
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHistorical and contemporary perspectives on metal–organic frameworks for gas sensing applications: a review
Gia Huy
Pham
and
Cerasela Zoica
Dinu
 *
*
Department of Chemical and Biomedical Engineering, Benjamin M. Statler College of Engineering and Mineral Resources, West Virginia University, Morgantown, WV 26505, USA. E-mail: hgpham@mix.wvu.edu; Cerasela-Zoica.Dinu@mail.wvu.edu
First published on 22nd June 2023
Abstract
Metal–organic frameworks (MOFs) are a class of crystalline compounds with porous characteristics and high specific surface area, featuring coordinated metal nodes and ligands formed through covalent and ionic bonds. In this review, we discuss selected research on MOF-based applications, specifically in the area of gas-sensing applications for ammonia (NH3), hydrogen (H2), and carbon dioxide (CO2). MOFs are highlighted both in the context of increasing the sensitivity and specificity of sensing towards such specific gases and defining essential design and functionalization parameters of the framework to allow its use over multiple cycles, while maintaining high efficiency and sustainability. Lastly, the outlook section of this review focuses on the next steps in the formation of MOFs and how their structure–function–performance relationships can help establish framework usability upon implementation, while under user control.
Sustainability spotlightMetal–organic frameworks (MOFs) have garnered significant attention as a promising class of green materials due to their porosity, tunable chemistry, and large surface area. These properties are achieved through coordination bonds between a chosen metal and an organic linker, as well as through the functionality of each component. We discuss the implementation of MOF in the context of United Nations' Sustainable Development Goals (SDG) for reducing deleterious impacts resulting from exposure to ammonia, hydrogen, and carbon dioxide, all known to induce environmental and logistical burden such as harm to human and aquatic life (SDG 14), asphyxiation in confined spaces (SDG 11) or global warming and climate changes (SDG 13), just to name a few. We provide examples of MOFs' design and characterization for sensing/monitoring/detection processes. We emphasize how high efficiency and sustainability could be achieved when using MOFs to reduce corrosiveness, volatility and parasitic energy consumption of currently used adsorbents. Lastly, we provide our perspective on how one can increase MOFs sensing performance through both rational design and functionality, for user controllled structure–function relationships and affordable, reliable, and sustainable energy (SDG 7) green technology development. |
A historical perspective into the discovery of metal–organic frameworks and their characterization
Metal–organic frameworks (MOFs), which are crystalline compounds with porous characteristics, are formed by binding inorganic polynuclear clusters [termed secondary building units (SBUs)] and organic linkers via strong bonds.1 One of the first MOFs was reported in 1999, specifically, Zn4O(BDC)3 (MOF-5); this MOF was synthesized by O'Keefe and Yaghi's group using a mixture of zinc(II) nitrate [Zn(NO3)] and 1,4-benzenedicarboxylic acid (H2BDC) in N,N-dimethylformamide (DMF)/chlorobenzene.2 The resulting framework displayed a tetranuclear supertetrahedral [Zn4O]6+ architecture. Later studies identified MOF-5 as a benzenedicarboxylate (BDC) dianion, with the oxygen (O) atoms of the carboxylate groups coordinated to different zinc (Zn) atoms in [Zn4O]6+ clusters.3 The reticular nature and coordination bond of the octahedral Zn–O–C clusters were later combined with various organic carboxylate linkers, resulting in isoreticular frameworks (IRMOF-n; where n = 1, 2, 3, …; Fig. 1a shows representative IRMOF-1).4–6 The different carboxylate linkers exhibited unique features, providing the frameworks different characteristics to be employed in a plethora of applications.7 For instance, studies showed that IRMOF-1 is an outstanding candidate as an adsorbent for gases (i.e., hydrogen (H2), carbon dioxide (CO2), and methane (CH4)); further, this framework possesses separation characteristics due to its (1) high surface area and pore volume and (2) tunability. IRMOF-1 was also reported to have the best CO2 sorption capacity,7,8 yet poorest water stability compared to other IRMOFs,9 namely, IRMOF-3 (ref. 10) and IRMOF-8.11 The poor stability was a result of the substitution of carboxylic groups by water molecules to subsequently coordinate with Zn2+ centers, with such coordination triggering an irreversible structural transformation within only a few minutes of exposure.12–14 | ||
| Fig. 1 Scanning electron microscopy (SEM) images (inset: framework representations) of selected MOFs: (a) IRMOF-1 (ref. 5 and 6) and (b) HKUST-1.16,17 Reprinted (adapted) with permission from ref. 16. Copyright 2019, the American Chemical Society. Reprinted (adapted) with permission from ref. 17. Copyright 2022, the American Chemical Society. (c) MIL-53.29,30 Reprinted (adapted) with permission from ref. 29. Copyright 2019, the American Chemical Society. Reprinted (adapted) with permission from ref. 30. Copyright 2010, the American Chemical Society.; (d) MIL-101.41,42 Reprinted (adapted) with permission from ref. 41. Copyright 2019, the American Chemical Society. (e) ZIF-8.65,66 Reproduced from ref. 65 with permission from The Royal Society of Chemistry. (f) UiO-66.72,73 Reprinted (adapted) with permission from ref. 72. Copyright 2008, the American Chemical Society. Reprinted (adapted) with permission from ref. 73. Copyright 2015, the American Chemical Society. | ||
In 1999, Chui et al.15 reported the synthesis of [Cu3(TMA)2(H2O)3]n, which is commonly known as HKUST-1 or MOF-199, a class of blue-cubic crystals (Fig. 1b (ref. 16 and 17)) obtained through the coordination of copper(II) (Cu2+) metal nodes with trimesic acid (TMA). The solvothermal synthesis involved heating cupric nitrate trihydrate in trimesic acid in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of water (H2O):ethyl alcohol (EtOH), at 180 °C for 12 h. The as-synthesized HKUST-1 was turquoise in color and composed of dimeric cupric tetra-carboxylate units, with a short Cu–Cu separation of 2.628(2) Å. Furthermore, the framework was neutral given that the 12 carboxylate oxygens from the two TMA ligands were bonded to four coordination sites of each of the three Cu2+ ions. Also, the framework had a higher dimensionality, presumably resulting from the loss of its terminal ancillary ligands.15 Each of the Cu metal nodes completed the pseudo-octahedral coordination sphere with an axial aqua TMA ligand opposite the Cu–Cu vector. The key SBUs of HKUST-1 were described as octahedral units with the Cu2 dimers occupying the six vertices of each such unit. The framework also had four trimesate ions tetrahedrally coordinated to each one of the four of its eight triangular faces, respectively. Thermal gravimetric analysis (TGA) indicated that this structure could hold up to ten to fifteen water molecules per unit.18 The compound was anhydrate, with analysis demonstrating that its dehydration leads to changes in color, i.e., from turquoise to dark blue.
:1 ratio of water (H2O):ethyl alcohol (EtOH), at 180 °C for 12 h. The as-synthesized HKUST-1 was turquoise in color and composed of dimeric cupric tetra-carboxylate units, with a short Cu–Cu separation of 2.628(2) Å. Furthermore, the framework was neutral given that the 12 carboxylate oxygens from the two TMA ligands were bonded to four coordination sites of each of the three Cu2+ ions. Also, the framework had a higher dimensionality, presumably resulting from the loss of its terminal ancillary ligands.15 Each of the Cu metal nodes completed the pseudo-octahedral coordination sphere with an axial aqua TMA ligand opposite the Cu–Cu vector. The key SBUs of HKUST-1 were described as octahedral units with the Cu2 dimers occupying the six vertices of each such unit. The framework also had four trimesate ions tetrahedrally coordinated to each one of the four of its eight triangular faces, respectively. Thermal gravimetric analysis (TGA) indicated that this structure could hold up to ten to fifteen water molecules per unit.18 The compound was anhydrate, with analysis demonstrating that its dehydration leads to changes in color, i.e., from turquoise to dark blue.
In 2002, Gérald Férey's group reported the synthesis of the MIL (MIL = Matériaux de l’Institute Lavoisier) frameworks, with chromium(III) (Cr3+) serving as the metal center and BDC-based ligands as the linkers.19 Generally, MILs are synthesized via either the solvothermal or hydrothermal route, by mixing trivalent (M3+) metal centers with BDC in DMF solvent. This process can be tuned by (1) substituting M3+ with divalent (M2+) or tetravalent (M4+) metal centers;20–22 (2) adding a mineralizer such as hydrochloric acid (HCl) or hydrofluoric acid (HF),23 or (3) by changing dicarboxylate-based ligands with different functional linkers,24–26 respectively. The uniqueness of the reported MIL-53, for instance, was the possibilities of assemblies of its inorganic chains in the trivalent metal centers and the terephthalate-based linkers, with MIL-53(Cr) shape-sharing trans-corners, alternating between Cr3+ and OH−. The BDC linker connected to four different metal centers by bridging each dicarboxylate functional group in a Z,Z-μ2-η1:η1 manner.23 Each metal center was also octahedrally coordinated by six O atoms, four of which originated from the four different carboxylate groups of the BDC linker, respectively. The remaining two O atoms were covalently bound to carbon (C) and linked to Cr3+via an ionocovalent bond.19,27 The resulting framework structure contained a one-dimensional diamond-shaped pore23 and had an overall topology of an sra net, with its morphology and histology being shown in Fig. 1c.28–30 The flexibility of the MIL-53 structure allowed its integration in applications such as liquid–gas sensing30–32 or water purification (e.g., for sensing nitrobenzene,33 ibuprofen,34 and dimethylphthalate35).
Recently, aluminum(III) (Al3+) was incorporated MIL MOFs36–39 as a metal node, resulting in the formation of frameworks with higher thermal stability.23 For instance, MIL-53 was shown to have “breathing effect”23,27 flexibility, in which the dihedral angle, α, between two planes was expanded to a maximum of 180°.27 The studies also showed that when water was present in the pores of the framework, such molecule formed strong hydrogen bonds with the MOF structure.
In 2005, Férey further synthesized MIL-101 through the hydrothermal reaction of Cr(NO3)3·9H2O and H2BDC in HF, for 8 h and at 220 °C.40 As presented in Fig. 1d,41,42 the structure of MIL-101 was comprised of M3+ trimers with each of the trimers forming octahedra with the metal atom located in the center. The analysis also showed that two of these trimers contained bound water molecules, while the third one had a halide or hydroxide ion present. Each octahedron was connected laterally to another octahedron through the carboxylic groups of two BDC molecules. These bonds led to four connections and a total of six bidentate carboxylic linkers.43 The trimers occupied the four vertices of the SBU tetrahedron, while the ligands were present at the six edges of the super tetrahedron. The resulting assembly displayed a complex microporous structure with two mesoporous cages. The smaller cage was shown to be connected through pentagonal windows, while the larger one was connected through both pentagonal and hexagonal windows, respectively.44
The development of zeolitic imidazolate frameworks (ZIFs) was first reported by Yaghi's group in 2006, with their analysis focusing on the synthesis of ZIF-1 to ZIF-12, as defined by the implementation of different imidazolate linkers (i.e., imidazole (IM), benzimidazole (H-PhIM), and 2-methyl imidazole (H-MeIM)) and three different solvents (i.e., DMF, N,N-diethylformamide (DEF), and N-methylpyrrolidine (NMP)), respectively.45 ZIFs have been synthesized either through solvent techniques (i.e., solvothermal,45,46 hydrothermal,47,48 microwave-assisted,49,50 and sol–gel synthesis51,52), solvent-free techniques (i.e., mechanochemical53,54 and accelerate aging55), all while using less-toxic solvent derivatives (i.e., methanol,46 ethanol,56 and water47) for a greener and more economical synthesis, or through controlled “link–link interaction”.57,58 For the synthesis of ZIFs, any transition metal can be used (M2+) (i.e., iron(II) (Fe2+), cobalt(II) (Co2+),59 Cu2+, and Zn2+60) to tetrahedrally bind in a self-assemble manner with the imidazolate (Im) linkers.46,58,61 Other modified synthesis methods involving the deprotonation of the imidazolate linker through the addition of additives or known imidazolate deprotonating agents (i.e., triethylamine (TEA), pyridine, sodium hydroxide, sodium formate, and n-butylamine) directly to the solvent were also introduced to ultimately increase the rate of reaction and thus improve the product yield.
ZIFs have been recognized for their high sorption capabilities for gases, with such capability being presumably due to their basic sites. Specifically, these frameworks have been implemented for gas conversion because of the presence of Lewis acid sites from their metal(II).62 Research also showed that ZIFs can be used for the separation and sequestration of gases,57 in catalysis,63 and as gases sensors.64 In this context, recently, ZIF-8 (Fig. 1e (ref. 65 and 66)) has attracted significant attention for extended sensing applications.67,68 The ∼145° bond angle of M–Im–M69,70 was analogous to the aluminosilicate (Si–O–Si) angle found in zeolites,45,57,71 while its SBU consisted of a Zn ion tetrahedrally linked to four 2-methyl imidazole groups.
Lastly (for the purpose of this review), in 2008, Karl Petter Lillerud et al. reported the synthesis of a Zr-MOF structure named UiO-66 through a solvothermal technique upon mixing zirconium tetrachloride (ZrCl4) with H2BDC in DMF at 120° for 24 h,72 as shown in Fig. 1f.72,73 Unlike MOF-5, UiO-66 is a Zr(IV)-based MOF with octahedral (∼11 Å) and tetrahedral (∼8 Å) cages connected by triangular windows (∼6 Å), yielding to the formation of a Zr6O4(OH)4(CO2)12 cluster. Early stages of synthesis studies showed a rapid reaction due to an accelerated nucleation process.74 Later on, optimization of Zr-based MOFs was attempted through the use of organic modulators,75 deprotonating agents,76 or a combination of both. Specifically, a single carboxylic acid bridged the carbon chains (R–COOH)77 competitively and reversibly and bound to the metal node to slow down the crystallization process. Deprotonating agents, such as TEA76,78 subsequently activated the ligands by removing the hydrogen group from H2BDC, thus promoting nucleation. Other reported modulators, such as (1) inorganic acids (e.g., HCl) were shown to slow down the rate of precipitation by inhibiting H2BDC dissociation,79 while (2) the addition of monocarboxylic acid resulted in the formation of larger crystals through the changes in the rate of precipitation.80 These optimizations are important factors to be considered when performing concentration and mass balances for reaction kinetics given that an excess of modulators or deprotonating agents inhibits the overall crystallization process.74,81
The above-mentioned examples provide insight into the historical evolution of some of the most well-known MOFs, while displaying evidence of the variety of shapes and structures imposed by the different synthesis conditions. To the best of our knowledge, currently, there are more than 90000 MOFs reported, with over 500000 more predicted.82
From design to implementation: motivation for the review's focus on MOFs for gas sensing
Presently, the sensing, monitoring, and detection of gases rely on the application of optical, acoustic, and gas chromatography systems; however, these methods are associated with challenges in miniaturization, flexibility, and portability, are expensive, and often sensitive to environmental changes (i.e., environmental noises).83 Furthermore, systems based on materials such as metal oxide semiconductors,84,85 conducting polymers,86,87 and carbon nanotubes88,89 have shown benefits when the sensitivity and specificity for a given sensing gas were considered, relying on the physico-chemical characteristics of the materials for changes in performance;90 however, they also show limited stability. Moreover, analysis showed that their thermodynamical decomposition leads to low sensitivity during the sensing measurements, as well as difficulties during their integration in the fabrication and reproduction of the sensor formed with these materials.91,92The porosity, tuneable sizes, high surface area to mass ratio, and chemical and structural stability of MOFs, as well as their user-controlled synthesis, have been recently explored for gas sensing. The focus on sensing using MOFs is motivated by both the need to help prevent explosions and/or help monitor exposure in surrounding environments, thus reducing the deleterious effects on human health and environmental burden. Further, the focus on sensing/monitoring/detection with MOFs is motivated by the need for processing controls and manufacturing steps to be implemented without logistical challenges, especially when considering the development and application of green technologies. In this context, the design and functionalization studies of MOFs for sensing generally aim to enhance sustainability of such processes using these frameworks and increasing their ability to be used over multiple cycles, while maintaining high efficiency. Thus, it is envisioned that MOFs applied in sensing can potentially reduce the high energy requirements associated with the use of adsorbents (for instance),93,94 the loss of adsorbent functionality,95,96 high corrosiveness,97,98 and volatility99,100 and parasitic energy consumption,101,102 all when considering large-scale implementation.103–105 Specific examples of MOFs applications in different gas sensing strategies are discussed below.
MOFs in ammonia sensing
Ammonia (NH3) is produced during the manufacturing of fertilizers, burning of biomass or landfill wastes, and unceasing volcanic eruptions. Consequently, the global emissions of NH3 have doubled to approximately 50 million tons in the past 50 years.106–108When considering MOFs for NH3 sensing, analysis showed that during its first step of detection (normally associated with the gas chemical adsorption process), NH3 behaves like a Lewis base/reducing agent.109,110 This discovery prompted the design and functionalization of MOFs with oxidation–reduction features. The specific modifications of MOFs included the integration of coordinative metal centers with custom functionalization on their porous surface through coordination bonds111,112 and/or acid–base interactions,113,114 as well as the implementation of ligands with certain functional groups (i.e., –OH, –O, –Br, –NH2, and urea).115 Furthermore, MOFs were also fine-tuned for adsorption or kinetic studies related to the detection of NH3, while still maintaining or even enhancing their thermal and chemical stability.116–118 Their open metal sites directed strong interactions, subsequently resulting in high affinity between the Lewis acidic centers and basic NH3 molecules.119
For instance, Kim et al. reported that high NH3 uptake was realized when divalent metal cations (M2) such as (1) magnesium(II) (Mg2+), (2) manganese(II) (Mn2+), (3) Co2+, (4) nickel(II) (Ni2+), and (5) Zn2+ and a tetradentate ligand called 4,4′-dioxidobiphenyl-3,3′-dicarboxylate (H4dobpdc)119 were used. Briefly, a microwave-assisted route led to the formation of MOFs containing metal cations (1) MgBr2·6H2O, (2) MnCl2·4H2O, (3) CoCl2·6H2O, (4) NiCl2·6H2O, and (5) ZnBr2·2H2O combined with H4dobpdc in a Pyrex cell, in a 1:1 mixture of DMF:EtOH solvent, for 20 min at 403 K.119,120 The resulting framework was iso-reticular with simulated pore size measurements in the range of 18–22 Å. At dry and low NH3 pressures, Mg2(dobpdc) showed a superior capacity of uptake of 7.82 and 8.25 mmol g−1 at 0.072 and 0.57 mbar, respectively. Increased uptake was also observed at 1 bar for the Mg2+ MOF, with a total reported value of 23.90 mmol g−1 (Fig. 2a). Regarding the recyclability, it was shown that the NH3 uptake by the Mg2+- and Ni2+-based MOFs decreased over three cycles of consecutive sorption, which is presumably due to the stronger Lewis acidities among the ions being tested.121 Under dry conditions (0% relative humidity (RH)), Mg2(dobpdc) showed an increased capacity of uptake of 8.37 mmol g−1. This value changed as the RH increased to 80%, with the adsorption capacity of Mg2(dobpdc) decreasing to 6.14 mmol g−1, presumably due to the competitive adsorption between NH3 and H2O respectively.122
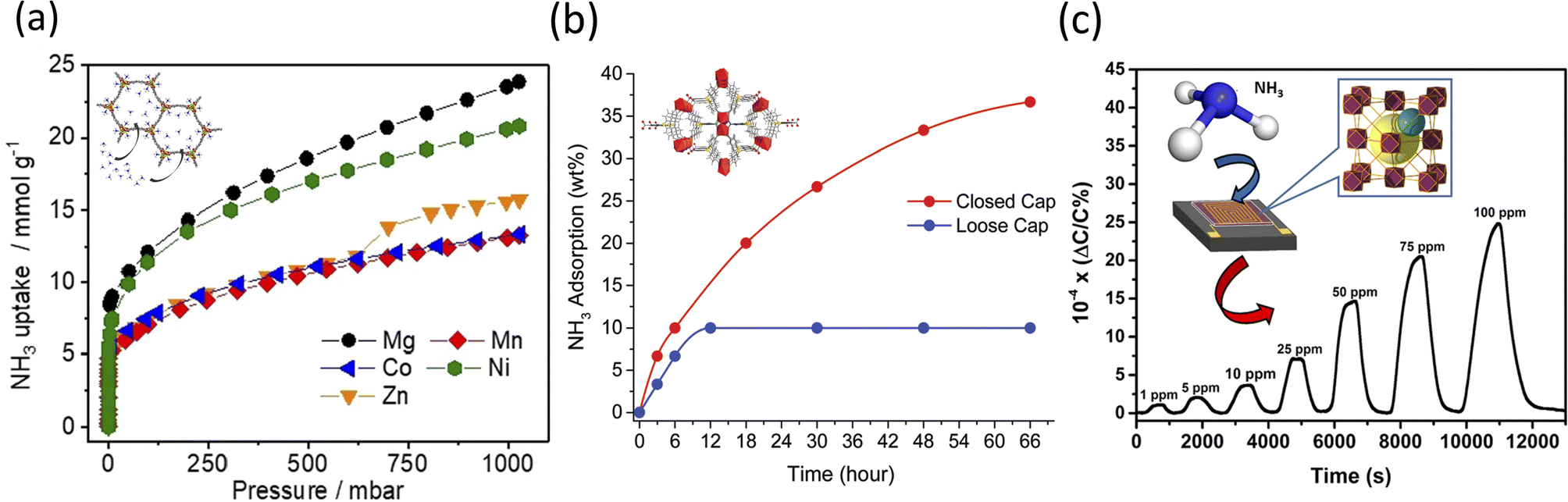 | ||
| Fig. 2 (a) Isotherms of NH3 adsorption of degassed M2(dobpdc) (M2+ = Mg2+, Mn2+, Co2+, Ni2+, and Zn2+) (structure shown in inset) samples at 298 K.119 (b) NH3 adsorption of SION105-Eu (structure shown in inset) as a function of time for both closed cap and open vapor sorption.126 Reproduced from ref. 126 with permission from The Royal Society of Chemistry. (c) NH3 detection at different ppm concentration ranges (i.e., 1–100 ppm) with a schematic representation of the NDC-Y-fcu-MOF thin film on interdigitated electron (IDE) substrate (shown in the inset).129 Reprinted (adapted) with permission from ref. 129. Copyright 2017, the American Chemical Society. | ||
Further, Matikolaei et al. presented a novel Zn-based framework composition, PFC-27,115 obtained via a post-synthetic modification (PSM) via anion insertion of three different types of derivatives, namely, (1) PFC-27/trifluoroacetate (CF3COO−), (2) PFC-27/trifluoromethane sulfonate (CF3SO3−), and (3) PFC-27/acetate (CH3COO−). Gray crystals of PFC-27 were synthesized by mixing Zn(NO3)2·6H2O, 4,4′-biphenyl dicarboxylic acid (BPDA) and tris(4-(4h-1,2,4-triazol-4-yl)phenyl) amine (TTPA) at 120 °C for 48 h. The post-synthetic modification involved the immersion of PCF-27 in aqueous solutions of anion salts, i.e., sodium trifluoroacetate (CF3COONa), sodium acetate (CH3COONa), and sodium trifluoromethane sulfonate (CF3SO3Na), at room temperature, for 24 h. Scanning electron microscopy (SEM) images showed that the pristine PFC-27, PFC-27/CF3SO3, and PFC-27/CF3COO crystals possessed a rod-thorn like morphology and smooth topology. Fourier transform infrared spectroscopy (FTIR) confirmed the presence of absorption bands at 1500, 1166, and 1260 cm−1, corresponding to the signatures of the COO−, CF3COO−, and CF3SO3− anions, respectively. The Brunauer–Emmett–Teller (BET) surface area analysis suggested that all the products followed a type I isotherm, with a decreasing trend for the measured surface area from PFC-27 = 1253 m2 g−1, to PFC-27/CF3SO3− = 877 m2 g−1, PFC-27/CF3COO− = 197 m2 g−1, and PFC-27/CF3COO− = 48 m2 g−1 respectively, most likely due to the anion insertion process, which affected the self-assembly of the framework.123
All the obtained crystals were further tested under static and dynamic conditions of NH3 exposure, as well as in dry and wet conditions respectively. Among them, PFC-27/CF3SO3− showed the highest NH3 gas uptake capacity of 177.85 mg g−1 (10.46 mmol g−1). Under static NH3 adsorption, PFC-27/CF3COO− and PFC-27/CF3SO3− showed a steep increase in their adsorption capabilities at a low relative pressure range (up to 0.1), with PFC-27/CF3COO− and PFC-27/CF3SO3− reaching 71.5% and 83.3% of the total uptake capacity, respectively, at P/P0 = 1.0 and 298 K. Furthermore, all the frameworks also showed the best fit with the Langmuir model, with the NH3 uptake decreasing rapidly from 298 to 345 K.
Under dynamic conditions, the adsorption by PFC-27, PFC-27/CF3COO−, PFC-27/CF3SO3−, and PFC-27/CH3COO− was 165, 307, 349, and 293 mg g−1, respectively. By manipulating the RH to 50%, a sharp decrease in the adsorption capacity was observed in all the products (to 73, 123, 160, and 98 mg g−1 respectively), with such decrease being presumably due to the additional adsorbed water molecules competing for the partially blocked MOF pores.124,125 The reusability studies showed that PFC-27 and PFC-27/CF3SO3− retained their characteristics after five cycles of gas uptake with no obvious changes in their adsorption capacity when different regeneration temperatures were considered.
Moreover, Moribe et al. reported the solvothermal synthesis of (1) Al-PMOF, (2) Ga-PMOF, and (3) In-PMOF performed using meso-tetra(4-carboxyphenyl porphine) (TCPP) in DMF and nitric acid, at 120 °C for 15 h. The resulting MOFs underwent two cycles of NH3 adsorption, both at 298 K, without thermal activation. At 1 bar, Al-PMOF adsorbed 7.67 and 7.34 mmol g−1 in the first and second cycle, respectively. Correspondingly, the uptake by Ga-PMOF was 10.50 and 7.71 mmol g−1, while In-PMOF adsorbed 9.41 and 7.83 mmol g−1, respectively. The higher uptake recorded for Ga-PMOF and In-PMOF in the second cycle was presumably due to the inability of the framework to be desorbing the initially adsorbed NH3 from their –OH sites, thus suggesting that Ga-PMOF and In-PMOF had stronger interactions with NH3.103 The crystal stability studies showed that the powder X-ray diffraction (PXRD) patterns of Al-PMOF remained unchanged pre- and post-exposure to NH3. However, Ga-PMOF and In-PMOF displayed two major peaks at higher angles indicating the limited stability toward multiple cycles of NH3 uptake.
Nguyen et al. reported the preparation of a targeted recyclable NH3 and highly stable MOF, ([Eu(tctb)3(H2O)]) or SION105-Eu.126 In this study, SION105-Eu was solvothermally synthesized by heating europium nitrate [Eu(NO3)3] and tris(p-carboxylic acid)tridurylborane (H3tctb) in a 2:1 DMF:H2O mixture, at 120 °C for 72 h.127 The authors showed that the NH3 gas adsorption at 1 bar was 5.7 mmol g−1 under dry conditions, and 5.9 mmol g−1 under wet conditions (Fig. 2b). Based on the Clausius–Clapeyron isotherm model, the isosteric heat adsorption was −28.7 kJ mol−1 for the coverage of 1.5 mmol g−1, which was believed to result from the specific interaction between NH3 and SION105-Eu. Post-exposure to NH3 was also performed for a total of 6 h. The PXRD analysis confirmed the preservation of the crystallinity of the MOF, although the presence of multiple split peaks was identified in the two regions of 2θ = 6.5–6.9° and 10.4–10.9°. A slight decrease in the structure original peaks intensity was also observed, thus indicating susceptibility to multiple-testing cycling. Further, the analysis suggested that there were only weak interactions (i.e., electrostatic and van der Waals) between SION105-Eu and NH3; specifically, the potential formation of a B–NH3 adduct and Lewis acidic boron–olefin interaction126,128 were observed and discussed. FTIR confirmed the presence of N–H stretching bands at 3300–3500 cm−1. Further studies on the stability assessment of SION105-Eu showed that the crystal was unaltered after 6 h of testing, while its topology slightly changed after 12 h, to fully degrade after 66 h of study, respectively.
Lastly, Assen et al. reported the synthesis of a rare earth (RE)-based MOF platform with face cubic unit (fcu) topology.129 In their solvothermal method, the authors heated yttrium(III) nitrate [Y(NO3)3] and 1,4-naphthalene dicarboxylic acid in 2-fluorobenzoic acid/deionized H2O/nitric acid/DMF solution at 115 °C for 48 h. The resulting MOFs were functionalized on an interdigitated electron (IDE) with a 11-mercatoundecanol (MUD) self-assembled monolayer (SAM) for a lab-on-open chip operation. SEM analysis showed a densely packed tetrahedral-shaped NDC-Y-fcu-MOF with the post-exposure adsorption of NH3 molecules on its surface. The lab-made device detected NH3 (Fig. 2c) in the range of 1 to 100 ppm, and had a detection limit of 92 ppb and a response time of around 250 s. The stability was demonstrated when evaluating steady and uniform sensing levels, with negligible variations during the reproducibility tests. The device detected two different NH3 concentrations (namely 10 and 25 ppm) over more than two weeks of testing at room temperature. However, a reduction in sensitivity was recorded when the temperature varied from 22 °C to 80 °C, most likely due to the reduction in the sorption capability of the framework.
The above-mentioned studies provide selected examples of MOFs with demonstrated functionality for NH3 adsorption; physical and chemical characteristics of these MOFs are highlighted in Table 1 to thus help correlate the structure and function of such individual frameworks. The above-mentioned studies also emphasize that while the synthesis, characteristics, and applications of MOFs are fully intertwined, it is necessary to improve their performance considering both their design and operation when NH3 adsorption is considered.
| Compound | Structure characteristics [powder X-ray diffraction (PXRD)/single crystal X-ray diffraction (SCXRD)] | Morphology/histology [scanning electron microscopy (SEM), transmission electron microscopy (TEM)] | Chemical characterization [X-ray photoelectron spectroscopy (XPS)/nuclear magnetic resonance (NMR)/Fourier transformed infrared (FTIR)/ultraviolet-visible spectroscopy (UV-Vis)] | Absorption properties [thermogravimetric analysis (TGA)/Brunauer–Emmet–Teller (BET)/Langmuir surface areas] |
|---|---|---|---|---|
| PFC-27 (ref. 115) | SCXRD | SEM | XPS | N 2 isotherm |
| (1) PFC-27/CF3COO− | − Trigonal (P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group) space group) |
- Rod-thorn like morphology | (1) 686.85 eV (F 1s) | Type I isotherm |
| (2) PFC-27/CF3SO3− | Asymmetrical with distorted trigonal pyramidal coordination geometry (τ4 = 0.69) | Smooth topology | (2) 164 eV and 163.3 eV (S 2p3/2 and S 2p1/2) and 677.03 eV (F 1s) | S BET (PFC-27) = 1253 m2 g−1 |
| (3) PFC-27/CH3COO− | (3) — | TGA | ||
| 13 C NMR | - Stable up to 260 °C | |||
| (1) 162.1 ppm (COO−) and 115.45 ppm (CF3) | - 11% weight loss | |||
| (2) — | ||||
| (3) 180 ppm (strong COO ester-type) and ∼22 ppm (strong CH3) | ||||
| FTIR | ||||
| (1) 1500 cm−1 (COO−) and 1166 cm−1 (CF3COO−) | ||||
| (2) 1166 cm−1 (CF3COO−) and 1260 cm−1 (CF3SO3−) | ||||
| (3) 1500 cm−1 (COO−) | ||||
| SION105-Eu126 | PXRD | — | — | N 2 isotherm |
| - Simulated: 2θ = 6.5–6.9°, 10–12.2° | S BET = 216 m2 g−1 | |||
| - SION105-Eu: 2θ = 6.5–6.9°, 10.5–12.2° | CO 2 adsorption | |||
| Uptake capacity = ∼1.9 mmol g−1 (195 K and 1 bar) | ||||
| M 2 (dobpdc)119 | PXRD | SEM (all) | ||
| (1) Mg2(dobpdc) | Isoreticular structure | Hexagonal rod morphology | — | — |
| (2) Mn2(dobpdc) | - Simulated: 2θ = 4.8–5°, 8–8.5° | |||
| (3) Co2(dobpdc) | - All: 2θ = 4.8–5°, 8–8.5° | |||
| (4) Ni2(dobpdc) | ||||
| (5) Zn2(dobpdc) | ||||
| M-PMOF103 | — | SEM | N 2 isotherms | |
| (1) Al-PMOF | (1) Plated-shaped (<1 μm) | At 77 K SBET | ||
| (2) Ga-PMOF | (2) Cubic (>1 μm) | (1) = 2060 m2 g−1 | ||
| (3) In-PMOF | (3) Oval-shaped (>1 μm) | (2) = 1600 m2 g−1 | ||
| (3) = 1610 m2 g−1 | ||||
| TGA | ||||
| >90% weight maintained up to 350 °C | ||||
Frameworks for effective and sustainable H2 detection
Hydrogen gas (H2) plays an integral role in energy production.130,131 The significant uses for H2 include NH3 synthesis132–134 and the production of methanol from carbon monoxide.135 H2 gas is also a sustainable fuel source that drives intense research for the transition from fossil fuels to renewable energy sources.136,137 Moreover, H2 has been proposed as a viable energy solution for the production and implementation of fuel cell vehicles (FCVs).138,139Elementally, H2 is colorless, odorless, and lighter than air (with a density of 1/14 of that of air), which makes it imperceptible to humans. Also, because of its light nature, H2 diffuses easily, and thus has a high tendency to leak, with its leakage in industrial settings leading to burns and respiratory problems (i.e., asphyxiation), especially at higher concentrations.140 Further, due to its flammable and combustible nature upon ignition, its rapid spread can lead to severe fires or explosions. Moreover, H2 leakage was shown to be a significant “indirect” contributor to the greenhouse effect because it interacts with CH4 in the atmosphere.141,142 These considerations have led to an increase in the interest in H2 safety directly applicable to the user safety for reducing the environmental burden associated with the implementation of this gas.
For instance, Weber et al. reported the preparation of a sensor comprised of ZnO nanowires (NW), palladium (Pd) nanoparticles (NPs), and ZIF-8.143 The ZIF-8/Pd/ZnO nanocomposite was chosen because of known (1) excellent crystallinity properties and high charge-carrier transport characteristics of ZnO; however, it was limited by its relatively low selectivity and response signal intensity,144–146 (2) the exceptional affinity of Pd towards H2, and thus presumed assumption to efficiently dissociate H2 into its atoms,147–149 and (3) the size of the micropores of ZIF-8 of ∼0.34 nm predicted to allow the passage of H2 molecules, while restricting that of larger gas molecules.150 In addition, upon combining ZnO NWs with ZIF-8, it was envisioned that the new structure can offer molecular sieving properties, to thus lead to a more efficient detection capability.147,148,151
The sensor was created by loading ZnO NWs on a patterned IDE, with the experiments occurring in a furnace, at 950 °C for 1 h, under constant Ar and O2 flows, respectively. The physical and chemical characterizations of the different elements of the sensor are shown in Table 2, which also contains specific structure-function related information for the listed frameworks. The sensing mechanism is shown in Fig. 3a, and was attributed to the different conductivities of the individual components used in the sensor set-up. Specifically, the analysis showed that the ZIF-8/Pd/ZnO sensor presented about ∼20% lower H2 sensitivity than the Pd/ZnO control. Further, analysis of the sensor response identified diffusion limitation of the H2 gas through the ZIF-8 membrane due to the favorable thermally activated transport mechanism of such gas at the ultra-microporous ZIF-8 network.
| Compound | Structure characteristics [powder X-ray diffraction (PXRD)/single crystal X-ray diffraction (SCXRD)] | Morphology/histology [scanning electron microscopy (SEM), transmission electron microscopy (TEM)] | Chemical characterization [X-ray photoelectron spectroscopy (XPS)/nuclear magnetic resonance (NMR)/Fourier transformed infrared (FTIR)/ultraviolet-visible spectroscopy (UV-Vis)] | Absorption properties [thermogravimetric analysis (TGA)/Brunauer–Emmet–Teller (BET)/Langmuir surface areas] |
|---|---|---|---|---|
| ZIF-8 (ref. 143) | PXRD | — | XPS | N 2 physisorption |
| 7.3° = (011) 10.4° = (200) | - C (1s) = 284.91 eV | - SBET = 1760 ± 260 m2 g−1 | ||
| 12.7° = (112) 18.0° = (222) | - O (1s) = 530.87 eV | |||
| - Zn (2p3) = 1021.66 eV | ||||
| - N (1s) = 399.13 eV | ||||
| HKUST-1 (ref. 152) | PXRD | AFM | FTIR | — |
| - Cubic crystal structure | - ∼240–330 nm | - Cu–O of BTC = 733 cm−1 | ||
| - Space group = Fmm |
- Not uniform in thickness | - C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (due to COO presence) = 1370 and 1649 cm−1 C (due to COO presence) = 1370 and 1649 cm−1 |
||
| - Wider peaks = finer crystal size | SEM | - Water-free = absence 3000–3500 cm−1 | ||
| - Cuboctahedron (chamfered cubic) structure | - Deprotonation of BTC18 = absence 1700 cm−1 | |||
| - Crystal size range = 800 nm to 2 μm | XPS | |||
| - Average crystal size = 855 ± 65 nm | - Cu (2p) = Cu(I) or Cu2O = 932.5 eV | |||
| - Showing presence of macropores (>50 nm) | - Cu (2p) = Cu(II) or CuO = 935 eV | |||
| - O (1s) = 530.1 eV | ||||
| - COO bond = 531.2 eV | ||||
| ZIF-8 (ref. 159) | PXRD | AFM | XPS | |
| - Match to other literature23,24 | - Ununiformed thicknesses | - Zn (2p1/2) = 1045.5 eV | ||
| 7.2° = (011) 10.3° = (002) | - Typical range = 4.4–70.7 nm | - Zn (2p3/2) = 1022.35 eV | ||
| 12.7° = (112) 14.6° = (022) | - Maximum = 300 nm | - N (1s) = 398.7 eV | — | |
| 16.4° = (013) 18.1° = (222) | ||||
| IRMOF-20 (ref. 168) | — | AFM | FTIR | N 2 physisorption |
| - Adding IRMOF-20 to Au-coated fiber increases the thickness by more than 1.2 μm | - Thio ring def = 412 cm−1 | - SBET = 5486 m2 g−1 | ||
| - CO def = 736 cm−1 | - Pore volume = 1.68 cm3 g−1 | |||
| - C–H def = 860 and 1012 cm−1 | ||||
| - C–O stretch = 1140 and 1572 cm−1 | ||||
| - CC in plane vib + –OH def = 1348 cm−1 |
||||
| - –OH def + C–O stretch + CC in plane = 1400 cm−1 |
||||
| - CO stretch = 1572 cm−1 |
||||
| XPS | ||||
| - Satisfied the 1.2:1:1.56 ratio of Zn:S:N |
||||
| - Zn(2p3/2) = 530.0 eV | ||||
| - S(2p) = 531.6 eV | ||||
 | ||
| Fig. 3 (a) (top) Schematic representation of the sensing mechanisms of ZIF-8-coated Pd/ZnO NW sensors with (bottom) sensing response of bare ZnO NW, Pd/ZnO NW, and ZIF-8-coated Pd/ZnO NW gas sensors, all recorded at 50 ppm for sensing of H2, C6H6, C7H8, C2H5OH, and CH3COCH3, in air and at 200 °C.143 Reprinted (adapted) with permission from ref. 143. Copyright 2018, the American Chemical Society. (b) (top) Cathodic electrodeposition of HKUST-1 on glassy carbon (GC) electrode with (bottom) cyclic voltammetry (CV) for 100% H2 oxidation on a GC (green), Pt (blue), and HKUST-1 on GC (red) in [C2 mim][NTf2] at a scan rate of 100 mV s−1 with the black dash line corresponding to the blank voltammogram of HKUST-1 on GC electrode in the absence of H2.152 Reprinted (adapted) with permission from ref. 152. Copyright 2020, the American Chemical Society. (c) (top) Schematic of the ZIF-8/Pd-IGZO sensor fabricated on a p-type Si substrate with finger-type Ti/SiO2 electrodes together with (bottom) sensing responses of ZIF-8/Pd-IGZO and Pd-IGZO sensors at different H2 concentrations.159 Reprinted (adapted) with permission from ref. 159. Copyright 2020, the American Chemical Society. (d) (left) Schematic representation of the fabrication and action of present H2 sensor design based on plasmon-active optical fiber surface decorated with IRMOF-20 film with (right) plasmon absorption bands in the H2/air mixture with different concentrations of H2.168 Reprinted (adapted) with permission from ref. 168. Copyright 2019, the American Chemical Society. | ||
In another study, Azhar et al. created a MOF electrode-based device for H2 sensing through cathodic-electrodeposition of HKUST-1 on glassy carbon (GC) electrodes with the addition of room-temperature ionic liquids (RTILs, i.e., 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([C2 mim]-[NTf2])).152 RTILs possess the necessary moieties for serving as electrolytes for H2 sensing when used on metal electrodes (i.e., Pt);153–155 further, there are additional benefits when using only a very small amount of this metal, thus overcoming the economical production cost of the overall device.
The full physical and chemical characterization of the sensing element is shown in Table 2, while an overall schematic of the electrosynthesis of the HKUST-1/GC electrode is included in Fig. 3b.156 The cathodic electrochemical synthesis deprotonates the OH− ions of the BTC, as facilitated by a potential of −1.4 V, NO3− and water. CTAB was extracted when the electrodeposited MOF film was placed in an EtOH/water mixture under moderate stirring. Hydrogen is a moderate soluble element in [C2 mim]-[NTf2], with a saturated concentration of 4.2 mM,157 with a diffusion coefficient of 5.5 × 10−6 cm2 s−1.157 As shown, no electroactivity was detected at the bare GC electrode. However, HKUST-1 displayed a large current response and a partial absorption-type CV shape, with the current dropping off rapidly after the peak, suggesting thin-layer-type behavior.158 In the same graph, the maximum current of HKUST-1 on GC was four-times higher than that for the Pt electrode alone, demonstrating the significant catalytic activity toward H2 oxidation of the framework. This higher sensitivity was attributed to the larger surface area as a result of the micro- and nano-porosity of the electrodeposited MOF.
In a different example, Kim et al. created a real-time photovoltaic sensor that contained a ZIF-8 layer with Pd-decorated n-type indium gallium zinc oxide (IGZO)/p-type silicon photovoltaic cell-covered sensor, which was termed ZIF-8/Pd-IGZO sensor.159 The sensor characterization with its physico-chemical properties is included in Table 2. Pd-based gas sensors are known to detect H2 in real-time;160–162 however, they were also shown to be prone to degradation under repeatable cycles presumably due to the volume expansion resulting from the diffusion of H2 into the inner side of the metal layers.163,164 The fusion of two materials, herein Pd and ZIF-8, was seen to overcome previously recorded small sensing signals, and thus expected to increase the reversible gas adsorption/desorption.165–167
In this design, the ZIF-8 layer was solvothermally synthesized by mixing Zn(NO3)2·6H2O and 2-methyl imidazole (HMIM) in methanol for 30 min. E-beam deposition of SiO2 and Ti on an Si substrate yielded finger-type Ti/SiO2 morphologies, as shown in Fig. 3c, while subsequent lift-off led to the formation of a Pd-IGZO layer through an RF magnetron sputtering system. ZIF-8 was subsequently coated on the top of the Pd-IGZO layer. Gold (Au) was deposited on top of all the finger-type Ti/SiO2 electrodes. The sensor response, denoted as Rgas, is the ratio of the photocurrent between H2 and air relative to only air. It was reported that at 1% H2, the Rgas for ZIF-8/Pd-IGZO was 1.57 × 104% with a low detection limit of ∼0.0035%, which was 8.13-times higher than 1.93 × 103% at the Pd-IGZO sensor interface. The response and recovery time of the ZIF-8/Pd-IGZO sensor were 14 and 7 s at 1% H2, respectively. The device accomplished what was reported to be irreproducible with Pd-based and photovoltaic sensors. Moreover, the sensor maintained long-term stability after three months of operation, while the Pd-IGZO control was completely inoperable after the same testing time, which is most likely due to the physical damage of the contained Pd nanoparticles.
Lastly, Miliutina et al. proposed the fabrication of an H2 plasmonic-fiber sensor through the combination of surface-grafted IRMOF-20 films.168 Specifically, photons excited the surface electrons of the sensor, leading to the collective oscillations and propagation of surface waves at the intersection between the metal nodes and dielectric nanofilms due to the energy transfer at the interfaces.169–171 Plasmonic optical fibers were shown to overcome the drawbacks of regular optical fiber sensors, namely their low sensitivity.172,173 IRMOF-20 was synthesized by dissolving thieno[3,2-b]thiophene-2,5-dicarboxylic acid and zinc nitrate tetrahydrate in DMF at 100 °C for 18 h. A thin film of Au was deposited on multimode plastic-clad silica optical (PCS) fibers via vacuum sputtering, and subsequently soaked in 4-carboxybenzenediazonium tosylate (ADT-COOH) solution for 15 min. The AFT–COOH–grafted optical fibers were then immersed in the IRMOF-20-based liquid for two days for surface-assisted growth of IRMOF-20 (characteristics shown in Table 2).
The performance pf the IRMOF-20 plasmon fiber-based sensor was evaluated at different H2 concentrations and at room temperature (Fig. 3d). The maximum H2 absorption band shifted towards lower wavelengths, with the intensity of the absorption band increasing with the H2 concentration. Even at a low concentration (2% H2/air), the plasmon band was shifted by 9 nm, and then increased by 22 nm when a 4% H2 concentration was used, thus demonstrating excellent detection capability. The reported sensor was stable in a temperature range of 0–50 °C presumably due to the lower dependency of IRMOF-20 on the mean polarizability. Further testing to determine the influence of humidity on the sensor performance (one of the most difficult problems when H2 detection is being considered174,175) revealed a shift in the plasmon absorption band wavelength for both 4 and 20% H2 exposures.
The above-mentioned examples show concrete advances in H2 sensing strategies when using MOF structures. However, for the considerable and impactful implementation of MOFs in H2 sensing platforms, the next generation of sensors for H2 detection should not only be tested in different temperature or gas concentration ranges, but also for leaks and permeation to thus account for the harmful potential of this gas when uncontrollably released.
Applications of frameworks for CO2 detection
Carbon dioxide (CO2), resulting from industrial products and by-products released upon burning materials, accounts for the majority of greenhouse gas emissions176 and can harm living species through heat-related illness, thermal absorption, climate change, and ocean water feedback,176,177 to name a few. Some of the current CO2 sensing methods rely on non-dispersive infrared (NDIR) sensors which use specific light wavelengths, with these systems offering long-term stability, high accuracy, and high gas specificity.178–180 However, NDIR sensors are affected by humidity and temperature fluctuations;180–183 moreover, many times their “bulky” design limits their suitability for field implementation.184,185 Alternatively, other CO2 detection systems use electrochemical sensors, which convert the chemical concentration of the gas to electrical current changes. Electrochemical impedance spectroscopy (EIS) devices overcome the limitations of NDIR sensors, although EIS sensors often encounter experimental “drifts”186 resulting from the imbalance of electrode surface reactions and electrode contaminations, respectively.187 These drifts were also observed when performing curve fitting and regression, leading to significant errors when processing non-linear and drifting data, respectively.188 Moreover, a metal oxide semiconductor (MOS) sensor that uses the resistivity of metal compounds to test for concentrations of CO2 in the air was also implemented for CO2 detection. The MOS has a simple design, and it was tested at higher, less common CO2 concentrations (>2000 ppm).189 The example provided by Shwetha et al. demonstrated the fabrication of an MEMS-based MOS sensor when adding Ag as an additive between the p-type semiconductor BaTiO3 and metal n-type CuO heterojunction to fabricate a 1% Ag-doped BaTiO3–CuO sensing film. The sensor had a sensing capability of 21% for a CO2 gas concentration of 400 ppm and 70% for 1000 ppm while the capacitive-based sensitivity was 54% for 400 ppm and 95% for 1000 ppm,190 respectively. However, this example and others have shown that at a lower ppm CO2 concentration (<1000 pm), MOS sensors do not exhibit good gas sensitivity thus leading to inaccurate responses.191To overcome the above-mentioned drawbacks, MOFs were introduced for CO2 sensing by Ling et al.; authors showed that the series of bimetallic MgxCu1−x-MOF-74 (x = 0.4, 0.2, and 0.17) obtained via one-pot synthesis could be used for CO2 adsorption under visible light illumination.192 Specifically, MgxCu1−x-MOF-74 was synthesized under ultrasonic stirring for 30 min using a mixture of 2,5-dihydroxyterephthalic acid (H4dhtp), magnesium nitrate hexahydrate (Mg(NO3)2·6H2O) and copper nitrate hydrate (Cu(NO3)2·3H2O) and a 15:1:1 solvent of DMF:EtOH:MeOH. The reaction mixture was maintained at 398 K for 24 h.
The pore structure of the resulting MgxCu1−x-MOF-74 exhibited a type I isotherm and H3-type hysteresis loop, thus indicating the presence of both micro and mesoporosity structures (Table 3). However, Mg-MOF-74 lacked stability when exposed to higher temperatures; specifically, ∼42.4% weight loss was observed for the framework exposed to 267 °C and ∼69.8% weight loss at 609 °C. However, the thermal properties were improved through the synergetic effects of the added Mg and Cu,193 which also enhanced the thermal resistance of the identical organic linker in response to changes in the Mg/Cu cation ratio.193 Overall, MgxCu1−x-MOF-74 was found to be stable at a temperature of less than 200 °C, with an increase in temperature leading to significant structural collapse of the framework. The authors also showed that as the Cu content increased, a red shift towards enhanced visible light absorption was observed. Mg-MOF-74 presented the highest band gap energy and the lowest visible light absorption capacity.
| Compound | Structure characteristics [powder X-ray diffraction (PXRD)/single crystal X-ray diffraction (SCXRD)] | Morphology/histology [scanning electron microscopy (SEM), transmission electron microscopy (TEM)] | Chemical characterization [X-ray photoelectron spectroscopy (XPS)/nuclear magnetic resonance (NMR)/Fourier transformed infrared (FTIR)/ultraviolet-visible spectroscopy (UV-Vis)] | Absorption properties [thermogravimetric analysis (TGA)/Brunauer–Emmet–Teller (BET)/Langmuir surface areas] |
|---|---|---|---|---|
| Mg x Cu 1−x -MOF-74 (ref. 192) | PXRD | SEM | FTIR | (1) SLangmuir = 1448 m2 g−1 |
| (1) Mg-MOF-74 | (1) and (5) | (1) Chrysanthemum-like | (2), (3), and (4) | Pore volume = 0.45 cm3 g−1 |
| (2) Mg0.4Cu0.6-MOF-74 | 6.8° = (2![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) 25.6° 0) 25.6° |
- Formed by polyhedral prism crystal branches |
ν(–CO) = 1522 cm−1 |
Pore size = 0.75 nm |
| (3) Mg0.2Cu0.8-MOF-74 | 11.8° = (300) 27.4° | (3) At 398 K | ν(–COO–) = 1420 cm−1 | (2) SLangmuir = 1278 m2 g−1 |
| (4) Mg0.17Cu0.83-MOF-74 | 17.3° 31.4° | - Spherical crystal structure | ν(C–H) = 890 and 823 cm−1 | Pore volume = 0.39 cm3 g−1 |
| (5) Cu-MOF-74 | 21.9° 42° | - Needle-like crystals (∼1 μm) | (C–O) stretch = sharp and weak at 1240 cm−1 | Pore size = 0.77 nm |
| 24.8° | At 418 K | (CO) conjugates (CC) = 1552 and 1191 cm−1 |
(3) SLangmuir = 1061 m2 g−1 | |
| - Non-spherical | Note: (2) ν(C–H) band disappeared at 145 °C synthesis | Pore volume = 0.32 cm3 g−1 | ||
| - High temperature → grain agglomeration | Pore size = 0.76 nm | |||
| (4) Average size (∼5 μm) | (4) SLangmuir = 978 m2 g−1 | |||
| (5) | Pore volume = 0.27 cm3 g−1 | |||
| - Formed by needle-like crystal branches | Pore size = 0.77 nm | |||
| - Average crystal (<5 μm) | (5) SLangmuir = 989 m2 g−1 | |||
| Pore volume = 0.28 cm3 g−1 | ||||
| Pore size = 0.77 nm | ||||
| (1) [Zn4O(PDC)3] (ref. 197) | PXRD | SEM | FTIR | N 2 adsorption |
| (2) [Zn4O(S-mPDC)3] | (1)–(4) confirms cubic system | (1) ∼60 nm aggregation small particles | (1) Stretching μ4-O–Zn = 522 cm−1 (ref. 26) | (1) SBET = 1525 m2 g−1 |
| (3) [Zn4O(R-mPDC)3] | Rietveld refinement | (2) 200–500 nm well-shaped cube-like particles | (2), (3), and (4) stretching μ4-O–Zn = 521–525 cm−1 | (2) SBET = 2366 m2 g−1 |
| (4) [Zn4O(dmPDC)3] | (1)–(4) Fmm space group with Z = 8 |
(3) 200–500 nm well-shaped cube-like particles | 1 H– 13 C CP-MAS SSNMR | (3) SBET = 1943 m2 g−1 |
| (1) a = 24.7739 Å | (4) 200–300 nm well-shaped cube-like particles | (1) Displayed 43.6 and 161.4 ppm | (4) SBET = 1270 m2 g−1 | |
| R p = 3.07%, Rwp = 3.90% | -NCO2− ascribed to 161.4 ppm | (1)–(4) pore sizes = 1.2 nm | ||
| (2) a = 24.8572 Å | 1 H– 13 C HETCOR NMR | Zero-coverage enthalpy of adsorption (Q st ) of H 2 | ||
| R p = 3.28%, Rwp = 4.36% | (1) PDC and –CH2– ascribed to 43.6 ppm | (1)–(4) at 77 and 87 K = 6.3–7.4 kJ mol−1 | ||
| (3) a = 24.8893 Å | XAS, XANES, EXAFS | → (4) present highest Qst = 7.4 kJ mol−1 | ||
| R p = 3.21%, Rwp = 4.11% | (1) Coordination number = 3.7 ± 0.2 | CO 2 adsorption | ||
| (4) a = 24.8597 Å | → Tetrahedral Zn–4O (ref. 31 and 32) | (1) CO2 content = 33.3 wt% | ||
| R p = 3.26%, Rwp = 4.28% | (2) CO2 content = 31.6 wt% | |||
| Full-width-half max. (fwhm) Bragg | (3) CO2 content = 31.6 wt% | |||
| (1) 0.205° | → Highest CO2 capacity = 429 mL g−1 | |||
| (2) 0.0406° | → Fully CO2-loaded = 68.8 wt% or 0.476 g cm−3 CO2 moieties (298 K and 2.6 MPa) | |||
| (3) 0.0617° | → high gravimetric CO2 = 1.562 g cm−1 (195 K and 0.1 MPa) | |||
| (4) 0.0540° | (4) CO2 content = 30.1 wt% | |||
| NTUniv-54 (ref. 205) | Single-crystal XRD | — | — | TGA |
| Crystal system = trigonal | ∼35.3 wt% loss from 25 °C to 280 °C | |||
| Space group = I4/m | Structure maintained before 300 °C | |||
| a = 32.327 Å | N 2 adsorption | |||
| R 1 = 13.9%, Rw1 = 31.6% | - Type-I adsorption isotherm | |||
| → Rhombihexahedron cage SBU, or (3,36)-connected net “nanoball”2,13,16,17 | - 636 cm3 g−1 N2 at 77 K (<1 bar) | |||
| - SBET = 2166 m2 g−1 | ||||
| - SLangmuir = 2903 m2 g−1 | ||||
| CO 2 adsorption | ||||
| - 171 cm3 g−1 at 273 K (<1 bar) | ||||
| - 84 cm3 g−1 at 298 K (<1 bar) | ||||
| - Qst CO2 = 23.0 kJ mol−1 | ||||
| Separation ratios (at 273 K and < 1 bar) | ||||
| - CO2/N2 = 23.8 | ||||
| - CO2/CH4 = 10.2 | ||||
| (1) ZIF-8 (ref. 208) | XRD | SEM | XPS | High pressure CO 2 adsorption (high vacuum to 40 bar) |
| (2) ZIF-8/PAN-30 | - Space group = cubic I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m 3m |
(1) Rhombic dodecahedral form | - Zn 2p3/2 = 1022.08 eV | (1) Pore size = 13.8 Å |
| (3) ZIF-8/PAN-60 | Average = 101 nm | - Zn 2p1/2 = 1044.88 eV | S BET = 1015 m2 g−1 | |
| (4) ZIF-8/PAN-90 | With PAN nanofiber | - N 1s = shift 400.8 eV (1) to 399.3 eV (4) | Pore volume = 0.70 cm3 g−1 | |
| *** Note: ZIF-8 loading = 57 wt% | (2) Average = 20 nm | - C 1s = shift 285.28 eV (1) to 287.08 eV (4) | (2) Pore size = 15.2 Å | |
| PAN-# | (3) Average = 36 nm | 13 C NMR | S BET = 495 m2 g−1 | |
| PAN = polyacrylonitrile nanofibers | (4) Average = 76 nm | - C1 (aromatic carbon) = 152.0 ppm | Pore volume = 0.38 cm3 g−1 | |
| # = Number of minutes | - C2 (–CHCH–) = shift 123.7 ppm (1) to 125.4 ppm (4) |
(3) Pore size = 13.0 Å | ||
| - C3 (–CH3) = 14.6 ppm | S BET = 862 m2 g−1 | |||
| Pore volume = 0.56 cm3 g−1 | ||||
| (4) Pore size = 11.2 Å | ||||
| S BET = 888 m2 g−1 | ||||
| Pore volume = 0.50 cm3 g−1 | ||||
| (1) MOF-5 (ref. 209) | XRD | SEM | Raman spectroscopy | TGA |
| (2) Carbonized MOF-5 (CMOF-5) | (1) 2θ = ∼7° = (002) 13.8° = (004) | (1) Cubic shape | (1) D band (vibration of C atoms with dangling bonds with termination by disordered graphite) = 1300 cm−1 | (2) CMOF |
| 9.8° = (022) 15.6° = (024) | Size = 3.75–5 μm | (2) G band (E2g mode in carbon with high graphitization degree and related to sp2-hybridized carbon atoms) = 1600 cm−1 | 2 wt% loss from 100 °C to 200 °C | |
| (2) 2θ = 20°, 25°, ∼45° | (2) Porous with additional cracks and cavities | → CMOF-5 have a lot of defects due to higher intensity in D-band than G-band | Some weight loss from ∼385 °C to 600 °C | |
| → Disorderly oriented carbon with low crystallinity | Size = 2.75–4.5 μm | → Thermal decomposition of CMOF-5 and formation of CO2 = ∼90 wt% at 1000 °C | ||
| EDS | N 2 adsorption/desorption | |||
| Confirmation of oxygen, carbon, and copper | (1) MOF-5 | |||
| - Pore size = <10 nm | ||||
| - SBET = 477 m2 g−1 | ||||
| - Pore volume = 0.33 cm3 g−1 | ||||
| - Micropore volume = 0.24 cm3 g−1 | ||||
| (2) CMOF-5 | ||||
| - Type-IV isotherms with H3 hysteresis loops = typical mesoporous materials | ||||
| → Not clearly seen in pristine MOF-5 | ||||
| - Pore size = <10 nm, but size distribution is bigger than MOF-5 | ||||
| - SBET = 1884 m2 g−1 | ||||
| - Pore volume = 1.84 cm3 g−1 | ||||
| - Micropore volume = 0.59 cm3 g−1 | ||||
| (1) MOF-808 (ref. 214) | SCXRD | SEM | FTIR | N 2 adsorption |
| (2) MOF-808-FR | (3) Cubic space group Fdm |
(1) 20–40 μm | (2) Absence of –COO stretches of formate ligands = 1582 and 1337 cm−1 | (1) SBET = 1680 m2 g−1 |
| (3) MOF-808-Gly | a = 35.1413(12) Å | (3) No change in morphology and size | (3) Appearance of C–H of glycinate ligands = ∼2960 cm−1 | Pore width = 14.2 Å (QSDFT) |
| 13 C NMR | (2) SBET = 1971 m2 g−1 | |||
| (3)- Carboxylate and aromatic carbons (BTC linker) = 170.3 and 133.6 ppm | Pore width = 13.0 Å (QSDFT) | |||
| - Glycine ligands = 179.5 and 45.2 ppm | (3) SBET = 1427 m2 g−1 | |||
| - Formation of carbamic acid (–NHCOOH) = 169.4 ppm | Pore width = 11.0 Å (QSDFT) | |||
| - Formation of carbamate (–NHCOO−) = 162.5 ppm | CO 2 adsorption | |||
| - Free CO2 = 121.4 ppm | (3) Uptake | |||
| - Ammonium bicarbonate (–NH3+–OCO2H) = 167.3 ppm | At 4 kPa = 0.247 mmol g−1 | |||
| 15 N NMR | At 15 kPa = 0.540 mmol g−1 | |||
| (3)- α-Amino of glycinate ligand = 34.3 and 27.1 ppm | ||||
| - Ammonium species (counterions of carbamate) and unreacted α-amines = 34.4 and 27.1 ppm | ||||
| Humidified (95% RH at 1 atm and 25 °C):- protonated α-ammonium = 31.1 ppm | ||||
Fig. 4a shows the CO2 adsorption–desorption isotherms of bimetallic Mg0.4Cu0.6-MOF-74 against a monometallic MOF-74, at 298 K and 1 bar. It was shown that Mg0.4Cu0.6-MOF-74 outperformed the standalone Cu-MOF-74 for CO2 detection presumably due to the synergistic effect from the adjacent Mg and Cu metal nodes and their role in the sensing process.194 Compared to monometallic Cu-MOF-74, all the bimetallic ratios of MgxCu1−x-MOF-74 were shown to adsorb better, with the highest uptake of 4.58 mmol g−1. Alternatively, MgxCu1−x-MOF-74 was shown to absorb less than Mg-MOF-74, presumably due to the CO2 adsorption capacity being influenced by the Mg content of the framework.195 Although Mg-MOF-74 showed the highest CO2 uptake in an experimental closed system (Micromeritics ASAP apparatus), Mg-MOF-74 also exhibited the largest decrease in CO2 uptake after exposing the system to a X-lamp for 48 h.
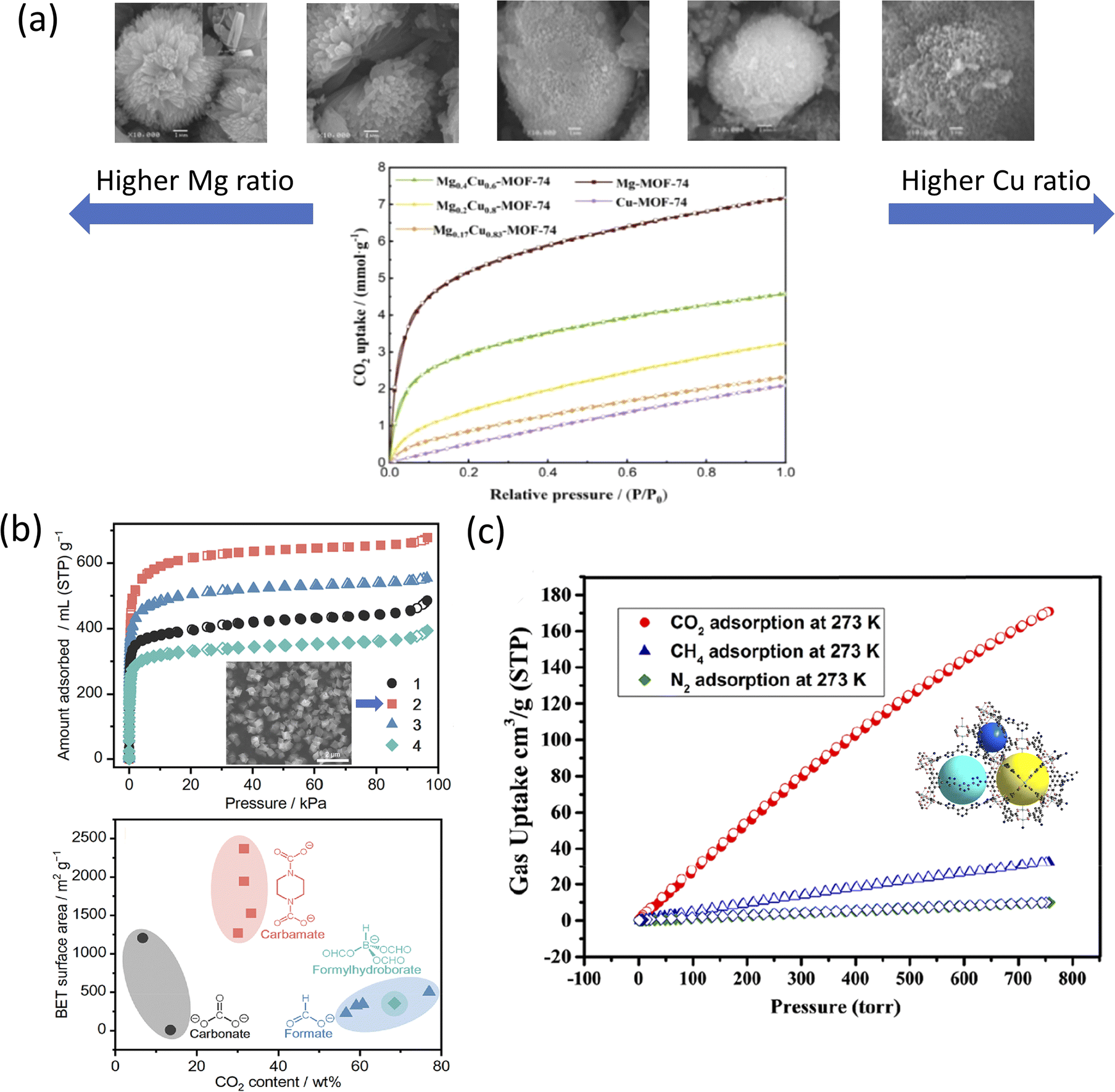 | ||
| Fig. 4 (a) (Top) SEM images from left to right: Mg–, Mg0.4Cu0.6–, Mg0.2Cu0.8–, Mg0.17Cu0.83–, Cu-MOF-74 and (bottom) CO2 adsorption–desorption isotherms of MgxCu1−x– at 298 K and 1 bar.192 (b) (Top) N2 adsorption isotherm of (1) [Zn4O(PDC)3], (2) [Zn4O(S-mPDC)3] with inset SEM image, (3) [Zn4O(R-mPDC)3], and (4) [Zn4O(dmPDC)3] and (bottom) CO2 BET surface areas as a function of CO2-derived linkers (black) carbonate, (red) carbamate, (blue) formate, and (cyan) formylhydroborate.197 Reprinted (adapted) with permission from ref. 197. Copyright 2021, the American Chemical Society. (c) CO2, CH4, and N2 adsorption isotherms of NTUniv-54 (crystallography shown in inset) conducted at 273 K.205 Reprinted (adapted) with permission from ref. 205. Copyright 2021, the American Chemical Society. | ||
Ling et al. presented the CO2 uptake of MgxCu1−x-MOF-74 treated for 12, 24, 36, and 48 h using an X-lamp, and how sensor responses led to changes in the CO2 adsorption rates.192 In particular, the CO2 uptake for Mg-MOF-74 was 7.18 mmol g−1, whereas the CO2 uptake for Cu-MOF-74 was the lowest recorded, reaching only up to 2.09 mmol g−1. The stability of Mg-MOF-74 was attributed to the formation of a metal–oxygen (M–O) bond.196 The distortion and deformation of the coordination environment of Cu2+via the Jahn–Teller effect were explained through the contraction of the M–O bonds, leading to improved photostability of the framework.195
Kadota et al. introduced a one-pot, room-temperature synthesis of mono-carbamates-MOF to convert atmospheric pressure CO2.197 The synthesis of (1) [Zn4O(PDC)3] was performed at 0.1 MPa CO2 (>99.99%) flow, at 25 °C and using a mixture of Zn(Oac)2·2H2O, piperazine (H2PZ), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in DMF and 2-propanol (iPrOH). The resulting MOF formed a white precipitate. To record the effect of the linker, the authors derived three extra carbamate linkers with the same metal core, namely, S-(+)-2-methylpiperazine dicarbamate (S-mPDC); (2) [Zn4O(S-mPDC)3], R-(−)-2-methylpiperazine dicarbamate (R-mPDC); (3) [Zn4O(R-mPDC)3] and trans-2,5-dimethylpiperazine dicarbamate (dmPDC) and (4) [Zn4O(dmPDC)3]. The DBU used in the mixture was described as a strong non-nucleophilic base, ultimately enhancing the reactivity of H2PZ toward CO2 through a deprotonation mechanism.198,199 The introduction of the hydrophobic methyl group in (2), (3), and (4) was expected to enhance the air/moisture stability of the resulting MOFs by preventing the diffusion of H2O molecules in their pores.200 However, in the N2 adsorption isotherm model, the porosity presented in (1)–(4) was poor presumably due to the undefined coordination orientation of [BH(OCHO)3]− and the short bridging distance of HCO2−.197 The increase in the N2 uptake in the low-pressure range is shown in Fig. 4b was due to the significantly higher surface area of the MOF when compared to previously reported CO2-derived coordination compounds such as carbonate, formate, and formylhydroborate.1,201,202 The smaller pore size of 0.7–1.2 nm was attributed to the tight corners formed between the linkers and [Zn4O(CO2)6] SBU.203
The N2 adsorption isotherm at 77 K showed moderately good uptake, with (4) [Zn4O(dmPDC)3] resulting in the highest zero-coverage enthalpy of adsorption (Qst) of 7.4 kJ mol−1. For CO2 adsorption capacity, the (3) [Zn4O(R-mPDC)3] MOF presented the highest CO2 adsorption capacity (i.e., 429 mL g−1) at 298 K and 2.6 MPa, corresponding to 68.8 wt% or 0.476 g cm−3 of CO2 moieties, as well as a high gravimetric CO2 curve tested at 195 K and 0.1 MPa, corresponding to 1.562 g cm−3, respectively. The TGA and temperature-programmed desorption (TPD) analysis indicated the release of CO2 from (2) [Zn4O(S-mPDC)3] at 331 °C.204
Moreover, Zhang et al. constructed a triazole-functionalized txt-type MOF for CO2/CH4 selective uptake at different pressures.205 [Cu2L(H2O)]·2DMF·11H2O (NTUniv-54) was solvothermally synthesized through click chemistry using an H4L linker and Cu(NO3)2·3H2O in DMF/nitric acid (HNO3) at 80 °C for 24 h. The resulting MOF displayed a blue block cube-shaped crystal, with its physico-chemical characteristics reported in Table 3. High-temperature activation of NTUniv-54 led to changes in its color, namely, from blue to deep purple-blue due to the generation of open Cu2+ sites.206,207 Under N2 and at 77 K (<1 bar), NTUniv-54 exhibited a type-I adsorption isotherm and yielded an SBET of 2166 m2 g−1 and SLangmuir of 2903 m2 g−1. As shown in Fig. 4c, the low-pressure CO2 (<1 bar) analysis showed that the uptake amount was 171 cm3 g−1 (@273 K) and 84 cm3 g−1 (@298 K) for CO2; meanwhile, the CH4 adsorption was 19 cm3 g−1 (@298 K), while the N2 adsorption was 6.0 cm3 g−1 (@298 K). These results indicated the selective adsorption of CO2 from a mixture of CH4 and N2. Further, the analysis indicated that high-pressure testing performed at 298 K enabled a CO2 uptake of 822 mg g−1 at 45 bar, with the CH4 total uptake of only 245 cm3 g−1 at the same pressure. The results also indicated that NTUniv-54 could sense and adsorb CO2 at different pressures and temperatures; contrary, it was only capable of CH4 uptake at high pressure.
Li et al. reported a phase-inversion method that allowed the growth of ZIF-8 on the surface of electrospun polyacrylonitrile (PAN) nanofibers to be used for CO2 capture.208 ZIF-8 was solvothermally synthesized by mixing zinc nitrate hexahydrate (Zn(NO3)2·6H2O) and 2-methyl imidazole (2-mim) for 1 h at room temperature; complete physical and chemical analysis of the resulting structures is shown in Table 3. PAN nanofibers were immersed in Zn(NO3)2·6H2O for 20 min, and then 2-mim were added and left to react for 30, 60, and 90 min, leading to formation of ZIF-8/PAN-30, ZIF-8/PAN-60, and ZIF-8/PAN-90, respectively. The goal of incorporating PAN was to limit the known temperature instability of the MOF material. It was reported that the longer the reaction time of ZIF-8/PAN, the higher the total pore volume of the resulting structures. Cyclic testing (Fig. 5a) showed that the MOF-based nanofiber had good cyclic stability, with the CO2 uptake at 1 bar and 40 bar of 7 and 130 cm3 g−1, respectively.
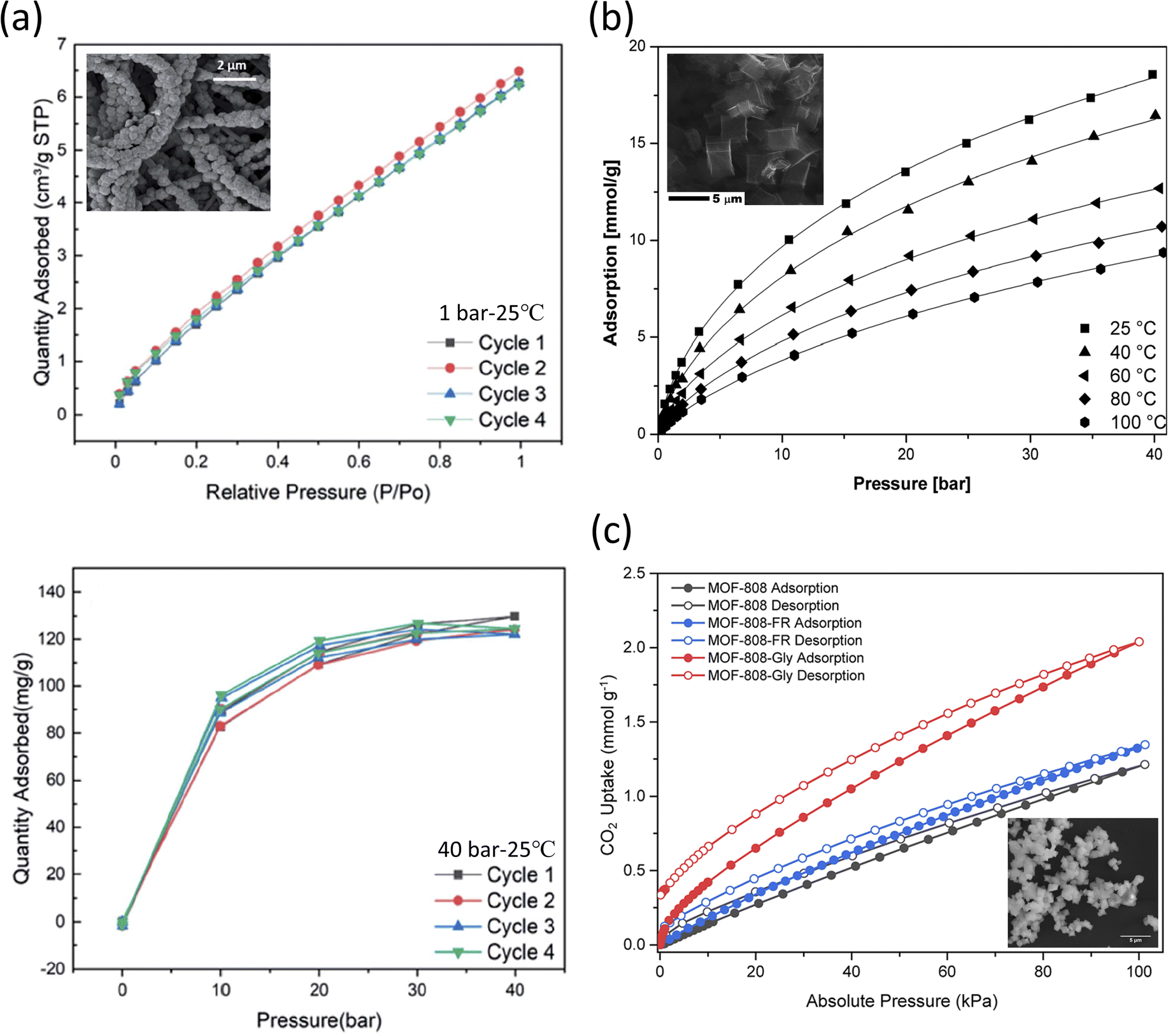 | ||
| Fig. 5 (a) ZIF-8/PAN-90 nanofibers tested at 25 °C with (top) cyclic adsorption isotherms up to 1 bar and (bottom) cyclic CO2 uptake at higher pressures (up to 40 bar).208 Reproduced from ref. 208 with permission from the Royal Society of Chemistry.; (b) CO2 sorption isotherms of carbonized MOF-5 (SEM shown in the inset) fitted through the Sips equation.209 Reproduced from ref. 209 with permission from the Royal Society of Chemistry.; (c) CO2 sorption isotherms evaluated at 25 °C for MOF-808, MOF-808-FR, and MOF-808-Gly (SEM shown in the inset).214 Reprinted (adapted) with permission from ref. 214. Copyright 2022 American Chemical Society. | ||
Kukulka et al. also reported a carbonized version of the stable MOF-5 for the adsorption of CO2 at various temperatures and pressures.209 Carbonization is the process of increasing the C content to make a structure with higher porosity, changes in surface area and larger pore volume.210,211 MOF-5 is known to accumulate traces of moisture, which leads to its faster decomposition,212 while carbonized MOF-5 became hydrophobic.211,213 MOF-5 was solvothermally synthesized by mixing zinc nitrate hexahydrate (Zn(NO3)2·6H2O) and terephthalic acid (C6H4(COOH)2) in DMF at 150 °C for 48 h, with the obtained product forming a white powder precipitate. The sample was subjected to the carbonization process under an Ar flow and heating at 1000 °C for 2 h. The resulting CMOF-5 exhibited a CO2 uptake of 2.43 mmol g−1 at 25 °C and 1 bar. The results also showed that the total pore volume after carbonization increased by five times, the specific surface area increased four times, while the micropore volume increased by two and a half times respectively. As shown in Fig. 5b, as the temperature and pressure decreased, the adsorption increased significantly. Further, the analysis showed that the CO2 adsorbed on the high-energy sites of the framework. It was also reported that the adsorption performance does not change for both pristine MOF-5 and CMOF-5, although CMOF-5 was found to adsorb 1.87 times higher (at 1 bar) than MOF-5 and was also reused post-15 cycles.
Lyu et al. reported on the functionalization of an amino acid (AA) on formate removed (FR)-MOF-808 for better CO2 sorption under different in vivo simulated humid flue gas conditions.214 The protonated amino groups (–NH2 or –NH–) on the alkyl chains of the AAs served as the primary sites to presumably encapsulate CO2 molecules from the exterior sites of the MOF-808, with the formate (HCOO−) removal step allowing the deprotonated carboxylate (–COO−) groups of AAs to be functionalized on MOF-808-FR. In the reported synthesis of MOF-808, zirconium oxychloride octahydrate (ZrOCl2·8H2O) was mixed with 1,3,5-benzenetricarboxylic acid (H3BTC) in DMF under heat activation at 130 °C for two days. Formate (HCOO−) was later removed by treatment with HCl (Table 3). The binding of the AA anions (i.e., glycine, sarcosine, alanine, and serine) yielded [Zr6O4(OH)4(BTC)2-(AA)N(OH)6−N(H2O)6−N], where N denotes the molar equivalence of amino acids per Zr6O4(OH)4 cluster.
In the gas sorption studies of MOF-808-AA, it was reported that MOF-808, -808-FR, and -808-AA presented a type I N2 gas adsorption isotherm. Among them, -FR revealed the highest surface area and increased porosity (SBET = 1971 m2 g−1); meanwhile, -Gly presented a reduced SBET (= 1427 m2 g−1) when compared to its precursor MOFs presumably due to the AA residues occupying the spaces inside the pores. As shown in Fig. 5c, the single-component CO2 sorption isotherms had a significant characteristic CO2 hysteresis between the adsorption and desorption branches for all the MOF-808s and their corresponding derivatives. MOF-808-Gly presented an outstanding CO2 uptake performance, with the uptake at 25 °C and at 4 kPa being 0.247 mmol g−1 (4% CO2 in 1 atm gas mixture, relevant to natural gas combined cycle flue gas capture) and 0.540 mmol g−1 at 15 kPa (15% CO2 in 1 atm gas mixture, relevant to coal flue gas capture) respectively. It was also shown that upon testing with other AA variants, DL-lysine presented the highest single-component CO2 uptake, which indicated the importance of the amine species in CO2 capture. In the simulated CO2/H2O binary adsorption study (RH ∼10%), a two-fold increase in CO2 uptake for -808-Gly was reported, with corresponding uptakes of 0.525 mmol g−1 at 4 kPa and 0.693 mmol g−1 at 15 kPa. Further dynamic breakthrough evaluation showed a significant delay in uptake for -808-Gly, indicating a change in uptake in the presence of humidity. This increased uptake was attributed to the thermodynamically favorable bicarbonate by-product (as studied extensively under NMR) formed upon exposure to both CO2 and H2O molecules known to lead to the formation of carbamic acid (–NHCOOH) and carbamate (–NHCOO− species).
The selected literature presented herein gave exclusive insights into how MOFs were developed either via membrane functionalization or ligand modification for the selective and sensitive detection of CO2. The CO2 physisorption and chemisorption binding nature reported showed the possibility to provide more active sites or better attraction for MOF-based sensors, including polymer- and membrane-MOF devices or the MOF itself. Regardless, the optimal outcome of any CO2 sorption studies is to reduce the environmental exposure to CO2 and to obtain a net-zero carbon footprint.
Outlook and new perspective for the design and implementation of MOFs
The above-mentioned studies, while being comprehensive and advancing the use of MOFs for sensing of NH3, H2, and CO2, are limited representations of how frameworks can be user-designed and customized for efficient, customizable MOF formation at reduced costs. Moreover, the studies highlighted here, while advancing the usage of MOFs, leave room for further developments especially in the context of understanding such frameworks structure–function–performance relationships and how they can be supported during the life cycle, operational, and implementation costs for sensor formation, respectively. For instance, although the SBU system was introduced in 2001 (ref. 215) and has been seen to bring the usage of molecular chemistry (both organic and inorganic) to extended solid-state structures,216 tuning the MOF topology by regulating the ligand and SBU geometry remains a challenge,217 mostly due to the available structural analysis methods in the different labs. Further, while the concept of the SBU system provides an essential key to decipher the capabilities of the synthesized frameworks, especially regarding predicted or directed applications with regard to both thermodynamic and architectural stability respectively, the advancement of these systems, especially in regard to the topological directionality of MOFs and the formation of stable structures is still limited.218Thus, we foresee the need to extend the “net” or topology of a to-be-developed MOF, where the “net”, or “underlying topology” refers to a “simple” and “connected” graph of edges and vertexes. These graphs are known to determine the combinatorial symmetry of the framework,219 thus explaining the structure–performance relationships through extended versions of TOPOS and SYSTRE219,220 algorithms, all in searchable databases performed in the context of the current restricted ones of the Reticular Chemistry Structure Resource (RCSR) (2000 entries221), EPINET project (15000 entries222,223) and TOPOS (70000 entries224,225). Thus, we anticipate that combining the structural diversity in MOF chemistry as a result of the wide variety of available SBU geometries will lead to specific structures to be designed by choosing appropriately shaped and sized building units.216
One can also envision complementing the experimental design of MOFs with atomistic-level characterizations, thus providing a mechanism-driven hypothesis for the efficient detection of gases under the performance-related rational design and synthesis of the framework. We foresee that by using both surface and bulk characterization of a given MOF, a full understanding of its physico-chemical properties as resolved with high-resolution secondary electron microscopy226 can be attained. Through this determination, a custom design can be undertaken to realize the high-sensitivity and sensibility detection of gases under the potentially physical observation of atomic-level phenomena associated with such processes. Specifically, regarding the three areas highlighted in this review, studies showed that coordinative metal centers can be custom functionalized on the porous surfaces of MOFs to enable an increase in their sensing performance through the formation of coordination bonds111,112 and/or acid–base interactions.113,114 It is thus anticipated that by choosing ligands with certain functional groups (i.e., –OH, –O, –Br, –NH2, and urea) which were previously shown to help increase the detection of NH3115 one can fine-tuned MOFs characteristics for the adsorption or kinetic analysis related to gases detection, while still maintaining or enhancing their thermal and chemical stability.116–118 Presently, the open metal sites in the frameworks are considered to provide strong interaction sites for NH3 for instace, even without a reduction in the pore size of the individual MOF, therefore are predicted to allow for high-affinity interactions between the Lewis acidic centers and the basic NH3 molecules.119 However, given that the studies in this field are currently focused on increasing the total NH3 uptake and affinity at low ppm levels,103–105 the complex characterization and custom design of the framework still need to be achieved.
Alternatively, in the case of H2, due to its known small size and low viscosity which can lead to its quick escape from pressurized systems, ensuring the proper design and characterization of such gas sensors when they are based on MOFs is essential, especially for these sensors integration in production or transportation of the H2 gas. For instance, we foresee that one will be able to formulate an atomic-level mechanism that will not only allow for the customized sensor design but also possibly reduce the rapid thermal and pressure changes known to affect H2-based MOF sensors upon their implementation. Moreover, given the ease of H2 permeation into sensors based on MOFs, this phenomenon can lead to false positives in the detection process. The full experimental detection of MOF-based systems through atomic and bulk structural inspection after a given operation or cycles of operation226 can be undertaken as a viable means for operational sensitivity assessment during the storage, handlining, and transportation of H2.
Lastly, one can envision the capability of atomic-level designed/characterized MOFs to also sense the smallest increments of emitted CO2 from a complex gas mixture, all while sustaining the original physical, chemical, and thermal capability of the sensor. These proposals can be complemented not only by enlarging the surface area of MOF-based sensors to realize a higher capacity for CO2 adsorption, but further experimentally benefit from the implementation of high-affinity ligands and/or metal against the testing adsorbate, as dictated through structure–function atomic-level analysis. Moreover, mechanisms can be derived for these specific MOF structures to become “next-generation” MOFs, achieving the reduction of CO2 into carbon monoxide (CO), a product employed as a precursor in several industrial processes ranging from methanol production227,228 to the hydroformylation of olefins.229,230 Therefore, we predict increased potential of user-synthesized MOFs to be included as a template for a carbon-neutral cycle positively offset by industrial emissions,231,232 with such frameworks possessing better transportation and recyclability,233 as well as potential to catalyze and reduce the carbon footprints emitted from non-industrial sites and transform them into green and clean molecules234 for a better and more sustainable environment.235,236
Conflicts of interest
There are no conflicts to declare.References
- O. M. Yaghi, M. O’Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423(6941), 705–714, DOI:10.1038/nature01650.
- H. Li, M. Eddaoudi, M. O’Keeffe and O. M. Yaghi, Nature, 1999, 402(6759), 276–279, DOI:10.1038/46248.
- E. Klontzas, A. Mavrandonakis, G. E. Froudakis, Y. Carissan and W. Klopper, J. Phys. Chem. C, 2007, 111, 13635–13640, DOI:10.1021/jp075420q.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O’Keeffe and O. M. Yaghi, Science, 2002, 295(5554), 469–472, DOI:10.1126/science.1067208.
- X. Xu, Y. Sun, Q. Zhang, S. Wang, L. Zhang, Z. Wu and G. Lu, ChemistrySelect, 2016, 1(8), 1763–1767, DOI:10.1002/slct.201600526.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41(6), 2308–2322, 10.1039/c1cs15221a.
- Z. Mai and D. Liu, Cryst. Growth Des., 2019, 19(12), 7439–7462, DOI:10.1021/acs.cgd.9b00879.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127(51), 17998–17999, DOI:10.1021/ja0570032.
- K. A. Cychosz and A. J. Matzger, Langmuir, 2010, 26(17), 17198–17202, DOI:10.1021/la103234u.
- J. G. Nguyen and S. M. Cohen, J. Am. Chem. Soc., 2010, 132(13), 4560–4561, DOI:10.1021/ja100900c.
- L. D. Salmi, M. J. Heikkilä, M. Vehkamäki, E. Puukilainen, M. Ritala and T. Sajavaara, J. Vac. Sci. Technol., A, 2015, 33(1), 01A121, DOI:10.1116/1.4901455.
- T. Wu, L. Shen, M. Luebbers, C. Hu, Q. Chen, Z. Ni and R. I. Masel, Chem. Commun., 2010, 46, 6120–6122, 10.1039/C0CC01170C.
- L. Huang, H. Wang, J. Chen, Z. Wang, J. Sun, D. Zhao and Y. Yan, Microporous Mesoporous Mater., 2003, 58(2), 105–114, DOI:10.1016/S1387-1811(02)00609-1.
- M. De Toni, R. Jonchiere, P. Pullumbi, F. X. Coudert and A. H. Fuchs, ChemPhysChem, 2012, 13(15), 3497–3503, DOI:10.1002/cphc.201200455.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283(5405), 1148–1150, DOI:10.1126/science.283.5405.1148.
- B. Liu, Y. Li, S. C. Oh, Y. Fang and H. Xi, RSC Adv., 2016, 6(66), 61006–61012, 10.1039/c6ra11917d.
- L. Liu, S. Wu, D. Li, Y. Li, H. Zhang, L. Li, S. Jin and Z. Yao, ACS Appl. Mater. Interfaces, 2022, 14(32), 36882–36889, DOI:10.1021/acsami.2c10346.
- V. V. Guerrero, Y. Yoo, M. C. McCarthy and H.-K. Jeong, J. Mater. Chem., 2010, 20(19), 3938–3943, 10.1039/b924536g.
- F. Millange, C. Serre and G. Ferey, Chem. Commun., 2002, 8, 822–823, 10.1039/b201381a.
- A. S. Munn, G. J. Clarkson, F. Millange, Y. Dumont and R. I. Walton, CrystEngComm, 2013, 15(45), 9679–9687, 10.1039/C3CE41268G.
- G. Xu, X. Zhang, P. Guo, C. Pan, H. Zhang and C. Wang, J. Am. Chem. Soc., 2010, 132(11), 3656–3657, DOI:10.1021/ja910818a.
- T. R. Whitfield, X. Wang, L. Liu and A. J. Jacobson, Solid State Sci., 2005, 7(9), 1096–1103, DOI:10.1016/j.solidstatesciences.2005.03.007.
- F. Millange and R. I. Walton, Isr. J. Chem., 2018, 58(9–10), 1019–1035, DOI:10.1002/ijch.201800084.
- T. Ahnfeldt, D. Gunzelmann, T. Loiseau, D. Hirsemann, J. Senker, G. Férey and N. Stock, Inorg. Chem., 2009, 48(7), 3057–3064, DOI:10.1021/ic8023265.
- J. Chen, K. Li, L. Chen, R. Liu, X. Huang and D. Ye, Green Chem., 2014, 16(5), 2490–2499, 10.1039/C3GC42414F.
- A. Comotti, S. Bracco, P. Sozzani, S. Horike, R. Matsuda, J. Chen, M. Takata, Y. Kubota and S. Kitagawa, J. Am. Chem. Soc., 2008, 130(41), 13664–13672, DOI:10.1021/ja802589u.
- G. Ferey and C. Serre, Chem. Soc. Rev., 2009, 38(5), 1380–1399, 10.1039/b804302g.
- A. Schoedel, M. Li, D. Li, M. O’Keeffe and O. M. Yaghi, Chem. Rev., 2016, 116(19), 12466–12535, DOI:10.1021/acs.chemrev.6b00346.
- L. Han, J. Zhang, Y. Mao, W. Zhou, W. Xu and Y. Sun, Ind. Eng. Chem. Res., 2019, 58(34), 15489–15496, DOI:10.1021/acs.iecr.9b02223.
- S. Bourrelly, B. Moulin, A. Rivera, G. Maurin, S. Devautour-Vinot, C. Serre, T. Devic, P. Horcajada, A. Vimont, G. Clet, M. Daturi, J.-C. Lavalley, S. Loera-Serna, R. Denoyel, P. L. Llewellyn and G. Férey, J. Am. Chem. Soc., 2010, 132(27), 9488–9498, DOI:10.1021/ja1023282.
- P. Freund, L. Mielewczyk, M. Rauche, I. Senkovska, S. Ehrling, E. Brunner and S. Kaskel, ACS Sustainable Chem. Eng., 2019, 7(4), 4012–4018, DOI:10.1021/acssuschemeng.8b05368.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132(15), 154104, DOI:10.1063/1.3382344.
- D. V. Patil, P. B. S. Rallapalli, G. P. Dangi, R. J. Tayade, R. S. Somani and H. C. Bajaj, Ind. Eng. Chem. Res., 2011, 50(18), 10516–10524, DOI:10.1021/ie200429f.
- D. T. C. Nguyen, H. T. N. Le, T. S. Do, V. T. Pham, D. Lam Tran, V. T. T. Ho, T. V. Tran, D. C. Nguyen, T. D. Nguyen, L. G. Bach, H. K. P. Ha and V. T. Doan, J. Chem., 2019, 2019, 1–11, DOI:10.1155/2019/5602957.
- Z. Li, Y. N. Wu, J. Li, Y. Zhang, X. Zou and F. Li, Chemistry, 2015, 21(18), 6913–6920, DOI:10.1002/chem.201406531.
- C. W. Ashling, D. N. Johnstone, R. N. Widmer, J. Hou, S. M. Collins, A. F. Sapnik, A. M. Bumstead, P. A. Midgley, P. A. Chater, D. A. Keen and T. D. Bennett, J. Am. Chem. Soc., 2019, 141(39), 15641–15648, DOI:10.1021/jacs.9b07557.
- N. Ahadi, S. Askari, A. Fouladitajar and I. Akbari, Sci. Rep., 2022, 12(1), 2649, DOI:10.1038/s41598-022-06518-8.
- K. H. Cho, J. W. Yoon, J. H. Lee, J. C. Kim, K. Kim, U. H. Lee, M. Choi, S. K. Kwak and J.-S. Chang, J. Mater. Chem. A, 2021, 9(25), 14593–14600, 10.1039/d1ta00366f.
- S. Kalhor, M. Zarei, M. A. Zolfigol, H. Sepehrmansourie, D. Nematollahi, S. Alizadeh, H. Shi and J. Arjomandi, Sci. Rep., 2021, 11(1), 19370, DOI:10.1038/s41598-021-97801-7.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309(5743), 2040–2042, DOI:10.1126/science.1116275.
- O. I. Lebedev, F. Millange, C. Serre, G. Van Tendeloo and G. Férey, Chem. Mater., 2005, 17(26), 6525–6527, DOI:10.1021/cm051870o.
- B. Rungtaweevoranit, C. S. Diercks, M. J. Kalmutzki and O. M. Yaghi, Faraday Discuss., 2017, 201, 9–45, 10.1039/c7fd00160f.
- M. Y. Zorainy, M. Gar Alalm, S. Kaliaguine and D. C. Boffito, J. Mater. Chem. A, 2021, 9(39), 22159–22217, 10.1039/d1ta06238g.
- S. Bhattacharjee, C. Chen and W.-S. Ahn, RSC Adv., 2014, 4(94), 52500–52525, 10.1039/c4ra11259h.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O’Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(27), 10186–10191, DOI:10.1073/pnas.0602439103.
- X. C. Huang, Y. Y. Lin, J. P. Zhang and X. M. Chen, Angew. Chem., Int. Ed. Engl., 2006, 45(10), 1557–1559, DOI:10.1002/anie.200503778.
- Y. Pan, Y. Liu, G. Zeng, L. Zhao and Z. Lai, Chem. Commun., 2011, 47(7), 2071–2073, 10.1039/c0cc05002d.
- S. Tanaka, K. Kida, M. Okita, Y. Ito and Y. Miyake, Chem. Lett., 2012, 41(10), 1337–1339, DOI:10.1246/cl.2012.1337.
- H. Bux, F. Liang, Y. Li, J. Cravillon, M. Wiebcke and J. Caro, J. Am. Chem. Soc., 2009, 131(44), 16000–16001, DOI:10.1021/ja907359t.
- V. V. Butova, A. P. Budnik, E. A. Bulanova and A. V. Soldatov, Mendeleev Commun., 2016, 26(1), 43–44, DOI:10.1016/j.mencom.2016.01.017.
- M. Shahsavari, P. Mohammadzadeh Jahani, I. Sheikhshoaie, S. Tajik, A. Aghaei Afshar, M. B. Askari, P. Salarizadeh, A. Di Bartolomeo and H. Beitollahi, Materials, 2022, 15(2), 447, DOI:10.3390/ma15020447.
- Y. Li, G. Wen, J. Li, Q. Li, H. Zhang, B. Tao and J. Zhang, Chem. Commun., 2022, 58(82), 11488–11506, 10.1039/D2CC04190A.
- J. F. Fernández-Bertrán, M. P. Hernández, E. Reguera, H. Yee-Madeira, J. Rodriguez, A. Paneque and J. C. Llopiz, J. Phys. Chem. Solids, 2006, 67(8), 1612–1617, DOI:10.1016/j.jpcs.2006.02.006.
- S. Tanaka, K. Kida, T. Nagaoka, T. Ota and Y. Miyake, Chem. Commun., 2013, 49(72), 7884–7886, 10.1039/C3CC43028F.
- C. Mottillo, Y. Lu, M.-H. Pham, M. J. Cliffe, T.-O. Do and T. Friščić, Green Chem., 2013, 15(8), 2121–2131, 10.1039/C3GC40520F.
- M. He, J. Yao, L. Li, K. Wang, F. Chen and H. Wang, ChemPlusChem, 2013, 78(10), 1222–1225, DOI:10.1002/cplu.201300193.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O’Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43(1), 58–67, DOI:10.1021/ar900116g.
- H. Hayashi, A. P. Cote, H. Furukawa, M. O’Keeffe and O. M. Yaghi, Nat. Mater., 2007, 6(7), 501–506, DOI:10.1038/nmat1927.
- A. Mittal, S. Gandhi and I. Roy, Sci. Rep., 2022, 12(1), 10331, DOI:10.1038/s41598-022-14630-y.
- X.-G. Yang, J.-R. Zhang, X.-K. Tian, J.-H. Qin, X.-Y. Zhang and L.-F. Ma, Angew. Chem., Int. Ed., 2023, 62(7), e202216699, DOI:10.1002/anie.202216699.
- J. Zhang, T. Wu, C. Zhou, S. Chen, P. Feng and X. Bu, Angew. Chem., Int. Ed. Engl., 2009, 48(14), 2542–2545, DOI:10.1002/anie.200804169.
- M. Zhu, D. Srinivas, S. Bhogeswararao, P. Ratnasamy and M. A. Carreon, Catal. Commun., 2013, 32, 36–40, DOI:10.1016/j.catcom.2012.12.003.
- C. H. Kuo, Y. Tang, L. Y. Chou, B. T. Sneed, C. N. Brodsky, Z. Zhao and C. K. Tsung, J. Am. Chem. Soc., 2012, 134(35), 14345–14348, DOI:10.1021/ja306869j.
- G. Lu and J. T. Hupp, J. Am. Chem. Soc., 2010, 132(23), 7832–7833, DOI:10.1021/ja101415b.
- Y. Jing, J. Wang, B. Yu, J. Lun, Y. Cheng, B. Xiong, Q. Lei, Y. Yang, L. Chen and M. Zhao, RSC Adv., 2017, 7(67), 42030–42035, 10.1039/c7ra08763b.
- Y.-R. Lee, M.-S. Jang, H.-Y. Cho, H.-J. Kwon, S. Kim and W.-S. Ahn, Chem. Eng. J., 2015, 271, 276–280, DOI:10.1016/j.cej.2015.02.094.
- S. Kouser, A. Hezam, M. J. N. Khadri and S. A. Khanum, J. Porous Mater., 2022, 29(3), 663–681, DOI:10.1007/s10934-021-01184-z.
- B. Chen, Z. Yang, Y. Zhu and Y. Xia, J. Mater. Chem. A, 2014, 2(40), 16811–16831, 10.1039/c4ta02984d.
- S. A. Moggach, T. D. Bennett and A. K. Cheetham, Angew. Chem., Int. Ed. Engl., 2009, 48(38), 7087–7089, DOI:10.1002/anie.200902643.
- D. Fairen-Jimenez, S. A. Moggach, M. T. Wharmby, P. A. Wright, S. Parsons and T. Düren, J. Am. Chem. Soc., 2011, 133(23), 8900–8902, DOI:10.1021/ja202154j.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O’Keeffe and O. M. Yaghi, Science, 2008, 319(5865), 939–943, DOI:10.1126/science.1152516.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130(42), 13850–13851, DOI:10.1021/ja8057953.
- J. Jiang and O. M. Yaghi, Chem. Rev., 2015, 115(14), 6966–6997, DOI:10.1021/acs.chemrev.5b00221.
- J. Winarta, B. Shan, S. M. McIntyre, L. Ye, C. Wang, J. Liu and B. Mu, Cryst. Growth Des., 2019, 20(2), 1347–1362, DOI:10.1021/acs.cgd.9b00955.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chemistry, 2011, 17(24), 6643–6651, DOI:10.1002/chem.201003211.
- Y. Zhao, Q. Zhang, Y. Li, R. Zhang and G. Lu, ACS Appl. Mater. Interfaces, 2017, 9(17), 15079–15085, DOI:10.1021/acsami.7b02887.
- S. Øien, D. Wragg, H. Reinsch, S. Svelle, S. Bordiga, C. Lamberti and K. P. Lillerud, Cryst. Growth Des., 2014, 14(11), 5370–5372, DOI:10.1021/cg501386j.
- B. Shan, J. B. James, M. R. Armstrong, E. C. Close, P. A. Letham, K. Nikkhah, Y. S. Lin and B. Mu, J. Phys. Chem. C, 2018, 122(4), 2200–2206, DOI:10.1021/acs.jpcc.7b11012.
- M. J. Katz, Z. J. Brown, Y. J. Colon, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49(82), 9449–9451, 10.1039/c3cc46105j.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135(28), 10525–10532, DOI:10.1021/ja404514r.
- J. Ren, H. W. Langmi, B. C. North, M. Mathe and D. Bessarabov, Int. J. Hydrogen Energy, 2014, 39(2), 890–895, DOI:10.1016/j.ijhydene.2013.10.087.
- S. M. Moosavi, A. Nandy, K. M. Jablonka, D. Ongari, J. P. Janet, P. G. Boyd, Y. Lee, B. Smit and H. J. Kulik, Nat. Commun., 2020, 11(1), 4068, DOI:10.1038/s41467-020-17755-8.
- X. Liu, S. Cheng, H. Liu, S. Hu, D. Zhang and H. Ning, Sensors, 2012, 12(7), 9635–9665, DOI:10.3390/s120709635.
- A. Dey, J. Mater. Sci. Eng. B, 2018, 229, 206–217, DOI:10.1016/j.mseb.2017.12.036.
- B. Saruhan, R. Lontio Fomekong and S. Nahirniak, Frontiers in Sensors, 2021, 2, 657931, DOI:10.3389/fsens.2021.657931.
- X. Liu, W. Zheng, R. Kumar, M. Kumar and J. Zhang, Coord. Chem. Rev., 2022, 462, 214517, DOI:10.1016/j.ccr.2022.214517.
- Y. C. Wong, B. C. Ang, A. S. M. A. Haseeb, A. A. Baharuddin and Y. H. Wong, J. Electrochem. Soc., 2019, 167(3), 037503, DOI:10.1149/2.0032003jes.
- C. Gabriel Kaufmann, R. Y. S. Zampiva, M. Rossi and A. K. Alves, in Environmental Applications of Nanomaterials, ed. A. Kopp Alves, Springer International Publishing, Cham, 2022, pp. 55–71, DOI: DOI:10.1007/978-3-030-86822-2_4.
- T. Han, A. Nag, S. Chandra Mukhopadhyay and Y. Xu, Sens. Actuators, A, 2019, 291, 107–143, DOI:10.1016/j.sna.2019.03.053.
- J. Rossignol, A. Harrabi, D. Stuerga, P. Pribetich, G. Bailly and T. Leblois, ACS Omega, 2020, 5(20), 11507–11514, DOI:10.1021/acsomega.0c00596.
- H. Chai, Z. Zheng, K. Liu, J. Xu, K. Wu, Y. Luo, H. Liao, M. Debliquy and C. Zhang, IEEE Sens. J., 2022, 22(6), 5470–5481, DOI:10.1109/jsen.2022.3148264.
- Y. Yan, G. Yang, J. L. Xu, M. Zhang, C. C. Kuo and S. D. Wang, Sci. Technol. Adv. Mater., 2021, 21(1), 768–786, DOI:10.1080/14686996.2020.1820845.
- K. K. Li, W. Leigh, P. Feron, H. Yu and M. Tade, Appl. Energy, 2016, 165, 648–659, DOI:10.1016/j.apenergy.2015.12.109.
- M. Stec, A. Tatarczuk, L. Wieclaw-Solny, A. Krotki, M. Sciazko and S. Tokarski, Fuel, 2015, 151, 50–56, DOI:10.1016/j.fuel.2015.01.014.
- S. A. Mazari, B. S. Ali, B. M. Jan, I. M. Saeed and S. Nizamuddin, Int. J. Greenhouse Gas Control, 2015, 34, 129–140, DOI:10.1016/j.ijggc.2014.12.017.
- P. Khakharia, L. Brachert, J. Mertens, A. Huizinga, B. Schallert, K. Schaber, T. J. H. Vlugt and E. Goetheer, Int. J. Greenhouse Gas Control, 2013, 19, 138–144, DOI:10.1016/j.ijggc.2013.08.014.
- Y. N. Li, J. Cheng, L. Q. Hu, J. Z. Liu, J. H. Zhou and K. F. Cen, Fuel, 2018, 216, 418–426, DOI:10.1016/j.fuel.2017.12.030.
- M. V. Diamanti, U. V. Velardi, A. Brenna, A. Mele, M. Pedeferri and M. Ormellese, Electrochimica Acta, 2016, 192, 414–421, DOI:10.1016/j.electacta.2016.02.003.
- C. N. Dai, W. J. Wei, Z. G. Lei, C. X. Li and B. H. Chen, Fluid Phase Equilib., 2015, 391, 9–17, DOI:10.1016/j.fluid.2015.02.002.
- Y. Du, Y. Yuan and G. T. Rochelle, Chem. Eng. Sci., 2016, 155, 397–404, DOI:10.1016/j.ces.2016.08.017.
- M. Wang, A. S. Joel, C. Ramshaw, D. Eimer and N. M. Musa, Appl. Energy, 2015, 158, 275–291, DOI:10.1016/j.apenergy.2015.08.083.
- S. F. Shen, Y. Y. Bian and Y. Zhao, Int. J. Greenhouse Gas Control, 2017, 56, 1–11, DOI:10.1016/j.ijggc.2016.11.011.
- S. Moribe, Z. Chen, S. Alayoglu, Z. H. Syed, T. Islamoglu and O. K. Farha, ACS Mater. Lett., 2019, 1(4), 476–480, DOI:10.1021/acsmaterialslett.9b00307.
- O. T. Wilcox, A. Fateeva, A. P. Katsoulidis, M. W. Smith, C. A. Stone and M. J. Rosseinsky, Chem. Commun., 2015, 51(81), 14989–14991, 10.1039/C5CC06209H.
- T. Kajiwara, M. Higuchi, D. Watanabe, H. Higashimura, T. Yamada and H. Kitagawa, Chem. - Eur. J., 2014, 20(47), 15611–15617, DOI:10.1002/chem.201403542.
- A. Bouwman, D. Lee, W. A. H. Asman, F. Dentener, K. Van der Hoek and J. Olivier, Global Biogeochem. Cycles, 1997, 11(4), 561–587, DOI:10.1029/97GB02266.
- S. Riddick, D. Ward, P. Hess, N. Mahowald, R. Massad and E. Holland, Biogeosciences, 2016, 13(11), 3397–3426, DOI:10.5194/bg-13-3397-2016.
- S. N. Behera, M. Sharma, V. P. Aneja and R. Balasubramanian, Environ. Sci. Pollut. Res. Int., 2013, 20(11), 8092–8131, DOI:10.1007/s11356-013-2051-9.
- W. Morris, C. J. Doonan and O. M. Yaghi, Inorg. Chem., 2011, 50(15), 6853–6855, DOI:10.1021/ic200744y.
- J. F. Van Humbeck, T. M. McDonald, X. Jing, B. M. Wiers, G. Zhu and J. R. Long, J. Am. Chem. Soc., 2014, 136(6), 2432–2440, DOI:10.1021/ja4105478.
- T. R. Cook, Y. R. Zheng and P. J. Stang, Chem. Rev., 2013, 113(1), 734–777, DOI:10.1021/cr3002824.
- S. R. Batten and N. R. Champness, Philos. Trans. R. Soc., A, 2017, 375(2084), 20160032, DOI:10.1098/rsta.2016.0032.
- A. Chołuj, R. Karczykowski and M. J. Chmielewski, Organometallics, 2019, 38(18), 3392–3396, DOI:10.1021/acs.organomet.9b00281.
- I. Strauss, A. Mundstock, M. Treger, K. Lange, S. Hwang, C. Chmelik, P. Rusch, N. C. Bigall, T. Pichler, H. Shiozawa and J. Caro, ACS Appl. Mater. Interfaces, 2019, 11(15), 14175–14181, DOI:10.1021/acsami.8b22002.
- M. Khanpour Matikolaei and E. Binaeian, ACS Appl. Mater. Interfaces, 2021, 13(23), 27159–27168, DOI:10.1021/acsami.1c03242.
- D. X. Xue, Y. Belmabkhout, O. Shekhah, H. Jiang, K. Adil, A. J. Cairns and M. Eddaoudi, J. Am. Chem. Soc., 2015, 137(15), 5034–5040, DOI:10.1021/ja5131403.
- D. X. Xue, A. J. Cairns, Y. Belmabkhout, L. Wojtas, Y. Liu, M. H. Alkordi and M. Eddaoudi, J. Am. Chem. Soc., 2013, 135(20), 7660–7667, DOI:10.1021/ja401429x.
- A. H. Assen, Y. Belmabkhout, K. Adil, P. M. Bhatt, D.-X. Xue, H. Jiang and M. Eddaoudi, Angew. Chem., Int. Ed., 2015, 54(48), 14353–14358, DOI:10.1002/anie.201506345.
- D. W. Kim, D. W. Kang, M. Kang, J. H. Lee, J. H. Choe, Y. S. Chae, D. S. Choi, H. Yun and C. S. Hong, Angew. Chem., Int. Ed. Engl., 2020, 59(50), 22531–22536, DOI:10.1002/anie.202012552.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134(16), 7056–7065, DOI:10.1021/ja300034j.
- A. J. Rieth, Y. Tulchinsky and M. Dinca, J. Am. Chem. Soc., 2016, 138(30), 9401–9404, DOI:10.1021/jacs.6b05723.
- Y. Chen, L. Li, J. Li, K. Ouyang and J. Yang, J. Hazard. Mater., 2016, 306, 340–347, DOI:10.1016/j.jhazmat.2015.12.046.
- X. Cui, Q. Yang, L. Yang, R. Krishna, Z. Zhang, Z. Bao, H. Wu, Q. Ren, W. Zhou, B. Chen and H. Xing, Adv. Mater., 2017, 29(28), 1606929, DOI:10.1002/adma.201606929.
- X. Zhang, Y. Yang, L. Song, J. Chen, Y. Yang and Y. Wang, J. Hazard. Mater., 2019, 365, 597–605, DOI:10.1016/j.jhazmat.2018.11.049.
- X. Zhang, X. Lv, X. Shi, Y. Yang and Y. Yang, J. Colloid Interface Sci., 2019, 539, 152–160, DOI:10.1016/j.jcis.2018.12.056.
- T. N. Nguyen, I. M. Harreschou, J. H. Lee, K. C. Stylianou and D. W. Stephan, Chem. Commun., 2020, 56(67), 9600–9603, 10.1039/d0cc00741b.
- F. M. Ebrahim, T. N. Nguyen, S. Shyshkanov, A. Gladysiak, P. Favre, A. Zacharia, G. Itskos, P. J. Dyson and K. C. Stylianou, J. Am. Chem. Soc., 2019, 141(7), 3052–3058, DOI:10.1021/jacs.8b11907.
- X. Zhao and D. W. Stephan, J. Am. Chem. Soc., 2011, 133(32), 12448–12450, DOI:10.1021/ja205598k.
- A. H. Assen, O. Yassine, O. Shekhah, M. Eddaoudi and K. N. Salama, ACS Sens., 2017, 2(9), 1294–1301, DOI:10.1021/acssensors.7b00304.
- C. Li, H. Zhang, M. Liu, F.-F. Lang, J. Pang and X.-H. Bu, Industrial Chemistry & Materials, 2023, 1(1), 9–38, 10.1039/D2IM00063F.
- M. Tahir, B. Ajiwokewu, A. A. Bankole, O. Ismail, H. Al-Amodi and N. Kumar, J. Environ. Chem. Eng., 2023, 11(2), 109408, DOI:10.1016/j.jece.2023.109408.
- A. Valera-Medina, F. Amer-Hatem, A. K. Azad, I. C. Dedoussi, M. de Joannon, R. X. Fernandes, P. Glarborg, H. Hashemi, X. He, S. Mashruk, J. McGowan, C. Mounaim-Rouselle, A. Ortiz-Prado, A. Ortiz-Valera, I. Rossetti, B. Shu, M. Yehia, H. Xiao and M. Costa, Energy Fuels, 2021, 35(9), 6964–7029, DOI:10.1021/acs.energyfuels.0c03685.
- S. Ghavam, M. Vahdati, I. A. G. Wilson and P. Styring, Front. Energy Res., 2021, 9, 580808, DOI:10.3389/fenrg.2021.580808.
- K. Tagawa, H. Gi, K. Shinzato, H. Miyaoka and T. Ichikawa, J. Phys. Chem. C, 2022, 126(5), 2403–2409, DOI:10.1021/acs.jpcc.1c09902.
- A. Kaithal, C. Werlé and W. Leitner, JACS Au, 2021, 1(2), 130–136, DOI:10.1021/jacsau.0c00091.
- S. Tang, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2020, 142(13), 5980–5984, DOI:10.1021/jacs.0c01592.
- H. Xiaowei, J. Zhijun, L. Quanyou, M. Qingqiang, Z. Dongya, L. Jiayi and L. Jinzhong, Front. Earth Sci., 2021, 9, 626111, DOI:10.3389/feart.2021.626111.
- Y. Manoharan, S. E. Hosseini, B. Butler, H. Alzhahrani, B. T. F. Senior, T. Ashuri and J. Krohn, Appl. Sci., 2019, 9(11), 2296, DOI:10.3390/app9112296.
- L. Fan, Z. Tu and S. H. Chan, Energy Rep., 2021, 7, 8421–8446, DOI:10.1016/j.egyr.2021.08.003.
- I. B. Ocko and S. P. Hamburg, Atmos. Chem. Phys., 2022, 22(14), 9349–9368, DOI:10.5194/acp-22-9349-2022.
- S. Timmerberg, M. Kaltschmitt and M. Finkbeiner, Energy Convers. Manage.: X, 2020, 7, 100043, DOI:10.1016/j.ecmx.2020.100043.
- N. Sánchez-Bastardo, R. Schlögl and H. Ruland, Ind. Eng. Chem. Res., 2021, 60(32), 11855–11881, DOI:10.1021/acs.iecr.1c01679.
- M. Weber, J. H. Kim, J. H. Lee, J. Y. Kim, I. Iatsunskyi, E. Coy, M. Drobek, A. Julbe, M. Bechelany and S. S. Kim, ACS Appl. Mater. Interfaces, 2018, 10(40), 34765–34773, DOI:10.1021/acsami.8b12569.
- H. T. Wang, B. S. Kang, F. Ren, L. C. Tien, P. W. Sadik, D. P. Norton, S. J. Pearton and J. Lin, Appl. Phys. Lett., 2005, 86(24), 243503, DOI:10.1063/1.1949707.
- O. Lupan, V. V. Ursaki, G. Chai, L. Chow, G. A. Emelchenko, I. M. Tiginyanu, A. N. Gruzintsev and A. N. Redkin, Sens. Actuators, B, 2010, 144(1), 56–66, DOI:10.1016/j.snb.2009.10.038.
- Ü. Özgür, D. Hofstetter and H. Morkoç, Proceedings of the IEEE, 2010, 98(7), 1255–1268, DOI:10.1109/jproc.2010.2044550.
- M. Drobek, M. Bechelany, C. Vallicari, A. Abou Chaaya, C. Charmette, C. Salvador-Levehang, P. Miele and A. Julbe, J. Membr. Sci., 2015, 475, 39–46, DOI:10.1016/j.memsci.2014.10.011.
- M. Drobek, J. H. Kim, M. Bechelany, C. Vallicari, A. Julbe and S. S. Kim, ACS Appl. Mater. Interfaces, 2016, 8(13), 8323–8328, DOI:10.1021/acsami.5b12062.
- J. Park, D. Song and S. Kim, Nanotechnology, 2008, 19, 105503, DOI:10.1088/0957-4484/19/10/105503.
- W. T. Koo, S. Qiao, A. F. Ogata, G. Jha, J. S. Jang, V. T. Chen, I. D. Kim and R. M. Penner, ACS Nano, 2017, 11(9), 9276–9285, DOI:10.1021/acsnano.7b04529.
- M. Drobek, J.-H. Kim, M. Bechelany, C. Vallicari, E. Leroy, A. Julbe and S. Kim, Sens. Actuators, B, 2018, 264, 410–418, DOI:10.1016/j.snb.2018.03.009.
- M. R. Azhar, G. Hussain, M. O. Tade, D. S. Silvester and S. Wang, ACS Appl. Nano Mater., 2020, 3(5), 4376–4385, DOI:10.1021/acsanm.0c00503.
- G. Hussain, M. V. Sofianos, J. Lee, C. Gibson, C. E. Buckley and D. S. Silvester, Electrochem. Commun., 2018, 86, 43–47, DOI:10.1016/j.elecom.2017.11.011.
- G. Hussain, L. Aldous and D. S. Silvester, Anal. Chim. Acta, 2019, 1048, 12–21, DOI:10.1016/j.aca.2018.09.055.
- G. Hussain and D. S. Silvester, Anal. Chem., 2016, 88(24), 12453–12460, DOI:10.1021/acs.analchem.6b03824.
- S. Alizadeh and D. Nematollahi, J. Am. Chem. Soc., 2017, 139(13), 4753–4761, DOI:10.1021/jacs.6b12564.
- D. S. Silvester, K. R. Ward, L. Aldous, C. Hardacre and R. G. Compton, J. Electroanal. Chem., 2008, 618(1–2), 53–60, DOI:10.1016/j.jelechem.2008.02.018.
- I. Streeter, G. G. Wildgoose, L. Shao and R. G. Compton, Sens. Actuators, B, 2008, 133(2), 462–466, DOI:10.1016/j.snb.2008.03.015.
- H. Kim, W. Kim, R. Lee, S. Cho, J. Park, Y. Pak and G. Y. Jung, ACS Sens., 2020, 5(4), 1050–1057, DOI:10.1021/acssensors.9b02565.
- S.-J. Choi, S. Chattopadhyay, J. J. Kim, S.-J. Kim, H. L. Tuller, G. C. Rutledge and I.-D. Kim, Nanoscale, 2016, 8(17), 9159–9166, 10.1039/C5NR06611E.
- H. Kim, Y. Pak, Y. Jeong, W. Kim, J. Kim and G. Y. Jung, Sens. Actuators, B, 2018, 262, 460–468, DOI:10.1016/j.snb.2018.02.025.
- M. Gao, M. Cho, H. J. Han, Y. S. Jung and I. Park, Small, 2018, 14(10), 1703691, DOI:10.1002/smll.201703691.
- L. Li, L. Hu, J. Li and Z. Wei, Nano Res., 2015, 8(2), 418–440, DOI:10.1007/s12274-014-0695-5.
- V. Saxena, N. Kumar and V. K. Saxena, Renewable Sustainable Energy Rev., 2017, 70, 563–588, DOI:10.1016/j.rser.2016.11.067.
- M. Chen, L. Zou, Z. Zhang, J. Shen, D. Li, Q. Zong, G. Gao, G. Wu, J. Shen and Z. Zhang, Carbon, 2018, 130, 281–287, DOI:10.1016/j.carbon.2018.01.013.
- D. Lee, H. Park, S. D. Han, S. H. Kim, W. Huh, J. Y. Lee, Y. S. Kim, M. J. Park, W. I. Park, C. Y. Kang and C. H. Lee, Small, 2019, 15(2), e1804303, DOI:10.1002/smll.201804303.
- H. Chen, M. Zhang, R. Bo, C. Barugkin, J. Zheng, Q. Ma, S. Huang, A. W. Y. Ho-Baillie, K. R. Catchpole and A. Tricoli, Small, 2018, 14(7), 1702571, DOI:10.1002/smll.201702571.
- E. Miliutina, O. Guselnikova, S. Chufistova, Z. Kolska, R. Elashnikov, V. Burtsev, P. Postnikov, V. Svorcik and O. Lyutakov, ACS Sens., 2019, 4(12), 3133–3140, DOI:10.1021/acssensors.9b01074.
- M. I. Stockman, Opt. Express, 2011, 19(22), 22029–22106, DOI:10.1364/OE.19.022029.
- C. Liu, L. Yang, X. Lu, Q. Liu, F. Wang, J. Lv, T. Sun, H. Mu and P. K. Chu, Opt. Express, 2017, 25(13), 14227–14237, DOI:10.1364/OE.25.014227.
- M. Wang, H. Li, T. Xu, H. Zheng, M. Yu, G. Li, J. Xu and J. Wu, Opt. Express, 2018, 26(22), 28277–28287, DOI:10.1364/OE.26.028277.
- S. Grassini, M. Ishtaiwi, M. Parvis and A. Vallan, Sensors, 2014, 15(1), 485–498, DOI:10.3390/s150100485.
- Y. Esfahani Monfared, Biosensors, 2020, 10(7), 77, DOI:10.3390/bios10070077.
- Z. Zhao, M. Knight, S. Kumar, E. T. Eisenbraun and M. A. Carpenter, Sens. Actuators, B, 2008, 129(2), 726–733, DOI:10.1016/j.snb.2007.09.032.
- L. Coelho, J. M. de Almeida, J. L. Santos and D. Viegas, Appl. Opt., 2015, 54(35), 10342–10348, DOI:10.1364/AO.54.010342.
- R. Cassia, M. Nocioni, N. Correa-Aragunde and L. Lamattina, Front. Plant Sci., 2018, 9, 273, DOI:10.3389/fpls.2018.00273.
- S. Manabe, Tellus, 2019, 71(1), 1620078, DOI:10.1080/16000870.2019.1620078.
- S. Neethirajan, D. S. Jayas and S. Sadistap, Food Bioprocess Technol., 2008, 2(2), 115–121, DOI:10.1007/s11947-008-0154-y.
- X. Jia, J. Roels, R. Baets and G. Roelkens, Sensors, 2019, 19(19), 4260, DOI:10.3390/s19194260.
- X. Tan, H. Zhang, J. Li, H. Wan, Q. Guo, H. Zhu, H. Liu and F. Yi, Nat. Commun., 2020, 11(1), 5245, DOI:10.1038/s41467-020-19085-1.
- T. Yasuda, S. Yonemura and A. Tani, Sensors, 2012, 12(3), 3641–3655, DOI:10.3390/s120303641.
- J. Park, H. Cho and S. Yi, Procedia Eng., 2010, 5, 303–306, DOI:10.1016/j.proeng.2010.09.108.
- R. Frodl and T. Tille, IEEE Sens. J., 2006, 6(6), 1697–1705, DOI:10.1109/jsen.2006.884440.
- C. Massie, G. Stewart, G. McGregor and J. R. Gilchrist, Sens. Actuators, B, 2006, 113(2), 830–836, DOI:10.1016/j.snb.2005.03.105.
- K. Thurmond, Z. Loparo, W. Partridge and S. S. Vasu, Appl. Spectrosc., 2016, 70(6), 962–971, DOI:10.1177/0003702816641261.
- L. Lin and X. Zeng, Anal. Bioanal. Chem., 2018, 410(19), 4587–4596, DOI:10.1007/s00216-018-1090-y.
- M. A. Ali, L. Dong, J. Dhau, A. Khosla and A. Kaushik, J. Electrochem. Soc., 2020, 167(3), 037550, DOI:10.1149/1945-7111/ab69fe.
- H. Ryu, D. Thompson, Y. Huang, B. Li and Y. Lei, Sens. Actuators Rep., 2020, 2(1), 100022, DOI:10.1016/j.snr.2020.100022.
- T. Nandy, R. A. Coutu Jr and C. Ababei, Sensors, 2018, 18(10), 3443, DOI:10.3390/s18103443.
- H. R. Shwetha, S. M. Sharath, B. Guruprasad and S. B. Rudraswamy, Micro Nano Eng., 2022, 16, 100156, DOI:10.1016/j.mne.2022.100156.
- A. Ghosh, C. Zhang, S. Shi and H. Zhang, Sens. Actuators, B, 2019, 301, 126958, DOI:10.1016/j.snb.2019.126958.
- J. Ling, A. Zhou, W. Wang, X. Jia, M. Ma and Y. Li, ACS Omega, 2022, 7(23), 19920–19929, DOI:10.1021/acsomega.2c01717.
- M. W. Kadi, H. M. A. El Salam, T. Zaki and R. M. Mohamed, J. Nanopart. Res., 2020, 22(6), 143, DOI:10.1007/s11051-020-04855-1.
- Z. Gao, L. Liang, X. Zhang, P. Xu and J. Sun, ACS Appl. Mater. Interfaces, 2021, 13(51), 61334–61345, DOI:10.1021/acsami.1c20878.
- R. Sanz, F. Martinez, G. Orcajo, L. Wojtas and D. Briones, Dalton Trans., 2013, 42(7), 2392–2398, 10.1039/c2dt32138f.
- J. D. Howe, C. R. Morelock, Y. Jiao, K. W. Chapman, K. S. Walton and D. S. Sholl, J. Phys. Chem. C, 2016, 121(1), 627–635, DOI:10.1021/acs.jpcc.6b11719.
- K. Kadota, Y. L. Hong, Y. Nishiyama, E. Sivaniah, D. Packwood and S. Horike, J. Am. Chem. Soc., 2021, 143(40), 16750–16757, DOI:10.1021/jacs.1c08227.
- D. J. Heldebrant, P. K. Koech, V. A. Glezakou, R. Rousseau, D. Malhotra and D. C. Cantu, Chem. Rev., 2017, 117(14), 9594–9624, DOI:10.1021/acs.chemrev.6b00768.
- L. Biancalana, G. Bresciani, C. Chiappe, F. Marchetti and G. Pampaloni, New J. Chem., 2017, 41(4), 1798–1805, 10.1039/c6nj03647c.
- J. Yang, A. Grzech, F. M. Mulder and T. J. Dingemans, Chem. Commun., 2011, 47(18), 5244–5246, 10.1039/c1cc11054c.
- R. Arai, K. Seto, A. Bell and H. Sugimoto, Polym. J., 2018, 50(4), 301–307, DOI:10.1038/s41428-017-0020-8.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed. Engl., 2004, 43(18), 2334–2375, DOI:10.1002/anie.200300610.
- L. K. Macreadie, E. J. Mensforth, R. Babarao, K. Konstas, S. G. Telfer, C. M. Doherty, J. Tsanaktsidis, S. R. Batten and M. R. Hill, J. Am. Chem. Soc., 2019, 141(9), 3828–3832, DOI:10.1021/jacs.8b13639.
- R. J. Young, M. T. Huxley, E. Pardo, N. R. Champness, C. J. Sumby and C. J. Doonan, Chem. Sci., 2020, 11(16), 4031–4050, 10.1039/d0sc00485e.
- M. Zhang, H. Cui, L. Zhang, G. Qin, P. Zhang, S. Wang, G. Jiang, J. Wang, M. Wang, M. Wang, T. Sun and Y. Tang, Inorg. Chem., 2021, 60(20), 15646–15652, DOI:10.1021/acs.inorgchem.1c02249.
- B. Zheng, J. Bai, J. Duan, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2010, 133(4), 748–751, DOI:10.1021/ja110042b.
- S. Ma, D. Sun, J. M. Simmons, C. D. Collier, D. Yuan and H.-C. Zhou, J. Am. Chem. Soc., 2008, 130(3), 1012–1016, DOI:10.1021/ja0771639.
- Z. Li, Z. Cao, C. Grande, W. Zhang, Y. Dou, X. Li, J. Fu, N. Shezad, F. Akhtar and A. Kaiser, RSC Adv., 2021, 12(2), 664–670, 10.1039/d1ra06480k.
- W. Kukulka, K. Cendrowski, B. Michalkiewicz and E. Mijowska, RSC Adv., 2019, 9(32), 18527–18537, 10.1039/c9ra01786k.
- K. Cendrowski, P. Skumial, P. Spera and E. Mijowska, Mater. Des., 2016, 110, 740–748, DOI:10.1016/j.matdes.2016.08.043.
- M. Hu, J. Reboul, S. Furukawa, N. L. Torad, Q. Ji, P. Srinivasu, K. Ariga, S. Kitagawa and Y. Yamauchi, J. Am. Chem. Soc., 2012, 134(6), 2864–2867, DOI:10.1021/ja208940u.
- Y. Ming, J. Purewal, J. Yang, C. Xu, R. Soltis, J. Warner, M. Veenstra, M. Gaab, U. Muller and D. J. Siegel, Langmuir, 2015, 31(17), 4988–4995, DOI:10.1021/acs.langmuir.5b00833.
- J. Tang and Y. Yamauchi, Nat. Chem., 2016, 8(7), 638–639, DOI:10.1038/nchem.2548.
- H. Lyu, O. I. Chen, N. Hanikel, M. I. Hossain, R. W. Flaig, X. Pei, A. Amin, M. D. Doherty, R. K. Impastato, T. G. Glover, D. R. Moore and O. M. Yaghi, J. Am. Chem. Soc., 2022, 144(5), 2387–2396, DOI:10.1021/jacs.1c13368.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O’Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34(4), 319–330, DOI:10.1021/ar000034b.
- M. J. Kalmutzki, N. Hanikel and O. M. Yaghi, Sci. Adv., 2018, 4(10), eaat9180, DOI:10.1126/sciadv.aat9180.
- X. Li, J. Liu, K. Zhou, S. Ullah, H. Wang, J. Zou, T. Thonhauser and J. Li, J. Am. Chem. Soc., 2022, 144(47), 21702–21709, DOI:10.1021/jacs.2c09487.
- J. Ha, J. H. Lee and H. R. Moon, Inorg. Chem. Front., 2020, 7(1), 12–27, 10.1039/C9QI01119F.
- M. O’Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112(2), 675–702, DOI:10.1021/cr200205j.
- L. Öhrström, Crystals, 2015, 5(1), 154–162, DOI:10.3390/cryst5010154.
- M. O’Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41(12), 1782–1789, DOI:10.1021/ar800124u.
- S. J. Ramsden, V. Robins and S. T. Hyde, Acta Crystallogr., Sect. A: Found. Crystallogr., 2009, 65, 81–108, DOI:10.1107/S0108767308040592.
- S. T. Hyde, O. Delgado Friedrichs, S. J. Ramsden and V. Robins, Solid State Sci., 2006, 8(7), 740–752, DOI:10.1016/j.solidstatesciences.2006.04.001.
- V. A. Blatov, Crystallogr. Rev., 2004, 10(4), 249–318, DOI:10.1080/08893110412331323170.
- V. A. Blatov, IUCr CompComm Newsletter, 2006, 7, 4–38 Search PubMed.
- J. Ciston, H. G. Brown, A. J. D’Alfonso, P. Koirala, C. Ophus, Y. Lin, Y. Suzuki, H. Inada, Y. Zhu, L. J. Allen and L. D. Marks, Nat. Commun., 2015, 6(1), 7358, DOI:10.1038/ncomms8358.
- T. Pinheiro Araújo, J. Morales-Vidal, T. Zou, M. Agrachev, S. Verstraeten, P. O. Willi, R. N. Grass, G. Jeschke, S. Mitchell, N. López and J. Pérez-Ramírez, Adv. Energy Mater., 2023, 13(14), 2204122, DOI:10.1002/aenm.202204122.
- N. Lawes, I. E. Gow, L. R. Smith, K. J. Aggett, J. S. Hayward, L. Kabalan, A. J. Logsdail, T. J. A. Slater, M. Dearg, D. J. Morgan, N. F. Dummer, S. H. Taylor, M. Bowker, C. R. A. Catlow and G. J. Hutchings, Faraday Discuss., 2023, 242, 193–211, 10.1039/D2FD00119E.
- M. R. Sakha, P. Halimitabrizi, S. Soltanali, F. Ektefa, Z. Hajjar and D. Salari, RSC Adv., 2023, 13(11), 7514–7523, 10.1039/d3ra00037k.
- M. Al-Samhan, J. Al-Fadhli, A. M. Al-Otaibi and R. Bouresli, Int. J. Chem. Eng., 2023, 2023, 7302409, DOI:10.1155/2023/7302409.
- J. Shen, R. Kortlever, R. Kas, Y. Y. Birdja, O. Diaz-Morales, Y. Kwon, I. Ledezma-Yanez, K. J. Schouten, G. Mul and M. T. Koper, Nat. Commun., 2015, 6, 8177, DOI:10.1038/ncomms9177.
- C. Chen, J. F. Khosrowabadi Kotyk and S. W. Sheehan, Chem, 2018, 4(11), 2571–2586, DOI:10.1016/j.chempr.2018.08.019.
- D. A. Gkika, A. C. Mitropoulos and G. Z. Kyzas, Science of The Total Environment, 2022, 822, 153612, DOI:10.1016/j.scitotenv.2022.153612.
- X. Si, R. Lu, Z. Zhao, X. Yang, F. Wang, H. Jiang, X. Luo, A. Wang, Z. Feng, J. Xu and F. Lu, Nat. Commun., 2022, 13(1), 258, DOI:10.1038/s41467-021-27919-9.
- K. Pobłocki, J. Drzeżdżon, B. Gawdzik and D. Jacewicz, Green Chem., 2022, 24(24), 9402–9427, 10.1039/D2GC03264C.
- Z. Yu, Z. Song, C. Lu, Y. Bai, J. Li, J. Liu, P. Liu and J. Peng, Appl. Organomet. Chem., 2022, 36(6), e6648, DOI:10.1002/aoc.6648.
| This journal is © The Royal Society of Chemistry 2023 |