
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hierarchical zeolite catalysed fructose dehydration to 5-hydroxymethylfurfural within a biphasic solvent system under microwave irradiation†
Huaizhong
Xiang
 a,
Shima
Zainal
a,
Henry
Jones
b,
Xiaoxia
Ou
ac,
Carmine
D'Agostino
ad,
Jesús
Esteban
*a,
Christopher M. A.
Parlett
*aefg and
Xiaolei
Fan
*ac
a,
Shima
Zainal
a,
Henry
Jones
b,
Xiaoxia
Ou
ac,
Carmine
D'Agostino
ad,
Jesús
Esteban
*a,
Christopher M. A.
Parlett
*aefg and
Xiaolei
Fan
*ac
aDepartment of Chemical Engineering, School of Engineering, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: jesus.estebanserrano@manchester.ac.uk; christopher.parlett@manchester.ac.uk; xiaolei.fan@manchester.ac.uk
bDepartment of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cNottingham Ningbo China Beacons of Excellence Research and Innovation Institute, University of Nottingham Ningbo China, 211 Xingguang Road, Ningbo 315100, China
dDipartimento di Ingegneria Civile, Chimica, Ambientale e dei Materiali (DICAM), Alma Mater Studiorum – Università di Bologna, Via Terracini, 28, 40131 Bologna, Italy
eUniversity of Manchester at Harwell, Diamond Light Source, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
fDiamond Light Source, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
gUK Catalysis Hub, Research Complex at Harwell, Rutherford Appleton Laboratory, Harwell, Oxfordshire OX11 0FA, UK
First published on 24th July 2023
Abstract
Realising sustainability within the chemical industry necessitates a shift from the traditional linear approach, based on crude oil, to a circular economy using alternative feedstock such as biomass, from which 5-hydroxymethylfurfural (HMF) is a potentially highly interesting platform chemical. While its production is relatively straightforward via the dehydration of fructose, derived from either saccharides or lignocellulosic biomass, its production is hindered by undesirable side reactions, which decrease the selectivity of the intended reaction to HMF, hence diminishing the overall yield. Here we report a green, highly selective approach to producing 5-hydroxymethylfurfural (HMF) from fructose based on the co-deployment of a biphasic reaction medium, microwave radiation, and a commercial solid acid catalyst (FAU Y zeolites). Following an initial evaluation of catalyst–solvent interactions and diffusion, a hierarchical mesoporous Y zeolite was chosen and deployed within a range of reaction media and process conditions for process optimisation, identifying a biphasic system consisting of ((6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 water:DMSO)/(7:3 MIBK:2-BuOH)) as the optimal reaction medium. This solvent combination facilitated an HMF yield of ∼73.9 mol% with an excellent selectivity of ∼86.1% at 160 °C after only 45 minutes under microwave irradiation. These, in turn, result in optimal energy efficiency and excellent green credentials relative to conventional heating.
:4 water:DMSO)/(7:3 MIBK:2-BuOH)) as the optimal reaction medium. This solvent combination facilitated an HMF yield of ∼73.9 mol% with an excellent selectivity of ∼86.1% at 160 °C after only 45 minutes under microwave irradiation. These, in turn, result in optimal energy efficiency and excellent green credentials relative to conventional heating.
Sustainability spotlightBiomass-derived 5-hydroxymethylfurfural is regarded as a versatile and key intermediate for the production of a range of sustainable bio-based chemicals, and thus it has drawn widespread academic and industrial attention. To further cement HMF as a sustainable platform chemical, synthetic routes with greater efficiency and reduced waste production are critical. The research presented here demonstrates a highly selective and energy-efficient HMF production route from fructose, based on the cooperation of a biphasic reaction media, microwave irradiation, and commercially available FAU Y zeolites, and aligns with the UN sustainable development goals: affordable and clean energy (SDG 7), responsible consumption and production (SDG 12), and climate action (SDG 13). |
Introduction
5-Hydroxymethylfurfural (HMF) is a key biomass-derived platform chemical with the potential to be used in the generation of an array of sustainable derivatives, including 2,5-dimethylfuran (biofuel), 5-ethoxymethylfurfural (biofuel), and 2,5-furandicarboxylic acid (polymer monomer).1 However, the market price of HMF ($8568 kg−1)2 is a significant barrier and requires addressing through more cost-effective production routes to increase its appeal to industry. With respect to this, Kazi et al.3 reported HMF from fructose produced using HCl as the catalyst could achieve a minimum selling price (MSP) of $1.07 kg−1, while Motagamwala et al.4 reached an MSP of $2.21 kg−1 by employing a niobium phosphate catalyst. HMF production from fructose5–14 and glucose15–18 (C6 monosaccharides), sucrose19 (disaccharides), starch,20 cellulose21 and raw biomass22 (polysaccharides), has been reported, with the dehydration of fructose the more commonly investigated given that it circumvents the need for prior depolymerisation and isomerisation.15–18,23 However, it should be pointed out that while fructose is found in nature, e.g., honey contains 40 g per 100 g while apples and pears range from 5–9 g per 100 g, the primary industrial sources of fructose is disaccharides and polysaccharides.24 The conversion of fructose to HMF proceeds via Brønsted acid catalysed dehydration (Scheme 1),5,23,25via the loss of three water molecules.23,25,26 However, the resulting HMF is prone to suffer undesirable side reactions, including rehydration to levulinic acid (LA) and formic acid (FA) and polymerisation to soluble and insoluble humins.13,23,25 Delivering operation conditions that minimise, and ideally eliminate, these side reactions to optimise HMF selectivity are critical and key to delivering the transition to a bio-based chemical economy. | ||
| Scheme 1 Reaction pathways for the production of HMF from fructose. | ||
HMF production (from fructose) has been reported for both homogeneous and heterogeneous catalytic species. Motagamwala et al.27 reported a yield of 95% through the deployment of 15 mM H2SO4 in an acetone:H2O mixed solvent system within 2 h at 120 °C. To aid catalyst isolation,5,12,28 a range of solid acids, including ion-exchange resins,11 metal oxides,29 and zeolites, specifically mordenite,30 H-β,31 H-ZSM5 (ref. 18) and H–Y,14,31 have been proposed and evaluated. HY zeolites, in particular, have been widely investigated and utilised in the chemical industry due to their large pore dimension (0.74 nm opening and 1.3 nm cavities) and high surface areas (>700 m2 g−1). Relative to other commercially deployed zeolites, the former should provide greater acid site accessibility for reactants, with relatively low cost as well. Given their capacity for tunability of acidity and porosity, and excellent hydrothermal stabilities, hierarchical mesoporous Y (i.e., USY) zeolites14,31,32 represent a further promising option. To date, however, the focus of such investigations typically probes porosity and acidity,5,14,31 with catalyst–solvent interactions often overlooked.5 The recent deployment of nuclear magnetic resonance (NMR) relaxation and pulsed-field gradient (PFG)-NMR studies, the latter being particularly suitable to study diffusion of liquids,33 opens up opportunities to rectify this knowledge-gap, through probing catalyst substrate interactions and molecular diffusion within heterogeneous catalysts, respectively.34–36
Complementary to catalyst design, tuning the reaction media can equally impact process performance towards HMF selectivity.5,6,13 Protic polar solvents (i.e., water), aprotic polar solvents (e.g., dimethylsulfoxide, DMSO6,9,37 and dimethylformamide, DMF7), and ionic liquids13,15 have been explored. From a green perspective, water represents an ideal solvent, i.e. non-toxic, high abundance, and sustainable, which results in its applications across numerous industries.38 However, its deployment for fructose dehydration is negatively impacted by the possibility of HMF rehydration and polymerisation, with yields typically under 50%.28,38 DMSO is frequently reported as a promising alternative,6,9,37 which in itself is catalytic towards the process;26,39–41 however, its inherent high boiling point hampers product isolation.1,28 Ionic liquids are another attractive alternative due to their low vapour pressure, non-flammability, and low toxicity;42 however, the economics of their use are typically prohibitive.11,43
Biphasic solvent systems represent an alternative approach to the conventional mono-solvent reaction media, with the potential for further benefits and refinement. These include facilitating the extraction of the product (HMF) from the (aqueous) reaction phase, to mitigate against further undesirable reactions32,44,45 while also aiding product isolation,45 by employing a low boiling point (organic) extraction phase. Bhaumik et al.46 reported high fructose conversion (89%) and HMF selectivity (88%) over silicoaluminophosphate (SAPO) catalysts in H2O/MIBK at 175 °C, while Román-Leshkov et al.47 employed ion-exchange resin catalysts in an H2O:DMSO (8:2 v/v)/MIBK:2-BuOH (7:3 v/v) biphasic reaction media to obtain the conversion of 74% and selectivity of 68% at 90 °C. Incorporating 2-BuOH within the extraction phase increases HMF solubility, escalating process selectivity by 20% without impacting the green credentials of the process.48,49
The integration of alternative technologies, especially the application of microwave (MW) irradiation, are gaining traction as options to elevate the green credentials of catalytic processes further. In particular, through the reduction in process time, enhanced energy efficiency, and promoted reaction rate.10,12,50 For example, MW irradiation induced a 53% increase in HMF yield over an ion-exchange catalyst relative to conventional heating,11 while a further elevation of 87% was reported for a sulfonated carbon catalyst.10 The latter represents a ∼19-fold increase in energy efficiency.
Here we report the use of hierarchical Y zeolites as the solid acid catalysts for HMF production from fructose under MW irradiation and the optimisation of a biphasic solvent system comprising a reaction phase of H2O and DMSO and an extraction phase of MIBK and 2-BuOH.
Experimental section
Materials and chemicals
D(−)-Fructose (biochemistry grade), D-(+)-glucose (≥99.5%, GC), levulinic acid (98%), formic acid (HPLC grade), furfural (ACS reagent, 99%), N,N-dimethylformamide (HPLC grade), isopropyl alcohol (HPLC grade), dimethylsulfoxide (ACS reagent, ≥99.9%), methyl isobutyl ketone (ACS reagent, ≥99.9%), 1-methyl-2-pyrrolidone (MNP) (ACS reagent, ≥99.9%), propylene carbonate (PC) (ACS reagent, ≥99.9%), cyrene (BioRenewable), and 2-butanol (anhydrous, 99.5%) were purchased from Sigma-Aldrich. HMF (99.9%) was purchased from Fisher Chemical. Commercial FAU Y zeolites (CBV300, CBV720, and CBV760) were purchased from Zeolyst International (UK). CBV300 (NH4+ form) has a silicon-to-aluminium (Si/Al ratio) of 2.6. Before use, CBV300 was calcined at 450 °C for 10 h (ramp rate 1 °C min−1) to generate the H+ form (denoted as HY-2.6).51 The Si/Al ratio of CBV720 and CBV760 are 15 and 30, respectively, and were supplied in their H+ forms and denoted as HY-15 and HY-30. HY-15 and HY-30 were manufactured by steam and acid treatment of CBV300 to introduce complementary mesoporosity in the hierarchical structure.52 The physicochemical properties of the Y zeolites used with the study are reported in our previous publication.51Catalysis
Fructose conversion was performed in 30 cm3 reaction vials within an Anton Paar Monowave 400 microwave reactor. In a typical procedure, fructose (0.2 g, 5 wt/v%) was dissolved in the reaction phase of H2O:DMSO (4 cm3) before adding the zeolite (0.08 g, 2 wt/v%) and agitating for 15 minutes at room temperature. MIBK:2-BuOH (12 cm3), as the extraction phase, was added, and the system was heated to 160 °C (ramp rate 160 °C min−1) and held for 45 min with stirring in the microwave reactor. Upon completion of the reaction, the vial was cooled to 70 °C in the air before being quenched in an ice-water bath. The catalyst was separated from the reaction media by centrifugation (at 3000g for 10 min), with aliquots (0.2 cm3) from the reaction and extraction phase prepared for HPLC analysis by dilution with deionised water (1:50 v/v) or isopropyl alcohol (IPA) (1:50 v/v), respectively, and filtration with a polyethersulfone syringe filter (0.2 μm). For the catalysts by conventional heating, reactions were either conducted in a 25 cm3 three-necked round bottom flask (equipped with magnetic stirring and a mercurial thermometer), heated by an oil bath, or using a 100 cm3 Parr 4598 autoclave reactor.
Quantitative analysis of reaction samples was performed by high-performance liquid chromatography (HPLC, Agilent Infinity 1260) equipped with a refractive index detector (RID) and UV detector at a wavelength of 277 nm. Product resolution was achieved on a Bio-Rad Aminex HPX-87H ion exclusion column (300 mm × 7.8 mm), using a 0.01 N H2SO4 mobile phase (flow rate 0.6 cm3 min−1) under isothermal conditions (column over at 65 °C). Humins were calculated from the carbon balance. The conversion of fructose (mol%), the product yield (mol%), product selectivity (%), and partition ratio (PR) of HMF are defined in the (ESI, eqn (S1)–(S4)†). PR is defined as the ratio of the weight fraction of HMF in the extraction phase to the weight fraction of HMF in the reaction phase.44 The total energy efficiency coefficient (η) of the reaction was also calculated (eqn (S5)†), which is the amount of HMF produced per unit of work.12 Sustainability metrics, including E-factor (eqn (S6) and (S7)†), mass intensity (MI) (eqn (S8)†), reaction mass efficiency (RME) (eqn (S9)†), and carbon economy (CE) (eqn (S10)†), have been evaluated to assess process efficiency to reduce waste in the environment.53,54 Details of the green metrics are presented in the ESI.†
NMR relaxation characterisation
The adsorption and diffusion properties of relevant probing molecules within different FAU Y zeolites were determined by nuclear magnetic resonance (NMR) relaxation measurements and pulsed-field gradient (PFG)-NMR, respectively. NMR measurements were performed using a Magritek Spinsolve benchtop NMR spectrometer operating at a 1H frequency of 43 MHz. The temperature was regulated and stabilised by a Magritek Spinsolver temperature control unit in a range between 18 °C to 28 °C. All measurements were conducted at room temperature. Prior to analysis, dry samples of the zeolites were soaked in deionised water for 48 hours. Excess liquid was then removed, and the samples were gently dried. Reference samples of each zeolite were also prepared by soaking in n-octane for 48 hours rather than water but were otherwise prepared by the same method. T1 spin–lattice relaxation times were measured using a standard inversion recovery pulse sequence. The T2 transverse relaxation times were measured using the Carr–Purcell–Meiboom–Gill (CPMG) sequence with a recycle delay of 5T1. The diffusion experiments were performed using the pulsed-field gradient stimulated echo (PGSTE) sequence, developed by Stejskal and co-workers.55,56Results and discussion
Dehydration of fructose over zeolites in pure water
Fructose dehydration over the three Y zeolites in water was first conducted at 160 °C under MW irradiation. The resulting conversions, selectivity, and product distribution profiles, benchmarked against a blank system (without a catalyst), are reported in Fig. 1 and S1, S2,† respectively. Background rates, i.e., in the absence of a catalyst, are comparable to previous studies,57–59 with humins contributing significantly to the lack of total product selectivity. Other observed by-products include glucose and furfural, arising from fructose isomerisation over Lewis acid sites within the zeolites,60 and C–C bond cleavage of acyclic hexoses to pentose,61,62 respectively. | ||
| Fig. 1 Fructose conversion and HMF yield (as a function of reaction time) for fructose dehydration in different aqueous systems under MW irradiation. Reaction conditions: 16 cm3 water, 5 wt% fructose, 2 wt% zeolites, at 160 °C and 800 rpm. | ||
Despite the lower acidity of HY-30, which possesses only a fraction of 1/70 and 1/12 of Brønsted acidity of HY-2.6 and HY-15,51 it proved favourable for fructose conversion. This increase in fructose conversion correlates with higher HMF yields, with an optimal HMF yield of ∼27% over HY-30 occurring within 45 minutes. This corresponds to a turnover frequency (TOF, eqn (S12)†) of 379.2 h−1, a dramatic increase compared to previous reports for zeolite catalysed fructose dehydration, as summarised in Table S1.† Rac et al.31 also employed HY-30, demonstrating the preferable performance of HY-30 when benchmarked against ZSM-5 and H-BEA, and reported a TOF of 17.4 h−1, albeit at the reduced temperature of 130 °C under conventional heating. However, the decrease in TOF is not solely a consequence of temperature, with HY-30 producing a TOF of 62.4 h−1 at 130 °C under MW irradiation. This is further verified through comparison to the performance of HY-30 at 160 °C in an autoclave batch reactor (Table S2†). The improved heating efficiency and heat transfer by MW heating,29,63,64 increases fructose conversion and process energy efficiency coefficient by a factor of ∼2. Jia et al.5 also investigated different zeolites for fructose dehydration. However, they observed optimal performance for H-BEA zeolites under equivalent reaction conditions to this study. This apparent contradiction can be attributed to the use of HY-2.6, rather than the optimal HY-30, and it is pointed out that the performance of HY-2.6 from both studies produced matching TOFs. HMF selectivity, as shown in Fig. S1,† reveals an optimal reaction length of 30–45 minutes for all the three catalysts. Further increases in the reaction time result in greater formation of humins (Fig. S2†) as the major by-product. The dominant minor by-products include glucose, favoured over catalysts with higher Lewis acid site loadings, and rehydration products (formic and levulinic acid),65 for catalysts with lower Lewis acidity.51
The surprisingly poor dehydration performance of HY-2.6 is justified by two factors: accessibility and hydrophilicity. The first relates to fructose diffusion and the apparent accessibility of the acid sites within the zeolite framework. The HY framework possesses micropores with ∼0.74 nm apertures diameter,51 present in all three catalysts, while HY-15 and HY-30 have the hierarchical structure possessing complementary mesoporosity as well resulting from the post-synthetic treatments (of steaming and acid treatments)66 to extract framework Al species. These mesopores span the range of 4–18 nm (from N2 and Hg measurements), with maxima at 14 and 16 nm, respectively,51 with the degree and size governed by the degree of Al extraction. HY-15 comprise a superior level of smaller mesopores of 2–5 nm (ca. 70.1% vs. 31.0% based on specific mesopores volume), whereas HY-30 has a greater degree of larger mesopores. These secondary mesopores give rise to a hierarchical porous framework, which has been shown to enhance active site accessibility.51,67,68
Water diffusivity within the two hierarchical zeolites (i.e., HY-15 and HY-30) was probed by PFG-NMR, as shown in Fig. S3 and summarised in Table S3.† A drop in water self-diffusivity within the hierarchical zeolite structures, relative to the bulk liquid, is observed, with the linear nature of the log-attenuation plots suggesting a homogeneous pore structure on the macroscopic length scale, which is also defined as quasi-homogeneous behaviour. Both zeolite samples show higher diffusion coefficients for water than for n-octane (a weakly-interacting non-polar hydrocarbon probe molecule). This suggests that water self-diffusion is less hindered in the hierarchical Y zeolites than anticipated,69 potentially due to disruption of the intermolecular hydrogen bonding network when confined within the pores.70
Hydrophilicity is the second potential factor,5 with NMR relaxation applied to explore the adsorption strengths of water and n-octane (Fig. S4, Table S4†). T1/T2 ratios confirm that water adsorption strength correlates with Brønsted acid site density, i.e., [HY-2.6] > [HY-15] > [HY-30], as expected. Increasing hydrophobicity has been shown beneficial in reactions yielding water as a by-product, through reducing the interaction between water and the Brønsted acid sites, and thus lowering hydrophilicity is at least partially responsible for the superior performance of HY-30. In contrast, T1/T2 ratios for weakly-interacting n-octane are comparable, indicating an identical weak interaction across all three catalysts.
Dehydration of fructose over HY-30 in biphasic systems
To enhance HMF production, a range of biphasic solvent systems consisting of an aqueous reaction phase and an organic extraction phase, the latter to isolate HMF and prevent further side reactions, were investigated with the results presented in Fig. 2. Adding DMSO, a known promotor for HMF production,39–41 to the reaction phase increases conversion but only positively impact HMF selectivity when HY-30 is present (compared to the relevant catalyst-free counterparts, as shown in Fig. 2a). Incorporating an extraction phase, through the addition of MIBK, enhances selectivity towards HMF (both with and without the catalyst and DMSO). This is consistent with previous studies,71–73 due to reducing polymerisation (humins formation), via in situ extraction, whilst simultaneously prohibiting rehydration to LA and FA. Furthermore, an HMF selectivity over HY-30 of 53.9% (Fig. 2b) compares favourably to other zeolites employing the water:MIBK biphasic reaction media, i.e., H-ZSM5 at 195 °C (∼49.0%)18 and H-MOR at 165 °C (∼44.0%).32 Substituting DMSO within the biphasic system with an alternative polar aprotic solvent, namely DMF, severely impacts the systems (both with and without the HY-30 catalyst) with reduced selectivity and activity when employing a catalyst. Interestingly, further diversifying the pool of polar aprotic solvents investigated as alternatives to DMSO to include NMP, PC, and cyrene, reveals all three increase the noncatalytic performance to levels akin to the solvent system containing DMSO, with PC and cyrene showing optimal performance. However, when evaluated in the presence of the catalyst, HMF selectivity dropped significantly for both relative to DMSO due to increased humin formation, while NMP negatively impacts fructose conversion by ∼45%. Thus, the promoting role of DMSO is unique, with it suggested to arise from the ability to suppress undesirable parallel reactions,41 through hydroxyl and carbonyl functional group solvation, which diminishes HMF susceptibility to nucleophilic attack (rehydration).25,74 Furthermore, in the presence of Brønsted acidity, protonation of DMSO to DMSOH+, can yield a more active catalytic species.26,39
 | ||
| Fig. 2 Comparison of the product distribution from fructose dehydration in different monophasic and biphasic systems: (a) catalyst free systems and (b) systems over HY-30. Reaction conditions: 16 cm3 solvent(s), 5 wt% fructose, 2 wt% HY-30 (for (b)), reaction phase:extraction phase (v/v) = 3, at 160 °C and 800 rpm for 45 min under MW irradiation. | ||
Optimisation of the biphasic reaction media, through identifying the optimal ratio of components, is presented in Fig. 3. There is a positive correlation between HMF yields (and fructose conversion) and DMSO content (Fig. 3a). However, this occurs at the expense of partition ratio (PR), i.e., the ratio of HMF in the extraction phase relative to the reaction phase, as MIBK becomes a less effective extraction solvent due to the high HMF solubility in DMSO.47,75 Furthermore, increasing the ratio further, so that DMSO is the dominant species, is counterproductive as the two-phase (reaction and extraction) converge into one. To combat the downswing in PR, 2-BuOH was added to the extraction phase to further increase HMF solubility within it,47,76 as shown in Fig. 3b. Increasing the proportion of 2-BuOH within the extraction phase improves extraction efficiency, with a maximum PR of 1.43 for a MIBK:2-BuOH ratio of 6:4. However, peak process performance occurs at a 7:3 ratio, which coincides with a decrease in E factor (solvent recycled), from 1.24 to 0.93, and a PR value of 1.30, being consistent with the findings from the relevant studies in the literature (shown in Table S5†).47,76 Having optimised both the reaction and extraction phases, the impact of the ratio of these on HMF production was evaluated (Fig. S5†). Increasing the volume ratio, so that the extraction phase is in excess, shows a significant improvement in overall performance, with HMF yields reaching ∼70% for a 1:3 ratio, whilst a further increase yielded no benefit to HMF yield or green metrics, with E factors (solvent recycled), of 1.94, 0.93, and 1.14 for 1:1, 1:3, and 1:4 volume ratios, respectively. Having established the optimal reaction and extraction media compositions and their relative ratio to each other, it is key to assess if any species leach from one phase to the other. After 45 min at 160 °C under MW irradiation and stirring at 200 rpm, HPLC revealed that 5.1% and 9.6% of MIBK and 2-BuOH are partitioned into the reaction phase, respectively. In contrast, no migration of DMSO or water into the extraction phase is detected.
 | ||
| Fig. 3 Impact of tuning composition of (a) reaction phase using MIBK as the extraction phase and (b) extraction phase using a 6:4 water:DMSO reaction phase on conversion, HMF yield, partition ratio. Reaction conditions: 16 cm3 solvents, 5 wt% fructose, 2 wt% HY-30, reaction phase:extraction phase (v/v) = 3, at 160 °C and 800 rpm for 45 min under MW irradiation. | ||
Having established the ideal reaction and extraction media composition, the impact of mixing (stirring rate) was investigated, with an initial aim to minimise external mass transfer limitations across the liquid–liquid interface,77 as shown in Fig. 4. An initial increase in stirring rate, up to 200 rpm, coincides with an escalation in fructose conversion and thus HMF yield, due to reduced bulk mass diffusion to the catalyst, and superior PR, also due to mass diffusion from the reaction phase to the extraction one, which is enhanced from the mixing of the two phases, shown in Fig. S6,† and the corresponding elevation in the interface surface area. Further amplifying the stirring rate shows no subsequent boost in either conversion or product yield. In fact, these decrease as does the PR. At stirring rates above 300 rpm, the mixing of the phases and catalyst distribution throughout them is more uniform, which may account for the decrease in catalyst performance, as a greater degree of HY-30 is suspended into the extraction phase. With an increasingly dispersed system, i.e., with greater uniformity and hence moving away from a biphasic layered reaction medium, we observe a greater pressure within the reactor and reduced power input requirements (Fig. S7†) for a constant measured temperature. It is pointed out that the temperature is recorded at the external wall of the glass microwave vial at 1 cm height from the bottom of the vial, which coincides with the extraction phase in an unagitated system. The fact that there is a significant pressure difference, as a function of stirring, strongly suggests a significant temperature gradient between the two phases at low stirring rates, which is only overcome at 400 rpm. Evaluation of the pressure generated from heating the two phases separately, shown in Fig. S8,† reveals the reaction phase to be more volatile, and thus is the contributing factor to the different pressure recorded during the reaction (Fig. S7†). At low stirring rates, the recorded temperature more closely reflects the temperature of the extraction phase (given the location of the heat detector), whereas higher stirring rates, and therefore more uniformity in the media, result in a more uniform temperature and recording of the bulk temperature. Specifically, there is an increase in the reaction phase temperature based on the increased pressure, i.e., greater evaporation. This temperature difference at the lower stirring rates also accounts for the PR, with a favourable diffusion gradient from the cooler reaction phase to the hotter extraction phase, although mixing is also critical, and thus there is a trade-off, with 200 rpm providing an optimal compromise. Furthermore, with greater uniformity of the reaction media, the MW power requirements diminish due to the greater microwave heating response of the reaction phase, i.e., at low stirring, the detected temperature and focus of the monomodal microwave is predominantly on the extraction phase, which requires a greater power input to reach an external temperature of 160 °C. While 200 rpm may not result in optimal mixing, this is offset by optimal fructose dehydration performance and PR and thus is considered as the optimal process condition.
 | ||
| Fig. 4 Influence of stirring rate on catalytic fructose dehydration and PR within the optimal reaction media 6:4 (v/v) water:DMSO/7:3 (v/v) MIBK:2-BuOH system. Reaction conditions: 16 cm3 solvents, 5 wt% fructose, 2 wt% HY-30, reaction phase:extraction phase (v/v) = 3, at 160 °C and different RPM for 5 min under MW irradiation. | ||
The influence of reaction temperature on fructose conversion and HMF yield is reported in Fig. 5. Both initially increase with temperature and time before decreasing at prolonged reaction times for 160 °C and 180 °C due to the increased side product formation as the reaction progresses.78,79 Intuitively, dropping the temperature to 140 °C slows the reaction rate of fructose dehydration but positively impacts HMF selectivity, reaching close to 100% at 1–2 h before again diminishing with extended reaction time. However, at 140 °C, the impact on conversion outweighs the increased selectivity and thus dominates the resulting HMF yield, which is considerably lower than those obtained at the higher reaction temperatures. Evaluation of process energy efficiency (Table S6†), based on HMF production over HY-30, confirms an optimal reaction temperature and time of 160 °C and 45 minutes, with a 2.5 and 4.5-fold increase against the cases at 180 °C and 140 °C, respectively. As observed for the water-only solvent system, microwave heating has a similarly beneficial impact on the biphasic system when assessed against conventional heating, shown in Fig. 6, with the increase in catalytic performance and reduced energy consumption resulting in a 6.7-fold increase in process energy efficiency. Furthermore, deploying MW irradiation obtains a lower E factor (solvent recycled) of 0.93 V s 2.77 for conventional heating.
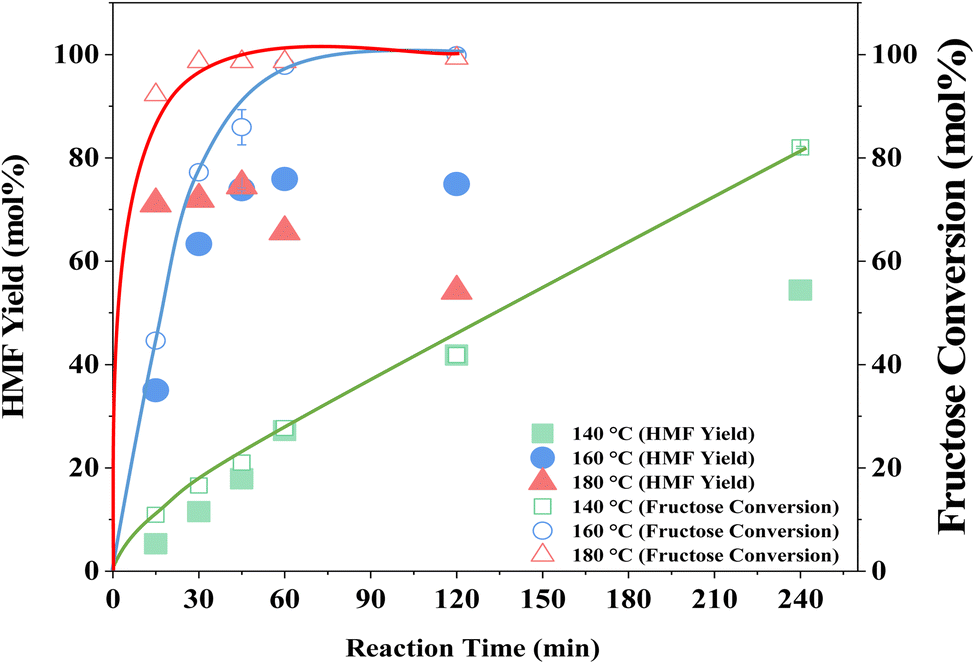 | ||
| Fig. 5 Effect of reaction temperature on reaction profiles for fructose conversion and HMF production. Reaction conditions: 16 cm3 solvents, 5 wt% fructose, 2 wt% HY-30, 6:4 (v/v) water:DMSO/7:3 (v/v) MIBK:2-BuOH with reaction phase:extraction phase (v/v) = 3, at 160 °C and 200 rpm under MW irradiation. The solid lines represent the trend of fructose conversion as a function of reaction time and are added to aid visualisation. | ||
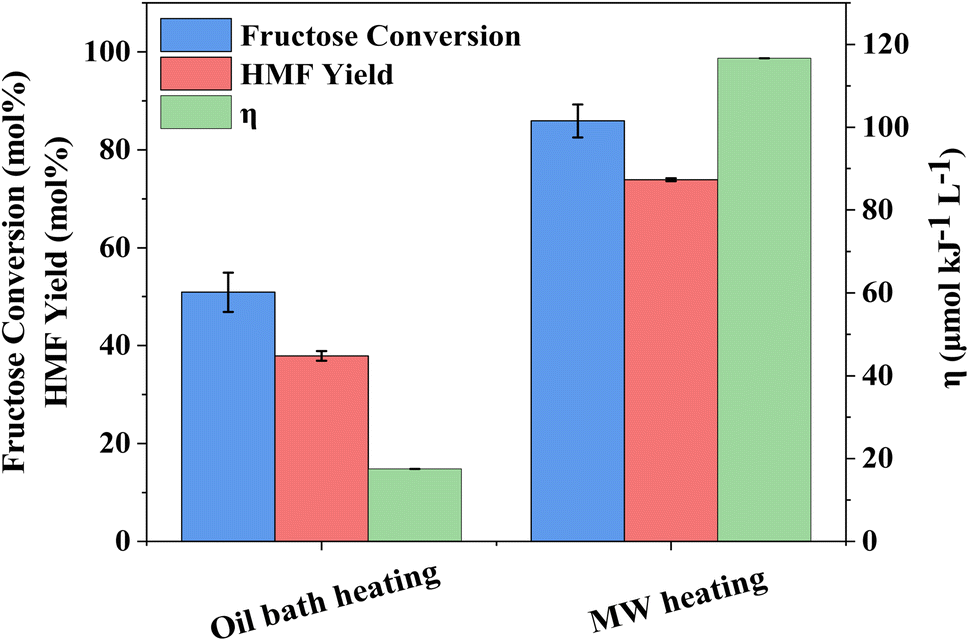 | ||
| Fig. 6 Comparison of microwave and conventional heating on fructose conversion, HMF yield and energy efficiency coefficient (η) in fructose dehydration over HY-30. Reaction conditions: 16 cm3 solvents, 5 wt% fructose, 2 wt% HY-30, 6:4 (v/v) water:DMSO/7:3 (v/v) MIBK:2-BuOH with reaction phase:extraction phase (v/v) = 3, at 160 °C and 200 rpm for 45 min. | ||
The capacity for recovery and reuse is a critical parameter to the overall success of any developed catalytic system. While the recovery of HY-30 is facile, via filtration, it is noted that the spent catalyst is discoloured (Fig. S9†), turning from pure white to a light brown, suggesting a degree of humin incorporation. That said, the impact appears to be minor, with good HMF selectivity and yields obtained for the subsequent runs (Fig. 7). After an initial drop of around 8% for selectivity and 10% for HMF yield after the first recycle, performance remained constant until the final recycle (run 5) which showed a further 5% decrease in yield (due to reduced conversion), but no further decrease in selectivity. These decreases are attributed to humin depositing on the catalyst, which may contribute to both pore and active site blockage. Regeneration of the catalyst by calcination (550 °C for 5 h in air), which consumed 3528 kJ of energy, recovered the catalyst to its initial bright white colour (Fig. S9c†) and reinstated the performance to that of the fresh catalyst. The capacity to return the catalyst activity to that of the fresh is clear evidence of a reversible catalyst deactivation mechanism, consistent with pore and site blockage by humins.
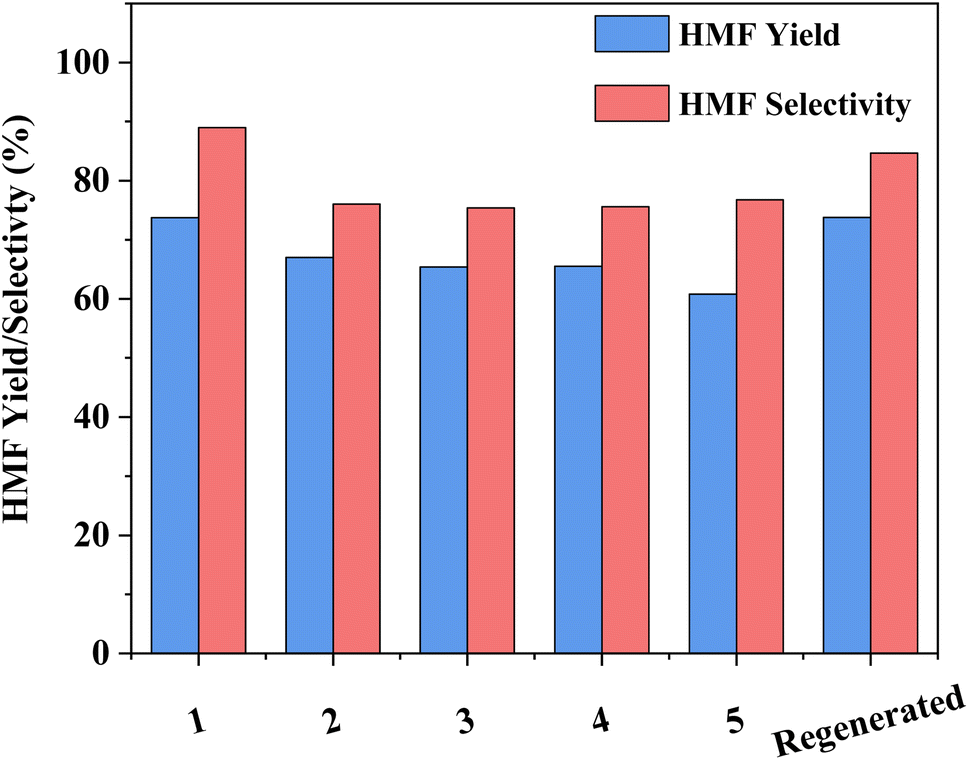 | ||
| Fig. 7 The reusability of HY-30 zeolite for HMF production from fructose, reaction 1 using the fresh catalyst and subsequent reactions 2–5 use the spent catalyst from the previous reaction recovered by centrifugation, washed with water, and dried at 80 °C in vacuum. The final reaction (regenerated) was conducted after catalyst reactivation through calcination at 550 °C. Reaction conditions: 16 cm3 solvents, 5 wt% fructose, 2 wt% HY-30, 6:4 (v/v) water:DMSO/7:3 (v/v) MIBK:2-BuOH with reaction phase:extraction phase (v/v) = 3, at 160 °C and 200 rpm for 45 min. | ||
Analysis of green metrics
Evaluation of the green credentials of our process in relation to the current state-of-the-art industrially relevant heterogeneous catalysts from the literature is depicted in Fig. 8, with a comparison to a more extensive list of literature-reported catalytic systems in Table S7.† Relative to other commercially available and industrially relevant zeolites and microporous zeo-type materials, including H-ZSM5,18 H-MOR zeolite30 and SAPO-44,46 performance is either comparable or enhanced under milder operating conditions and reduced reaction timescales. Comparison to heteropoly acids is less favourable, and these appear to be interesting candidates,72,73,80 however, they are plagued by their inherent solubility in polar solvents, which hinders their recovery.73,80 Other reported approaches have employed sulphonic acid functionality, typically supported on SiO2 or C.81,82 While having potential, these suffer lower hydrothermal stability, while using mesopore templating agents, common for SiO2 supports, can reduce the catalyst green credentials. | ||
| Fig. 8 Comparison of green metrics of industrially relevant heterogeneous catalysts for catalytic fructose dehydration to HMF employing biphasic reaction media. | ||
Conclusions
The union of a biphasic reaction medium, microwave irradiation, and a commercial hierarchical zeolite catalyst (HY-30) results in an optimal reaction process for highly selective and energy-efficient fructose dehydration to HMF. A study of a biphasic system to perform the conversion of fructose to HMF with its in situ extraction led to a media consisting of ((6:4 water:DMSO)/(7:3 MIBK:2-BuOH)) as the optimal composition. The catalyst displays both lower hydrophilicity and greater in-pore diffusion (through its hierarchical framework), relative to the two other HY zeolites initially investigated, resulting in a peak HMF yield (∼73.9%) in only 45 minutes at 160 °C, with the catalyst demonstrating good recyclability and facile regeneration. An analysis on the effect of stirring rate on reaction and extraction performance and potential limitations to mass transfer identified 200 rpm as the best one for operation. Simultaneously, the employment of energy-efficient microwave heating provides a 6.7-fold increase in the energy efficiency of HMF formation, with further evaluation through several green metrics validating the improved process sustainability. Further enhancements with regard to sustainability and green metric performance are likely to arise from the transition from a small-scale lab batch process to a continuous one, e.g., using a microwave continuous flow liquid phase reactor,83 rather than through increasing reactor size due to limited penetration depth of microwaves in absorbing reaction media.84 From these promising results, the commercially available hierarchical HY-30 zeolite can be considered an economical and robust solid acid catalyst for dehydrating fructose to HMF.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No. 872102. XO thanks the Zhejiang Provincial Natural Science Foundation for funding (LQ23B060005).References
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- G. K. Parshetti, M. S. Suryadharma, T. P. T. Pham, R. Mahmood and R. Balasubramanian, Bioresour. Technol., 2015, 178, 19–27 CrossRef CAS PubMed.
- F. K. Kazi, A. D. Patel, J. C. Serrano-Ruiz, J. A. Dumesic and R. P. Anex, Chem. Eng. J., 2011, 169, 329–338 CrossRef CAS.
- E. G. L. de Carvalho, F. d. A. Rodrigues, R. S. Monteiro, R. M. Ribas and M. J. da Silva, Biomass Convers. Biorefin., 2018, 8, 635–646 CrossRef CAS.
- X. Jia, I. K. M. Yu, D. C. W. Tsang and A. C. K. Yip, Microporous Mesoporous Mater., 2019, 284, 43–52 CrossRef CAS.
- S. L. Barbosa, M. d. S. Freitas, W. T. P. dos Santos, D. L. Nelson, S. I. Klein, G. C. Clososki, F. J. Caires, A. C. M. Baroni and A. P. Wentz, Sci. Rep., 2021, 11, 1919 CrossRef CAS.
- G. Sampath and S. Kannan, Catal. Commun., 2013, 37, 41–44 CrossRef CAS.
- S. F. Mayer, H. Falcón, R. Dipaola, P. Ribota, L. Moyano, S. Morales-delaRosa, R. Mariscal, J. M. Campos-Martín, J. A. Alonso and J. L. G. Fierro, Mol. Catal., 2020, 481 Search PubMed.
- L. Wang, L. Zhang, H. Li, Y. Ma and R. Zhang, Composites, Part B, 2019, 156, 88–94 CrossRef CAS.
- X. H. Kong, S. V. Vasudevan, M. J. Cao, J. Cai, H. P. Mao and Q. Bu, ACS Sustain. Chem. Eng., 2021, 9, 15344–15356 CrossRef CAS.
- X. H. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Green Chem., 2008, 10, 799–805 RSC.
- X. Lyu, H. Li, H. Xiang, Y. Mu, N. Ji, X. Lu, X. Fan and X. Gao, Chem. Eng. J., 2022, 428 Search PubMed.
- X. Guo, Q. Cao, Y. Jiang, J. Guan, X. Wang and X. Mu, Carbohydr. Res., 2012, 351, 35–41 CrossRef CAS.
- A. Pande, P. Niphadkar, K. Pandare and V. Bokade, Energy Fuels, 2018, 32, 3783–3791 CrossRef CAS.
- L. Hu, Z. Wu, J. Xu, Y. Sun, L. Lin and S. Liu, Chem. Eng. J., 2014, 244, 137–144 CrossRef CAS.
- X. Wang, H. Zhang, J. Ma and Z.-H. Ma, RSC Adv., 2016, 6, 43152–43158 RSC.
- M. Nahavandi, T. Kasanneni, Z. S. Yuan, C. C. Xu and S. Rohani, ACS Sustainable Chem. Eng., 2019, 7, 11970–11984 CAS.
- M. Moreno-Recio, J. Santamaría-González and P. Maireles-Torres, Chem. Eng. J., 2016, 303, 22–30 CrossRef CAS.
- X. Tian, B. Qi, S. Zhang, J. Luo and Y. Wan, Biomass Convers. Biorefin., 2021, 11, 1931–1941 CrossRef CAS.
- S. Roy Goswami, M.-J. Dumont and V. Raghavan, Ind. Eng. Chem. Res., 2016, 55, 4473–4481 CrossRef CAS.
- K. Y. Nandiwale, N. D. Galande, P. Thakur, S. D. Sawant, V. P. Zambre and V. V. Bokade, ACS Sustainable Chem. Eng., 2014, 2, 1928–1932 CrossRef CAS.
- P. H. Hoang, N. M. Dat, T. D. Cuong and D. T. Tung, RSC Adv., 2020, 10, 13489–13495 RSC.
- P. Wrigstedt, J. Keskiväli, M. Leskelä and T. Repo, ChemCatChem, 2015, 7, 501–507 CrossRef CAS.
- W. Wach, in Ullmann's Encyclopedia of Industrial Chemistry, 2004, https://doi.org/10.1002/14356007.a12_047.pub2 Search PubMed.
- X. Fu, Y. Hu, Y. Zhang, Y. Zhang, D. Tang, L. Zhu and C. Hu, ChemSusChem, 2020, 13, 501–512 CrossRef CAS.
- L.-K. Ren, L.-F. Zhu, T. Qi, J.-Q. Tang, H.-Q. Yang and C.-W. Hu, ACS Catal., 2017, 7, 2199–2212 CrossRef CAS.
- A. H. Motagamwala, K. Huang, C. T. Maravelias and J. A. Dumesic, Energy Environ. Sci., 2019, 12, 2212–2222 RSC.
- X. Zhang, K. Wilson and A. F. Lee, Chem. Rev., 2016, 116, 12328–12368 CrossRef CAS PubMed.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Catal. Commun., 2008, 9, 2244–2249 CrossRef CAS.
- C. Moreau, R. Durand, S. Razigade, J. Duhamet, P. Faugeras, P. Rivalier, P. Ros and G. Avignon, Appl. Catal., A, 1996, 145, 211–224 CrossRef CAS.
- V. Rac, V. Rakić, D. Stošić, O. Otman and A. Auroux, Microporous Mesoporous Mater., 2014, 194, 126–134 CrossRef CAS.
- V. V. Ordomsky, J. van der Schaaf, J. C. Schouten and T. A. Nijhuis, J. Catal., 2012, 287, 68–75 CrossRef CAS.
- Q. Zhu, G. D. Moggridge and C. D'Agostino, Chem. Eng. Sci., 2015, 132, 250–258 CrossRef CAS.
- C. D'Agostino, M. R. Feaviour, G. L. Brett, J. Mitchell, A. P. E. York, G. J. Hutchings, M. D. Mantle and L. F. Gladden, Catal. Sci. Technol., 2016, 6, 7896–7901 RSC.
- Y. Jiao, L. Forster, S. Xu, H. Chen, J. Han, X. Liu, Y. Zhou, J. Liu, J. Zhang, J. Yu, C. D'Agostino and X. Fan, Angew. Chem., Int. Ed., 2020, 59, 19478–19486 CrossRef CAS PubMed.
- C. D'Agostino, G. L. Brett, P. J. Miedziak, D. W. Knight, G. J. Hutchings, L. F. Gladden and M. D. Mantle, Chemistry, 2012, 18, 14426–14433 CrossRef.
- G. Morales, M. Paniagua, J. A. Melero and J. Iglesias, Catal. Today, 2017, 279, 305–316 CrossRef CAS.
- T. Zhang, H. Wei, H. Xiao, W. Li, Y. Jin, W. Wei and S. Wu, Mol. Catal., 2020, 498 Search PubMed.
- A. S. Amarasekara, L. D. Williams and C. C. Ebede, Carbohydr. Res., 2008, 343, 3021–3024 CrossRef CAS PubMed.
- X. Yi, I. Delidovich, Z. Sun, S. Wang, X. Wang and R. Palkovits, Catal. Sci. Technol., 2015, 5, 2496–2502 RSC.
- S. H. Mushrif, S. Caratzoulas and D. G. Vlachos, Phys. Chem. Chem. Phys., 2012, 14, 2637–2644 RSC.
- G. Portillo Perez, A. Mukherjee and M.-J. Dumont, J. Ind. Eng. Chem., 2019, 70, 1–34 CrossRef CAS.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- J. Esteban, A. J. Vorholt and W. Leitner, Green Chem., 2020, 22, 2097–2128 RSC.
- N. Sweygers, J. Harrer, R. Dewil and L. Appels, J. Clean. Prod., 2018, 187, 1014–1024 CrossRef CAS.
- P. Bhaumik and P. L. Dhepe, RSC Adv., 2013, 3, 17156–17165 RSC.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- A. Mohammad, Green Solvents I: Properties and Applications in Chemistry, Springer Science & Business Media, 2012 Search PubMed.
- J. González-Rivera, I. R. Galindo-Esquivel, M. Onor, E. Bramanti, I. Longo and C. Ferrari, Green Chem., 2014, 16, 1417–1425 RSC.
- R. Zhang, S. Xu, D. Raja, N. B. Khusni, J. Liu, J. Zhang, S. Abdulridha, H. Xiang, S. Jiang and Y. Guan, Microporous Mesoporous Mater., 2019, 278, 297–306 CrossRef CAS.
- M. J. Remy, D. Stanica, G. Poncelet, E. J. P. Feijen, P. J. Grobet, J. A. Martens and P. A. Jacobs, J. Phys. Chem., 1996, 100, 12440–12447 CrossRef CAS.
- F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752–768 RSC.
- V. Hessel, M. Escribà-Gelonch, J. Bricout, N. N. Tran, A. Anastasopoulou, F. Ferlin, F. Valentini, D. Lanari and L. Vaccaro, ACS Sustainable Chem. Eng., 2021, 9, 9508–9540 CrossRef CAS.
- F. Stallmach and J. Kärger, Adsorption, 1999, 5, 117–133 CrossRef CAS.
- F. Stallmach and P. Galvosas, in Annual Reports on NMR Spectroscopy, ed. G. A. Webb, Academic Press, 2007, vol. 61, pp. 51–131 Search PubMed.
- A. Ranoux, K. Djanashvili, I. W. C. E. Arends and U. Hanefeld, ACS Catal., 2013, 3, 760–763 CrossRef CAS.
- R. Montgomery and I. F. Wiggins, J. Soc. Chem. Ind., 1947, 66, 31–32 CrossRef CAS.
- B. M. Kabyemela, T. Adschiri, R. M. Malaluan and K. Arai, Ind. Eng. Chem. Res., 1999, 38, 2888–2895 CrossRef CAS.
- A. E. Beers, J. A. van Bokhoven, K. De Lathouder, F. Kapteijn and J. Moulijn, J. Catal., 2003, 218, 239–248 CrossRef CAS.
- J. Cui, J. Tan, T. Deng, X. Cui, Y. Zhu and Y. Li, Green Chem., 2016, 18, 1619–1624 RSC.
- Y. Wang, X. Yang, H. Zheng, X. Li, Y. Zhu and Y. Li, Mol. Catal., 2019, 463, 130–139 CrossRef CAS.
- M. Möller, F. Harnisch and U. Schröder, Biomass Bioenergy, 2012, 39, 389–398 CrossRef.
- K. W. Omari, J. E. Besaw and F. M. Kerton, Green Chem., 2012, 14 Search PubMed.
- S. Karwa, V. M. Gajiwala, J. Heltzel, S. K. R. Patil and C. R. F. Lund, Catal. Today, 2016, 263, 16–21 CrossRef CAS.
- M. Remy, D. Stanica, G. Poncelet, E. Feijen, P. Grobet, J. Martens and P. Jacobs, J. Phys. Chem., 1996, 100, 12440–12447 CrossRef CAS.
- S. Jiang, W. Xiong, B. He and M. Li, Cellulose, 2020, 28, 59–69 CrossRef.
- C. M. A. Parlett, P. Keshwalla, S. G. Wainwright, D. W. Bruce, N. S. Hondow, K. Wilson and A. F. Lee, ACS Catal., 2013, 3, 2122–2129 CrossRef CAS.
- C. D'Agostino, T. Kotionova, J. Mitchell, P. J. Miedziak, D. W. Knight, S. H. Taylor, G. J. Hutchings, L. F. Gladden and M. D. Mantle, Chemistry, 2013, 19, 11725–11732 CrossRef PubMed.
- C. D'Agostino, J. Mitchell, L. F. Gladden and M. D. Mantle, J. Phys. Chem. C, 2012, 116, 8975–8982 CrossRef.
- V. V. Ordomsky, J. C. Schouten, J. van der Schaaf and T. A. Nijhuis, Chem. Eng. J., 2012, 207–208, 218–225 CrossRef CAS.
- G. Lv, L. Deng, B. Lu, J. Li, X. Hou and Y. Yang, J. Clean. Prod., 2017, 142, 2244–2251 CrossRef CAS.
- Q. Zhao, L. Wang, S. Zhao, X. Wang and S. Wang, Fuel, 2011, 90, 2289–2293 CrossRef CAS.
- G. Tsilomelekis, T. R. Josephson, V. Nikolakis and S. Caratzoulas, ChemSusChem, 2014, 7, 117–126 CrossRef CAS PubMed.
- J. N. Chheda and J. A. Dumesic, Catal. Today, 2007, 123, 59–70 CrossRef CAS.
- J. N. Chheda, Y. Román-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- F. Borzeix, F. Monot and J.-P. Vandecasteele, Enzym. Microb. Technol., 1992, 14, 791–797 CrossRef CAS.
- S. K. R. Patil, J. Heltzel and C. R. F. Lund, Energy Fuels, 2012, 26, 5281–5293 CrossRef CAS.
- S. K. R. Patil and C. R. F. Lund, Energy Fuels, 2011, 25, 4745–4755 CrossRef CAS.
- C. Fan, H. Guan, H. Zhang, J. Wang, S. Wang and X. Wang, Biomass Bioenergy, 2011, 35, 2659–2665 CrossRef CAS.
- A. J. Crisci, M. H. Tucker, J. A. Dumesic and S. L. Scott, Top. Catal., 2010, 53, 1185–1192 CrossRef CAS.
- A. J. Crisci, M. H. Tucker, M.-Y. Lee, S. G. Jang, J. A. Dumesic and S. L. Scott, ACS Catal., 2011, 1, 719–728 CrossRef CAS.
- M. Baker-Fales, T. Y. Chen and D. G. Vlachos, Chem. Eng. J., 2023, 454, 13 CrossRef.
- T. N. Glasnov and C. O. Kappe, Macromol. Rapid Commun., 2007, 28, 395–410 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00162h |
| This journal is © The Royal Society of Chemistry 2023 |