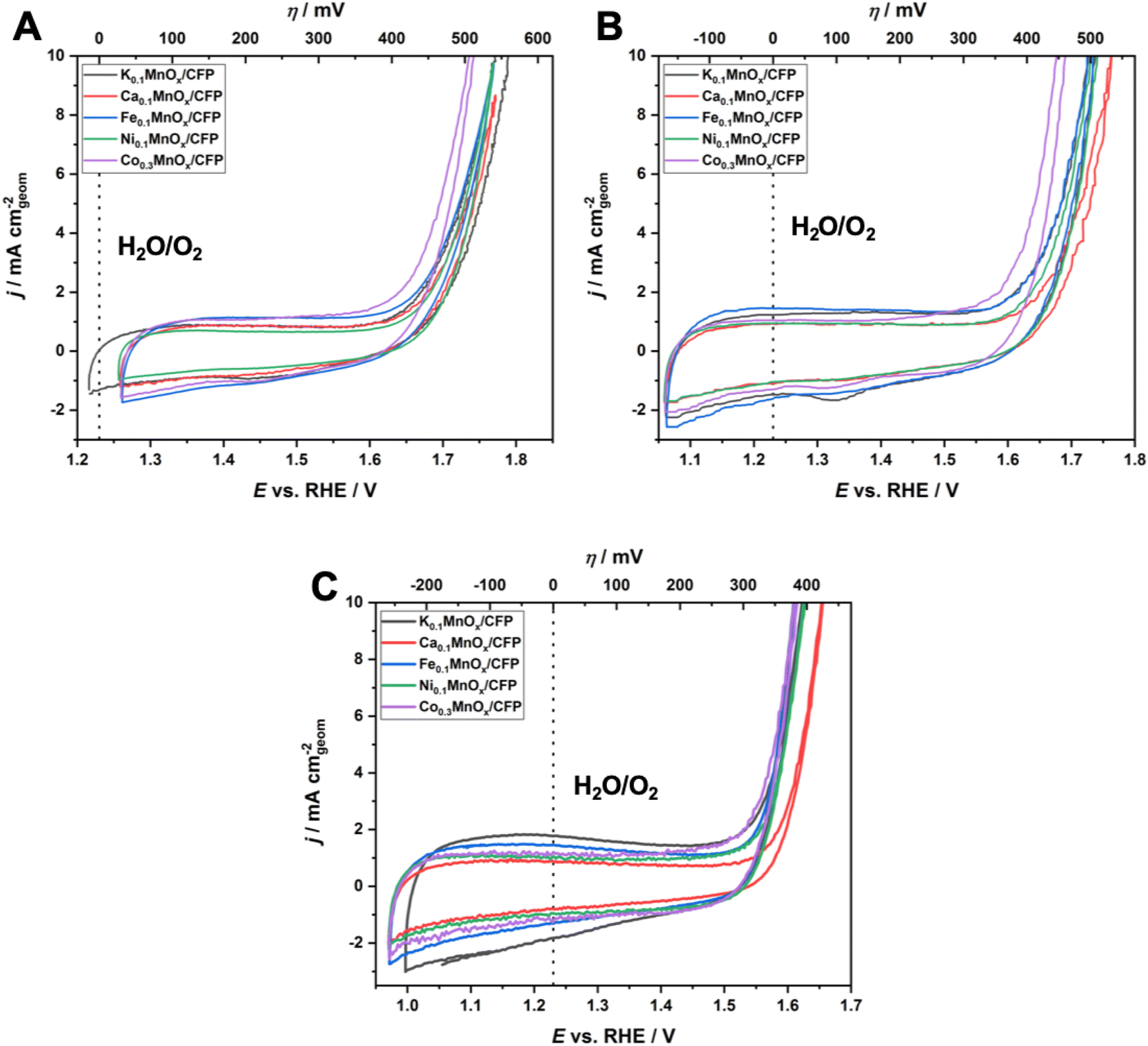 | ||
| Fig. 5 Cyclic voltammograms for MyMnOx/CFP electrodes in 1 M KPi-buffer at pH 2.5 (A) and pH 7.0 (B) and in 1 M KOH at pH 14.0 (C). In each case the 2nd CV cycle is plotted; scan rate: 20 mV s−1. All shown traces are the average of at least three individual measurements. | ||
(1) from around 1.0–1.6 V, broad, unresolved pseudo-capacitive redox waves are discernible. As all electrodes are mainly composed of manganese, these are most likely attributable to Mn3+ ⇌ Mn4+ transitions in combination with insertion/removal of H+ or K+ ions into/from the electrolytes to compensate for the differences in charge.11,14,77 Nevertheless, a contribution of redox transitions from Co, Ni or Fe ions cannot be excluded, although no additional redox peaks, e.g. indicating a Ni3+ ⇌ Ni4+ transition typically observable in NiOx, are present;78–81
(2) an irreversible oxidation feature with a steep slope above potentials E of ∼1.6 V for pH 2.5 and 7.0 and 1.5 V for pH 14.0 is indicative of the beginning of the water oxidation catalysis. The fact that this feature is visible for all electrodes at all tested pH values demonstrates their ability to catalyze the OER over the entire pH range. In general, earlier onset and higher current densities can be found for the electrodes operated under alkaline conditions. This is typical of first-row transition metal electrodes and has also been observed before.14,24
To further evaluate the electrochemical performances of MyMnOx/CFP electrodes, Tafel analyses were conducted using data generated from quasi-stationary stepwise chronoamperometry experiments as shown in Fig. S5.† In all cases, a nearly ideal linear dependence of log(j) vs. η was found for the current-density range from 0.1–10 mA cm−2, indicating that the catalytic reaction is kinetically controlled under these conditions (vs. mass-transport limitations).
For a comparison of the electrode activities, the overpotentials η needed to reach current densities of 1 and 5 mA cm−2, extracted from the Tafel plots in Fig. S5,† are shown as bar graphs in Fig. 6A and are additionally listed in Table 1. All electrodes reached a benchmark current density of j = 1 mA cm−2 at overpotentials from 450–480 mV (pH 2.5), 420–450 mV (pH 7.0) and 310–360 mV (pH 14.0), respectively. Additionally, the difference Δη between the overpotential needed by the K0.1MnOx/CFP electrode to reach 1 mA cm−2 and that needed by all other types of electrodes is shown in Fig. S6 in the ESI† (for values see Table 1). At pH 2.5, a minor activity enhancement can be found for the Co0.3MnOx/CFP electrode, while all other electrodes are worse than K0.1MnOx/CFP. For pH 7.0, the results are quite different: all tested electrodes show lower overpotentials of up to 30 mV indicating that at this pH the activity can be increased by metal doping. This is also the case at pH 14.0 for the Co- and Ni-doped electrodes, whereas Ca-doping leads to a pronounced deactivation.
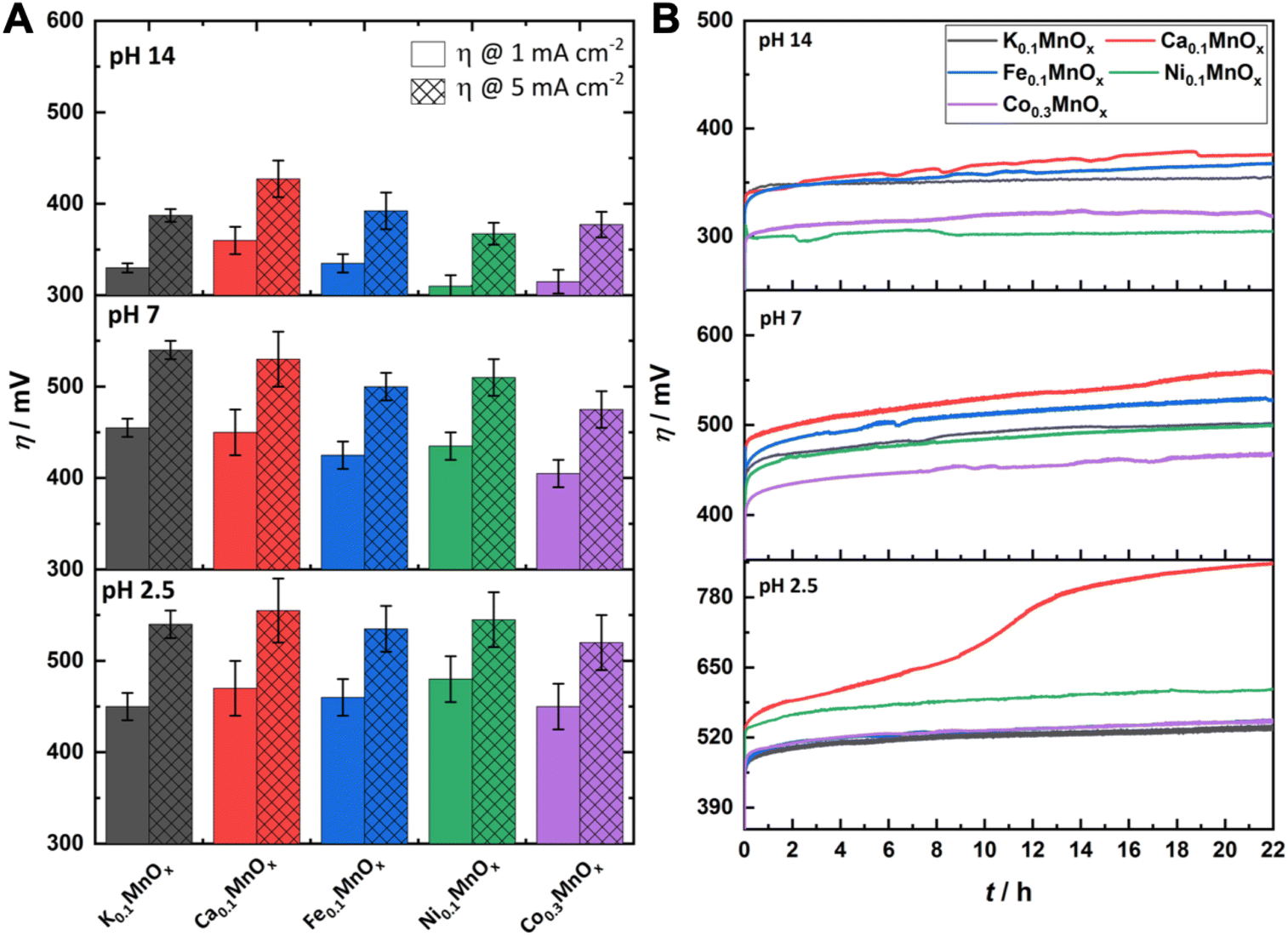 | ||
| Fig. 6 (A) Overpotentials η needed to achieve WOC current-densities of 1 and 5 mA cm−2 at MyMnOx/CFP electrodes. Values are extracted from Tafel plots (see Fig. 5) generated from quasi-stationary stepwise chronoamperometry data. (B) η needed to maintain a current density of 2 mA cm−2 over a time of 22 h. All measurements were conducted in 1 M potassium phosphate buffer or 1 M potassium hydroxide solution at three different pH values (pH 2.5/7.0/14.0). All shown traces are the average of at least three individual measurements. The error bars represent one standard deviation. | ||
| Tafel slopes [mV dec−1] | η @ 1 (5) mA cm−2 [mV] | Δη [mV] | η @ 2 mA cm−2 after 22 h [mV] | n(M)Q [nmol cm−2] | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pH 2.5 | pH 7.0 | pH 14.0 | pH 2.5 | pH 7.0 | pH 14.0 | pH 2.5 | pH 7.0 | pH 14.0 | pH 2.5 | pH 7.0 | pH 14.0 | pH 2.5 | pH 7.0 | pH 14.0 | |
| K0.1MnOx | 130 | 120 | 45 | 450 (540) | 450 (540) | 330 (360) | 0 | 0 | 0 | 530 | 500 | 355 | 235 | 335 | 340 |
| Ca0.1MnOx | 115 | 120 | 60 | 470 (555) | 450 (530) | 360 (400) | +20 | −5 | +30 | 840 | 560 | 380 | 140 | 280 | 245 |
| Fe0.1MnOx | 110 | 110 | 50 | 460 (535) | 425 (500) | 335 (365) | +10 | −30 | +5 | 540 | 530 | 360 | 215 | 390 | 380 |
| Ni0.1MnOx | 110 | 105 | 40 | 480 (545) | 435 (510) | 310 (340) | +30 | −20 | −20 | 610 | 500 | 300 | 205 | 390 | 390 |
| Co0.3MnOx | 100 | 105 | 45 | 450 (520) | 405 (475) | 315 (350) | 0 | −50 | −15 | 540 | 460 | 320 | 235 | 385 | 350 |
Tafel slopes, listed in Table 1, were obtained by linear regression. For pH 2.5 and 7.0, the Tafel slopes are almost constant and close to 2 × 59 = 118 mV dec−1, which is typical of multi-step reactions, where the first oxidation is turnover-limiting.82,83 Nevertheless, some minor differences can be discovered: slightly faster reaction kinetics are observed for doped electrodes as lower slopes than for the K0.1MnOx/CFP electrode are found.
One explanation for this behavior could be that the transfer of electrons can be increased by the incorporation of metal ions that increase the conductivity of the oxide backbone. Manganese oxides have a lower conductivity compared to Ni or Co oxides, a factor which has been shown in previous studies to limit the catalytic activity.13–15 Nevertheless, a lowering of the Tafel slope is not in all cases accompanied by an improvement of the activity as shown by the ηs needed to reach a distinct reaction rate (see Table 1 and Fig. 6A).
For electrodes operated at pH 14.0, a change of the rate determining step can be assumed which is indicated by a pronounced decrease of the Tafel slope from ∼120 mV dec−1 to values as low as 40 mV dec−1, the latter being in line with multi-step reactions where the second step is potential-independent.82,84 Under alkaline conditions, the Ni- and Co-doped electrodes show the fastest reaction kinetics. Overall, the incorporation of Ni or Co has a positive effect on the activity of MyMnOx/CFP, whereas the effect of Ca or Fe is only small. The general trend described here for the Co-doped electrode Co0.3MnOx/CFP can also be observed for an electrode, where the molar ratio of Mn:Co is only 1:0.1 (see Fig. S5†).
For a possible use as OER electrodes in technical applications, the long-term stabilities/activities of the anodes are of great importance. Therefore, the MyMnOx/CFP electrodes were also tested by chronopotentiometric measurements at a constant current density of 2 mA cm−2 over 22 h under the three pH conditions and the resulting η vs. t traces are shown in Fig. 6B.
For pH 2.5 the trend from the Tafel analyses is retained and no improvement of the electrode activity can be found. The behavior of Co0.3MnOx/CFP and Fe0.1MnOx/CFP is very similar to that of K0.1MnOx/CFP, whereas the Ni- and Ca-doped birnessites are less active. K, Ni, Co and Fe electrodes stay intact over the whole experiment and only a slight increase of η can be observed. In contrast, Ca-doping leads to an inactivation and a doubling of the overpotential after ∼10 h. Although such an immense difference is only observable for pH 2.5, Ca0.1MnOx/CFP generally exhibits the worst long-term activity and stability of all tested electrodes at all pHs. This result was rather unexpected as Ca2+-doping has been proven to enhance the catalytic activity of MnOx in experiments with Ce4+ (at pH ∼ 2) and also thin-film MnOx electrodes operated at neutral pH. Furthermore, a Ca2+ cation is also an essential component of the OEC.44,65,85,86 For synthetic CaMnOx, the presence of Ca2+ leads to amorphization, which is accompanied by an increase of the surface area and assumed to be responsible for the higher catalytic activity. However, in K0.1MnOx/CFP, this increase in disorder is already achieved by the annealing step at 400 °C carried out in this study for all electrodes.14 Furthermore, the additional Ca2+-induced amorphization can have a destabilizing effect on the layered birnessite structure as for example shown by Suib and coworkers.47 They concluded that this is caused by the larger ionic radius of Ca2+ causing stress on the Mn–O backbone leading to an easier breakdown of the ordered stacking of the layers. The combination of annealing and Ca2+ incorporation may here account for the destabilization of the structure and for the lower catalytic activity and stability of Ca0.1MnOx/CFP.
Under neutral conditions, Co-doping leads to a 40 mV improvement of the overpotential compared to all other electrodes, showing that the synergy of these metals, whose oxides belong to the most active earth-abundant OER catalysts under neutral conditions, is a promising approach.15,87,88 However, no improvements can be found for Ni- and Fe-ion exchanged electrodes at pH 7.0.
At pH 14.0, the Ni- and Co-doped electrodes are able to catalyze the OER at overpotentials which are up to 60 mV lower compared to K0.1MnOx/CFP. Again, both NiOx and CoOx are themselves highly stable and active catalysts under alkaline conditions.16–21 In the case of Ni0.1MnOx/CFP, an activation of the catalyst most likely caused by the formation of an active phase at the surface is observed during the experiment (see Fig. 7). Such a behavior is well known for Ni oxides and has been attributed to the incorporation of Fe impurities, which might also be accounted for here.89
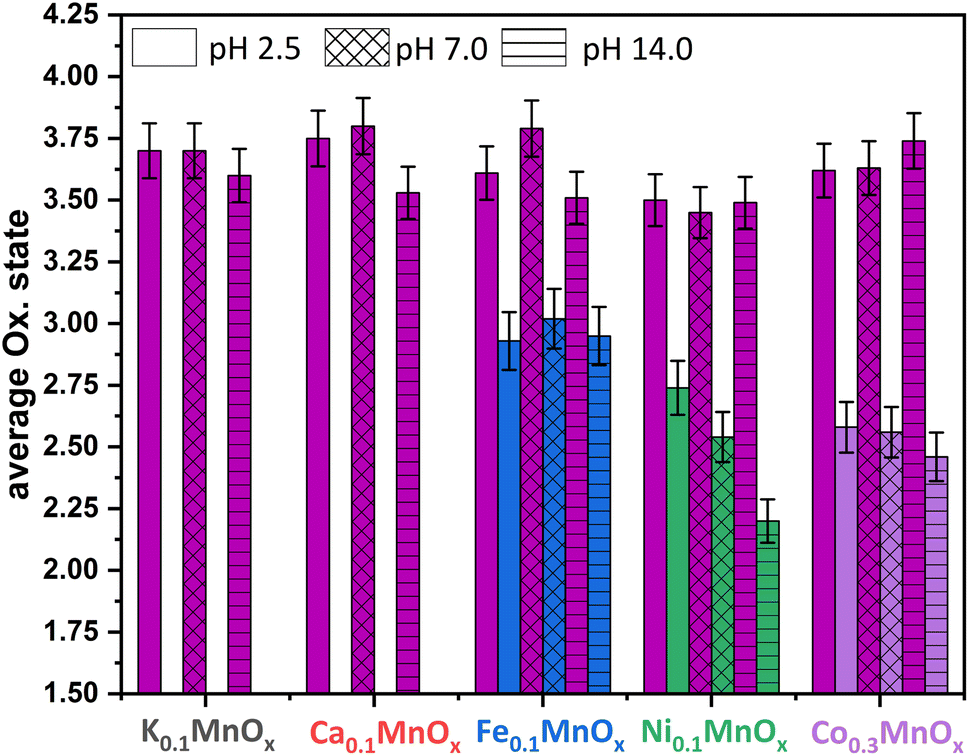 | ||
| Fig. 7 Average oxidation states of Mn (plum), Fe (blue), Ni (green) and Co (purple) for MyMnOx/CFP electrodes after electrochemical operation under different pH conditions. The oxidation states are derived from a linear fit of the data extracted at a normalized X-ray absorption intensity of 0.5 for reference compounds. See also Fig. S19–S26 in the ESI† for more information. The error bars represent one standard deviation. | ||
Recently, our groups found a correlation between the catalytic activity and the number of manganese ions undergoing redox reactions during the catalytic cycle.14 Such a correlation has also been found for Co-based electrodes.90 In order to determine the influence of the cation doping on the molar amount of redox active metal ions, the cathodic scan of CVs recorded subsequent to the determination of the Tafel plots was integrated. As a result, the reduction charge Q is obtained, which divided by the Faraday constant F leads to the molar amount of redox active metal ions n(M)Q (see Table 1). From the n(M)Q data, it can be concluded that the number of redox active metal ions can be influenced by the incorporation of additional OER-active cations into the manganese oxide backbone. The previously observed activity trend is resembled here as the Ca0.1MnOx/CFP-electrode show the smallest n(M)Q values. As expected, a slight increase can be observed for pH 7.0 and pH 14.0, whereas a decrease for pH 2.5 is discernible for Fe-, Ni- and Co-doped electrodes compared to K0.1MnOx/CFP. For pH 2.5, the incorporated ions most likely do not participate in OER catalysis. Furthermore, the decrease (pH 2.5) or increase (pH 7/14) of the redox active sites for all doped electrodes additionally indicates that an electronic interaction between the Mn and M ions influencing their activity occurs. Thus, the increase in n(M)Q cannot be explained solely by the increased number of redox-active metal sites due to ion exchange. However, the extent of this interaction and its influence on the activity cannot be determined. Hence, the determination of n(M)Q is a very useful instrument to compare the activities of MnOx or other metal oxide electrodes.
To further evaluate whether the higher activity, especially of the Co-doped electrode, is only due to the higher loading of OER-active cations, the long-term stability of a Co0.3MnOx/CFP electrode during constant application of j = 2 mA cm−2 is compared to that of a K0.1MnOx/CFP electrode exhibiting nearly an identical number of Mn/M ions (n(Co + Mn)AAS = 3.9 μmol cm−2vs. n(Mn)AAS = 4.0 μmol cm−2, Fig. S9†). Interestingly, the Co-doped electrode needed an app. 50 mV lower overpotential after 24 h of operation in 1 M KPi at pH 7.0. Consequently, the higher activity of Co0.3MnOx/CFP electrodes cannot simply be explained by the increase in the total number of OER active metal sites. Thus, a synergistic effect between the incorporated ions and manganese seems more likely.
In conclusion, the electrochemical activity and stability of K0.1MnOx/CFP electrodes can be improved by the incorporation of Fe, Ni, and Co cations by an ion-exchange reaction. The incorporation has a positive influence on the electrode kinetics, the overall activities and the number of active redox sites. However, the effect is not the same for all ions tested and additionally depends on the pH at which the electrochemical tests are carried out. Thus, large improvements are especially found for Co under neutral and Co and Ni under strongly alkaline conditions.
2.3 Post operando characterization of electrodes
In order to determine morphological or structural changes that could explain the different WOC behavior of the MyMnOx/CFP catalysts, ex situ post operando studies after 22 h of continuous operation were conducted at the same three pH values as studied before.First, possible changes of the surface morphologies due to long-term electrochemical water oxidation were examined by SEM (Fig. S8†). Only minor changes are visible for the electrodes operated under different pH conditions, indicating that the basic birnessite structure of the parent MyMnOx/CFP electrodes is retained. Only for the electrodes operated at pH 2.5, a broadening of the threads building a sponge-like structure can be detected, which has also been observed before for K0.1MnOx/CFP electrodes.14
To evaluate whether the electrochemical treatment led to a leaching of the incorporated metal ions from the manganese oxide backbone, the atomic ratios were determined by EDX. From Fig. S10–S14,† it can be seen that even under acidic conditions no leaching occurred. This points towards a reasonable stabilization especially of Co and Ni ions by incorporation into the birnessite backbone as these oxides normally dissolve in the acid according to their Pourbaix diagrams.91 Additionally, an incorporation of potassium (atomic ratio of K:Mn ∼ 0.1:1) and phosphate (atomic ratio of P:Mn ∼ 0.1:1) ions from the electrolyte can be observed.
Powder XRD patterns (Fig. S15†) show no additional reflexes compared to the electrodes after preparation, which confirms the SEM findings and additionally shows that the carbon support remains intact. ATR-IR-spectra reveal no additional bands for the MyMnOx/CFP electrodes that have been exposed to anodic potentials, except for electrodes operated in phosphate buffers (pH 2.5 and pH 7.0; Fig. S16†). Here, two broad features between 1150 and 950 cm−1 can be observed, which have been observed before and are most likely attributable to the incorporated phosphate ions (HnPO(3−n)−4).14 Such features are not visible for the electrodes operated in 1 M KOH, confirming the interpretation as interactions between the phosphate anions and the manganese oxide.
All results obtained by these mainly bulk sensitive measurement techniques are typical of amorphous birnessites. In order to reveal details on the chemical states and the local atomic environment of the metal centers, synchrotron-based XAS studies were again carried out.
The XAS-data based oxidation states for Mn, Fe, Ni and Co are visualized in Fig. 7. The average manganese oxidation states vary within a rather narrow range from +3.4 to +3.8 and are generally higher compared to their pre operando values (see Fig. 3). This confirms our interpretation of the CV that the Mn ions are oxidized, a fact that was repeatedly observed before.11–14 However, the highest oxidation states for Mn are generally found for the least active catalysts. This supports the importance of stable Mn3+ states within the mixed valent MnOx-based OER catalysts proposed by several authors.46,69,70,92
No change of the average oxidation state of the Fe ions is visible pointing to relatively high inertness of the +III state within the manganese oxide framework. Nevertheless, only in situ XAS or Mössbauer spectroscopy measurements could show whether other oxidation states, e.g. Fe4+, are also present at catalytic potentials, as these have recently been found for Ni–Fe oxyhydroxides,10,93 and see also ref. 94.
The likely contribution of Ni to the higher activity under neutral and especially under alkaline conditions can be recognized by a lowering of the average oxidation state of the Ni ions from ∼2.6 (as-prepared, pH 2.5) via ∼2.5 (pH 7.0) to ∼2.2 for pH 14.0. This eventually leads to the assumption that Ni2+ is, besides Mn, also an OER active species in Ni0.1MnOx/CFP under alkaline conditions. In addition, the lowering of the Ni oxidation state at pH 14.0 might be the reason for the lowering of η (catalyst activation) in the first minutes of the chronopotentiometry measurements discussed above. A stabilization of Ni in lower oxidation states than +IV (the usually dominant Ni oxidation state in Ni-only oxides) has recently been observed for Fe containing Ni oxides.81,95
For the incorporated Co ions, a slight oxidation after operation in 1 M phosphate buffers of pH 2.5 and 7.0 can be observed, whereas the average oxidation state is retained after operation at pH 14.0. Fractional oxidation states of ∼+2.6 have also been found for phosphate containing cobalt oxide in its resting state.96,97 Furthermore, Co oxide OER catalysts are highly dynamic systems able to reversibly switch between the oxidation states +2/+3/+4 during catalysis.96,97 A contribution of the Co ions to the OER activity in Co0.3MnOx/CFP seems therefore likely, but again has to be proven by in situ techniques.
A detailed look at the XAS data in Fig. S19–S26† reveals that the M–O and M–M distances are well conserved for all metals (Mn, Fe, Co, and Ni) under all conditions (as prepared, operated at pH 2.5, 7.0, and 14.0). Only above 3 Å changes larger than 0.1 Å appear. Shell populations typically vary in the range of roughly ±0.3 for the first ligand sphere, ±0.5 for the second and ±0.7 for far distant shells. Considering this, a tendency for higher M–M shell populations after operation can be detected for all metal types except Fe, suggesting a higher degree of ordering in most cases.
In conclusion, no major structural changes of the basic birnessite phase as well as the local coordination environments of the tested metal-doped electrodes were uncovered by ATR-IR, XRD, XAS, EDX and SEM measurements after long-term WOC operation at pH 2.5/7.0/14.0. Therefore, the studied anodes can be characterized as stable and catalytically active OER anodes over a wide pH range. EDX measurements revealed that no leaching occurred during operation. Furthermore, it can be concluded that the increase of the activity and stability, especially for Ni- and Co-doped electrodes, is due to a synergistic effect between the manganese oxide backbone and the incorporated metal ions.
2.4 Comparison with other state-of-the-art OER catalysts
For a better comparison of the metal doped manganese oxide catalysts, we immobilized IrOx, CoPi and FeNiOx on CFP following well-established literature procedures.87,98–101 To confirm the deposition, the SEM micrographs of the metal oxide coated CFP can be found in Fig. S14† and again carbon supports uniformly covered by the catalyst layers are visible.As before for MyMnOx/CFP, the electrocatalytic OER activities and stabilities were evaluated by Tafel analyses (Fig. S17†) and long-term chronopotentiometry measurements (Fig. S18†). The overpotentials needed after 22 h of constant operation are schematically presented in Fig. 8 in comparison to the results obtained for the mixed metal oxide electrodes. Overpotentials, Tafel slopes and long-term stability parameters are listed in Table 2.
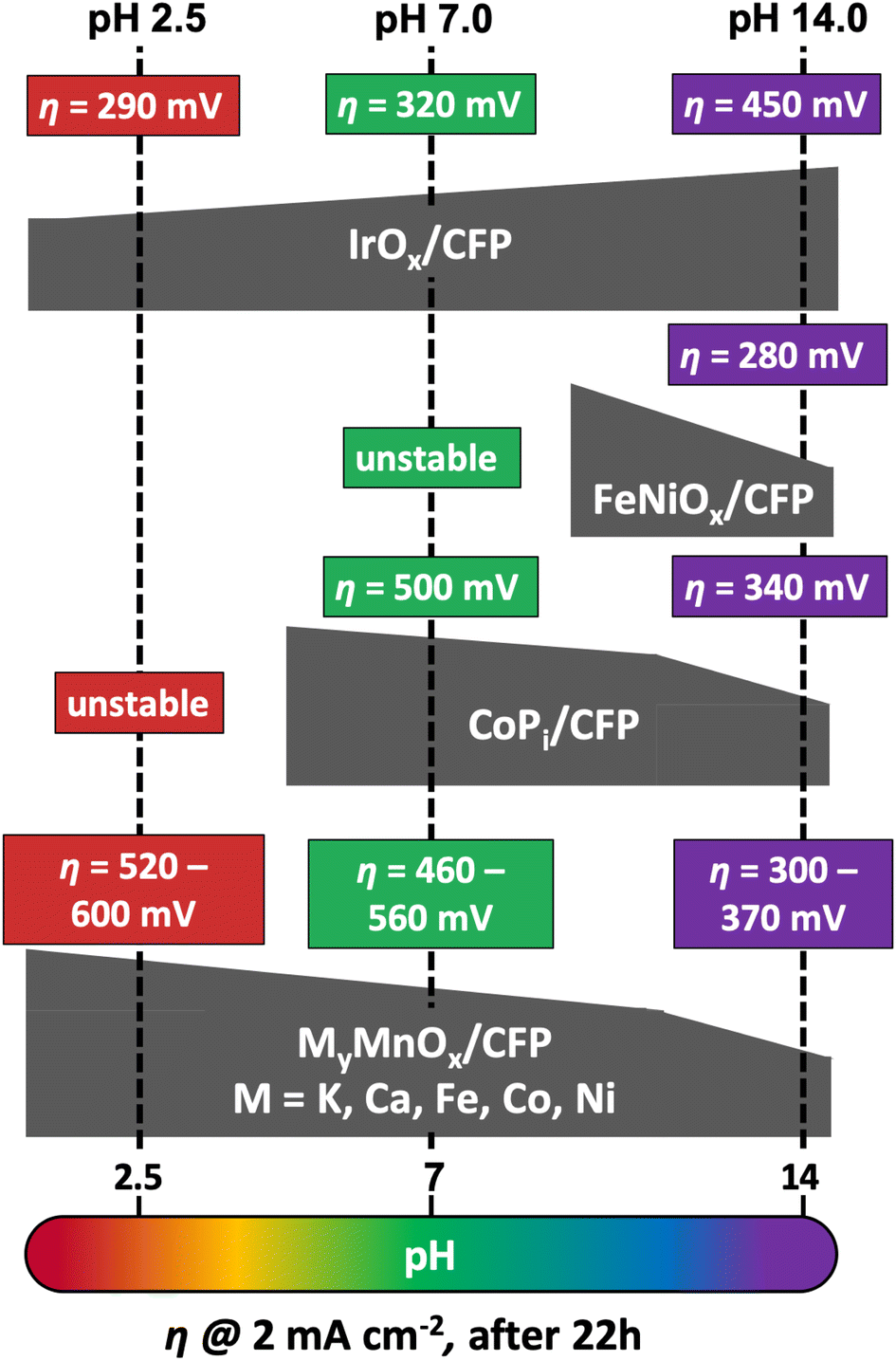 | ||
| Fig. 8 Comparison of overpotentials η needed to reach 2 mA cm−2 after 22 h of operation at pH 2.5, 7.0 or 14.0 by different MOx/CFP electrodes. | ||
| Tafel slope [mV dec−1] | η @ 1 (5) mA cm−2 [mV] | η @ 2 mA cm−2 after 24 h [mV] | |||||||
|---|---|---|---|---|---|---|---|---|---|
| pH 2.5 | pH 7.0 | pH 14.0 | pH 2.5 | pH 7.0 | pH 14.0 | pH 2.5 | pH 7.0 | pH 14.0 | |
| IrOx | 80 | 70 | 60 | 260 (300) | 230 (280) | 250 (290) | 290 | 320 | 450 |
| CoPi | * | 120 | 60 | * | 420 (510) | 310 (360) | * | 500 | 340 |
| NiFeOx | * | * | 40 | * | * | 280 (310) | * | * | 280 |
As predicted from the Pourbaix diagram, FeNiOx/CFP and CoPi/CFP electrodes are unstable at pH 2.5, leading to a complete dissolution of the catalyst layer from the carbon support within minutes of operation.91 On the other hand, IrOx/CFP exhibits good stability and overpotentials, which are more than 200 mV lower compared to the best MyMnOx/CFP electrodes in acidic media, highlighting the remarkable catalytic abilities of iridium oxides at low pH.16,102 Nevertheless, the manganese oxide-based electrodes are the only stable alternative among the tested electrodes under acidic conditions. Furthermore, the activity of the MyMnOx/CFP electrodes is still very good compared to other earth-abundant metal oxide catalyst systems and ranks amongst the best that have so far been reported.15,22,24,54,102–105
At pH 7.0, IrOx/CFP still exhibits the highest activity and stability, whereas FeNi oxide remains unstable. The behavior of CoPi/CFP, one of the most active earth-abundant catalysts under neutral conditions, is quite similar to that of K0.1MnOx/CFP, showing that both metal oxides are nearly equally active. However, the mixture of Co and Mn reaches higher activities, again indicating that the synergy of these two elements can be a promising approach to yield even more active OER electrocatalysts.
Interestingly, under alkaline conditions, IrOx behaves contrary to the other transition metal oxides studied, as a fast increase of η in the first hours of the experiments was observed as reported before.106–108 On the other hand, CoPi and FeNiOx behave like MnOx and show higher stabilities and activities at pH 14.0.14 The FeNiOx/CFP electrodes show remarkable activities similar to the Ni-doped MnOx electrode at pH 14.0, under the tested catalyst systems. Hence, the MyMnOx/CFP electrodes and especially the Ni0.1MnOx/CFP electrode can easily compete with the CoPi and the IrOx electrode as well as with other Mn-based catalysts.15,54,103,109 However, the exceptional activities of FeNiOx at pH ≥ 13 and of that reported in the literature for operation in strongly alkaline media are so far out of reach for MnOx-based electrocatalysts.17,80,110,111
3 Conclusions
In this study, we presented the facile preparation of metal-doped (Co, Ca, Ni, and Fe) layered manganese oxides coated on carbon fiber paper substrates for electrocatalytic water oxidation. The preparation was based on a well-established, easily scalable redox-deposition approach followed by an ion-exchange reaction using metal nitrate solutions. The additional metal ions were incorporated in both the interlayer space and the MnO6 sheets and had only a minor effect on the morphology and the structure of the basic birnessite backbone. XAS measurements, on the other hand, showed that the oxidation states of manganese can be lowered by ion exchange, leading to the introduction of a larger number of WOC-active Mn3+ ions. Overall, Fe doped MnOx layers exhibited the lowest average manganese oxidation state.Electrochemical measurements revealed that MyMnOx/CFP are efficient and stable anodes for electrocatalytic water oxidation over nearly the entire aqueous pH regime (pH 2.5–14.0). Differences were especially found for Co- and Ni-doped electrodes, whose WOC activities and stabilities were clearly improved compared to the undoped K0.1MnOx/CFP electrode. The reasons for these improvements might be due to a synergistic effect between the OER-active metal centers, a higher electrical conductivity and/or better charge transfer properties. The exact causes have to be addressed in future studies, for example by in situ XAS- or four-point probe resistivity measurements.
Ex situ post operando spectroscopy analysis confirmed the stability of MyMnOx electrodes. Slight changes of the average Mn, Co and Ni oxidation states were observed, indicating the participation of these elements in the catalytic process. However, a clear proof (for example by in situ XAS measurements) has so far been missing, but will be tackled in subsequent studies.
Overall, (doped) manganese oxides stand out as WOC materials over a wide pH range, especially due to their universal applicability. In addition, under intermediate pH conditions, together with cobalt oxides, they show one of the highest activities of earth-abundant catalysts. They thus represent a promising catalyst alternative at pHs below 7 with possible applications like artificial leaves or photoelectrochemical cells (PECs), which are normally operated at intermediate pHs and at relatively low current densities (j ∼ 10 mA cm−2).112,113 Similarly, microbial electrosynthesis cells (MESCs) might be an alternative application for MnOx anodes.
4 Experimental section
4.1 Materials
Graphitized carbon fibre paper (CFP, TGP-H-60) was purchased from Toray Carbon Inc., cut into pieces of 1 × 2 cm2 and pre-treated by sonication for 15 min each in 0.1 M HNO3, ethanol and water, respectively. Please see the ESI† for further details concerning the materials used for this study.4.2 Preparation of MyMnOx/CFP electrodes
The preparation of K0.1MnOx/CFP electrodes using the in situ redox reaction between permanganate and carbon has been described in detail elsewhere.13,14 In short, 2.5 mL of HNO3 (69%) was added to a beaker containing a KMnO4 solution (0.1 M, 100 mL) and the strongly acidic mixture (pH 0.4) was then heated to 70 °C. Subsequently, four pre-treated pieces of CFP were dipped into the slowly stirred solution simultaneously for 90 min. To ensure that only 1 cm2 of the carbon material was exposed to the solution, the rest of the area was covered with adhesive tape (tesafilm®, “matt-unsichtbar”, thickness ∼ 70 μm). The electrodes were then taken out of the KMnO4 solution, washed thoroughly with water and dried in air at 60 °C for 1 h. In order to obtain ion exchanged MyMnOx/CFP electrodes, the parent K0.1MnOx/CFP electrodes were then dipped into a beaker containing a solution of 1 M M2+/3+(NO3)2/3 (with M = Fe3+, Ni2+ or Ca2+) or 0.1 M Co2+(NO3)2. Ion exchange was carried out for different times (15 min to 24 h) in order to obtain various numbers of incorporated metal ions. Finally, the MyMnOx/CFP electrodes were washed with water, dried in air at 60 °C for 1 h and calcined in a muffle furnace in air at a temperature of 400 °C for 1 h.4.3 Preparation of MOx/CFP electrodes
For a better comparison with current state-of-the-art OER catalysts, IrOx, FeNiOx and CoPi were electrodeposited on CFP following published procedures:IrOx/CFP was prepared via a method developed by Petit et al.:98 2 mM K3IrCl6 was dissolved in water and 15 mM oxalic acid and 100 mM K2CO3 were added (pH 10.5). This solution was then stirred for 10 days at room temperature until the color turned from pale yellow to light blue. During the aging time, it is assumed that an [Ir(C2O4)(OH)4]2− complex is formed, which can be anodically electrodeposited on CFP.99 The electrodeposition was conducted in a typical three electrode setup (see below) by applying a constant current density of 350 μA cm−2 until a charge Q = 5 mC cm−2 had passed through the electrode.
FeNiO x /CFP was prepared following a synthesis route developed by Lu et al.:100 3 mM Fe3+(NO3)3·9H2O and 3 mM Ni2+(NO3)2·6H2O were dissolved in water. This mixture was then used to cathodically deposit mixed NiFeOx on CFP in a typical three electrode setup (see below). The electrodeposition was carried out by applying a constant potential of −1.0 V vs. Ag/AgCl for 300 s. It is assumed that the electrodeposition occurs via the reduction of NO3− at the electrode surface leading to the generation of hydroxides, which then react with Fe3+ and Ni2+ to form bimetallic hydroxide deposits on the surface.
CoPi/CFP was prepared following a well-established route developed by Nocera and co-workers:87,101 0.5 mM Co2+(NO3)2·6H2O was dissolved in a 0.1 M potassium phosphate buffer (pH 7.0). The electrodeposition of CoPi on CFP was conducted in a typical three electrode setup by applying a constant potential of 1.71 V vs. RHE until a charge Q = 40 C cm−2 passed through the WE.
In order to be able to compare the activities per geometric electrode area of each type of catalyst, we tried to deposit approximately the same mass loading on the conductive substrate.
4.4 Instrumental methods
Details of the instrumentation and measurement conditions used to record vibrational spectra (ATR-FT-IR), electron micrographs (SEM), energy dispersive X-ray analyses (EDX), and X-ray absorption spectra (XAS) and for the determination of metal ion fractions by flame atomic absorption spectroscopy (fAAS) are provided in the ESI.†4.5 General electrochemical details
All electrochemical experiments were conducted in a typical three-electrode setup using a BioLogic VSP potentiostat/galvanostat with an integrated module for electrochemical impedance spectroscopy (EIS). The electrochemical cell (volume 100 mL) typically consisted of an Ag/AgCl reference electrode (Metrohm, 3 M KCl, +0.207 V vs. NHE at 25 °C), a Pt rod as the counter electrode separated by a porous glass frit (Metrohm, diameter ∼ 2 mm) and MyMnOx/CFP as the working electrodes (WE). All measurements were conducted in 50 mL of the respective air-saturated electrolyte (1 M potassium phosphate buffer at pH 2.5/7.0 or 1 M KOH at pH 14.0) at ambient temperature (∼23 °C). WE potentials were converted to the RHE according to eqn (1).| ERHE = EAg/AgCl + 0.207 + 0.059pH (in V) | (1) |
To evaluate the electrocatalytic behaviour of the MyMnOx/CFP anodes for water oxidation in different electrolytes, a multi-step protocol consisting of the determination of the uncompensated resistance Ru (by EIS) followed by “staircase” chronoamperometry measurements (CA, for the determination of Tafel plots) and terminated by cyclic voltammetry sweeps (CV) over a wide potential range was used (see the ESI for details†). To minimize the possibility of mass transport limitations and the formation of a pH gradient in the pores of the catalyst, the solutions were stirred at a constant speed (600 rpm) during the CA measurements. In staircase CA protocols, potentials were held constant for 5 min at each step in order to exclude a large contribution of the capacitive current to the overall current density. The average current density over the last 30 s of each step was then used for the Tafel analyses. To evaluate the long-term stability of MyMnOx/CFP under different electrolyte conditions, chronopotentiometry measurements (CP) at current densities of 2 mA cm−2 were performed for a duration of 24 h. All CA, CP and CV measurements were iR corrected at 85%.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support of the Bundesministerium für Bildung und Forschung (BMBF cluster project MANGAN, FKZ 03SF0511A; BMBF project Operando-XAS). The work (of SM and HD) has also been funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008/1 – 390540038 (Gefördert durch die Deutsche Forschungsgemeinschaft (DFG) im Rahmen der Exzellenzstrategie des Bundes und der Länder – EXC 2008/1 – 390540038.). We thank the Helmholtz-Zentrum Berlin (HZB) for synchrotron beamtime allocation at KMC-3/BESSY (Berlin-Adlershof) and Dr Ivo Zizak as well as other HZB staff for experimental support. We would also like to thank Prof. Dr Anna Fischer at the ALU Freiburg for the possibility to carry out electron microscopy at a BMBF founded HR-SEM instrument (project EDELKAT, FKZ 03X5524).Notes and references
- D. G. Nocera and N. S. Lewis, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef.
- R. Schlögl, ChemSusChem, 2010, 3, 209–222 CrossRef.
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- G. P. Gardner, Y. B. Go, D. M. Robinson, P. F. Smith, J. Hadermann, A. Abakumov, M. Greenblatt and G. C. Dismukes, Angew. Chem., Int. Ed., 2012, 51, 1616–1619 CrossRef CAS.
- M. Dincă, Y. Surendranath and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10337–10341 CrossRef.
- D. K. Bediako, B. Lassalle-Kaiser, Y. Surendranath, J. Yano, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6801–6809 CrossRef CAS.
- Y. Wu, M. Chen, Y. Han, H. Luo, X. Su, M. T. Zhang, X. Lin, J. Sun, L. Wang, L. Deng, W. Zhang and R. Cao, Angew. Chem., Int. Ed., 2015, 54, 4870–4875 CrossRef CAS.
- J. Y. C. Chen, L. Dang, H. Liang, W. Bi, J. B. Gerken, S. Jin, E. E. Alp and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 15090–15093 CrossRef CAS PubMed.
- I. Zaharieva, P. Chernev, M. Risch, K. Klingan, M. Kohlhoff, A. Fischer and H. Dau, Energy Environ. Sci., 2012, 5, 7081–7089 RSC.
- S. Y. Lee, D. González-Flores, J. Ohms, T. Trost, H. Dau, I. Zaharieva and P. Kurz, ChemSusChem, 2014, 7, 3442–3451 CrossRef CAS PubMed.
- J. Melder, W. L. Kwong, D. Shevela, J. Messinger and P. Kurz, ChemSusChem, 2017, 10, 4491–4502 CrossRef CAS.
- J. Melder, S. Mebs, P. A. Heizmann, R. Lang, H. Dau and P. Kurz, J. Mater. Chem. A, 2019, 7, 25333–25346 RSC.
- J. Melder, P. Bogdanoff, I. Zaharieva, S. Fiechter, H. Dau and P. Kurz, Z. Phys. Chem., 2020, 234(5), 925–978 CrossRef CAS.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- E. L. Miller and R. L. Rocheleau, J. Electrochem. Soc., 1997, 144, 3072 CrossRef CAS.
- A. Ursua, L. Gandia and P. Sanchis, Proc. IEEE, 2012, 100, 410–426 CAS.
- A. M. Smith, L. Trotochaud, M. S. Burke and S. W. Boettcher, Chem. Commun., 2015, 51, 5261–5263 RSC.
- B. M. Hunter, J. D. Blakemore, M. Deimund, H. B. Gray, J. R. Winkler and A. M. Müller, J. Am. Chem. Soc., 2014, 136, 13118–13121 CrossRef CAS PubMed.
- S. Loos, I. Zaharieva, P. Chernev, A. Lißner and H. Dau, ChemSusChem, 2019, 12, 1966–1976 CrossRef CAS PubMed.
- K. Sun, I. A. Moreno-Hernandez, W. C. Schmidt, X. Zhou, J. C. Crompton, R. Liu, F. H. Saadi, Y. Chen, K. M. Papadantonakis and N. S. Lewis, Energy Environ. Sci., 2017, 10, 987–1002 RSC.
- A. Minguzzi, F.-R. F. Fan, A. Vertova, S. Rondinini and A. J. Bard, Chem. Sci., 2012, 3, 217 RSC.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS.
- P. C. K. Vesborg and T. F. Jaramillo, RSC Adv., 2012, 2, 7933–7947 RSC.
- R. E. Smalley, MRS Bull., 2005, 30, 412–417 CrossRef.
- K. Rabaey and R. A. Rozendal, Nat. Rev. Microbiol., 2010, 8, 706–716 CrossRef CAS PubMed.
- R. Ganigué, S. Puig, P. Batlle-Vilanova, M. D. Balaguer, J. Colprim, M. Mikkelsen, M. Jørgensen, F. C. Krebs, R. S. Haszeldine, K. Rabaey, R. A. Rozendal, K. P. Nevin, T. L. Woodard, A. E. Franks, Z. M. Summers, D. R. Lovley, C. W. Marshall, D. E. Ross, E. B. Fichot, R. S. Norman, H. D. May, C. W. Marshall, D. E. Ross, E. B. Fichot, R. S. Norman, H. D. May, M. Sharma, N. Aryal, P. M. Sarma, K. Vanbroekhoven, B. Lal, X. D. Benetton, D. Pant, K. J. J. Steinbusch, H. V. M. Hamelers, J. D. Schaap, C. Kampman, C. J. N. Buisman, M. C. A. A. V. Eerten-Jansen, A. T. Heijne, T. I. M. Grootscholten, K. J. J. Steinbusch, T. H. J. A. Sleutels, H. V. M. Hamelers, C. J. N. Buisman, M. Dwidar, J.-Y. Park, R. J. Mitchell, B.-I. Sang, P. Sánchez, R. Ganigue, L. Bañeras, J. Colprim, J. Daniell, M. Köpke, S. D. Simpson, A. S. Gössner, F. Picardal, R. S. Tanner, H. L. Drake, R. K. Thauer, K. Jungermann, K. Decker, P. P. H. Pi, M. T. Agler, B. A. Wrenn, S. H. Zinder, L. T. Angenent, P. Batlle-Vilanova, S. Puig, R. Gonzalez-Olmos, A. Vilajeliu-Pons, L. Bañeras, M. D. Balaguer, J. Colprim, A. W. Jeremiasse, H. V. M. Hamelers, C. J. N. Buisman, M. C. A. A. V. Eerten-jansen, A. T. Heijne, C. J. N. Buisman, H. V. M. Hamelers, I. Bechthold, K. Bretz, S. Kabasci, R. Kopitzky, A. Springer, S. J. Andersen, T. Hennebel, S. Gildemyn, M. Coma, J. Desloover, J. Berton, J. Tsukamoto, C. V. Stevens, K. Rabaey, R. S. Tanner, L. M. Miller and D. Yang, Chem. Commun., 2015, 51, 3235–3238 RSC.
- K. P. Nevin, T. L. Woodard and A. E. Franks, mBio, 2010, 1, e00103–e00110 CrossRef PubMed.
- N. Aryal, F. Ammam, S. A. Patil and D. Pant, Green Chem., 2017, 5748–5760 RSC.
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 430–433 CrossRef.
- A. Marshall, S. Sunde, M. Tsypkin and R. Tunold, Int. J. Hydrogen Energy, 2007, 32, 2320–2324 CrossRef CAS.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588–1596 CrossRef CAS PubMed.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- H. Dau and I. Zaharieva, Acc. Chem. Res., 2009, 42, 1861–1870 CrossRef CAS PubMed.
- C. E. Frey and P. Kurz, Chem.–Eur. J., 2015, 21, 14958–14968 CrossRef CAS.
- I. Zaharieva, M. M. Najafpour, M. Wiechen, M. Haumann, P. Kurz and H. Dau, Energy Environ. Sci., 2011, 4, 2400–2408 RSC.
- R. K. Hocking, R. Brimblecombe, L.-Y. Chang, A. Singh, M. H. Cheah, C. Glover, W. H. Casey and L. Spiccia, Nat. Chem., 2011, 3, 461–466 CrossRef CAS.
- A. Ramírez, D. Friedrich, M. Kunst and S. Fiechter, Chem. Phys. Lett., 2013, 568–569, 157–160 CrossRef.
- Y. Meng, W. Song, H. Huang, Z. Ren, S.-Y. Chen and S. L. Suib, J. Am. Chem. Soc., 2014, 136, 11452–11464 CrossRef CAS PubMed.
- D. M. Robinson, Y. B. Go, M. Mui, G. Gardner, Z. Zhang, D. Mastrogiovanni, E. Garfunkel, J. Li, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2013, 135, 3494–3501 CrossRef CAS PubMed.
- M. Wiechen, I. Zaharieva, H. Dau and P. Kurz, Chem. Sci., 2012, 3, 2330 RSC.
- D. González-Flores, I. Zaharieva, J. Heidkamp, P. Chernev, E. Martínez-Moreno, C. Pasquini, M. R. Mohammadi, K. Klingan, U. Gernet, A. Fischer and H. Dau, ChemSusChem, 2016, 9, 379–387 CrossRef.
- J. E. Post, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3447–3454 CrossRef CAS.
- A. Li, H. Ooka, N. Bonnet, T. Hayashi, Y. Sun, Q. Jiang, C. Li, H. Han and R. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 5054–5058 CrossRef CAS.
- J. Cai, J. Liu and S. L. Suib, Chem. Mater., 2002, 14, 2071–2077 CrossRef CAS.
- I. G. McKendry, A. C. Thenuwara, S. L. Shumlas, H. Peng, Y. V. Aulin, P. R. Chinnam, E. Borguet, D. R. Strongin and M. J. Zdilla, Inorg. Chem., 2018, 57, 557–564 CrossRef CAS.
- I. G. Mckendry, L. J. Mohamad, A. C. Thenuwara, T. Marshall, E. Borguet, D. R. Strongin and M. J. Zdilla, ACS Energy Lett., 2018, 3, 2280–2285 CrossRef CAS.
- A. C. Thenuwara, S. L. Shumlas, N. H. Attanayake, Y. V. Aulin, I. G. Mckendry, Q. Qiao, Y. Zhu, E. Borguet, M. J. Zdilla and D. R. Strongin, ACS Catal., 2016, 6, 7739–7743 CrossRef CAS.
- B. Dutta, S. March, L. Achola, S. Sahoo, J. He, A. Amin Shirazi, Y. Wu, S. Poges, S. P. Alpay and S. L. Suib, Green Chem., 2018, 20, 3180–3185 RSC.
- S. Biswas, B. Dutta, A. Mannodi-Kanakkithodi, R. Clarke, W. Song, R. Ramprasad and S. L. Suib, Chem. Commun., 2017, 53, 11751 RSC.
- X. Li, T. Lunkenbein, V. Pfeifer, M. Jastak, P. K. Nielsen, F. Girgsdies, A. Knop-Gericke, F. Rosowski, R. Schlögl and A. Trunschke, Angew. Chem., Int. Ed., 2016, 55, 4092–4096 CrossRef CAS PubMed.
- M. Huynh, T. Ozel, C. Liu, E. C. Lau and D. G. Nocera, Chem. Sci., 2017, 8, 4779–4794 RSC.
- K. Fujimoto, T. Okada and M. Nakayama, J. Phys. Chem. C, 2018, 122, 8406–8413 CrossRef CAS.
- K. Fujimoto, Y. Ueda, D. Inohara, Y. Fujii and M. Nakayama, Electrochim. Acta, 2020, 354, 136592 CrossRef CAS.
- M. Nakayama, K. Fujimoto, T. Kobayakawa and T. Okada, Electrochem. Commun., 2017, 84, 24–27 CrossRef CAS.
- A. Li, S. Kong, C. Guo, H. Ooka, K. Adachi, D. Hashizume, Q. Jiang, H. Hongxian, J. Xiao and R. Nakamura, Nat. Catal., 2022, 109–118 CrossRef CAS.
- J. Murray and P. Brewer, in Mar. Manganese Depos., ed. G. Glasby, Elsevier, Amsterdam, 1977 Search PubMed.
- R. G. Burns, Geochim. Cosmochim. Acta, 1976, 40, 95–102 CrossRef CAS.
- R. G. Burns, Nature, 1965, 999 CrossRef.
- J. Hanawalt, H. Rinn and L. Frevel, Anal. Chem., 1938, 10, 457 CrossRef CAS.
- H. Yin, W. Tan, L. Zheng, H. Cui, G. Qiu, F. Liu and X. Feng, Geochim. Cosmochim. Acta, 2012, 93, 47–62 CrossRef CAS.
- H. Yin, H. Li, Y. Wang, M. Ginder-Vogel, G. Qiu, X. Feng, L. Zheng and F. Liu, Chem. Geol., 2014, 381, 10–20 CrossRef CAS.
- C. E. Frey, M. Wiechen and P. Kurz, Dalton Trans., 2014, 43, 4370–4379 RSC.
- C. M. Julien, M. Massot and C. Poinsignon, Spectrochim. Acta, 2004, 60, 689–700 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds Part A : Theory and Applications, John Wiley & Sons, New Jersey, 6th edn, 2009 Search PubMed.
- C. E. Frey, F. Kwok, D. González-Flores, J. Ohms, K. Cooley, H. Dau, I. Zaharieva, T. Walter, H. Simchi, S. Mohney and P. Kurz, Sustainable Energy Fuels, 2017, 1, 1162–1170 RSC.
- T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 1519–1527 CrossRef CAS PubMed.
- A. Yamaguchi, R. Inuzuka, T. Takashima, T. Hayashi, K. Hashimoto and R. Nakamura, Nat. Commun., 2014, 5, 4256 CrossRef CAS.
- T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 18153–18156 CrossRef CAS PubMed.
- U. Maitra, B. S. Naidu, A. Govindaraj and C. N. R. Rao, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11704–11707 CrossRef CAS PubMed.
- J. W. Murray and J. G. Dillard, Geochim. Cosmochim. Acta, 1979, 43, 781–787 CrossRef CAS.
- C. L. Peacock and D. M. Sherman, Chem. Geol., 2007, 238, 94–106 CrossRef CAS.
- J. Cai, J. Liu, W. S. Willis and S. L. Suib, Chem. Mater., 2001, 13, 2413–2422 CrossRef CAS.
- N. Wiberg and A. F. Hollemann, Lehrbuch der anorganischen Chemie, Walter de Gruyter, 102nd edn, 2007 Search PubMed.
- S.-L. Kuo and N.-L. Wu, J. Electrochem. Soc., 2006, 153, A1317 CrossRef CAS.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 16501–16509 CrossRef CAS.
- M. Risch, K. Klingan, F. Ringleb, P. Chernev, I. Zaharieva, A. Fischer and H. Dau, ChemSusChem, 2012, 5, 542–549 CrossRef CAS PubMed.
- M. Görlin, J. F. De Araujo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2017, 139, 2070–2082 CrossRef.
- M. Görlin, P. Chernev, J. F. De Araújo, T. Reier, S. Dresp, B. Paul, R. Krähnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef PubMed.
- E. Gileadi, Electrode Kinetics for Chemists, Chemical Engineers and Materials Scientists, VCH Publishers, Inc., Weinheim, 1st edn, 1993 Search PubMed.
- R. L. Doyle, I. J. Godwin, M. P. Brandon and M. E. Lyons, Phys. Chem. Chem. Phys., 2013, 15, 13737–13783 RSC.
- L. R. Faulkner and A. J. Bard, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, 2nd edn, 2001 Search PubMed.
- M. Wiechen, I. Zaharieva, H. Dau and P. Kurz, Chem. Sci., 2012, 3, 2330 RSC.
- H. Simchi, K. A. Cooley, J. Ohms, L. Huang, P. Kurz and S. E. Mohney, Inorg. Chem., 2018, 57, 785–792 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- Y. Surendranath, D. A. Lutterman, Y. Liu and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6326–6336 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- K. Klingan, F. Ringleb, I. Zaharieva, J. Heidkamp, P. Chernev, D. Gonzalez-Flores, M. Risch, A. Fischer and H. Dau, ChemSusChem, 2014, 7, 1301–1310 CrossRef CAS PubMed.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, Pergamon Press Ltd., 1st edn, 1966 Search PubMed.
- A. Ramírez, P. Hillebrand, D. Stellmach, M. M. May, P. Bogdanoff and S. Fiechter, J. Phys. Chem. C, 2014, 118, 14073–14081 CrossRef.
- D. Wang, J. Zhou, Y. Hu, J. Yang, N. Han, Y. Li and T.-k. Sham, J. Phys. Chem. C, 2015, 119, 19573–19583 CrossRef CAS.
- D. González-Flores, K. Klingan, P. Chernev, S. Loos, M. Mohammadi, C. Pasquini, P. Kubella, I. Zaharieva, R. Smith and H. Dau, Sustainable Energy Fuels, 2018, 2, 1986–1994 RSC.
- M. K. Bates, Q. Jia, H. Doan, W. Liang and S. Mukerjee, ACS Catal., 2016, 6, 155–161 CrossRef CAS.
- M. Risch, F. Ringleb, M. Kohlhoff, P. Bogdanoff, P. Chernev, I. Zaharieva and H. Dau, Energy Environ. Sci., 2015, 8, 661–674 RSC.
- A. Bergmann, E. Martinez-Moreno, D. Teschner, P. Chernev, M. Gliech, J. F. de Araújo, T. Reier, H. Dau and P. Strasser, Nat. Commun., 2015, 6, 8625 CrossRef CAS PubMed.
- M. A. Petit and V. Plichon, J. Electroanal. Chem., 1998, 444, 247–252 CrossRef CAS.
- M. Yagi, E. Tomita and T. Kuwabara, J. Electroanal. Chem., 2005, 579, 83–88 CrossRef CAS.
- X. Lu and C. Zhao, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- Y. Surendranath, M. Dinca and D. G. Nocera, J. Am. Chem. Soc., 2009, 8, 2615–2620 CrossRef.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 197–203 CrossRef.
- M. Huynh, C. Shi, S. J. L. Billinge and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 14887–14904 CrossRef CAS PubMed.
- L. Han, P. Tang, Á. Reyes-Carmona, B. Rodríguez-García, M. Torréns, J. R. Morante, J. Arbiol and J. R. Galan-Mascaros, J. Am. Chem. Soc., 2016, 138, 16037–16045 CrossRef CAS PubMed.
- M. Huynh, D. K. Bediako and D. G. Nocera, J. Am. Chem. Soc., 2014, 136, 6002–6010 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J.-P. Grote, A. Savan, B. Ratna, S. Merzlikin, B. Breitbach, A. Ludwig and K. J. J. Mayrhofer, Catal. Today, 2016, 262, 170–180 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, A. Mingers and K. J. Mayrhofer, J. Electroanal. Chem., 2016, 773, 69–78 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, A. Mingers and K. J. Mayrhofer, J. Electroanal. Chem., 2016, 774, 102–110 CrossRef CAS.
- B. Zhang, Y. Li, M. Valvo, L. Fan, Q. Daniel, P. Zhang, L. Wang and L. Sun, ChemSusChem, 2017, 10, 1–8 CrossRef.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, ChemSusChem, 2016, 9, 962–972 CrossRef CAS PubMed.
- F. Urbain, V. Smirnov, J.-P. Becker, A. Lambertz, F. Yang, J. Ziegler, B. Kaiser, W. Jaegermann, U. Rau and F. Finger, Energy Environ. Sci., 2016, 9, 145–154 RSC.
- B. Turan, J.-P. Becker, F. Urbain, F. Finger, U. Rau and S. Haas, Nat. Commun., 2016, 7, 12681 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2se01401g |
| ‡ Present address: German Aerospace Center, Institute of Combustion Technology, Pfaffenwaldring 38-40, 70569 Stuttgart, Germany. |
| This journal is © The Royal Society of Chemistry 2023 |