
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, biocompatibility, and antimicrobial properties of glucose-based ionic liquids†
Stefan
Jopp‡
 *a,
Tabea
Fleischhammer
b,
Antonina
Lavrentieva
b,
Selin
Kara
b and
Johanna
Meyer‡
*b
*a,
Tabea
Fleischhammer
b,
Antonina
Lavrentieva
b,
Selin
Kara
b and
Johanna
Meyer‡
*b
aInstitute of Chemistry, University of Rostock, Albert-Einstein-Str. 3a, 18059, Rostock, Germany. E-mail: stefan.jopp@uni-rostock.de; johanna.meyer@iftc.uni-hannover.de
bInstitute of Technical Chemistry, Leibniz University Hannover, Callinstraße 5, 30167, Hannover, Germany
First published on 8th August 2023
Abstract
The diversity in structure, the variety in chirality, as well as the large occurrence of carbohydrates in nature, led to the development of the next generation of ionic liquids (ILs). These carbohydrate-based ionic liquids, also known as CHILs, are expected to overcome limitations such as aquatic ecotoxicity and poor biodegradability. In this work, we present the glucosyl imidazolium ILs, obtained via a simple two to three-step synthesis with total yields up to 90%. These compounds were obtained with several variations in alkyl- and aromatic side chains, glycosidic groups, anions, and protecting groups to study the influence of these variations on the biocompatibility and antimicrobial properties of the CHILs. The in vitro studies confirmed the biocompatibility of most of the CHILs for L929 mouse fibroblasts at 10−2 mol L−1, a feat not achieved by commercial imidazolium ILs. We could confirm observed trends, like increased cytotoxicity with increasing alkyl chain length, as well as higher fluorinated anions. Additionally, some of the here reported novel CHILs had significantly higher IC50 values than comparable imidazolium, pyridinium, and pyrrolidinium-based ILs. Additionally, antibiotic resistance is an increasingly serious threat to global health. Consequently, the development of new substances with antibiotic properties is of high priority. The before-synthesized CHILs were investigated in their overall antimicrobial behavior towards a Gram-negative strain (Escherichia coli K-12), a Gram-positive strain (Bacillus subtilis), as well as a common yeast (Candida auris WT) via the disk diffusion test. The minimum inhibition concentration (MIC), minimum bactericidal concentration (MBC), and minimum fungicidal concentration (MFC) were determined from the active substances. Similar to the biocompatibility experiments, correlations can be found between the length of the alkyl chain, the non-polarity of the structure, as well as the amount of fluorine in the counterion. For Candida auris, GMIM-NTf2 as well as GOIM-I show the strongest effect, with a MIC and MFC of 1 mmol L−1 and 5 mmol L−1, respectively.
Sustainability spotlightIonic liquids (ILs) possess diverse and tuneable properties, enabling them for a diverse field of applications, such as organic synthesis, biocatalysis, as well as biomedical applications. Especially for biomedical applications, ILs need to exert low toxicity, which starkly contrasts the usually high toxicity, aquatic ecotoxicity, and poor biodegradability of classical ILs. The next generation of ILs overcomes this problem, containing more biocompatible cations and anions, originating from inexpensive raw materials such as amino acids or choline, which are known to be non-toxic. In this work, we present carbohydrate-based ILs from renewable resources (SDG 15). Additionally, most of these new materials had significantly better biocompatibility, compared to imidazolium, pyridinium, and pyrrolidinium-based ILs (SDG 6). |
Introduction
Ionic liquids (ILs), defined as salts with a melting point below 100 °C, are a topic of broad interest due to their diverse and tunable properties as well as their wide field of applications.1 These applications include organic synthesis,2 biocatalysis,3 electrochemistry,4 extraction, and separation processes,5 analytical chemistry,6 as well as biomedical and pharmaceutical applications,7 among many others. Especially for biomedical applications, ILs need to exert low toxicity, which starkly contrasts the usually high toxicity, aquatic ecotoxicity, and poor biodegradability of classical ILs based on imidazolium and pyridinium cations and fluorated anions such as triflate and bistriflylimide.8,9 Additionally, the synthesis of these ILs of the first and second generation is neither cost-effective nor environmentally benign.10 The third generation of ILs is ought to overcome these problems, and is defined differently by various sources. On the one hand, the third generation is described by containing more biologically active ions, like pharmaceuticals or agrochemicals,11 on the other hand the generation is stated to contain more biocompatible cations and anions, originating from inexpensive raw materials such as amino acids or choline, which are known to be non-toxic.10,12 Another type of renewable resource that might be converted into ILs is carbohydrates. In recent years, these carbohydrate-based ionic liquids (CHILs)13–15 have found several potential applications, like a positive influence on the rate and selectivity of Diels–Alder cycloadditions,16 the possibility to adsorb metal cations like Pb2+ and Cd2+ from an aqueous solution,17,18 as precursors for N-doped carbon materials19 or as supported ionic liquid phase for the immobilized Candida antarctica lipase B biocatalyst (Novozym 435).20Jopp et al. recently developed several synthetic processes to produce ILs based on carbohydrates such as riboses,21 glucosamine,22 and glucose,20 and has furthermore proven remarkable biocompatibility of glucose and pentose-based pyridinium ILs.23 However, many syntheses towards these CHILs include several reaction steps, including insertion and removal of protecting groups and inserting a leaving group to enable following quaternization with N-heterocycles or amines. This might be followed by an additional anion exchange. Thus, the synthesis of CHILs often suffers from low total yields due to the multi-step procedure. In some cases, even when only a straightforward three-step synthesis is employed, the total yields are still below 40–50%, due to the overall complexity of downstreaming when working with carbohydrates.16
This work expands on our group's recently developed synthetic procedure in which simple glucosyl imidazolium ILs have been prepared in a two to three-step synthesis with total yields of up to 90%.20 These compounds will be synthesized to include several variations in alkyl- and aromatic side chains, varying glycosidic groups, different anions, and protecting groups to study the influence of these variations on the biocompatibility and antimicrobial properties of the CHILs.
According to the overall approved definition of biocompatibility published by Williams et al., it is the ability of a material to perform with an appropriate host response in a specific application.24 The selection of the suitable assay, international standard, or even the correct cell line should be well-considered. In most cases, the initial screening of biocompatibility is based on cell culture methods due to high sensitivity, reliability, and reproducibility, compared to other methods using algae and higher plants,25 invertebrate animals,26 vertebrate animals,27 or even bacteria.28 In this work, L929 mouse fibroblasts were used since this cell line is recommended by several biocompatibility regulation norms (e.g. DIN EN ISO 10993-1:2021-05). The mouse fibroblasts L929 are a reliable choice for materials and samples with skin contact, due to their important role in the wound healing process, for example around implants or drug delivery systems.29
Furthermore, we investigated the antimicrobial activity of these compounds with the disc diffusion method. The disk diffusion method, established by Bauer and Kirby et al., is the gold standard for testing antimicrobial susceptibility and was performed according to the Clinical Laboratory Standard Institute (CLSI) – Performance Standards for Antimicrobial Disk Susceptibility Tests.30,31 To obtain the minimum inhibition concentration (MIC) and minimum bactericidal concentration (MBC), the XTT assay was performed.
Experimental
Synthetic procedures and characterization data
The NMR spectra were recorded on a Bruker AVANCE 300 III or 500. CDCl3 was calibrated as 7.27 (1H) and 77.00 (13C). DMSO-d6 was calibrated as 2.49 (1H) and 39.50 (13C). D2O was calibrated as 4.80 (1H). ESI-MS was measured in an Agilent 1200/6210 Time-of-Flight LC-MS or a Thermo Scientific Exactive ESI/DART FTMS. The specific rotations were measured with a Dr Kernchen Gyromat-HP Digital Automatic Polarimeter with concentrations given in mg per mL. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements were performed on a Setaram Labsys TGA-DSC 1600 with a heating program from 25 to 500 °C and a heating rate of 10 °C min−1 under an argon flow of 100 mL min−1. Scanning electron microscopy (SEM) measurements were done using a JEM-ARM200F (JEOL), additional EDX investigations performed with the Dry SGD60 SDD-EDXS-Detector (JEOL). Acceleration voltage was between 10 and 20 kV. The silver content of the catalysts was measured by inductively coupled plasma optical emissions spectroscopy (ICP-OES) using the IRIS Intrepid II XPS device.The chemicals N-ethylimidazole (>98%), N-butylimidazole (>98%), N-octyllimidazole (>98%), and N-benzylimidazole (>98%) have been supplied by TCI. N-Methylimidazole (99%) was supplied by Alfa Aesar. N-Vinylimidazole (99%), silver methanesulfonate (99%), methyl-α-D-glucopyranoside (99%), phenyl-β-D-glucopyranoside (98%), triphenylphosphine (99%), and imidazole (99%) were supplied by Thermo Scientific. N-Phenylimidazole (>98%) and octyl-β-D-glucopyranoside (99%) were supplied by Apollo Scientific. Iodine (99.5%) and silver bis-(trifluoromethanesulfonyl)-imide (96%) were supplied by Carbolution. N-Mesitylimidazole (purity not given) was supplied by ABCR. The solvents THF (99.9%) and ethyl acetate (99.7%) were supplied by Honeywell Riedel-de-Haën and the solvents chloroform (99.8%), methanol (99.8%) and DMF (99.5) were supplied by Fisher. Column chromatography was performed with silica gel (230–400 mesh particle size) supplied by Supelco.
General procedure for the synthesis of 6-iodo-glucopyranosides
Glucopyranoside 1a–d (10 mmol), triphenylphosphine (15.5 mmol), iodine (14.5 mmol), and imidazole (20 mmol) were refluxed in THF (60 mL) for 4 h. The resulting solid was filtered off, the solvent was removed and the product was obtained after column chromatography.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
T m = 148–149 °C. [α]25D = +94.2 (c = 1.0, H2O). 1H-NMR (300 MHz, DMSO-d6): δ = 2.87–2.95 (m, 1H); 3.16–3.27 (m, 3H); 3.31 (s, 3H, CH3); 3.34–3.42 (m, 1H); 3.50–3.57 (m, 1H); 4.54 (d, 1H, 3J = 3.65 Hz, H-1); 4.78 (d, 1H, 3J = 6.43 Hz, OH); 4.86 (d, 1H, 3J = 4.99 Hz, OH); 5.17 (d, 1H, 3J = 5.83 Hz, OH). 13C-NMR (75 MHz, DMSO-d6): δ = 9.5 (C-6); 54.6 (CH3); 70.9, 71.9, 72.7, 74.1 (C-2, C-3, C-4, C-5); 99.8 (C-1).
:1 to 15:1.
T m = 72–74 °C. [α]24D = −10.7 (c = 1.3, H2O). 1H-NMR (300 MHz, DMSO-d6): δ = 0.82–0.87 (m, 3H, CH3); 1.25–1.32 (m, 10H, 5CH2); 1.48–1.55 (m, 2H, CH2); 2.89–3.04 (m, 3H); 3.11–3.19 (m, 1H); 3.24 (dd, 1H, 2J = 10.49 Hz, 3J = 7.35 Hz, H-6a); 3.42–3.47 (m, 1H, OCH2a); 3.51 (dd, 1H, 2J = 10.53 Hz, 3J = 2.26 Hz, H-6b); 3.69–3.77 (m, 1H, OCH2b); 4.16 (d, 1H, 3J = 7.83 Hz, H-1); 5.01 (t, 2H, 3J = 4.96 Hz, OH); 5.18 (d, 1H, 3J = 5.29 Hz, OH). 13C-NMR (75 MHz, DMSO-d6): δ = 9.1 (C-6); 13.9 (CH3); 22.1, 25.5, 28.7, 28.8, 29.3, 31.3, 68.8 (CH2); 73.4, 73.7, 74.7, 76.1 (C-2, C-3, C-4, C-5); 102.7 (C-1). HRMS (ESI, m/z): calculated for C14H27IO5+Na+, 425.0801; measured 425.0805.
:1.
T m = 157–159 °C. [α]25D = −82.1 (c = 1.9, H2O). 1H-NMR (500 MHz, DMSO-d6): δ = 3.02–3.06 (m, 1H); 3.24–3.35 (m, 4H); 3.56 (dd, 1H, 2J = 10.39 Hz, 3J = 1.90 Hz, H-6a); 4.93 (d, 1H, 3J = 7.66 Hz, H-1); 5.18 (d, 1H, 3J = 5.02 Hz, OH); 5.34 (d, 1H, 3J = 5.42 Hz, OH); 5.37 (d, 1H, 3J = 5.09 Hz, OH); 6.98–7.01 (m, 1H, HAr); 7.07–7.09 (m, 2H, HAr); 7.27–7.30 (m, 2H, HAr). 13C-NMR (125 MHz, DMSO-d6): δ = 8.5 (C-6); 73.3, 73.5, 75.0, 75.8 (C-2, C-3, C-4, C-5); 100.1 (C-1); 116.3, 121.9, 129.3 (CHAr); 157.2 (CAr). HRMS (ESI, m/z): calculated for C12H15IO5+, 365.9959; measured 365.9956.
:1 to 4:1.
[α]27D = +142.5 (c = 2.7, CHCl3). 1H-NMR (300 MHz, CDCl3): δ = 2.94–3.00 (m, 1H); 3.20 (dd, 1H, 3J = 9.62 Hz, 3J = 3.62 Hz, H-2); 3.29–3.39 (m, 2H); 3.46 (s, 3H, CH3); 3.50–3.56 (m, 2H); 3.52 (s, 3H, CH3); 3.61 (s, 3H, CH3); 3.62 (s, 3H, CH3); 4.82 (d, 1H, 3J = 3.62 Hz, H-1). 13C-NMR (75 MHz, CDCl3): δ = 7.5 (C-6); 55.5, 59.0, 60.8, 60.9 (CH3); 69.3, 81.8, 83.1, 83.4 (C-2, C-3, C-4, C-5); 97.5 (C-1). HRMS (ESI, m/z): calculated for C10H19IO5+Na+, 369.0169; measured 369.0175.
General procedure for the quarternization with imidazoles
6-Iodo-glucopyranoside 2a–d (3 mmol) and the N-substituted imidazole (5 mmol) were dissolved in DMF (5 mL) and stirred at 110 °C for 20 hours. After cooling down, ethyl acetate (40 mL) was added and the flask was stored in a fridge overnight. The solvent was decanted and the precipitated solid or oil was repeatedly washed with ethyl acetate and dried under a high vacuum to achieve the product.T m = 172–173 °C. Td = 246 °C. [α]25D = +63.4 (c = 1.8, H2O). 1H-NMR (300 MHz, D2O): δ = 3.26–3.32 (m, 1H); 3.31 (s, 3H, OCH3); 3.63 (dd, 1H, 3J = 9.77 Hz, 3J = 3.76 Hz, H-2); 3.72–3.78 (m, 1H); 3.94–3.98 (m, 1H); 4.00 (s, 3H, NCH3); 4.50 (dd, 1H, 2J = 14.60 Hz, 3J = 7.30 Hz, H-6a); 4.69 (dd, 1H, 2J = 14.60 Hz, 3J = 2.54 Hz, H-6b); 4.89 (d, 1H, 3J = 3.74 Hz, H-1); 7.55 (d, 1H, 3J = 1.95 Hz, HAr); 7.64 (d, 1H, 3J = 1.95 Hz, HAr); 8.89 (s, 1H, HAr). 13C-NMR (75 MHz, D2O): δ = 36.1 (NCH3); 49.9 (C-6); 55.2 (OCH3); 69.4, 70.5, 71.0, 72.9 (C-2, C-3, C-4, C-5); 99.3 (C-1); 123.2, 123.6, 137.0 (CHAr). HRMS (ESI, m/z): calculated for C11H19N2O5+, 259.1294; measured 259.1299. Calculated for I−, 126.9040; measured 126.9045.
T m = 99–101 °C. Td = 248 °C. [α]26D = +46.5 (c = 1.5, H2O). 1H-NMR (500 MHz, D2O): δ = 1.53 (t, 3H, 3J = 7.37 Hz, CH3); 3.22–3.26 (m, 1H); 3.25 (s, 3H, OCH3); 3.57 (dd, 1H, 3J = 9.78 Hz, 3J = 3.83 Hz, H-2); 3.68–3.71 (m, 1H); 3.92 (ddd, 1H, 3J = 10.05 Hz, 3J = 7.66 Hz, 3J = 2.44 Hz, H-5); 4.29 (q, 2H, 3J = 7.37 Hz, NCH2); 4.44 (dd, 1H, 2J = 14.60 Hz, 3J = 7.64 Hz, H-6a); 4.64 (dd, 1H, 2J = 14.59 Hz, 3J = 2.46 Hz, H-6b); 4.84 (d, 1H, 3J = 3.81 Hz, H-1); 7.57–7.60 (m, 2H, HAr); 8.90 (s, 1H, HAr). 13C-NMR (125 MHz, D2O): δ = 14.5 (CH3); 45.0 (NCH2); 49.9 (C-6); 55.0 (OCH3); 69.4, 70.5, 71.0, 72.9 (C-2, C-3, C-4, C-5); 99.3 (C-1); 122.0, 123.2, 136.0 (CHAr). HRMS (ESI, m/z): calculated for C12H21N2O5+, 273.1455; measured 273.1461. Calculated for I−, 126.9040; measured 126.9047.
T m = 185 °C. Td = 236 °C. [α]24D = +30.3 (c = 2.6, H2O). 1H-NMR (250 MHz, D2O): δ = 3.25 (s, 3H, OCH3); 3.24–3.28 (m, 1H, H-4); 3.58 (dd, 1H, 3J = 9.77 Hz, 3J = 3.77 Hz, H-2); 3.66–3.75 (m, 1H, H-3); 3.95 (ddd, 1H, 3J = 9.96 Hz, 3J = 7.47 Hz, 3J = 2.46 Hz, H-5); 4.50 (dd, 1H, 2J = 14.55 Hz, 3J = 7.38 Hz, H-6a); 4.70 (dd, 1H, 2J = 14.55 Hz, 3J = 2.55, H-6b); 4.85 (d, 1H, 3J = 3.77 Hz, H-1); 5.49 (dd, 1H, 3J = 8.68 Hz, 2J = 2.84 Hz, vinyl-CH2); 5.86 (dd, 1H, 3J = 15.58 Hz, 2J = 2.86 Hz, vinyl-CH2); 7.2 (dd, 1H, 3J = 15.58 Hz, 3J = 8.70 Hz, vinyl-CH); 7.70 (d, 1H, 3J = 2.09 Hz, HAr); 7.86 (d, 1H, 3J = 2.11 Hz, HAr); 9.16 (s, 1H, HAr). 13C-NMR (125 MHz, D2O): δ = 50.2 (C-6); 55.1 (OCH3); 69.2 (C-5); 70.5 (C-4); 71.0 (C-2); 72.8 (C-3); 99.3 (C-1); 109.8 (vinyl-CH2); 119.4, 123.8 (CHAr); 123.8 (vinyl-CH); 135.0 (CHAr). HRMS (ESI, m/z): calculated for C12H19N2O5+, 271.1299; measured 271.1306. Calculated for I−, 126.9040; measured 126.9045.
T d = 249 °C. [α]26D = +48.2 (c = 3.2, H2O). 1H-NMR (300 MHz, D2O): δ = 0.95 (t, 3H, 3J = 7.37 Hz, CH3); 1.26–1.39 (m, 2H, CH2); 1.85–1.94 (m, 2H, CH2); 3.21–3.27 (m, 1H); 3.25 (s, 3H, OCH3); 3.57 (dd, 1H, 3J = 9.77 Hz, 3J = 3.79 Hz, H-2); 3.67–3.74 (m, 1H); 3.92 (ddd, 1H, 3J = 10.18 Hz, 3J = 7.76 Hz, 3J = 2.58 Hz, H-5); 4.27 (t, 2H, 3J = 7.02 Hz, NCH2); 4.44 (dd, 1H, 2J = 14.57 Hz, 3J = 7.67 Hz, H-6a); 4.66 (dd, 1H, 2J = 14.52 Hz, 3J = 2.56 Hz, H-6b); 4.84 (d, 1H, 3J = 3.78 Hz, H-1); 7.57–7.61 (m, 2H, HAr); 7.97 (s, 1H, HAr). 13C-NMR (75 MHz, D2O): δ = 12.6 (CH3); 18.7, 31.2, 49.4 (CH2); 49.9 (C-6); 55.0 (OCH3); 69.4, 70.6, 71.0, 72.8 (C-2, C-3, C-4, C-5); 99.3 (C-1); 122.3, 123.2, 136.3 (CHAr). HRMS (ESI, m/z): calculated for C14H25N2O5+, 301.1768; measured 301.1763. Calculated for I−, 126.9040; measured 126.9050.
T d = 250 °C. [α]27D = +50.1 (c = 1.5, H2O). 1H-NMR (500 MHz, D2O): δ = 0.88 (t, 3H, 3J = 7.01 Hz, CH3); 1.27–1.34 (m, 10H, 5CH2); 1.88–1.93 (m, 2H, CH2); 3.22–3.26 (m, 1H); 3.23 (s, 3H, OCH3); 3.56 (dd, 1H, 3J = 9.77 Hz, 3J = 3.82 Hz, H-2); 3.68–3.71 (m, 1H); 3.91 (ddd, 1H, 3J = 10.17 Hz, 3J = 7.87 Hz, 3J = 2.48 Hz, H-5); 4.26 (t, 2H, 3J = 6.88 Hz, NCH2); 4.43 (dd, 1H, 2J = 14.58 Hz, 3J = 7.80 Hz, H-6a); 4.65 (dd, 1H, 2J = 14.55 Hz, 3J = 2.48 Hz, H-6b); 4.82 (d, 1H, 3J = 3.79 Hz, H-1); 7.57–7.61 (m, 2H, HAr); 8.91 (s, 1H, HAr). 13C-NMR (125 MHz, D2O): δ = 13.4 (CH3); 22.0, 25.2, 27.9, 28.1, 29.1, 30.9 (CH2); 49.7, 50.0 (CH2, C-6); 55.0 (OCH3); 69.4, 70.6, 71.0, 72.8 (C-2, C-3, C-4, C-5); 99.3 (C-1); 122.3, 123.2, 136.4 (CHAr). HRMS (ESI, m/z): calculated for C18H33N2O5+, 357.2394; measured 357.2397. Calculated for I−, 126.9040; measured 126.9051.
T m = 48–51 °C. Td = 239 °C. [α]24D = +28.3 (c = 3.0, H2O). 1H-NMR (500 MHz, D2O): δ = 3.28 (s, 3H, OCH3); 3.30–3.34 (m, 1H); 3.60 (dd, 1H, 3J = 9.79 Hz, 3J = 3.82 Hz, H-2); 3.71–3.75 (m, 1H); 4.01 (ddd, 1H, 3J = 10.00 Hz, 3J = 7.59 Hz, 3J = 2.45 Hz, H-5); 4.57 (dd, 1H, 2J = 14.56 Hz, 3J = 7.56 Hz, H-6a); 4.77 (dd, 1H, 2J = 14.56 Hz, 3J = 2.51 Hz, H-6b); 4.87 (d, 1H, 3J = 3.80 Hz, H-1); 7.66–7.68 (m, 5H, HAr); 7.81 (d, 1H, 3J = 2.09 Hz, HAr); 7.96 (d, 1H, 3J = 2.09 Hz, HAr). 13C–13C-NMR (125 MHz, D2O): δ = 50.4 (C-6); 55.1 (OCH3); 69.3, 70.5, 71.0, 72.9 (C-2, C-3, C-4, C-5); 99.4 (C-1); 121.8, 122.3, 123.9, 130.3 (CHAr); 134.6 (CAr). HRMS (ESI, m/z): calculated for C16H21N2O5+, 321.1455; measured 321.1453. Calculated for I−, 126.9040; measured 126.9042.
T m = 67–69 °C. Td = 255 °C. [α]24D = +37.8 (c = 2.9, H2O). 1H-NMR (300 MHz, D2O): δ = 3.09 (s, 3H, CH3); 3.24 (dd, 1H, 3J = 10.01 Hz, 3J = 8.90 Hz); 3.54 (dd, 1H, 3J = 9.77 Hz, 3J = 3.80 Hz, H-2); 3.65–3.71 (m, 1H); 3.86 (ddd, 1H, 3J = 10.36 Hz, 3J = 8.00 Hz, 3J = 2.47 Hz, H-5); 4.42 (dd, 1H, 2J = 14.52 Hz, 3J = 8.00 Hz, H-6a); 4.74 (dd, 1H, 2J = 14.51 Hz, 3J = 2.51 Hz, H-6b); 4.77 (d, 1H, 3J = 3.78 Hz, H-1); 5.46 (s, 2H, NCH2); 7.45–7.53 (m, 5H, HAr); 7.60–7.62 (m, 2H, HAr); 8.95 (s, 1H, HAr). 13C-NMR (75 MHz, D2O): δ = 50.1 (C-6); 53.0 (NCH2); 54.9 (CH3); 69.3, 70.6, 71.0, 72.8 (C-2, C-3, C-4, C-5); 99.2 (C-1); 122.4, 123.5, 128.5, 129.4, 129.4 (CHAr); 133.7 (CAr). HRMS (ESI, m/z): calculated for C17H22N2O5+, 335.1612; measured 335.1613. Calculated for I−, 126.9040; measured 126.9042.
T m = 155–157 °C. Td = 244 °C. [α]24D = +44.1 (c = 2.2, H2O). 1H-NMR (300 MHz, D2O): δ = 2.07 (s, 3H, CH3); 2.09 (s, 3H, CH3); 2.38 (s, 3H, CH3); 3.29 (dd, 1H, 3J = 9.87 Hz, 3J = 9.04 Hz); 3.31 (s, 3H, OCH3); 3.58 (dd, 1H, 3J = 9.78 Hz, 3J = 3.79 Hz, H-2); 3.72–3.78 (m, 1H); 4.03 (ddd, 1H, 3J = 10.11 Hz, 3J = 7.65 Hz, 3J = 2.59 Hz, H-5); 4.59 (dd, 1H, 2J = 14.46 Hz, 3J = 7.65 Hz, H-6a); 4.81 (dd, 1H, 2J = 14.36 Hz, 3J = 2.63 Hz, H-6b); 4.86 (d, 1H, 3J = 3.76 Hz, H-1); 7.19 (s, 2H, HAr); 7.64 (t, 1H, 3J = 1.76 Hz, HAr); 7.88 (t, 1H, 3J = 1.77 Hz, HAr); 9.09 (t, 1H, 3J = 1.56 Hz, HAr). 13C-NMR (75 MHz, D2O): δ = 16.4, 20.2 (CH3); 50.4 (C-6); 55.2 (OCH3); 69.2, 70.9, 71.1, 72.8 (C-2, C-3, C-4, C-5); 99.4 (C-1); 123.9, 124.1, 129.3 (CHAr); 130.7, 134.6 (CAr); 137.6 (CHAr); 141.5 (CAr). HRMS (ESI, m/z): calculated for C19H27N2O5+, 363.1925; measured 363.1927. Calculated for I−, 126.9040; measured 126.9046.
T d = 240 °C. [α]24D = −19.2 (c = 3.4, H2O). 1H-NMR (250 MHz, D2O): δ = 0.86–0.91 (m, 3H, CH3); 1.25–1.36 (m, 10H, 5CH2); 1.53–1.64 (m, 2H, CH2); 3.23–3.31 (m, 2H); 3.50–3.57 (m, 1H); 3.63–3.83 (m, 3H); 3.95 (s, 3H, NCH3); 4.39 (dd, 1H, 2 J = 14.47 Hz, 3J = 7.74 Hz, H-6a); 4.46 (d, 1H, 3J = 8.01 Hz, H-1); 4.69 (dd, 1H, 2J = 14.69 Hz, 3J = 2.41 Hz, H-6b); 7.51–7.54 (m, 2H, HAr); 8.81 (s, 1H, HAr). 13C-NMR (63 MHz, D2O): δ = 13.4 (CH3); 22.0, 25.0, 28.4, 28.7, 31.1 (CH2); 35.9 (NCH3); 50.0 (C-6); 70.9 (OCH2); 70.6, 73.0, 73.1, 75.5 (C-2, C-3, C-4, C-5); 102.2 (C-1); 122.2, 123.5, 136.8 (CHAr). HRMS (ESI, m/z): calculated for C18H33N2O5+, 357.2394; measured 357.2388. Calculated for I−, 126.9040; measured 126.9040.
T m = 195 °C. Td = 244 °C. [α]24D = −70.7 (c = 1.0, H2O). 1H-NMR (300 MHz, D2O): δ = 3.43–3.49 (m, 1H); 3.60–3.72 (m, 2H); 3.80 (s, 3H, NCH3); 3.94 (td, 1H, 3J = 9.35 Hz, 3J = 2.53 Hz, H-5); 4.37 (dd, 1H, 2J = 14.55 Hz, 3J = 8.97 Hz, H-6a); 4.74 (dd, 1H, 2J = 14.55 Hz, 3J = 2.48 Hz, H-6b); 5.15 (d, 1H, 3J = 7.51 Hz, H-1); 6.91–6.95 (m, 2H, HAr); 7.17–7.22 (m, 1H, HAr); 7.37–7.42 (m, 2H, HAr); 7.47–7.50 (m, 2H, HAr); 8.60 (s, 1H, HAr). 13C-NMR (75 MHz, D2O): δ = 35.8 (NCH3); 50.1 (C-6); 70.8, 72.4, 73.4, 75.3 (C-2, C-3, C-4, C-5); 99.1 (C-1); 116.3, 123.0, 123.4, 123.5, 129.8, 136.9 (CHAr); 155.8 (CAr). HRMS (ESI, m/z): calculated for C16H21N2O5+, 321.1455; measured 321.1461. Calculated for I−, 126.9040; measured 126.9042.
T m = 40–41 °C. Td = 309 °C. [α]24D = +55.6 (c = 3.4, H2O). 1H-NMR (300 MHz, D2O): δ = 3.21 (dd, 1H, 3J = 9.93 Hz, 3J = 9.00 Hz); 3.23 (s, 3H, CH3); 3.43 (dd, 1H, 3J = 9.82 Hz, 3J = 3.65 Hz, H-2); 3.51 (s, 3H, CH3); 3.56–3.62 (m, 1H); 3.64 (s, 3H, CH3); 3.65 (s, 3H, CH3); 3.89–3.95 (m, 1H); 3.96 (s, 3H, NCH3); 4.46 (dd, 1H, 2J = 14.51 Hz, 3J = 8.13 Hz, H-6a); 4.66 (dd, 1H, 2J = 14.49 Hz, 3J = 2.50 Hz, H-6b); 5.05 (d, 1H, 3J = 3.62 Hz, H-1); 7.52 (t, 1H, 3J = 1.79 Hz, HAr); 7.62 (d, 1H, 3J = 1.81 Hz, HAr); 8.86 (s, 1H, HAr). 13C-NMR (75 MHz, D2O): δ = 35.9 (NCH3); 49.7 (C-6); 54.8, 58.0, 60.0, 60.1 (OCH3); 68.5, 79.6, 79.9, 82.1 (C-2, C-3, C-4, C-5); 96.6 (C-1); 123.1, 123-6 (CHAr). HRMS (ESI, m/z): calculated for C14H25N2O5+, 301.1768; measured 301.1761. Calculated for I−, 126.9040; measured 126.9044.
General procedure for the anion exchange via silver iodide precipitation
The silver salt silver methanesulfonate or silver bis-(trifluoromethanesulfonyl)-imide (1 mmol) and 3a (1 mmol) were suspended in water (5 mL) and stirred for 20 h under absence of light. The product was obtained after filtration and removal of water. In some cases, black particles will form in the product after its storage under light. A further dissolution in methanol, filtration and drying leads to the full removal of these remaining silver particles.T m = 36–38 °C. Td = 244 °C. [α]25D = +76.7 (c = 1.3, H2O). 1H-NMR (300 MHz, D2O): δ = 2.84 (s, 3H, CH3); 3.22 (dd, 1H, 3J = 9.95 Hz, 3J = 9.05 Hz); 3.27 (s, 3H, OCH3); 3.56 (dd, 1H, 3J = 9.78 Hz, 3J = 3.79 Hz, H-2); 3.67–3.73 (m, 1H); 3.89–3.93 (m, 1H); 3.95 (s, 3H, NCH3); 4.45 (dd, 1H, 2J = 14.60 Hz, 3J = 7.35 Hz, H-6a); 4.64 (dd, 1H, 2J = 14.60 Hz, 3J = 2.51 Hz, H-6b); 4.84 (d, 1H, 3J = 3.77 Hz, H-1); 7.50–7.51 (m, 1H, HAr); 7.59–7.60 (m, 1H, HAr); 8.83 (s, 1H, HAr). 13C-NMR (75 MHz, D2O): δ = 35.8 (NCH3); 38.4 (CH3); 49.8 (C-6); 55.0 (OCH3); 69.4, 70.4, 71.0, 72.9 (C-2, C-3, C-4, C-5); 99.3 (C-1); 123.2, 123.5, 137.0 (CHAr). HRMS (ESI, m/z): calculated for C11H19N2O5+, 259.1294; measured 259.1298. Calculated for CH3O3S−, 94.9798; measured 94.9802.
T d = 250 °C. [α]25D = +55.8 (c = 2.1, H2O). 1H-NMR (300 MHz, D2O): δ = 3.23 (dd, 1H, 3J = 10.01 Hz, 3J = 8.97 Hz); 3.27 (s, 3H, OCH3); 3.57 (dd, 1H, 3J = 9.78 Hz, 3J = 3.80 Hz, H-2); 3.67–3.74 (m, 1H); 3.89–3.93 (m, 1H); 3.95 (s, 3H, NCH3); 4.45 (dd, 1H, 2J = 14.61 Hz, 3J = 7.36 Hz, H-6a); 4.64 (dd, 1H, 2J = 14.60 Hz, 2J = 2.55 Hz, H-6b); 4.84 (d, 1H, 3J = 3.78 Hz, H-1); 7.50–7.51 (m, 1H, HAr); 7.59–7.60 (m, 1H, HAr); 8.82 (s, 1H, HAr). 13C-NMR (75 MHz, D2O): δ = 35.7 (NCH3); 49.8 (C-6); 55.0 (OCH3); 69.4, 70.5, 71.0, 72.9 (C-2, C-3, C-4, C-5); 99.3 (C-1); 119.2 (q, 1J = 319.8 Hz, CF3); 123.2, 123.5, 136.9 (CHAr). 19F-NMR (282 MHz, D2O): δ = −79.2. HRMS (ESI, m/z): calculated for C11H19N2O5+, 259.1294; measured 259.1293. Calculated for C2F6NO4S2−, 279.9168; measured 279.9174.
In vitro biocompatibility
Antimicrobial activity testing
The examinations were performed in Mueller–Hinton agar (for fungal cultures 2% v/v glucose was added), prepared according to the manufacturer's instructions, and poured into 100 mm plates. The obtained bacteria solutions were adjusted for each organism to 1–2 × 107 CFU mL−1 (0.5 McFarland Standard, OD600 ≈ 0.120) and poured over the whole Mueller Hinton agar plate, using a sterilized bacteria solution-soaked cotton swab. 10 μL of the test solutions (10−1 mol L−1 in ultrapure H2O) were transferred to the Whatman No. 1 filter paper discs (6 mm in diameter) and placed with sterile forceps on the agar plates after the H2O was evaporated. As negative controls served filter paper discs with H2O only, and the positive controls were Gentamicin (Roti®Antibiotic Discs Gentamicin (GEN) 10 μg, 6 mm, 50 Units, Carl Roth, Karlsruhe, Germany) for the bacterial strains and Amphotericin B (Roti®Antibiotic Discs Amphotericin B (AP) 100 Units, Carl Roth, Karlsruhe, Germany) for the yeast. The bacteria agar plates were incubated for 18 h, and the yeast agar plates were incubated for 24 h at 35 °C ± 2 °C and the zones of inhibition (ZOI, diameter) were measured in mm.
Results
Synthesis of CHILs
The first step in the synthesis of the glucosylimidazolium ILs is an Appel reaction of the glucopyranosides 1a–d (Scheme 1). This reaction enables the direct conversion of a hydroxy group into a halogen, which will function as a leaving group for the following quarternization. Due to the higher reactivity in both this step as well as the following quarternization, we chose to work with iodine. Furthermore, the reaction has a very high selectivity towards the primary hydroxy group in the C-6 position of the carbohydrate. These advantages of the Appel reaction are a key component in this overall strategy, as they allow us to skip several steps of protecting group chemistry prior to the introduction of the leaving group, leading to overall fewer steps and higher total yields. Starting materials 1a–c are commercially available and the synthesis of starting material 1d has been previously described by Jopp et al.23 Interestingly, while the reaction protocol leads to yields of 91% and 89%, respectively, for the protected and unprotected α-methyl-glycosides 2a and 2d, both β-glycosides 2b and 2c were achieved in significantly lower yields of 41 and 51%, respectively. These reactions had remaining starting material; however, an extension of the reaction time from 4 to 20 hours did not increase the yields. Furthermore, the reaction with β-methylglucopyranoside as starting material leads to no product formation, leading us to conclude that the β-anomers are generally less reactive for this kind of Appel reaction.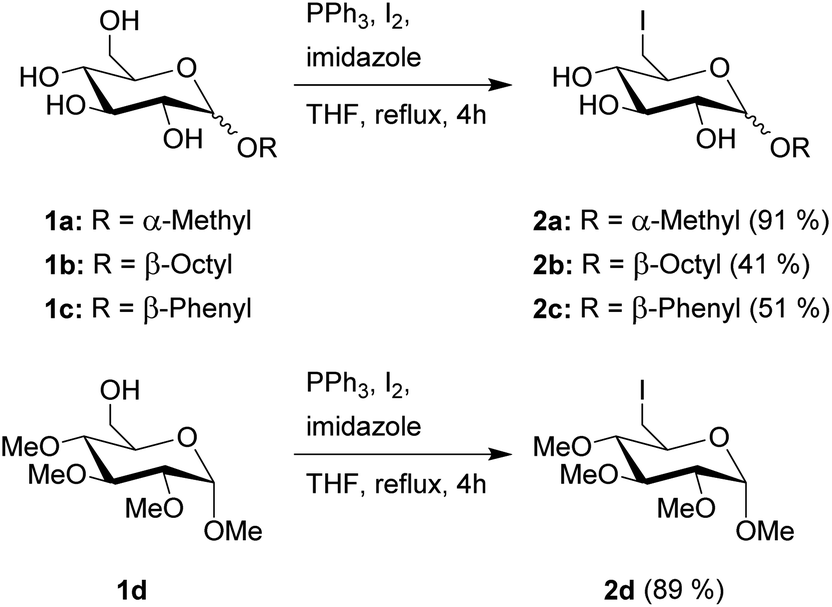 | ||
| Scheme 1 Synthesis of 6-iodo-glucopyranosides 2a–d. | ||
Next, we converted 2a into the CHILs 3a–h. This reaction is performed with varying N-substituted imidazoles in DMF (Scheme 2). After the reaction, most of the products can be obtained through precipitation after the addition of ethyl acetate to the reaction mixture. Only 3g needs a short column chromatography using ethyl acetate and methanol for purification since N-benzylimidazole is not sufficiently removable through washing after precipitation of 3g from ethyl acetate.
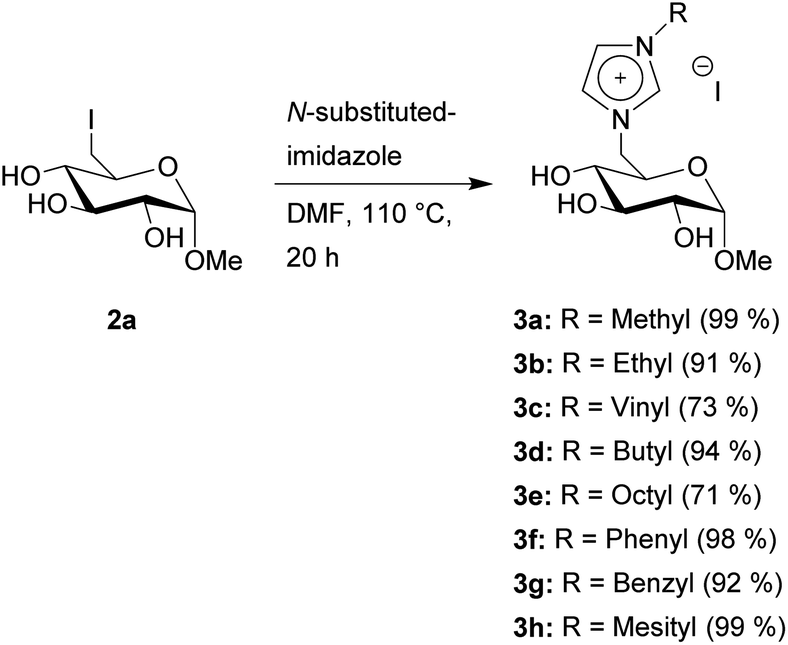 | ||
| Scheme 2 Synthesis of glucosyl imidazolium CHILs with varying side chains on the imidazole. | ||
Most of the products are obtained with yields of ≥90%. The vinyl product 3c needs a lowered reaction temperature of 95 °C, otherwise, the product will turn from a light-brown solid to a dark-brown oil, with no visible change in the NMR data however. This lowered reaction temperature also leads to a lower yield of 73%. The octyl-substituted product 3e was obtained in a yield of 71% due to the partial solubility of 3e in ethyl acetate, which was used for precipitation as described before.
To achieve two products directly comparable to 3a, but with different glycosidic groups, we converted 2b and 2c with N-methylimidazole to 3i–j (Scheme 3). Furthermore, the permethylated 2d was also converted with N-methylimidazole to 3k. These three products were synthesized and purified in the same manner as before and obtained with yields ranging from 81 to 94%.
 | ||
| Scheme 3 Synthesis of glucosylimidazolium CHILs with varying glycosidic groups and with permethylated hydroxy groups. | ||
As the last synthetic step, we produced two additional products through anion exchange. 3a and a silver salt were suspended in water, leading to the precipitation of silver iodide. The products 4a–b bearing a mesylate (OMs) and a bistriflylimide (NTf2) anion were both achieved in full conversion (Scheme 4). The full removal of the silver particles, which are formed during the anion exchange, was confirmed by SEM-EDX and ICP-OES measurements (Fig. S37 to S60†).
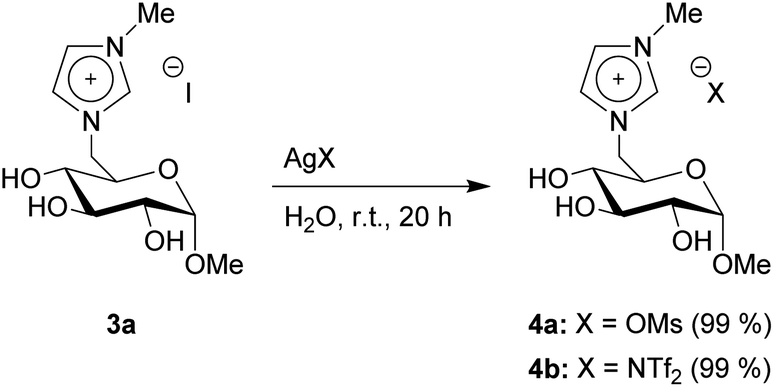 | ||
| Scheme 4 Anion exchange reactions. | ||
Since our novel ionic liquids are produced from natural resources and aimed to achieve a high biocompatibility, we were also interested to optimize our reactions to be as green as possible. For this, we performed a First Pass Green Chemistry Metrics test as published by Clark et al. for the two-step synthesis of GMIM-I 3a.34 In this toolkit, each reaction is given green, amber or red flags for several parameters such as yield and selectivity, but also for the toxicity of the used solvents or reagents, the use of catalysts and much more. Unsurprisingly, the first reaction from 1a to 2a fails this test in many parameters. The Appel reaction uses an excess of reagents, which leads to a low atom economy, was performed under reflux and was worked up by column chromatography with the highly hazardous chloroform, all of which are red flags. The reaction however has a high yield and high selectivity, which are major green flags. We made many efforts to address these red flag concerns. In case of the work-up with column chromatography, no other mixtures of ethyl acetate, acetonitrile or methanol were able to achieve a good separation on TLC. We also tried to move away from the column chromatography in general. We found that in case of product 2a, the extraction of an aqueous reaction mixture with toluene leads to the full removal of triphenylphosphine(oxide), leaving only product 2a and the remaining imidazole in the aqueous phase. This mixture however still needs a chloroform/methanol column chromatography.
Lastly, we tried to replace the imidazole with other bases like triethylamine and di-iso-propylethylamine or with other imidazoles like N-methyl-, N-butyl-, or N-benzylimidazole, which all might lead to an easier separation of 2a. Unfortunately, this proved to be unsuccessful as well, with all of these bases leading to either no full conversion of 1a or having no positive influence on the separation process. Finally, we also tried to perform the Appel reaction under catalytic conditions, as published by Werner et al. for organocatalytic chlorination, but this was also not possible for the carbohydrate starting material.35 As such, this reaction can unfortunately not be performed in a greener way. Other pathways to the same product 2a would most likely need multi-step reactions including the introduction and removal of protecting groups, as no known reaction is as selective as the Appel reaction. It should however be noted, as these points are not addressed in the green chemistry evaluation, that all side products can be fully removed from the reaction and would thus be available for other processes. Imidazolium hydroiodide is collected via filtration, and both the triphenylphoshine oxide and the remaining excess of imidazole can be fully separated by column chromatography without any mix phases.
On a more positive note, the second reaction from 2a to GMIM-I 3a passes the green chemistry evaluation in most of the parameters. This reaction has green flags for its nearly 100% yield as well as 100% selectivity and atom economy and an amber flag for the 87% reaction mass efficiency, since only a small excess of N-methylimidazole is used. The reaction is performed at medium–high temperatures (110 °C), which is an amber flag, but not under reflux, which is a green flag. The work-up is a filtration/washing with the green solvent ethyl acetate, which are again major green flags. Overall this reaction has only one concern, which is the red flag solvent DMF. This reaction however needs a highly polar solvent and a temperature of at least 110 °C, thus leaving not many alternatives. One less problematic solvent would be DMSO, however, the desirable work-up via filtration does not work from DMSO.
Thermal analysis of CHILs
Next, we performed the measurements of melting points (Tm) and decomposition temperatures (Td) for 3a–k and 4a–b (Table 1). The comparison of the melting points of 3a to 3h gives insight into the effect of the different side chains on the imidazolium cation. The melting points fall with increasing alkyl chain length, with the methyl-substituted salt 3a starting with a high melting point of 172–173 °C, the ethyl-substituted product 3b already bordering the ionic liquid definition with 99–101 °C and both butyl- and octyl-substituted 3c–d appearing as viscous liquids at room temperature. This follows the established trend for ionic liquids, in which bulky groups on either cation or anion lead to a lower ion interaction and thus to a lower melting point.36 The β-octyl glycoside 3i is a constitutional isomer of 3e. However, whether the octyl chain is bound to the imidazolium cation (3e) or to the anomeric center (3i), in both cases the products are viscous liquids at room temperature.| Product | Abbreviation | T m [°C] | T d [°C] |
|---|---|---|---|
| 3a | GMIM-I | 172–173 | 246 |
| 3b | GEIM-I | 99–101 | 248 |
| 3c | GVIM-I | 185 | 236 |
| 3d | GBIM-I | n.a. | 249 |
| 3e | GOIM-I | n.a. | 250 |
| 3f | GPhIM-I | 48–51 | 239 |
| 3g | GBnIM-I | 67–69 | 255 |
| 3h | GMesIM-I | 155–157 | 244 |
| 3i | OctO-GMIM-I | n.a. | 240 |
| 3j | PhO-GMIM-I | 195 | 244 |
| 3k | PerMe-GMIM-I | 40–41 | 309 |
| 4a | GMIM-OMs | 36–38 | 244 |
| 4b | GMIM-NTf2 | n.a. | 250 |
In contrast, the β-phenyl glycoside 3j, a constitutional isomer of 3f (Tm = 50 °C), exhibits the highest melting point of all products at 195 °C. Lastly, the anion exchange from iodide 3a to mesylate 4a and bistriflylimide 4b leads to a drastically lowered melting point. 4a has a melting point of 36–38 °C and 4b appears as a viscous liquid at room temperature. This again follows the established trends known for ionic liquids, in which sulfonate anions often have fewer interactions with the cations, thus reducing the melting point. While the melting points of all our CHILs allow the assessment of certain trends, all decomposition temperatures fall into the same range of around 240 to 250 °C, indifferent from the existing variations. This behavior has previously also been observed by our group for protected riboses21 as well as for trimethylammonium-glucosamines.37 Both, interestingly, fall into similar ranges with 230–250 °C and 240–260 °C, respectively. Thus, the overall thermal stability of these CHILs, including the aforementioned riboses and glucosamines, might be dictated by a function existing unchanged in all of these products, such as the anomeric center. Only one product exhibits a notably higher decomposition temperature: the permethylated glucosyl imidazolium product 3k.
In vitro biocompatibility analyses of CHILs
L929 cells are standard for biocompatibility testing by the international organization for standardization and, therefore, are commonly used. This work assessed the potential cytotoxicity of the CHILs to L929 cells using the CTB cell viability assay (Fig. 1) and live/dead staining with calcein-AM and propidium iodide (Fig. 2). As shown in Fig. 1, the CTB assay reveals that the CHILs, as well as the commercial ILs, are all cytotoxic at the highest tested concentration of 10−1 mol L−1. The trend of increasing toxicity to L929 mouse fibroblasts with increasing hydrophobicity, corresponding to an increasing alkyl chain length of the substituted groups on the imidazolium rest was observed (Fig. 1A). Thus, those CHILs with longer alkyl chains on carbohydrate cations were more toxic. In addition, cell viability is significantly decreased for CHILS with longer alkyl chains even after reducing the concentration to 10−2 mol L−1, which leads to the biocompatibility of most of the other substances. These two trends are also confirmed by the light microscopy images and the live/dead staining (Fig. 2). At the highest concentration of GMIM-I and GOIM-I, cells show a rounded morphology in both samples and a reduced growth compared to the control in the brightfield microscopy pictures, indicating that both compounds are cytotoxic at this concentration. However, the live/dead staining revealed a strong difference between the two substances. Whereas with GMIM-I some strongly rounded cells were stained with calcein-AM, there are no viable cells stained with the live stain at all with GOIM-I. The same correlation of chain lengths can also be observed in Fig. 1B. Longer chain lengths or generally a higher hydrophobicity, leads to lower cell viability. This can best be observed in the OctO-GMIM-I and GOIM-I data, as the cell viability similarly low in both cases. It can therefore be stated that the position of the alkyl chains is mostly insignificant. | ||
| Fig. 1 Cell viability after cultivation for 48 h of L929 cells in different concentrated (10−1 mol L−1; 10−2 mol L−1; 10−3 mol L−1; 10−4 mol L−1) CHIL solutions ((A) different imidazolium substituents; (B) different substituents at the anomeric center; (C) different counter ions and (D) commercial IL samples). The mean value of the wells without cells (background fluorescence) was subtracted from the fluorescence values of the rest of the wells, and the values of treated cultures were normalized to the mean fluorescence of the control cultures. Three biological replicates with six technical replicates each were analyzed, except for OctO-GMIM-I (3i; 10−2 mol L−1), PerMe-GMIM-I (3k; 10−2 mol L−1), GMIM-NTf2 (4b; 10−2 mol L−1) and EMIM-I (10−3 mol L−1). These four CHILs had one biological replication significantly different from the other two (Fig. S36 to S38†) and were defined as outliers. Every cell viability of each biological replicate is demonstrated in Fig. S36 to S38.† | ||
 | ||
| Fig. 2 (A) Microscopic images after 0 h and 48 h (scale bar 200 μm) and (B) calcein-AM/PI staining (scale bar 1000 μm) of L929 cells cultivated for 48 h in different concentrated (10−1 mol L−1; 10−2 mol L−1; 10−3 mol L−1; 10−4 mol L−1) CHIL solutions. One representative of each concentration of every CHIL is demonstrated in Fig. S39 to S48.† | ||
Additionally, we investigated anion-dependant patterns in the new CHILs (Fig. 1C), as well as in the studied commercial ILs (Fig. 1D). The influence of GMIM-OMs and GMIM-NTf2 to L929 fibroblasts lead to a lower cell viability than GMIM-I, which can already be considered biocompatible at a concentration of 10−2 mol L−1. It can be concluded that the counterions increase the toxicity of the overall CHIL with greater non-polarity or are more fluorinated. With the common imidazolium-ILs as well as for the commercially available EMIM-I and EMIM-NTf2 samples, the same trend can be observed. The HO-EMIM-I, which was synthesized in one of our previous works,20 to show the influence of hydroxy groups, which are of course also present in the CHILs, was an interesting comparison. It already showed significantly higher cell viability, especially at a concentration of 10−2 mol L−1. This could be due to the hydrophilic character of the structure. These findings could be confirmed by the calculated IC50 values (Fig. 3).
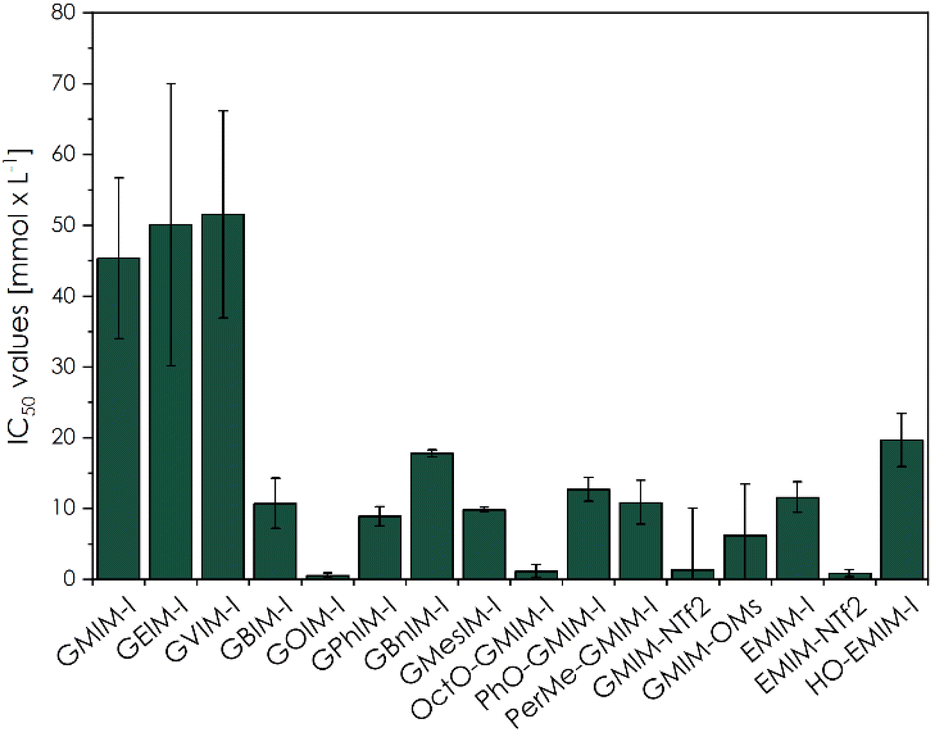 | ||
| Fig. 3 Comparison of IC50 values for L929 mouse fibroblasts towards the investigated CHILs (13 entries from left) and commercial IL samples (last three entries) during a 48 h incubation (CellTiter-Blue fluorescence). | ||
Antimicrobial activity
The initial antimicrobial screening of the new substances towards the Gram-positive strain B. subtilis, the Gram-negative strain E. coli K-12, as the yeast C. auris (WT) was carried out by disk diffusion assays (Fig. 4).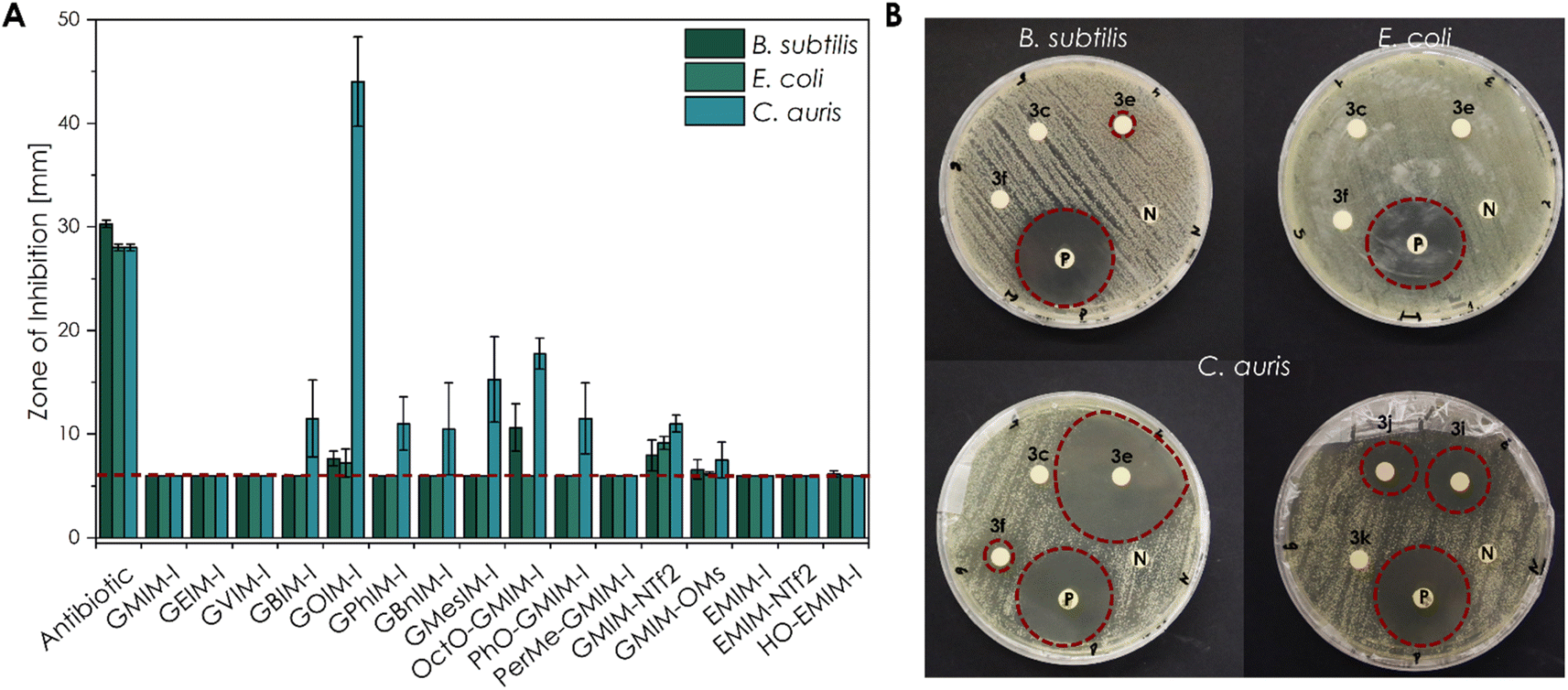 | ||
| Fig. 4 Antimicrobial activity of different CHIL samples against E. coli K-12, B. subtilis, and C. auris (WT) obtained by the disk diffusion method (10 μL of an aqueous 10−1 mol L−1 solution). (A) The mean diameter of the ZOI (in mm, including the 6 mm diameter of the disc) of all CHILs performed in n = 4 experiments, error bars indicate + SD. The red dashed line indicates the size of the filter disk itself so that only a ZOI with a bigger size showed an antimicrobial effect. Only a ZOI above this line showed an antimicrobial effect. (B) Representative sample agar plates showing the ZOI formed by the CHILs and the antibiotics (illustrated by the red dashed line; 3c GVIM-I; 3e GOIM-I; 3f GPhIM-I; 3i OctO-GMIM-I; 3j PhO-GMIM-I and 3k PerMe-GMIM-I). Complete overviews of the disk diffusion tests are given in Fig. S49 to S51.† | ||
Antimicrobial susceptibility disks, loaded with 10 μL of 10−1 mol L−1 aqueous CHIL solution, showed a high sensitivity of several substances. The obtained zones of inhibitions (ZOI) are clearly related to the structure of the investigated CHIL samples.
Based on the results, a clear structure–property relationship could be obtained. For all organisms, the most nonpolar compounds, as well as the most fluorinated compounds, exhibited the most pronounced effect. In particular, the CHIL with the longest alkyl chain GOIM-I and the CHIL GMIM-NTf2 with fluorinated anion showed the strongest effect on nearly all three organisms. In addition to the disc diffusion tests, broth microdilution experiments were performed to determine the respective MIC, as well as the MBC and MFC values (Table 2).
| Micro-organism | Product | MIC | MBC/MFC | |
|---|---|---|---|---|
| B. subtilis | 3e | GOIM-I | 2.5 | 2.5 |
| 3i | OctO-GMIM-I | 2.5 | 2.5 | |
| 4b | GMIM-NTf2 | 1 | 1 | |
| 4a | GMIM-OMs | 37.5 | 37.5 | |
| HO-EMIM-I | >125 | >125 | ||
| E. coli | 3e | GOIM-I | 2.5 | 5 |
| 4b | GMIM-NTf2 | 1 | 2.5 | |
| 4a | GMIM-OMs | 12.5 | 12.5 | |
| C. auris | 3d | GBIM-I | >125 | >125 |
| 3e | GOIM-I | 5 | 5 | |
| 3f | GPhIM-I | 75 | 75 | |
| 3g | GBnIM-I | 125 | 125 | |
| 3h | GMesIM-I | 37.5 | 37.5 | |
| 3i | OctO-GMIM-I | 10 | 10 | |
| 3j | PhO-GMIM-I | 125 | >125 | |
| 4b | GMIM-NTf2 | 1 | 1 | |
| 4a | GMIM-OMs | 37.5 | 37.5 | |
It can be concluded that the investigated substances show the most effect on C. auris compared to E. coli and B. subtilis. Similar to the biocompatibility experiments, correlations can be found between the length of the alkyl chain, the non-polarity of the structure, as well as the amount of fluorine in the counterion. In each organism, GMIM-NTf2 as well as GOIM-I show the strongest effect.
Discussion
As the main investigations of this work, after the synthesis of the novel CHILs, in total, the 13 glucosyl imidazolium ILs as well as three additional commercial imidazolium ILs have been tested for their in vitro biocompatibility and antimicrobial properties. The biocompatibility tests, performed with L929 mouse fibroblasts, showed higher cell viability for most of the CHIL-treated samples than the commercial imidazolium ILs. At 10−2 mol L−1 most of the CHILs can be considered biocompatible, a feat not achieved by commercial imidazolium ILs. Additionally, the antimicrobial properties of the CHILs were investigated with the disk diffusion test. The glucosyl imidazolium ILs exhibited a stronger effect on the yeast C. auris compared to the Gram-negative and Gram-positive bacteria E. coli and B. subtilis. Here, the octyl-substituted CHIL was able to reach zones of inhibition higher than the control antibiotic.We have furthermore defined trends, like increased cytotoxicity with increasing alkyl chain length. These correlations are in accordance with the data of ILs based on imidazolium from the literature.38 Docherty et al. investigated the antibacterial behavior of 1-alkyl-3-methyl g-imidazolium ILs towards different alkyl chain lengths against E. coli, S. aureus, and B. subtilis. When the alkyl chain of 1-hexyl-3-methyl imidazolium bromide was extended to 1-octyl-3-methyl imidazolium bromide, the EC50 value increased from 1.17 ppm to 6.44 ppm.39 The log EC50 values of Docherty et al. indicated a clear trend with 4.01 ± 0.05 μmol L−1 (BMIM-Br), 1.42 ± 0.05 μmol L−1 (HexMIM-Br), and 0.63 ± 0.05 μmol L−1 (OMIM-Br) with increasing chain length at the imidazolium cation against Vibrio fischeri.39,40 As well as the LC50 values against Artemia salina, published by Gouveia et al., with 0.092 ± 0.005 mmol L−1 (BuMIM-Br) and 0.079 ± 0.005 mmol L−1 (HexMIM-Br).26
According to the literature, the anions of ILs do not affect physical and chemical properties as much as the structure of the cation as well as the length of the alkyl side chain.25,27,41 However, biocompatibility can still be influenced by anions. In this study, we verified the dependency between the cytotoxicity of the investigated compounds and the fluorination of the counter ion. This is already described with different organisms by Cho et al. They defined the influences of counterions on the growth of Selenastrum capricornutum, indicating a decreased toxicity in the following order: SbF6− > PF6− > BF6− > NTf2− > Ts− > Br− ≈ Cl−.25 This would indicate higher toxicity if the anions are fluorinated. Other tendencies differ immensely through the literature. While Biczak et al. investigated different EMIM IL concentrations in soil towards common radish, resulting in a clear trend of the anions: DMP− (dimethyl phosphate) > OMs− > Ts− > NO3− > Br−.42 Rebros et al. could only find minor differences between OMs− and I− counterions on the different investigated imidazolium ILs.43 In our biocompatibility experiments as well as antibacterial testings, we could observe similar patterns: NTf2− > OMs− > I−.
The calculated the IC50 values from the L929 mouse fibroblasts towards the CHIL samples reached from 0.55 ± 0.31 mmol L−1 to 51.50 ± 14.61 mmol L−1, GOIM-I and GVIM-I, respectively. Compared to previously published CHILs by Reiß et al., these were within the expected range.23 When comparing the IC50 values of here described CHILs to other common ILs investigated by Dzida et al., the typical imidazolium-based ILs were reported with IC50 values from 0.17 ± 0.02 mmol L−1 of 1-methyl-3-octylimidazolium trifluoromethanesulfonate (OMIM-TFO) to 30.50 ± 2.50 mmol L−1 of 1-ethyl-3-methylimidazolium dimethylphosphate (EMIMDMP).44 These IC50 values are in the range of our samples, whereas the CHILs with shorter alkyl chains, such as GMIMI, GEIMI and GVIMI showed a significant higher value. The other by Dzida et al. reported IC50 values from pyridinium- and pyrrolidinium-based ILs to normal human dermal fibroblasts (NHDF) were even in lower ranges. Other more vaguely comparable studies, with EC50 values based on the decrease of light emission by P. phosphoreum, the reported values range from 0.70 ± 0.16 μmol L−1 (OMIM-PF6) to 4.02 ± 0.14 μmol L−1 (EMIMESO4).40
Conclusion
Synthetically this work features a total of 13 glucosyl imidazolium ILs, which have been prepared from commercially available glucosides in a straightforward two- or three-step synthesis with total yields up to 90%. Six of these CHILs have not been described in the literature before.The melting points as well as the decomposition temperatures of these CHILs have been put in perspective to confirm certain trends, like the reduction of the melting point, from 172–173 °C with a methyl group (GMIM-I) to the viscous liquid of GOIM-I at room temperature, with increasing bulk of the alkyl- or aromatic side chain. On the other hand, the decomposition temperatures, ranging from 236 °C (GVIM-I) to 255 °C (GBnIM-I), of the glucosyl imidazolium ionic liquids change only slightly with structural variations, confirming a trend found through several works of our group, indicating that most likely the anomeric center is the main influence on the thermal stability of the CHILs.
We have observed trends, like increased cytotoxicity with increasing alkyl chain length (GMIM-I ≈ GEIM-I ≈ GVIM-I < GBnIM-I < GBuIM-I < GPhIM-I ≈ GMesIM-I < GOIM-I), as well as higher fluorinated anions (NTf2− > OMs− > I−). Even though the organisms in the literature described in the discussion, are mostly different in the literature, these tendencies we observed with the here-described new CHILs were confirmed and extended in our experiments. In future studies, the mechanism of these cytotoxicity trends with longer chain lengths will be studied.
Additionally, some of the here reported novel CHILs had significantly higher IC50 values, than comparable imidazolium-, pyridinium-, and pyrrolidinium-based ILs. GVIM-I, the CHIL with the highest IC50 value of 51.50 ± 14.61 mmol L−1, showing a significantly higher biocompatibility compared to 0.17 ± 0.02 mmol L−1 of OMIM-TFO against P. phosphoreum, 0.392 mmol L−1 of 1-butylpyridinium chloride against S. vacuolatus or 2.43 mmol L−1 of 1-butyl-1-methylpyrrolidinium chloride against S. vacuolatus.44,45 This could path the way to novel applications of these compounds, such as an environmentally friendly solvent or a polymerizable monomer for biomaterials.
Author contributions
Stefan Jopp: conceptualization, methodology, formal analysis, investigation & writing – original draft; Tabea Fleischhammer: methodology, investigation, writing – review & editing; Antonina Lavrentieva: funding acquisition, writing – review & editing; Selin Kara: funding acquisition, writing – review & editing; Johanna Meyer: conceptualization, methodology, formal analysis, investigation & writing – original draft. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SJ thanks the German Research Center (Deutsche Forschungsgemeinschaft, DFG) for the financial support (NFDI4Cat; DFG grant no. 441926934). AL and TF thank the German Research Center (Deutsche Forschungsgemeinschaft, DFG) for the financial support (DFG grant no 398007461 488). SK and JM thank the Ministry for Science and Culture for Lower Saxony for the Holen & Halten starting grant (grant no. 12.5-76251-17-9/20). We also like to thank Martin Paehler, Martina Weiß, Caroline Mueller, Laura Schmitz from the Institute of Technical Chemistry TCI (Leibniz University Hannover), and Sandra Diederich from the Institute of Chemistry (University of Rostock) for their support on the day-to-day laboratory routine. Furthermore, we thank Sina Lambrecht from the Institute of Chemistry (University of Rostock), Tom Kunde and Riekje Biermann from the TCI (Leibniz University Hannover) for their assistance in the lab. Lastly, we thank Dr Carsten Kreyenschulte (Leibniz Institute for Catalysis, Rostock) and Dr Fanny Langschwager (Faculty of Mechanical Engineering and Marine Technologies, University of Rostock) for the measurements of SEM-EDX and ICP-OES, respectively.Notes and references
- G. Kaur, H. Kumar and M. Singla, Diverse applications of ionic liquids: A comprehensive review, J. Mol. Liq., 2022, 351, 118556, DOI:10.1016/j.molliq.2022.118556.
- Z. S. Qureshi, K. M. Deshmukh and B. M. Bhanage, Applications of ionic liquids in organic synthesis and catalysis, Clean Technol. Environ. Policy, 2014, 16, 1487–1513, DOI:10.1007/s10098-013-0660-0.
- L. E. Meyer, J. von Langermann and U. Kragl, Recent developments in biocatalysis in multiphasic ionic liquid reaction systems, Biophys. Rev., 2018, 10, 901–910, DOI:10.1007/s12551-018-0423-6.
- G. A. O. Tiago, I. A. S. Matias, A. P. C. Ribeiro and L. M. D. R. S. Martins, Application of Ionic Liquids in Electrochemistry-Recent Advances, Molecules, 2020, 25(24), 5812, DOI:10.3390/molecules25245812.
- S. P. M. Ventura, F. A. E Silva, M. v. Quental, D. Mondal, M. G. Freire and J. A. P. Coutinho, Ionic-Liquid-Mediated Extraction and Separation Processes for Bioactive Compounds: Past, Present, and Future Trends, Chem. Rev., 2017, 117, 6984–7052, DOI:10.1021/acs.chemrev.6b00550.
- T. D. Ho, C. Zhang, L. W. Hantao and J. L. Anderson, Ionic Liquids in Analytical Chemistry: Fundamentals, Advances, and Perspectives, Anal. Chem., 2014, 86(1), 262–285, DOI:10.1021/ac4035554.
- J. M. Gomes, S. S. Silva and R. L. Reis, Biocompatible ionic liquids: fundamental behaviours and applications, Chem. Soc. Rev., 2019, 48, 4317–4335, 10.1039/C9CS00016J.
- M. Petkovic, K. R. Seddon, L. P. N. Rebelo and C. S. Pereira, Ionic liquids: a pathway to environmental acceptability, Chem. Soc. Rev., 2011, 40, 1383–1403, 10.1039/C004968A.
- K. J. Kulacki and G. A. Lamberti, Toxicity of imidazolium ionic liquids to freshwater algae, Green Chem., 2008, 10, 104–111, 10.1039/B709289J.
- R. A. Sheldon, Biocatalysis in ionic liquids: state-of-the-union, Green Chem., 2021, 23, 8406–8427, 10.1039/D1GC03145G.
- W. L. Hough, M. Smiglak, H. Rodrìguez, R. P. Swatloski, S. K. Spear, D. T. Daly, J. Pernak, J. E. Grisel, R. D. Carliss, M. D. Soutullo, J. H. Davis and R. D. Rodgers, The third evolution of ionic liquids: active pharmaceutical ingredients, J. Chem., 2007, 31, 1429–1436, 10.1039/b706677p.
- K. Kuroda, A simple overview of toxicity of ionic liquids and designs of biocompatible ionic liquids, New J. Chem., 2022, 46, 20047–20052, 10.1039/D2NJ02634A.
- S. Jopp, Carbohydrate Based Ionic Liquids (CHILs): Synthesis and Applications, Eur. J. Org. Chem., 2020, 2020(41), 6418–6428, DOI:10.1002/ejoc.202000714.
- B. Gaida and A. Brzęczek-Szafran, Insights into the Properties and Potential Applications of Renewable Carbohydrate-Based Ionic Liquids: A Review, Molecules, 2020, 25(14), 3285, DOI:10.3390/molecules25143285.
- V. Zullo, A. Iuliano and L. Guazzelli, Sugar-Based Ionic Liquids: Multifaceted Challenges and Intriguing Potential, Molecules, 2021, 26(7), 2052, DOI:10.3390/molecules26072052.
- K. Erfurt, I. Wandzik, K. Walczak, K. Matuszek and A. Chrobok, Hydrogen-bond-rich ionic liquids as effective organocatalysts for Diels–Alder reactions, Green Chem., 2014, 16, 3508–3514, 10.1039/C4GC00380B.
- R. Jayachandra, R. Lakshmipathy and S. R. Reddy, Adsorption of Rare Earth Ce3+ and Pr3+ Ions by Hydrophobic Ionic Liquid, J. Mol. Liq., 2016, 219, 1172–1178, DOI:10.1155/2021/6612500.
- R. Jayachandra, S. R. Reddy and R. Lakshmipathy, D-Galactose based hydrophobic ionic liquid: A new adsorbent for the removal of Cd2+ ions from aqueous solution, Environ. Prog. Sustainable Energy, 2019, 38, S139–S145, DOI:10.1002/ep.12948.
- A. Brzeczek-Szafran, K. Erfurt, A. Blacha-Grzechnik, M. Krzywiecki, S. Boncel and A. Chrobok, Carbohydrate Ionic Liquids and Salts as All-in-One Precursors for N-Doped Carbon, ACS Sustainable Chem. Eng., 2019, 7, 19880–19888, DOI:10.1021/acssuschemeng.9b05297.
- P. Lehmann and S. Jopp, Novel Glucosylimidazolium Ionic-Liquid-Supported Novozym 435 Catalysts – A Proof of Concept for an Acrylation Reaction, ChemistryOpen, 2022, 11, e202200135, DOI:10.1002/open.202200135.
- M. Komabayashi, T. Stiller and S. Jopp, Structure-property relationships of ribose based ionic liquids, J. Mol. Liq., 2021, 325, 115167, DOI:10.1016/j.molliq.2020.115167.
- M. Komabayashi, S. Okushiba, T. Nokami and S. Jopp, Scope and Limitations in the Synthesis of Glucosamine-based Ionic Liquids, Asian J. Org. Chem., 2023, e202300093, DOI:10.1002/ajoc.202300093.
- M. Reiß, A. Brietzke, T. Eickner, F. Stein, A. Villinger, C. Vogel, U. Kragl and S. Jopp, Synthesis of novel carbohydrate based pyridinium ionic liquids and cytotoxicity of ionic liquids for mammalian cells, RSC Adv., 2020, 10, 14299–14304, 10.1039/D0RA01370F.
- D. F. Williams, There is no such thing as a biocompatible material, Biomaterials, 2014, 35, 10009–10014, DOI:10.1016/j.biomaterials.2014.08.035.
- C. W. Cho, T. P. T. Pham, Y. C. Jeon and Y. S. Yun, Influence of anions on the toxic effects of ionic liquids to a phytoplankton Selenastrum capricornutum, Green Chem., 2008, 10, 67–72, 10.1039/B705520J.
- W. Gouveia, T. F. Jorge, S. Martins, M. Meireles, M. Carolino, C. Cruz, T. v. Almeida and M. E. M. Araújo, Toxicity of ionic liquids prepared from biomaterials, Chemosphere, 2014, 104, 51–56, DOI:10.1016/j.chemosphere.2013.10.055.
- K. S. Egorova and V. P. Ananikov, Toxicity of ionic liquids: eco (cyto) activity as complicated, but unavoidable parameter for task-specific optimization, ChemSusChem, 2014, 7, 336–360, DOI:10.1002/cssc.201300459.
- J. Claus, A. Jastram, E. Piktel, R. Bucki, P. A. Janmey and U. Kragl, Polymerized ionic liquids-based hydrogels with intrinsic antibacterial activity: Modern weapons against antibiotic-resistant infections, J. Appl. Polym. Sci., 2021, 138, e50222, DOI:10.1002/app.50222.
- J. Claus, A. Brietzke, C. Lehnert, S. Oschatz, N. Grabow and U. Kragl, Swelling characteristics and biocompatibility of ionic liquid based hydrogels for biomedical applications, PLoS One, 2020, 15, e0231421, DOI:10.1371/journal.pone.0231421.
- A. W. Bauer, W. M. M. Kirby, J. C. Sherris and M. Turck, Antibiotic susceptibility testing by a standardized single diffusion method, Am. J. Clin. Pathol., 1966, 45, 493–496 CrossRef CAS PubMed.
- F. R. Cockerill, M. A. Wikler, J. Alder, M. N. Dudley, G. M. Eliopoulos, M. J. Ferraro, D. J. Hardy, D. W. Hecht, J. A. Hindler, J. B. Patel, M. Powell, J. M. Swenson, R. B. Thomson, M. M. Traczewski, J. D. Turnidge, M. P. Weinstein and B. L. Zimmer, Performance Standards for Antimicrobial Disk Susceptibility Tests; Approved Standard, 11th edn, 2012 Search PubMed.
- S. Winkler, K. V. Meyer, C. Heuer, C. Kortmann, M. Dehne and J. Bahnemann, In vitro biocompatibility evaluation of a heat-resistant 3D printing material for use in customized cell culture devices, Eng. Life Sci., 2022, 22(11), 699–708, DOI:10.1002/elsc.202100104.
- L. Raddatz, M. Kirsch, D. Geier, J. Schaeske, K. Acreman, R. Gentsch, S. Jones, A. Karau, T. Washington, M. Stiesch, T. Becker, S. Beutel, T. Scheper and A. Lavrentieva, Comparison of different three dimensional-printed resorbable materials: In vitro biocompatibility, In vitro degradation rate, and cell differentiation support, J. Biomater. Appl., 2018, 33(2), 281–294 CrossRef CAS PubMed.
- C. R. McElroy, A. Constatinou, L. C. Jones, L. Summerton and J. H. Clark, Towards a holistic approach to metrics for the21st century pharmaceutical industry, Green Chem., 2015, 17, 3111–3121, 10.1039/c5gc00340g.
- L. Longwitz, S. Jopp and T. Werner, Organocatalytic Chlorination of Alcohols by P(III)/P(V) Redox Cycling, J. Org. Chem., 2019, 84, 7863–7870, DOI:10.1021/acs.joc.9b00741.
- J. O. Valderrama and R. E. Rojas, Data selection and estimation of the normal melting temperature of ionic liquids using a method based on homologous cations, C. R. Chimie, 2012, 15, 693–699, DOI:10.1016/j.crci.2012.04.011.
- M. Komabayashi, T. Nokami and S. Jopp, From Chitin to CHILs: First Glucosamine based Ionic Liquids, Asian J. Org. Chem., 2020, 9(12), 2092–2094, DOI:10.1002/ajoc.202000569.
- M. Petkovic, J. L. Ferguson, H. Q. N. Gunaratne, R. Ferreira, M. C. Leitão, K. R. Seddon, L. P. N. Rebelo and C. S. Pereira, Novel biocompatible cholinium-based ionic liquids—toxicity andbiodegradability, Green Chem., 2010, 12, 643–664, 10.1039/b922247b.
- K. M. Docherty and C. F. Kulpa, Toxicity and antimicrobial activity of imidazolium and pyridinium ionic liquids, Green Chem., 2005, 7, 185–189, 10.1039/B419172B.
- A. Romero, A. Santos, J. Tojo and A. Rodríguez, Toxicity and biodegradability of imidazolium ionic liquids, J. Hazard. Mater., 2008, 151, 268–273, DOI:10.1016/j.jhazmat.2007.10.079.
- S. P. M. Ventura, A. M. M. Gonçalves, T. Sintra, J. L. Pereira, F. Gonçalves and J. A. P. Coutinho, Designing ionic liquids: the chemical structure role in the toxicity, Ecotoxicol, 2013, 22, 1–12, DOI:10.1007/s10646-012-0997-x.
- R. Biczak, B. Pawłowska, P. Bałczewski and P. Rychter, The role of the anion in the toxicity of imidazolium ionic liquids, J. Hazard. Mater., 2014, 274, 181–190, DOI:10.1016/j.jhazmat.2014.03.021.
- M. Rebros, H. Q. N. Gunaratne, J. Ferguson, K. R. Seddon and G. Stephens, A high throughput screen to test the biocompatibility of water-miscible ionic liquids, Green Chem., 2009, 11, 402–440, 10.1039/B815951C.
- M. Musiał, E. Zorębski, K. Malarz, M. Kuczak, A. Mrozek-Wilczkiewicz, J. Jacquemin and M. Dzida, Cytotoxicity of Ionic Liquids on Normal Human Dermal Fibroblasts in the Context of Their Present and Future Applications, ACS Sustainable Chem. Eng., 2021, 9(22), 7649–7657, DOI:10.1021/acssuschemeng.1c02277.
- S. P. M. Ventura, M. Gurbisz, M. Ghavre, F. M. M. Ferreira, F. Gonçalves, I. Beadham, B. Quilty, J. A. P. Coutinho and N. Gatherhood, Imidazolium and Pyridinium Ionic Liquids from Mandelic AcidDerivatives: Synthesis and Bacteria and Algae Toxicity Evaluation, ACS Sustainable Chem. Eng., 2013, 1, 393–402, DOI:10.1021/sc3001299.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00191a |
| ‡ The authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |