
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnabling a rapid SnO2 chemical bath deposition process for perovskite solar cells†
Darrell Jun Jie
Tay
 ab,
Benny
Febriansyah
a,
Teddy
Salim
c,
Zi Sheng
Wong
c,
Herlina Arianita
Dewi
a,
Teck Ming
Koh
a and
Nripan
Mathews
*ac
ab,
Benny
Febriansyah
a,
Teddy
Salim
c,
Zi Sheng
Wong
c,
Herlina Arianita
Dewi
a,
Teck Ming
Koh
a and
Nripan
Mathews
*ac
aEnergy Research Institute @ NTU (ERI@N), Nanyang Technological University, Singapore 637553, Singapore. E-mail: nripan@ntu.edu.sg
bInterdisciplinary Graduate Programme (IGP), Graduate College, Nanyang Technological University, Singapore 637460, Singapore
cSchool of Materials Science and Engineering, Nanyang Technological University, Singapore 639798, Singapore
First published on 16th January 2023
Abstract
Chemical bath deposition (CBD) is a common method to fabricate SnO2 electron-transport layers in perovskite solar cells. However, this typically requires long deposition times, which is a significant drawback for the eventual commercialisation of perovskite solar cells. By applying ultrasonication during the chemical bath deposition process, higher rates of heterogenous nucleation were triggered, which accelerated the CBD process without sacrificing coverage. Deposition times of 15 min, 30 min and 45 min were investigated and 30 minutes was determined to be the optimal duration for the ultrasonication assisted chemical bath deposition with the current parameters.
Introduction
Tin(IV) oxide (SnO2) is one of the few common metal oxides that can be used as an electron-transport layer (ETL) in perovskite solar cells (PSCs). Its optimal conduction band level relative to lead-halide perovskite,1 high electron mobility,2 and low-temperature processing (180 °C)3 put it at an advantageous position against other common metal oxides, such as titanium dioxide (TiO2). A variety of techniques for its thin-film fabrication exist in the current literature, namely the spin-coating of an alcoholic-sol,3 spin-coating or large-scale coating of a nanoparticle suspension,4,5 atomic-layer deposition,1 combustion synthesis,6 and chemical bath deposition.7,8 Among the variety of techniques reported thus far, chemical bath deposition (CBD) is an attractive option due to its simple and coating homogeneity at lower capital costs.9 The CBD process involves the dipping of substrates into a precursor solution, where thin film nucleation and growth are initiated by the reaction taking place on the surface of the substrates. These processes can be modulated via the control of various factors, such as precursor solution concentration, pH, and temperature. First used in the manufacturing of PbS and PbSe thin-films for infrared detectors,9 CBD has been now most commonly associated with the fabrication of CdS films as the n-type material for second-generation solar panels.10 Ortega-Borges and Lincot,11 for example, distinguished between two stages of growth: (i) an atom-by-atom growth process, where Cd(OH)2 in the precursor solution adsorbs onto the surface of the substrate and reacts there to form CdS; and (ii) a colloidal-growth stage, where homogeneous nucleation occurs and large precipitates are deposited onto the films.The use of CBD to fabricate the SnO2 ETL layer for PSCs was first demonstrated by Anaraki and co-workers back in 2016.7 It was shown that a mixture of urea, hydrochloric acid, water, and tin(II) chloride, with thioglycolic acid as the complexing agent, could be used as a precursor bath for SnO2 thin-film growth. For high-quality thin films to be obtained, a much lower precursor concentration relative to that utilised in CBD of the CdS layer11 was required due to the propensity of high SnCl2 concentrations to accelerate the reaction, causing the formation of large agglomerates and uneven film growth. The low concentration solution, however, resulted in an extended deposition time of 3 h. In addition, it could not produce a layer of SnO2 that completely covered the substrate within a single CBD process. Thus, a combination of spin-coating and CBD was needed to obtain the best coverage for the SnO2 films, and they achieved a high efficiency of 20.8% with 92% of their devices working. In contrast, Ko et al.12 demonstrated a process that utilised a concentration of up to 0.12 M, ten times that of the concentration used by Anaraki et al. (0.012 M). It was reasoned that increasing the concentration promotes the formation of more uniform SnO2 colloid sizes in the precursor solution due to competing growth processes amongst the particles, leading to smoother and pinhole-free SnO2 films. However, their initial CBD step required the dissolution of SnCl2 in ethanol, followed by the storage of the resulting solution in a freezer. The role of this additional step, however, remains unclear and it introduces an additional factor that needs to be controlled, e.g., the growth rate of the colloids in the alcoholic precursor and possible aging effects.
More recently, Yoo et al.13 proposed that the pH of the precursor solution played a major role in the quality of SnO2 produced. A reaction route was proposed, showing how the SnCl2 species could be oxidised to SnO2 by the dissolved oxygen in the solution. It was believed that heterogenous nucleation occurred at the beginning of the process, allowing for conformal growth at lower concentration, justifying the use of a low concentration of precursor solution in their case. Although it contradicts the claim made by Ko et al.,12 who posited that higher concentrations are required to avoid heterogenous nucleation for high quality films, good SnO2 coverage and highly efficient PSCs could still be achieved with the method devised by Yoo et al. However, the champion devices were fabricated by performing the CBD process three times, with a fresh precursor solution used each time. This avoids the homogeneous nucleation growth stage, although the total duration required for the entire process was 6 h. While the technique was similar to that used by Zhang et al.,14 where the CBD was done twice for the fabrication of efficient large-area PSCs, the need for repeated CBD cycles increases the time for the already slow process, and as such, hampers commercialisation.
Spurred by the aforementioned studies, we sought an extrinsic approach to accelerate the CBD of SnO2 films without multiple deposition processes. Specifically, ultrasonication, which utilises low-frequency ultrasound, was chosen because it can facilitate the thin film formation through the generation of cavitation bubbles15 capable of serving as nucleation spots, especially in the presence of a heterogenous surface16,17 (Fig. 1a). While being beneficial in terms of increasing the rate of heterogenous nucleation (and in turn, the speed of the reaction), ultrasonication concurrently maintains the circulation of reactants within the precursor solution.18 This eventually can contribute to a high degree of SnO2 film coverage. Although the sonochemical synthesis of SnO2 nanoparticles has been demonstrated before,19 the synthesis of SnO2 thin films for the purposes of fabricating an ETL for perovskite solar cells has not been demonstrated in the literature. We note that ultrasonication has been utilised for CdS films by Choi et al., where they showed that the ultrasonication prevented large agglomerations of CdS from attaching to the substrate surface, whilst promoting the formation of a compact, homogenous layer of CdS.20
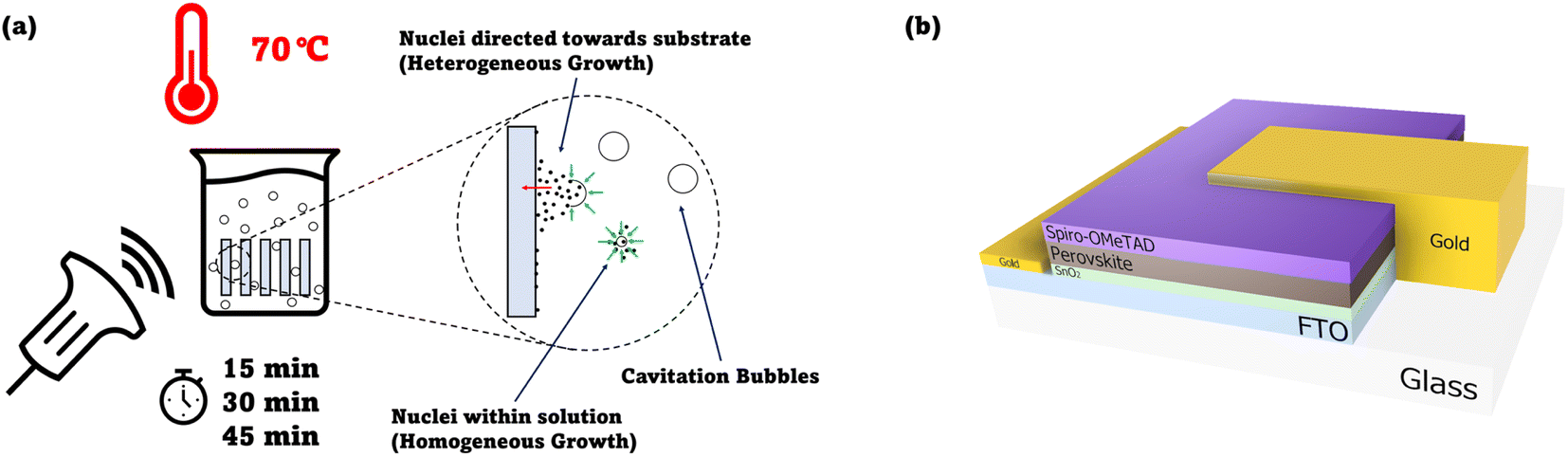 | ||
| Fig. 1 (a) Suggested mechanism of cavitation improving the heterogenous crystallisation occurring on the substrate, leading to improved SnO2 coverage. Diagram shows a beaker containing the chemical bath precursor with substrates immersed in it (placed vertically). An ultrasound probe signifies the application of ultrasound waves to the beaker. A detailed diagram of the methodology employed can be found in Fig. S1.† (b) Cross-section schematic of the n-i-p device architecture employed in this study. | ||
In this work, a comparative study was performed between SnO2 films made via the ultrasonication-assisted CBD and standard CBD. Devices based on the n-i-p architecture (Fig. 1b) were then fabricated from these films, and the device performances were compared.
Results and discussion
Characterisation of SnO2 film coverage
To assess the viability of using ultrasonication to speed up the CBD process, we first investigated the coverage of SnO2 on FTO substrates using the cyclic voltammetry method suggested by Kavan et al. First pioneered by Kavan et al. in 2014 (ref. 21) to investigate TiO2 ETLs in dye-sensitized solar cells, and later applied to SnO2 atomic layer deposited films,22 the technique relies on the fact that the SnO2–electrolyte junction is diode-like, blocking current flow in one direction, whereas the FTO–electrolyte junction does not share this property. This difference can be observed by performing cyclic voltammetry measurements with a standard redox couple, as long as the redox potential is lower than the flatband potential of SnO2.22 We investigated three different sonication durations – 15 min (15-SnO2), 30 min (30-SnO2), and 45 min (45-SnO2), and compared them with bare FTO. We note that several different recipes for CBD have been adopted by various groups. Anaraki et al. added 0.5 g of urea, 0.108 g SnCl2·2H2O, 10 μL of thioglycolic acid, and 500 μL of hydrochloric acid in 40 mL of water, and the deposition was done at 70 °C, although they claimed that a diluted version (by 6 times) of their recipe had better reproducibility.7 On the other hand, Yoo et al. used similar concentrations, but the CBD was done at a higher temperature of 90 °C.13 We have therefore decided to base our studies on our current optimised recipe for CBD, except that a slightly higher concentration of SnCl2·2H2O was used (0.0033 M).23Fig. 2b shows the cyclic voltammograms of the sonicated SnO2 samples – 15-SnO2, 30-SnO2, and 45-SnO2. The SnO2–electrolyte junction blocks the flow of electrons from the electrolyte to SnO2. Therefore, a complete absence of the oxidation peak suggests complete coverage of SnO2 on FTO. The cyclic voltammograms suggest that all three sonicated samples have SnO2 coated on them, since the oxidation peaks are lower than that of FTO. An additional reduction peak is noted for 30-SnO2 and 45-SnO2, whose origin is unclear and may be attributed to residual oxygen in the electrolyte being used. However, our coverage analysis focuses on the oxidation peak in line with the approach by Kavan et al.21
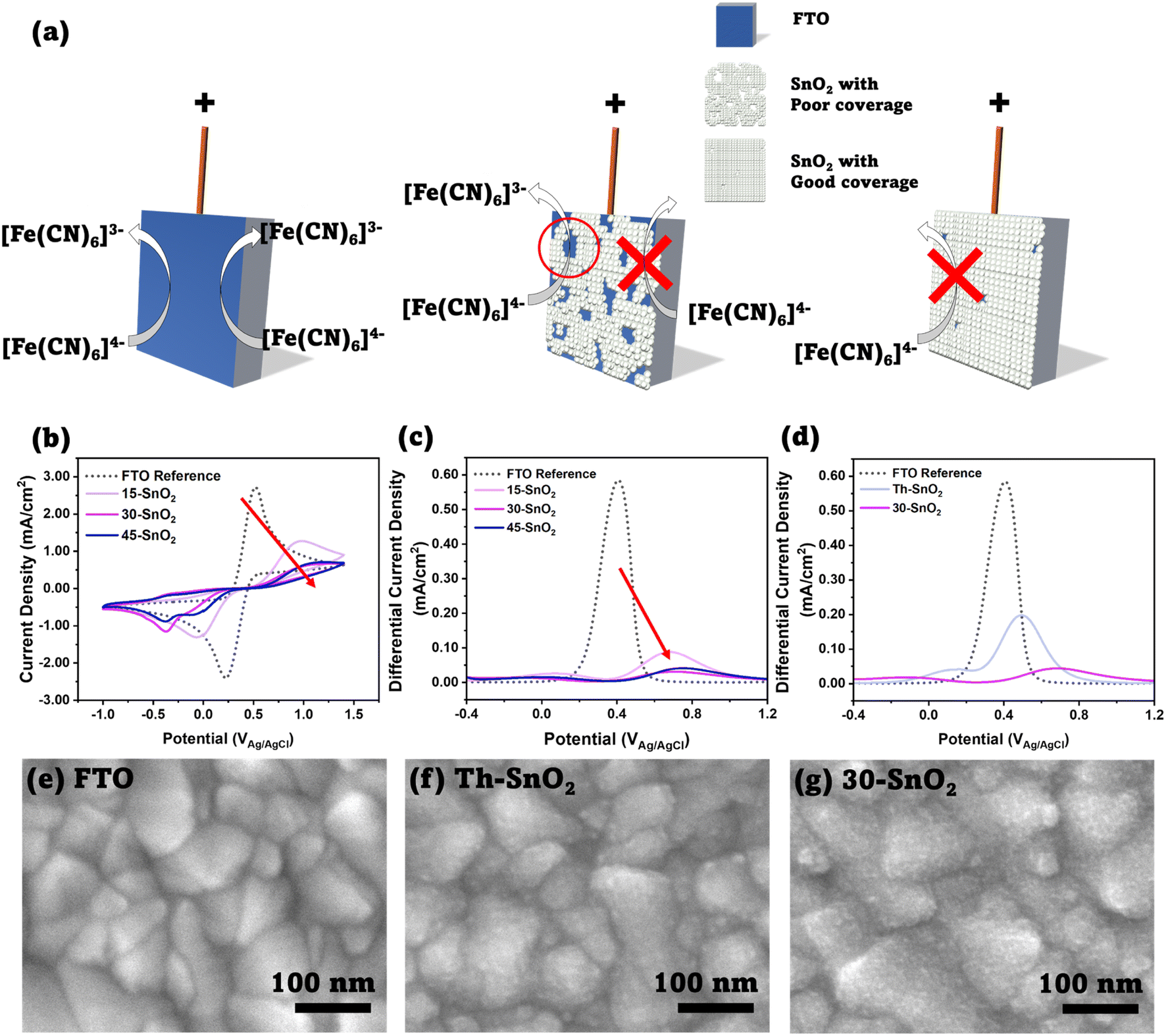 | ||
| Fig. 2 (a) Schematic showing the principle behind the cyclic voltammetry method, as discussed by Kavan et al.20 Left image shows a bare FTO substrate, where oxidation of [Fe(CN)6]4− to [Fe(CN)6]3− can occur. Middle image shows an FTO substrate partially covered by SnO2; the SnO2 blocks the reaction, but the exposed FTO allows the reaction, thus an increase in faradaic current is expected in the cyclic voltammogram. Right image shows complete coverage, where almost no oxidation reaction is expected to occur. (b) Cyclic voltammograms of FTO, 15-SnO2, 30-SnO2, and 45-SnO2. The decreasing oxidation peaks (IUPAC convention) with increased sonication time, as shown with the red arrow, indicates improved SnO2 coverage with increasing sonication time. (c) Differential pulse voltammograms for (a). A similar trend showing a decrease in the main peak can be observed with increasing sonication time. (d) Differential pulse voltammograms for FTO, Th–SnO2, and 30-SnO2, showing improved coverage from the sonication method, as opposed to a conventional CBD process at the same temperature and duration (30 minutes). (e)–(g) Top-view SEM images of FTO, Th–SnO2 and 30-SnO2. 30-SnO2 shows a rougher surface as compared to Th–SnO2, indicating the presence of more SnO2 due to sonication, and therefore illustrating the effect of sonication. Scale bar is 100 nm. | ||
Additionally, it shows that while 30-SnO2 and 45-SnO2 have better coverage than 15-SnO2, there is little improvement in coverage between 30-SnO2 and 45-SnO2. SEM images of the top surface (Fig. S2†) also reveal that 45-SnO2 has an uneven morphology with large agglomerations on the surface compared to 30-SnO2, which may be detrimental to device performance. This suggests that the maximum coverage possible with the sonication technique is likely obtained around 30 minutes, reducing the necessity for extending the CBD process by an additional 15 minutes.
Although the oxidation peak heights can be normalised to the FTO peak to provide a numerical comparison of the ratio of coverage for the sonicated samples, the peak heights in a typical cyclic voltammogram can be influenced by non-faradaic charging of the double layer that exists between the FTO electrode and the electrolyte. Thus, differential pulse voltammetry was also carried out on the same samples, and the corresponding voltammograms are presented in Fig. 2c. Differential pulse voltammetry removes the non-faradaic contribution to the peak height by measuring the current before and at the end of a pulse, and subtracting one from the other.24 From the peaks, the coverage of the SnO2 was calculated with the following equation:
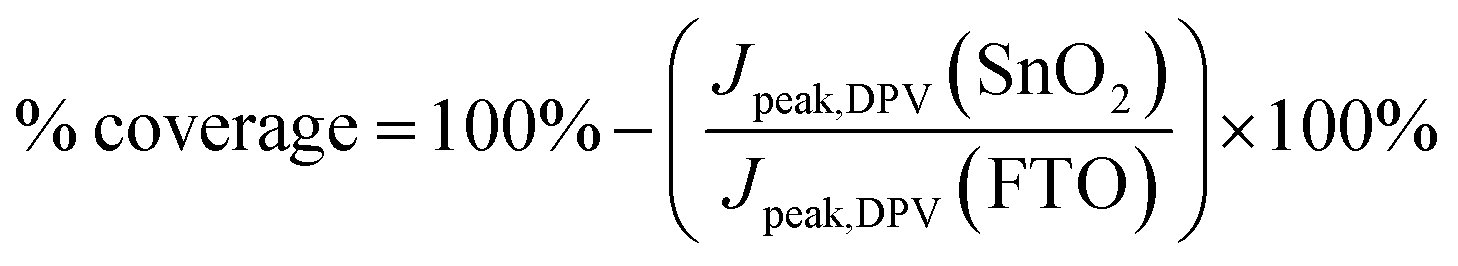 | (1) |
Based on the differential pulse voltammogram peaks, the percentages of coverage for 30-SnO2 (94.6%) and 45-SnO2 (92.9%) are also shown to be higher than the coverage for 15-SnO2 (84.9%). Therefore, this suggests that devices based on 30-SnO2 and 45-SnO2 would likely have better efficiencies than 15-SnO2-based devices. The slight decrease in coverage for 45-SnO2 may be attributed to large agglomerations that have fallen off the surface of the film during the washing process (see Experimental section below) before annealing. Combining the data collected from different characterisation techniques, we demonstrate that sonication of the CBD bath is able to improve the coverage of SnO2 on FTO, with a shorter duration of reaction as compared to the standard CBD technique.
Noting the upper limit of coverage by 30 minutes of ultrasonication, another experiment was conducted to see whether the ultrasonication contributes to obtaining coverage of SnO2 faster than the conventional CBD method (Th–SnO2, “Thermal”), where the CBD bath is placed in an oven set to 70 °C, following Anaraki et al.7 For a fair comparison, the conventional method is done for only 30 minutes to isolate the effect of the ultrasonication. Fig. 2d shows the differential pulse voltammogram of 30-SnO2 and Th–SnO2, with the associated cyclic voltammogram shown in Fig. S3.† The coverage for 30-SnO2 is about (92.4%), compared to the coverage of Th–SnO2 at (66.1%). The oxidation peak height in the cyclic voltammogram of 30-SnO2 is also lower than that of Th–SnO2. This shows that the ultrasonication speeds up the CBD process, and allows for better coverage in a shorter amount of time. The difference in coverage could also be visualized via scanning electron microscopy (SEM) technique. Fig. 2e–g show the top-view images of bare FTO, 30-SnO2, and Th–SnO2. The bare FTO image shows grains of various sizes with sharp, well-defined boundaries. In contrast, the other two images show several bumps on these FTO grains, blurring the boundaries. The bumps are indicative of a conformal layer of SnO2 covering the FTO, similar to the visual appearance of the CBD-based SnO2 reported by Yoo et al.13 Furthermore, 30-SnO2 appears peppered with more bumps than Th–SnO2, suggesting once more the improved coverage brought about by sonication.
X-ray photoelectron spectroscopy
Next, we sought to understand the effects of the sonication on the extent of the deposition reaction. In particular, avoiding excessive reaction is crucial. Yoo et al. showed that Sn2+, which is detrimental to device performance, forms when the chemical bath deposition time was extended beyond 6 hours.13 Thus, X-ray Photoelectron Spectroscopy (XPS) was also done for Th–SnO2, 15-SnO2, 30-SnO2, 45-SnO2, and as references, SnO2 powder, and the uncoated substrate. These substrates are FTO substrates on glass, with a region where FTO was etched away, leaving behind a non-conductive glass region. The XPS spectra were measured on this non-conductive region, with the intention to prevent the peaks from crystalline FTO from influencing the XPS data. Fig. 3a shows the survey spectra for the uncoated, bare substrate, 15-SnO2, 30-SnO2, and 45-SnO2. Regions of interests are the Sn 3d peaks and the O 1s peaks, for which high-resolution XPS scans were done (Fig. 3b and c). The presence of the Si 2p peaks in the survey spectra suggests the presence of pinholes in the sonicated SnO2 films, which is in agreement with the cyclic voltammetry and differential pulse voltammetry analysis. The survey spectra for all samples, along with the full high-resolution scans (with Th–SnO2 and the reference SnO2) of the Sn 3d peaks and O 1s peaks, can be found in Fig. S4 and S5,† respectively.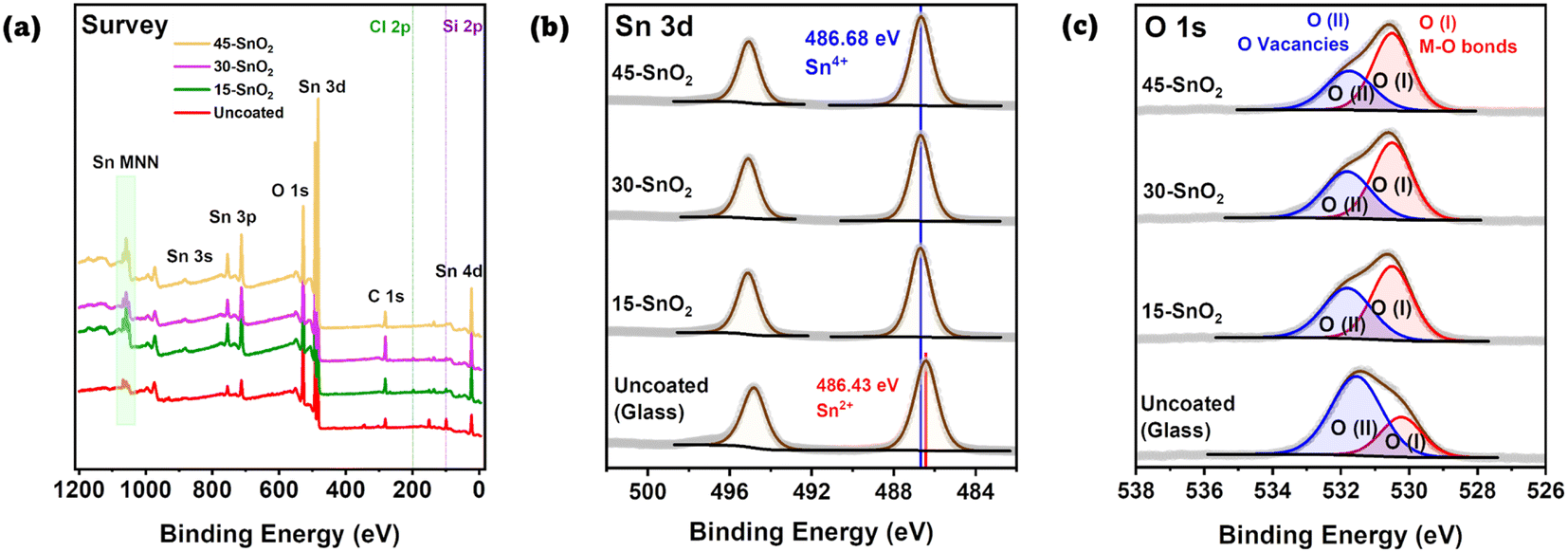 | ||
| Fig. 3 (a) Survey spectra for all samples. The uncoated sample refers to bare glass substrate. (b) High-resolution XPS data for Sn 3d. Except for the uncoated glass substrate, all SnO2 samples had similar binding energies corresponding to Sn4+. (c) High-resolution XPS data for O 1s. O(I) corresponds to lattice oxygen, while O(II) corresponds to oxygen vacancies. | ||
Fig. 3b shows the Sn 3d peaks for the three sonicated samples along with an uncoated substrate. Quantification values are provided in Table S2.† No deconvolution was needed for the CBD samples (15-SnO2, 30-SnO2, 45-SnO2). All of the peaks have binding energies close to 486.6 eV,25 likely suggesting the full conversion of Sn2+ from the precursor into Sn4+. To confirm this, XPS of commercial SnO2 powder was also performed, which showed similar peak energies (Fig. S5†). Since the presence of Sn2+ in the films is detrimental to device performance,13 this suggests that thus far, the entire process can lead to films without Sn2+. The fact that 45-SnO2 did not contain Sn2+, which Yoo et al. showed is also an indicator of excessive CBD, shows the possibility of increasing the sonication time or sonication intensity to bring about better coverage. Interestingly, the uncoated substrate also showed Sn 3d peaks, which suggests that during the preparation of the bare substrates, not all of the FTO was etched away. Instead, only enough was etched to ensure that region is non-conductive. Fortunately, these peaks have binding energies closer to Sn2+ (486 eV)25 rather than Sn4+, so the fact that the coated samples seem to comprise only Sn4+ shows that Sn 3d contributions from the uncoated glass are likely to be negligible, despite the lack of 100% SnO2 coverage on all sonicated films (as shown in Fig. 2b).
The O 1s spectra (Fig. 3c) for the films could be deconvoluted into 2 regions – marked as O(I) and O(II), where O(I) refers to the oxygen bonded to metal (M–O) and O(II) likely refers to the presence of oxygen vacancies.26 The contribution for O(II) appears to decrease monotonically from uncoated > 15-SnO2 > 30-SnO2 > 45-SnO2. The trend suggests that the monotonic decrease in the contribution for O(II) from 15-SnO2 to 45-SnO2 can be attributed to a decrease in oxygen vacancies, and the incorporation of sonication may lead to films with lower oxygen defect concentrations.
Overall, the cyclic voltammetry and XPS data seem to support the hypothesis that ultrasonication of the CBD solution speeds up the reaction, allowing for improved coverage without causing detrimental effects attributed to the excessive reaction. No Sn2+ peaks were present in the sonicated SnO2 samples, and the amount of oxygen vacancies decreased with sonication time for at least up to 45 minutes of sonication time. This was not the case for the typical CBD process, where Yoo et al. showed an increase in oxygen vacancies and Sn2+ contribution with increasing CBD time. Therefore, this shows the effectiveness of using ultrasonication to promote faster deposition times. Ultrasonication of the CBD bath promotes heterogenous nucleation due to lower contact angles favouring nucleation,27 increases the diffusion coefficients of precursors,27 and induces asymmetric cavitation, which enable the precursor and nuclei to be directed to the substrates.16 While Fig. S6† shows the eventual homogeneous nucleation occurring after 30 minutes, Fig. 2b shows that a film of SnO2 has already been formed on the substrate, highlighting the preference for heterogenous nucleation over homogeneous nucleation.
Device characterisations
While cyclic voltammetry was able to quantify the extent of coverage of SnO2, it does not show the locations of the SnO2-covered areas. Therefore, as a simple and practical test for SnO2 homogeneity, and with evidence showing the improved coverage of SnO2 after using ultrasonication to speed up the sonication process, perovskite solar cell devices were then fabricated to see if the improved coverage also translates to better cell efficiencies. We expect that a more uniform nucleation would translate into a greater number of working devices since we have four cells on one substrate. Furthermore, fewer pinholes on the SnO2 layer would mean better charge extraction, and lower possibility of recombination between the FTO and perovskite where there is no SnO2.22Firstly, solar cells based on the “triple cation” perovskite composition with n-i-p architecture (where planar SnO2 and Spiro-OMeTAD were used as electron- and hole-transporting materials, respectively) were fabricated. The SnO2 layers were deposited via CBD using both sonication method and the conventional method (Th–SnO2). More details of the experiment can be found in the Experimental section. Similar durations were used for both sonicated and oven (Th–SnO2) methods, allowing for an equivalent comparison of the rate of the CBD reaction to be made.
Fig. S7† shows the distributions of VOC, JSC, FF, and PCE for all cells, revealing the failed cells associated primarily with Th–SnO2. These failed cells are either shunted, or have suppressed VOC and FF likely due to high recombination (Fig. S8†). Assuming the same quality of perovskite and Spiro-OMeTAD layers, the results suggest that use of ultrasonication in the CBD process contributes to the formation of a better quality SnO2 layer in the device, which ultimately results in PSCs with higher PCEs. The higher PCE values for the sonicated samples are brought about by an increase across the average open-circuit potential (VOC) and the fill factor (FF) of the cells. IPCE data are provided in Fig. S9.† The similar transmission spectra in Fig. S10† also show that the degree of coverage did not significantly affect the light reaching the perovskite layer, so optical properties are unlikely a factor in the observed device performance trends. The FF value is associated with the series and shunt resistances within the devices. Any pinholes in the SnO2 layer would mean that the perovskite layer may come in contact with the transparent conducting oxide layer, facilitating recombination and decreasing the shunt resistance value.7 As the sonicated-based devices exhibit higher shunt resistances relative to the oven ones (Fig. 4e), it can be concluded that ultrasonication helps improve the SnO2 coverage during the coating process.
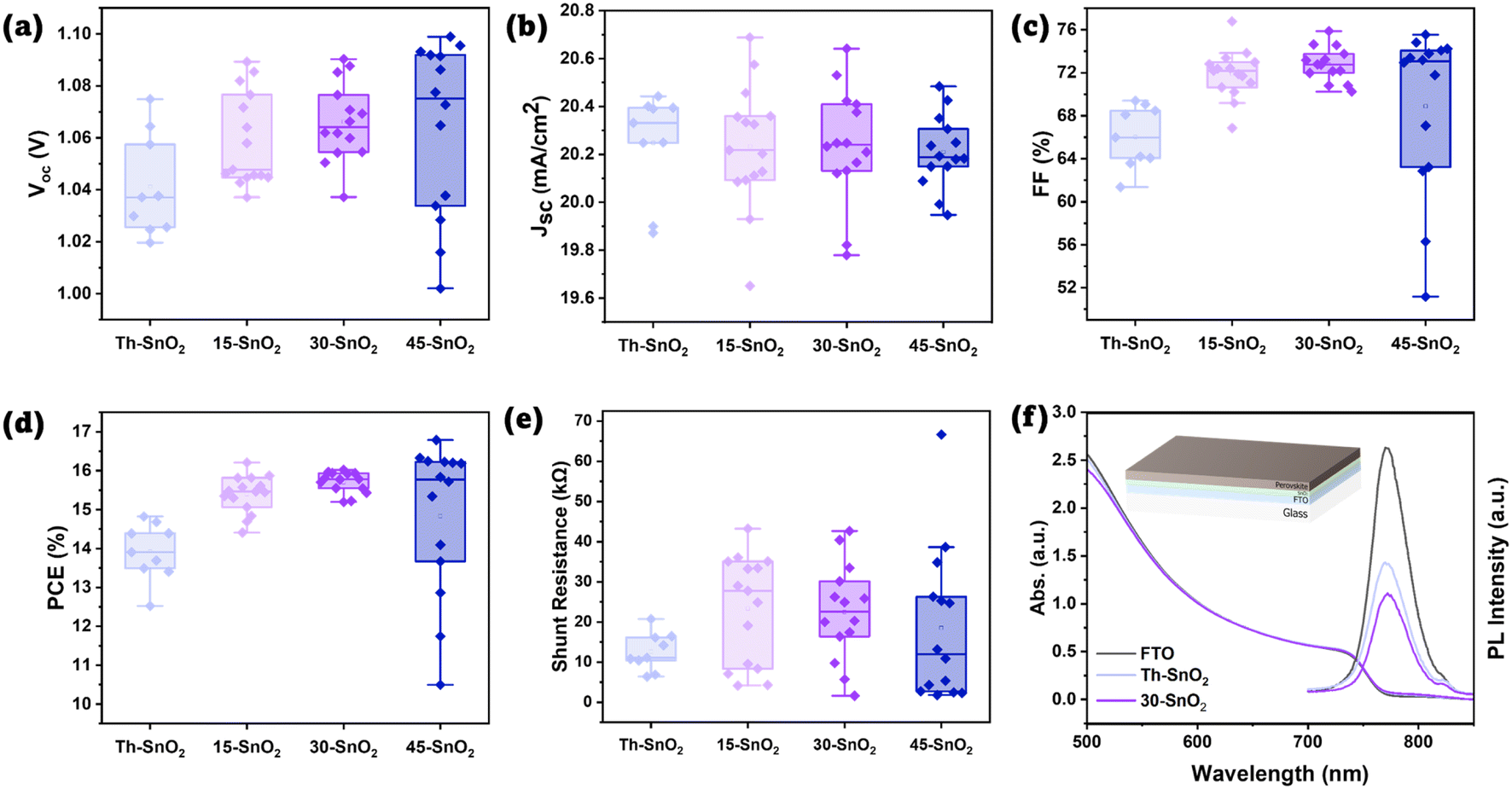 | ||
| Fig. 4 (a–e) Boxplots of device properties (VOC, JSC, FF, PCE, and estimated shunt resistance, respectively). (f) UV-vis and PL spectra of perovskite coated on FTO, Th–SnO2, and 30-SnO2. Inset diagram shows the tested device architecture. Colour scheme is the same as that for Fig. 1b. | ||
The greater number of working devices for the sonicated samples compared to Th–SnO2 also suggests that more uniform nucleation occurs due to ultrasonication. However, the negligible difference in yields amongst the devices made from 15-SnO2, 30-SnO2, and 45-SnO2 films suggests that increasing the sonication time does not help increase the nucleation rate. As mentioned previously, there are two stages in the deposition process for CBD of CdS films.11 The first stage is an ion-by-ion deposition of precursors on the substrate, which eventually grows into a compact, adherent layer. The second stage is the physical deposition of large agglomerations of CdS particles, which forms a poorly adhering rough layer on top of the compact layer. While we do not have evidence of the ion-by-ion deposition process in the case of SnO2, we do have evidence of the agglomeration from the cross-sectional SEM image of one of the devices (Fig. S11†). The presence of the poorly adhering layer suggests that homogeneous nucleation (and growth) has occurred. This layer breaks up easily when the substrates are sonicated in deionised water after the CBD process. Therefore, it does not improve the hole-blocking function of the electron-transporting layer in devices. Yet, because the rate of homogeneous nucleation is now comparable to heterogenous nucleation, this also means that the rate of heterogeneous nucleation and growth will be reduced, as both homogeneous and heterogeneous processes will compete for precursors (conservation of mass), which would therefore affect the final coverage of the compact, adherent SnO2 layer. Therefore, a possible way to further increase the yields would be to increase the concentration or ultrasonication intensity, which would help to increase the nucleation rate at the beginning27 without triggering homogeneous nucleation too early in the process.
Fig. 4a–e show the distribution of the various photovoltaic parameters after excluding failed devices (defined as <10% PCE), for a fairer comparison of the median values. A more detailed discussion can be found in the ESI.† Fig. S12a and b† show the boxplots for estimated series resistance and hysteresis index for working cells. From Fig. 4a–e, it can be observed that even after 15 min, the device properties of 15-SnO2 were better than that of Th–SnO2. At 30 and 45 min, it appears that the coverage is roughly similar, although both are poorer than that for 15-SnO2. The large difference in percentage is caused by 1 additional cell failing in 30-SnO2 and 45-SnO2, suggesting that the difference may be negligible. The apparent contradiction with Fig. 2b may be explained by the fact that the devices do not cover every area of the substrate. The median device PCEs for 30-SnO2 (η = 15.78%) is still better than that for 15-SnO2 (η = 15.45%), suggesting that even if the difference in yield (via percentage) is significant, the higher PCE comes about as a trade-off, and 30-SnO2 is the optimal point according to this data. This trend can also be explained by the competing homogeneous and heterogeneous processes, as the degree of SnO2 coverage will affect the device efficiency due to recombination at the FTO–perovskite interface.22 Larger precipitates were spotted through cross-sectional SEM for 45-SnO2 as mentioned earlier, although not spotted in the others (Fig. S11 and S13†), suggesting that the sonication step after CBD may need to be extended to ensure that more of these large precipitates are removed.
Correspondingly, we observe a larger spread of data for 45-SnO2, with the highest and lowest VOC, FF, and therefore PCE. Yet, the median PCE for 45-SnO2 is lower than that for 30-SnO2 despite the highest VOC obtained. This is due to the spread of VOC and FF values. The large precipitates can exceed 100 nm as seen in Fig. S11,† thus creating possible shunting pathways (if the grains were larger) or possible increased local defects near the precipitate, leading to the reduced VOC. Conversely, in other areas of 45-SnO2 without the large precipitates, coverage may be almost complete, leading to the highest VOC. Such a spread of device efficiencies over small areas is undesirable, and it appears that unless the onset of the rapid increase in homogeneous nucleation can be delayed, 30-SnO2 provides the best trade-off in terms of surface coverage and duration of the CBD process.
Corroborating with the FF trend, lower VOC values in the oven samples compared to the sonicated ones can also be attributed to the poorer ETL coverage. As mentioned, poor coverage implies that some of the FTO is in direct contact with the perovskite, providing a region for non-radiative electron–hole recombination to occur.22 To test this hypothesis, steady-state photoluminescence measurement was done on perovskite samples coated on glass, on bare FTO, on FTO/30-SnO2/PVK and FTO/Th–SnO2/PVK, and the spectra are presented in Fig. 4f. The suppressed PL peak of the FTO/30-SnO2/PVK suggests a large degree of charge transfer from the perovskite to the ETL. On the other hand, while the PL peak for FTO/Th–SnO2/PVK is lower than that from FTO/PVK, which is indicative of some degree of ETL coverage, the peak height is still lower than that of FTO/30-SnO2/PVK, again suggesting that coverage is poorer with the oven-based, non-sonicated ETL compared to the sonicated SnO2. Fig. S14 and S15† show the XRD and GIWAXS data for the perovskite coated on both Th–SnO2 and 30-SnO2 to highlight that the perovskites are similar, and thus the UV-vis and PL data are comparable.
Finally, an additional experiment was performed to compare the performances of devices made with the ultrasonication-assisted CBD method against those based on a spin-coated colloidal SnO2 precursor (Fig. S16†). The ultrasonication-assisted CBD-based devices had higher reproducibility, hence showing the viability of the ultrasonication-assisted CBD, with speed and reproducibility as advantages over other methods for SnO2 deposition.
Conclusions
The above data suggest that a 30 minute sonicated-CBD process would be ideal for the fabrication of ETL films for perovskite solar cells, assuming that conditions are similar to the ones used in this paper. Several devices made from 45-SnO2 have efficiencies that outperform those fabricated with 30-SnO2, which suggests that further optimisation is necessary. A combination of methods used in the existing literature to improve CBD can be used in conjunction with sonication to further improve the quality of the SnO2 films. However, for the purpose of this study, it has been demonstrated as proof-of-concept that sonication can help speed up the CBD reaction without sacrificing the quality of the SnO2 films made. The optimal duration for CBD sonication is dependent on maximising the rate of heterogeneous nucleation, whilst reducing the rate of homogeneous nucleation. Tweaking the various other factors that affect the chemical bath deposition in general (pH, temperature) are likely to help improve the quality of SnO2 films formed by the ultrasonication method. In conclusion, our work has demonstrated that ultrasonication is able to speed up the CBD process without sacrificing the quality of the SnO2 films, thus aligning the eventual goal of commercialisation. More work on incorporating the various other strategies employed by other research groups can also be done, all with the eventual aim of rapid fabrication of SnO2 ETLs.Experimental
SnO2 film fabrication
An amount of 0.250 g of urea (Sigma-Aldrich, ACS reagent, ≥99%) was dissolved in 200 mL of DI water in a clean glass beaker, and shaken until fully dissolved. A volume of 5 μL of thioglycolic acid (Sigma-Aldrich, anhydrous 99%) and 250 μL of 37 wt% HCl (Sigma-Aldrich) were then added to the solution, and shaken. An amount of 0.150 g of SnCl2·2H2O (Sigma-Aldrich, ≥99.995% trace metals basis), corresponding to 0.0033 M, was then added to the solution and shaken. Other concentrations were also attempted (0.012 M, 0.12 M). A detailed discussion can be found in the ESI (Fig. S17 and S18†). The precursor solution was then sonicated at room temperature for 40 minutes for mixing (Elma EH120), with the temperature of the water never rising above 34 °C. Afterwards, the beaker was removed, and substrates were placed vertically in the solution using a Teflon holder. For the oven control, the beaker was placed in a convection oven at 70 °C for 30 minutes. For the sonicated samples, the beaker was placed in another ultrasonicator (Soltec Sonica 5300, 500 W) with the water bath maintained at 70 °C. The sonicator was then turned on for 15, 30, or 45 minutes. After the CBD process, the substrates were removed, rinsed with DI water, and then sonicated again in DI water for 3 minutes to remove the poor adherent SnO2 precipitates on the FTO substrates. Finally, the substrates were dried and annealed at 180 °C for 1 h.Device fabrication
Pre-etched FTO substrates were washed in Decon soap, DI water, acetone, and ethanol for 15 minutes each. The substrates were then UV-ozone treated for 30 minutes at 100 °C before SnO2 ETL deposition, as described above. After the deposition, the substrates were UV-ozone treated again for 20 minutes before perovskite deposition. Mass amounts of 18 mg CsI (99.99% trace metals basis), 25.4 mg MABr (GreatCell Solar), 84.7 mg PbBr2 (TCI, 99.99% trace metals basis), 198.5 mg FAI (GreatCell Solar) and 585.1 mg PbI2 (TCI, 99.99% trace metals basis) were dissolved in 1 mL of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 DMF:DMSO (DMF: Sigma-Aldrich, anhydrous 98.8%, DMSO: Sigma-Aldrich, anhydrous, ≥99.9%) prior to deposition, and left on the hotplate at 50 °C for 2 h before use. A two-step spin-coating process was used. After addition of the perovskite precursor on the substrate, the substrate was spun at 1000 rpm for 10 s and 6000 rpm for 20 s, with chlorobenzene dripped onto the substrate 5 s before the end of the entire spin-coating process. The substrates were then annealed at 100 °C for 1 h. Spiro-OMeTAD solution was then spin-coated onto the perovskite film (72.3 mg Spiro-OMeTAD (Lumtech), 28.5 μL tBP (Sigma-Aldrich, 98%), 17.5 μL Li-TFSI (99.95% trace metals basis) in ACN (Sigma-Aldrich), in 1 mL chlorobenzene) at 4000 rpm for 30 s. Finally, 100 nm of gold electrical contacts were evaporated onto the devices using a thermal evaporator.
:2 DMF:DMSO (DMF: Sigma-Aldrich, anhydrous 98.8%, DMSO: Sigma-Aldrich, anhydrous, ≥99.9%) prior to deposition, and left on the hotplate at 50 °C for 2 h before use. A two-step spin-coating process was used. After addition of the perovskite precursor on the substrate, the substrate was spun at 1000 rpm for 10 s and 6000 rpm for 20 s, with chlorobenzene dripped onto the substrate 5 s before the end of the entire spin-coating process. The substrates were then annealed at 100 °C for 1 h. Spiro-OMeTAD solution was then spin-coated onto the perovskite film (72.3 mg Spiro-OMeTAD (Lumtech), 28.5 μL tBP (Sigma-Aldrich, 98%), 17.5 μL Li-TFSI (99.95% trace metals basis) in ACN (Sigma-Aldrich), in 1 mL chlorobenzene) at 4000 rpm for 30 s. Finally, 100 nm of gold electrical contacts were evaporated onto the devices using a thermal evaporator.
Characterisations
JV scans were obtained with a Keithley 2612B SourceMeter and Newport 94043A Solar Simulator (450 W xenon), which provided an illumination intensity of 1000 W m−2, AM 1.5 G spectral distribution. A shadow mask of size 0.086 cm2 was used, with a scan rate of approximately 300 mV s−1.For cyclic voltammetry and differential pulse voltammetry, the substrates were first taped up such that the exposed surface area is 1.7 cm2 (based on the width of the pre-etched FTO substrate). The substrates were then dipped into a 10 mM [Fe(CN)6]3−/[Fe(CN)6]4− redox couple (K3Fe(CN)6: >99.0%, K4Fe(CN)6: 98.5–102.0%, both from Sigma-Aldrich) with 1 M of KCl (Sigma-Aldrich, 99.0–100.5%). A platinum wire was used as the counter-electrode, and a 1 M Ag/AgCl electrode was used as the reference electrode. The electrolyte was purged with N2 gas for 40 minutes before starting the measurement. A Metrohm Autolab PGSTAT302N was used. For cyclic voltammetry, 3 cycles were scanned from 1.4 V to −1 V or −0.2 V, with a scan rate of 100 mV s−1, and the third cycle was used. For differential pulse voltammetry, the substrate was scanned from −0.2 V or −1 V to 1.4 V, with a step size of 0.005 V, modulation amplitude of 0.02 V, at 25 Hz.
SEM images of films were all obtained using a field emission scanning electron microscope (FESEM), model JOEL JSM 6700F.
Steady-state photoluminescence was performed with a Horiba FluoroMax Spectrometer. The samples were excited at 600 nm and scanned from 700 nm to 850 nm (increment of 1 nm), with a slit width of 10 nm for both source and detector.
UV-vis absorbance spectra were measured with a Shimadzu UV2600 spectrometer, and scanned from 850 nm to 300 nm with a step size of 0.1 nm.
GIWAXS spectra were obtained with a Xenocs Nano-inXider with a PILATUS3 3 × 100 K detector and Cu Kα1 radiation. For the perovskite-coated half-stack device (FTO + ETL + perovskite only), the normal out-of-plane configuration (φ = 0°) was adopted at a grazing angle of 4°. Data were analysed with the Foxtrot programme that was bundled with the Nano-inXider.
X-ray photoelectron spectroscopy (XPS) analysis was performed using an AXIS Supra spectrometer (Kratos Analytical Inc., UK) equipped with a hemispherical analyser and a monochromatic Al K-alpha source (1487 eV) operated at 15 mA and 15 kV. The XPS spectra were acquired from an area of 700 × 300 μm2 with a take-off angle of 90°. Pass energies of 160 eV and 20 eV were used for the survey and high-resolution scans, respectively. A 3.1 volt bias was applied to the sample to neutralise charge build-up on the sample surface. The binding energies (BEs) were charge-corrected based on the C 1s of adventitious carbon at 284.8 eV, and verified with the O 1s of the metal oxide peak at 530.5 eV. As reference, XPS measurements of commercially-available SnO2 (Sigma-Aldrich, ∼325 mesh, 99.9% trace metals basis) powder were also performed.
Conflicts of interest
The authors declare the following competing financial interest(s): N. M. is the director of Prominence Photovoltaics Pte Ltd., a perovskite solar cell commercialisation company.Acknowledgements
This research was funded by the National Research Foundation, Prime Minister's Office, Singapore through the Intra-CREATE Collaborative Grant (NRF2018-ITC001-001), and Ministry of Education Tier 2 Project (MOE2019-T2-2-097). The X-ray Photoelectron Spectroscopy work was performed at the Facility for Analysis, Characterisation, Testing and Simulation (FACTS), Nanyang Technological University, Singapore.References
- J. P. C. Baena, L. Steier, W. Tress, M. Saliba, S. Neutzner, T. Matsui, F. Giordano, T. J. Jacobsson, A. R. S. Kandada, S. M. Zakeeruddin, A. Petrozza, A. Abate, M. K. Nazeeruddin, M. Grätzel and A. Hagfeldt, Energy Environ. Sci., 2015, 8, 2928–2934 RSC.
- L. Xiong, Y. Guo, J. Wen, H. Liu, G. Yang, P. Qin and G. Fang, Adv. Funct. Mater., 2018, 28, 1802757 CrossRef.
- W. Ke, G. Fang, Q. Liu, L. Xiong, P. Qin, H. Tao, J. Wang, H. Lei, B. Li, J. Wan, G. Yang and Y. Yan, J. Am. Chem. Soc., 2015, 137, 6730–6733 CrossRef CAS PubMed.
- Q. Jiang, L. Zhang, H. Wang, X. Yang, J. Meng, H. Liu, Z. Yin, J. Wu, X. Zhang and J. You, Nat. Energy, 2016, 2, 1–7 Search PubMed.
- J. A. Smith, O. S. Game, J. E. Bishop, E. L. K. Spooner, R. C. Kilbride, C. Greenland, R. Jayaprakash, T. I. Alanazi, E. J. Cassella, A. Tejada, G. Chistiakova, M. Wong-Stringer, T. J. Routledge, A. J. Parnell, D. B. Hammond and D. G. Lidzey, ACS Appl. Energy Mater., 2020, 3, 5552–5562 CrossRef CAS PubMed.
- X. Liu, K.-W. Tsai, Z. Zhu, Y. Sun, C.-C. Chueh and A. K.-Y. Jen, Adv. Mater. Interfaces, 2016, 3, 1600122 CrossRef.
- E. H. Anaraki, A. Kermanpur, L. Steier, K. Domanski, T. Matsui, W. Tress, M. Saliba, A. Abate, M. Grätzel, A. Hagfeldt and J.-P. Correa-Baena, Energy Environ. Sci., 2016, 9, 3128–3134 RSC.
- J. Barbé, M. L. Tietze, M. Neophytou, B. Murali, E. Alarousu, A. E. Labban, M. Abulikemu, W. Yue, O. F. Mohammed, I. McCulloch, A. Amassian and S. Del Gobbo, ACS Appl. Mater. Interfaces, 2017, 9, 11828–11836 CrossRef PubMed.
- G. Hodes, Phys. Chem. Chem. Phys., 2007, 9, 2181–2196 RSC.
- I. Kaur, D. K. Pandya and K. L. Chopra, J. Electrochem. Soc., 1980, 127, 943 CrossRef CAS.
- R. Ortega-Borges and D. Lincot, J. Electrochem. Soc., 1993, 140, 3464 CrossRef CAS.
- Y. Ko, Y. Kim, C. Lee, T. Kim, S. Kim, Y. J. Yun, H. Gwon, N.-H. Lee and Y. Jun, ChemSusChem, 2020, 13, 4051–4063 CrossRef CAS PubMed.
- J. J. Yoo, G. Seo, M. R. Chua, T. G. Park, Y. Lu, F. Rotermund, Y.-K. Kim, C. S. Moon, N. J. Jeon, J.-P. Correa-Baena, V. Bulović, S. S. Shin, M. G. Bawendi and J. Seo, Nature, 2021, 590, 587–593 CrossRef CAS PubMed.
- J. Zhang, C. Bai, Y. Dong, W. Shen, Q. Zhang, F. Huang, Y.-B. Cheng and J. Zhong, Chem. Eng. J., 2021, 425, 131444 CrossRef CAS.
- K. S. Suslick, Science, 1990, 247, 1439–1445 CrossRef CAS PubMed.
- J. R. G. Sander, B. W. Zeiger and K. S. Suslick, Ultrason. Sonochem., 2014, 21, 1908–1915 CrossRef CAS PubMed.
- W. Peng, L. Wang, B. Murali, K.-T. Ho, A. Bera, N. Cho, C.-F. Kang, V. M. Burlakov, J. Pan, L. Sinatra, C. Ma, W. Xu, D. Shi, E. Alarousu, A. Goriely, J.-H. He, O. F. Mohammed, T. Wu and O. M. Bakr, Adv. Mater., 2016, 28, 3383–3390 CrossRef CAS PubMed.
- M. Kim, S. Lee and S. Sohn, Thin Solid Films, 2011, 519, 1787–1793 CrossRef CAS.
- J. Zhu, Z. Lu, S. T. Aruna, D. Aurbach and A. Gedanken, Chem. Mater., 2000, 12, 2557–2566 CrossRef CAS.
- J. Y. Choi, K.-J. Kim, J.-B. Yoo and D. Kim, Sol. Energy, 1998, 64, 41–47 CrossRef.
- L. Kavan, N. Tétreault, T. Moehl and M. Grätzel, J. Phys. Chem. C, 2014, 118, 16408–16418 CrossRef CAS.
- L. Kavan, L. Steier and M. Grätzel, J. Phys. Chem. C, 2017, 121, 342–350 CrossRef CAS.
- L. Xie, P. Vashishtha, T. M. Koh, P. C. Harikesh, N. F. Jamaludin, A. Bruno, T. J. N. Hooper, J. Li, Y. F. Ng, S. G. Mhaisalkar and N. Mathews, Adv. Mater., 2020, 32, 2003296 CrossRef CAS PubMed.
- B. J. Venton and D. J. DiScenza, in Electrochemistry for Bioanalysis, ed. B. Patel, Elsevier, 2020, pp. 27–50 Search PubMed.
- H. I. Bang, H. B. Seo, B. S. Bae and E.-J. Yun, Phys. Status Solidi A, 2019, 216, 1800863 CrossRef.
- J.-C. Dupin, D. Gonbeau, P. Vinatier and A. Levasseur, Phys. Chem. Chem. Phys., 2000, 2, 1319–1324 RSC.
- Z. Guo, A. G. Jones, H. Hao, B. Patel and N. Li, J. Appl. Phys., 2007, 101, 054907 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: XPS quantification, XPS data for Th–SnO2 and powdered SnO2; full device data; cross-sectional SEM images of devices; XRD and GIWAXS data for perovskite film coated on the SnO2 layers; IPCE, transmission and absorption data for SnO2 layers, study on concentration effect on device properties, comparison of device data with spin-coated colloidal SnO2. See DOI: https://doi.org/10.1039/d2se01475k |
| This journal is © The Royal Society of Chemistry 2023 |