
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cholinium-based ionic liquid catalysts for polyethylene terephthalate glycolysis: understanding the role of solvent and a reappraisal of the cation contribution†
Diana
Bura
 a,
Lorenzo
Pedrini
a,
Cristina
Trujillo
*ab and
Stephen J.
Connon
*a
a,
Lorenzo
Pedrini
a,
Cristina
Trujillo
*ab and
Stephen J.
Connon
*a
aSchool of Chemistry, Trinity Biomedical Sciences Institute, Trinity College Dublin, 152-160 Pearse St., Dublin 2, Ireland. E-mail: connons@tcd.ie
bDepartment of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: cristina.trujillodelvalle@manchester.ac.uk
First published on 19th October 2023
Abstract
A combined computational and experimental investigation has revealed a new pathway for the degradation of PET using cholinium-based ionic liquid catalysis, which led to a reappraisal of the role of the cholinium unit in catalysis and the development of more efficacious related systems.
Sustainability spotlightThere is a growing interest in utilising choline derivatives as ionic liquid catalysts for the glycolysis-based recycling of polyethylene terephthalate. This study employs DFT methods to reveal that the cholinium ion's role in the reaction mechanism has been previously overstated in the literature. Building on this mechanistic understanding, we have developed more efficient ionic liquid catalysts for plastic recycling that do not contain the cholinium component. These novel catalysts deliver higher yields with reduced loading, aligning perfectly with the objectives of ‘UN SDG 12: ensure sustainable consumption and production patterns.’ This research underscores the importance of considering the impact of biodegradable catalyst units on activity when designing environmentally friendly catalysts for plastic recycling. |
Petroleum-derived plastics are a central feature of modern life. The burgeoning generation1,2 and disposal of these products are contributors to the current climatological and ecological emergency. For instance, it has been estimated that there will be greater mass of plastic than fish in the oceans by 2050.3 Polyethylene terephthalate (PET, 1) is a fossil fuel-derived constituent of beverage bottles, fibres, and food packaging, which in 2021 represented ca. 45% of single-serve small drink packages in the United States, and 12% of global solid waste.4
PET waste has characteristics distinct from virgin polymer due to degradation and absorption of chemicals, which complicates mechanical/physical recycling processes (which dominate the commercial recycling landscape and can further degrade the PET) challenged with producing recycled PET of appropriate purity for a variety of applications.4–7 As a result, recycled PET in consumer products is often a blend comprising virgin polymer and recycled material; which further drives demand for fossil fuel-derived virgin PET.6
Chemical recycling by catalytic depolymerisation to a monomeric species which can be purified and repolymerised to generate material with properties identical to virgin polymer is considerably less often applied, yet holds potential (if efficient) to obviate the need for either downcycling or blending recycled with virgin PET polymer in the recycled plastic.8 Various modalities exist by which depolymerisation can be accomplished, e.g. aminolysis, hydrolysis (including biocatalytic variants), alcoholysis and glycolysis;8 the latter methodology holding particular promise due to its amenability to reaction at atmospheric pressure and the generation of bis(2-hydroxyethyl) terephthalate (BHET 2) – which can be readily repolymerised to 1 (Fig. 1A). An array of catalysts have emerged including metal salts,9 metal complexes,10 heterogeneous materials,11 nanocatalysts12 organocatalysts,13 deep eutectic solvents14 and ionic liquids (ILs).15,16 In an attempt to design IL-based catalysts of lower concern from an environmental toxicity perspective,17 several studies have recently disclosed the use of cholinium-based metal-free ILs 3–5 in the degradation of PET (Fig. 1B).18 Excellent activity and selectivity have been reported.
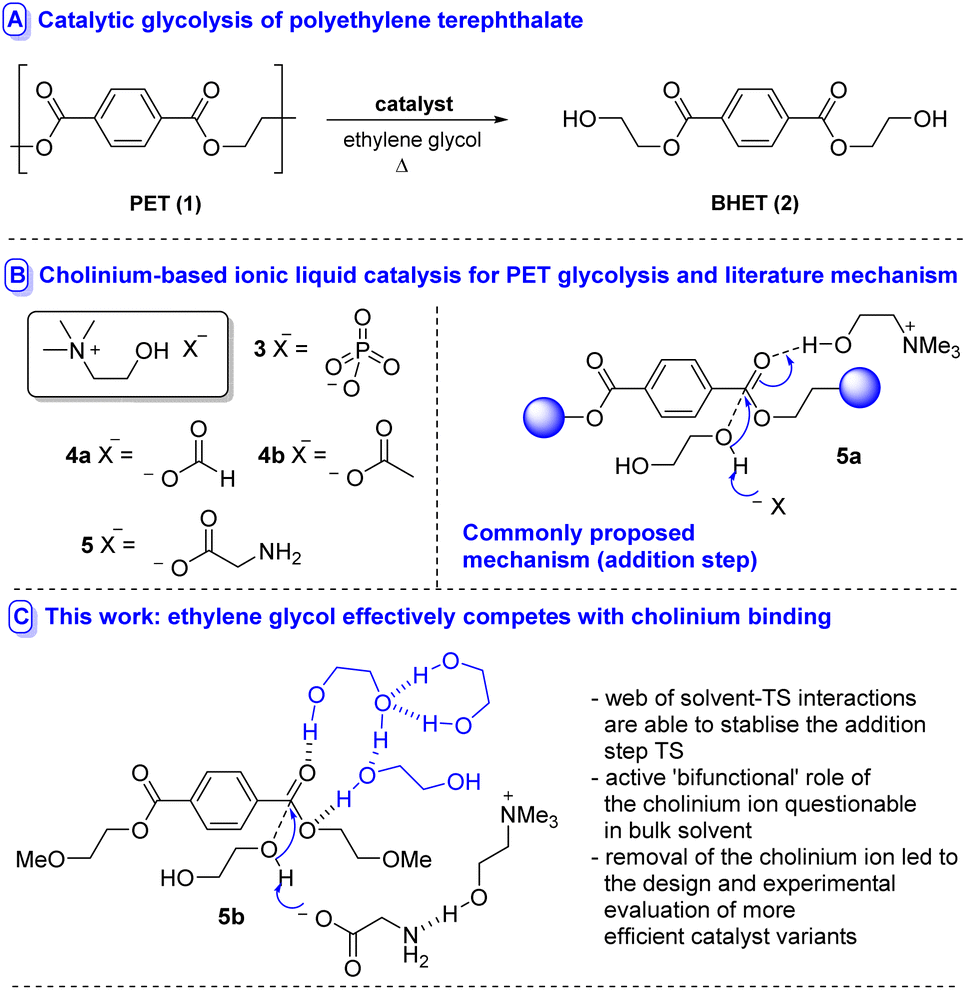 | ||
| Fig. 1 Catalytic glycolysis of PET. | ||
Where mechanistic details have been proposed based on calculation/spectroscopic evidence,18a,b a bifunctional catalysis mode of action has been advanced (i.e.5a, Fig. 1b) in which the cholinium ion stabilises developing negative charge (through either the cation or the hydroxyl unit) in the addition step transition state while the anion acts as a general base. Similar modes of catalysis have been advanced using another IL-based catalysis for PET-recycling.13h,19,20
While this mode of action is an intuitive concept, it raises questions regarding the ability of the cholinium moiety to stabilise developing negative charge in the face of competition from ethylene glycol – a hydrophilic strong H-bond donor – solvent. In order to better understand the modes by which cholinium cations can participate in these reactions and to understand the role of the glycol and base, with a view to guiding the course of future catalyst design, we studied the glycolysis of 1 by explicit inclusion of solvent molecules mediated by cholinium glycinate (5)18c at 180 °C using DFT.
The investigation began with an examination of the literature mechanism for PET glycolysis using cholinium-based catalysts. This mode of activation involves the interaction of the cholinium cation with the PET carbonyl moiety via hydrogen bonding to stabilise the charge aggregation in the addition step transition state (Fig. 2, pathway I). Since the reaction occurs in ethylene glycol solvent, 3 explicit glycol molecules (in addition to the glycol which serves as the attacking nucleophile) were included in the computational scenario. An alternative mechanism was considered in which glycol molecules (and not cholinium ion) stabilise developing charge by H-bonding to the ester carbonyl unit, while the cholinium cation interacts primarily with the glycinate anion as the carboxylate group associated with the latter acts as a general base in the addition step (Fig. 2, pathway II). To ensure valid comparison between the literature and newly proposed pathways, the same number of explicit stabilising solvent molecules were considered for both calculations. The conformational space was thoroughly examined; in addition, calculations were performed to investigate the possibility of the amino moiety acting as the base (Fig. 2, pathway III). In the interests of brevity, only lower energy orientations will be discussed in detail here. The remaining are provided in the ESI (Fig. S1).†
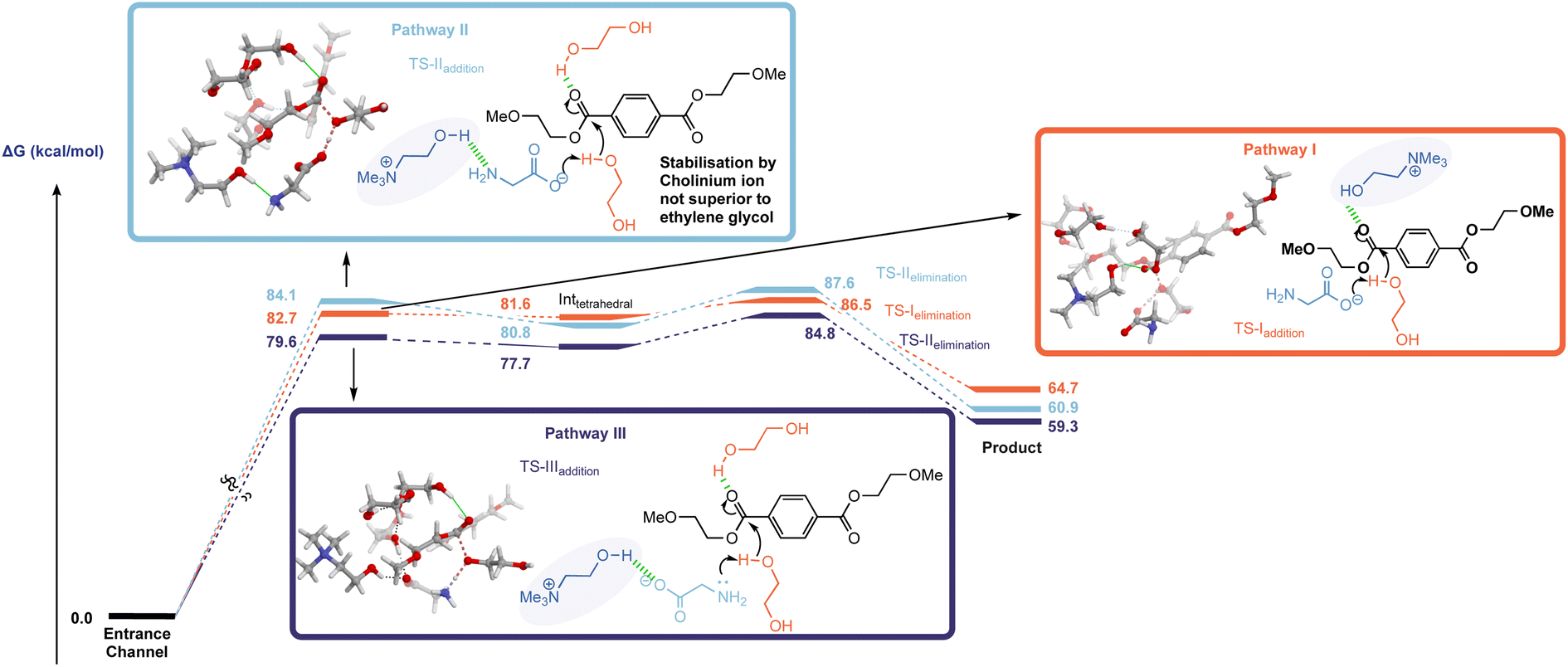 | ||
| Fig. 2 Free energy profile of PET glycolysis for pathway I (orange), pathway II (light blue) in which OH of cholinium ion acts as a base, and pathway III, in which amine associated with the glycinate ion acts as a base (dark blue). | ||
The free energy profiles of both activation modes (Fig. 2), pathway I and II, indicate that both TSaddition and TSelimination are energetically accessible in the literature- and newly-proposed modes of action. The elimination step appears to be marginally rate determining, and it is clear that while cholinium ion H-bonding is a viable activation mode, the difference between it and simple solvolytic stabilisation of developing charge is within the margin of error – even with the inclusion of only three glycol molecules to represent the solvent contribution to transition state stabilisation.
Analysis of the non-covalent interactions (Fig. S2†) of the two mechanisms by QTAIM (Quantum Theory of Atoms in Molecules) allowed the identification of a web of interactions established upon complexation which underpin the proposed solvolytic hydrogen bond-assisted catalysis (i.e. pathway II).
An interesting discrepancy between pathways I and II relates to the mobility of the cholinium ion. In pathway I this plays a key role (vide supra) in developing charge stabilisation, hence is it perhaps unsurprising that along the reaction coordinate it's position relative to the other reacting partners changes as the reaction progresses through the addition and elimination steps – with the cation moving from the ‘back’ to the ‘front’ faces of PET as it is cleaved (shown in Fig. S3(b)†). Meanwhile, solvent-mediated charge stabilisation in pathway II does not necessitate such a flexible system wherein bulky catalyst fragments migrate between different faces of the ester – the extent of physical reorganisation here is comparatively small (Fig. S3(b)†).
General base catalysis involving the secondary amine moiety (Fig. 2 pathway III) was calculated to be marginally more energetically favourable than either pathway I or II. This mechanism appears to have been neglected in the literature despite the known ability of amines to catalyse the process.13
The interest in cholinium-based ILs as catalysts in these reactions stems from a desire to use ‘greener’ catalysts which also can actively participate in accelerating the reaction. While the green credentials associated with cholinium-based amino acid catalysts are not in question here, the almost isoenergetic nature of solvolytic stabilisation raises questions regarding the consequences of using cholinium ions from catalyst activity/efficacy perspectives. With this in mind, subsequent calculations involving a hypothetical mechanism with the cation removed from the system (Fig. S4†) revealed that its exclusion from the system lowers the energy of TSaddition, Inttetrahedral and TSelimination.
The mechanism in the absence of cholinium ion mimics that associated with pathway II; a glycol molecule stabilises the charge accumulation on the PET carbonyl moiety in the addition step (Fig. S5(b)†). It was also found that the presence of the choline introduces another potential mechanistic complication – i.e. the cation's hydroxyl group acting as the proton donor; with the deprotonation of choline by glycinate being potentially competitive with deprotonation of glycol (Fig. S6†). This is supported by literature pKa (H2O) values associated with choline and ethylene glycol of 13.9 (ref. 21a) and 15.1 (ref. 21b) respectively.
From an analysis of the computational data, a mechanistic advantage associated with the use of cholinium ions was difficult to discern. To further probe the influence of both the cholinium and glycinate components on catalysis, an experimental study was carried out involving glycolysis of 1 to 2 under conditions used in the DFT calculations (vide supra) using a variety of known and novel IL-based catalysts 4b, 5 and 6–9. To facilitate comparisons, a low catalyst loading of 2 mol% was initially utilised (Table 1). The literature cholinium glycinate catalyst 5 (ref. 18c) exhibited significant efficacy; mediating the formation of 2 with appreciable conversion and yield (entry 1). The difference between the two is ascribable to the formation of partially glycolysed oligomers. The corresponding acetate 4b18b was more active (entry 2), indicating that while the amino acid-derived component is useful from a biodegradability standpoint, it does not add value catalytically relative to a simple carboxylate species. A novel analogue of 5 incorporating a conformationally restricted (and presumably less prone to eliminative degradation) cation (i.e.6, entry 3) proved superior to 5 yet did not outperform the simple cholinium acetate system 4b, indicating – in agreement with the DFT studies outlined above – that the contribution of the cholinium cation to catalysis may have been previously overestimated in literature.18 This was confirmed when catalyst 7 – a glycinate system with a more lipophilic phosphonium cation22 – promoted glycolysis with outstanding conversion and significantly improved yield (entry 4).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Loading (mol%) | Conv.a (%) | Yielda,b (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Conditions: PET (1.00 g), catalyst (1–2 mol%), ethylene glycol (3.6 mL). Conversion based on residual 2. Results determined from an average of 2 experiments within 5% agreement. b Isolated yield after precipitation. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 5 | 2 | 59 | 49 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 4b | 2 | 75 | 63 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 6 | 2 | 67 | 56 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 7 | 2 | 94 | 77 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 8 | 2 | 72 | 61 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 9 | 2 | 93 | 74 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 7 | 1 | 63 | 54 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 9 | 1 | 73 | 63 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
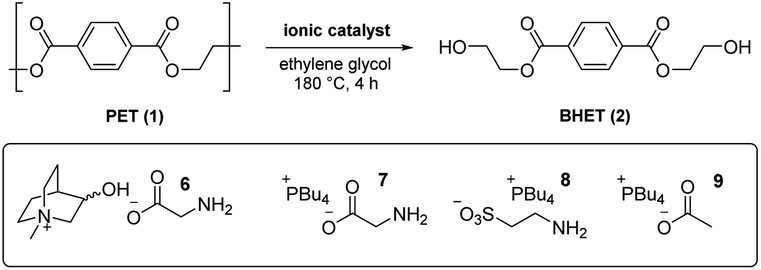
To probe the possibility of the computationally identified amine-mediated catalysis (pathway III) while retaining an IL system we prepared and evaluated a phosphonium taurinate variant (i.e.8, entry 5) devoid of the basic carboxylate unit.23 This proved to be a superior catalyst to the parent cholinium glycinate species 5 yet inferior to its phosphonium glycinate analogue 7. While this demonstrates that catalysis by the amine the certainly possible, the origins of the experimental superiority of 7 over 8 are unclear at this point and could be related to either solvation or catalyst stability issues at 180 °C. Finally, the simpler phosphonium acetate 9 (incorporating the most advantageous anion and cation combination among those evaluated) was anticipated to therefore serve as an outstanding catalyst (entry 6). Conversion and yield were comparable to those associated with the use of the corresponding glycinate 7 at 2 mol% catalyst loading – however, at 1 mol% level, the enhanced efficacy is more clearly discernible (entries 7–8).
In summary, it has been found that while the use of cholinium-based catalysts in the glycolytic deploymerisation of PET has been of interest from a sustainable chemistry perspective, the mechanistic advantages purportedly associated with this cation could not be identified in either DFT or experimental studies. Competitive stabilisation of developing negative charge by solvent glycol molecules is possible with as little as three explicit molecules in the transition state, and catalysts devoid of the cholinium ion outperformed their choline-based analogues experimentally. When designing amino acid-based carboxylate anions for catalysis of these reactions, general base catalysis by the amine needs to be considered from a mechanistic standpoint. Removal of the cholinium ion and amino acid unit gave rise to a highly active phosphonium acetate catalyst 9. Further investigations regarding the influence of the solvent, cation and anion on catalyst activity are underway.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to extend their gratitude and thanks to the Irish Research Council (GOIPG/2022/1037) and Science Foundation Ireland (19/FFP/6527) for supporting this research, as well as the Irish Centre for High-End Computing (ICHEC) for computational resources. The authors acknowledge the assistance provided by Research IT and the use of the Computational Shared Facility at The University of Manchester.Notes and references
- R. Geyer, J. R. Jambeck and K. L. J. S. A. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- K. Beydoun and J. Klankermayer, Sci. Adv., 2020, 13, 488 CAS.
- The New Plastics Economy – Rethinking the Future of Plastics, World Economic Forum, Ellen MacArthur Foundation, and McKinsey & Company, 2016 Search PubMed.
- P. Benyathiar, P. Kumar, G. Carpenter, J. Brace and D. K. Mishra, Polymers, 2022, 14, 2366 CrossRef CAS PubMed.
- (a) N. Torres, J. J. Robin and B. Boutevin, Eur. Polym. J., 2000, 36, 2075 CrossRef CAS; (b) N. Dimitrov, L. K. Krehula, A. P. Siročić and Z. Hrnjak-Murgić, Polym. Degrad. Stab., 2013, 98, 972 CrossRef CAS.
- (a) F. A. Chacon, M. T. Brouwer and E. U. Thoden van Velzen, Packag. Technol. Sci., 2020, 33, 347 CrossRef; (b) C. Stärker, F. Welle and E. U. Thoden van Velzen, Packag. Technol. Sci., 2020, 33, 347–359 CrossRef; (c) M. T. Brouwer, F. A. Chacon and E. U. Thoden van Velzen, Packag. Technol. Sci., 2020, 33, 373 CrossRef CAS.
- B. Geueke, K. Groh and J. Muncke, J. Cleaner Prod., 2018, 193, 491 CrossRef CAS.
- Selected recent reviews: (a) S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. de los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084 RSC; (b) J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041 CrossRef CAS PubMed; (c) E. Barnard, J. J. Rubio Arias and W. Thielemans, Green Chem., 2021, 23, 3765 RSC; (d) K. Ghosal and C. Nayak, Mater. Adv., 2022, 3, 1974 RSC; (e) C. Jehanno, M. M. Pérez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Polym. Chem., 2019, 10, 172 RSC.
- Selected references: (a) S. Baliga and W. T. Wong, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2071 CrossRef CAS; (b) C.-H. Chen, C.-Y. Chen, Y.-W. Lo, C.-F. Mao and W.-T. Liao, J. Appl. Polym. Sci., 2001, 80, 943 CrossRef CAS; (c) C.-H. Chen, J. Appl. Polym. Sci., 2003, 87, 2004 CrossRef CAS; (d) K. Troev, G. Grancharov, R. Tsevi and I. Gitsov, J. Appl. Polym. Sci., 2003, 90, 1148 CrossRef CAS; (e) S. R. Shukla and A. M. Harad, J. Appl. Polym. Sci., 2005, 97, 513 CrossRef CAS; (f) N. D. Pingale and S. R. Shukla, Eur. Polym. J., 2008, 44, 4151 CrossRef CAS; (g) N. D. Pingale, V. S. Palekar and S. R. Shukla, J. Appl. Polym. Sci., 2010, 115, 249 CrossRef CAS; (h) N. D. Pingale, V. S. Palekar and S. R. Shukla, J. Appl. Polym. Sci., 2010, 115, 249 CrossRef CAS; (i) R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, S. Arnaiz and J. I. Gutiérrez-Ortiz, Polym. Degrad. Stab., 2010, 95, 1022 CrossRef; (j) R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, L. Flores-Giraldo and J. I. Gutiérrez-Ortiz, Chem. Eng. J., 2011, 168, 312 CrossRef; (k) S. Wang, C. Wang, H. Wang, X. Chen and S. Wang, Polym. Degrad. Stab., 2015, 114, 105 CrossRef CAS.
- R. Esquer and J. J. García, J. Organomet. Chem., 2019, 902, 120972 CrossRef CAS.
- (a) S. R. Shukla, V. Palekar and N. Pingale, J. Appl. Polym. Sci., 2008, 110, 501 CrossRef CAS; (b) M. Zhu, Z. Li, Q. Wang, X. Zhou and X. Lu, Ind. Eng. Chem. Res., 2012, 51, 11659 CrossRef CAS; (c) M. Zhu, S. Li, Z. Li, X. Lu and S. Zhang, Chem. Eng. J., 2012, 185–186, 168 CrossRef CAS; (d) M. Imran, D. H. Kim, W. A. Al-Masry, A. Mahmood, A. Hassan, S. Haider and S. M. Ramay, Polym. Degrad. Stab., 2013, 98, 904 CrossRef CAS; (e) F. Chen, F. Yang, G. Wang and W. Li, J. Appl. Polym. Sci., 2014, 131, 41053 CrossRef; (f) Y. Geng, T. Dong, P. Fang, Q. Zhou, X. Lu and S. Zhang, Polym. Degrad. Stab., 2015, 117, 30 CrossRef CAS; (g) F. Chen, Q. Zhou, R. Bu, F. Yang and W. Li, Fibers Polym., 2015, 16, 1213 CrossRef CAS; (h) Q. Suo, J. Zi, Z. Bai and S. Qi, Catal. Lett., 2017, 147, 240 CrossRef CAS; (i) M. R. Nabid, Y. Bide, N. Fereidouni and B. Etemadi, Polym. Degrad. Stab., 2017, 144, 434 CrossRef CAS; (j) P. Fang, B. Liu, J. Xu, Q. Zhou, S. Zhang, J. Ma and X. Lu, Polym. Degrad. Stab., 2018, 156, 22 CrossRef CAS; (k) Y. Zhao, M. Liu, R. Zhao, F. Liu, X. Ge and S. Yu, Res. Chem. Intermed., 2018, 44, 7711 CrossRef CAS; (l) I. Yunita, S. Putisompon, P. Chumkaeo, T. Poonsawat and E. Somsook, Chem. Pap., 2019, 73, 1547 CrossRef CAS; (m) A. Bahramian, Polym. Degrad. Stab., 2020, 179, 109243 CrossRef CAS; (n) S. Shirazimoghaddam, I. Amin, J. A. F. Albanese and N. R. Shiju, ACS Eng. Au, 2023, 3, 37 CrossRef CAS PubMed.
- (a) G. Park, L. Bartolome, K. G. Lee, S. J. Lee, D. H. Kim and T. J. Park, Nanoscale, 2012, 4, 3879 RSC; (b) A. M. Al-Sabagh, F. Z. Yehia, D. R. K. Harding, G. Eshaq and A. E. ElMetwally, Green Chem., 2016, 18, 3997 RSC; (c) F. R. Veregue, C. T. P. da Silva, M. P. Moises, J. G. Meneguin, M. R. Guilherme, P. A. Arroyo, S. L. Favaro, E. Radovanovic, E. M. Girotto and A. W. Rinaldi, ACS Sustainable Chem. Eng., 2018, 6, 12017 CrossRef CAS; (d) G. R. Lima, W. F. Monteiro, C. M. Scheid, R. A. Ligabue and R. M. C. Santana, Catal. Lett., 2019, 149, 1415 CrossRef CAS.
- (a) K. Fukushima, O. Coulembier, J. M. Lecuyer, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. Mcneil, P. Dubois, R. M. Waymouth, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1273 CrossRef CAS; (b) Q. Wang, X. Yao, S. Tang, X. Lu, X. Zhang and S. Zhang, Green Chem., 2012, 14, 2559 RSC; (c) K. Fukushima, D. J. Coady, G. O. Jones, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1606 CrossRef CAS; (d) C. Jehanno, I. Flores, A. P. Dove, A. J. Müller, F. Ruipérez and H. Sardon, Green Chem., 2018, 20, 1205 RSC; (e) E. Sert, E. Yilmaz and F. S. Atalay, J. Polym. Environ., 2019, 27, 2956 CrossRef CAS; (f) K. R. Delle Chiaie, F. R. McMahon, E. J. Williams, M. J. Price and A. P. Dove, Polym. Chem., 2020, 11, 1450 RSC; (g) L. Wang, G. A. Nelson, J. Toland and J. D. Holbrey, ACS Sustainable Chem. Eng., 2020, 8, 13362 CrossRef CAS; (h) C. Jehanno, J. Demarteau, D. Mantione, M. C. Arno, F. Ruipérez, J. L. Hedrick, A. P. Dove and H. Sardon, Angew. Chem., Int. Ed., 2021, 60, 6710 CrossRef CAS PubMed.
- (a) Q. Wang, X. Yao, Y. Geng, Q. Zhou, X. Lu and S. Zhang, Green Chem., 2015, 17, 2473 RSC; (b) E. Sert, E. Yilmaz and F. S. Atalay, J. Polym. Environ., 2019, 27, 2956 CrossRef CAS.
- (a) H. Wang, Y. Liu, Z. Li, X. Zhang, S. Zhang and Y. Zhang, Eur. Polym. J., 2009, 45, 1535 CrossRef CAS; (b) Q. F. Yue, C. X. Wang, L. N. Zhang, Y. Ni and Y. X. Jin, Polym. Degrad. Stab., 2011, 96, 399 CrossRef CAS; (c) Q. Wang, X. Yao, S. Tang, X. Lu, X. Zhang and S. Zhang, Green Chem., 2012, 14, 2559 RSC; (d) A. M. Al-Sabagh, F. Z. Yehia, A. M. F. Eissa, M. E. Moustafa, G. Eshaq, A. M. Rabie and A. E. ElMetwally, Ind. Eng. Chem. Res., 2014, 53, 18443 CrossRef CAS; (e) Y. Liu, X. Yao, H. Yao, Q. Zhou, J. Xin, X. Lu and S. Zhang, Green Chem., 2020, 22, 3122 RSC.
- For a summary of studies involving metal salt-containing ionic liquids see ref. 8a.
- Recent reviews on IL-toxicity: (a) J. Flieger and M. Flieger, Int. J. Mol. Sci., 2020, 21, 6267 CrossRef CAS PubMed; (b) A. R. P. Gonçalves, X. Paredes, A. F. Cristino, F. J. V. Santos and C. S. G. P. Queirós, Int. J. Mol. Sci., 2021, 22, 5612 CrossRef PubMed.
- (a) J. Sun, D. Liu, R. P. Young, A. G. Cruz, N. G. Isern, T. Schuerg, J. R. Cort, B. A. Simmons and S. Singh, ChemSusChem, 2018, 11, 781 CrossRef CAS PubMed; (b) Y. Liu, X. Yao, H. Yao, Q. Zhou, J. Xin, X. Lu and S. Zhang, Green Chem., 2020, 22, 3122 RSC; (c) S. Marullo, C. Rizzo, N. T. Dintcheva and F. D'Anna, ACS Sustainable Chem. Eng., 2021, 9, 15157 CrossRef CAS.
- (a) X. Zhou, X. Lu, Q. Wang, M. Zhu and Z. Li, Pure Appl. Chem., 2012, 84, 789 CAS; (b) Q. Wang, X. Lu, X. Zhou, M. Zhu, H. He and X. Zhang, J. Appl. Polym. Sci., 2013, 3574 CrossRef CAS; (c) Q. Wang, Y. Geng, X. Lu and S. Zhang, ACS Sustainable Chem. Eng., 2015, 3, 340 CrossRef CAS.
- For an alternative mechanistic rationale involving imidazolium-ion based ILs see: H. Wang, Z. Li, Y. Liu, X. Zhang and S. Zhang, Green Chem., 2009, 11, 1568 RSC.
- (a) R. M. C. Dawson, D. C. Elliott, W. H. Elliott and K. M. Jones, Data for Biochemical Research, Clarendon Press, Oxford, 1959 Search PubMed; (b) P. Ballinger and F. A. Long, J. Am. Chem. Soc., 1959, 82, 795 CrossRef.
- Interestingly, a lipophilic phosphonium bromide have been shown to serve as a phase transfer catalyst for alkaline PET hydrolysis: R. López-Fonseca, J. R. González-Velasco and J. I. Gutiérrez-Ortiz, Chem. Eng. J., 2009, 146, 287 CrossRef.
- In preliminary experiments the corresponding tetrabutylphosphoniun mesylate catalyst did not promote glycolysis under these conditions; so catalysis by sulphonate ion can be discounted.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00336a |
| This journal is © The Royal Society of Chemistry 2023 |