
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of solvent on pyrolysis-assisted catalytic hydrogenolysis of softwood lignin for high-yield production of monomers and phenols, as studied using coniferyl alcohol as a major primary pyrolysis product†
Jiaqi
Wang
 ,
Eiji
Minami
and
Haruo
Kawamoto
*
,
Eiji
Minami
and
Haruo
Kawamoto
*
Department of Socio-Environmental Energy Science, Graduate School of Energy Science, Kyoto University, Yoshida-honmachi, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: kawamoto@energy.kyoto-u.ac.jp
First published on 16th June 2023
Abstract
Pyrolysis-assisted catalytic hydrogenolysis over Pd/C in anisole (phenyl methyl ether) at relatively high temperatures (>300 °C) can convert softwood lignin into aromatic monomers in >60 mol% yield (based on lignin aromatic rings). In this process, lignin is pyrolytically degraded to soluble intermediates prior to catalytic conversion, therefore the pyrolysis stage plays an important role in determining the yield and monomer composition. In this study, pyrolysis-assisted hydrogenolysis of coniferyl alcohol, which is a major pyrolysis product, and milled wood lignin isolated from Japanese cedar was investigated in various solvents, including water, methanol, toluene, hexane, and anisole, to clarify the solvent effects. The effects of the solvent on undesired side reactions were also explored. The results show that anisole is the best solvent for aromatic monomer production, but hexane is the best solvent for phenol production via demethoxylation. These findings provide insights that will facilitate the development of efficient methods for monomer production from lignin.
Sustainability spotlightLignin is an aromatic component of wood and other lignocellulosic biomass that is abundant and does not compete with food. Accordingly, the use of lignin for aromatic chemicals to replace petroleum-based chemicals is highly desirable and in line with UN Sustainable Development Goal 9 (industry, innovation and infrastructure), Goal 11 (sustainable cities and communities), Goal 12 (responsible consumption and production) and Goal 13 (climate action). The challenge is how to convert lignin polymer into monoaromatic compounds. We have proposed “pyrolysis-assisted catalytic conversion” as a potential method for this purpose. The present paper provides insight into the solvent selection in this process by reporting the solvent-characteristic effects for various protic, aprotic, and hydrophobic solvents. |
Introduction
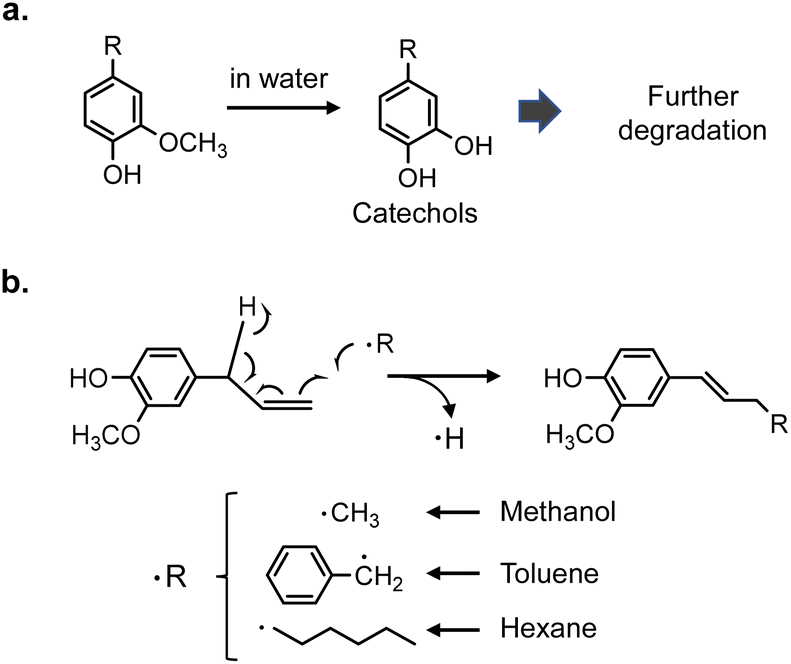
Lignin is an aromatic polymer composed of phenylpropanes and it accounts for 20–35% of lignocellulosic biomasses such as wood and herbaceous plants. Accordingly, lignin has attracted attention as a renewable resource that can replace petroleum-derived aromatic chemicals. The development of efficient methods for converting lignin to aromatic monomers in high yields and with high selectivity is therefore important. Catalytic hydrogenolysis is a potential method.Most catalytic hydrogenolysis approaches have been investigated at relatively low temperatures, namely 200 °C or lower, to suppress undesired benzene-ring saturation.1 However, the monomer yields are usually low, especially below 20% for softwood lignin. Our previous study2 showed that the yield of monomers from softwood lignin can be increased to over 60% by catalytic hydrogenolysis at higher temperatures, preferably >300 °C, in the aprotic solvent anisole (phenyl methyl ether). In this process,2,3 pyrolytic degradation of insoluble lignin to soluble intermediates occurs first, therefore subsequent catalytic reactions are efficient (Fig. 1). In addition, thermally stable 4-O-5 and condensed C–C linkages are catalytically cleaved at such high temperatures. Formation of α-aryl bonds by condensation via a quinone methide (QM) intermediate, which occurs during pyrolysis and pulping processes, suppresses monomer production. α-Aryl bonds are efficiently cleaved by pyrolysis-assisted catalytic hydrogenolysis, therefore this method is also effective for the organosolv lignin which is a technical lignin. Undesired aromatic-ring saturation is also effectively suppressed at such high temperatures.
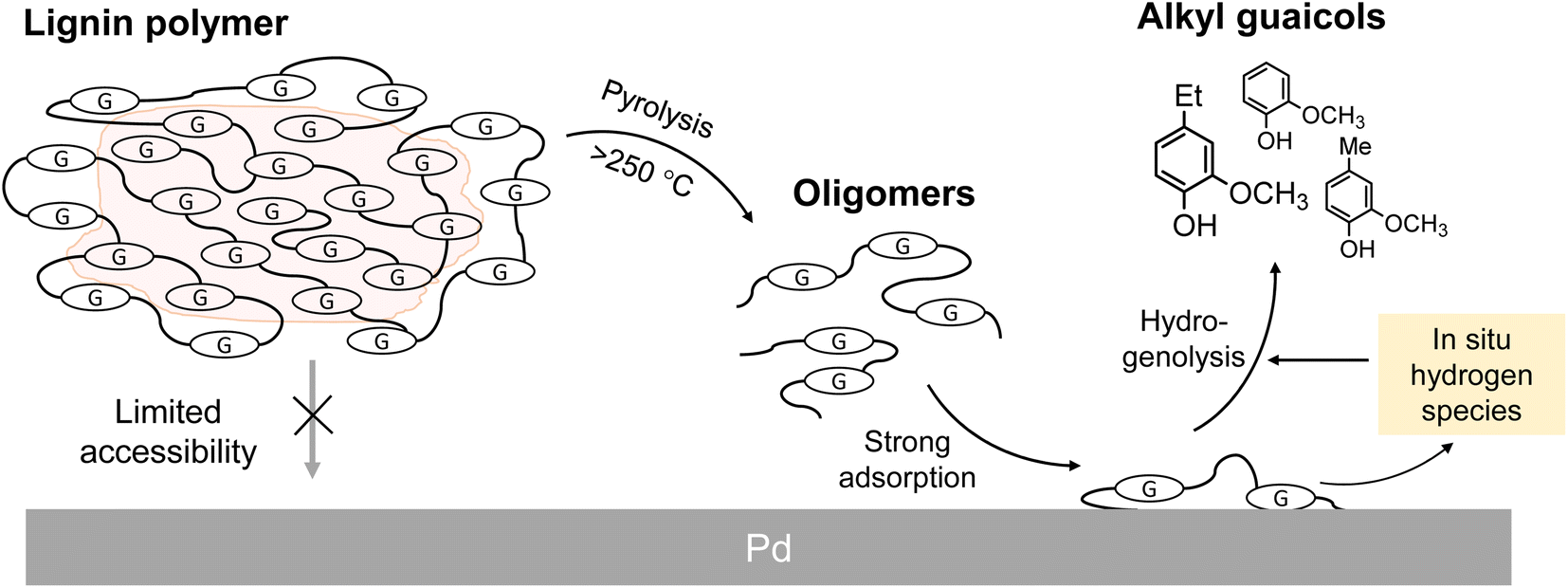 | ||
| Fig. 1 Proposed scheme for production of monomers from lignin polymers under pyrolysis-assisted catalytic hydrogenolysis conditions. G: guaiacyl units. | ||
We previously reported that the composition of lignin-derived monomers depends on the chemical structures of the intermediates formed by pyrolysis.3 However, the solvent type may affect the pyrolysis and subsequent catalytic hydrogenolysis of lignin. Various studies have investigated the yields and compositions of monomers in the high-temperature pyrolysis of softwood lignin in water,4 methanol,5 dioxane,6 and 1,3-diphenoxybenzene7 under catalyst-free conditions. To the best of our knowledge, there have been no systematic studies of solvent effects in the pyrolytic conversion of lignin to monomers, dimers, and oligomers, and their subsequent catalytic conversion.
In conventional catalytic hydrogenolysis approaches, various solvents, e.g., methanol,8,9 dioxane,10 water,11,12 and their binary mixtures,13–16 have been used, but there are only a few reports on the effects of the solvent. Schutyser et al.17 compared the monomer yields in the catalytic hydrogenolysis of birchwood lignin in hexane, dioxane, tetrahydrofuran, alcohols, or water at 200 °C. The monomer yield was solvent dependent and varied from 1.8% in hexane to 43.8% in water. However, the temperature, i.e., 200 °C, was too low for effective lignin pyrolysis. Héroguel et al.18 conducted the hydrogenolysis of aldehyde-stabilized beechwood lignin in isooctane at 250 °C. The major products were cyclohexanes and cyclohexanones, and the total yield was only 13.0%.
On the other hand, in the temperature range above 300 °C, where the pyrolysis of lignin proceeds efficiently, solvent effects are expected to be different from those previously reported, which, with a few exceptions, have been investigated in the temperature range below 250 °C. Some research has been conducted at temperatures higher than 300 °C in supercritical alcohols,19,20 but high-yield aromatic monomer production has not been achieved. Therefore, in the present study, the solvent effects on the pyrolysis-assisted catalytic hydrogenolysis of milled wood lignin (MWL) isolated from Japanese cedar (Cryptomeria japonica) wood were studied by performing the reaction with Pd/C in various solvents at 350 °C. Lignin is rapidly degraded at 350 °C,21 therefore this temperature was used in all experiments. Model compound studies using coniferyl alcohol (CA) were conducted under pyrolytic (no Pd/C in N2) and catalytic (Pd/C in N2 or H2) conditions because CA is an important primary pyrolysis product, and is formed by cleavage of the most abundant linkage in lignin, i.e., the β-ether bond.22 The MWL results were compared with the CA data. The solvents used are protic (water and methanol), aprotic (anisole, toluene, and 1,4-dioxane), and hydrophobic (hexane).
Results and discussion
Pyrolysis of coniferyl alcohol (CA)
CA, which is a major primary product of softwood lignin pyrolysis, was heated with a solvent at 350 °C for 60 min in a closed batch-reaction vessel under pyrolytic (no Pd/C in N2) and catalytic (Pd/C in N2 or H2) conditions. The amount of H2 (3 mL) introduced into the vessel at 0.1 MPa corresponds to 2.4 times the number of moles of CA under the catalytic hydrogenolysis conditions. GPC profiles of the reaction mixtures are shown in Fig. 2. The total yields and compositions of the identified monomers are summarized in Fig. 3 and 4, respectively.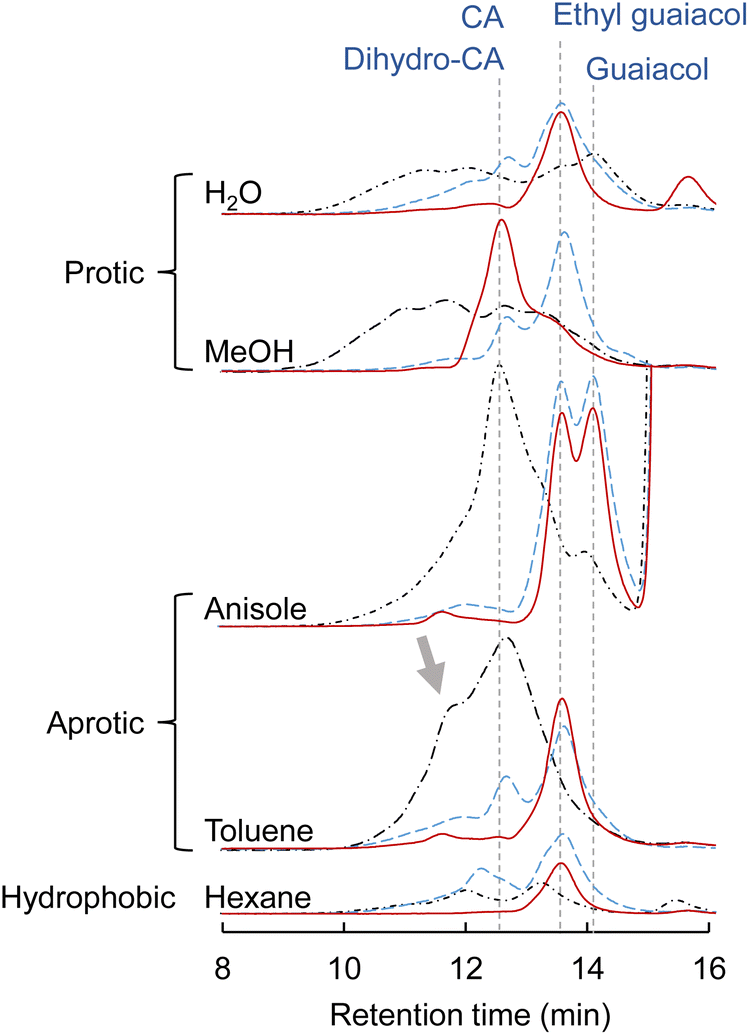 | ||
| Fig. 2 Gel-permeation chromatograms of reaction mixtures obtained from coniferyl alcohol (CA) after treatment at 350 °C for 1 h in different solvents. Dotted and dashed black lines: N2; dashed blue line: Pd/C, N2; solid red line: Pd/C, H2. The retention time indicated by the dashed line is the retention time of the signal peak when CA, dihydro-CA, guaiacol, or ethyl guaiacol was analyzed separately. | ||
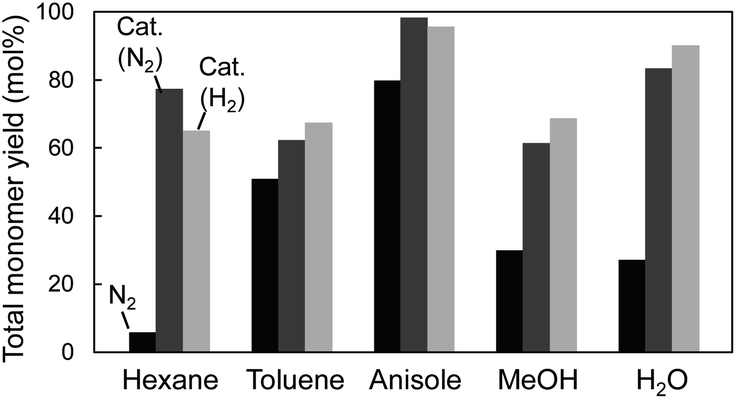 | ||
| Fig. 3 Total monomer yields from coniferyl alcohol after treatment at 350 °C for 1 h in different solvents. Black column: N2; dark-gray column: Pd/C, N2; light-gray column: Pd/C, H2. | ||
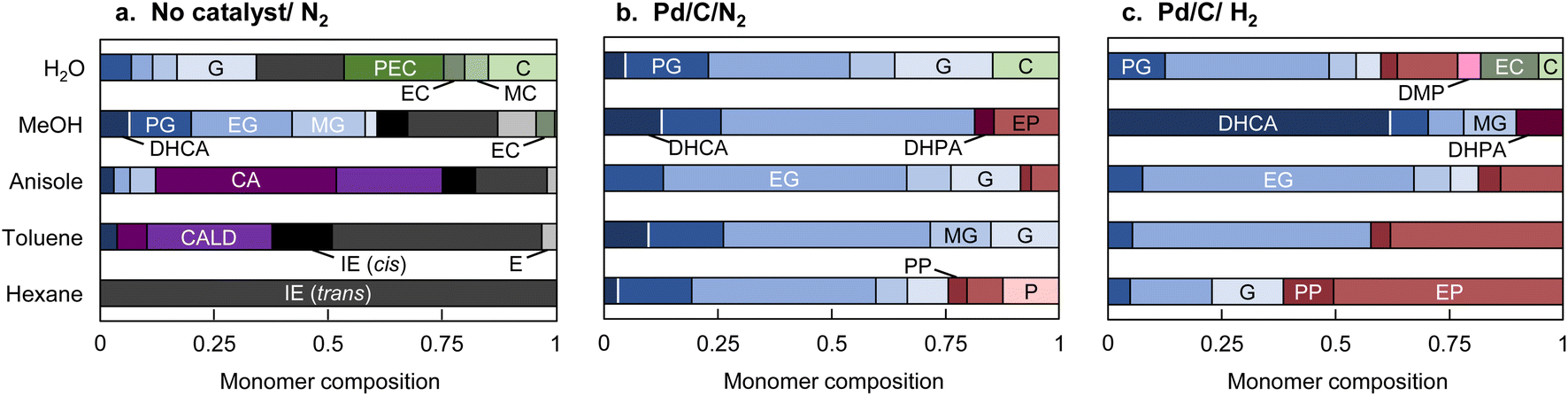 | ||
| Fig. 4 Monomer compositions of products obtained from coniferyl alcohol after treatment at 350 °C for 1 h in different solvents. (a): N2; (b): Pd/C, N2; and (c): Pd/C, H2. DHCA: dihydroconiferyl alcohol, PG: 4-propyl guaiacol, EG: 4-ethyl guaiacol, MG: 4-methyl guaiacol, G: guaiacol, CA: coniferyl alcohol, CALD: coniferyl aldehyde, IE (cis): isoeugenol (cis), IE (trans): isoeugenol (trans), E: eugenol, PEC: 4-propenyl catechol, EC: 4-ethyl catechol, MC: 4-methyl catechol, C: catechol, DHPA: p-coumaryl alcohol, PP: 4-propyl phenol, EP: 4-ethyl phenol, MP: 4-methyl phenol, P: phenol, o-MP: 2-methyl phenol, m-MP: 3-methyl phenol, DMP: dimethyl phenol. | ||
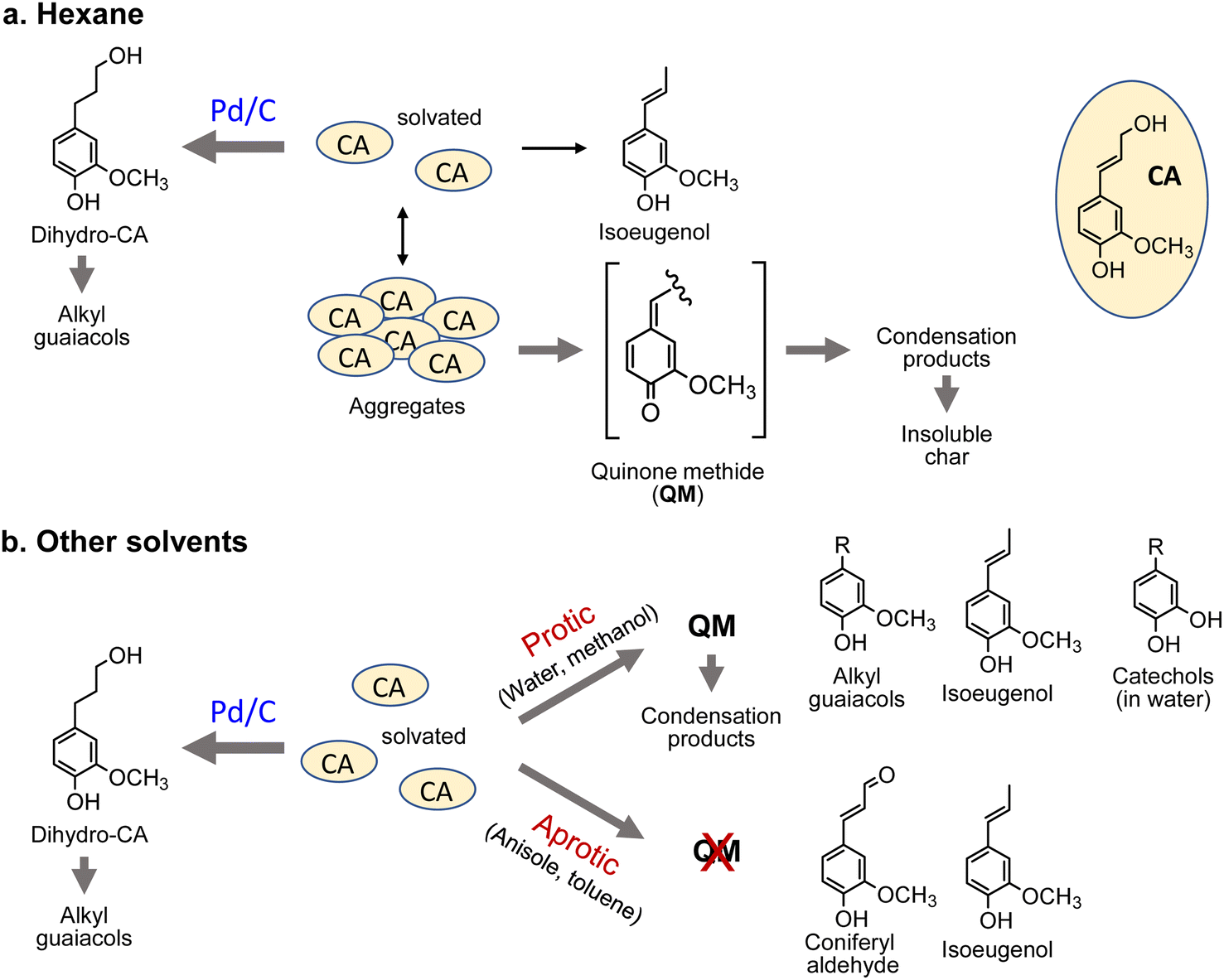
Under the pyrolysis conditions (no Pd/C in N2), the product compositions varied greatly with solvent type. In the GPC profiles (Fig. 2), broad signals at retention times shorter than that of CA were clearly observed when water or methanol was used as the solvent. This indicates that CA tended to condense in these protic solvents, and this resulted in relatively low monomer yields (27.2 mol% for water and 30.0 mol% for methanol; Fig. 3). Fig. 5 shows that QM intermediates would be produced from CA as condensation precursors in water and methanol, in which proton transfer from phenolic OH to Cβ or Cγ–oxygen,23 which is required for QM formation, occurs by solvation with these protic solvents.
 | ||
| Fig. 5 Proposed solvent-dependent conversion pathways for coniferyl alcohol (CA) at 350 °C. | ||
Kotake et al.7 found that QM formation from CA was effectively suppressed in diphenoxybenzene, an aprotic solvent. When CA or lignin was pyrolyzed in this solvent, condensation was effectively inhibited, which increased the monomer yield. In aprotic solvents, the proton transfer required for QM formation is inhibited by solvation. The GPC profiles of the reaction mixtures obtained in anisole and toluene can be explained by this inhibitory effect. This is supported by the higher monomer yields obtained with anisole (79.8 mol%) and toluene (50.9 mol%) than with water and methanol. The monomer yield in toluene was lower than that in anisole because of coupling of radical species formed from monomers with those from toluene, as explained below. The radical-coupling products would be observed as a shoulder at 11.7 min in the GPC profile (indicated by an arrow in Fig. 2).
For hexane, the GPC signals were comparatively small, and the monomer yield was only 5.9 mol%. A black residue (char) was generated in the pyrolysates only when hexane was used as the solvent. This indicates that CA tended to condense and was converted into solid carbonized substances. As shown in Fig. 5, the solubility of CA in hydrophobic hexane is limited. Consequently, CA molecules aggregate, and their condensation via QM intermediates can occur, as the proton transfer required for QM formation is enabled by the interactions of CA molecules,7 similar to the action of protic solvents. Only a small portion of CA was dissolved in hexane and converted to monomeric products.
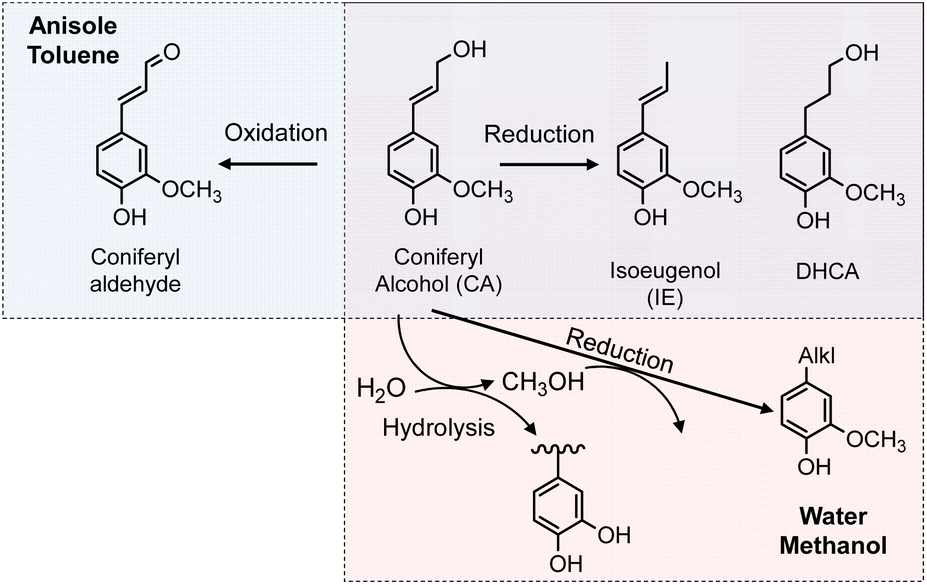
The chemical structures of the monomers also depended on the solvent type under the pyrolysis conditions (Fig. 4a). The CA side chain can be transformed into oxidized and reduced structures,23 therefore redox-type reactions occur during pyrolysis. The monomer compositions obtained in anisole and toluene can be explained by redox-type reactions (Fig. 6). Some CA was oxidized to coniferyl aldehyde, but some was reduced to isoeugenol, eugenol, and DHCA. In hexane, the only detected monomer was isoeugenol, and the yield was only 5.9 mol%. The reason for this is currently unknown, but is probably related to the low solubility of CA and CA-derived products in hydrophobic hexane. Polar products other than isoeugenol would aggregate and be converted to char, as described above.
 | ||
| Fig. 6 Proposed effects of solvents on redox reactions of coniferyl alcohol during pyrolysis. | ||
The monomer compositions obtained in water and methanol differed from those obtained in other solvents. Hydrolysis products (catechol, and methyl, ethyl, and propenyl catechol) and a methanolysis product (methyl catechol) were detected in water and methanol in 46.7 and 4.6 mol% yields, respectively.
No CA oxidation products were detected in water and methanol. All the detectable monomers were reduction products, namely isoeugenol, eugenol, DHCA, and alkyl guaiacols (Fig. 4a). The yields of alkyl guaiacols were 51.6 mol% in methanol, 16.8 mol% in water, and 9.1 mol% in anisole. These results suggest that methanol has the ability to reduce CA and other intermediates (Fig. 6). In our previous study,3 the hydroxypropyl side chain of DHCA was converted to an ethyl group via deformylation, with release of reactive hydrogens, which were used for catalytic hydrogenolysis. This suggests that methanol could be converted to formaldehyde and reactive hydrogens. The formaldehyde could be further degraded to CO and reactive hydrogens. Further study is necessary to confirm this proposal. The methoxyl groups of the guaiacyl units are hydrolyzed in water to form methanol, although it is not certain whether water has a reducing ability. Anisole has a methyl ether structure, and could serve as a source of methanol, but there is no evidence of methanol formation from anisole.
Catalytic hydrogenolysis of coniferyl alcohol (CA)
When Pd/C was used, the monomer compositions were similar regardless of the solvent type (Fig. 4b and c). Ethyl guaiacol was the major component along with other hydrogenated products (DHCA, guaiacol, and methyl and propyl guaiacol). The reaction atmosphere (N2 or H2) had little effect on the monomer composition, with a few exceptions (DHCA in methanol, phenols, and propyl guaiacol). These exceptions will be discussed below. These observations support our previous proposal that H2 is not required because reactive hydrogen species are generated in situ.The pathways for transformation of the pyrolysis products in Fig. 4a to guaiacol, and methyl, ethyl, and propyl guaiacol were reported in a previous paper3 (isoeugenol and eugenol → propyl guaiacol, coniferyl aldehyde and DHCA → ethyl guaiacol). However, saturated alkyl groups are stable under the present reaction conditions, therefore the monomer compositions can be mostly explained by catalytic conversion of the pyrolysis products in Fig. 4a.
The monomer yields in water, methanol, and hexane significantly improved on addition of Pd/C (27.2 mol% → 83.5 mol% for water, 30.0 mol% → 61.5 mol% for methanol, and 5.9 mol% → 77.4 mol% for hexane). These results can be explained by competition between pyrolytic condensation and catalytic conversion. Catalytic conversion is more efficient than pyrolytic condensation. This is supported by the GPC profiles obtained in water and methanol (Fig. 2), which show almost no condensation product signal in the presence of Pd/C. The significant improvement in monomer yield in hexane is worth noting. Although hexane is a poor solvent for CA, the Pd/C-mediated catalytic transformation proceeded efficiently. The monomer yields in anisole and toluene did not change significantly. This is because of effective inhibition of pyrolytic condensation in these aromatic (aprotic) solvents.
Changing the reaction atmosphere from N2 to H2 tended to decrease the contribution of propyl guaiacol. This can also be explained in terms of competition between pyrolysis and catalytic conversion. As mentioned above, propyl guaiacol is mainly produced from the pyrolysis products isoeugenol and eugenol, whereas CA is directly converted to ethyl guaiacol via DHCA via the catalytic process. The contributions of propyl guaiacol decreased in all solvents when the atmosphere was changed from N2 to H2. This suggests that the reaction conditions favor catalytic hydrogenolysis.
As reported in the literature, the conversion of guaiacol to phenol via demethoxylation is more efficient under H2 than under N2.3 The conversion rates from guaiacols to phenols were inversely related to the solvent polarity, except in water [phenols/guaiacols molar ratio: 1.6 (hexane) > 0.7 (toluene) > 0.2 (anisole) > 0.1 (methanol), Fig. 4c]. In water, phenols were produced more efficiently than in methanol, presumably via catechol formed by hydrolysis. In a model experiment using guaiacol or catechol under similar reaction conditions in H2 (Fig. S1†), catechol was converted to phenol in 67 mol% yield. Phenol was obtained from guaiacol in 36.7 mol% yield, but catechol was not produced. These results indicate that the conversion of catechol to phenol occurred rapidly once catechol was formed.
Research on catalytic demethoxylation suggests that the selectivity for ring saturation or demethoxylation is governed by the type of association between the compound and the catalytic surface.24Fig. 7 shows that an association in which the aromatic compounds are arranged perpendicular to the catalyst surface favors demethoxylation. In a non-polar solvent, the methyl, ethyl, and propyl side chains are more efficiently solvated, whereas polar hydroxyl and methoxyl groups are forced to face the polar Pd surface, which facilitates such associations.
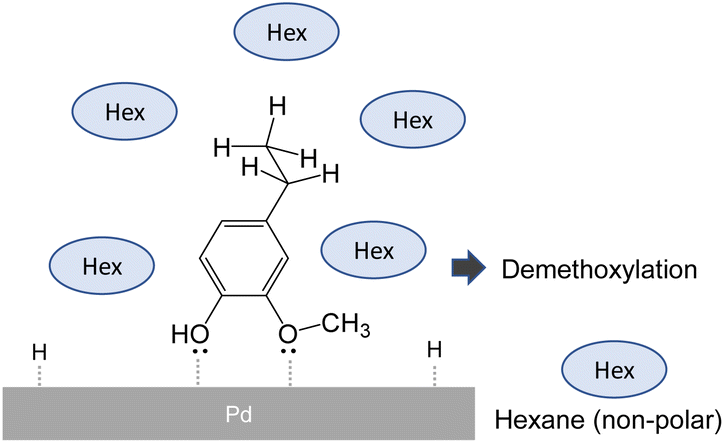 | ||
| Fig. 7 Possible effect of hexane on adsorption mode of monomer products in pyrolysis-assisted hydrogenolysis. | ||
Another important effect of the reaction atmosphere is the yield of DHCA in methanol. Under catalytic hydrogenolysis conditions, DHCA is converted to ethyl guaiacol, but H2 inhibits this conversion to some extent. Similar results were observed with dioxane (GPC profiles in Fig. S2†). In other solvents, this reaction was promoted by H2 and DHCA was not detected under H2. The solvents must therefore be involved in these unexpected results. A rational explanation for the inhibition of DHCA hydrogenolysis in methanol and dioxane is currently elusive, but may involve association of DHCA, methanol, and dioxane on the Pd surface (Fig. S3†). In our previous paper,3 we proposed that the deformylation of DHCA proceeds via a four-centered transition state on the Pd surface, even in the absence of H2. The effects of H2 on these competitive association modes may be involved, but further studies are needed to explain these unexpected results.
Monomer formation from milled wood lignin (MWL)
Other less important pyrolysis products such as vanillin are also produced from the pyrolysis of MWL25 along with CA. Radical species make a greater contribution to the MWL reactions because pyrolytic cleavage of the ether bond occurs mainly via a homolytic mechanism,26 with formation of radical species as the primary pyrolysis products. These differences should be considered when discussing the MWL reactivity in terms of the CA results. The monomer yields and compositions obtained from Japanese cedar MWL in different solvents at 350 °C for 30 and 60 min are summarized in Fig. 8. The yield (60 min)/yield (30 min) ratios are included in this figure to clarify the effect of the treatment duration. The anisole results were reported in our previous paper.2 Since anisole formed small amounts of phenol, phenol is not included in the yield of lignin-derived monomers obtained in anisole. Therefore, the yield of monomers in anisole was underestimated.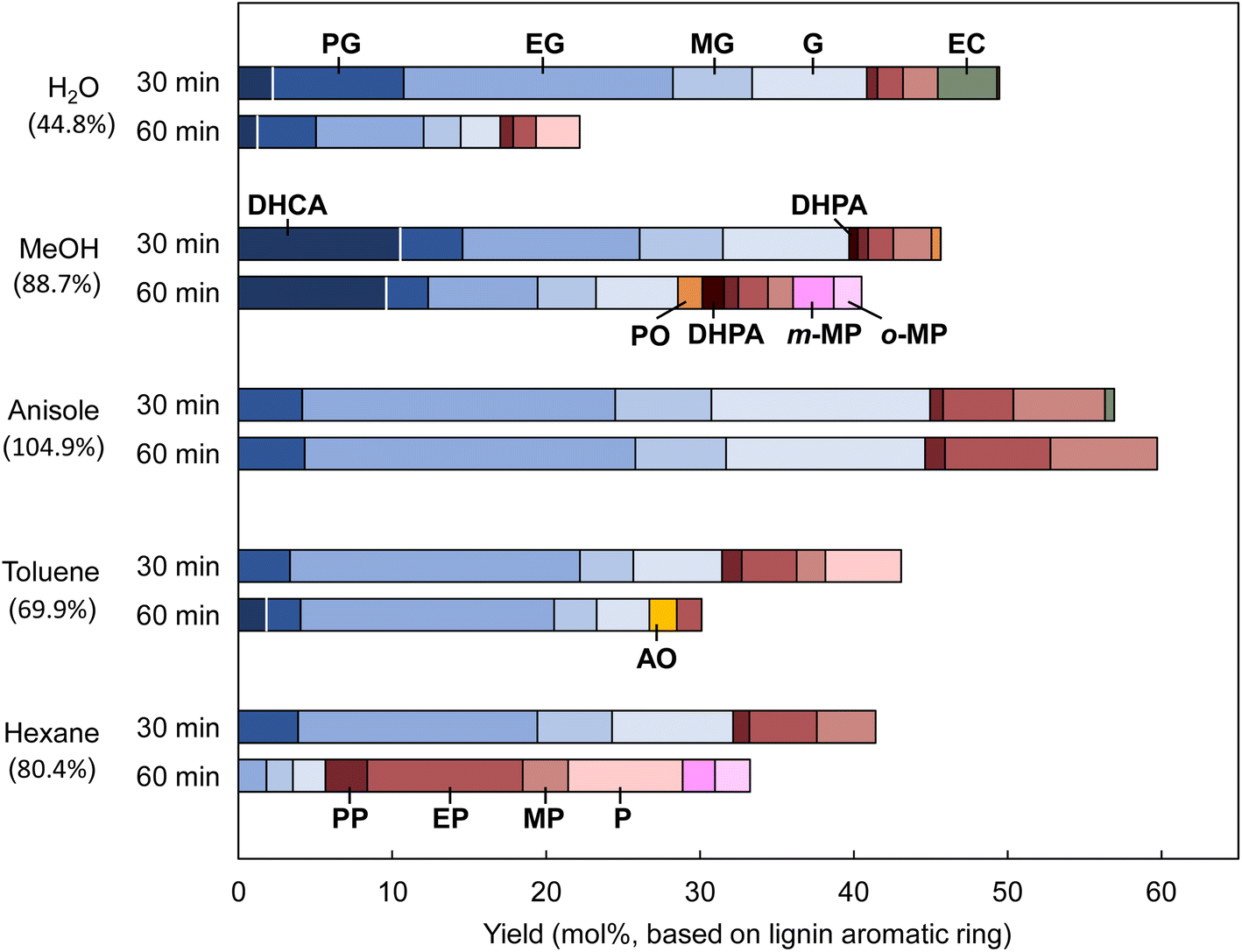 | ||
| Fig. 8 Monomers obtained from pyrolysis-assisted catalytic hydrogenolysis of Japanese cedar milled wood lignin (MWL) in different solvents at 350 °C (MWL: 10 mg; Pd/C: 10 mg; solvent: 2 mL; H2: 3 mL/0.1 MPa). Percentages in parentheses are total monomer yields at 60 min compared with those at 30 min. PO: propiovanillone. AO: acetovanillone. | ||
The monomer yields at 30 min follow the same order as that observed for CA (60 min) [anisole (56.9 mol%) > water (49.5 mol%) > methanol (45.7 mol%) > toluene (43.1 mol%) > hexane (41.4 mol%), values in parentheses: monomer yields from MWL]. However, increasing the reaction time from 30 to 60 min decreased the yields in all the solvents except anisole; the yield increased slightly in anisole. In particular, the reduction rate in water was as high as 44.8%. This can be explained by hydrolysis of the methoxyl group of the guaiacyl unit.27 In a model experiment (Fig. S1†), only 74.8 mol% and 66.7 mol% of aromatic monomers were recovered from guaiacol and catechol, respectively, and phenol was the sole monomer product. These results indicate that the monomeric products formed from MWL in water undergo further degradation via catechol-type intermediates (Fig. 9a). In water, ethyl catechol was produced in 3.9 mol% yield at 30 min, but no catechol derivatives were detected at 60 min.
 | ||
| Fig. 9 Side reactions expected to occur during pyrolysis-assisted catalytic hydrogenolysis of coniferyl alcohol (a) in water, (b) in methanol, toluene, and hexane. | ||
Another type of side reaction that occurred in methanol, toluene, and hexane explains the yield loss with extended reaction time. Careful analysis of the products from CA detected the formation of various radical-coupling products via addition of solvent-derived radicals to the side-chain C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of CA and its pyrolytic intermediates (Fig. S4, S5† and 9b). Methyl, benzyl, and hexyl radicals would be produced from methanol, toluene, and hexane, respectively, under the pyrolysis-assisted catalytic hydrogenolysis conditions. Ortho- and meta-methyl-substituted phenols detected from MWL in methanol (60 min, Fig. 8) could be formed by the coupling of methyl radicals to phenol. Methylation products were also detected from CA in water (Fig. S4 and S5†). This indicates that methyl radicals were formed from the methanol produced by hydrolysis of the methoxyl group. These radical-coupling reactions are more favorable for MWL than for CA because the lignin ether bonds in MWL are homolytically cleaved,26 which results in more radicals in the reaction medium.
C bonds of CA and its pyrolytic intermediates (Fig. S4, S5† and 9b). Methyl, benzyl, and hexyl radicals would be produced from methanol, toluene, and hexane, respectively, under the pyrolysis-assisted catalytic hydrogenolysis conditions. Ortho- and meta-methyl-substituted phenols detected from MWL in methanol (60 min, Fig. 8) could be formed by the coupling of methyl radicals to phenol. Methylation products were also detected from CA in water (Fig. S4 and S5†). This indicates that methyl radicals were formed from the methanol produced by hydrolysis of the methoxyl group. These radical-coupling reactions are more favorable for MWL than for CA because the lignin ether bonds in MWL are homolytically cleaved,26 which results in more radicals in the reaction medium.
Such undesired reactions were not detected in anisole, therefore the monomer yield and stability were high. Anisole is therefore the solvent of choice for monomer production from MWL. The monomer yield in anisole was the highest among the yields obtained in the solvents used in this study. Prolonging the treatment did not decrease the yield.
The characteristic monomer compositions observed in various solvents for CA were also observed for MWL (Fig. 8). The DHCA contribution was higher in methanol and it could not be completely hydrogenated to ethyl guaiacol, even at 60 min. The smaller contribution of DHCA to the monomers obtained from MWL than that to the monomers obtained from CA could be attributable to less H2 being available for the MWL reactions. Hydrogen would be consumed to stabilize radical species formed by homolysis of lignin ether bonds. Vanillin and other carbonyl compounds formed through other lignin pyrolysis reactions also consume hydrogen.3
As observed for CA, phenol formation via demethoxylation was prominent in hexane, and the phenol content in the monomers obtained from MWL reached 83.0% in hexane at 60 min. In contrast, the yield of phenols in toluene was much lower than that expected from the CA results. This is probably because of radical coupling of the produced phenols with the benzyl radicals formed from toluene. Unlike the case for the model reaction with CA, the lignin ether bonds are homolytically cleaved,26 which results in more radicals in the reaction medium. Under such conditions, radical-coupling reactions predominate. Phenols are more reactive in radical coupling than guaiacols because they have another ortho-carbon as a coupling site. Radical formation from toluene is easier than that from hexane because of formation of the more-stable benzyl radical. These observations could explain the differences among the yields of phenols. Hexane is the best solvent for phenol production from lignin among the solvents used in this study. Phenols are potential commodity chemicals in the chemical industry.
Experimental
Materials
CA was synthesized by the reduction of coniferyl aldehyde (98%, Sigma-Aldrich, Germany) with NaBH4, and purified by thin-layer chromatography. Nuclear magnetic resonance spectroscopic analysis of these synthesized compounds showed that their purities were approximately 99%.MWL was prepared from the extractive-free sapwood flour from the Japanese cedar according to a procedure previously reported by Björkman.28 The MWL contained the hydrolysable sugars glucose (0.6 wt%), xylose (0.7 wt%), mannose (0.3 wt%), and arabinose (0.2 wt%), as reported in our previous work.2
Thermal reactions
CA was subjected to thermal reactions with different solvents in a batch reactor. In each experiment, solid CA (10 mg) was placed in a sealed 5 mL batch-reaction vessel, as described in our previous work,2 together with the solvent (2 mL), namely deionized water, methanol (GR, Nacalai Tesque, Kyoto, Japan), anisole (GR, Nacalai Tesque), toluene (GR, Nacalai Tesque), hexane (EP, Nacalai Tesque), benzene (GR, Nacalai Tesque), or 1,4-dioxane (GR, Nacalai Tesque). The remaining space in the vessel (approximately 3 mL) was filled with N2 at 0.1 MPa. The reactor was then immersed and oscillated in a salt bath that had been preheated to 350 °C. After treatment for 30 or 60 min, the reaction was quenched by using a water bath.After the thermal reaction, the contents of the reactor were washed several times with methanol. The extracts were combined to give a solution of volume approximately 20 mL, which included the reaction solvent. When the solvent was deionized water, the reaction mixture was extracted several times with ethyl acetate to give a solution of volume 20 mL.
Catalytic hydrogenolyses of CA and MWL were conducted by using the same procedure. In each case, 5% Pd/C (10 mg; EP, Nacalai Tesque) was loaded with the sample before addition of the reaction solvent (2 mL). The remaining space in the vessel was filled with N2 or H2 at 0.1 MPa. After the reaction, the contents of the reactor were washed by using the previously described procedure. A turbid mixture containing the reaction mixture and solid Pd/C was obtained. A portion of the suspension was centrifuged to remove Pd/C before analysis.
All solvents were tested for the blank tests to know the stability in the presence of Pd/C under H2 at high temperatures. Toluene and anisole were quite stable and no ring saturation products such as cyclohexane were formed but very small amounts of dimers were detected. Small amounts of phenol and benzyl alcohol were obtained from anisole. Therefore, phenol formed by the conversion of lignin in anisole was not quantified as a lignin-derived product.2 Hexane formed very small amounts of dodecane. Methanol produced perceptible dimethyl ether, while dioxane produced perceptible ethylene glycol.
Product analysis
Gel-permeation chromatography (GPC) was used to determine the molecular-weight distribution of the methanol-soluble and ethyl acetate-soluble (in the case of deionized water) products in the reaction mixture. The analysis was performed by using a LC-10A system (Shimadzu Corporation, Kyoto, Japan) with a Shodex KF801 column (exclusion limit 1500 Da as the polystyrene equivalent; Showa Denko K.K., Tokyo, Japan), tetrahydrofuran at 0.6 mL min−1 as the eluent, a column oven temperature of 40 °C, and an ultraviolet detector operated at 280 nm. The retention times of the signal peaks for guaiacol (14.1 min), ethyl guaiacol (13.6 min), CA (12.6 min), and dihydro-CA (12.6 min) were obtained by analyzing the authentic compounds.Gas chromatography-mass spectrometry was used to analyze the methanol-soluble and ethyl acetate-soluble portions after trimethylsilyl derivatization. The analysis was performed by using a GCMS-QP2010 Ultra instrument (Shimadzu Corporation) with a CPSil 8CB column (length: 30 m, inner diameter: 0.25 mm, film thickness: 0.25 μm; Agilent Technologies, Inc., Santa Clara, CA, USA), He at 2.09 mL min−1 as the carrier gas, an injector temperature of 250 °C, and a split ratio of 1/10. The column oven temperature was held at an initial temperature of 70 °C for 2 min, ramped at 4 °C min−1 to 150 °C and held for 1 min, and then ramped at 10 °C min−1 to 300 °C and held for 1 min. Prior to analysis, 1,3,5-triphenylbenzene (an internal standard) was added to the methanol-soluble portion, and methanol was removed by using evaporator at 30 °C under vacuum. This relatively low temperature was used to avoid the evaporation of some products with relatively low boiling point, e.g., phenol. Trimethylsilylation was performed by addition of pyridine (100 μL), hexamethyldisilazane (150 μL), and trimethylchlorosilane (80 μL), and then heating at 60 °C for 30 min. The products were quantified by using calibration curves prepared from analysis of standards.
The molar yield, Mi, of each monomeric product, i, was calculated as:
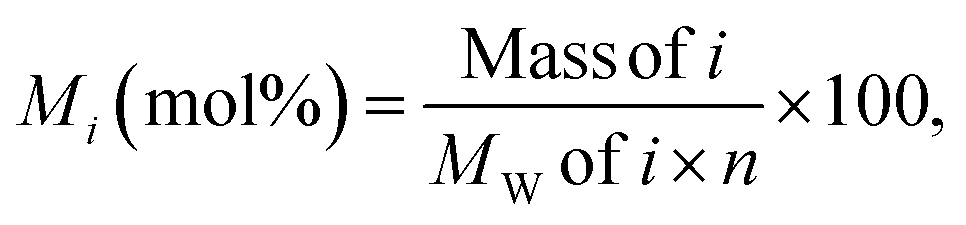
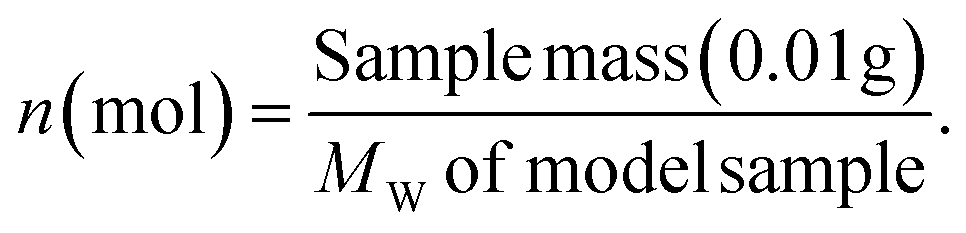
The value, 182 Da, was used as the average molecular weight of the phenylpropanoid units in the Japanese cedar (softwood) MWL according to literature.29
Conclusions
The effects of solvents on pyrolysis-assisted catalytic hydrogenolysis of CA (the major pyrolysis product of lignin) and MWL isolated from Japanese cedar were investigated at 350 °C by using a Pd/C catalyst and various solvents. The conclusions were as follows. Under pyrolysis (no catalyst in N2) conditions, CA was condensed in protic solvents (water and methanol) but not in aprotic solvents (anisole and toluene). In hydrophobic hexane, the CA solubility was low and condensation occurred to give insoluble aggregates, which formed char. Hydrolysis of the methoxyl group of CA occurred in water. Redox reactions of CA occurred in anisole and toluene, but no oxidation products were detected in methanol and water, probably because of the reducing ability of methanol.These characteristic features of CA pyrolysis almost disappeared on addition of Pd/C. This indicates that catalytic hydrogenolysis proceeded efficiently even under N2. The production of phenols by demethoxylation also occurred over Pd/C, particularly under H2, and the efficiency of this production from CA was inversely related to the solvent polarity. Unexpectedly, hydrogenolysis of DHCA to ethyl guaiacol was inhibited by changing the atmosphere from N2 to H2 only in methanol and dioxane. The transformations of MWL were basically explained by the CA results, except for some side reactions that were caused by the higher number of radicals in the MWL reaction medium. These radicals were formed by homolytic cleavage of the lignin ether bonds. Degradation via catechols formed by hydrolysis (water) and radical-coupling reactions with radicals formed from solvents (methanol, toluene, and hexane) decreased the monomer yields, especially in prolonged treatments. These results show that anisole is the best solvent for monomer production from MWL, and hexane is the best solvent for the production of phenols from MWL.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was funded by the Japan Society for the Promotion of Science (Grant Number JP19H03019) and the JST-Mirai Program (Grant Number JPMJMI20E3), Japan. We thank the China Scholarship Council (CSC) for supporting Jiaqi Wang to conduct this research at Kyoto University. We thank Helen McPherson, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.References
- Z. Sun, B. Fridrich, A. De Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed
.
- J. Wang, E. Minami and H. Kawamoto, Green Chem., 2023, 25, 2583–2595 RSC
- J. Wang, E. Minami and H. Kawamoto, J. Anal. Appl. Pyrolysis, 2023, 105930 CrossRef CAS
- N. Phaiboonsilpa, K. Yamauchi, X. Lu and S. Saka, J. Wood Sci., 2010, 56, 331–338 CrossRef CAS
- E. Minami and S. Saka, J. Wood Sci., 2003, 49, 73–78 CrossRef CAS
- E. Dorrestijn, M. Kranenburg, D. Poinsot and P. Mulder, Holzforschung, 1999, 53, 611–616 CAS
- T. Kotake, H. Kawamoto and S. Saka, J. Anal. Appl. Pyrolysis, 2014, 105, 309–316 CrossRef CAS
- T. Parsell, S. Yohe, J. Degenstein, T. Jarrell, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. I. Kim, H. Choudhari, B. Saha, R. Meilan, N. Mosier, F. Ribeiro, W. N. Delgass, C. Chapple, H. I. Kenttämaa, R. Agrawal and M. M. Abu-Omar, Green Chem., 2015, 17, 1492–1499 RSC
- J. Sun, H. Li, L. P. Xiao, X. Guo, Y. Fang, R. C. Sun and G. Song, ACS Sustainable Chem. Eng., 2019, 7, 4666–4674 CrossRef CAS
- J. M. Pepper and H. Hibbert, J. Am. Chem. Soc., 1948, 70, 67–71 CrossRef CAS PubMed
- J. Zhang, J. Teo, X. Chen, H. Asakura, T. Tanaka, K. Teramura and N. Yan, ACS Catal., 2014, 4, 1574–1583 CrossRef CAS
- C. Li, M. Zheng, A. Wang and T. Zhang, Energy Environ. Sci., 2012, 5, 6383–6390 RSC
- M. V. Galkin, A. T. Smit, E. Subbotina, K. A. Artemenko, J. Bergquist, W. J. J. Huijgen and J. S. M. Samec, ChemSusChem, 2016, 9, 3280–3287 CrossRef CAS PubMed
- Y. Li, B. Demir, L. M. Vázquez Ramos, M. Chen, J. A. Dumesic and J. Ralph, Green Chem., 2019, 21, 3561–3572 RSC
- W. Lan, Y. P. Du, S. Sun, J. Behaghel De Bueren, F. Héroguel and J. S. Luterbacher, Green Chem., 2021, 23, 320–327 RSC
- X. Ouyang, X. Huang, J. Zhu, M. D. Boot and E. J. M. Hensen, ACS Sustainable Chem. Eng., 2019, 7, 13764–13773 CrossRef CAS
- W. Schutyser, S. Van Den Bosch, T. Renders, T. De Boe, S. F. Koelewijn, A. Dewaele, T. Ennaert, O. Verkinderen, B. Goderis, C. M. Courtin and B. F. Sels, Green Chem., 2015, 17, 5035–5045 RSC
- F. Héroguel, X. T. Nguyen and J. S. Luterbacher, ACS Sustainable Chem. Eng., 2019, 7, 16952–16958 CrossRef
- K. Barta, T. D. Matson, M. L. Fettig, S. L. Scott, A. V. Iretskii and P. C. Ford, Green Chem., 2010, 12, 1640–1647 RSC
- X. Huang, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ChemSusChem, 2014, 7, 2276–2288 CrossRef CAS PubMed
- T. Nakamura, H. Kawamoto and S. Saka, J. Anal. Appl. Pyrolysis, 2008, 81, 173–182 CrossRef CAS
- H. Kawamoto and S. Saka, J. Wood Chem. Technol., 2007, 27, 113–120 CrossRef CAS
- T. Kotake, H. Kawamoto and S. Saka, J. Anal. Appl. Pyrolysis, 2013, 104, 573–584 CrossRef CAS
- J. Zhang, J. Sun and Y. Wang, Green Chem., 2020, 22, 1072–1098 RSC
- S. Wang, B. Ru, H. Lin, W. Sun and Z. Luo, Bioresour. Technol., 2015, 182, 120–127 CrossRef CAS PubMed
- H. Kawamoto, T. Nakamura and S. Saka, Holzforschung, 2008, 62, 50–56 CrossRef CAS
- T. L. K. Yong and M. Yukihiko, Ind. Eng. Chem. Res., 2013, 52, 9048–9059 CrossRef CAS
- A. Björkman, Sven. Papperstidn., 1956, 59, 477–485 Search PubMed
-
S. Y. Lin and C. W. Dence, Methods in Lignin Chemistry, Springer Science & Business Media, 2012 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00128h |
| This journal is © The Royal Society of Chemistry 2023 |