
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New insights into orthophosphoric acid assisted rapid aqueous processing of NMC622 cathodes†
Julian R.
Tolchard
a,
Per E.
Vullum
 bd,
Bjørnar
Arstad
c and
Nils P.
Wagner
*ae
bd,
Bjørnar
Arstad
c and
Nils P.
Wagner
*ae
aDept. of Sustainable Energy Technology, SINTEF Industry, Trondheim, Norway. E-mail: nils.peter.wagner@sintef.no
bDept. of Materials and Nanotechnology, SINTEF Industry, Trondheim, Norway
cDept. of Process Technology, SINTEF Industry, Oslo, Norway
dDept. of Physics, Norwegian University of Science and Technology, Trondheim, Norway
eDept. of Material Science and Engineering, Norwegian University of Science and Technology, Trondheim, Norway
First published on 15th February 2023
Abstract
The use of orthophosphoric acid to stabilize aqueous processing of high nickel NMC622 electrodes was assessed under rapid processing conditions, in which the active material powder was in suspension for less than 1 hour. It was found, as previously reported, that corrosion of the aluminum current-collector was suppressed, and capacity retention greatly improved relative to aqueous-processed material without orthophosphoric acid addition. The rapid processing conditions led to incomplete reaction of the orthophosphoric acid with the surface of the electrode active material however, with concomitant formation of orthophosphate precipitates upon drying. Reaction of these with organic carbonate electrolytes resulted in rapid cell death typically occurring between 50 and 100 cycles, characterized by formation of a thick degradation film on the NMC622 cathode. Washing with ethanol was found to remove the components responsible for the degradation process, yielding an electrode that demonstrated almost 400 cycles with 78% capacity retention. It is concluded that whilst the addition of orthophosphoric acid aids aqueous processing of NMC materials, close attention must be paid to the quantities and reaction times used.
Sustainability spotlightAqueous Li-ion cathode production is aligned with: SDG7. Affordable and clean energy – LIBs can mitigate issues associated with the intermittency of energy harvesting (e.g., solar cells and wind turbines). Aqueous processing can further decrease the price of cathode processing and hence of LIBs. By replacing the fluorinated cathode binder with a biopolymer, the price and environmental footprint will be further improved. SDG13. Climate action – aqueous cathode processing will mitigate the use and recovery of the harmful organic pyrrolidone solvent used in the LIB industry. Lower energy consuming and more environmentally friendly battery production will contribute to improving our climate. |
Introduction
With electrification of the transport sector being viewed as a key component in combatting climate change, Li-ion battery (LIB) manufacturing is in a period of exponential growth and rapid technological development.1,2 A shift in chemistry towards nickel-rich cathodes is driven by a need for higher energy- and power-densities and a desire to move away from cobalt on ethical and supply grounds.3 Manufacturing is also an energy-intensive process, which negatively impacts the cost and carbon-footprint of electrified transportation. Compounding this is the use of n-methyl-2-pyrrolidone (NMP) as a solvent, which is necessitated by the use of PVDF binders at the cathode and must be recovered and recycled during manufacturing.For graphitic- and LTO-type anodes the use of aqueous processing routes using SBR and Na-CMC is long known and is commercially implemented.4–6 The use of water as a solvent eliminates the need for solvent capture, though it does not eliminate a time-consuming drying step in the electrode manufacture. Wood et al. already reported major cost benefit arising from equipment and infrastructure savings in 2015 and a recent study by Yuan et al. concluded energy savings of up to 43% could be made by using aqueous electrode processing.7,8 Although the use of water-based binders has been shown to be feasible for cathode chemistries such as LiFePO4, its implementation for high energy layered oxide cathodes such as NMC (LiNixMnyCozO2) remains a challenge, as these materials have a tendency to leach transition metals and lithium on contact with water.9,10 As well as negatively impacting available capacity, the formation of LiOH(aq) also raises the pH of the aqueous slurry significantly and destabilizes the passivating oxide layer on the aluminium current collector used for Li-ion cathodes. The amphoteric character of trivalent aluminium causes the passivation layer to be dissolved as Al(OH)4− by the alkaline slurry upon casting, leaving a bare aluminium surface which will subsequently undergo anodic dissolution to evolve hydrogen and cause pitting of the foil and pinholes in the active electrode.10–12 Often overlooked is the fact that aluminium hydroxide, likely in the form of a hydrated lithium aluminate, remains in the cast upon drying. In an assembled LIB this may cause further unwanted side reactions.10 A clear trend towards cathodes with higher Ni content has been evident over the last years, addressing both the need for higher energy density as well as the cost and ethical aspects related to the use of Co.3,13 The pH, and hence the level of Li leaching, is however also directly related to the Ni content of layered oxides. Nonetheless very promising results have been achieved with aqueously processed Nickel-rich layered oxides such as NMC811 (LiNi0.8Mn0.1Co0.1O2).14
The alkaline nature of layered oxides is discussed in literature to be caused by a Li-proton exchange forming LiOH, but also the formation of a rock salt species can generate LiOH on the materials' surface when exposed to water.14–17 A logical means to alleviate aluminium corrosion caused by the high pH is the addition of acids to the slurry formulation to maintain a pH within the passivation window of aluminium,12,18–20 though Bauer et al. highlighted that care has to be taken as the pH as well as the acid additive can have profound effects on the adhesion of the electrode laminate.19 Furthermore, lowering the pH does not address the fundamental problems related to the leaching of lithium and transition metals. Passerini and co-workers described a possible solution to this issue by adding small amounts of orthophosphoric acid to keep the pH within the passivation window of aluminium, between 4.5 and 8.5. Orthophosphoric acid simultaneously hinders excessive transition metal leaching by the formation of a protective nanometre-thick layer of transition metal orthophosphates on the active material via precipitation of leached metal ions.10,21 If successful, such an approach could enable aqueous processing of high energy cathode materials for Li-ion batteries. At the manufacturing level, a good dispersion of the active materials, conductive additives and binder has a major effect on the electrochemical properties of the resulting electrodes.22 Although the mixing is not the most energy demanding process in the production of electrodes, it is time demanding and hence can be seen as a constriction of the coating process.23,24 Continuous slurry processing such as high shear extrusion, have been reported to be capable of reducing the homogenization time by a factor of 6, while also increasing the solid content of the electrode slurry, and hence can have a profound influence on the electrode drying.22,25,26 For non-reactive traditional electrode preparation this is a logical route to cost reductions, but for preparations involving a reactive slurry component, as proposed by Loeffler et al.,21 it adds an important variable to the slurry preparation and casting. In this work then, we report on the fast aqueous processing, with mixing times of 20 minutes, of nickel-rich NMC622 cathode slurries with orthophosphoric acid addition and compare with additive-free aqueous processing and a standard PVDF binder formulation.
Experimental
Electrode and cell fabrication
Commercial LiNi0.6Mn0.2Co0.2O2 (NMC622) was provided by SAFT. The electrode blend consisted of 92 wt% active material, 5 wt% carbon black (Super C65, Imerys) and 3 wt% binder (Na CMC, average Mw ∼ 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Aldrich or PVDF, Kynar). Binder solutions were made by dissolution of CMC in water or PVDF in n-methyl pyrrolidone (NMP), with the carbon black homogenized into these using a mixer mill before the active material was added and homogenized for 20 minutes. In the case of orthophosphoric acid addition, 1 wt% or 0.5 wt% orthophosphoric acid (with respect to the NMC622 amount) was added to the binder solution prior to the addition of the conductive agent and NMC622 powder. Electrodes were formed by casting the slurries onto 25 μm thick aluminium foil (battery grade) using a fixed gap applicator (100 μm wet film deposit), with pre-drying at 60 °C. Final electrode drying was carried out at 90 °C for 15 hours under vacuum. After drying electrodes were roll-pressed to ensure good contact between the active materials, conductive additive and the current collector. A second drying step at 170 °C for 15 hours was performed for some orthophosphoric acid containing samples, to evaluate the thermal stability of secondary phases. Some samples were rinsed in 100% ethanol in order to remove soluble secondary phases. From the dried laminates, circular electrodes were punched out (Ø = 12 and 16 mm) and transferred to an Ar filled glove box (MBraun, O2/H2O < 0.1 ppm) for battery assembly, with additional drying for ∼8 h at 90 °C under vacuum in the glovebox transfer chamber. The typical active material loading of the electrodes was 6 mg cm−2. CR2016 coin cells were assembled in the glove box using a circular Li counter electrode, 40 μL electrolyte (1 M LiPF6 in EC/DEC 1:1, Sigma-Aldrich, battery grade) and a polypropylene separator (Celgard 2400).
000, Aldrich or PVDF, Kynar). Binder solutions were made by dissolution of CMC in water or PVDF in n-methyl pyrrolidone (NMP), with the carbon black homogenized into these using a mixer mill before the active material was added and homogenized for 20 minutes. In the case of orthophosphoric acid addition, 1 wt% or 0.5 wt% orthophosphoric acid (with respect to the NMC622 amount) was added to the binder solution prior to the addition of the conductive agent and NMC622 powder. Electrodes were formed by casting the slurries onto 25 μm thick aluminium foil (battery grade) using a fixed gap applicator (100 μm wet film deposit), with pre-drying at 60 °C. Final electrode drying was carried out at 90 °C for 15 hours under vacuum. After drying electrodes were roll-pressed to ensure good contact between the active materials, conductive additive and the current collector. A second drying step at 170 °C for 15 hours was performed for some orthophosphoric acid containing samples, to evaluate the thermal stability of secondary phases. Some samples were rinsed in 100% ethanol in order to remove soluble secondary phases. From the dried laminates, circular electrodes were punched out (Ø = 12 and 16 mm) and transferred to an Ar filled glove box (MBraun, O2/H2O < 0.1 ppm) for battery assembly, with additional drying for ∼8 h at 90 °C under vacuum in the glovebox transfer chamber. The typical active material loading of the electrodes was 6 mg cm−2. CR2016 coin cells were assembled in the glove box using a circular Li counter electrode, 40 μL electrolyte (1 M LiPF6 in EC/DEC 1:1, Sigma-Aldrich, battery grade) and a polypropylene separator (Celgard 2400).
Electrochemical and physical characterization
Galvanostatic cycling experiments were carried out at 20 °C (±1 °C) using a LANHE CT 2001A battery cycler between voltage limits of 3.0 V and 4.3 V vs. Li/Li+. The cells underwent three slow cycles at 14 mA g−1, before the cyclability of the cells was analysed at 70 mA g−1 in cc-cv (cv until the current dropped to 14 mA g−1) charge mode and cc discharge mode.Electron imaging and X-ray microanalysis of fresh and cycled electrodes was performed using Hitachi S-3400N and FEI Apreo scanning electron microscopes (SEM) equipped with Oxford Instruments Aztec Energy Dispersive X-ray Spectroscopy (EDS) systems. The nature of the orthophosphoric acid residues in the electrodes were further analysed by Scanning Transmission Electron Microscopy (STEM) using a double Cs aberration corrected cold FEG Jeol ARM200FC, operated at 200 kV. This TEM is equipped with a 100 mm2 Centurio EDS detector (covering a solid angle of 0.98 sr) and a GIF Quantum ER for EELS. Cross section lamellae were prepared with a Helios G4 UX focused ion beam (FIB). Protection layers were made of carbon, and the first part of the protection layer was deposited by electron beam assisted deposition to avoid ion beam damage and implantation into the top of the sample. All coarse thinning was performed at 30 kV acceleration voltage for the Ga ions. Final thinning was done at 5 kV and then 2 kV on either side of the lamellae to minimize surface damage. To gain further insights of the chemical nature of the phosphorus species the cast were analysed by solid state nuclear resonance spectroscopy (NMR) and X-ray diffraction (XRD). NMR 31P magic angle spinning (MAS) NMR spectra were collected on a Bruker Avance III 500 MHz WB spectrometer at SINTEF, Oslo (NO), using a 3.2 mm MAS probe at room temperature. The MAS frequency was 15 or 20 kHz. The applied 31P resonance frequency was 202.46 MHz. Single pulse experiments with π/2 pulses were conducted on the samples, with a 5 s recycle delay. 4000 free induction decays (FIDs, number of scans, NS) were accumulated for each spectrum. The spectra were referenced to 85% H3PO4 acid by the substitution method.44 Before Fourier transform of the averaged FIDs zero filling and apodization were applied to improve line shape definitions and signal to noise. The apodization were done by multiplying the FIDs with a decaying exponential window function with a processing line broadening (LB) factor of 250 Hz. All NMR spectra were then adjusted by proper signal phasing and baseline corrections.
X-ray diffraction (XRD) analysis of the electrodes was performed using a Bruker D8 Advance DAVINCI working in Bragg–Brentano (Θ/2Θ) geometry and utilizing Cu-Kα radiation. Crystalline phases were identified via reference to the ICDD PDF4+ database (2021) and Crystallographic Online Database (COD), and Rietveld analysis performed using the Bruker Topas V5 fitting software. The solubility of the phosphate residues in aprotic organic electrolytes was measured via immersion into battery grade Propylene Carbonate (PC) of electrodes incorporating CMC and 1% PA, and dried at 90 °C. PC was chosen to be representative of a Li-ion battery electrolyte as it has a high dielectric constant and is liquid at room temperature. Electrodes (16 mm diameter) were immersed in 5 ml of PC for 1 hour, 24 hours and 168 hours before the PC solution was filtered, diluted with 5% HNO3 and the phosphorous content analysed using an Agilent 8800 Triple Quadrupole Inductively Coupled Mass Spectrometer (ICP-MS) with SPS 4 Autosampler.
Results
Electrochemistry
Fig. 1 and 2 compare the cycling performance of NMC622 electrodes cast using PVDF as binder with equivalent electrodes cast using pure CMC, and CMC with 0.5% and 1% orthophosphoric acid (PA) additions. The cells underwent three slow cycles at 14 mA g−1 before cyclability was analysed at 70 mA g−1. Fig. 1a shows the first cycle voltage evolution for the best tested cells of each composition, with first and second cycle statistics presented in Table 1. It can be seen that the CMC-containing samples demonstrate a higher polarization upon first de-lithiation, particularly the electrodes incorporating PA, which echoes the observations of Bichon et al.27 Similar first charge capacities are observed for cells utilizing the PVDF reference binder and the aqueous-cast CMC binder, with lower initial capacities observed for cells using CMC binder with PA additions. The electrode utilizing aqueous-cast CMC without PA shows the lowest 1st cycle efficiency though, and for the second cycle (Fig. 1b) the polarization of the CMC + 1% PA sample closely resembles that of the reference sample utilizing PVDF as a binder. | ||
| Fig. 1 Voltage-capacity curves showing the first (a) and second (b) cycles of rapid-processed NMC622 electrodes using binders of PVDF, CMC and CMC with orthophosphoric acid. | ||
 | ||
| Fig. 2 Cycling stability at 70 mA g−1 for electrodes utilizing binders of: (a) PVDF and CMC, (b) CMC with 0.5% orthophosphoric acid, and (c) CMC with 1.0% orthophosphoric acid. For PVDF and CMC the values given are an average of 3 cells, for electrodes using orthophosphoric acid additions the data from individual cells are shown. Note the different x axis range for (b). | ||
| Binder | Cycle 1 charge (mA h g−1) | Cycle 1 discharge (mA h g−1) | Cycle 2 reversible (mA h g−1) | Cycle 1 coulombic eff. (%) |
|---|---|---|---|---|
| PVDF | 186 | 168 | 167 | 90 |
| CMC | 188 | 159 | 159 | 85 |
| CMC + 0.5% PA | 179 | 160 | 162 | 89 |
| CMC + 1.0% PA | 180 | 165 | 168 | 92 |
The cycling stability of the four electrode formulations is shown in Fig. 2, with associated coulombic efficiencies given in the ESI (Fig. S1†). It can be seen that the reference sample using PVDF binder demonstrates a stable and linear capacity loss, with a capacity retention of 93.5% over 100 cycles. The sample utilizing CMC without PA addition shows a similar linear capacity loss behaviour, but degrades much faster to retain only 50% of its starting capacity after 100 cycles. The respective average coulombic efficiencies for the samples were 99.5% and 98.7%.
For the CMC-based electrodes, a link is also observed between PA content and the second cycle reversible capacity, with reversible capacity increasing with increasing acid addition.
The electrodes incorporating PA with CMC demonstrate, in contrast, relatively poor and extremely inconsistent cycling behaviour. Fig. 2b and c show the cycling stability of parallel half-cell tests for cells using CMC + PA as electrode binder. All cells are seen to cycle stably initially, though marked differences in the initial reversible capacities are evident, with values lying in the range 150–160 mA h g−1 at a 70 mA g−1 cycling rate. Again, a rough trend is observed with respect to PA content, with the electrode formulation utilizing 0.5% PA showing generally better cycle life than the 1.0% formulation. An extreme level of cell-to-cell scatter in the cycle life is observed for both formulations though, with a stable cycling regime generally evident for several 10's of cycles before the onset of sudden and rapid fading. The coulombic efficiencies (ESI Fig. S1†) for these cells highlight how the use of half-cell testing can mask the presence of secondary degradation reactions, as despite showing stable capacities they also demonstrate considerably lower coulombic efficiencies than the PVDF reference, averaging below 99% in the stable cycling regime with quite a high level of scatter, which is also seen for the pure CMC formulation.
Fig. S2 in the ESI† presents the voltage profiles of the 5th, 25th, 50th and 100th cycles as well as the median discharge voltage of electrodes incorporating PVDF, CMC and CMC + 1% PA. A notable contrast is observed between the electrode using pure CMC and that using CMC and PA additive: The characteristic shape of the NMC capacity vs. voltage response is maintained during the stable cycling regime for the PA-containing electrode, but not for the formulation using pure CMC, despite both displaying similarly poor coulombic efficiencies below 99%. This is also reflected in the median discharge potential behaviour. It is also seen that the median discharge potential of the PVDF-containing electrode falls approx. 90 mV on the transition from 14 mA g−1 formation cycling to 70 mA g−1 cyclability testing, whilst the CMC containing electrodes maintain their potential at the higher current density and maintain a considerably higher median discharge potential during cycling (∼100 mV higher for CMC + 1% PA). It is speculated that the water-based route yields a more homogeneous distribution of the conducting additive, and thus a higher overall electronic conductivity for the as-made electrode.
Following observation of surface precipitates by SEM analysis (discussed below) two further tests were performed. The first was with a CMC + 1% PA electrode sheet dried at a higher temperature of 170 °C, the second was with a CMC + 0.5% PA electrode washed in 100% ethanol to remove soluble residues. The cycling performance of these are respectively shown in the ESI (Fig. S3 and S4†). It can be seen from Fig. S3† that drying at higher temperature did not lead to an improvement in electrochemical performance. A first cycle efficiency of 85% and first cycle discharge capacity of 148 mA h g−1 are substantially worse than the equivalent values obtained with drying at 90 °C. Most notably though, the sample is seen to demonstrate the same rapid degradation phenomenon as the 90 °C dried electrodes. The sample washed with ethanol, in contrast, cycles extremely well (Fig. S4†). A first cycle efficiency of 87% and second cycle discharge capacity of 159 mA h g−1 are comparable with the same CMC + 0.5 wt% electrode without washing, and a capacity at 140 mA g−1 of 137 mA h g−1 is measured. In stark contrast to the equivalent unwashed samples, the best of which only completed 175 cycles before the onset of rapid death, a capacity retention of 78% over 396 cycles is recorded at this rate with a relatively linear degradation. The averaged degradation rate of 5.53% per 100 cycles is better than that observed for the non-aqueous processed electrode using PVDF as a binder. Higher and much more stable coulombic efficiencies (in excess of 99%) for the washed electrode (Fig. S4 vs. S2b†) reflect the lower degradation rate and very different degradation mechanism. The washed electrode also demonstrates much more stable coulombic efficiency values than the untreated CMC sample.
Physical characterization
Fig. 3 compares secondary electron images of the surface of electrodes in the pristine state (images a–c) with the same electrodes after 100 cycles. Major differences in microstructure are clearly evident, with the electrode using pure CMC binder demonstrating serious cracking and pinhole formation. Similar effects are well reported in literature for aqueous processing of cathodes and are a consequence of the reaction of alkaline slurries with the aluminium current collector to form hydrogen gas.11,28 The addition of PA to the slurry suppresses this reaction, resulting in a homogeneous electrode microstructure with few cracks and no pinholes, similar to that observed for the PVDF-based electrode. A rougher surface is observed for the electrode incorporating PVDF, suggesting some possible inhomogeneity in the distribution of carbon additive and active material. | ||
| Fig. 3 Secondary electron images before and after cycling of surfaces of electrodes using binders of PVDF (images (a) and (d)), CMC (images (b) and (e)), and CMC + 1% orthophosphoric acid (images (c) and (f)). | ||
After 100 cycles testing, the PVDF and CMC-based electrodes exhibit similar microstructures as when pristine, with a slight apparent smoothing of the PDVF-containing electrode surface probably caused by compression within the test cell and the formation of a thin Cathode Electrolyte Interphase (CEI) layer.
The cycled electrode incorporating PA, however, exhibits a thick film covering a large portion of the electrode surface. SEM-EDS analysis of this film (Fig. 4) confirms it to be fluorine- and carbon-rich. It is also expected to contain phosphorus and oxygen, but even at the low emission edge energies used here for EDS mapping (7 kV electron beam accelerating voltage) there is considerable interference from the electrode underneath, making this difficult to definitively determine. Also observable in Fig. 4 is that phosphorous is present in “hotspots” in the EDS maps, rather than being evenly distributed across the electrode as might be expected for a surface reaction on the NMC electrode active materials.
 | ||
| Fig. 4 Secondary electron image and associated X-ray emission maps taken after cycling of an electrode incorporating CMC + 1% PA binder. Data were collected at 7 kV electron accelerating voltage. | ||
High resolution electron imaging and mapping of the surface of electrodes incorporating CMC + 1% PA binder are presented in Fig. 5 and S5 (ESI†), for an electrode vacuum-dried at 90 °C. This electrode sheet is that for which cycling data is presented in Fig. 2c. Fig. S6 (ESI†) shows a section of the same electrode cast after vacuum drying at 170 °C.
 | ||
| Fig. 5 Secondary electron image of an electrode incorporating CMC and 1% PA binder, after drying at 90 °C. | ||
The presence of phosphorous rich areas in the low magnification mapping of an electrode dried at 90 °C is explained by the observation of a “fluffy” phosphorus-rich precipitate across the entire surface of the electrode (Fig. 5 and S5†).
Cross-sectional HAADF STEM imaging of this sample (Fig. 6 and S7†) confirms the low density precipitate to be phosphorus- and oxygen-rich, and predominantly located close to the electrode surface. The expected ultra-thin surface phosphate layer on the surface of the NMC active material is also evident via the phosphorus EDS map. No spatial correlation of the phosphorous-rich surface precipitate with transition metal species is observed in the EDS and EELS measurements. Compositional analysis by EELS also shows no measurable levels of lithium in the surface precipitate, though a very low level of sodium, in the range of 1–2 at%, is suggested by EDS. From the low brightness level of the precipitate phase in the HAADF imaging, the presence of a light counter cation is however inferred.
 | ||
| Fig. 6 Cross-section, High Angle Annular Dark Field (HAADF) scanning transmission electron microscopy (STEM) image of the surface of an electrode containing 1% orthophosphoric acid, with associated element maps collected via EDS or EELS (as labelled). | ||
When vacuum-dried at 170 °C this phosphorous-rich phase is seen to change to a dense polyhedral morphology (Fig. S6†), suggesting that the “fluffy” phase melts and crystallizes, possibly also reacting with other components of the electrode. X-ray diffraction data supports this premise, with the observation of a secondary Li3PO4 phase becoming more crystalline after drying at 170 °C, compared to drying at 90 °C (ESI Fig. S8 and S9†). Volume weighted crystallite size measurements for the Li3PO4 phases after 90 °C and 170 °C drying are respectively calculated as 16 nm ± 2 nm and 150 nm ± 35 nm following the Integral Breadth method, with calculated weight fractions between 2 and 3 wt% Li3PO4 for both samples. It is noted that the extremely low phase fraction and correspondingly weak diffraction pattern tends these numbers to have a relatively high associated error. Drying at the higher temperature was also observed to increase the apparent crystallinity of the CMC binder phase, with the primary characteristic diffraction line of CMC becoming visibly sharper and more prominent.
It was determined that the phosphate phases present in the electrodes after drying at 90 °C show a limited solubility in Propylene Carbonate (PC), which can be considered a proxy for typical Li-ion battery electrolytes. Table 2 presents the concentration of dissolved phosphorus obtained from a 16 mm diameter electrode soaked in 5 ml of PC for different times. Dissolution is clearly slow, with a maximum concentration of approx. 639 μg L−1 (0.64 ppm) of phosphorus detected in the PC by ICP-MS after 168 h dissolution time. For reference, full dissolution of the 1% PA included in the electrode formulation would yield a phosphorus concentration of approx. 7 ppm.
| Duration in PC (hours) | Phosphorous concentration (μg L−1) | Relative standard deviation RSD (%) |
|---|---|---|
| 1 | 181.1 | 6.1 |
| 24 | 401.3 | 3.7 |
| 168 | 638.8 | 3.1 |
Solid state 31P MAS NMR measurements of the CMC + 0.5% PA, CMC + 1% PA, and the CMC + 1% PA electrodes after 100 cycles were performed to further investigate the chemical environment and nature of the phosphorous compounds in the samples. The spectra for these are shown in Fig. 7. MAS variations (15 and 20 kHz) showed that the isotropic shift peak is at a chemical shift value of 10 ppm. This is a typical chemical shift value for [PO4]3−, albeit at the high frequency end of the region. The spectra are thus most easily attributed to orthophosphate [PO4]3− moieties, since pyrophosphates [P2O7]4− and polyphosphates typically have an isotropic shift at lower frequencies. Counterions/ligands will change chemical shift values, however, meaning that whilst oligo- and polyphosphates are more unlikely, they cannot be ruled out. After 100 lithium intercalation/de-intercalation cycles the main 31P signal can be still attributed to the same moieties as pre-cycling. However, a shoulder to the main peak is evident at a lower frequency which might be attributable to new orthophosphate compounds formed from the degradation of the electrolyte salt LiPF6, or to an increase in amount of pyrophosphate [P2O7]4− units.
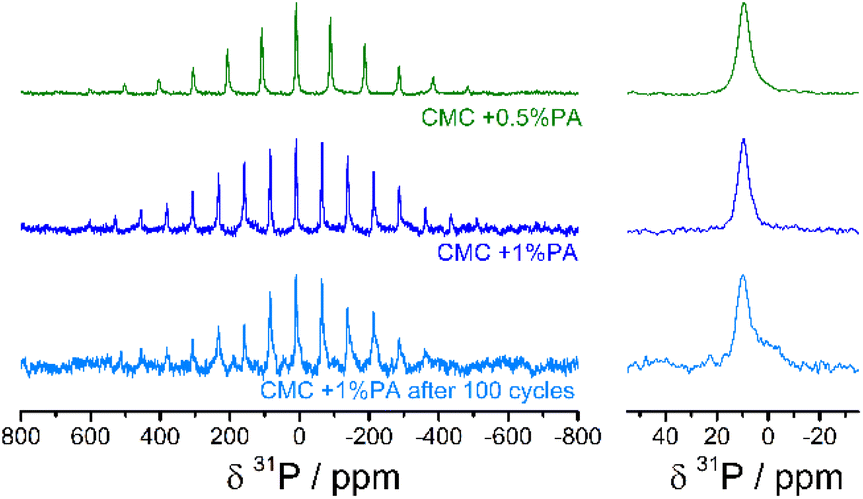 | ||
| Fig. 7 31P MAS NMR spectra of electrodes incorporating (a) CMC + 0.5% PA (top), (b) CMC + 1% PA (middle), and (c) CMC + 1% PA after 100 electrochemical cycles (bottom). The isotropic shift is shown to the right. The peak at the isotropic chemical shift is identified at about 10 ppm, with a shoulder at 0 ppm appearing in the cycled sample. | ||
A noticeable feature of these spectra are the large spinning sideband manifolds. This feature suggests interaction of the P nuclei with paramagnetic centres, since diamagnetic materials with P atoms four-fold coordinated to O-atoms, as in phosphate compounds, typically have minor (or no) spinning sidebands at MAS rates of 15 kHz and above. The paramagnetism will also tend to quench the proton signal, and accordingly the 1H signal was not strong enough to adequately characterise.
Discussion
It is well established that unbuffered aqueous processing of NMC materials results in shifts in slurry pH beyond the stability window of oxide-passivated aluminium driven by leaching of lithium from the active material, and that this occurs to a substantial degree within seconds of exposing NMC to water.14 The resulting high pH slurry causes current collector corrosion, and surface chemistry changes on the NMC which can be expected to impede full re-lithiation and degraded cycling performance.10 If aluminium species cross over to the anode, they can undergo electrochemical reduction followed by the formation of a lithium aluminium alloy, causing further destabilization and side reactions such SEI build up.29–31 This is clearly observed in this work also, with characteristic defects observed in the electrode cast using unbuffered CMC, and the observation of lowered reversible capacity and a generally linear capacity loss with cycle number. It is still not fully understood if Li leaching from layered oxides is accompanied by oxidation of the transition metals, proton intercalation or oxygen loss, and this might also be dependent on the chemical composition of the layered oxide.14–16,32,33 Whether the addition of small amounts of an acid will have a strong influence on the Li leaching is controversial in literature. In an earlier study, Loeffler et al. reported a strong increase in Li concentration when adding 5 or 10 wt% orthophosphoric acid to an aqueous dispersion of NMC 111,21 and Bichon et al. observed a similar behaviour with NMC532.27 A more recent study by Wood et al. found the amount of Li leaching to be less affected by the starting pH of the solution when comparing different NMC compositions dispersed at pH 2, pH 6.6 and pH 12.14 As the current study utilized only half cell testing regimes with an intrinsic excess of lithium however, it cannot be determined to what extent PA promotes leaching of Li from the active material. The higher first cycle efficiencies and second cycle reversible capacities demonstrated here by the electrodes incorporating PA do broadly support the thesis that addition of PA stabilizes and protects the surface and hinders the dissolution of transition metals.21 The observation of stable cycling over nearly 400 cycles with low degradation rates for the electrode washed in alcohol also confirms the efficacy of the approach of using PA to stabilize the NMC surface. An additional overpotential observed both here and by Bichon et al. for the first cycle charging also suggests an initial impeding behaviour that differs from the PVDF based system.In common with the results of Bichon et al., cathodes using an aqueous-processed pure CMC binder demonstrate a steady and essentially linear loss of capacity with cycle number. This was attributed by these authors to aluminium corrosion products formed during electrode processing then reacting further within the cell during cycling to create, amongst other things, a thick SEI on the lithium anode. This is presumed to also occur here. In contrast to their results, however, in the current work the addition of PA in the aqueous slurry mixture does not necessarily result in improved cyclability. A more stable cycling, as most studies report,21,27,34–38 is also observed initially when PA is included in the formulation, but for most electrodes tested in this work it is succeeded by rapid-onset catastrophic cell failure. This occurred anywhere between 50 and 175 cycles for electrodes cut from the same sheet and cycled to the same program, and associates with substantial CEI build-up on the cathode surface. It is confirmed that the rapidly processed electrodes contain a phosphoric acid residue with significant solubility in carbonate electrolyte. This compound is also ethanol-soluble, as washing with absolute alcohol before cycling yielded an electrode with extended cycle life and a degradation rate similar to that reported previously21,27,34 and better than that of an equivalent non-aqueous processed (PVDF-based) electrode. Different degradation mechanisms are then at play in the systems based on pure CMC and those including PA additive. With PA addition, a relatively rapid process is likely occurring between the electrolyte and a residue within the cathode left from processing. The scatter observed in the data relating to cycle life reflects variation in the amount of electrolyte added to the cells, and differing concentrations of the residue arising from drying effects. Without PA addition, aluminium corrosion products likely degrade the cycling performance.
The observations from SEM and TEM confirm the presence of a phosphorous-rich precipitate, solid state NMR suggests orthophosphate moieties, and XRD confirms the presence of Li3PO4 at the surface, but no other crystalline compounds. The calculated weight fractions by XRD are high though, at approx. 2–2.5× those expected if the entire phosphate loading was converted to Li3PO4. Even though these are subject to some error, it is indicated that substantial reaction of phosphate occurs with lithium from the sample, and that this phase is concentrated at the surface. This correlates with the electron microscopy observation of a “fluffy” precipitate that looks to have formed during drying, and the observed nano-morphology for the primary particles in this are broadly in agreement with the sub-20 nm crystallite size calculated by XRD. The XRD and SEM also agree with respect to the observation of increased crystallinity with higher temperature processing. The exact nature of the precipitate is still not completely clear however, as EELS measurements do not detect any lithium content in this phase and the melting point of Li3PO4 (1205 °C) is not consistent with the substantial change in morphology observed when drying at 170 °C rather than 90 °C. Furthermore, the dramatic improvement in cycle life observed for an ethanol-washed electrode is not consistent with the idea that Li3PO4 is the sole impurity: As with other metal phosphates that may precipitate at the NMC surface, it has extremely limited solubility in water (0.04 g/100 ml) and would be expected to have similarly low solubility in other polar protic solvents such as ethanol. Li3PO4 is also known as a solid electrolyte in Li-ion batteries, and other reports of Li3PO4 coating on high nickel NMC indicate it to be performance-enhancing, not performance degrading.37 It is then concluded that an additional residue, likely a hydrogen phosphate of some form, is present after aqueous processing in a form that is not easily detectable by the techniques employed here. The paramagnetic quenching of the 1H NMR signal is particularly unfortunate in this respect. A simple explanation is the presence of not fully reacted H3PO4, but this is thought unlikely as H3PO4 has a melting point ∼ 40 °C and would then be expected to be dense and highly crystalline after electrode drying at 90 °C, and thus easily detected by the techniques applied here. Partial reaction of such a residue with electrode components during the drying step is not ruled out however.
A question also remains with respect to the differing results between this work and that previously reported. There are notable differences with respect to processing which may explain this. Although similar additive concentrations of PA are used, in this work the electrode slurry was prepared and processed rapidly, in line with commercial trends. So that the “wet” time for the powder from slurry making to cast drying is estimated at approximately 45 minutes, whereas Bichon et al.27 estimate a wet time of approx. 3 hours, based on 1 hour mixing plus powder filtering/drying, and Loeffler et al.21 quote 2 hours mixing plus casting and drying. Neither of these report the sudden failure characteristics observed in this work. Loeffler et al. do report a strong time dependence for pH with time in acidified slurries, which suggests a time dependency of metal phosphate formation and qualitatively supports the hypothesis that the short reaction times employed in this study may have resulted in unreacted residues from the PA that contain protons which in turn can interact with either the LiPF6 salt or the ethylene carbonate (EC) electrolyte component, and explain the exacerbated electrolyte degradation on the cathode surface.39–43 These phosphate residues can however be washed away easily with ethanol, indicating that long reaction times with PA may not be necessary to stabilize NMC622 in this manner.
Conclusions
The effect of using low levels of phosphoric acid additions to stabilize the aqueous processing of NMC type cathodes has been evaluated from the standpoint of rapid electrode processing. It is found that in common with previous studies utilizing long reaction times, slurry processing with a short “wet” time of ∼45 min yields a protective phosphate layer on the active material, and suppresses corrosion reactions on the cathode current collector. In contrast to previous studies, however, it was found that rapid processing may result in incomplete reaction of the acidic additive and the consequent formation of reactive orthophosphate residues which are soluble in organic carbonate electrolytes and cause lower coulombic efficiency, accelerated degradation, excessive CEI build-up and result in rapid and sudden cell death. These residues could not be fully characterized by the techniques available, but were found to be easily removed by washing with ethanol, which yielded an aqueous processed cathode with a capacity retention of 78% over 396 cycles. It is then concluded that the use of phosphoric acid as an additive remains a promising route to aqueous processing of high nickel NMC materials, but that care must be taken to ensure that processing conditions and slurry stoichiometry are tailored to avoid the formation of performance-degrading by-products.Author contributions
Conceptualization: N. P. Wagner; supervision and project administration: N. P. Wagner; investigation and formal analysis: all co-authors; writing – original draft: J. R. Tolchard and N. P. Wagner. Writing – review and editing: all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Marianne Kjos and Pablo J Lebed for performing the ICP-MS analyses, and Dr Artur Tron for assistance with cathode laminate processing and cell assembly. The work was performed within MoZEES, a Norwegian Centre for Environment friendly Energy Research (FME), co-sponsored by the Research Council of Norway (project number 257653) and 40 partners from research, industry, and the public sector. Furthermore, the authors gratefully acknowledge the Research Council of Norway for the provision of strategic funding via the strategic institute project Oxipath through the basic grant received by SINTEF Industry. The TEM work was carried out on NORTEM infrastructure, Grant 197405, TEM Gemini Centre, Norwegian University of Science and Technology (NTNU), Norway. Support from NorFab [245963/F50] is gratefully acknowledged.References
- S. Dühnen, J. Betz, M. Kolek, R. Schmuch, M. Winter and T. Placke, Small Methods, 2020, 4, 2000039, DOI:10.1002/smtd.202000039.
- R. Zhang and S. Fujimori, Environ. Res. Lett., 2020, 15, 034019, DOI:10.1088/1748-9326/ab6658.
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278, DOI:10.1038/s41560-018-0107-2.
- D. Bresser, D. Buchholz, A. Moretti, A. Varzi and S. Passerini, Energy Environ. Sci., 2018, 11, 3096–3127, 10.1039/C8EE00640G.
- J. Drofenik, M. Gaberscek, R. Dominko, F. W. Poulsen, M. Mogensen, S. Pejovnik and J. Jamnik, Electrochim. Acta, 2003, 48, 883–889, DOI:10.1016/S0013-4686(02)00784-3.
- J.-H. Lee, S. Lee, U. Paik and Y.-M. Choi, J. Power Sources, 2005, 147, 249–255, DOI:10.1016/j.jpowsour.2005.01.022.
- D. L Wood, J. Li and C. Daniel, J. Power Sources, 2015, 275, 234–242, DOI:10.1016/j.jpowsour.2014.11.019.
- C. Yuan, H. Cao, K. Shen, Y. Deng, D. Zeng, Y. Dong and M. Hauschild, CIRP Ann., 2021, 70, 25–28, DOI:10.1016/j.cirp.2021.04.038.
- W. Porcher, P. Moreau, B. Lestriez, S. Jouanneau and D. Guyomard, Electrochem. Solid-State Lett., 2008, 11, A4, DOI:10.1149/1.2795833.
- I. Doberdò, N. Löffler, N. Laszczynski, D. Cericola, N. Penazzi, S. Bodoardo, G.-T. Kim and S. Passerini, J. Power Sources, 2014, 248, 1000–1006, DOI:10.1016/j.jpowsour.2013.10.039.
- K. Ishii, R. Ozaki, K. Kaneko, H. Fukushima and M. Masuda, Corros. Sci., 2007, 49, 2581–2601, DOI:10.1016/j.corsci.2006.12.015.
- C.-C. Li, J.-T. Lee, Y.-L. Tung and C.-R. Yang, J. Mater. Sci., 2007, 42, 5773–5777, DOI:10.1007/s10853-006-1172-7.
- Y. Ding, Z. P. Cano, A. Yu, J. Lu and Z. Chen, Electrochem. Energy Rev., 2019, 2, 1–28, DOI:10.1007/s41918-018-0022-z.
- M. Wood, J. Li, R. E. Ruther, Z. Du, E. C. Self, H. M. Meyer, C. Daniel, I. Belharouak and D. L. Wood, Energy Storage Mater., 2020, 24, 188–197, DOI:10.1016/j.ensm.2019.08.020.
- X. J. Zhu, H. H. Chen, H. Zhan, D. L. Yang and Y. H. Zhou, J. Mater. Sci., 2005, 40, 2995–2997, DOI:10.1007/s10853-005-2392-y.
- N. V. Faenza, L. Bruce, Z. W. Lebens-Higgins, I. Plitz, N. Pereira, L. F. J Piper and G. G. Amatucci, J. Electrochem. Soc., 2017, 164, A3727, DOI:10.1149/2.0921714jes.
- R. Jung, R. Morasch, P. Karayaylali, K. Phillips, F. Maglia, C. Stinner, Y. Shao-Horn and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, A132, DOI:10.1149/2.0401802jes.
- S. F. Lux, F. Schappacher, A. Balducci, S. Passerini and M. Winter, J. Electrochem. Soc., 2010, 157, A320, DOI:10.1149/1.3291976.
- W. Bauer, F. A. Çetinel, M. Müller and U. Kaufmann, Electrochim. Acta, 2019, 317, 112–119, DOI:10.1016/j.electacta.2019.05.141.
- P. Zhu, J. Han and W. Pfleging, Nanomaterials, 2021, 11, 1840, DOI:10.3390/nano11071840.
- N. Loeffler, G.-T. Kim, F. Mueller, T. Diemant, J.-K. Kim, R. J. Behm and S. Passerini, ChemSusChem, 2016, 9, 1112–1117, DOI:10.1002/cssc.201600353.
- H. Dreger, H. Bockholt, W. Haselrieder and A. Kwade, J. Electron. Mater., 2015, 44, 4434–4443, DOI:10.1007/s11664-015-3981-4.
- J.-H. Schünemann, H. Dreger, H. Bockholt and A. Kwade, ECS Trans., 2016, 73, 153–159, DOI:10.1149/07301.0153ecst.
- A. Kwade, W. Haselrieder, R. Leithoff, A. Modlinger, F. Dietrich and K. Droeder, Nat. Energy, 2018, 3, 290–300, DOI:10.1038/s41560-018-0130-3.
- M. Haarmann, W. Haselrieder and A. Kwade, Energy Technol., 2020, 8, 1801169, DOI:10.1002/ente.201801169.
- J. Seeba, S. Reuber, C. Heubner, A. Müller-Köhn, M. Wolter and A. Michaelis, Chem. Eng. J., 2020, 402, 125551, DOI:10.1016/j.cej.2020.125551.
- M. Bichon, D. Sotta, N. Dupré, E. De Vito, A. Boulineau, W. Porcher and B. Lestriez, ACS Appl. Mater. Interfaces, 2019, 11, 18331–18341, DOI:10.1021/acsami.9b00999.
- Z. Szklarska, -Smialowska, 1999, 41, 1743–1767, DOI:10.1016/S0010-938X(99)00012-8.
- H. Li, T. Yamaguchi, S. Matsumoto, H. Hoshikawa, T. Kumagai, N. L. Okamoto and T. Ichitsubo, Nat. Commun., 2020, 11, 1584, DOI:10.1038/s41467-020-15452-0.
- H. Wang, H. Tan, X. Luo, H. Wang, T. Ma, M. Lv, X. Song, S. Jin, X. Chang and X. Li, J. Mater. Chem. A, 2020, 8, 25649–25662, 10.1039/D0TA09762D.
- X. Chang, Z. Xie, Z. Liu, X. Zheng, J. Zheng and X. Li, Energy Storage Mater., 2020, 25, 93–99, DOI:10.1016/j.ensm.2019.10.027.
- X. Zhang, W. J. Jiang, X. P. Zhu, A. Mauger, Qilu and C. M. Julien, J. Power Sources, 2011, 196, 5102–5108, DOI:10.1016/j.jpowsour.2011.02.009.
- I. A. Shkrob, J. A. Gilbert, P. J. Phillips, R. Klie, R. Haasch, J. Bareño and D. P. Abraham, J. Electrochem. Soc., 2017, 164, A1489, DOI:10.1149/2.0861707jes.
- Z. Chen, G.-T. Kim, D. Chao, N. Loeffler, M. Copley, J. Lin, Z. Shen and S. Passerini, J. Power Sources, 2017, 372, 180–187, DOI:10.1016/j.jpowsour.2017.10.074.
- M. Kuenzel, D. Bresser, T. Diemant, D. V. Carvalho, G.-T. Kim, R. J. Behm and S. Passerini, ChemSusChem, 2018, 11, 562–573, DOI:10.1002/cssc.201702021.
- M. Kuenzel, R. Porhiel, D. Bresser, J. Asenbauer, P. Axmann, M. Wohlfahrt-Mehrens and S. Passerini, Batteries Supercaps, 2020, 3, 155–164, DOI:10.1002/batt.201900140.
- C.-H. Jo, D.-H. Cho, H.-J. Noh, H. Yashiro, Y.-K. Sun and S. T. Myung, Nano Res., 2015, 8, 1464–1479, DOI:10.1007/s12274-014-0631-8.
- A. Kazzazi, D. Bresser, A. Birrozzi, J. von Zamory, M. Hekmatfar and S. Passerini, ACS Appl. Mater. Interfaces, 2018, 10, 17214–17222, DOI:10.1021/acsami.8b03657.
- U. Heider, R. Oesten and M. Jungnitz, J. Power Sources, 1999, 81–82, 119–122, DOI:10.1016/S0378-7753(99)00142-1.
- P. G. Kitz, P. Novák and E. J. Berg, ACS Appl. Mater. Interfaces, 2020, 12, 15934–15942, DOI:10.1021/acsami.0c01642.
- K. Edström, T. Gustafsson and J. O. Thomas, Electrochim. Acta, 2004, 50, 397–403, DOI:10.1016/j.electacta.2004.03.049.
- J. Kalhoff, G. G. Eshetu, D. Bresser and S. Passerini, ChemSusChem, 2015, 8, 2154–2175, DOI:10.1002/cssc.201500284.
- J. L. Tebbe, T. F. Fuerst and C. B. Musgrave, ACS Appl. Mater. Interfaces, 2016, 8, 26664–26674, DOI:10.1021/acsami.6b06157.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Solid State Nucl. Magn. Reson., 2002, 22, 458–483, DOI:10.1006/snmr.2002.0063.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00031a |
| This journal is © The Royal Society of Chemistry 2023 |