
Graphitic carbon nitride (g-C3N4) based heterogeneous single atom catalysts: synthesis, characterisation and catalytic applications
Suja
P
ac,
Jubi
John
 bc,
T. P. D.
Rajan
ac,
Gopinathan M
Anilkumar
d,
Takeo
Yamaguchi
d,
Suresh C.
Pillai
e and
U. S.
Hareesh
*ac
bc,
T. P. D.
Rajan
ac,
Gopinathan M
Anilkumar
d,
Takeo
Yamaguchi
d,
Suresh C.
Pillai
e and
U. S.
Hareesh
*ac
aMaterials Science and Technology Division, CSIR-National Institute for Interdisciplinary Science and Technology, Thiruvananthapuram-695019, India. E-mail: hareesh@niist.res.in
bChemicals Science and Technology Division, CSIR-National Institute for Interdisciplinary Science and Technology, Thiruvananthapuram-695019, India
cAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
dLaboratory for Chemistry and Life Sciences, Tokyo Institute of Technology, R1-17, 4259 Nagatsuta, Midori-Ku, Yokohama 226-8503, Japan
eNanotechnology and Bio-Engineering Research Group and Health and Bio-Medical Research Centre (HEAL), Atlantic Technological University, ATU Sligo, Ash Lane, Sligo, F91 YW50, Sligo, Ireland
First published on 25th March 2023
Abstract
Graphitic carbon nitride (g-C3N4), the well-known visible light active, two-dimensional organic semiconductor is gaining further prominence as a potential matrix for developing heterogeneous single atom catalysts. g-C3N4 by virtue of its abundant and periodically separated nitrogen atoms effectively stabilises single atoms of metal through the electron lone pairs of nitrogen acting as anchoring sites. Herein, the synthetic strategies adopted for the development of g-C3N4 based single atom catalysts (SACs) are summarised in detail for an understanding of the state-of-the-art designing of g-C3N4 SACs. Advanced characterisation techniques like extended X-ray absorption fine structure (EXAFS), X-ray absorption near-edge structure (XANES), electron energy-loss spectroscopy (EELS), X-ray photoelectron spectroscopy, and aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) offer valuable inputs to determine the electronic and geometric structure of g-C3N4 based SACs are discussed. Furthermore, the experimental and computational efforts carried out in demonstrating the potential applications of g-C3N4 SACs in the field of photocatalysis, organic reaction catalysis and electrocatalysis are reviewed. The challenges associated with the practical utility of g-C3N4 based SACs and their future perspectives in heterogeneous catalysis are outlined.
 Suja P | Suja P is currently a UGC-Senior Research Fellow at CSIR-National Institute for Interdisciplinary Science and Technology (CSIR-NIIST), Thiruvananthapuram. She received her B.Sc. in Chemistry from NSS college, Ottapalam and M.Sc. in Chemistry from Sri Vyasa NSS College, Wadakkancherry. She is currently pursuing her doctoral research on the design and development of g-C3N4 based single atom catalysts and semiconductor heterostructures for photocatalytic applications and for organic reaction catalysis. She has authored 2 patents, 1 research article, and 1 book chapter. |
 Jubi John | Dr Jubi John obtained his PhD in synthetic organic chemistry from CSIR-NIIST under the supervision of Dr K. V. Radhakrishnan. Soon after he joined as a CEA-Eurotalents postdoctoral fellow with Dr Eric Doris at CEA Saclay, France. In 2011, he joined as an Alexander von Humboldt fellow with Prof. Henning Hopf at TU Braunschweig, Germany and from 2013, with Prof. Wim Dehaen at the KU Leuven, Belgium. He then joined CSIR-NIIST in 2015 and is presently working as a senior scientist at the same institute. His research interests include development of novel synthetic methodologies, homogenous and heterogeneous catalyzed organic transformations and new antiviral drug candidates. |
 T. P. D. Rajan | Dr T. P. D. Rajan is currently a Senior Principal Scientist in Materials Science and Technology Division of CSIR – National Institute for Interdisciplinary Science and Technology (NIIST), Thiruvananthapuram. He obtained B.Sc. and M.Sc. in Chemistry, M.Tech. in Metallurgy from NITK, Surathkal and PhD in Metallurgical Engineering from CSIR-NIIST. His research interests are composite materials, light metals, surface technology and nanomaterials. He had published 146 research papers, 10 book chapters and filed 3 patents. He is recipient of Award for Excellence in Corrosion Science and Technology (NACE), IIM Nalco Gold Medal, IIF Industrial Research Award, INAE Innovation Award and IIM Metallography Awards. |
 Anilkumar G. M | Dr Gopinathan M Anilkumar is currently a visiting professor at the Institute of Innovative Research (IIR), Tokyo Institute of Technology, Japan. He is employed as a senior research manager at the R&D Center of Noritake Co., Ltd., Aichi, Japan. Dr Anilkumar obtained his PhD in Chemistry from Mahatma Gandhi University, Kerala for the work carried out at CSIR-NIIST, India. He was a post-doctoral researcher at the Korea Advanced Institute of Science and Technology (KAIST), Korea (S), and the University of Tokyo, Japan. He is currently engaged in the development of advanced materials for fuel cells and water electrolyzers. He has authored more than 70 peer-reviewed research papers and holds 25 granted patents. |
 Takeo Yamaguchi | Prof. Takeo Yamaguchi is a Professor at the Laboratory for Chemistry and Life Sciences, Institute of Innovative Research (IIR), Tokyo Institute of Technology, Japan. He also serves as the Director of the Tokyo Tech Academy for Convergence of Materials and Informatics (TAC-MI). Prof. Yamaguchi received his Doctor of Engineering degree from the Department of Chemical Engineering, The University of Tokyo, followed by post-doctoral research at the University of Colorado at Boulder, USA. He also holds several positions in various scientific societies, such as Vice President of the membrane society of Japan, Council member of the Aseanian Membrane Society (AMS), and a steering committee member of the World Association of Membrane Societies. He has authored more than 230 publications and holds more than 30 granted patents. His research interests include the systematic design of energy materials and systems for fuel cells and water electrolysis, stimuli-responsive membranes inspired by biosystems, and the utilization of computational techniques for the efficient design of materials. |
 Suresh C. Pillai | Suresh C. Pillai obtained his PhD from Trinity College Dublin and completed his postdoctoral research at California Institute of Technology (Caltech, USA). He currently heads the Nanotechnology and Bio-Engineering Research Group at the Institute of Technology Sligo, Ireland. His research interests include the synthesis of nanomaterials for energy and environmental applications. He is the recipient of several awards including the Boyle-Higgins Award 2019, Linus-Pauling Lecture Award 2020 etc. Suresh is also a recipient of the ‘Industrial Technologies Award 2011’ for licensing functional coatings to Irish companies. He was also the recipient of the ‘Hothouse Commercialisation Award 2009’ from the Minister of Science, Technology and Innovation and also the recipient of the ‘Enterprise Ireland Research Commercialization Award 2009’. He has also been nominated for the ‘One to Watch’ award 2009 for commercialising R&D work (Enterprise Ireland). |
 U. S. Hareesh | Dr U. S. Hareesh is currently a Senior Principal Scientist at the Materials Science and Technology division of CSIR-National Institute for Interdisciplinary Science and Technology (CSIR-NIIST), Thiruvananthapuram. He obtained his PhD in Chemistry from Mahatma Gandhi University, Kerala for the work carried at CSIR-NIIST, India and Technical University of Hamburg-Harburg (TUHH), Germany. Subsequently, he worked as a visiting scientist at the Institute for New Materials, Saarbruecken, Germany and as a scientist at the Centre for Ceramic Processing, Advanced Research Centre International (ARCI), Hyderabad, India. He is currently engaged in the development of advanced functional materials for energy and environmental applications. He has authored more than 90 publications in peer reviewed journals, 10 granted patents and 5 book chapters. He is the recipient of the German Academic Exchange Service (DAAD) fellowship, Malaviya award of the Indian Ceramic Society and Technology award of ARCI, Hyderabad. |
1 Introduction
Heterogeneous catalysts comprising metal nanoparticles on supports are widely employed for accelerating the kinetics of industrially important reactions. Nevertheless, the full potential of such systems is hitherto unexploited as the atoms exposed only on the metal nanoparticle surface are active and the atomic utilisation of metal is low. Moreover, the predominant use of precious metals like Pt, Rh imparts exorbitant costs for heterogeneous catalysts. Single Atom Catalysts (SACs) or Atomically Dispersed Metal Catalysts (ADMCs) refer to an incipient class of materials where active centers of metals are individually dispersed on appropriate support through coordination with atoms like nitrogen, oxygen, sulphur, etc. The partial positive charges on metal sites due to decreased electron density offer modified interactions thereby enhancing the rate of conversion of substrate into product. Furthermore, metal atoms in SACs/ADMCs are spatially isolated to influence the adsorption characteristics of reactive intermediates, thereby preventing the unwanted side reactions that proceed through adjacent metal sites. The development of SACs and ADMCs provides the most ideal strategy for creating catalysts that are cost-effective for a multitude of applications including photocatalysis, electrocatalysis and organic transformations.One of the essential requirements for realisation of SACs is the effectiveness of coordination of metal centers with matrix/support atoms. The free energy of metal increases as it tends to manifest itself in the finest form and a strong coordination with support atoms is imperative to counter aggregation tendencies. In this context, 2D materials owing to their surface area and weak van der Waals forces possess significant advantages, to be employed as the matrices for SACs and ADMCs. Consequently, a host of supports like N-doped carbon, graphene, and high surface area metal oxides are explored. More recently, 2D layered g-C3N4, an organic semiconductor was established as an effective candidate for visible-light induced photocatalysis by virtue of its band gap of 2.7 eV. g-C3N4 is constructed of layered hexagonal building blocks of tri-s-triazine units and forms sheet-like pi conjugated structures composed of sp2 C and sp2 N.1
The abundant nitrogen atoms in g-C3N4 facilitate the anchoring of single metal atoms through the effective coordination between empty (or partially empty) orbitals of metal atoms with the electron lone pairs of nitrogen. g-C3N4 has thus been demonstrated to be an excellent host for the development of SACs, despite its below average surface area. Atomic level dispersion of metal species over g-C3N4 results in the development of 6 major varieties of catalyst systems (Fig. 1) depending upon the metal distribution and loading. The conventional terminology of g-C3N4 based single atom catalysts (SACs) refers to the ideal condition of uniformly dispersed, individual, isolated metal atoms coordinated to g-C3N4 matrix. In g-C3N4 based atomically dispersed metal catalysts (ADMCs), metal atoms are dispersed in the form of clusters containing dimers, trimers, etc along with isolated single metal atoms. More recently g-C3N4 based dual metal single atom catalysts emerged where two different metals are coordinated to the matrix as single atoms (DM-SACs). Ultra-high density single atom catalysts (UHD-SACs) refer to the atomic level dispersion of metal atoms at very high metal loading (>20 wt%) and with ultrahigh atom density (5–15 atoms nm−2 at a spacing of 0.2–0.5 nm).2 Depending upon the positioning of single atoms on g-C3N4 matrix two more classifications are reported. In edge confined single atom catalysts (EC-SACs) the metal atoms are preferentially located at the edges of g-C3N4 sheets through unsaturated vacancies along the edges. On the other hand, in interlayer coordinated SACs (ILC-SACs), confinement of the single atoms occurs in between the layers of g-C3N4 through coordination with appropriate functional groups.
 | ||
| Fig. 1 Various configurations of metal atom dispersions over g-C3N4 matrix. | ||
The chronology of developments leading to the establishment of SACs based on g-C3N4 is provided in the timeline below (Fig. 2).
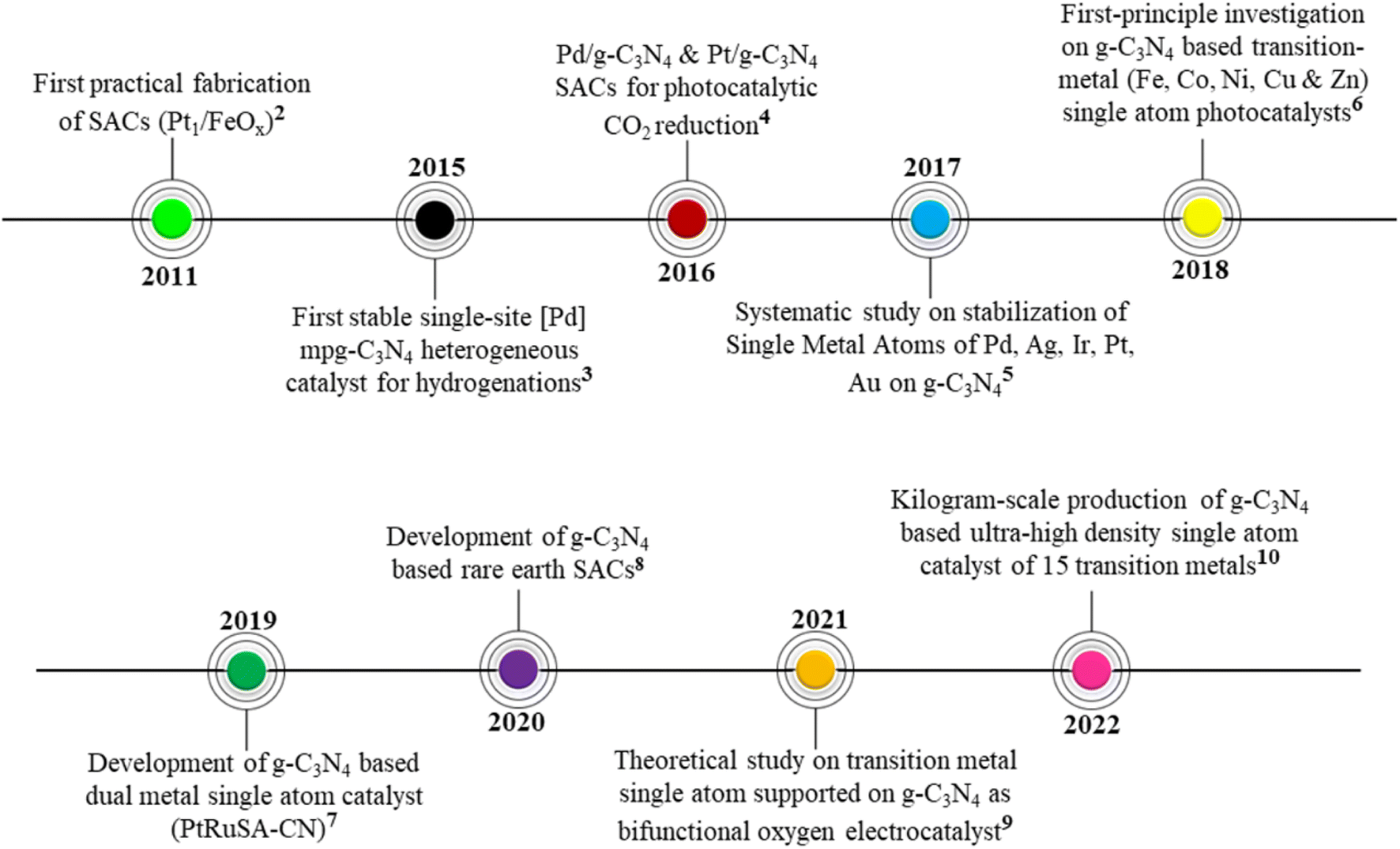 | ||
| Fig. 2 Timeline representing the chronology of advancements in SACs based on g-C3N4.3–11 | ||
In this review, we attempt to comprehensively analyse the developments based on g-C3N4 supported single atom catalysts (SACs) and cover its applications in the areas of photo, electro and thermal catalysis. A tutorial like description of the fundamental aspects related to the synthesis and characterisation of g-C3N4 SACs is included. DFT studies in the reported works are also incorporated primarily to throw light on the prospective experimental works that can be pursued. To the best of our knowledge, this review is the first attempt to cover all the applications thus far explored on g-C3N4 SACs.
2 Graphitic carbon nitride as support for SACs
g-C3N4 consists of s-triazine or tri-s-triazine rings as the basic constituent units that offer uniformly placed, abundant nitrogen atoms to confine single metal atoms effectively and with high loading. The terminal tri-s-triazine unit of g-C3N4 comprises different types of nitrogen12 as presented schematically in Fig. 3. The various N-species in g-C3N4 are demonstrated to be very effective for catalytic reactions as they impart H-bonding, tunable basicity, Lewis acidity. Strong metal–support interaction is essential to stabilise the well-dispersed metal species as isolated single metal atoms for imparting selectivity and catalytic activity. This will also prevent the diffusion and aggregation of single atoms at the time of synthesis as well as during the utilisation of SACs at various reaction conditions. This is primarily facilitated by the substantially greater surface energy of metal single atoms compared to their nanoclusters and nanoparticles.13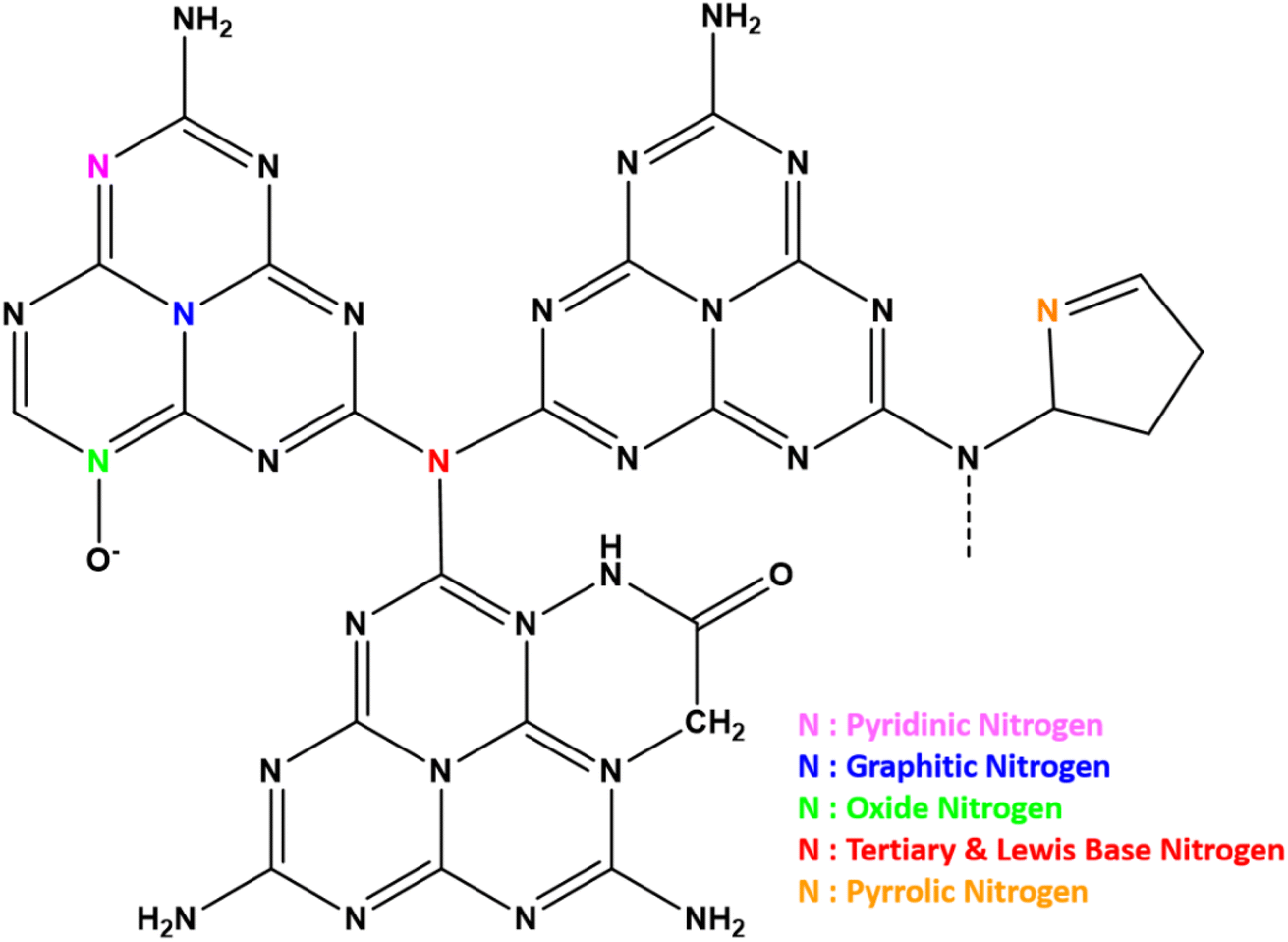 | ||
| Fig. 3 Types of nitrogen functionalities possible in g-C3N4. | ||
Synthesis of g-C3N4 is carried out by thermal decomposition and polymerisation of N-rich precursors like melamine, cyanamide, dicyandiamide, urea, thiourea and their mixtures at 550 °C in air,14 as shown schematically in Fig. 4. The bulk material formed at 550 °C is similar irrespective of the atmosphere of firing but may differ in the degree of condensation and product yield. Generally, all the precursors first condense to melamine at 235 °C followed by a self-condensation leading to the formation of a dimer called melam (N2-(4,6-diamino-1,3,5-triazin-2-yl)-1,3,5-triazine-2,4,6-triamine). Essentially melamine (triazine) based structures are formed up to a temperature of 350 °C. At 390 °C melam units are converted into tri-s-triazine units called melem by the rearrangement of melamine. Finally, the networked polymeric g-C3N4 is formed by the successive condensation of melem units at 520 °C. The material is unstable above 600 °C and complete decomposition to volatile components occurs at temperatures >700 °C in air.15 Template assisted thermal polymerisation of g-C3N4 precursors results in the formation of mesoporous polymeric g-C3N4 (mpg-C3N4) with enhanced surface area. Thermal exfoliation of bulk g-C3N4 (BCN) leads to the formation of g-C3N4 nanosheets/exfoliated g-C3N4 (ECN) with improved surface area. All the 3 forms of g-C3N4i.e., BCN, mpg-C3N4, ECN can perform as a support for SACs.
 | ||
| Fig. 4 Synthesis of g-C3N4 from various nitrogen abundant precursors. | ||
3 Synthesis of g-C3N4 based single atom catalysts
Pérez-Ramírez et al. proposed direct and post-synthesis approaches for g-C3N4 based SACs. Direct synthesis involving the pyrolysis of the g-C3N4 precursors with metal salts is a bottom-up approach. During the polymerisation of g-C3N4 from precursors, single metal atoms are anchored on to the g-C3N4 support. Post synthesis is a strategy employed to disperse single atoms on synthesised g-C3N4 types like bulk g-C3N4, mpg-C3N4, ECN, etc which are prepared either through template free or template assisted synthesis strategy.6Fig. 5 depicts the multiple approaches involved in both the above categories for the preparation of g-C3N4 SACs.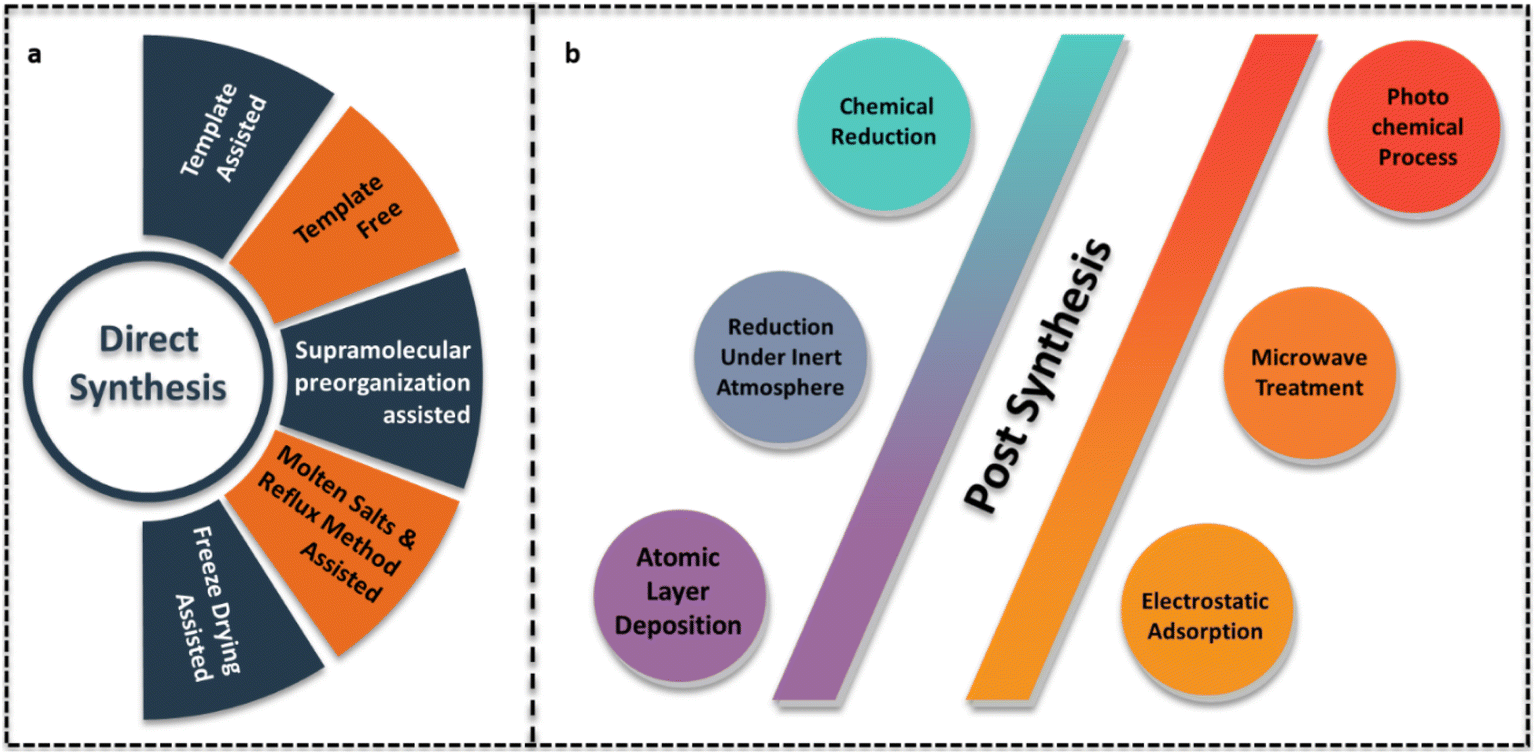 | ||
| Fig. 5 Various strategies employed during (a) direct and (b) post synthesis of g-C3N4 SACs. | ||
3.1 Direct synthesis
 | ||
| Fig. 6 (a) Template free direct synthesis of La single-atoms dispersed carbon nitride (O/La–CN). Reproduced with permission from ref. 16. Copyright 2020 American Chemical Society. (b) Direct synthesis of single Cu atoms supported crystalline g-C3N4 (Cu–CCN) through molten salts-reflux method. Reproduced with permission from ref. 18. Copyright 2020 American Chemical Society. (c) Mechanism of supramolecular preorganisation of g-C3N4 based cobalt SAC. Reproduced with permission from ref. 19. Copyright 2020 Elsevier. (d) Freeze drying assisted direct synthesis of erbium single atoms decorated g-C3N4 (Er1/CN-NT). Reproduced with permission from ref. 9. Copyright 2020 John Wiley and Sons. | ||
3.2 Post synthesis
Use of template like silica, SBA-15, etc during the pyrolysis of g-C3N4 precursors, forms mpg-C3N4 with enhanced surface area. Exfoliated g-C3N4 is obtained through thermal or chemical exfoliation. Post synthesis strategy involves the mixing of metal salts in g-C3N4 dispersion and its conversion to single metal atoms using techniques like chemical reduction, reduction under an inert atmosphere, photochemical reduction, microwave treatment, electrostatic adsorption, and atomic layer deposition (ALD).Similarly, Zbořil and co-workers successfully employed microwave heating assisted post synthesis strategy for the development of ruthenium single atom dispersed mesoporous g-C3N4 catalyst (Fig. 7(a)).21
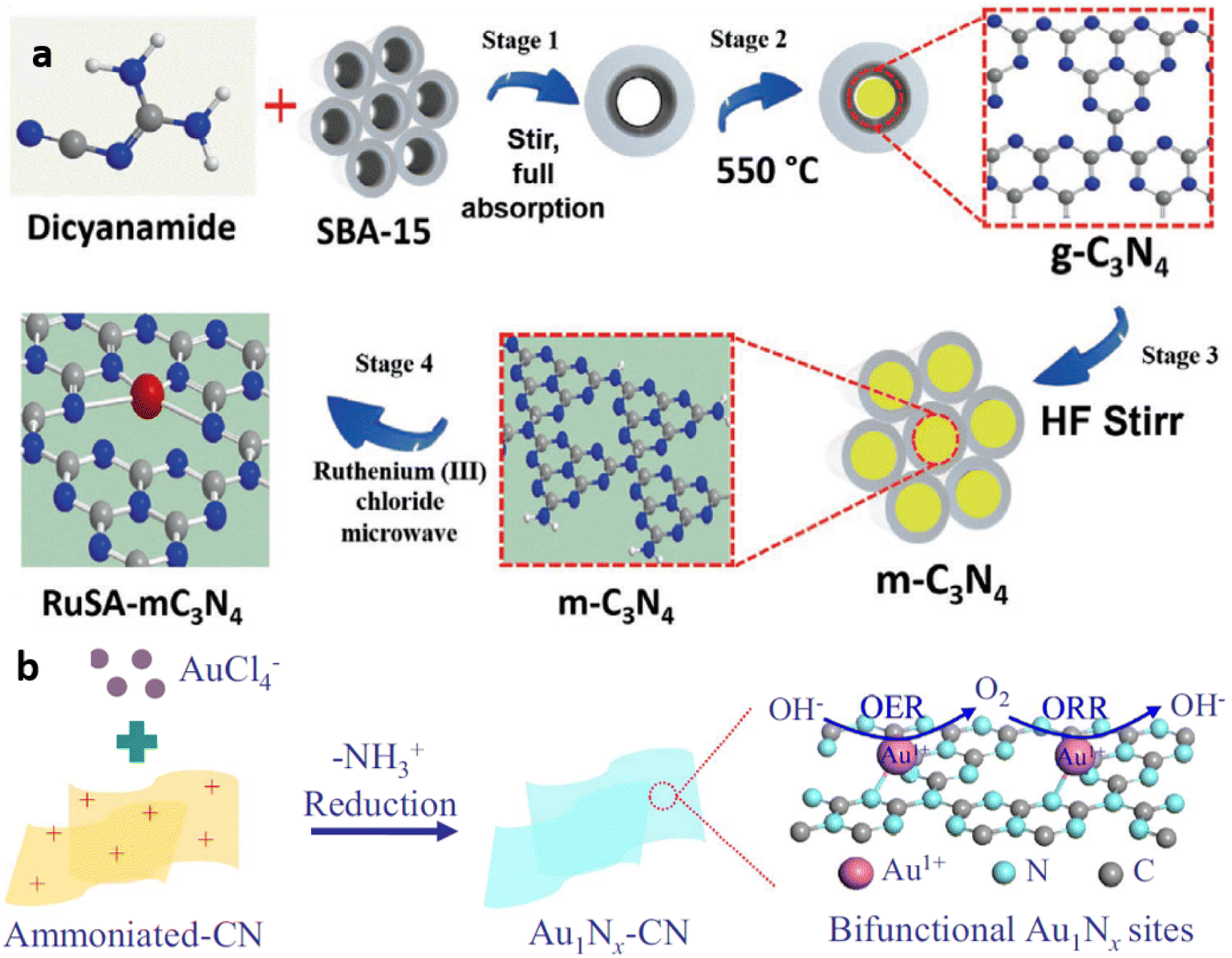 | ||
| Fig. 7 (a) Schematic of microwave assisted post synthetic strategy for the development of ruthenium single atoms decorated mesoporous C3N4 (RuSA-mC3N4) catalyst. Reproduced with permission from ref. 21. Copyright 2021 John Wiley and Sons. (b) Schematic of electrostatic adsorption mediated post synthetic strategy for Au1Nx single-site/C3N4 catalyst synthesis. Reproduced with permission from ref. 22. Copyright 2018 Elsevier. | ||
Ultra-high density single atom catalysts (UHD-SACs) by atomic level dispersion of 15 transition metals on suitable supports having different structural and chemical characteristics were illustrated recently by Lu and co-workers. Consequently, UHD-SACs on supports like N doped carbon (NC), polymeric C3N4 (PCN) and metal oxide (CeO2) were developed through a post synthetic strategy involving the impregnation of aqueous solutions of metal salts on supports followed by two-step annealing under inert atmosphere to realise a metal loading up to 23 wt%. The two-step annealing process facilitated the removal of ligands from metal precursors in a controlled manner and selectively coordinated metal ions to all the available anchoring sites present on the support surface to realise ultra-high metal loading. The first annealing step was done at temperatures (T1) lower than the decomposition temperatures of the metal salts under an inert atmosphere. The unbound metal species were removed by washing to prevent the aggregation and sintering of the metal species in the subsequent annealing step. Further, the second stage of annealing at a higher temperature (T2) completely removed the remaining ligands and transformed the chemisorbed metal species into highly stable UHD-SAC systems. The above two-step annealing strategy enabled the kilogram-scale production through robot assisted-automatic synthesis taking Ni UHD-SAC on PCN as a case study.11
Table 1 summarises the synthesis strategies with their attributes. In comparison, the direct synthesis route is more beneficial than post synthesis route to achieve g-C3N4 SACs with high metal content. Post synthetic methods suffer from the drawback of promoting aggregation leading to nanoparticle formation at high concentrations of the localised metal species. On the other hand, direct synthesis methodologies enable uniform distribution of single atoms but are susceptible to reduced atom efficiency owing to the entrapment of metal in the support matrices.
| Synthesis | Techniques | Remarks |
|---|---|---|
| Direct synthesis | • Template free synthesis | • Effective stabilisation of single atoms as coordination occurs along with polymerisation of g-C3N4 precursors |
| • Template assisted synthesis | • High & ultra-high metal loading can be realised | |
| • Molten salts & reflux method assisted synthesis | • Easily scalable & low cost | |
| • Supramolecular preorganisation assisted synthesis | • Necessarily involves high temperature pyrolysis (550–600 °C) | |
| Post synthesis | • Chemical reduction | • Better control of the textural properties like surface area |
| • Reduction under an inert atmosphere | • Low metal loading in general | |
| • Photochemical reduction | • Metal–support interaction is weak compared to direct synthesis | |
| • Electrostatic adsorption | • MW & ALD are hardware intense | |
| • Microwave treatment (MW) | ||
| • Atomic layer deposition (ALD) |
4 Analytical tools for understanding g-C3N4 based single atom catalysts
The atomically dispersed metal species over g-C3N4 matrix, act as a catalytically active centre for facilitating various catalytic reactions. It is imperative to cognise the atomic and electronic structure of the single atom sites of SACs to elucidate the mechanism governing its catalytic activity. Various analytical tools have thus been employed to substantiate the occurrence of metal species in single atom configuration, to determine its distribution over g-C3N4 matrix and its coordination environment. In this section, we examine the various characterisation techniques used for analysing g-C3N4 SACs and the elucidation of data from those techniques (Fig. 8).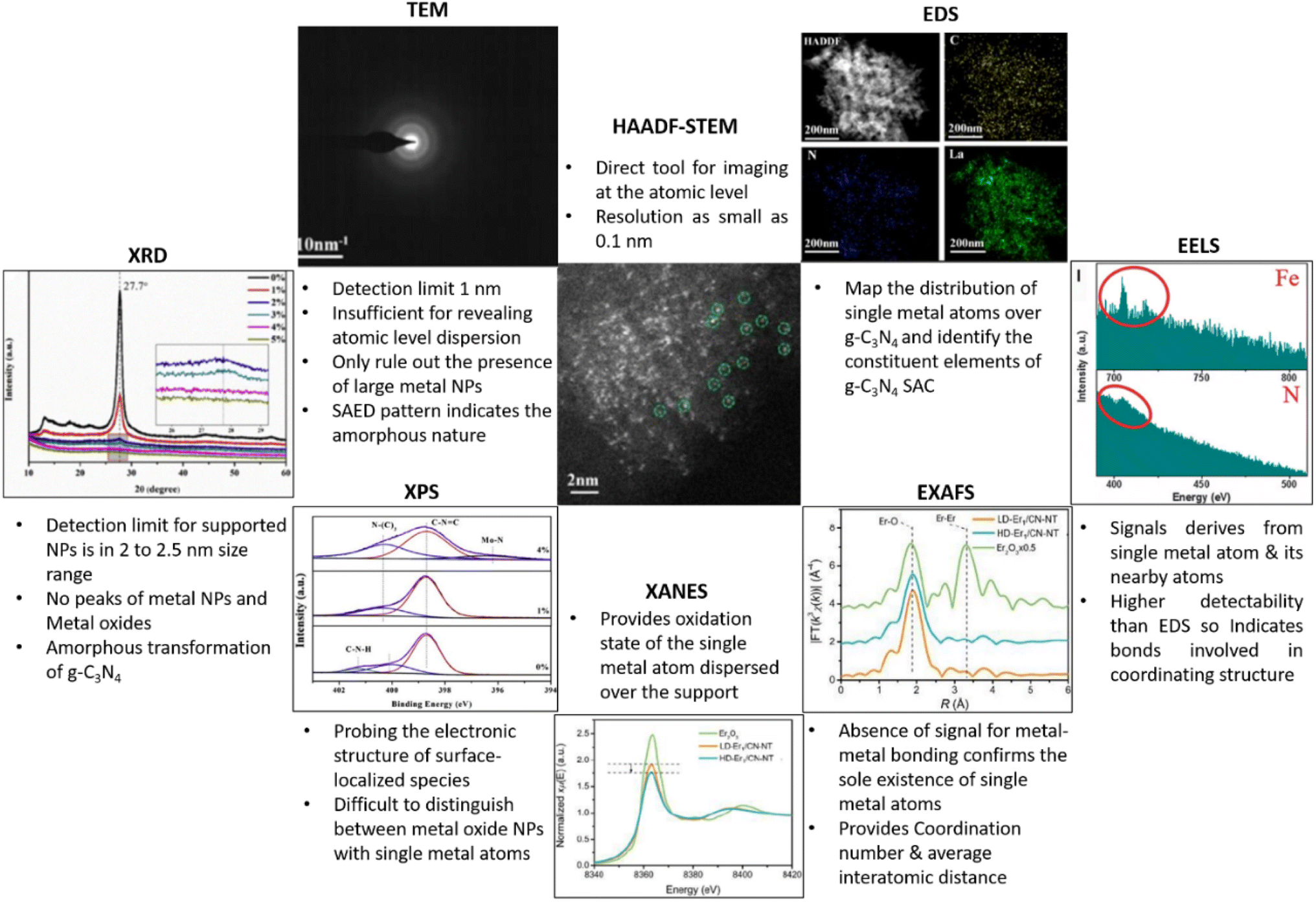 | ||
| Fig. 8 Various analytical tools for the characterisation g-C3N4 based SACs. Reproduced with permission from ref. 9. Copyright 2020 John Wiley and Sons. Reproduced with permission from ref. 16. Copyright 2020 American Chemical Society. Reproduced with permission from ref. 27. Copyright 2019 Elsevier. Reproduced from ref. 28. | ||
4.1 XRD analysis
The detection limit of conventional X-ray powder diffraction for supported nanoparticles is often set at 2–2.5 nm size range, below which low signal to noise ratios make size detection challenging.29 Absence of peaks in the XRD pattern of g-C3N4 based SACs merely rules out the existence of metal NPs and metal oxides, but not confirming the formation of metal single atoms. g-C3N4 exhibit two characteristic XRD diffraction peaks at 2θ values of 13.3° and 27.8° are attributed respectively to the in-planar structural packing of (100) crystallographic plane and the (002) lattice plane corresponding to the interplanar stacking of aromatic systems.30 Due to the atomic level dispersion of single atoms over g-C3N4 matrix, the above characteristic XRD peaks diminish. For e.g., Zhou and co-workers reported the complete disappearance of peaks when the Mo loading is increased to 4% (Fig. 9(a)). The long-range order of g-C3N4 is thus disturbed inducing the amorphous transformation of g-C3N4.27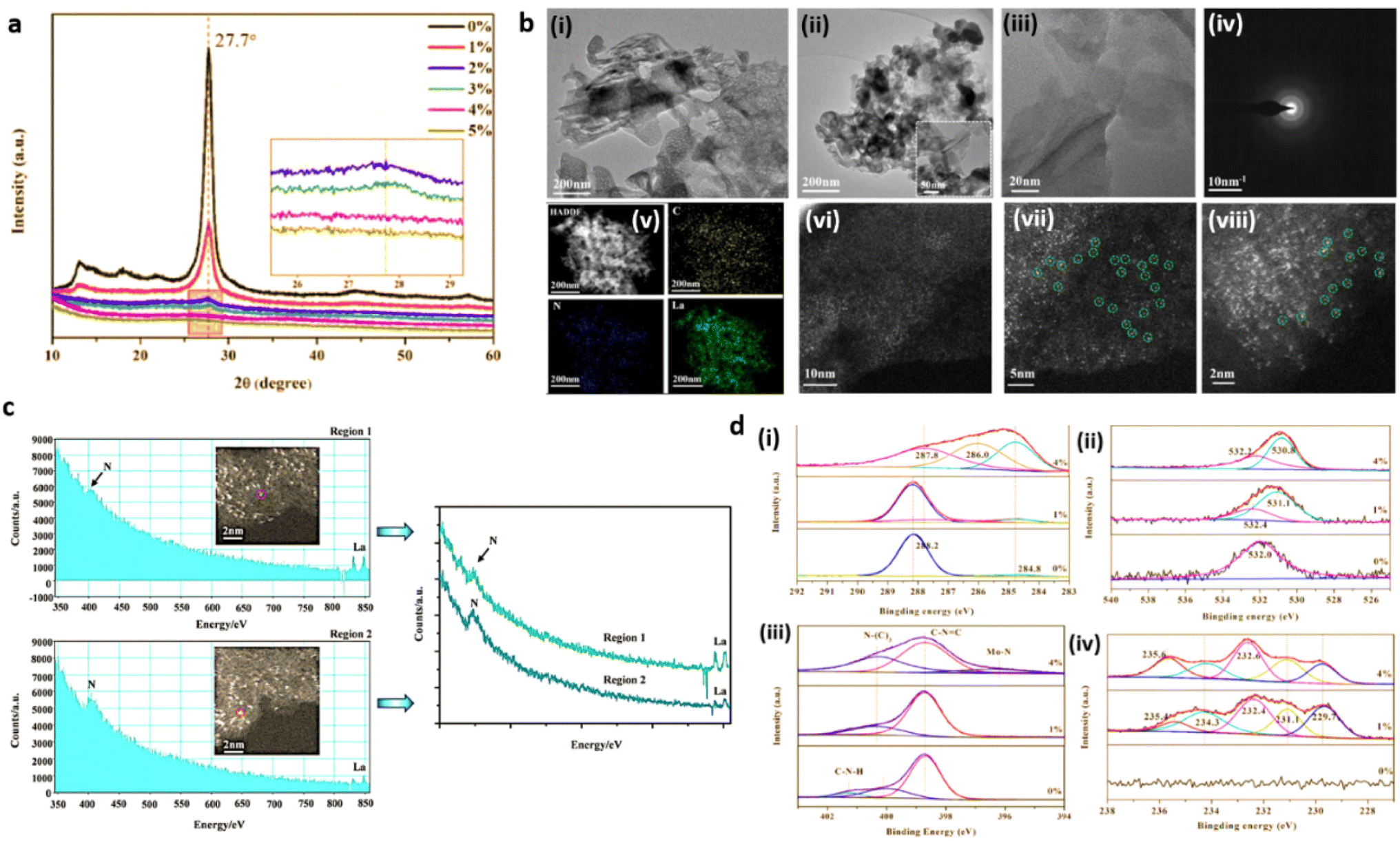 | ||
| Fig. 9 (a) XRD patterns of g-C3N4 (0%) and Mo/C3N4 (1–5%). Reproduced with permission from ref. 27. Copyright 2019 Elsevier. (b) Morphological analysis of O/La–CN catalyst; (i and ii) TEM images, (iii) HAADF-STEM image of the thinner region, (iv) SAED pattern, (v) HAADF-STEM EDS mapping (yellow, blue, and green color represents carbon, nitrogen and lanthanum), (vi–viii) spherical aberration-corrected HAADF-STEM images, (c) EELS spectra of two separate labelled regions. Reproduced with permission from ref. 16. Copyright 2020 American Chemical Society. (d) XPS spectra; (i) C 1s, (ii) O 1s; (iii) N 1s, (iv) Mo 3d. Reproduced with permission from ref. 27. Copyright 2019 Elsevier. | ||
4.2 Transmission electron microscopy (TEM)
Transmission electron microscopy (TEM) is extensively employed for the morphological and structural identification of material. However, the conventional TEM of nanoscale resolution is insufficient for imaging single-atoms as its normal detection limit is ≈1 nm. A combination of high-resolution TEM imaging and SAED imaging is commonly used to rule out the presence of large metal NPs in a given area of the SAC sample.13,31 Nevertheless, the mentioned tools cannot be used to confirm the absence of NPs in the bulk sample. Additionally, the absence of lattice fringes and diffractions in HRTEM image and the selected area electron diffraction (SAED) pattern confirm the amorphous nature, as has been demonstrated by Dong et al. in g-C3N4 system with atomic level dispersion of La (O/La–CN) (Fig. 9(b)(i, ii) and (iv)).164.3 Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM)
Recent developments in the field of electron microscopy have led to the emergence of aberration-corrected high-angle annular dark-field imaging scanning TEM (HAADF-STEM), with a resolution as small as 0.1 nm providing atomic scale information.32 Single metal atoms are identified on supports with the aid of HAADF-STEM with aberration correctors, provided the atomic number of the metal atom of interest is much higher than the constituent element of the supports. The single-atoms dispersed on the g-C3N4 matrix and its spatial distribution can be directly imaged using aberration corrected HAADF-STEM analysis.33 For example, HAADF-STEM imaging and the associated analysis of rare-earth La single atoms on carbon nitride (O/La–CN) (Fig. 9(b)(iii) and (vi–viii)) revealed the existence of single La atoms over g-C3N4 matrix.164.4 Energy-dispersive X-ray spectroscopy (EDS) and electron energy-loss spectroscopy (EELS)
Energy-dispersive X-ray spectroscopy (EDS) and/or electron energy-loss spectroscopy (EELS) are always used in conjunction with STEM for validating the composition of the synthesised g-C3N4 SACs.34,35 Energy-dispersive X-ray spectroscopy (EDS/EDX) together with HAADF-STEM is used to map the distribution of single metal atoms over g-C3N4 matrix and to identify the constituent elements. The energy-dispersive spectroscopy (EDS) mapping of O/La–CN confirmed the uniform distribution of La, N and C elements in the g-C3N4 framework (Fig. 9(b)(v)). EELS spectra derived from the positioning of an electron probe directly over a single atom can deliver information about the coordination environment by nearest neighbouring atoms. EELS spectra of O/La–CN from two selected locations are compared in (Fig. 9(c)). The presence of N and La atoms in this location, with no evident O, suggests La–N coordination structure.16HAADF-STEM in combination with EDS/EELS allows the direct visualisation of single atoms only in a limited area and cannot provide conclusive evidence for the absence of metal nanoparticles in the bulk of the material. So, it's critical to analyse microscopic observations in conjunction with spectroscopic tools that could provide overall information about SACs such as chemical and electronic structure, coordination environment of g-C3N4 based SACs. X-rays, owing to their short wavelength (0.01–1 nm), are the most suitable source for atomic scale structural information, X-ray Absorption Spectroscopy (XAS) is thus used to detect the detailed coordination environment of single atoms. Spectroscopic techniques, in comparison with microscopic tools, can better resolve elements of similar atomic weights (e.g., N and C, Co and Mo) and can thus be utilised to derive qualitative and quantitative information about the valence state of single atoms.36In situ and operando characterisations are enabled in XAS as it can operate without ultrahigh vacuum requirements. Similarly, X-ray photoelectron spectroscopy (XPS) measurements can identify the variations in surface composition and valence state.37
4.5 X-ray photoelectron spectroscopy (XPS)
XPS is an essential characterisation tool for probing the electronic structure of surface-localised species and for determining the valence states of surface elements. Single metal atoms supported on 2D matrices normally possess partial (or full at times) positive charge due to the strong covalent coordination or ionic interaction with neighbouring surface atoms or ligands. The heterogeneity/homogeneity of single atom species is mostly identified through a singlet peak in the XPS analysis which indicates a unified positive charge for the metal elements.38,39XPS analysis of a single Mo atom dispersed on g-C3N4 matrix (a-Mo/C3N4, where a = (0–4%)) revealed the amorphous transformation of g-C3N4 due to atomic level dispersion of Mo atoms over the matrix. The C 1s spectra of pure g-C3N4 exhibit peaks at 284.8 eV and 288.2 eV which are attributed to the adventitious C contamination and C–N bonds respectively. For a-Mo/C3N4, the peak at 284.8 eV related to the C–C bond has grown significantly, and the peak of the C–N bond has shifted to 287.8 eV. The “outward twisting of melon units” and the hydrogen bond cleavage of polymeric strands of melon units are responsible for these changes in the XPS spectra, confirming that g-C3N4 has undergone an amorphous transformation. The high-resolution N 1s XPS spectra of g-C3N4 displayed peaks corresponding to C–N–C bond and N–(C)3 bond, and C–N–H bond at 398.7 eV, 400.1 eV, and 401.3 eV respectively. The dispersion of Mo single atoms induces the disappearance of C–N–H bond peak due to the above-mentioned H-bond cleavage. A new peak for a-Mo/C3N4 can be seen at 396.2 eV, which is derived from the Mo–N bond, demonstrating the creation of the Mo–N bond after the insertion of Mo atoms. The O 1s spectra of pure g-C3N4 have a peak at 532.0 eV due to the C–O bond, and a second peak at 531.1 eV appears due to the –OH bond during the formation of Mo single atoms from ammonium molybdate precursor. Furthermore, Mo(VI) and Mo(V) 3d3/2 orbitals are evidenced by peaks at 235.4 and 234.3 eV respectively, while Mo(VI), Mo(V), and Mo(IV) 3d5/2 orbitals are confirmed from peaks at 232.4, 231.1, and 229.7 eV, revealing the abundant valence state of Mo in a-Mo/C3N4 (Fig. 9(d)(i–iv)).27
4.6 X-ray absorption spectroscopy
X-ray Absorption Spectroscopy (XAS) is an element-specific technique for probing the local coordination environment by detecting fluctuations in the absorption of an ejected photoelectron due to multiple scattering pathways.37,40 XAS analysis mainly consists of extended X-ray absorption fine structure (EXAFS) and X-ray absorption near-edge structure (XANES) analyses. The atoms surrounding the single atom produce outgoing and backscattered waves and their interference yields EXAFS spectra. EXAFS analysis of SAC provides essential information regarding the coordination number of single atom site(s) and its interatomic distance with backscatter atoms within its vicinity, in comparison with standard metal foil and metal oxides. The structural disorder and the type of neighbouring atoms present at a specific bond length are also deduced from EXAFS. The absence of signal corresponding to metal–metal interaction in EXAFS spectra rules out the formation of metal NPs and metal oxides.38XANES analysis further helps in determining the oxidation state of the single metal atom dispersed over the support. XAS analysis techniques establish the identity of single metal atoms through EXAFS and derive quantitative data related to coordination. Further, XAS analysis proposes suitable geometric arrangements for single metal atoms through the curve-fitting of XANES data based on predictable coordination environments. Fig. 10(a–e) shows the XAS analysis details of low-density (2.5 wt% Er loading) and high-density (20.1 wt% Er loading) Er single atom anchored on g-C3N4 nanotubes (Er1/CN-NT). Fourier transform-extended X-ray absorption fine structure (FT-EXAFS) reveals radial distance resolution (R-space) while wavelet transform-extended X-ray absorption fine structure (WT-EXAFS) can discriminate the backscattering atoms to provide resolution in both R-space and k-space. FT-EXAFS spectra, of LD-Er1/CN-NT and HD-Er1/CNNT samples, exhibited only one main peak at around 1.8 Å. The absence of a peak at about 3.5 Å, corresponding to Er–Er interaction for LD-Er1/CN-NT and HD-Er1/CNNT samples confirmed the formation of a single Er atom over CNNT without the formation of metallic crystalline Er species. WT-EXAFS plots also supplemented the above observation due to the presence of only one intensity maximum at around 4.0 Å−1 corresponding to Er–N bond for the LD-Er1/CN-NT and HD-Er1/CN-NT samples.
 | ||
| Fig. 10 (a) The k3-weighted Fourier transform spectra from EXAFS for Er2O3, LD-Er1/CN-NT, HD-Er1/CN-NT in R space (b) wavelet transform (WT) contour plots of Er2O3, LD-Er1/CN-NT, HD-Er1/CN-NT (c), the normalised XANES spectra at the Er L3 edge of Er2O3, LD-Er1/CN-NT, HD-Er1/CN-NT, The EXAFS R space fitting curves and corresponding model for (d) LD-Er1/CN-NT and (e) HD-Er1/CN-NT respectively Er, N, C atoms are represented by yellow, blue, green spheres respectively. Reproduced with permission from ref. 9. Copyright 2020 John Wiley and Sons. | ||
The white line intensity reduction of Er L3-edge XANES spectra with the increase in Er loading from 2.5 wt% to 20.1 wt%, indicate a decrease in the oxidation state of Er in the order Er2O3 > LD-Er1/CN-NT > HD-Er1/CN-NT. In comparison to LD-Er1/CN-NT, HD-Er1/CN-NT has a flattened white-line peak at 8360 eV, implying an interaction between central and surrounding Er atoms resulting in the charge redistribution due to the shortening of spatial distance between neighbouring Er atoms. Quantitative least-squares EXAFS analysis demonstrated that both LD-Er1/CN-NT, and HD-Er1/CN-NT exhibited one main peak at around 1.8 Å arising from the first shell of Er–N scattering. EXAFS results also indicated that the single Er atoms have a first shell coordination number of six in both LD-Er1/CN-NT and the HD-Er1/CN-NT samples.9
5 Catalytic applications of single atom confining on g-C3N4 matrix
5.1 Photocatalysis
Visible light-driven semiconductor photocatalysis is widely pursued as an environmentally benign and sustainable solution for pollution control and energy production. Photocatalyst alters the kinetics of a chemical reaction upon light irradiation through the creation of highly effective reactive oxygen species (ROS) and has been demonstrated for applications like the degradation of organic pollutants in wastewater, creating self-cleaning surfaces, splitting water for hydrogen evolution, etc.41 The photocatalyst possesses a favourable combination of electronic structure, excited-state life time, light absorption properties and charge transport characteristics.42g-C3N4, because of its flexible indirect band edge positions, excellent thermal stability, resistance to acids and bases, and insolubility in common solvents like water, ethanol, acetone, etc., is found suitable for a wide range of photocatalytic applications. These properties originate from the van der Waals force that holds together the stacked graphitic layers.43,44 Nevertheless, the application of g-C3N4 is limited as it utilises only a small region of the visible spectra. A low specific surface area in the range of 10–30 m2 g−1 for bulk g-C3N4 is also an impediment to widespread applications owing to the reduction in reactive sites. The high rate of recombination of excitons, poor light harvesting, and limited generation of radical species are the factors primarily responsible for its low catalytic efficiency during the redox reactions.45
The recombination process in g-C3N4 is perceived to be controlled by facilitating separation between photogenerated electron–hole pairs and various strategies such as heterostructure formation, incorporation of metal co-catalyst, impurity doping, etc are demonstrated thus far. Among them, the incorporation of a metal co-catalyst is a viable strategy to reduce exciton recombination as the metal Fermi level is positioned in between the band edge positions of C3N4. The transfer of photogenerated electrons from the conduction band (CB) to the metal enables the separation of the exciton pairs leading to improved photocatalytic efficiency.43
The usage of noble metals such as Au, Pt, Pd as co-catalyst is effective but is limited due to their high cost. Replacement of noble metals with cost-effective transition metals is an affordable solution for the applications of g-C3N4 based photocatalysts. However, very low atom utilisation efficiency, availability of minimal catalytically active species and poor selectivity of the metal nanoparticles are the bottlenecks for its practical application. Metal particles of appropriate sizes can only act as catalytically active species while those with non-optimal sizes may facilitate various undesired secondary reactions which further reduce the catalytic efficiency of the catalyst. To overcome these limitations, g-C3N4 based catalysts with atomic level dispersion of metal are recently developed.46
SACs comprising of spatially isolated, individual metal atoms stabilised on C3N4 matrix, show unusual potential due to unsaturated coordination sites for realising maximum atom-utilisation efficiency. Consequently, C3N4 based on SACs and ADMCs with unique electronic structures serve as potential candidates for catalysing various reactions by virtue of their advantages such as remarkably high catalytic activity, reduced catalyst amount, and abundance of single active sites.13
The past few years have seen significant research efforts leading to the development of a research domain of g-C3N4 based single atom photocatalysts for various applications (Fig. 11).
 | ||
| Fig. 11 Various reactions catalysed by g-C3N4 based SACs in the area of photocatalysis. | ||
Zhang et al. employed density functional theory (DFT) to study the improved photocatalytic activity of g-C3N4 based transition-metal (Fe, Co, Ni, Cu and Zn) single atom catalysts. To identify the adsorption sites of single atoms of various transition metals on multi-layered g-C3N4 sheets, a three-layer g-C3N4 structure was constructed by cleaving the (001) g-C3N4 surface. The structure comprising 24 N atoms and 18 C atoms and with the optimised lattice parameter values of a = b = 7.13 Å, c = 21.54 Å contained numerous six-fold cavities capable of trapping metal single atoms (Fig. 12).7
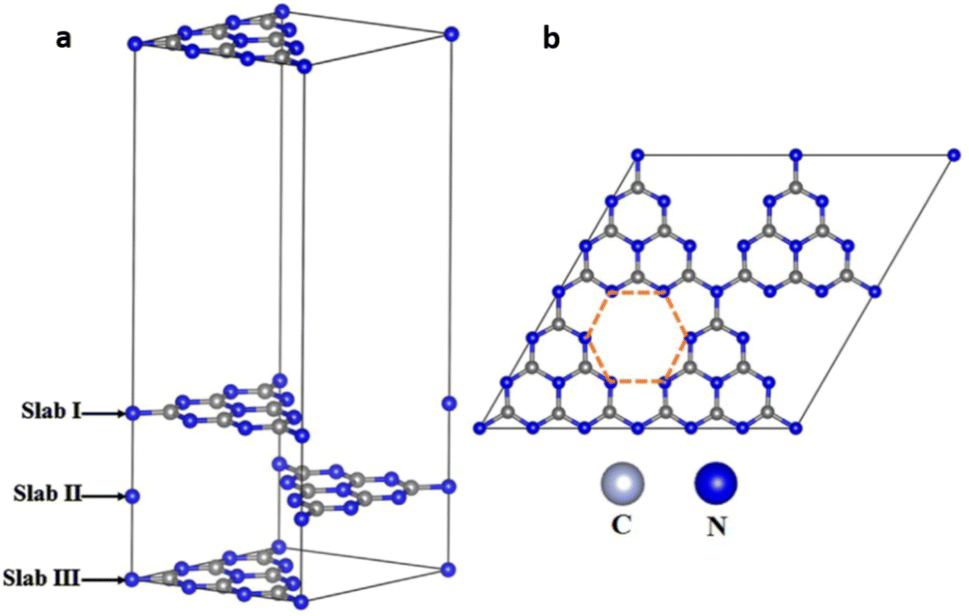 | ||
| Fig. 12 (a) Schematic representation of three-layer g-C3N4 model, (b) 2 × 2 × 1 supercell monolayer g-C3N4. The highlighted ring composed of six numbers of bi-coordinated nitrogen atoms is referred to as six-fold cavity. Reproduced with permission from ref. 7. Copyright 2018 Elsevier. | ||
Due to the presence of empty d orbitals in transition metals, single atoms of Fe, Co, and Ni were inserted into the layers of g-C3N4 sheets through strong coordination with their neighbouring C, N atoms. On the other hand, Cu and Zn atoms with completely filled d orbitals stay at the six-fold cavities present in the g-C3N4 surface through coordination with N atoms present at the edge of the cavities. The charge carrier transfer between the interlayers of g-C3N4 is reduced due to the high interlayer distance of 3.27 Å resulting in a high rate of exciton recombination. Incorporating single atoms of metal altered the interlayer spacing of g-C3N4. The loading of Fe, Co, and Ni single atoms significantly shortens the interlayer distance between slabs I and II. The value decreased from 3.27 to 2.25, 2.41 and 2.39 Å for Fe, Co and Ni respectively. The interlayer charge transfer was thus accelerated for aforementioned SACs. However, the incorporation of Cu and Zn atoms hardly affected the interlayer spacing with minimal reduction in values to 2.90 Å and 2.75 Å respectively. Similar reductions in the interlayer spacing between slab II and slab III were also observed in Fe/g-C3N4, Co/g-C3N4 and Ni/g-C3N4 with interlayer distances of 2.47 Å, 2.48 Å and 2.45 Å respectively. For Cu/g-C3N4 and Zn/g-C3N4, the interlayer spacing between slab II and III remains almost unchanged at 3.28 Å.7
The bond population is an effective measure of the nature of a chemical bond and a low value indicates that the ionic nature of the bond increases and the covalent nature decreases. The covalent nature of C–N bond in g-C3N4 is justified by its bond population value of >0.7. The bond population values for all the bonds between metal atoms and adjacent non-metal atoms (C or N) present in the catalysts are significantly lower than 0.7, which indicates greater electron delocalisation leading to accelerated charge transfer between g-C3N4 layers. The formations of Cu–N and Zn–N (metal–nitrogen bonds) in Cu/g-C3N4 and Zn/g-C3N4 are not sufficient to improve the charge transfer between layers while the presence of both metal–carbon and metal–nitrogen bonds in Fe/g-C3N4, Co/g-C3N4 and Ni/g-C3N4 systems exhibited enhanced charge transfer properties.7
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O dissociation energy of ∼750 kJ mol−1 and the optically inert nature of CO2 molecule in the wavelength range of 200–900 nm, render the reduction process extremely difficult and complex with multiple proton and electron transfer pathways.48 It is therefore imperative to develop photocatalytic systems with appropriate band structures that are responsive to solar radiation. The resulting photogenerated electrons facilitate the CO2 reduction process to various value-added products. The one-electron reduction of CO2 to CO2˙− is thermodynamically unfavourable due to the highly negative reduction potential value of −1.90 V vs. NHE, at pH 7.0.49 The multiple proton–electron assisted CO2 reduction reactions occur at lower redox potential values as listed in Fig. 13.
O dissociation energy of ∼750 kJ mol−1 and the optically inert nature of CO2 molecule in the wavelength range of 200–900 nm, render the reduction process extremely difficult and complex with multiple proton and electron transfer pathways.48 It is therefore imperative to develop photocatalytic systems with appropriate band structures that are responsive to solar radiation. The resulting photogenerated electrons facilitate the CO2 reduction process to various value-added products. The one-electron reduction of CO2 to CO2˙− is thermodynamically unfavourable due to the highly negative reduction potential value of −1.90 V vs. NHE, at pH 7.0.49 The multiple proton–electron assisted CO2 reduction reactions occur at lower redox potential values as listed in Fig. 13.
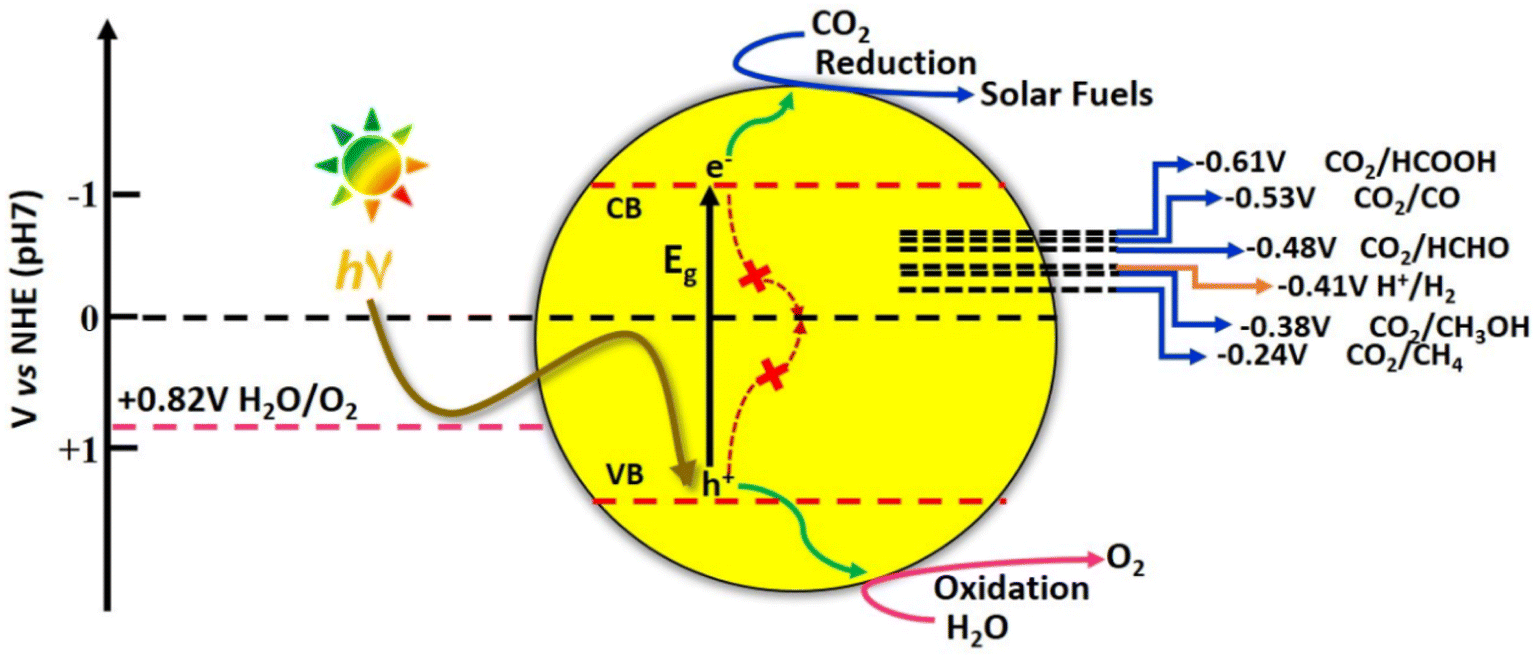 | ||
| Fig. 13 Schematic illustration of photocatalytic carbon dioxide reduction. | ||
However, the reaction pathways are all uphill reduction processes (ΔG > 0) and the conversion efficiencies are not yet appreciable for the reduction of CO2 to hydrocarbon fuels using H2O as a reductant.50 Moreover, due to the very close matching of redox potentials of possible products, the selectivity is very poor, particularly for C2 products. Furthermore, the stability of the intermediate formed during multiple proton–electron transfer pathways involved in the reduction reaction determines the product selectivity.51
In the early emerging period of SACs, Du and co-workers explored the potency of Pd and Pt single metal atoms supported on g-C3N4 towards photocatalytic CO2 reduction by density functional theory (DFT) calculations. Pd and Pt single atoms are stably confined into the six-fold cavity of g-C3N4 through the interaction of electron lone pairs present in the neighbouring pyridinic nitrogen atoms. The calculated binding energies for Pd and Pt were respectively −2.17 eV & −2.95 eV. In Pd/g-C3N4 & Pt/g-C3N4 systems, the noble metal atoms act as active sites, while the g-C3N4 matrix serves as a source of hydrogen which is generated through hydrogen evolution reaction. The evaluation of reaction pathways suggests HCOOH, with a reaction barrier of 0.66 eV, as the product expected for Pd/g-C3N4. On the other hand, Pt/g-C3N4 system was expected to reduce CO2 to CH4 with a reaction barrier of 1.16 eV. This study established the correlation between the metal and the product selectivity during the photoreduction process. The distribution of Pd or Pt metal as single atoms over g-C3N4 framework led to enhanced visible light absorption, thereby making it a potential candidate for visible light driven photocatalytic CO2 reduction. This theoretical study paved the way for developing various g-C3N4 based SACs for CO2 photoreduction reaction and its experimental validation.5
Li and co-workers synthesised visible light active non-noble metal Co2+@C3N4 catalyst for CO2 reduction by stabilising cobalt single atoms on g-C3N4 matrix through Co–N coordination. Photocatalysts containing single Co2+ sites on C3N4 exhibited excellent CO2 reduction capacity and product selectivity towards CO formation. The effect of metal loadings on CO production and product selectivity was studied by varying cobalt content between 0.004 and 0.430 μmol Co2+ per 1 mg C3N4. A significant amount of CO formation was observed with cobalt loading lower than even 0.010 μmol mg−1. The CO formation increased linearly until a cobalt loading of 0.128 μmol mg−1 was reached. Further increase in metal loading led to a slight decline in CO production due to the formation of inactive cobalt oxides. The product selectivity of the reduction reaction was however unaffected by the presence of dormant cobalt oxide formed at relatively high Co content.52
Zhou and co-workers successfully demonstrated the amorphous transformation of g-C3N4 by atomically dispersing Mo atoms over g-C3N4 matrix. The Mo–C and Mo–N bonds altered the long-range order of g-C3N4 and the retention of each melon unit led to a crystalline to an amorphous phase transition. This transformation to amorphous g-C3N4 enabled significant enhancement in its light absorption in the visible range with a strong band tail and an absorption edge up to 750 nm. Additionally, the newly formed Mo–C and Mo–N bonds enhanced the charge separation dynamics by acting as pathways for photogenerated excitons. The excellent photocatalytic CO2 reduction capacity of Mo/C3N4 systems was attributed to the enhanced visible light activity of the catalyst along with the reduced recombination rate of photo excitons. Under visible light irradiation, Mo/C3N4 systems exhibited the CO and H2 production rates of 18 and 37 μmol g−1 h−1 respectively which is 10.6- and 4-folds greater than that of crystalline g-C3N4.27
Wang et al. developed atomically dispersed rare earth erbium (Er) atoms on carbon nitride nanotubes for CO2 reduction through a strategy involving “atom confinement and coordination (ACC)”. The methodology of confining Er precursor and urea on melamine sponge, achieved at liquid N2 temperature, facilitated coordination between dispersed Er ions and N atoms of the g-C3N4 precursor. The ACC strategy helped to fine tune the metal content from 2.5 wt% to 20.1 wt% and could also be utilised for a variety of rare earth metals of lanthanide series. Photocatalytic CO2 reduction by high-density Er single-atom catalyst (HD-Er1/CN-NT) under simulated solar light produced CH4 and CO at the rates of 2.5 μmol g−1 h−1 and 47.1 μmol g−1 h−1 respectively which is higher than that of low-density Er single-atom catalyst (LD-Er1/CN-NT) and bare g-C3N4 nanotube catalyst (CN-NT). The presence of Er active sites in large numbers is presumed to be the cause for improved CO2 conversion rates. Moreover, the availability of multiple basic sites for CO2 adsorption, as corroborated by CO2-TPD measurements, promoted enhanced reduction capacity in HD-Er1/CN-NT samples. DFT calculations revealed that the gaseous CO formation has a low theoretical limiting potential (0.3 V) than CH4 (0.39 V). Advantageously, the limiting potential for H2 evolution reaction, calculated to be 0.82 V, was much greater than that of CO and CH4. Hence unfavourable H2 production could be successfully alleviated by Er atom catalysts.9
Another prominent school of thought practiced in SACs is on the synthesis of crystalline g-C3N4, primarily to overcome the demerits associated with the bulk morphology and excessive defects of amorphous g-C3N4. Xiang and co-workers reported the development of copper single atoms incorporated crystalline g-C3N4 nanorods (Cu–CCN) by molten salt and reflux method. The prepared Cu–CCN photocatalyst showed 100% CO selectivity with an improved generation rate of 3.086 μmol h−1 g−1 compared to 1.68 μmol h−1 g−1 and 0.895 μmol h−1 g−1 for crystalline and bulk counterparts. The CO2 adsorption capacity of Cu–CCN samples was enhanced as single Cu atoms served as sorption sites for CO2. Theoretical calculations revealed that the reduction of CO2 to CH4 is an entropy increasing process on Cu–CCN samples, while the reduction of CO2 to CO is a process of decreasing entropy, thereby facilitating 100% product selectivity towards CO (Fig. 14(a)).18
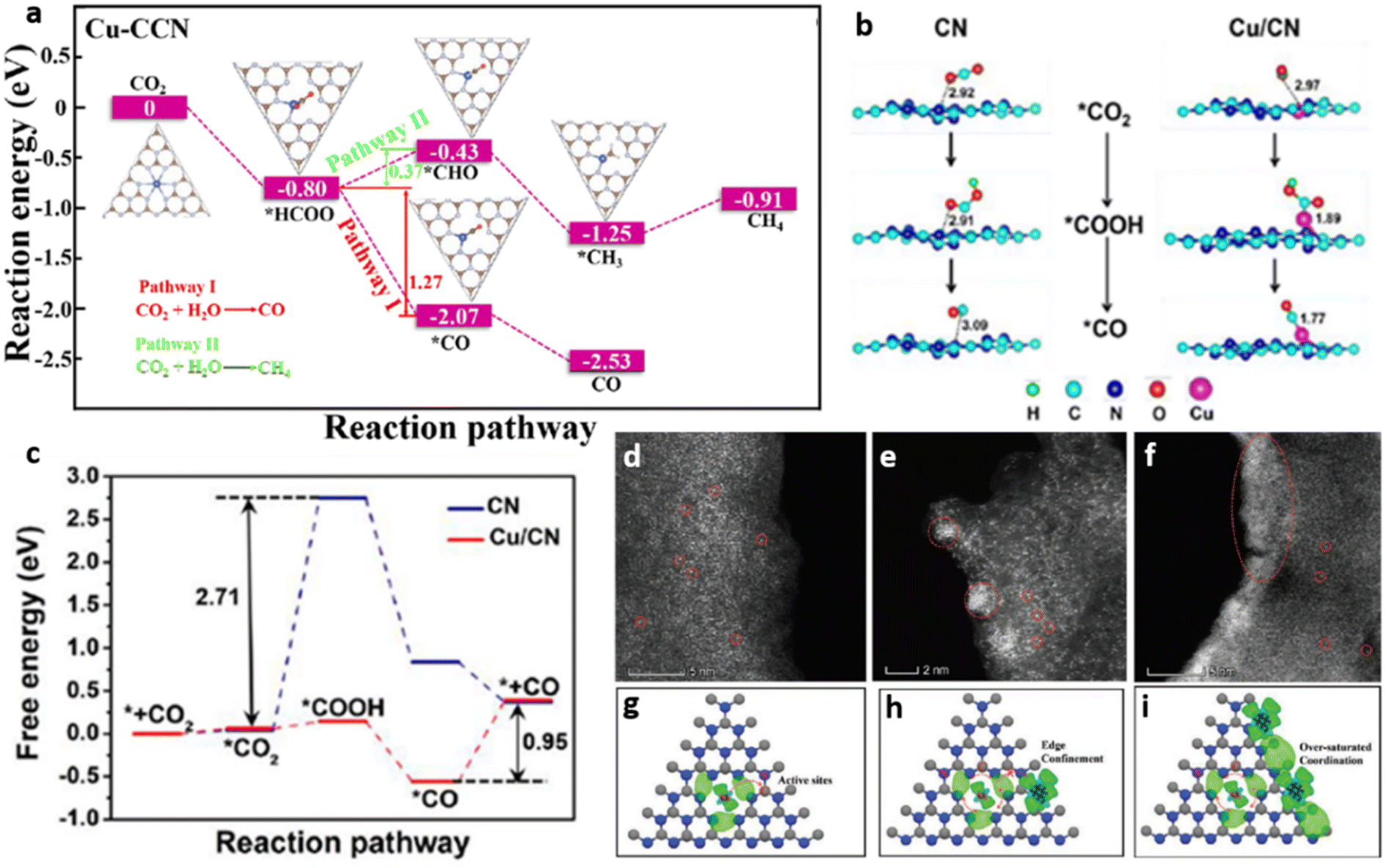 | ||
| Fig. 14 (a) Schematic of the photocatalytic CO2 reduction pathway on Cu–CCN derived from DFT calculation. Reproduced with permission from ref. 18. Copyright 2020 American Chemical Society. (b) Proposed structures and CO2 reduction pathway corresponding to CN & Cu/CN photocatalyst (c) free energy diagram for the conversion of CO2 to CO on CN and Cu/CN photocatalysts. Reproduced with permission from ref. 53. Copyright 2020 American Chemical Society. Aberration-corrected HAADF-STEM images of (d) Ni1–CN, (e) Ni5–CN, (f) and Ni10–CN, models showing a difference in the charge distribution of (g) Ni1–CN, (h) Ni5–CN, and (i) Ni10–CN. Reproduced with permission from ref. 54. Copyright 2020 John Wiley and Sons. | ||
In a noteworthy contribution, Cao et al. developed a g-C3N4 based photocatalyst with atomically fused-Cu atoms through C–Cu–N2 four-center bond formation. An artificial enzyme like configuration, similar to hemocyanin, is created where the single atom copper exhibited a mixed valence state of Cu+/Cu0 at the oxidation potential of +1.5 V. Composition with 0.25 wt% copper produced CO, CH3OH and CH4 at the rates of 11.21, 1.75 and 0.61 μmol g−1 h−1 respectively. Further increase in copper content led to the decline of photocatalytic activity owing to the surplus metal sites acting as charge recombination centres. CO was the major product of CO2 reduction, as the desorption of CO molecules was made easier at single Cu atom active centres. The DFT studies revealed that the terminal C–Cu–N2 four-centre active sites present in g-C3N4 nanosheets served as a sink for photogenerated electrons while providing reactive binding sites for CO2 activation. The proposed reaction pathway (Fig. 14(b)) followed the sequences of CO2 adsorption followed by its hydrogenation to form *COOH. Subsequently, OH group dissociates from *COOH forming adsorbed *CO followed by the desorption of CO molecule from the catalyst surface. The enhanced CO2 reduction performance of Cu/CN system over CN is attributed to the low free energy change (ΔG) of 0.95 eV associated with the rate limiting step (desorption of CO) on Cu/CN compared to a large ΔG of 2.71 eV associated with rate limiting step (hydrogenation of adsorbed *CO2) on bare CN (Fig. 14(c)).53
In a recent contribution, Dong et al. converted CO2 to CO efficiently by employing single-atoms of La on C3N4 by creating La–N charge bridges for efficient electron transport. The photocatalyst (O/La–CN), synthesised by one-step calcination involving the precursors of urea and lanthanum carbonate, exhibited complete amorphisation characterised by the absence of XRD peaks of g-C3N4. O/La–CN exhibited 80.3% selectivity at the high yielding rate of 92 μmol g−1 h−1 for CO. The La–N bridge, formed by the combination of various electronic states originating from 4f and 5d orbitals of La and the p–d orbital hybridisation, facilitated the CO2 activation, COOH* formation and CO desorption steps.16
The Ni-anchored g-C3N4 photocatalyst was designed by anchoring single atoms of Ni on a few-layer g-C3N4 synthesised through a self-limiting process. Such a few-layer g-C3N4 structure with high porosity offers ligands for trapping Ni atoms, thereby realising a high density of single atoms. The single Ni atoms were distributed on the edges of the g-C3N4 sheets through an “active unsaturated edge confinement strategy” providing maximum atom-utilisation efficiency for high CO2 adsorption capacity. The highly unsaturated Ni–N coordination offered pathways for the electron transfer from g-C3N4 to single Ni atoms realising improved photocatalytic activity for CO2 conversion to CO. The prepared photocatalysts contained 2.54, 7.95, and 12.18 wt% of Ni content respectively for Ni1–CN, Ni5–CN, and Ni10–CN compositions. The Ni5–CN composition produced 8.6 μmol g−1 of CO and 0.5 μmol g−1 of CH4 generation in the first 1 h, and was much higher than those of bare CN (CO: 1.1 μmol g−1 h−1, CH4: 0.1 μmol g−1 h−1), and Nip + CN (CO: 2.7 μmol g−1 h−1, CH4: none), and NiCl2+CN (CO: 3.3 μmol g−1 h−1, CH4: none) compositions (Fig. 14(d–i)).54
Combining experimental and computational studies, Hao et al. evaluated photocatalytic reduction of CO2 to CO using water as a reductant and g-C3N4 containing single-atom Fe as a catalyst. The photo-physical and photo-chemical processes associated with CO2 reduction mechanisms were analysed in detail. The reduction mechanism involved the activation of CO2 molecules, formation of COOH radicals through water oxidation (proton transfer from H2O to CO2) and cleavage of C–O bond in COOH radicals. It is proposed that the activation process initiates through hydrogen-bonded complexes between CO2, H2O, and photocatalysts i.e., g-C3N4 or Fe-g-C3N4. As represented in Fig. 14(j), there are two hydrogen bonds, namely HB-1(H-bond between O atom of CO2 and the H atom of H2O) and HB-2 (H-bond between H atom of H2O and the N atom of g-C3N4) formed when g-C3N4 is used as a catalyst. In the photocatalyst with single atom Fe, in addition to the formation of HB-1, CO2 and H2O molecules, are coordinated with Fe atom through Fe–C and Fe–O coordination bonds, resulting in a four-coordination structure for Fe (Fig. 14(k)).55 Fe single atoms dispersed in g-C3N4 matrix serve as catalytically active centres and facilitate the activation of CO2. The reaction barrier for the cleavage of the C–O bond in the COOH radical was reduced from 32.3 kcal mol−1 to 15.3 kcal mol−1. The change of catalytically active sites from N atom to single atom Fe accelerates the photochemical processes, thereby enhancing the CO evolution rate from 2.7 μmol gcat−1 and 5.1 μmol gcat−1.55
Zbořil and coworkers reported ruthenium-based SACs on mpg-C3N4 support for the efficient photocatalytic conversion of CO2 using water as a reductant. Mesoporous C3N4, obtained through a silica template assisted pyrolysis of dicyanamide, is used as a matrix for the microwave assisted precipitation and dispersion of Ru single atoms. Catalyst with Ru loading as low as 0.4 wt% yielded methanol as the conversion product, at a yield of 1500 μmol g−1 within 6 h. Single atoms of Ru bonded to N/C sites facilitated enhanced electron transfer by virtue of increased charge density around it.56
SACs based on g-C3N4 support exhibited superior photocatalytic carbon dioxide reduction efficacy compared to conventional metal–semiconductor heterostructures and the developments are summarised below in Table 2.
| System | Single atom & loading | Synthesis method | Products & rate | Coordination site or structure & oxidation state | Ref. |
|---|---|---|---|---|---|
| Pd/g-C3N4 & Pt/g-C3N4 | Pd, Pt | DFT study | HCOOH for Pd/g-C3N4 & CH4 for Pt/g-C3N4 | Nitrogen | 5 |
| Co2+@C3N4 | Co-0.128 μmol mg−1 | Microwave assisted post synthesis | CO 1.056 μmol mg−1 for 2 h | Nitrogen, +1 | 52 |
| a-Mo/C3N4 | Mo 3.3% | Template free direct synthesis | CO & H2,18 & 37 μmol g−1 h−1 | Mo–C & Mo–N | 27 |
| Er1/CN-NT | Er 18.4 wt% | Freeze drying assisted direct synthesis | CH4 & CO 2.5 & 47.1 μmol g−1 h−1 | Er–N3, +1 | 9 |
| Cu–CCN | Cu atomic fraction 10.95% | Molten salts & reflux method (direct synthesis) | CO 3.086 μmol g−1 h−1 | Nitrogen | 18 |
| Cu/CN | Cu 0.25% | Supramolecular pre-organisation (direct synthesis) | CO, CH3OH, CH4 11.21, 1.75 & 0.61 μmol g−1 h−1 | C–Cu–N2 | 53 |
| O/La–CN | La | Template free direct synthesis | CO 92 μmol g−1 h−1 | La–N | 16 |
| Ni/C3N4 | Ni 2.54 wt% | Template free direct synthesis | CO & CH4 1.1 μmol g−1 h−1 & 0.1 μmol g−1 h−1 | Ni–N | 54 |
| Fe/C3N4 | Fe 1.4 wt% | Post synthesis | CO 5.1 μmol gcat−1 in 10 h | Fe–N | 55 |
| Ru/mpg-C3N4 | Ru 0.4% | Microwave assisted post synthesis | CH3OH 1500 μmol g−1 within 6 hours | Ru–N/C | 56 |
Fujishima and Honda introduced photocatalytic water splitting using TiO2 photoelectrode under UV light irradiation60 and since then, numerous studies on H2 generation from photocatalytic water splitting using a variety of semiconductor photocatalysts have been conducted. The positive Gibbs free energy change (ΔG) of 237 kJ mol−1, associated with the energetics of splitting water into H2 and O2, indicates it to be an uphill reaction pathway thermodynamically.61
The photocatalytic water splitting proceeds through the three steps (i) exciton generation on the absorbance of energy equivalent to the band gap energy of the semiconductor (ii) separation and migration of photogenerated excitons (e− and h+ pairs) to the semiconductor surface and (iii) electrons mediated reduction of H+ ions to H2 and holes mediated oxidation of H2O to O2 (Fig. 15). Among oxygen evolution and hydrogen evolution reactions (OER and HER), OER is the more complicated half-reaction as it involves the release of 4e− from two H2O molecules to form the double bond between two oxygen atoms.62
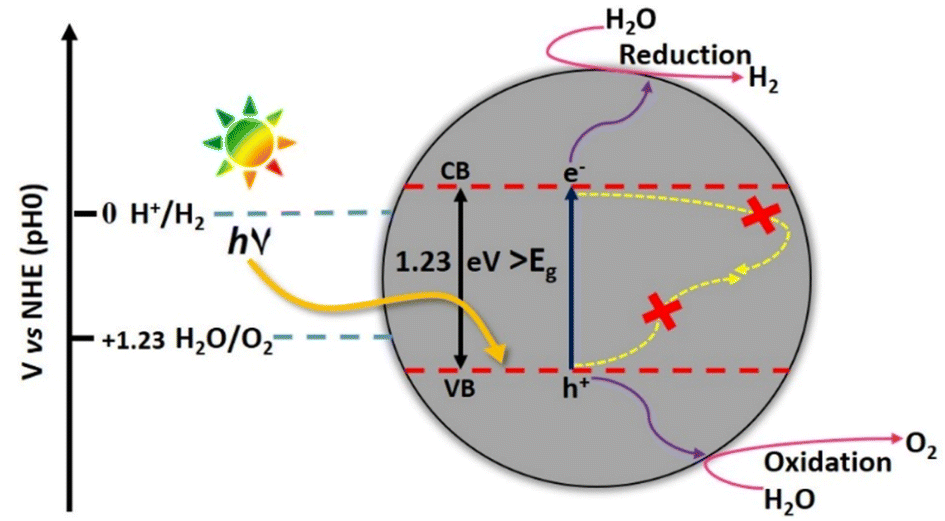 | ||
| Fig. 15 Schematic diagram of photocatalytic water splitting. | ||
The pre-requisite condition that a semiconductor must satisfy for facilitating the photocatalytic water splitting reaction, is that the valence band (VB) maxima should be more positive than the oxidation potential of water (+1.23 V vs. NHE) for OER, while the conduction band (CB) minima should be more negative with respect to the H+ reduction potential (0 V vs. NHE). The formation of water by the recombination of H2 and O2 is a detrimental step to catalytic efficiency and should be prevented.63
Copolymerising silver tricyanomethanide (AgTCM) along with mesoporous g-C3N4 precursors was demonstrated to be advantageous for the synthesis of Ag/mpg-C3N4 SACs. The uniform dispersion of Ag species in the semiconductor matrix was realised by inducing negative charges to C and N of C3N4 matrix. This approach was demonstrated to be superior to the conventional impregnation-chemical reduction method that often results in the formation of metal nanoparticles. The AgTCM-mpg-CN catalysts with up to 10 wt% of well dispersed Ag species displayed much better performance for Pt assisted photocatalytic water reduction (39.5 μmol H2 h−1 generation for 2 wt% AgTCM compared to a value of 9.8 μmol H2 h−1 for mpg-CN).17
Wu et al. synthesised highly dispersed, isolated single Pt atoms anchored on g-C3N4 (Pt–CN) as a stable photocatalyst for HER. Achieving atomic level dispersions of Pt cocatalyst over g-C3N4 enhances the overall photocatalytic HER activity by facilitating the charge transfer as well as by providing an increased number of proton reduction sites. HAADF-STEM analysis revealed that >99% of Pt species are less than 0.2 nm in size corroborating the presence of isolated single atoms of Pt over g-C3N4 matrix. The cluster formation was avoided up to a Pt loading of 0.16 wt%. As the Pt loading was increased to 0.38 wt% Pt nanoparticle formation was observed. The Pt–CN with Pt loading of 0.16 wt% exhibited the highest HER activity with a turnover frequency (TOF) of 775 h−1, compared to a TOF of 83 h−1 obtained for Pt-NPs-CN (Fig. 16(a and b)). Ultrafast transient absorption (TA) spectroscopy was employed to elucidate the unique role of Pt single atom in enhancing the HER activity. Average recovery life time values, a measure of the extent of separation of photogenerated excitons, of g-C3N4, Pt–CN, and Pt-NPs-CN were found to be ≈237 ps, ≈433 ps, and ≈117 ps respectively (Fig. 16(c and d)). The two-fold and four-fold enhancement in average recovery lifetime for Pt–CN compared to bare g-C3N4 and Pt–NPs–CN was attributed to the incorporation of Pt single atoms in g-C3N4 framework. The incorporation of isolated single Pt atoms modifies the near band-edge electron trap states of g-C3N4 favouring the H+ reduction process by photogenerated electrons (Fig. 16(e and f)).64
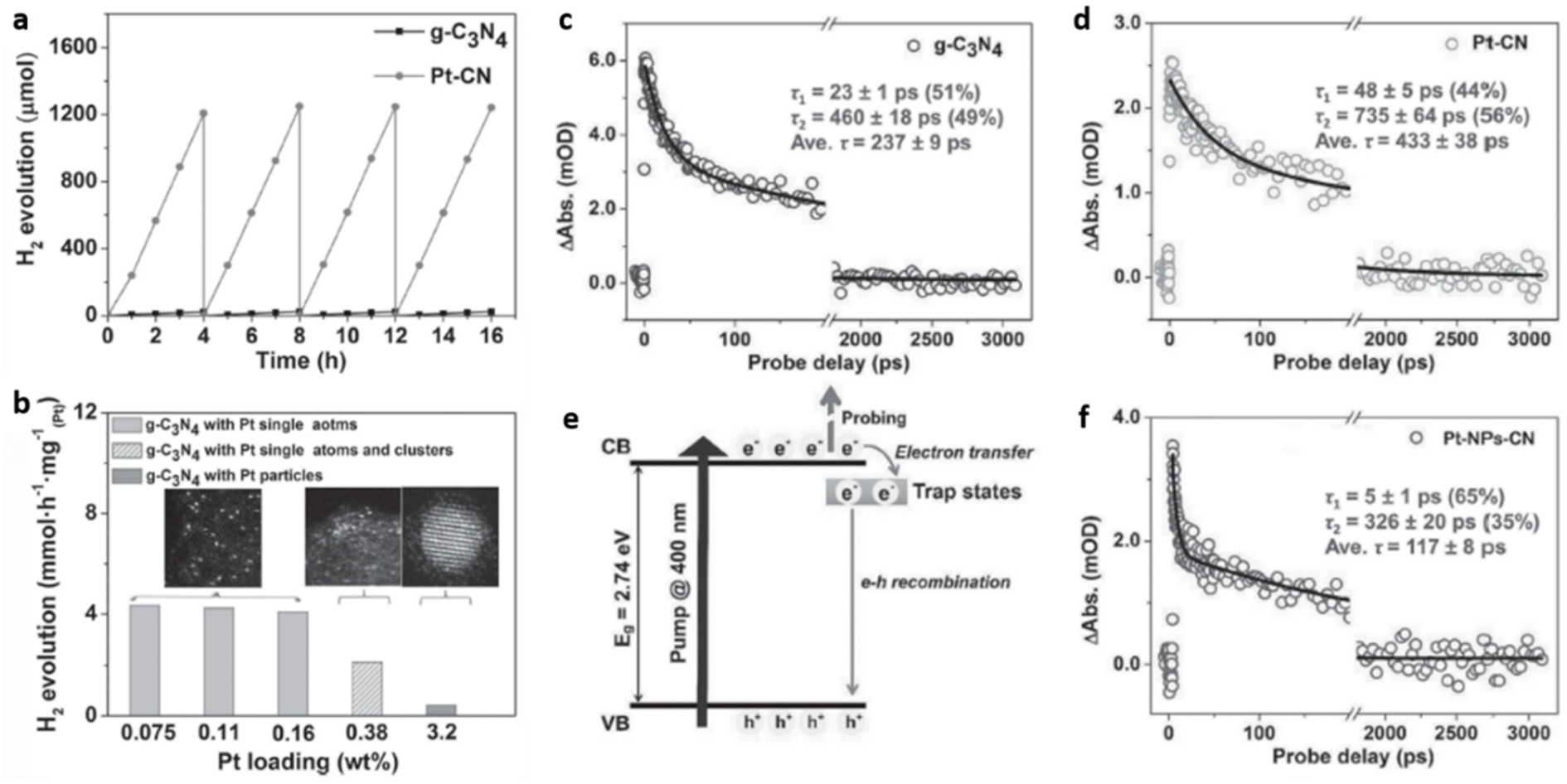 | ||
| Fig. 16 (a) Photocatalytic H2 evolution with time for g-C3N4 and Pt–CN, (b) photocatalytic H2 evolution with varying Pt content (normalised per Pt atom), average recovery life time values in mOD (optical density) of (c) g-C3N4, (d) Pt–CN, (e) an illustration of the various mechanisms involved, (f) data on TA kinetics for Pt-NPs-CN. Reproduced with permission from ref. 64. Copyright 2016 John Wiley and Sons. | ||
Yao et al. employed phosphidation followed by atomic-layer-deposition (ALD) to atomically disperse single-site Co atoms on g-C3N4 nanosheets (Co1/PCN) for the photocatalytic HER reaction using triethanolamine (TEOA) as the sacrificial electron donor. The grafting of Co atoms over C3N4 nanosheet is achieved through the formation of a covalently bonded Co1–N4 structure. Co–N coordination is confirmed with the aid of XANES, EXAFS and XPS. Co1/PCN exhibited XANES spectrum which is different in shape from that of cobalt oxides (CoO and Co3O4) presumably due to Co–C/N coordination. Similarly, a single peak observed at 1.58 Å in Fourier-transformed (FT) k3-weighted χ(k) function of Co1/PCN is close to Co–N bond present in the reference material (CoPc) ruling out the formation of Co–Co, Co–P and Co–O bonds. XPS spectrum of Co1/PCN further confirmed the existence of Co–N bond with a Co 2p3/2 peak located at a significantly higher binding energy value of 781.1 eV compared to that of Co–O bond at 780 eV. Additionally, the presence of N vacancies in Co1/PCN sample is substantiated by XPS analysis as the pyridinic N content observed in the high-resolution N 1s spectra is decreased. To understand the structural configuration of the Co single site in Co1/PCN, various possible atomic configurations are proposed with specific deposition sites/coordination sites. Accordingly, Co single atoms may be at the corner and center of the six-N cavity (Co–N2 and Co–N6 respectively), top of the five-membered ring (Co N3C2), and unsaturated coordination site containing four in-plane N atoms (Co N4). The Co–N bond length value of 2.02 Å obtained from EXAFS spectra ruled out the existence of Co–N6 structure where the Co–N bond length value should be 2.40 Å. The experimental Co K-edge XANES spectra of Co1/PCN exactly matched with calculated XANES spectra for Co–N4 configuration which confirmed the formation of Co1–N4 structure (Fig. 17(a–d)). Co1/PCN with a metal mass loading of 1.0% exhibited an improved H2 production rate of 10.8 μmol h−1 which is approximately an order of magnitude higher than the PCN with a H2 production rate of 1.0 μmol h−1 confirming the unique role of Co1–N4 site in improved activity (Fig. 17(e)). The coordinated nitrogen atom present in Co1–N4 structure enhanced the electron density around the Co atom. The energy barrier was lowered for the formation of Co hydride intermediate with 3H atom co-adsorbed configuration promoting H–H coupling for accelerated H2 evolution (Fig. 17(f)).24
 | ||
| Fig. 17 (a) XANES (Co K-edge) spectra of Co1/PCN in comparison with standards and possible atomic configurations (b) Fourier spectra of Co1/PCN in comparison with standards (c) XPS Co 2p spectra of Co1/PCN and C3N4, (d) model structures of “Co1–N4”, “Co1–N2”, “Co1–N3C2” with blue, yellow and grey spheres representing N, Co and C atoms, respectively, (e) H2 evolution with time under simulated solar irradiation, (f) energy diagram of Co1–N4 for H2 evolution on single-sites with the transition state of the reaction TS-(H + H → H2). N (blue), Co (green) and H (white) spheres. Reproduced with permission from ref. 24. Copyright 2017 John Wiley and Sons. (g) The Tauc plot of the g-C3N4 and Co1 phosphide/PCN catalysts (h) electronic band structure of Co1-phosphide/PCN catalyst. Reproduced with permission from ref. 65. Copyright 2017 John Wiley and Sons. | ||
Wei et al. designed HER and OER phosphorus-doped g-C3N4 (PCN) photocatalysts with active Co1-phosphide species. Co1-phosphide/PCN composite photocatalyst was synthesised by the phosphidation of Co1 oxo/C3N4 composite obtained through the pyrolysis of cobalt precursor and g-C3N4. The phosphidation of Co1-Oxo/C3N4 using sodium hypophosphite altered the coordination environment around Co metal atoms from oxygen to phosphorus to form Co1–P4 structure. Co1-phosphide/PCN photocatalyst with a Co mass loading of 0.4% exhibited superior photocatalytic activity than most of the g-C3N4 and oxynitride based materials. H2 production rate of 410.3 μmol h−1 g−1 and O2 production rate of 204.6 μmol h−1 g−1 were achieved without using noble metal co-catalysts, sacrificial reagents, or other additives and by employing pure water under simulated sunlight. The superior photocatalytic water splitting capacity of Co1-phosphide/PCN can be attributed to the enhanced light absorption in UV to visible wavelength range and to the introduction of a new mid-gap state within the band structure of Co1-phosphide/PCN. The band-gap energy (Eg) and transition energy (Et) from VB to the mid-gap states for Co1-phosphide/PCN system were estimated to be 2.97 and 2.15 eV, respectively (Fig. 17(g)). Enhanced light harvesting capacity of Co1-phosphide/PCN was ascribed to the small energy gap between the mid-gap state and VB, favouring the excitation of an electron from VB to the mid-gap states (Fig. 17(h)).65
Zhong et al. prepared Pt single atoms dispersed on g-C3N4 nanosheets (Pt-SA-CN) by a copolymerisation strategy involving melamine, cyanuric acid and 2,4-diamino-6-methyl-1,3,5-triazine. Pt loading less than 0.3 wt% has been realised by the wet impregnation method using H2PtCl6. The immobilisation and atomic level dispersion of Pt atoms over g-C3N4 was established through Pt–N coordination bonds. Pt-SA-CN catalyst with a Pt loading of 0.20 wt% displayed enhanced H2 production of 4875.0 μmol g−1 in 4 h. A correlation is established with the Pt single atom size on the metal loading and its corresponding photocatalytic activity. The size of Pt atoms in Pt-SA-CN with 0.2 and 0.3 wt% Pt loading was found to be 0.21 and 0.5 nm respectively and a subsequent reduction in the H2 evolution was observed with increasing Pt size.66
Liu et al. successfully developed PtII–C3N4 single atom catalyst for efficient photocatalytic water splitting by employing high-valence metal single-atom confinement strategy. PtII–C3N4 exhibited H2 evolution at the rate of 140 μmol g−1 h−1, which is ∼10 times higher compared to that of Pt NP–C3N4 (15 μmol g−1 h−1). Upon visible light irradiation, the PtII–C3N4 system shows a quantum efficiency of 1.5% at 420 nm. Anchoring PtII single atoms on g-C3N4 matrix via PtII–N bond formation facilitated higher absorption of visible light and effectively suppressed exciton recombination. The Eg values for g-C3N4, PtII–C3N4, Pt nanoparticle-C3N4, derived from Tauc plot were found to be 2.77, 2.72, and 2.6 eV respectively. The flat band potential of PtII–C3N4 (−1.01 V vs. RHE) is appreciably shifted to a more positive value compared to Pt NP–C3N4 (−1.25 V) and g-C3N4 (−1.44 V) (Fig. 18(a)). Ultraviolet photoelectron spectra (UPS) revealed that the VB maxima for PtII–C3N4 were shifted downward by ∼0.27 V compared to g-C3N4, which may be due to the hybridisation of 5d orbital of PtII & 2p orbital of N atom present in the g-C3N4 framework (Fig. 18(b)). The modified electronic band structure of PtII–C3N4 effectively promoted the overall photocatalytic water splitting reaction (Fig. 18(c)). Band structure calculations based on the first principles were performed to understand the correlation between the electronic structure of PtII–C3N4 photocatalyst and its improved charge carrier transfer kinetics. Electron density of state (DOS) analysis on g-C3N4 (Fig. 18(d)) revealed that the Fermi level (middle of the band gap) is devoid of mobile electrons but an intersection of the valence band and Fermi level is evident in the DOS of PtII–C3N4 photocatalyst (Fig. 18(e)). In addition, the strong electrophilic nature of the valence band of PtII–C3N4 formed through the hybridisation of PtII 5d and N 2p orbitals promotes the electron transfer to CB catalysing HER and leaving behind the holes at the VB to facilitate OER (Fig. 18(f)).23
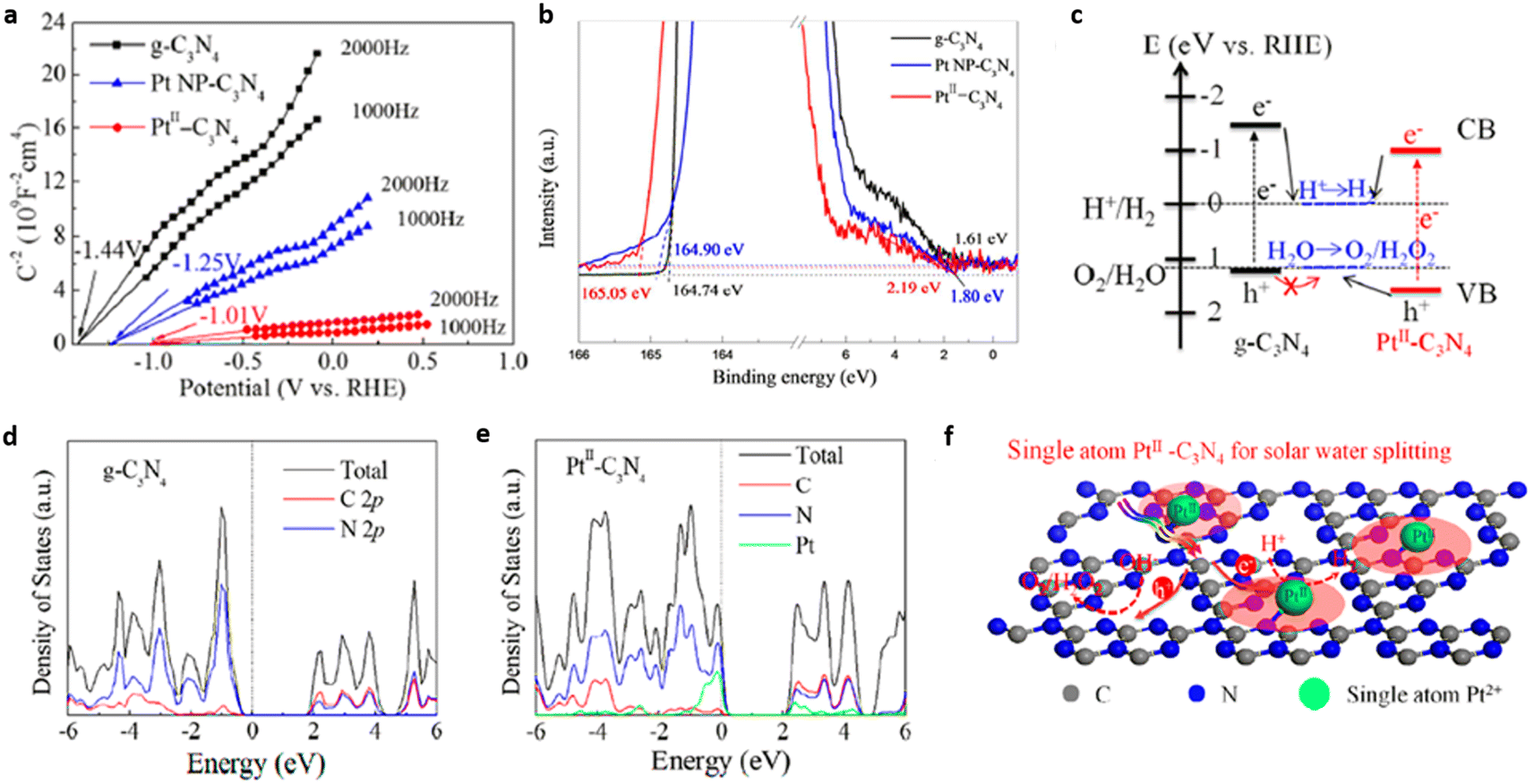 | ||
| Fig. 18 (a) Mott–Schottky plot of PtII–C3N4 in comparison with g-C3N4 and Pt NP–C3N4, (b) UPS spectra of g-C3N4, PtII–C3N4 & Pt NP-C3N4, (c) schematic band structure alignment, DOS of (d) g-C3N4 and (e) PtII–C3N4, (f) mechanistic pathways (schematic) of the photocatalytic water splitting by PtII–C3N4. Reproduced with permission from ref. 23. Copyright 2018 American Chemical Society. | ||
g-C3N4 functionalised with amino groups at the edges were utilised to develop atomically dispersed Fe SACs for solar driven photocatalytic hydrogen generation. The spontaneous coordination of dicyandiamidine nitrate with Fe ions led to the formation of porous crimpled g-C3N4. The incorporation of Fe single atoms altered the band and electron structures enabling faster charge transfer kinetics realising hydrogen evolution rate of 3390 μmol h−1 g−1 at a quantum efficiency of 6.89% at 420 nm.67
A promising approach to optimise catalytically active sites is by tuning the reactive metal–support interaction (RMSI) which is conventionally realised by the high-temperature reactions (T > 550 °C) of reducible oxides. Guo et al. reported strong RMSI in Pt single atom containing g-C3N4 obtained by the in situ photocatalytic reduction of (H2PtCl6) at liquid nitrogen temperatures. The strong RMSI, induced by rich N vacancies in C3N4 provided a record single atom coverage density of 0.35 mg m−2 leading to 174.5 mmol g−1 h−1 of hydrogen evolution. Simulation studies indicated that RMSI stabilises single atoms of Pt through chemical bonds with two coordinated C (C2C) formed by the N vacancies.25
A photoactive, single atom cobalt loaded g-C3N4 catalyst obtained through a supramolecular polycondensation process based on dicyandiamide is demonstrated for photocatalytic hydrogen evolution using TEOA as a sacrificial agent and 3 wt% Pt as cocatalyst. The synthesised Co@g-C3N4 composite exhibited improved photocatalytic hydrogen evolution at a rate of 2481 μmol h−1 g−1. The incorporation of Co single atoms strengthened the reducing capability of the conduction band (CB) electrons due to a shift in the CB edge minima to a more negative value compared to that of g-C3N4 (0.83 V and −0.95 V vs. SCE respectively).19
Employing 2D confinement strategy, Quan et al. developed Pt single atoms confined on carbon nitride photocatalyst with ultrahigh Pt loading of 8.7 wt% for the H2 evolution reaction. The photocatalytic performance of the synthesised samples was dependent on the location of the Pt atoms at either the surface or within the inner layers of g-C3N4 (Fig. 19(a–d)). The interlayer interactions altered the electronic structure by delocalising the charge density of the Pt atom which in turn facilitated the adsorption of protons and caused the reduction of the energy barrier for HER (Fig. 19(e and f)). The developed photocatalyst exhibited H2 evolution rate of 22.65 mmol g−1 h−1 with an apparent quantum yield (AQY) of 22.5% at 420 nm.68
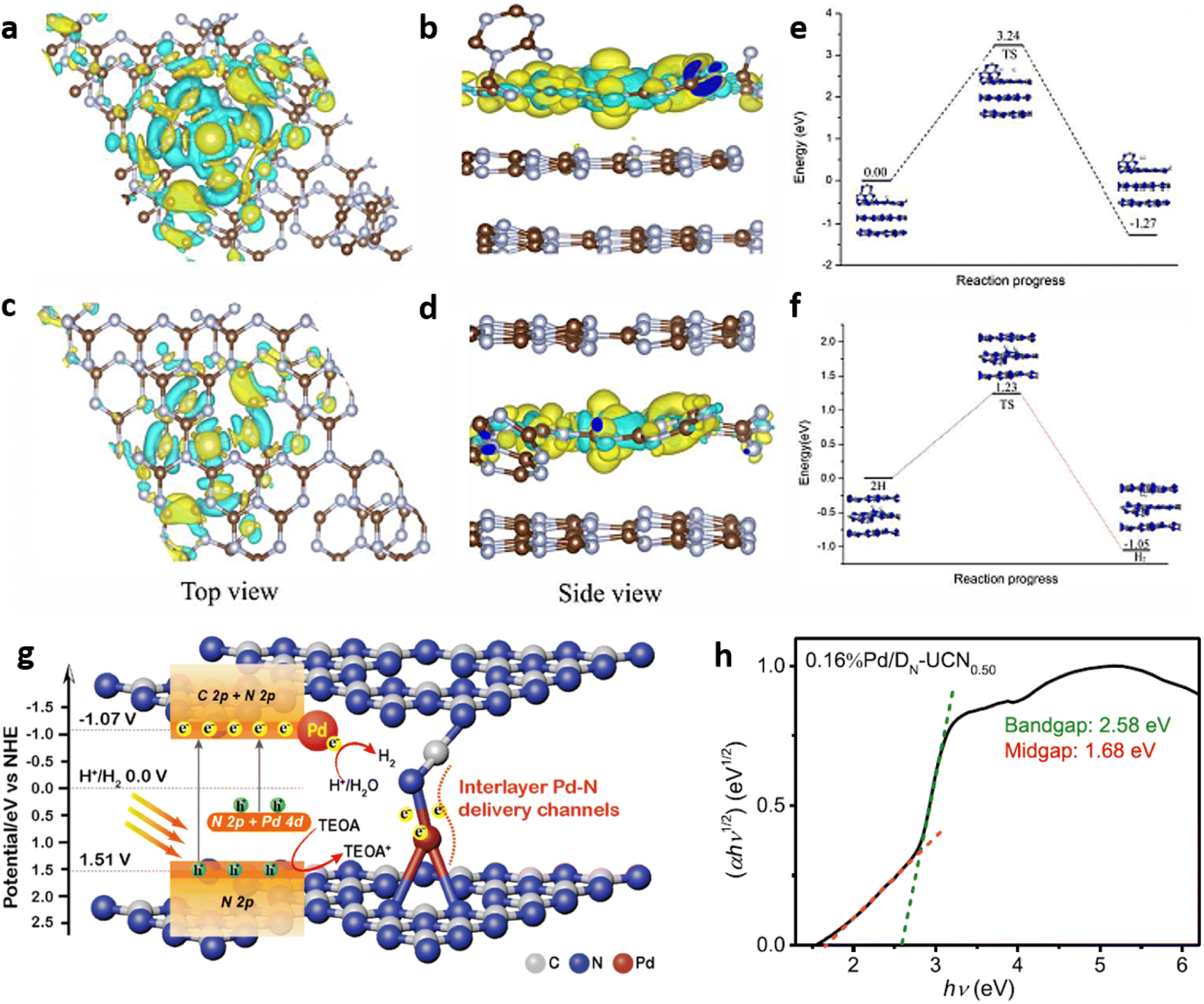 | ||
| Fig. 19 Charge distributions (top and side view) on Pt single atoms (a and b) on C3N4 surface (SA-Pt/g-C3N4-Sur), (c and d) within the inner layer of C3N4 (SA-Pt/g-C3N4-In), energy profiles of HER over Pt atom (e) on C3N4 surface and (f) within the inner layer of C3N4. Reproduced with permission from ref. 68. Copyright 2020 Elsevier.68 (g) Mechanism of charge separation, transfer and H2 evolution in the photocatalyst 0.16%Pd/DN-UCN0.50, (h) Kubelka–Munk plot of 0.16%Pd/DNUCN0.50. Reproduced with permission from ref. 70. Copyright 2022 American Chemical Society. | ||
C3N4 hollow spheres dispersed with single atoms of Cu were developed by a molecular assembly involving melamine (ME) and cyanuric acid (CA) with copper nitrate. Single atoms of Cu were either embedded within g-C3N4 sheets (Cu1@HCNS) or dispersed on its surface (Cu1/HCNS) as Cu1N3 species through Cu–N coordination. The copper single atoms embedded within the sheets were more effective in promoting interfacial charge transfer as well as tuning the electronic band structure. The Cu1@/HCNS exhibited improved HER activity (using 3 wt% Pt cocatalyst) with AQY of 7.1% which is 1.6, 2.7 35.4 times higher compared to Cu1/HCNS, HCNS, and bulk PCN respectively.69
Pd single atoms dispersed on ultrathin nanosheets of cyano group rich C3N4 (Pd/DN-UCN), obtained by the copolymerisation of urea and NH4Cl followed by wet impregnation was demonstrated to possess improved photoinduced hydrogen evolution rate. Pd atom coordinated with one N of the cyano group and two sp2 hybridised N atoms of the adjacent g-C3N4 layers was firmly stabilised in g-C3N4 interlayers inducing a mid-gap state in the band structure (Fig. 19(g and h)). SACs with 0.16 wt% of Pd (0.16%Pd/DNUCN0.50) produced noticeably higher hydrogen evolution rate, compared to Pd nanoparticle coordinated C3N4 based catalysts, at an apparent quantum yield of 15.8% at 400 nm light irradiation.70
A two-component synergistic photocatalyst with atomic dispersions of Co and nanoparticles of Pt–Co alloy on C3N4 nanosheets exhibited hydrogen production at the rate of 300.9 mmol h−1 g−1 during overall water splitting (OWS). The single Co atoms efficiently activated HER while the alloy particles of PtCo were active sites for OER. The synergistic combination enabled high atom utilisation and the presence of different reaction centres permitted the transport of reaction intermediates with efficient exciton separation.71 The salient features of the reported works are summarised in Table 3.
| System | Single atom & loading | Synthesis method | HER rate | Coordination site or structure & oxidation state | Ref. |
|---|---|---|---|---|---|
| AgTCM-mpg-CN | Ag 2 wt% | Template assisted direct synthesis | 39.5 μmol H2 h−1 | C/N, +1 | 17 |
| Pt–CN | Pt 0.16 wt% | Post synthesis-photo chemical reduction | 318 μmol h−1 | N/C | 64 |
| Co1/PCN | Co 1 wt% | Post synthesis-atomic layer deposition (ALD) | 10.8 μmol h−1 | Co1–N4 | 24 |
| Co-phosphide/PCN | Co 0.4 wt% | Post synthesis | H2 410.3 μmol h−1 g−1, O2 204.6 μmol h−1 g−1 | Co1–P4 | 65 |
| Pt-SA-CN | Pt 0.11 wt% | Post synthesis | 4875.0 μmol g−1 in 4 h | Nitrogen | 66 |
| Pt/C3N4 | Pt | Post synthesis-electrostatic deposition | 140 μmol g−1 h−1 | Nitrogen, +2 | 23 |
| Fe@g-C3N4 | Fe 0.5 at% | Supramolecular preorganisation assisted direct synthesis | 3390 μmol h−1 g−1 | Nitrogen, +2 | 67 |
| Pt-SA-CN | Pt 1.72 wt% | Post synthesis -photo chemical reduction | 3.02 mmol g−1 h−1 | Nitrogen, carbon | 25 |
| Co@g-C3N4 | Co 0.305 wt% | Supramolecular preorganisation assisted direct synthesis | 2481 μmol h−1 g−1 | Nitrogen oxidation state between +3 & +2 | 19 |
| SA-Pt/g-C3N4 | Pt 8.7 wt% | Post synthesis-photo chemical reduction | 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 650 μmol g−1 h−1 650 μmol g−1 h−1 |
Pt–N4 oxidation state <+2 | 68 |
| Cu1/HCNS | Cu | Post synthesis | 3261 μmol g−1 h−1 | Cu1N3, +1 | 69 |
| Pd/DN-UCN | Pd 0.16% | Post synthesis-Photochemical reduction | 441.6 μmol in 4 h | Pd–N3, +2 | 70 |
| CoSAs/PtCo@CNN | Co | Post synthesis-photo chemical reduction | 300.9 μmol h−1 g−1 | Co(II)Nx, +2 | 71 |
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond with a bond energy of −941 kJ mol−1, high ionisation potential, and non-polarity. Since nitrogen is indispensable for the sustenance of life, living organisms take N2 in the form of ammonia or nitrates. This is mostly realised through 3 important pathways of biological nitrogen fixation by azobacteria, high energy nitrogen fixation by geochemical processes such as lightning and industrial nitrogen fixation through the well-known Haber–Bosch process. However, the contributions from biological and geochemical processes are extremely low and most of the demand is met from the energy intensive Haber–Bosch process that operates at both high temperatures (400–500 °C) and pressures (150–250 atm). Moreover, in the industrial synthesis of ammonia, 1.87 tons of CO2 is released for every ton of NH3 produced. Photocatalytic nitrogen fixation thus assumes larger significance and is hence pursued widely as a sustainable process for ammonia production in an environmentally benign manner.72
N bond with a bond energy of −941 kJ mol−1, high ionisation potential, and non-polarity. Since nitrogen is indispensable for the sustenance of life, living organisms take N2 in the form of ammonia or nitrates. This is mostly realised through 3 important pathways of biological nitrogen fixation by azobacteria, high energy nitrogen fixation by geochemical processes such as lightning and industrial nitrogen fixation through the well-known Haber–Bosch process. However, the contributions from biological and geochemical processes are extremely low and most of the demand is met from the energy intensive Haber–Bosch process that operates at both high temperatures (400–500 °C) and pressures (150–250 atm). Moreover, in the industrial synthesis of ammonia, 1.87 tons of CO2 is released for every ton of NH3 produced. Photocatalytic nitrogen fixation thus assumes larger significance and is hence pursued widely as a sustainable process for ammonia production in an environmentally benign manner.72
Photocatalytic nitrogen fixation proceeds by the rate determining step of NN bond cleavage through electron mediated activation at the reducing sites of semiconductor photocatalysts. However, till date no reported photocatalyst possesses the appropriate electronic band structure to facilitate the one-electron transfer reduction of N2. The defect type activation centers utilised by most semiconductors are not adequate to make the process efficient and commercially viable. In this context single atom photocatalysts by virtue of their abundant, defect-free surface-active sites for effective N2 adsorption and activation, offer promise for efficient photocatalytic N2 fixation.13
In one of the earlier works on ‘single atom like’ copper-doped g-C3N4 containing Cu(I)–N active sites, the charge density differences were used to simulate the electron transfer process between Cu+ and N2 molecules leading to electron depletion and accumulation on Cu+ and N2 respectively. Further successive electron transfer processes lead to NH4+ through high-energy intermediates like –N2−, –N2H, or HNNH, etc. DOS results confirmed the delocalisation of σg2p orbital (highest occupied molecular orbital (HOMO) of N2) upon adsorption on Cu centres as well as the shifting of πg*2p orbital (lowest unoccupied molecular orbital (LUMO) of N2) near to Fermi level. This led to a decrease in the electron states of the LUMO orbital from 6.9 eV for free N2 to 1.0 eV for MS–Cu–CN facilitating the activation of N2 molecule and subsequent H+ attack leading to the formation of NH4+.73
Wang and co-workers demonstrated, by first-principles calculations, the potential use of a metal-free photocatalyst with boron (B) single atoms decorated on g-C3N4 (B/g-C3N4) for efficient N2 reduction. The sp3 hybridised B with three half-filled and one empty orbital form two B–N bonds with g-C3N4 matrix (utilising two of the half-filled sp3 orbitals), facilitating the atomic level distribution of B atoms. During the N2 fixation process, one half-filled and the empty sp3 orbitals remaining in the B atom interact with N2 through the “acceptance–donation” process facilitating active sites for N2 adsorption and subsequent reduction Fig. 20(a–f).74 The high binding energies of −1.04 and −1.28 eV can thus be realised in B/g-C3N4 SACs through side-on and end-on bonding with N2 respectively. Photoreduction of N2 to NH3 over B/g-C3N4 catalyst may follow any of the three typical mechanisms of distal, alternating, and enzymatic mechanisms (Fig. 20(g)). The low onset potential of 0.20 V obtained by B/g-C3N4 SACs supported the enzymatic pathway for N2 reduction Fig. 20(h–j). This theoretical study also predicted low formation energy for B/g-C3N4 SACs, enhanced light absorption in the visible and infrared region, and high thermal stability up to1000K enabling sustainable NH3 production through photocatalytic N2 fixation. The B/g-C3N4 SACs showed enhanced visible light and infrared light absorbance, high thermal stability and ease of preparation enabling them to be effective for photocatalytic nitrogen reduction.74
 | ||
| Fig. 20 (a) DOS of free N2 molecule, (b) N2 molecule adsorbed on pure GCN and (c) N2 molecule adsorbed on MS-Cu-CN. Reproduced with permission from ref. 73. Copyright 2017 American Chemical Society. (d) Illustration of N2 bonding to transition metals, (e) formation of sp3 orbitals in B atom, (f) N2 binding to the B atom stabilised on g-C3N4, (g) distal, alternating, and enzymatic mechanisms for N2 reduction, free energy diagrams for (h) distal, (i) alternating, (j) enzymatic pathways for N2 reduction on B/g-C3N4. Reproduced with permission from ref. 74. Copyright 2018 American Chemical Society. | ||
Mo single-atoms on in situ formed g-C3N4 were prepared by the calcination of urea and Na2MoO4·2H2O by establishing coordination with two N donors of g-C3N4 to form MoN2 structures. The active sites for N2 chemisorption provided by low-coordinated Mo centers in 3-Mo-PCN catalyst, with a metal loading of 0.346 wt%, facilitated high photocatalytic activity towards NH3 evolution in pure water at a rate of 50.9 μmol gcat−1 h−1. The use of ethanol as an electron scavenger increased the rate to 830 μmol gcat−1 h−1 at a quantum efficiency of 0.70% at 400 nm. DFT calculations supplemented experimental observations and predicted that coordinatively unsaturated Mo single atom centres strongly adsorb N2 through an end-on configuration resulting in the elongation of the NN bond from 1.11 Å to 1.15 Å. The photogenerated electrons are thus transferred to the weakened NN bond providing efficient N2 reduction at ambient conditions.75
Chen and co-workers successfully synthesised multifunctional single atom cobalt loaded g-C3N4 (Co@g-C3N4) photocatalyst for N2 fixation reaction in a methanol/H2O medium. Co@g-C3N4-1 with a metal loading of 0.305 wt% selectively reduced N2 to NH3 at the rate of 50.2 μmol h−1 and without any by-products like hydrazine hydrate and nitrates. In comparison bulk g-C3N4 and g-C3N4 obtained through the supramolecular route exhibited a production rate of 8.2 μmol h−1 and 19.5 μmol h−1 respectively.19
Single atoms of ruthenium (Ru) dispersed on the g-C3N4 surface were obtained by the pyrolysis of a precursor mix containing dicyandiamide and RuCl3·xH2O in an argon atmosphere. SAC containing 0.05 atomic % of Ru exhibited photocatalytic nitrogen fixation at the rate of 2.26 mg L−1 gcat−1 h−1 in pure water and without hole-scavengers. The rate was 1.5 times greater than that of pure g-C3N4 (Ru–CCN-0). The improved catalytic activity was ascribed to the high N2 adsorption capacity combined with increased light absorbance in the spectral region of 200 to 800 nm owing to a redshift in the absorption edge of Ru–CCN-0.05 compared to that of Ru–CCN-0.76 The SAC systems demonstrated for photocatalytic nitrogen fixation are listed in Table 4.
| System | Single atom & loading | Synthesis method | NH3 evolution rate | Coordination site or structure & oxidation state | Ref. |
|---|---|---|---|---|---|
| MS-Cu-CN | Cu 0.25 wt% | Direct synthesis | 8.8 mg L−1 h−1 gcat−1 | Cu(I)–N, +1 | 73 |
| B/g-C3N4 | B | DFT study | — | Nitrogen | 74 |
| Mo–PCN | Mo 0.346 wt% | Template free direct synthesis | 50.9 μmol gcat−1 h−1 in pure water & without using electron scavenger, 830 μmol gcat−1 h−1 using ethanol as electron scavenger | Mo–N2, oxidation state in between 0 & +4 | 75 |
| Co@g-C3N4 | Co 0.305 wt% | Chemical reduction assisted Post synthesis | 50.2 μmol h−1 | Nitrogen, +2 | 19 |
| Ru–CCN | Ru 0.34 wt% | Direct synthesis | 2.26 mg L−1 gcat−1 h −1 in pure water | Nitrogen | 76 |
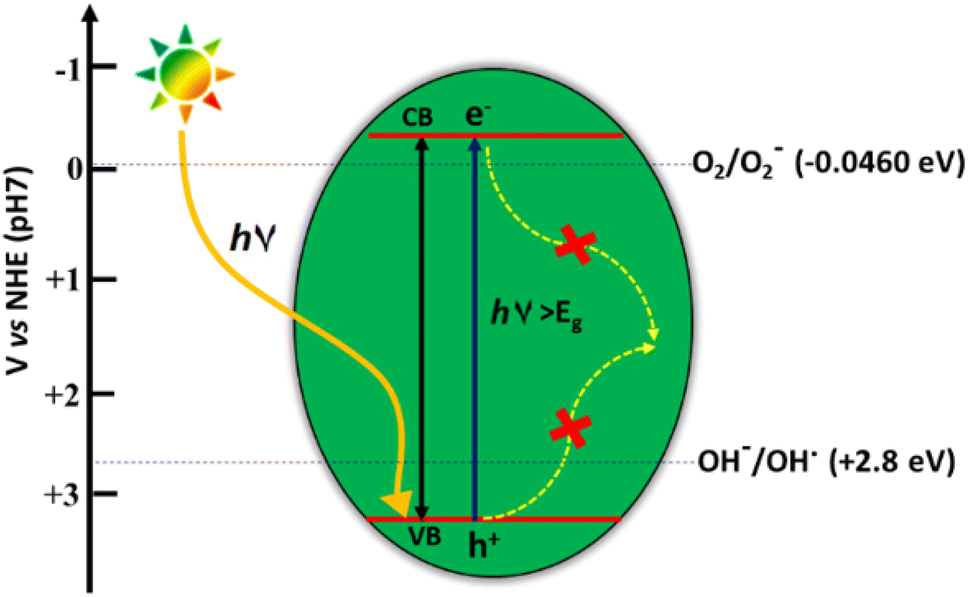 | ||
| Fig. 21 Schematic of the photocatalytic process. | ||
Reactions:
| photocatalyst + hv → e− + h+ |
| h+ + H2O → H+ + OH˙ |
| O2 + e− → O2−˙ |
| O2−˙ + H+ → HO2 |
| 2HO2 + 2H+ → H2O2 + 2OH˙ |
| OH˙/H2O2/O2−˙ + pollutant → degradation products |
Zhao and co-workers developed single-atom Ag supported mpg-C3N4 (Ag/mpg-C3N4) by a method involving the co-condensation of dicyandiamide and silver tricyanomethanide (AgTCM) through a silica templating approach. The photocatalytic activity was demonstrated by peroxymonosulfate (PMS) mediated bisphenol A (BPA) degradation by 10% Ag/mpg-C3N4 catalyst, where in 100% activity was achieved within 1 h of visible light illumination. The improved catalytic activity was realised due to the synergistic effect of atomically dispersing Ag atoms on high surface area g-C3N4 leading to a reduction in band gap from 2.5 eV (mpg-C3N4) to 2.09 eV (10% Ag/mpg-C3N4).82
Single Pt atom catalyst on g-C3N4 described earlier66 for photocatalytic HER has been successfully employed for the selective photocatalytic NO oxidation to NO3−. The oxidation proceeded progressively over 150 min with a removal ratio of 96.2% for Pt 0.2-SA-CN. In comparison, pure g-C3N4 and g-C3N4 with 0.1 and 0.3 wt% of single atom Pt (Pt 0.1-SA-CN, Pt 0.3-SA-CN) exhibited 78.9%, 84.7%, and 92.0% of NO removal respectively. Additionally, g-C3N4 with photo-deposited Pt nanoclusters of 4 nm size (Pt 0.2-VL-CN) samples exhibited 90.9% NO removal. The negative shift of CB and the increased generation of ROS (h+, ˙OH or ˙O2−) expedited the photo-induced catalytic activity.66
Liu et al. synthesised Ag loaded SAC on ultrathin g-C3N4 sheets (AgTCM/UCN) via co-polymerisation of dicyandiamide and silver tricyanomethanide (AgTCM) in the presence of NH4Cl. AgTCM/UCN with 1 wt% Ag loading photocatalytically degraded 86.4% of sulfamethazine (SMT) in the presence of PMS during the 21 min of visible light irradiation. Aberration corrected HAADF-STEM analysis revealed that Ag single atoms of ∼0.25 nm diameter were dispersed over the g-C3N4 matrix. The photodegradation of SMT was shown to proceed via any of the three pathways involving sulfonamide bond cleavage, SO2 extrusion, and aniline oxidation.83
g-C3N4 based catalyst containing Fe single atoms and ultra-small clusters with Fe(II)–Nx active sites was prepared by pyrolysing a precursor mix containing melamine (MA) and iron imidazole complex (Fe-ICC). High Fe content up to 18.2 wt% was realised in a composition (I-FeNx/g-C3N4-5) with an optimum ratio of MA/Fe-ICC to be 5. DFT calculations revealed that Fe single atoms preferably occupy the interlayers of g-C3N4via bonding with nitrogen located above and below it and with a binding energy (BE) of −3.92 eV compared to a low BE of −2.77 eV in Fe atom at the center of the 6-fold cavity. Additionally, ultra-small Fe clusters (Fe2 dimer as a model) are stable only at the interlayers with a BE of −2.50 to −2.83 eV. A high binding energy value (−3.92 eV) further revealed that Fe atoms remain isolated preferably over its cluster formation (Fig. 22(a–d)). The developed I-FeNx/g-C3N4-5 catalyst are shown to exhibit rapid photo-fenton degradation (100% within 15 min at pH 7) of organic contaminants like methylene blue (MB), methyl orange (MO), rhodamine B (RhB), and phenol. The degradation process proceeds via the rapid generation of HO˙ radicals from H2O2 at the Fe(II)–Nx active sites.84
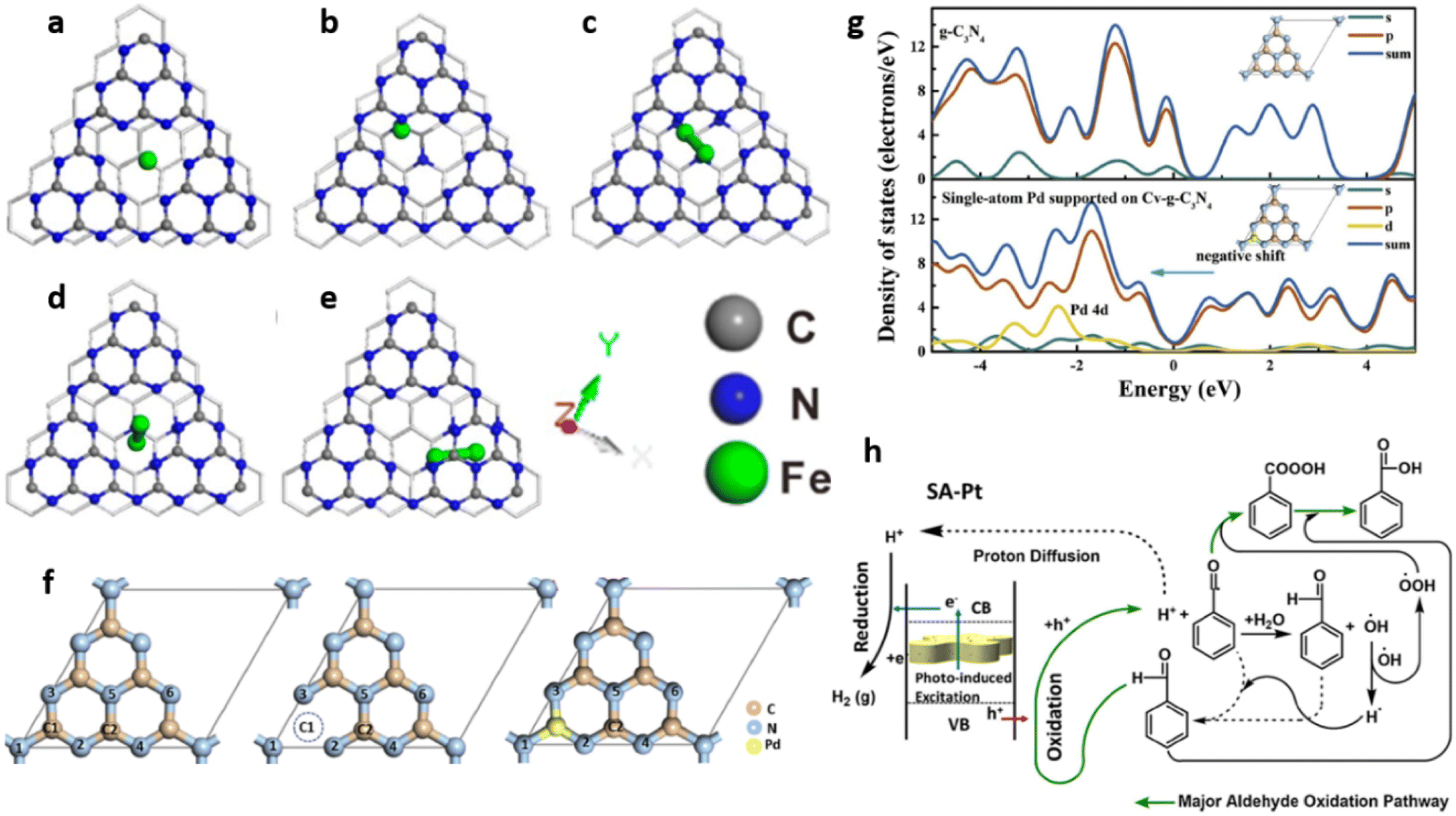 | ||
| Fig. 22 Structures depicting possible locations for Fe single atom and cluster over g-C3N4; Fe single atom in (a) 6-fold cavity centre, (b) interlayer. Fe cluster sites in the interlayer (c), Site 1, (d) Site 2 (e) Site 3. Reproduced with permission from ref. 84. Copyright 2018 American Chemical Society. (f) The carbon vacancies and single atom site of Pd in g-C3N4 and (g) corresponding DOS. Reproduced with permission from ref. 90. Copyright 2021 Elsevier. (h) Schematic diagram representing the mechanism of benzaldehyde oxidation and associated H2 evolution with photoinduced electron and hole transfer. Reproduced with permission from ref. 91. Copyright 2021 Elsevier. | ||
Silver single-atom catalyst based on amorphous g-C3N4 was developed by Tong et al. through a supramolecular gel formation involving precursors of melamine and silver nitrate. The thermal polymerisation yielded a high density of silver atoms that selectively broke H-bonds of layered g-C3N4 inducing amorphisation. An optimised MA composition with Ag atomic ratio of 11.6% exhibited a narrow Eg value of 2.54 eV, improved visible light absorption, and accelerated charge transfer leading to the effective degradation of naproxen (NPX).85
Ammonia-assisted selective catalytic reduction (SCR-NH3) is the conventional methodology to reduce NO emissions but suffers from the disadvantages of particulate formation, hardware corrosion and direct release of unreacted NH3. Direct reduction of NO with CO, without NH3 injection, is an environmentally benign approach to circumvent the disadvantages of the SCR-NH3 process. Ni Single atoms on monolayer g- C3N4 (Ni-g-C3N4) were demonstrated to be an efficient and stable catalyst for the photocatalytic reduction of NO to N2 at about 350 °C and space velocity of 15000 h−1. Ni-g-C3N4 adsorbed multiple numbers of NO molecules simultaneously and the N2O intermediate was rapidly reduced to N2 with an energy barrier of 0.50 eV. The oxidised catalyst was regenerated rapidly by CO through OR2 mechanism with an energy barrier of 0.16 eV.86
Zeng et al. employed an in situ growth process to develop single-atom cobalt based polymeric C3N4 (pCN) catalyst and demonstrated its catalytic efficacy for the degradation of oxytetracycline (OTC). The anchoring of single-atom Co on pCN was realised through Co–O and Co–N covalent bond formation. The catalyst with the cobalt loading of 1.28% (Co(1.28%)-pCN) displayed an excellent rate constant of 0.038 min−1 for OTC degradation which was nearly 4 times more than that of pCN.87
Gawande et al. evaluated the photodegradation of pharmaceutical pollutants utilising single-atom Ni dispersed on C3N4 nanosheets (saNi-nC3N4). The pyrrolic N mediated Ni–N coordination confirmed by spectrometric analyses, is responsible for the improved interfacial charge transfer via N–Ni–N bridges. Gemfibrozil, a model pollutant was degraded up to 75.9% in 30 minutes of visible light irradiation. The degradation efficiency of saNi-nC3N4 was better than the widely used TiO2 photocatalyst.88
Chen et al. prepared single atoms of various transition metals (SA-TM, TM = Cr, Mn, Fe, Co, Cu) on g-C3N4 containing abundant pyrrolic N (PN-g-C3N4) through an aqueous self-assembly process involving the precursor mix of xanthine, cyanuric acid, melamine. Transition metal ions were adsorbed on the self-assembled MCAXT through binding sites provided by imidazole groups and subsequent calcination in N2 atmosphere formed SA-TM/PN-g-C3N4 catalysts that exhibited excellent performances for Heterogeneous Fenton-Like Reaction (HFLR). Cr dispersed SACs were found effective for visible light induced photocatalytic degradation of bisphenol A, in the pH range of 3.0–11.0 with outstanding cyclic stability. The enhanced charge carrier production as well as their separation along with the cycling of the Cr3+/Cr2+ couple boosted the HFLR performance. Theoretical studies predicted that Cr(II)–N4 active sites with its structure analogous to metalloporphyrin mimicked peroxidase nanozymes for the effective homolysis of peroxide O–O in H2O2.89
Wang et al. designed Pd SACs (Pd-Cv-CN) for photocatalytic NO conversion, by anchoring Pd atoms on carbon vacancies present in modified g-C3N4 (Cv-CN) via a photo-reduction process. The theoretical simulation predicted the possibility of isolated Pd–N3 active site formation. The DOS plot of Pd-Cv-CN revealed a negative shift in the peak positions on the negative energy side due to the modification of 4d orbital of Pd (Fig. 22(g and f)). This has led to an increase in band levels and a decrease in the band gap for Pd-Cv-CN compared to that of g-C3N4. The developed Pd-Cv-CN effected 56.3% of NO conversion in 30 min illumination time, while pristine CN and Pd nano-Cv-CN converted only 12.7% and 46.0% of NO respectively.90
Polymeric g-C3N4 containing Pt dispersions in the form of single atoms SA-Pt (0.2 nm), nanoclusters CL-Pt (1 nm), and nanoparticles NP1–Pt and NP2–Pt (4 and 7 nm diameter respectively) was demonstrated for the simultaneous benzaldehyde oxidation to benzoic acid and photocatalytic hydrogen production. The catalytic efficiency was highest for Pt single atoms compared to nanoclusters and nanoparticles of Pt. For SA-Pt catalyst, the benzaldehyde conversion and H2 evolution reached a value of 70.8 mmol gPt−1 and 34 mmol gPt−1 respectively in 3 h. Upon visible light excitation, SA-Pt catalyst promoted the transfer of photo-generated holes for benzaldehyde oxidation while CB electrons promoted the proton reduction leading to H2 evolution (Fig. 22 (h)).91 The SACs explored for photocatalytic environmental remediation with the applications demonstrated are presented in Table 5.
| System | Single atom & loading | Synthesis method | Catalytic application, degradation % or rate & time | Coordination site or structure & oxidation state | Ref. |
|---|---|---|---|---|---|
| Ag/mpg-C3N4 | Ag | Template assisted direct synthesis | PMS mediated bisphenol A degradation, 100%, 60 min | Nitrogen, 0 | 82 |
| Pt–C3N4 | Pt 0.2 wt% | Post synthesis reduction under an inert atmosphere | NO oxidation, 96.2%, 150 min | Nitrogen, +2 | 66 |
| AgTCM/UCN | Ag 1 wt% | Template assisted direct synthesis | PMS mediated sulfamethazine degradation, 86.4%, 21 min | Nitrogen | 83 |
| I-FeNx/g-C3N4 | Fe 18.2 wt% | Template free direct synthesis | Photo-fenton degradation; methylene blue, methyl orange, rhodamine B, & phenol, 100%, 15 min | Fe–Nx, +2 (64.3%), +3 (35.7%) | 84 |
| MA (Ag/g-C3N4) | Ag 12.0%, (atomic ratio) | Direct synthesis | Degradation of naproxen, 1.92 × 10−1 min−1 | Carbon | 85 |
| Ni/g-C3N4 | Ni | Post synthesis | Photocatalytic degradation of NO with 100% yield rate-15000 h−1 | Nitrogen | 86 |
| Co-pCN | Co 1.28% | Template free direct synthesis | Degradation of oxytetracycline, 75.7%, 40 min | Nitrogen, oxygen | 87 |
| saNi-nC3N4 | Ni 8 wt% | Post synthesis- microwave treatment | Degradation of gemfibrozil, 75.9%, 30 min | Ni–Nx, +2 | 88 |
| SA-TM/PN-g-C3N4 | TM = Cr, Mn, Fe, Co, Cu | Direct synthesis | Degradation of bisphenol A, SA-Cr/PN-g-C3N4 exhibited highest activity: 98.8%, 70 min | Cr(II)–N4, +2 | 89 |
| Pd-Cv-CN | Pd | Post synthesis-photochemical reduction | NO oxidation, 56.3%, 30 min | Pd–N3, +2 | 90 |
| Pt/g-C3N4 (SA-Pt) | Pt 0.35 wt% | Post synthesis-photochemical reduction | Benzaldehyde oxidation: 70.8 mmol gPt−1 & H2 production: 34 mmol gPt−1 in 3h | Nitrogen | 91 |
H2O2, being an excellent hole scavenging material is prone to oxidation at these potential values.96 On the other hand, photosynthesis of H2O2 by 2e− ORR pathway has been hitherto achieved albeit with a low efficiency of less than 8% for non-sacrificial H2O2 production eqn (3).97 In order to enhance the efficiency of non-sacrificial H2O2 synthesis, concurrent promotion of both 2e− ORR and 4e− WOR eqn (4) is necessary.98 O2, generated in situ as a product of 4e− WOR, is also a reactant for the 2e− ORR and hence the rapid consumption of O2 by ORR will accelerate the kinetics of WOR.96 Therefore, the presence of active sites, in SACs with O2 selectivity for the 2e− ORR promotes artificial H2O2 synthesis via photocatalysis.99
| 2e− ORR O2 + 2H+ + 2e− → H2O2 (0.695 V versus NHE) | (1) |
| 2e− WOR 2H2O → H2O2 + 2H+ + 2e− (1.76 V versus NHE) | (2) |
| Non-sacrificial H2O2 production 2H2O + O2 → 2H2O2 | (3) |
| 4e− WOR 2H2O + 4h+ → O2 + 4H+ (1.23 V versus NHE) | (4) |
| 4e− ORR O2 + 4H+ + 4e− → 2H2O (1.23 V versus NHE) | (5) |
The adsorption of O2 on the metal surface occurs via end on Pauling-type and side on Griffiths and Yeager types (side-on) (Fig. 23(a)).99,100 The Pauling type end-on O2 adsorption minimises O–O bond fission and the 4e− ORR eqn (5) can be suppressed.101 End-on and side-on adsorption of O2 molecules occur on metal particles leading to O–O bond splitting. In SACs O2 molecules adsorb on atomically dispersed sites through the end-on type, reducing the breaking of O–O bond (Fig. 23(b)).102–106
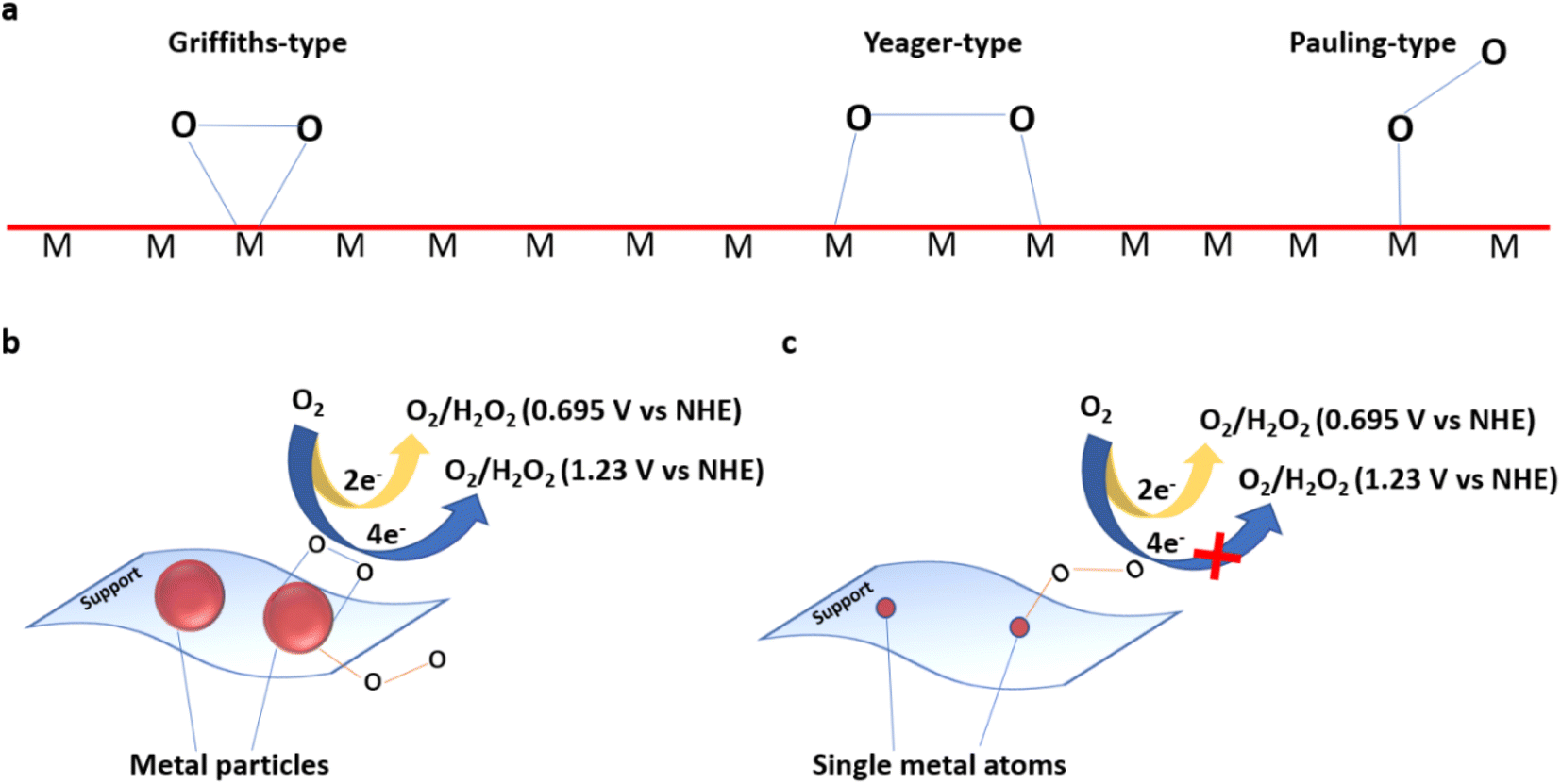 | ||
| Fig. 23 (a) O2 adsorption modes on a metal surface, ORR pathways on (b) metal nanoparticles and (c) single metal atom sites. | ||
Single atom photocatalysts (SAPCs) with metals of d10 electronic configuration favour efficient charge separation through the formation of intermediate bands and form reactive centres with a high density of charge carriers.56,107 Such SACs are considered as potential catalysts for the photocatalytic H2O2 synthesis through the 2e− ORR.108 Ohno et al. developed Sb dispersed g-C3N4 (Sb-SAPC) where single atoms of Sb enabled the photoreduction of O2 through the 2 electron ORR, generating H2O2 at the rate of 12.4 mg L−1 in 120 min. The N atoms of C3N4 surrounding the Sb single atoms, on the other hand, enhanced the kinetics of water oxidation. The apparent quantum yield (ΦAQY, 17.6% at 420 nm) and conversion efficiency (solar to chemical), 0.61% were better than the best performing photocatalysts reported. The analysis of the products of photocatalysis suggested that the H2O2 generation was through the two-electron ORR pathway.100,101
In another noteworthy contribution, SACs with single atoms of (Fe, Ni, Co, In and Sn) were prepared through a wet-chemical synthetic route followed by thermal treatment. Invoking time dependent DFT (TDDFT) calculations, it was demonstrated that the inclusion of metal single atoms on melem units significantly altered the charge separation process. The adsorption energy (EabX) of melem was similar for single atoms of In and Sn suggesting lesser tendencies for charge recombination while Fe (2+ and 3+), Co and Ni (2+) increased the EabX due to dominant recombination. Moreover, iso surfaces created due to the incorporation of In and Sn single atoms favoured the adsorption of oxygen accelerating oxygen reduction at the sites.101Table 6 lists the reported systems for photocatalytic H2O2 synthesis.
| System | Single atom & loading | Synthesis method | H2O2 production rate & time | Coordination site or structure & oxidation state | Ref. |
|---|---|---|---|---|---|
| Sb-SAPC | Sb 10.9 wt% | Template free direct synthesis | 12.4 mg L−1, 120 min | Nitrogen, +3 | 100 |
| M-SAPC | Fe, Ni, In, Co, Sn, 0.5–0.6 mmol per 1 g catalyst | Template free direct synthesis | In-SAPC & Sn-SAPC exhibited superior H2O2 production | Nitrogen, oxidation state of Fe is between II & III, and for Co, Ni, In & Sn is close to II, II, III & IV respectively | 101 |
5.2 Thermal catalysis
Catalysts play a vital role in numerous industrial processes for the synthesis of chemicals.102 The development of efficient catalysts that demonstrate product selectivity in appreciable yields is an essential requirement for the bulk synthesis of chemicals.103 Consequently, the majority of industrial catalysts are based on noble metals on appropriate supports as they are capable of activating important molecules like CO, H2, O2, H2O, etc. Nevertheless, the widespread practical application of noble metal-based catalysts in bulk manufacture is significantly hampered by the scarcity and exorbitant cost of precious metals.104,105 Transition metal catalysed organic reactions, which can be classified under thermal catalysis are now actively employed in the fields of pharmaceutical synthesis, materials development, and biological sciences.106 However, such catalyst systems suffer from shortcomings like poor selectivity, low utilisation efficiency, minimal catalytically active species and unwanted side reactions during their practical application.107 SACs are currently receiving significant attention by virtue of their high conversion efficiency in thermal catalysis for organic transformations.56 The beneficial aspects of both homogeneous and heterogeneous catalysts are integrated in SACs.108SACs are shown to catalyse organic reactions efficiently owing to the presence of unsaturated metal centres, unique electronic structure and 100% atom utilisation efficiency. Electron transfer between metal single atoms and the coordinating atoms of the support induces positive charges (partial or full) on the metal atoms of SACs and offers an enhanced rate of conversion of substrate into product. Furthermore, metal atoms are spatially isolated in SACs due to which the reactive intermediates of the catalytic reactions are adsorbed differently to prevent undesirable side reactions for which the presence of adjacent metal sites is mandatory.109 Development of SACs and ADMCs provides the most ideal strategy for creating cost-effective catalysts for organic transformations (Fig. 24).110
 | ||
| Fig. 24 Various reactions catalysed by g-C3N4 based SACs in the area of thermal catalysis. | ||
Single-site [Pd] mpg-C3N4 heterogeneous catalyst obtained by silica templated thermal condensation of cyanamide, was demonstrated as the first stable single-site heterogeneous catalyst for the hydrogenation of alkynes and nitroarenes. Atomically dispersed [Pd] mpg-C3N4 with 0.5 wt% of Pd and with an average particle size of 0.3–0.4 nm displayed excellent product selectivity (>90%) and significantly improved catalytic activity over conventional nanoparticle based heterogeneous catalysts. For 1-hexyne hydrogenation, the reaction rate at 30 °C and 1 bar pressure was 3 orders higher in magnitude than the conventional catalysts like Ag, Au, and CeO2. The performance was further improved at 60 °C and 2 bar pressure where 100% selectivity for 1-hexene was realised. The [Pd] mpg-C3N4 single-site catalyst also displayed excellent chemoselectivity and stereoselectivity (cis/trans ratio >20) as has been shown in the hydrogenation reaction of 3-hexyne to cis-3-hexene and 2-methyl-3-butyn-2-ol to 2-methyl-3-buten-2-ol. [Pd] mpg-C3N4 catalyst is also capable of catalysing the hydrogenation reaction of nitrobenzene to aniline. The improved catalytic activity of [Pd] mpg-C3N4 single-site catalyst was rationalised by DFT calculations which confirmed hydrogen activation and alkyne adsorption on single-site Pd atoms. g-C3N4 as a matrix facilitates the homogeneous distribution of Pd atoms, promotes H2 activation and inhibits CO poisoning of metal active sites.4
Dual functional catalyst Ag/mpg-C3N4 SAC as mentioned earlier for photocatalytic HER, was also demonstrated to be very effective for alkyne hydrogenation with near 100% selectivity for the conversion of 1-hexyne at a reaction rate exceeding 100 molalkyne h−1 molAg−1 at 303 K and 10 bar compared to the rates of 60 and 40 molalkyne h−1 molAg−1 for catalysts prepared by impregnation–reduction and spray deposition respectively.17
SAC with extremely low loading (519 ppm) of Au on mpg-C3N4 (Au1/mpg-C3N4) demonstrated high turnover frequency (50200 h−1) for the oxidation of silane in the presence of water and oxygen. The coordination of AuI with three N atoms or C atoms in the repeating tri-s-triazine units of g-C3N4 provides atomic level dispersion of highly active AuI. In comparison with Au1/mpg-C3N4, HAuCl4 and Au nanoparticles showed inferior performance for the oxidation of diphenylmethylsilane with water. Au1/mpg-C3N4 also displayed excellent catalytic activity for the oxidation of various organosilanes with greater than 95% yield. It was proposed that the AuI single atoms activate the Si–H bond through oxidative insertion forming a Si–AuIII–H intermediate which further reacted with H2O to form silanol and H2 with the regeneration of the AuI active sites (Fig. 25(a)).26
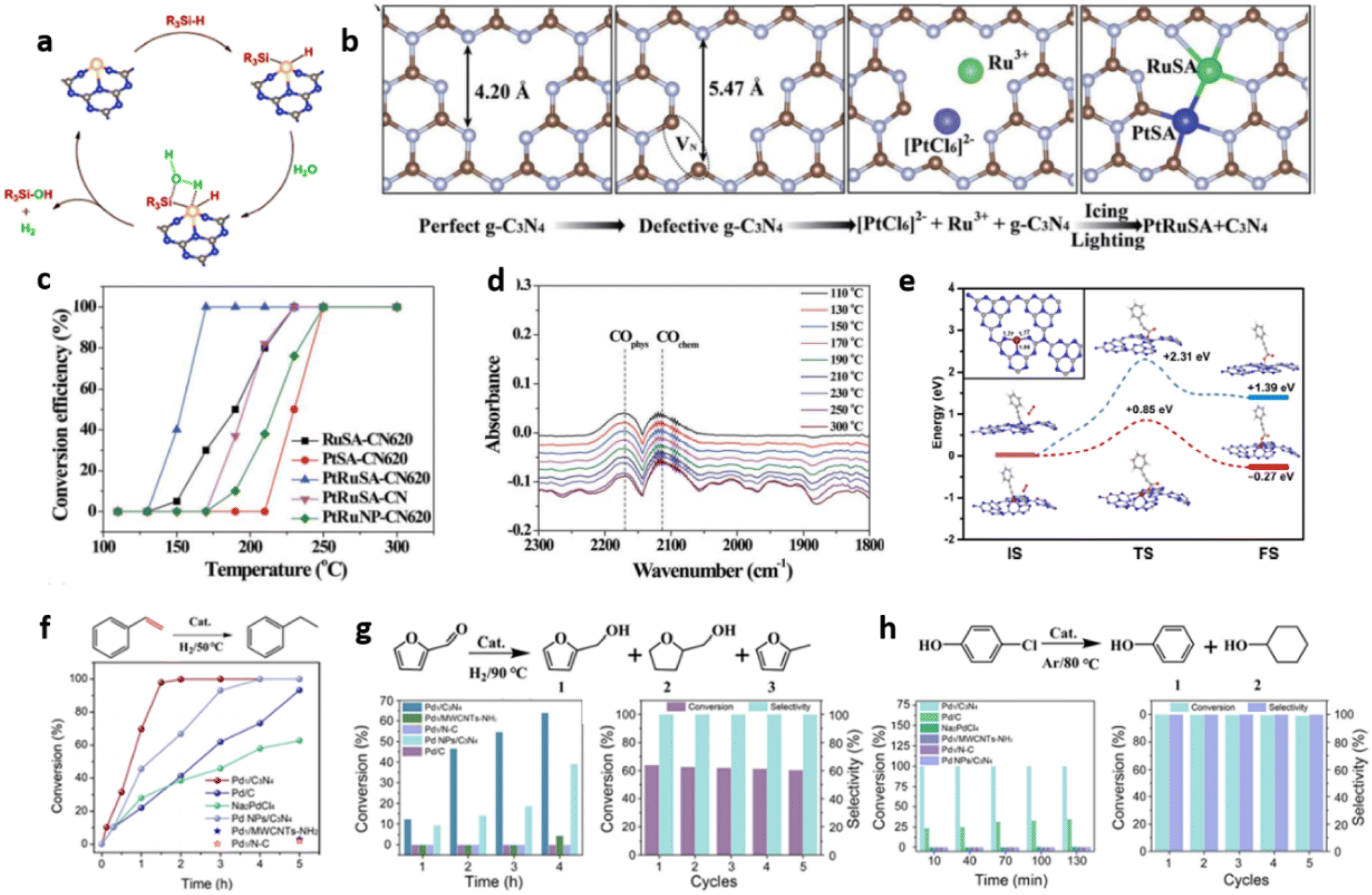 | ||
| Fig. 25 (a) The plausible mechanism of Au1/mpg-C3N4 catalysed water-based silane oxidation. Reproduced with permission from ref. 26. Copyright 2017 John Wiley and Sons. (b) Scheme for the synthesis of PtRuSA-CN620 by the photoreduction method, (c) CO conversion over synthesised catalysts at CO concentration of 1 vol%, (d) CO oxidation monitored by in situ infrared spectra using PtRuSA–CN620. Reproduced from ref. 8. Under the terms of Creative Commons License, published 2019 Royal Society of Chemistry. (e) Reaction profile for CO2 insertion over Cu–CN-8.0 & CN catalysts (inset: the optimised structure of Cu–CN-8.0). Reproduced with permission from ref. 112. Copyright 2020 American Chemical Society. Reaction scheme and catalytic performance of Pd1/C3N4 for (f) hydrogenation of styrene, (g) hydrogenation of furfural (h) hydrodechlorination of 4-chlorophenol. Reproduced with permission from ref. 113. Copyright 2020 American Chemical Society. | ||
Single atom palladium catalysts (Pd-ECN) on C3N4, obtained by the thermal exfoliation, is demonstrated to outperform conventional catalysts (homogeneous and nanoparticle based heterogeneous systems) for the Suzuki coupling between bromobenzene and phenylboronic acid pinacol ester. The ECN matrix not only provides appropriate coordination of Pd atoms in catalyst formation but also participates in the catalytic process through adsorption, stabilisation and activation of substrates and intermediates. The Pd-ECN SACs displayed 63% conversion with 90% selectivity towards biphenyl. The rate of product formation increased linearly from 0.22 to 0.57 mmol product min−1 gcat−1 with the loading of Pd (from 0.25 to 0.66 wt%). However, as the concentration is increased to 1.25 wt%, the reaction rate is decreased to 0.32 mmol product min−1 gcat−1. The turnover frequency (TOF) varied between 558 and 549 h−1, respectively, for (0.25 and 0.66 wt% Pd) but was reduced significantly to 163 h−1 as Pd content increased to 1.25 wt%.111 The electron density around the N-sites of ECN matrix activated bromobenzene while the lattice flexibility of ECN facilitated multiple coordination patterns for enhanced stability. This mechanism is further verified by performing the reaction with single atom Pd on mesoporous C3N4 (Pd-MCN) which displayed less activity albeit high stability and selectivity. The mediocre catalytic performance of Pd-MCN is ascribed to the enhanced structural disorder present in MCN relative to ECN.111
Guo et al. designed SACs with neighbouring Pt and Ru (Pt–Ru) monomers in N deficient g-C3N4 (PtRuSA-CN620) by photoreduction at liquid nitrogen temperature. Conventionally the structure of g-C3N4 contains a C atom coordinated to three N atoms while a few N atoms (denoted as N2C sites) are coordinated only with two C atoms. The creation of nitrogen vacancy leads to the formation of two neighbouring two-coordinated C atoms (denoted as C2C sites) in the g-C3N4 framework. The structure simulation revealed that the creation of N vacancies in g-C3N4 increases the largest vertical dimension from 4.20 Å to 5.47 Å which facilitates the incorporation of Pt and Ru atoms of diameter 2.2 Å as two neighbouring monomers. During photoreduction, the photogenerated electrons concentrate on the N vacancies of CN, facilitating the reduction of noble metal ions into monomer atoms.8
Due to the electronegativity difference, C2C sites are positively charged while N2C sites are negatively charged. During the photoreduction of Pt and Ru metal ions to Pt–Ru monomers, negatively charged [PtCl6]2− ions preferentially get adsorbed on C2C sites while the positively charged Ru3+ ions are adsorbed on N2C sites to form C–Pt–Ru–N coordination in PtRuSA-CN620 (Fig. 25(b)). Pt–Ru monomers with the above coordination structure contains two Pt–C, one Pt–N and three Ru–N bonds with a more negative adsorption energy (Eads) value of −9.40 eV. They are found to be more stable compared to Pt–Pt/Ru–Ru monomers, Pt–Ru monomers with C–Ru–Pt–N, N–Pt–Ru–N coordination structure, and isolated Pt/Ru atoms.8
The PtRuSA-CN620 catalyst with 0.45 wt% Ru and 0.51 wt% Pt exhibited 100% CO conversion into CO2 at the lowest conversion temperature of 150 °C by Eley–Rideal (E–R) mechanism through preferential adsorption of O2 followed by activation on the catalyst surface (Fig. 25(c)). The feeble IR absorption peaks for CO molecule at 2171 and 2111 cm−1 in the temperature range of 110 to 300 °C, ruled out the possibility of Langmuir–Hinshelwood (L–H) mechanistic pathway for CO oxidation in which both CO and O2 adsorption take place on catalyst surface (Fig. 25(d)).8
Photocatalytic non-oxygenative coupling of amines to imines employing H2O as an oxidant is a prospective approach, by virtue of its ease of handling, for the bulk synthesis of imines. Single atoms of copper (0.85 wt%) embedded within g-C3N4 sheets (Cu1@HCNS) exhibited superior benzylamine oxidation (BOR) to imine at the rate of 10583 μmol g−1 h−1 and with 96% selectivity. The catalytic activity was found to be 3.8 and 2.5 times better than Cu single atoms on the surface of g-C3N4 (Cu1/HCNS) and hollow C3N4 spheres comprising of C3N4 nanosheets (HCNS) respectively. The BOR activity of Cu1@HCNS was much higher than the most performing precious metal Pt/MOF and was comparable with commonly employed Ni/CdS catalyst which is toxic and photocorrosive.69
mpg-C3N4 containing single-atom Cu (Cu–CN-x) up to 26.6 wt% was synthesized by a one-pot two step strategy using urea and CuCl2. The first heat treatment at 180 °C led to the coordination and precondensation between urea derived N-containing species and Cu2+ ions through the formation of Cu–N bond. Subsequent calcination at 550 °C resulted in the confinement of Cu single atom in CN matrix through the replacement of one C atom and coordination with three N atoms. The synthesized Cu–CN-x (x indicates the Cu wt%) displayed excellent catalytic activity towards carboxylation of terminal alkynes, utilizing atmospheric CO2. Cu–CN-8.0 aided the carboxylation of phenylacetylene with 97% yield of phenylpropiolic acid and with highest TOF of 9.7 h−1. The mechanism involves the formation of an alkynyl carboxylic intermediate via the addition of deprotonated phenylacetylene to CO2. The reaction further proceeds through CO2 insertion in copper acetylide to result in Cu(I)–propiolate complexes, leading to phenylpropiolic acid. DFT calculations supplemented the experimental observations as the above addition reaction on Cu–CN is exothermic in nature with a reaction energy of −0.27 eV and an energy barrier of +0.85 eV. On the contrary, pure CN was shown to be catalytically inactive due to the high endothermic reaction energy of +1.39 eV coupled with a high activation energy barrier of +2.31 eV (Fig. 25(e)).112
Atomically dispersed palladium on g-C3N4 (Pd1/C3N4) was synthesised by confining them into the six-fold cavity via N-coordination employing a spatial confinement-reduction strategy involving urea and Na2PdCl4. The catalytic performance of Pd1/C3N4 SACs with Pd loading of 0.18 wt% was demonstrated for various selective hydrogenation reactions and the hydrodechlorination reaction of 4-chlorophenol. Pd1/C3N4 exhibited a high TOF value of 834 h−1 with 98% conversion for hydrogenating styrene to ethylbenzene. In comparison catalysts with nanoparticles of Pd (Pd NPs/C3N4) up to 11 wt% metal loading displayed the TOF values of 476 h−1 only. Alternatively, g-C3N4, single atom Pd dispersed in MWCNTs-NH2 (Pd-MWCNTs-NH2, 0.32 wt%) and nitrogen doped carbon (Pd1/N–C, 0.82 wt%) showed negligible activity. The control sample of commercial Pd/C (5 wt%) and Na2PdCl4 salt exhibited TOF values of 333 h−1.
Pd1/C3N4 was also utilised for selectively hydrogenating furfural using H2O as a solvent at 90 °C temperature and 1 atm H2 pressure. Pd1/C3N4 exhibited 64% conversion of furfural to furfuryl alcohol with a TOF of 146 h−1 and 99% product selectivity. In comparison Pd NPs/C3N4 exhibited only 39% of conversion with a TOF of 67 h−1 and 99% product selectivity. Furthermore, Pd1/C3N4 was also utilised for the hydrodechlorination of 4-chlorophenol at 80 °C using ammonium formate as the source of hydrogen. 99% conversion and 99% product selectivity towards phenol were achieved within 10 min and with an exceptionally high TOF of 13333 h−1. In contrast, Pd NPs/C3N4, Pd1/MWCNTs-NH2, Na2PdCl4 salt, and Pd1/N–C yielded no product while Pd/C exhibited 35% of conversion along with 99% selectivity and a TOF value of 1527 h−1.113
Au SACs (Au1/g-C3N4) synthesised by the calcination of HAuCl4/g-C3N4 in N2 atmosphere at 300 °C, was demonstrated for the acetylene hydrochlorination reaction to produce vinyl chloride which is an important monomer in polyvinyl chloride industry. Au exists in +1 oxidation state and forms AuI–N3 active sites through N coordination. DFT study indicated that AuI–N3 active sites preferentially coordinate with HCl than acetylene inhibiting AuI–N3 to Au0 reduction and thereby sustaining the catalytic active sites for hydrochlorination reaction. In contrast, the conventional catalyst, AuCl3 on carbon preferentially coordinates with acetylene leading to the reduction of AuIII ion to Au0 thereby losing its catalytic activity. Au atom occupies the Cv2 defect site present in g-C3N4. The electron localisation function (ELF) maps revealed that the Au–N3 sites are having a T-shape structure with lone-pair electrons (Fig. 26(a–c)). Au1/g-C3N4 single atom catalyst exhibited 70% conversion of acetylene in 600 min without any loss of activity. In comparison, HAuCl4/g-C3N4 catalyst exhibited a maximum conversion of 72.1%, but got significantly decreased to 14.5% in just over 500 min as a result of the reduction of AuIII to Au0. However, the Au NPs/g-C3N4 catalyst showed only very weak activity confirming that Au0 is catalytically inactive towards acetylene conversion. Au–N3 active sites preferentially adsorb HCl by a dissociative mechanism with an adsorption energy (Eads) of −2.25 eV which is higher than the Eads of acetylene (−1.17 eV), preventing the reduction of AuI to Au0 by C2H2. But in the case of AuCl3/C catalyst, C2H2 has a higher adsorption energy of −1.04 eV than HCl (−0.74 eV). The interaction of HCl with Au1/g-C3N4 weakens the Au–N bond as evident from the shifting of the d band center, associated with the Au 5d orbital, farther from the Fermi level in PDOS of HCl adsorbed catalyst. However, in the case of acetylene adsorbed catalyst, there is no shift of band center associated with the 5d and 6s orbital in the PDOS (Fig. 26(d–f)). The competitive HCl adsorption prevents AuI to Au0 reduction rendering enhanced stability for the catalyst. g-C3N4 as a support contributes towards the enhanced stability of the catalyst through π–π interaction of its electron-deficient heptazinic center with electron-rich C2H2. The HCl adsorbed on the active site interact with the acetylene following Langmuir–Hinshelwood (LH) mechanism. The DFT calculation supplements the improved performance of Au1/g-C3N4 by predicting a small energy barrier for C–H and C–Cl bond formation (0.98 eV) along with small desorption energy of 0.87 eV for the product.114
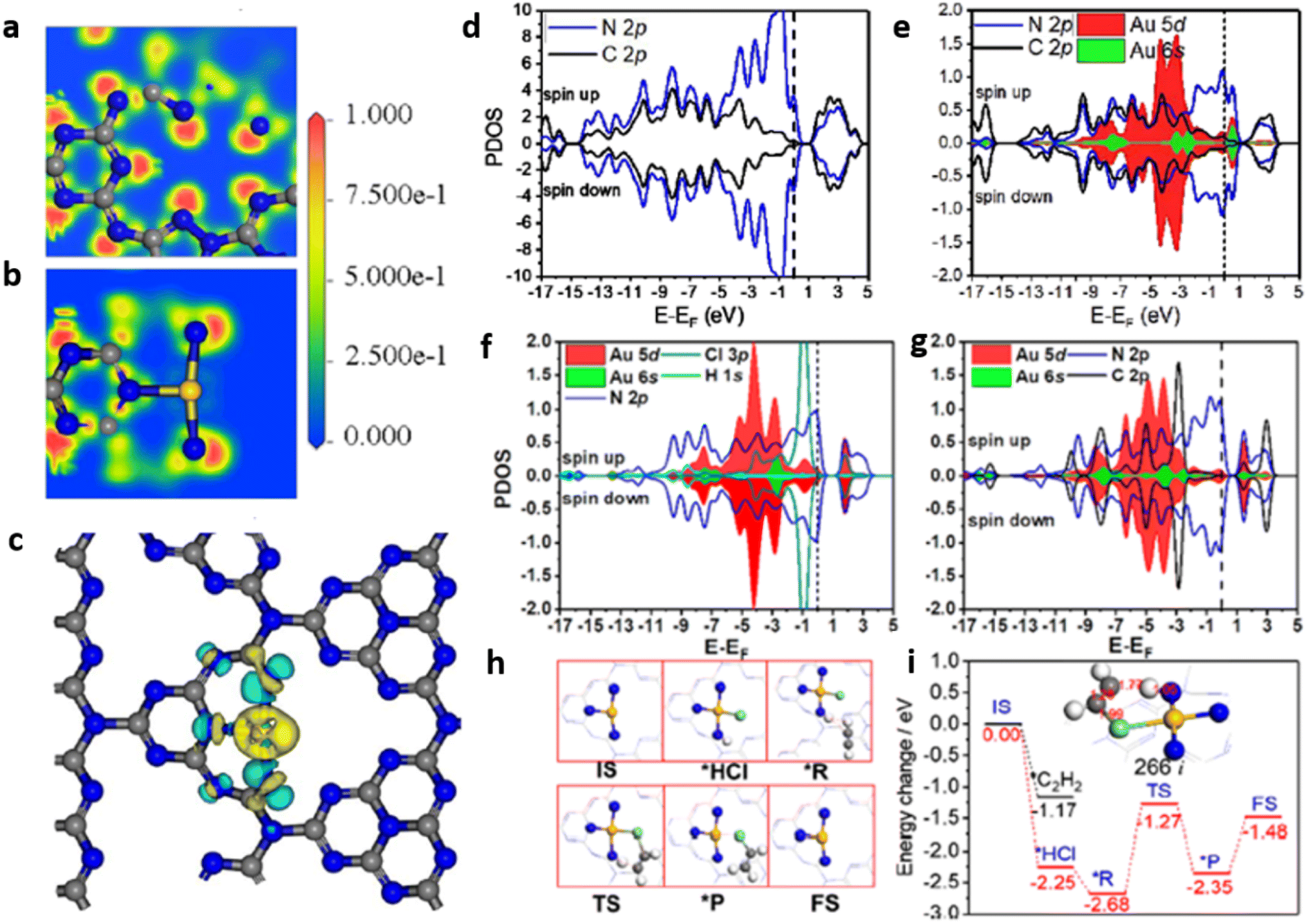 | ||
| Fig. 26 ELF (electron localisation function) map of (a) carbon deficient g-C3N4, (b) Au1/g-C3N4, (c) charge density differences for Au atom on the g-C3N4 (yellow for increasing and blue for decreasing charge density), PDOS of (d) one carbon deficient g-C3N4, (e) Au1/g-C3N4, Au1/g-C3N4 (f) after HCl adsorption (g) after acetylene adsorption, (h) reaction species configuration, (i) the reaction profile of hydrochlorination reaction on Au1/g-C3N4 catalyst. Reproduced with permission from ref. 114. Copyright 2020 American Chemical Society. | ||
The non-oxidative dehydrogenation of propane (denoted as PDH) is an effective synthetic route for the economical production of propylene. The mechanistic pathway for PDH involves C–H bond scission followed by propylene adsorption on the catalysts. Hence low values for C–H bond activation energy (ΔE) and propylene adsorption energies (Ead) on catalysts are essential attributes. Additionally, the desorption energy of propylene should also be low compared to the energy required for breaking the C–H bond to realise higher selectivity towards propylene. Li et al. demonstrates that the sum of ΔE and Ead can be used as a theoretical indicator to elucidate the catalyst efficiency. Thus, a positive value suggests that C–H bond dissociation is slower than propylene desorption while a negative value is an indicator of propylene being strongly adsorbed on TM/g-C3N4. Employing DFT calculations, the group screened the potential of 12 transition metals supported on g-C3N4 (V, Cr, Mn, Zr, Nb, Ru, Rh, Pd, Os, Ir, Pt, Au) as SACs for PDH. The study predicts that a single atom transition metal catalyst can be active and selective for PDH by virtue of its highly exposed d states that enable C–H bond activation and rapid desorption of propylene through weak propylene-π adsorption mode.
g-C3N4, being thermally stable up to T ∼550 °C, can effectively perform the role of stable support for PDH catalysis which conventionally operates at 530 °C. Moreover, the support facilitates firm anchoring of single atoms, preventing agglomeration at high-temperature operating conditions. The adsorption energies of single metal atom should be similar to or higher than their cohesive energies for them to bond on g-C3N4. This selection rule is satisfied by five transition metals of V, Cr, Mn, Zr, and Nb. Together with conventionally active metals like Ag, Ru, Rh, Pd, Os, Ir, Pt, and Au, atomically dispersed SACs based on g-C3N4 were employed for PDH catalysis. Of the 12 systems studied, Ag1/g-C3N4 displayed poor PDH activity owing to its high reaction energy (ΔE) for the first C–H bond cleavage and high propylene adsorption energy (Ead). On the other hand, the rate-limiting step of PDH was characterised by a low activation barrier of 1.11 eV for V1/g-C3N4 catalyst. Moreover, high selectivity was also displayed by V1/g-C3N4 as the adsorption energy of propylene (−0.59 eV) was much lower compared to the activation barrier for deep dehydrogenation (1.97 eV).
In SACs based on C3N4, the lattice constant of the planar structure (7.14 Å) is slightly higher than the buckled C3N4 structure (7.00 Å). The buckled structure is more stable than the planar structure by 2.05 eV. The four possible adsorption sites (A, B, C, D in Fig. 27(a)) of the single metal atoms move from their initial positions on structural relaxations. The adsorption energies of various single metal atoms on the g-C3N4 surface are shown in Fig. 27(b). V, Cr, Mn, and Zr are g-anchored firmly on g-C3N4 as the cohesive energies are lower than their adsorption energies. Moreover, the Pd single atoms of synthesised Pd1/g-C3N4 had adsorption energy smaller than the binding energies of above-mentioned TMs enabling the practical viability of TM based SACs for PDH.115
 | ||
| Fig. 27 (a) Possible adsorption sites for single metal atoms in g-C3N4 layer, (b) adsorption energies of various metal single atoms on available adsorption sites in the g-C3N4 surface. Reproduced with permission from ref. 115. Copyright 2020 American Chemical Society. | ||
g-C3N4 containing single atom Cobalt (Co SACs) up to 23.58 wt% loading was demonstrated to be an excellent catalyst for oxidation of ethylbenzene in the air with a high turn-over frequency of 19.6 h−1 and 97% selectivity for acetophenone formation. The catalytic pathway involves the formation of Co–O bond through O2 adsorption while ethylbenzene adsorbs on the catalyst surface. The subsequent abstraction of the H atom from the CH2 group of ethylbenzene produced acetophenone through the intermediates of phenylethyl radical and hydroperoxyl radical.116
The potential of g-C3N4 based ultra-high density copper single atom catalyst (UHD-Cu1/PCN SAC) was demonstrated for catalysing the azide–alkyne cycloaddition reaction between benzyl azide and 4-ethynylanisole at 60 °C using water-tert-butanol (1:1) mixture as solvent. The effect of Cu content on the catalytic activity to yield 1-benzyl-5-(4-methoxyphenyl)-1H- 1,2,3-triazole was investigated. As the Cu loading increased, the number of active sites as well as the site-specific activity for UHD-Cu1/PCN SAC also enhanced remarkably yielding 92% of 1,5-disubstituted triazole in 12 h. However, no explanation was provided for the selective formation of the 1,5-disubstituted triazole rather than the 1,4-regioisomer.11
Recently, single atom copper catalysts on mesoporous graphitic nitride (saCu-x@mpgC3N4), obtained by the template assisted direct synthesis involving sodium tricyanomethanide, CuCl2·2H2O, cyanamide and SiO2 particles, were demonstrated to catalyse click cycloaddition between various phenylacetylene derivatives and in situ formed azides (from benzyl bromide, and sodium azide) (Fig. 28(a)). Cu-2@mpgC3N4 catalyst with 1.6 wt% of Cu loading exhibited a yield of 45% for 1-benzyl-4-phenyl-1H-1,2,3-triazole in DMF solvent at 130 °C for 30 min (Fig. 28(b and c)). The reaction mechanism evolved from the three possible coordinating structures (Fig. 28(e–g)), involved acetylene activation by adsorption on Cu(ads)/C3N4 with a minimal thermodynamic barrier of +0.09 eV relative to Cu(sub)/C3N4 (+0.48 eV) and Cu(ads)/NG (+0.66 eV). Subsequently, sodium azide reacts with the C atom of acetylene forming a stable linear intermediate over Cu(ads)/C3N4 with a free energy change of −4.19 eV compared to Cu(ads)/NG and Cu(sub)/C3N4 with more positive free energy values of −1.10 eV and −2.73 eV respectively. A triazolate pentacyclic ion attached to the Cu SAC was formed on the completion of cyclisation with free energies of −3.79 eV, −5.34 eV, −5.47 eV for Cu(ads)/NG, Cu(sub)/C3N4, and Cu(ads)/C3N4 respectively. The final step of catalyst regeneration was by the desorption of sodium triazolate molecule where the desorption energy barrier was highest for Cu(ads)/C3N4 (2.27 eV) compared to Cu(ads)/NG (0.59 eV) and Cu(sub)/C3N4 (2.14 eV) (Fig. 28(h and i)).117
 | ||
| Fig. 28 (a) Reaction scheme of cycloaddition, (b and c) the comparison of catalytic performance of mpgC3N4, CuCl2, CuCl, and saCu-2@mpgC3N4 with respect to (b) time and (c) temperature, (e–g) the structural model for the Cu coordination in Cu-based SACs (e) Cu adsorbed in the heptazinic pore of C3N4 (Cu(ads)/C3N4), (f) Cu substituted the carbon in C3N4 (Cu(sub)/C3N4), and (g) Cu adsorbed on N-doped graphene at its dicarbon vacancy (Cu(ads)/NG), (h) proposed reaction mechanism for click cycloaddition over supported Cu1 active sites, (i) reaction free-energy profile for (Cu(ads)/C3N4), (Cu(sub)/C3N4), and (Cu(ads)/NG) at 130 °C (inset: structures of intermediates formed over (Cu(ads)/C3N4)). Reproduced with permission from ref. 117. Copyright 2022 the authors. Published by American Chemical Society. | ||
Sleim et al. demonstrated the complete conversion of CO to CO2 by thermal oxidation at 184 °C utilising single atoms of Cu incorporated on ultra-thin sheets of porous C3N4 (Cu–P-g-C3N4). The SAC developed was characterised by a high surface area of 240 m2 g−1 and contained 1.8 wt% of Cu atoms isolated through coordination with the nitrogen of C3N4. The Cu–N coordination sites bond weakly to the intermediates and products enabling faster reaction kinetics at relatively low temperature.118
The potential of metal embedded C3N4 catalysts (M@gt-C3N4, where M stands for Mo, Ni, Co, Cu, Mn and Fe) were evaluated through first principles calculations and ab initio molecular dynamics (AIMD) simulations, for the catalytic hydrogenation of CO2 to HCOOH, CO, and CH3OH. The p orbitals of pyridinic N atoms in s-triazine-based graphitic C3N4 (gt-C3N4), bond strongly with the d (p) orbitals of single metal atoms activating H2 and CO2. The co-adsorbed state, thus formed, acts as intermediates to the hydrogenation of CO2 to different C1 products, with the metals Mn, Fe, Mn and Co providing superior catalytic performance compared to others. The binding energy (ΔEb) values of a metal single atom anchored on gt-C3N4 and the ΔEb values for all the M@gt-C3N4 vary from −2.25 to −3.72 eV confirming the thermodynamic feasibility of metal incorporation on gt-C3N4. Furthermore, the thermal stability of the SACs was evaluated from AIMD simulations and estimating the energy barrier values for surface migration of metal atoms. Considering Co@gt-C3N4 as a representative example, it was established that the framework structure was retained without bond fission and metal migration even after 10 ps AIMD stimulation. A high energy barrier of 2.11 eV ruled out the possibility of Co atom migration. Additionally, the evaluation of electronic properties through band structure analysis, charge density difference and electron spin density calculations indicated that Cu@gt-C3N4 possessed metallic nature whereas all other metal atoms induced semiconducting properties. This was attributed to the availability of more valence electrons in Cu compared to the rest of the metals as well as to the strong hybridisation of Cu orbitals with neighbouring N orbitals resulting in a high Fermi level value of −2.02 eV relative to the range of −2.98 to −2.15 eV for other metal SACs. The hydrogenation of CO2 starts with the adsorption of CO2 and H2 molecules in bidentate configuration. The adsorption energies (Eads) of CO2 over the metal atom, vary between −0.33 to −1.25 eV whereas Eads of H2 lies between −0.70 and −1.21 eV. The co-adsorption of CO2 and H2 is energetically more favourable for all M@gt-C3N4, except for that of Mn. For the representative Co@gt-C3N4 system, H2 adsorption proceeds exothermic route with an Eads value of 1.13 eV followed by the adsorption of CO2 whereas co-adsorption occurs with the release of energy of 0.19 eV. CO2 hydrogenation follows the Langmuir–Hinshelwood (L–H) mechanism which starts with the coadsorption of CO2 and H2. The CO2 hydrogenation process follows various pathways to produce various products. The HCOO* pathway led to HCOOH formation. A reverse water-gas shift (RWGS) reaction pathway produces CO, whereas a mixed reaction pathway involving CO intermediate hydrogenation (CO-hydro) and RWGS results in CH3OH formation. The hydrogenation of CO2 led to the formation of CO intermediate with high Eads ranging from −2.07 to −2.61 eV which makes the desorption of CO difficult from the catalyst surface and thereby facilitates further hydrogenation to produce CH3OH. In the case of Co@gt-C3N4 which was theoretically predicted to be the most efficient system for CO2 hydrogenation, HCOOH formation occurs with the lowest energy barrier of 0.58 eV, whereas the formation of CO and CH3OH occurs by overcoming an energy barrier of 0.67 eV and 0.95–1.19 eV respectively.119 The SACs demonstrated for thermal catalysis with the salient outputs are presented in Table 7.
| System | Single atom & loading | Synthesis method | Catalytic application, product yield/rate/turnover frequency (TOF) & selectivity | Coordination site or structure & oxidation state | Ref. |
|---|---|---|---|---|---|
| [Pd]mpg-C3N4 | Pd 0.5 wt% | Post synthesis-chemical reduction | Hydrogenation of alkynes & nitroarenes 1-hexyne to 1-hexene: rate 1.41 × 103 molproduct molPd−1 h−1 & selectivity 90%, 2-methyl-3-butyn-2-ol to 2-methyl-3-buten-2-ol, 3-hexyne to cis-3-hexene, nitrobenzene to aniline | Nitrogen | 4 |
| AgTCM-mpg-CN | Ag 1 wt% | Direct synthesis | Hydrogenation of 1-hexyne to 1-hexene, rate 100 molalkyne h−1 molAg−1, selectivity 100% | Carbon or nitrogen, +1 | 17 |
| Au1/mpg-C3N4 | Au 519 ppm | Post synthesis-reduction under an inert atmosphere | Silane oxidation with water, PhMe2SiH to PhMe2SiOH, yield 99%, 30 min, selectivity >99.9%, TOF 50200 h−1 | Carbon or nitrogen, +1 | 26 |
| Pd-ECN | Pd 0.66 wt% | Microwave treatment (post synthesis) | Suzuki coupling of bromobenzene with phenylboronic acid pinacol ester to form biphenyl, 63% conversion, 90% selectivity, rate 0.57 mmolproduct min−1 gcat−1 | Nitrogen, Pd2+/Pd4+ ratio 0.82 | 111 |
| PtRuSA-CN | Pt 0.51 wt% Ru 0.45 wt% | Post synthesis-photochemical reduction | CO oxidation 100% conversion at 150 °C | C–Pt–Ru–N | 8 |
| Cu1@HCNS | Cu 0.85 wt% | Supramolecular preorganisation assisted direct synthesis | Non-oxygenative coupling of amines to imines using H2O & light, benzylamine to imine: 10583 μmol g−1 h−1, 96% selectivity |
Cu1N3, +1 | 69 |
| Cu–CN-x | Cu 8 wt% | Template free direct synthesis | Carboxylation of terminal alkynes using atmospheric CO2; phenylacetylene to phenylpropiolic acid: yield 97%, TOF 9.7 h−1 | Nitrogen, +1 | 112 |
| Pd1/C3N4 | Pd 0.18 wt% | Post synthesis-reduction under an inert atmosphere | (a) Hydrogenation; (i) styrene to ethylbenzene: 98% conversion in 1.5 h, TOF: 834 h−1, (ii) furfural to furfuryl alcohol: 64% conversion in 4 h, TOF: 146 h−1, (b) hydrodechlorination of 4-chlorophenol to phenol: 99% conversion in 10 min, TOF: 13333 h−1 |
Nitrogen, +1 | 113 |
| Au1/g-C3N4 | Au 0.414 wt% | Post synthesis | Hydrochlorination of acetylene to vinyl chloride: 70% conversion in 600 min | Au–N3, +1 | 114 |
| TM/g-C3N4 | V, Cr, Mn, Zr, Nb, Ru, Rh, Pd, Os, Ir, Pt, Au | DFT study | Non-oxidative dehydrogenation of propane to propylene, predicted highest activity & selectivity for V1/g-C3N4 | — | 115 |
| Co SAC (Co SACs/CN) | Co 23.58 wt% | Direct synthesis | Aerobic oxidation of ethylbenzene to acetophenone: 62% conversion, 40 h, 99% selectivity, TOF: 19.6 h−1 | Co–N2, +2 | 116 |
| UHD-Cu1/PCN | Cu 23 wt% | Post synthesis – reduction under an inert atmosphere | Azide–Alkyne cycloaddition reaction: cycloaddition between benzyl azide & 4-ethynylanisole to form 1-benzyl-5-(4-methoxyphenyl)-1H- 1,2,3-triazole, yield 92%, 12h | Nitrogen, +1 | 11 |
| saCu@mpg C3N4 | Cu1.6 wt% | Template assisted direct synthesis | Click cycloaddition between phenylacetylene and in situ formed benzyl azide (from benzyl bromide, & sodium azide) to form 1-benzyl-4-phenyl-1H-1,2,3-triazole, yield 45%, 30 min | Nitrogen, +1 | 117 |
| Cu–P-g-C3N4 | Cu 1.8 wt% | Template free direct synthesis | Oxidation of CO to CO2 100% conversion | Nitrogen, 0 | 118 |
| M@gt-C3N4 | M = Mn, Fe, Co, Ni, Cu, and Mo | DFT study | CO2 hydrogenation to HCOOH, CO, & CH3OH, Co@gt-C3N4 was theoretically predicted to be the most efficient system | Nitrogen | 119 |
5.3 Electrocatalytic applications of g-C3N4 based SACs
Electrocatalysis is perceived to be a promising approach to meet the continuously increasing energy demand by employing various technologies, such as the electrolysis of water, CO2 reduction, fuel cells, etc. The underlying electrochemical processes enable the sustainable utilisation of renewable electrical energy with appreciable energy conversion efficiencies. Precious metals (Pt, Ir, Ru, Rh, Pd, etc.) and their metal oxide-based catalysts are widely used in the field of electrocatalysis.120 However less availability and high cost hinder their widespread application. The utilisation of single atom catalysts of precious & non-precious metals with high intrinsic activity, unique electronic structure, and high atom utilisation efficiency help to improve the electrocatalytic performance for various reactions (Fig. 29).121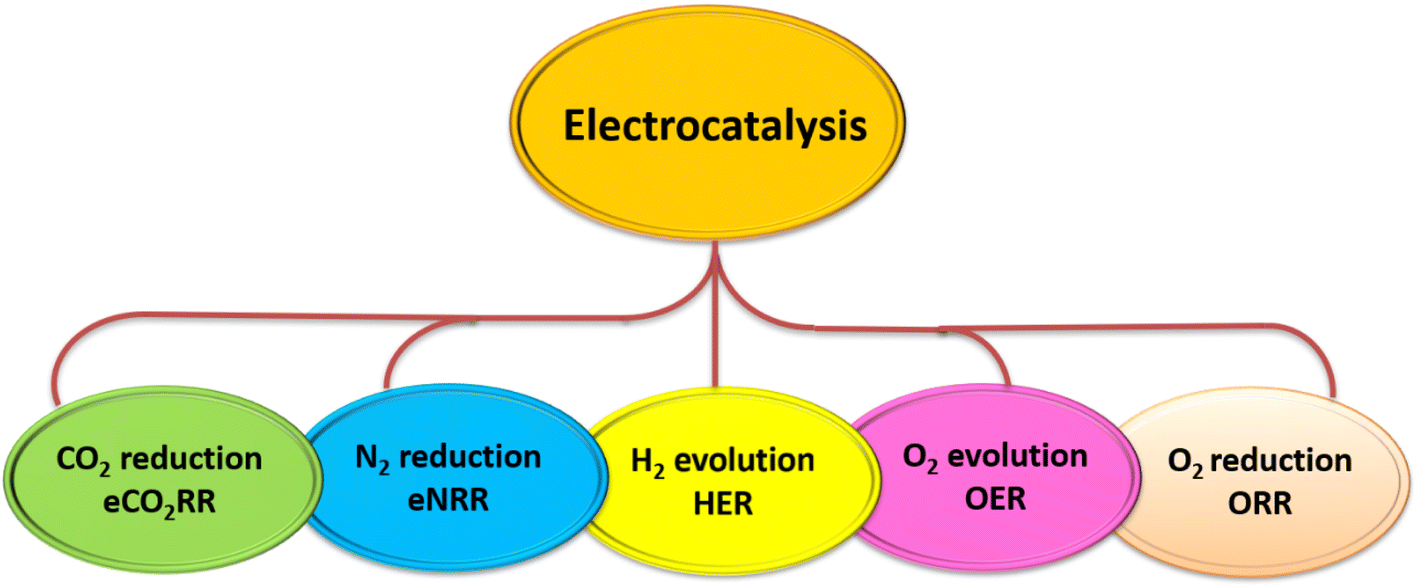 | ||
| Fig. 29 Various electrocatalytic reactions catalysing by g-C3N4 based SACs. | ||
| CO2 + 2H+ + 2e− → HCOOH |
| CO2 + 2H+ + 2e− → CO + H2O |
| CO2 + 6H+ + 6e− → CH3OH + H2O |
| CO2 + 8H+ + 8e− → CH4 + 2H2O |
| 2CO2 + 12H+ + 12e− → C2H4 + 4H2O |
| 2CO2 + 12H+ + 12e− → C2H5OH + 3H2O |
Zhang et al. developed Mn based SACs through thermal treatment of a precursor mix containing manganese acetate, dicyandiamide (DCD), and CNT under an inert atmosphere of N2 at 600 °C. SACs with Mn–N3 sites embedded in the mixed matrix of g-C3N4 with carbon nanotubes (Mn–C3N4/CNT) exhibited excellent eCO2RR in aqueous electrolyte with 98.8% of faradaic efficiency (FE) and CO partial current density (jCO) value of 14.0 mA cm−2, at a low overpotential of 0.44 V. jCO further improved to 18.6 and 29.7 mA cm−2 at an overpotential of 0.42 and 0.62 V, respectively by the usage of ionic liquid (IL) electrolyte. Theoretical studies and XAS analysis revealed that CO2 can easily get adsorbed on Mn–N3 active site which facilitated a low free energy barrier for COOH* intermediate formation.123
The potential of atomically dispersed Er atoms on g-C3N4 nanotubes described earlier for photocatalytic CO2 reduction, has been evaluated for eCO2RR. HD-Er1/CN-NT exhibited enhanced cathodic current compared to pure CN-NT and LD-Er1/CN-NT.9
Yan Jiao et al. used DFT calculations to predict different C–C coupling pathways for the eCO2RR to C2H4 over Cu–C3N4 SACs utilising Cu/C or Cu/N dual active sites present in it. The reduction proceeded via 12 e− and 12 proton transfer pathways at a potential of −0.764 V vs. SHE. The first in the process is the protonation of a CO2 molecule to *COOH at any of the three active sites of Cu, C, N present in the catalyst. DFT study and associated ΔG calculations divulged that the Cu active site with a ΔE of 0.37 eV is preferred to N active site with 1.47 eV. Even though active sites of C possess the lowest ΔE of 0.37 eV, it is difficult to facilitate C–C coupling as the reaction proceeds. Based on the sites and intermediates involved in the formation of C2H4, 14 different pathways are suggested in two categories involving the Cu/N dual active sites and Cu/C dual active sites. The Cu/C pathway exhibited a better synergistic effect than Cu/N which is beneficial for possessing catalytic activity comparable to Cu (100).124
Electrochemical CO2 reduction to formate was demonstrated in exfoliated g-C3N4 (GCN) containing atomic dispersions of Cu (Cu-GCN). XANES analysis indicated the presence of both Cu2+ and Cu+ with an average oxidation state of +1.4 confirming different Cu–N co-ordinations, coupled with DFT calculations, the authors predicted a partially polymerised GCN that retained a few amino functional groups coordinated to Cu as the Cu atoms are expected to interact preferentially with pyridinic N atoms. The catalyst Cu-GCN(20) exhibited CO2RR activity in bicarbonate solution through the formation of formate as the only product in the liquid phase, with a faradaic efficiency of 12% and a current density of 14 mA mgcat−1. However, in the phosphate solution HER was predominant over CO2RR and hydrogen was obtained as a result of HER rather than CO2RR.125
Ge et al. successfully developed an efficient electrocatalyst (ZnSA–CN–G) for CO2RR by dispersing zinc single atoms (ZnSA) on graphitic carbon nitride nanosheets (CN) and graphene (G) through the pyrolysis of a precursor mix containing DCDA, graphene-oxide nanosheets (GO nanosheets) and ZnCl2. During the pyrolysis process at 700 °C, the conversion of DCDA to CN facilitated the atomic level dispersion of Zn single atoms to CN matrix with Zn–N coordination and GO was reduced to graphene. ZnSA–CN–G catalyst with a metal loading of 1.2 wt%, exhibited excellent conversion of CO2 to CO with 87.1% of FE at a potential of −0.5 V vs. RHE. The enhanced electrocatalytic activity is derived from the availability of abundant Zn single atoms as well as from the enhanced surface area & electrical conductivity of ZnSA–CN–G.126
The electrocatalytic CO2 reduction capabilities demonstrated for g-C3N4 based SACs are presented in Table 8.
| System | Single atom & loading | Synthesis method | Products, FE, current density (j) & overpotential | Coordination site or structure & oxidation state | Ref. |
|---|---|---|---|---|---|
| Mn–C3N4/CNT | Mn 0.17 wt% | Template free direct synthesis | CO, FE 98.8%, jCO 14.0 mA cm−2 at 0.44 V | Mn–N3, +2 | 123 |
| Er1/CN-NT | Er 18.4 wt% | Freeze drying assisted direct synthesis | HD-Er1/CN-NT exhibited higher cathodic current than LD-Er1/CN-NT & CN-NT | Er–N3, +1 | 9 |
| Cu–C3N4 | Cu | DFT study | C2H4, Predicted Cu/C active sites favours C2H4 formation than Cu/N | — | 124 |
| Cu-GCN(20) | Cu 20 wt% | Chemical reduction assisted post synthesis | Formate (HCOO−), FE 12% | Nitrogen, +1 & +2 | 125 |
| ZnSA–CN–G | Zn, 1.2 wt% | Freeze drying assisted direct synthesis | CO, FE 87.1% at −0.5 V | Nitrogen, oxidation state in between 0 & +2 | 126 |
eNRR is a six-electron reaction progressing in both associative and dissociative mechanisms. The former involves the release of NH3 simultaneously with NN bond cleavage and proceeds through the distal, alternating, and enzymatic pathways. In the dissociative route, NN cleaves into two separate N atoms before hydrogenation. The reduction process is initiated by the adsorption of N2 to metal atoms and the adsorption energy calculations suggest that it proceeds preferably via an end on configuration.130 Computing ΔG associated with the formation of possible species (N2H*, 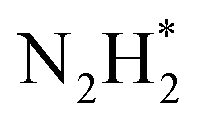 ,
,  ,
,  , N*, NH*, and
, N*, NH*, and  ) and identification of the potential determining step (PDS) through different pathways facilitate mechanisms to evaluate the efficiency of the catalyst.131
) and identification of the potential determining step (PDS) through different pathways facilitate mechanisms to evaluate the efficiency of the catalyst.131
In one of the initial reports on SACs based on g-C3N4, Zhao and Chen employed DFT based computational studies to investigate the effectiveness of a variety of single atoms (Ag, Ru, Pd, Rh, Au, Pt, W, Ni, Ti, Mo Cr, Mn, Co, and Fe) on C3N4 for eNRR (Fig. 30(a)). The structural stabilities of the catalysts were computed from the energy of the adsorption of the metal atoms on g-C3N4 (Fig. 30(b)). Single atoms of Ti, Mn, Pd, Ag, Au and W were adsorbed on the six-fold cavity of g-C3N4 with metal–N bond lengths varying in the range of 2.28–2.51 Å. The group comprising of Cr, Ni, Fe, Co, Cu, Mo, Ru, Rh, and Pt were anchored favourably at the corner of the same cavity with metal–N bond lengths of 1.90–2.11 Å. The difference in adsorption sites was ascribed to the differences in atomic radii and electronegativity values. Electron transfer from metal atoms to C3N4 induces positive charges on metals facilitating eNRR and suppressing the competitive HER.132
 | ||
| Fig. 30 (a) g-C3N4 monolayer structure with possible coordination sites for single metal atoms and their (b) adsorption energies (calculated) HER. Reproduced with permission from ref. 132. Copyright 2018 John Wiley and Sons. (c) Optimised monolayer g-C3N4 structure with V, Nb, Ta single atoms. Reproduced with permission from ref. 133. Copyright 2020 American Chemical Society. (d) Structural transformation of h-C3N4 to h-C4N3, (e) −ΔG1versus group number for 4d and 5d TMs (f) ΔG1 for the screened TM-SACs. Reproduced with permission from ref. 134. Copyright 2021 American Chemical Society. (g) Alternating mechanism of NRR on Au1/C3N4 with optimised structures of various intermediates, (h) free energy profile of NRR using Au1/C3N4 and Au (211), (i) change in electron density induced by anchored Au atom on g-C3N4 (cyan and pink colour indicate accumulation and depletion of electron density respectively), (j) limiting potential difference in NRR and HER. Reproduced with permission from ref. 135. Copyright 2018 Elsevier. | ||
Ding et al. compared the suitability of g-C3N4 and graphene, as support layers with differences in their coordination environment, for incorporating single atoms of V, Nb, and Ta (Fig. 30(c)). Their subsequent use as electrocatalysts for nitrogen reduction was evaluated based on the ability of the catalyst to adsorb nitrogen for the activation of NN, the creation of a stable N2H* adsorption on the catalyst and the destabilisation of  . The reactivity screening revealed that V, Nb, and Ta anchored onto graphene support, could only weakly stabilise the
. The reactivity screening revealed that V, Nb, and Ta anchored onto graphene support, could only weakly stabilise the  species and hence were considered to be unsuitable for eNRR. The binding energy of single atoms on C3N4 was used to ascertain the structural stability of SACs. The values ranging from −6.48 to −7.58 eV confirmed that the C3N4 layers with the corner vacancy are best suited to facilitate the anchoring of TM atoms. The band gap and spin density distribution calculations indicated strong N2 adsorption capacity for TMs on C3N4. The Eg of C3N4 lowered significantly from 2.84 eV to 1.36 eV (Ta/C3N4) and 1.35 eV (Nb/C3N4). Among the three TMs evaluated, SACs based on Nb/C3N4 were shown to possess the highest catalytic efficiency with high selectivity for NRR compared to HER by virtue of the ΔG max of 0.05 eV realised through a distal mechanism.133
species and hence were considered to be unsuitable for eNRR. The binding energy of single atoms on C3N4 was used to ascertain the structural stability of SACs. The values ranging from −6.48 to −7.58 eV confirmed that the C3N4 layers with the corner vacancy are best suited to facilitate the anchoring of TM atoms. The band gap and spin density distribution calculations indicated strong N2 adsorption capacity for TMs on C3N4. The Eg of C3N4 lowered significantly from 2.84 eV to 1.36 eV (Ta/C3N4) and 1.35 eV (Nb/C3N4). Among the three TMs evaluated, SACs based on Nb/C3N4 were shown to possess the highest catalytic efficiency with high selectivity for NRR compared to HER by virtue of the ΔG max of 0.05 eV realised through a distal mechanism.133
Singh et al. employed density functional theory for the design of transition metal (TM) anchored heptazine-derived graphitic C4N3 (h-C4N3) based SACs. The catalytic efficiency was gauged by the ability of a metal to facilitate HER through the adsorption and protonation of nitrogen. The number of valence electrons of TM was identified to be an indicator to predict the catalytic efficiency of SACs. Among the carbon nitrides, h-C4N3 formed by the replacement of two N atoms with C atoms was identified to be a better support than C3N4, as the former is metallic offering a free flow of electrons (Fig. 30(d)). 27 transition metals anchored SACs were investigated and the evaluation of N2 adsorption energies qualified Sc, Co, Ni, Zr, Nb, Mo, Ru, Ta, W, and Os for eNRR (Fig. 30(e and f)). Subsequently, considering the feasibility of protonation of 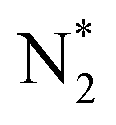 to N2H* intermediate, only 3 of the 27 TMs namely Mo, W and Ta were found comparatively more effective. Among the three qualified, only Mo and W showed higher selectivity for eNRR and the highest efficiency was accorded to Mo based SACs by virtue of their extremely low overpotential of 0.02 eV versus SHE. The estimated faradaic efficiency was 97.3% for Mo-SAC. Thermodynamic analysis of the reduction pathways (distal, alternating and mixed) suggested that the most favourable mechanism for the conversion of N2 to NH3 by Mo-SAC is the mixed mode with a limiting potential of −0.18 eV vs. SHE. The potential limiting step (PLS) for this mechanism is the conversion of N2H2* to
to N2H* intermediate, only 3 of the 27 TMs namely Mo, W and Ta were found comparatively more effective. Among the three qualified, only Mo and W showed higher selectivity for eNRR and the highest efficiency was accorded to Mo based SACs by virtue of their extremely low overpotential of 0.02 eV versus SHE. The estimated faradaic efficiency was 97.3% for Mo-SAC. Thermodynamic analysis of the reduction pathways (distal, alternating and mixed) suggested that the most favourable mechanism for the conversion of N2 to NH3 by Mo-SAC is the mixed mode with a limiting potential of −0.18 eV vs. SHE. The potential limiting step (PLS) for this mechanism is the conversion of N2H2* to 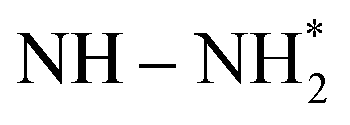 intermediate.134
intermediate.134
Wang et al. evaluated the catalytic performance of atomically dispersed Au on C3N4 for the electrochemical reduction of N2 to NH4+ in an aqueous sulphuric acid solution. A high ammonium yield rate of 1305 μg h−1 mgAu−1 had been realised and the yield was roughly 22.5 times more than that obtained by employing Au nanoparticles as catalysts. The direct eNRR to NH4+ was thus demonstrated with an energy utilisation rate of 4.02 mmol kJ−1. DFT calculations revealed that the mechanistic pathway involves an alternating mechanism with the reduction of N2 to *NNH as the rate determining step with a positive ΔG value of 1.33 eV for Au–C3N4 SACs compared to a higher ΔG value of 2.01 eV for Au nanoparticles (Fig. 30(g and h)). The electron density difference due to the anchoring of Au on g-C3N4 also favoured stronger binding interaction between Au and *NNH (Fig. 30(i)). Bader charge population analysis confirmed charge transfer from Au to C3N4 leading to the shifting of the d-orbital position towards the Fermi level enabling enhanced interaction with intermediates. The selectivity of Au–C3N4 SACs towards NRR, compared to HER, was ascertained based on the thermodynamic limiting potentials which was computed to be −0.88 V for SACs relative to −1.67 for Au nanoparticles-based catalysts (Fig. 30(j)).135Table 9 summarises the g-C3N4 based SACs demonstrated for eNRR applications.
| System | Single atom & loading | Synthesis method | Rate, FE, limiting potential | Coordination site or structure & oxidation state | Ref. |
|---|---|---|---|---|---|
| TM@C3N4 | TM = Ag, Ru, Pd, Rh, Au, Pt, W, Ni, Ti, Mo Cr, Mn, Co, Fe | DFT study | W@C3N4 is predicted to exhibit the highest NRR activity with a limiting potential of −0.35 V | Nitrogen | 132 |
| TM/CN | TM = V, Nb, Ta | DFT study | Nb/CN is predicted to exhibit highest NRR activity with a low ΔGmax (0.05 eV) | Nitrogen | 133 |
| TM-SAC (TM h−1-C4N3) | TM = Sc, Co, Ni, Zr, Nb, Mo, Ru, Ta, W, and Os | DFT study | Mo-SAC is predicted to be the most effective eNRR catalyst with an ultralow overpotential value of 0.02 V & high FE | Nitrogen | 134 |
| Au–C3N4 | Au 0.15% | Post synthesis – reduction under inert atmosphere | Rate: 1305 μg h−1 mgAu−1, FE: 11.1% at −0.10 V | Nitrogen, +1 | 135 |
Dai et al. employed computational studies to explore the potential of transition metal (TM = Ti, V, Cr, Mn, Fe, Co and Ni) based SACs on holey g-C3N4 (TM/g-CN) for overall water splitting (HER & OER). Among the various TM/g-CN SACs designed, Co1/g-CN and Ni1/g-CN were predicted to perform as the most efficient bifunctional electrocatalysts owing to the strong interaction between the intermediates and catalysts as governed by the d band centre of the metal atoms. The calculations indicated that Co1/g-CN and Ni1/g-CN SACs can attain overpotential values of 0.15 V and 0.12 V respectively for HER. Values are comparable with that of Pt reference catalyst with 0.09 V overpotential value.137
Single Atom Co dispersed over g-C3N4 matrix coupled with RGO and having a metal loading of 6 wt% (Co-g-C3N4/rGO SACs, Co-CNG) are shown to perform (10 mA cm−2; ∼47 mV) at par with commercial Pt/C catalysts for electrocatalytic HER (10 mA cm−2 at ∼48 mV). The Co-CNG was better than the commercial Pt/C catalyst with 4 times higher mass activity with 500 h durability (Fig. 31(a–e)). DFT studies supplemented the experimental observations and the superior HER activity of Co-CNG was ascribed to the presence of 20% and 80% of Co–N and Co–3N type coordination structures respectively. Coordination engineering is thus promoted as a pathway to boost HER for Co–N site than Co–3N model by virtue of the unique electronic structure, downshift of d-band centres and low free energy barriers (Fig. 31(f–i)).138
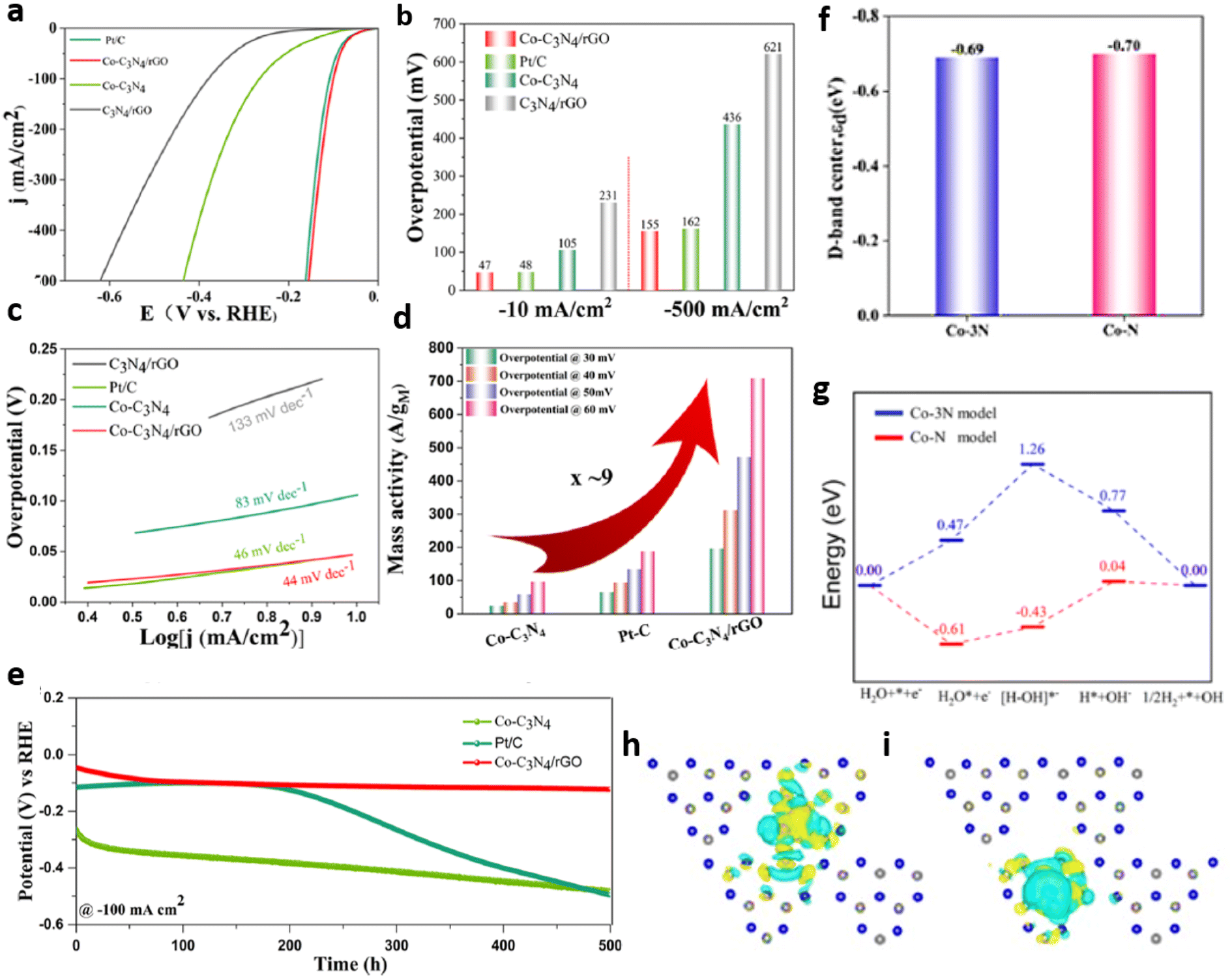 | ||
| Fig. 31 Data elucidation of HER activity, (a) LSV curves in 1.0 m KOH, at a scan rate of 5 mV s−1 for Co–C3N4/rGO, Co–C3N4, C3N4/rGO, and Pt/C, (b) overpotential values of the developed catalysts for a current density of −10 and −500 mA cm−2, (c) Tafel plots, (d) mass activity of the developed catalyst in comparison with Pt/C, at different overpotentials (e) time-dependent overpotential curves of Co–C3N4/rGO, Co–C3N4, in comparison with Pt/C. Co–N and Co–3N systems with (f) d-band centers, (g) energy profiles, charge density differences in structures of (h) Co–N3, (i) Co–N1. Reproduced with permission from ref. 138. Copyright 2022 American Chemical Society. | ||
The electrocatalytic hydrogen evolution reported for g-C3N4 based SACs is summarised in Table 10.
| System | Single atom & loading | Synthesis method | Current density (j) & overpotential | Coordination site or structure & oxidation state | Ref. |
|---|---|---|---|---|---|
| TM/g-CN | TM = Ti, V, Cr, Mn, Fe, Co, Ni | DFT study | Co1/g-CN & Ni1/g-CN SACs are predicted to be the most effective HER catalysts with overpotential values of 0.15 V and 0.12 V respectively | Nitrogen | 137 |
| Co-gC3N4/rGO (Co-CNG) | Co 6 wt% | Template free direct synthesis | 10 mA cm−2, 47 mV | 20% Co–N & 80% Co–3N type coordination | 138 |
In one of the earlier works, Park et al. developed molecularly dispersed nickel on C3N4 matrix (Ni–CN) by employing a direct synthetic methodology involving the precursors of melamine and Ni(II) chloride hexahydrate precursors. The optimised catalyst displayed OER activity with an onset potential of 1.54 V (vs. RHE) which was 80 mV less positive than g-C3N4. Atomic dispersions of Ni species on C3N4 through Ni–N bonding were confirmed by EXAFS analysis.142
Qiao et al. developed organometallic complexes of g-C3N4 with transition metals (M−C3N4) as electrocatalysts for both OER & ORR activities. Theoretical studies predicted that among the 3d transition metal centres, (M−C3N4, where M = Cr, Mn, Fe, Co, Ni, Cu, Zn) Fe, Ni and Co possess the potential to exhibit bifunctional activity toward reversible ORR/OER processes due to high stability of structures. The representative catalyst of Co–C3N4 with multi-walled carbon nanotubes (CNT) exhibited higher electrode conductivity exposing all the electrocatalytically active sites of Co–C3N4. An onset potential of 0.9 V and a diffusion limited current density of ∼5 mA cm−2 in O2-saturated 0.1 M KOH solution was obtained for the Co–C3N4/CNT catalyst via 4e− dominated ORR pathway. The catalyst also showed an onset potential of ∼1.5 V and an anodic current density of 10 mA cm−2 at 1.61 V for OER activity in N2-saturated 1 M KOH solution. The excellent bifunctional performance of Co–C3N4/CNT catalysts was at par with precious metal benchmarks and was attributed to the presence of Co–N2 active sites.143
Liu et al. developed SAC based electrocatalyst by stabilising Au1Nx single-sites on g-C3N4 (Au1Nx single-site/C3N4) via a post synthesis strategy involving electrostatic adsorption. Au1Nx single-site/C3N4 exhibited excellent catalytic activity with good durability for ORR and OER with mass activity values of ∼9000 A gAu−1 and ∼1500 A gAu−1 at a half-wave potential (E1/2) of 0.76 V and at an overpotential 0.45 V respectively. The catalytic performances were 20 and 26 times higher than the conventional Pt/C and RuO2 benchmarks. DFT calculations coupled with analysis of the electronic structure revealed the injection of electrons from Au1Nx site to g-C3N4 enabling the formation of an additional energy level near the Fermi level. This promotes faster redox kinetics for highly efficient OER and ORR reactions. Moreover, the presence of mobile electrons near the Fermi level facilitated surface adsorption of the key intermediates *OO and *OH on Au1Nx single-sites with a theoretical binding energy of 1.3 and 1.5 eV, close to that Pt and RuO2 (1.4 eV and 1.7 eV).22
In a noteworthy contribution, Sun et al. prepared Fe, N doped hollow carbon catalyst (C-FeHZ8@g-C3N4) by the pyrolysis of a precursor mixture containing ZIF-8, C3N4 and ferric acetyl acetonate. The Fe–N4 atomic level coordination was affected by C3N4 and ferric acetyl acetonate as N and Fe precursors respectively. The optimised catalyst exhibited high ORR activity with E1/2 values of 0.18 and 0.845 V in acidic and alkaline conditions. The use of C3N4 prevented agglomeration of Fe during high-temperature pyrolysis and facilitated the formation of Fe–Nx sites through its cyano products formed during decomposition at 950 °C. The incorporation of g-C3N4 during the synthesis provides an effective pathway for the stabilisation of single atoms in Fe–C–N catalyst.144
Wang and co-workers developed a hybrid ORR–OER electrocatalyst (NGM–CN–Fe) with atomic Fe–Nx between g-C3N4 and graphene and were utilised as a material for air electrode cathode in Zn–air battery (ZAB). Fe is atomically dispersed over g-C3N4 (bulk CN–Fe) through template free direct synthesis involving dicyandiamide (DCDA) and ferric chloride precursors. Exfoliated porous CN–Fe nanosheets derived from bulk CN–Fe through sonication were subjected to hydrothermal treatment in the presence of graphene nanomesh and annealed at 600 °C to form hybrid NGM–CN–Fe SAC. The catalyst exhibited superior ORR activity with a current density of −6.42 mA cm−2 at an improved onset potential of nearly 0.00 V which is 13 times larger than that of atomic Fe dispersed g-C3N4 (CN–Fe). Both pure g-C3N4 (CN) and CN–Fe displayed an onset potential around −0.26 V and the current density was −0.45 mA cm−2 at −0.5 V. In comparison with commercial Pt–C (20 wt%) catalyst, NGM–CN–Fe exhibited a comparable onset potential, better half-wave potential with 20 mV improvement & enhanced current density. The superior activity of NGM–CN–Fe SAC is attributed to the existence of Fe–N4.1 coordination sites via atomic Fe-bridged coordination between g-C3N4 and nitrogen-doped graphene nanomesh.145
Li et al. demonstrated the development of OER catalyst with highly dispersed Co atoms over the hierarchical structure of g-C3N4 and carbon spheres (C3N4@CS), where ultrathin g-C3N4 aided the atomic level dispersion of Co atoms. As the electrocatalytic performance of the SACs is influenced by the chemical bonding between the support and single atoms, catalysts with Co–O coordination (Co–O–C3N4@CS), and Co–N coordination (Co–C3N4@CS), were precisely synthesised by changing the calcination atmosphere from air to argon. Co–O–C3N4@CS and Co–C3N4@CS catalysts exhibited appreciable OER activity and delivered overpotentials of 0.23 V at 10 mA cm−2 and 0.47 V at 50 mA cm−2, respectively in alkaline media. The Co–O–C3N4@CS exhibited higher OER performance, at low overpotentials, as the higher electronegativity of O, compared to N, accelerated the adsorption of oxygen species. However, at high overpotentials the activity worsened due to unfavourable desorption energies.146
Employing first principle calculations, Guo et al. established the suitability of a single transition metal atom dispersed g-C3N4 catalyst for ORR. The adsorption energy of the key intermediate OH (ΔE*OH) and the d-band centres form the basis of predicting the overpotential for ORR. The study predicted that among the various TM/g-C3N4 catalyst, Pd/g-C3N4 exhibited a low overpotential value of 0.46 V qualifying it to be an effective substituent for the conventional Pt catalyst.147
N-doped porous carbon containing Fe atoms (Fe-g-C3N4/HPNCPs, 2.15 wt%) coordinated to C3N4 matrix was demonstrated to be an effective ORR electrocatalyst, with an E1/2 of 0.902 V, outperforming the commercial Pt/C catalysts. The hierarchical structure characterised by a micro–mesoporous architecture enabled increased mass transport to the catalytically active single atomic Fe–N2 sites favouring enhanced ORR activity with appreciable durability.148
The potential of Co1/g-CN and Ni1/g-CN SACs described earlier for overall water splitting, have been evaluated for OER. The theoretical calculations predicted that Co1/g-CN and Ni1/g-CN SACs can exhibit overpotential values of 0.61 V and 0.40 V respectively for OER reactions that are comparable with that of IrO2 reference catalysts with 0.55 V overpotential value.137
By combining DFT with machine learning Guo et al. predicted the possibility of developing highly efficient OER and ORR bifunctional catalysts based on single atom transition metal (TM) dispersed over g-C3N4 (Fig. 32(a and b)). The defects induced by the nitrogen vacancies in g-C3N4 (VN–CN) facilitate a strong RMSI with TM single atoms providing stability for TM/VN-CN. Among various TMs, Rh/VN-CN indicated low overpotentials of 0.43 and 0.32 V for ORR and OER respectively (Fig. 32(c–h)).10
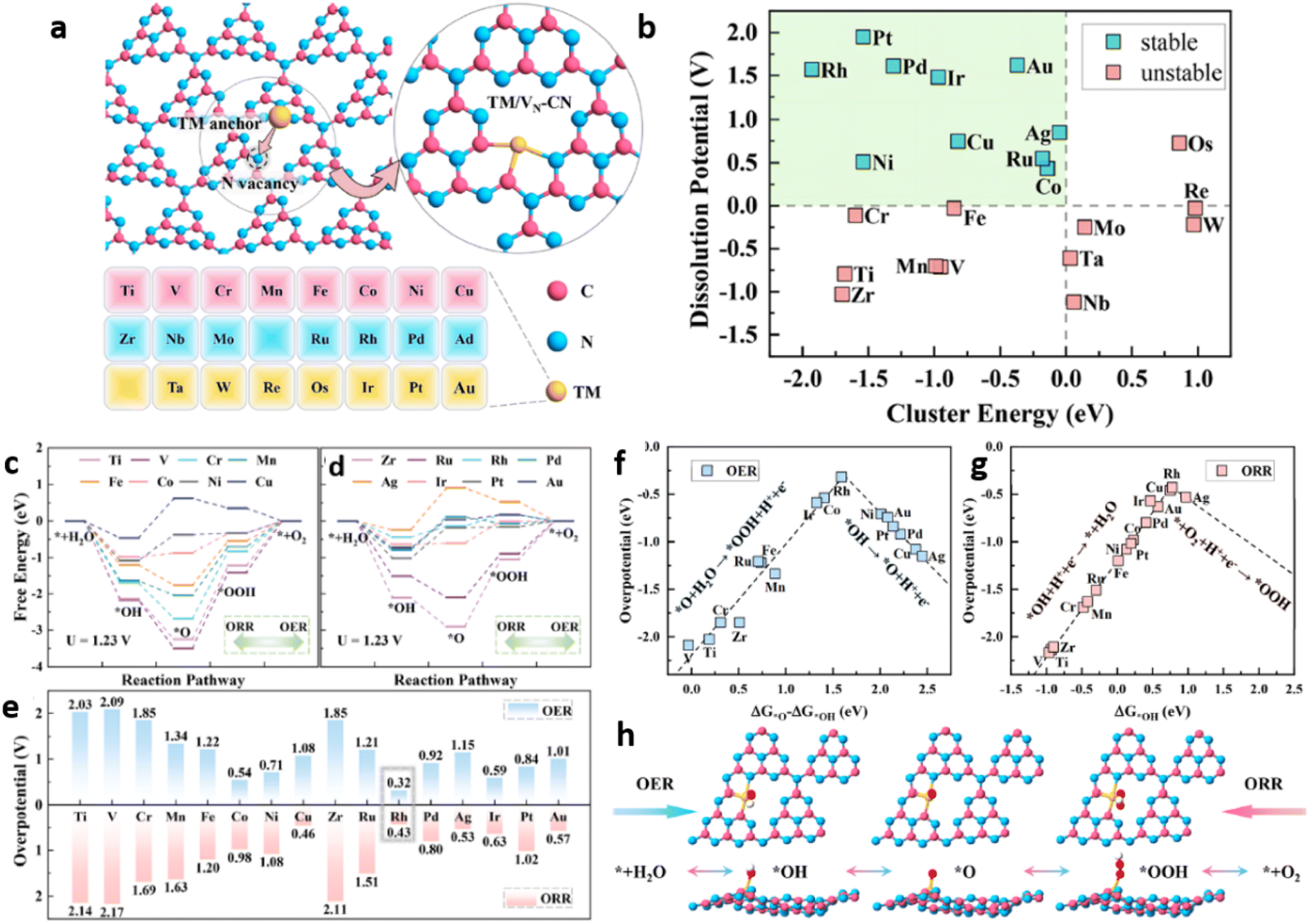 | ||
| Fig. 32 (a) Schematic representation of various transition metals (TM) based SACs on VN-CN, (b) stability of TM/VN-CN based on cluster energy (EClus < 0 eV) and dissolution potential (Udiss > 0 eV), the free energy diagrams of TM/VN-CN (c) from Ti to Cu and (d) from Zr to Au related to both OER and ORR at the equilibrium potential of 1.23, (e) The OER and ORR overpotentials of TM/VN-CN (from Ti to Au), the volcano plots of (f) OER (−ηOER vs. ΔG*O − ΔG*OH) and (g) ORR (−ηORR vs. ΔG*OH) on TM/VN-CN, (h) OER and ORR intermediate structures adsorbed on Rh/VN-CN. Reproduced with permission from ref. 10. Copyright 2021 American Chemical Society. | ||
Peng et al. demonstrated the development of Fe, N co-doped carbon (Fe/N/C) using Fe coordinated g-C3N4 and glucose as precursors. The intense mixing of urea-derived g-C3N4, FeCl3·6H2O and glucose under ultrasonication resulted in the formation of Fe coordinated g-C3N4 matrix. The adsorbed glucose on hydrothermal treatment, formed linear and/or branchlike oligosaccharides. Carbonisation at 1100 °C resulted in the conversion of g-C3N4-coordinated Fe embedded in glucose-derived carbon to porous Fe/N/C. The pyridinic N atoms of heptazine rings (known as N pots) in the g-C3N4 matrix can prevent the aggregation of the metal species during high-temperature pyrolysis by realising coordination between lone pairs of electron and metal species. The negative adsorption energy values calculated via DFT studies for Fe3+ ion (−38.26 eV) and Fe atom (−3.54 eV) on the N pots further substantiated the role of g-C3N4 in realising atomic level dispersion of Fe on carbon matrix. Fe/N/C-575 A (Fe/N/C-575) sample after the ORR catalytic activation steps such as acid leaching and second annealing exhibited good ORR activity with large onset protectional (Eonset = 0.972 V) and half-wave potential (E1/2 = 0.890 V) with a mass specific activity (jm) of 9.103 mA mg−1 at 0.9 V. The availability of a large number of Fe–N4 active sites over the high surface area matrix, synergistic activity of Fe single atoms with N–C nanosheets, and the enhanced mass transfer due to high porosity collectively contributed towards the enhanced ORR performance of Fe/N/C-575-A catalyst.149
Xu et al. demonstrated a facile template strategy for the development of ORR electrocatalyst based on N-doped graphene containing up to 14.0 wt% of Co single atoms and atomic clusters. The catalyst CoSAs/AC@NG was obtained by the pyrolysis of dicyandiamide with the formation of layered g-C3N4 as the sacrificial template with abundant N anchoring sites to realise the single Co atom loading. The catalyst was synthesised by the low-temperature pyrolysis (550 °C) of a precursor mix of glucose, dicyandiamide (DCDA) and CoCl2·6H2O followed by a high-temperature annealing at 800 °C for the conversion of g-C3N4 to porous carbon. The numerous defective sites and high N content acted as the anchoring sites for Co single atoms and the transformation of intermediate products to conductive carbon led to the in situ formation of high-density cobalt single atoms in nitrogen-doped graphene (Co SAs/AC@NG). XANES analysis revealed that the valence state of cobalt is close to +2 as the Co K-edge absorption position of Co SAs/AC@NG varied with that of Co foil but was similar to that of CoO. The EXAFS spectral analysis of the catalyst confirmed Co–N(O) coordination through a major peak at ≈1.41 Å, and a weak peak at 2.22 Å, indicating the presence also of Co clusters. The EXAFS fittings suggested an average coordination number of 3.7 ± 0.3 for Co–N(O). It was inferred from the XPS and XAS analysis that the most dominant active sites in Co SAs/AC@NG are likely to be the Co–N4 moiety (Fig. 33 (a–g)). Co40SAs/AC@NG with E1/2 of 0.890 V, kinetic current density (Jk) of 17.070 mA cm−2 and mass activity of 305 A g−1 at 0.85 V outperformed the commercial Pt/C (Jk of 4.845 and mass activity of 60.5 A g−1) for ORR reaction (Fig. 33(h and i)). Additionally, Co40SAs/AC@NG exhibited excellent performance in Zn–air battery as an air electrode material with a high-peak power density value of 221 mW cm−2 and with high cycling stability.150
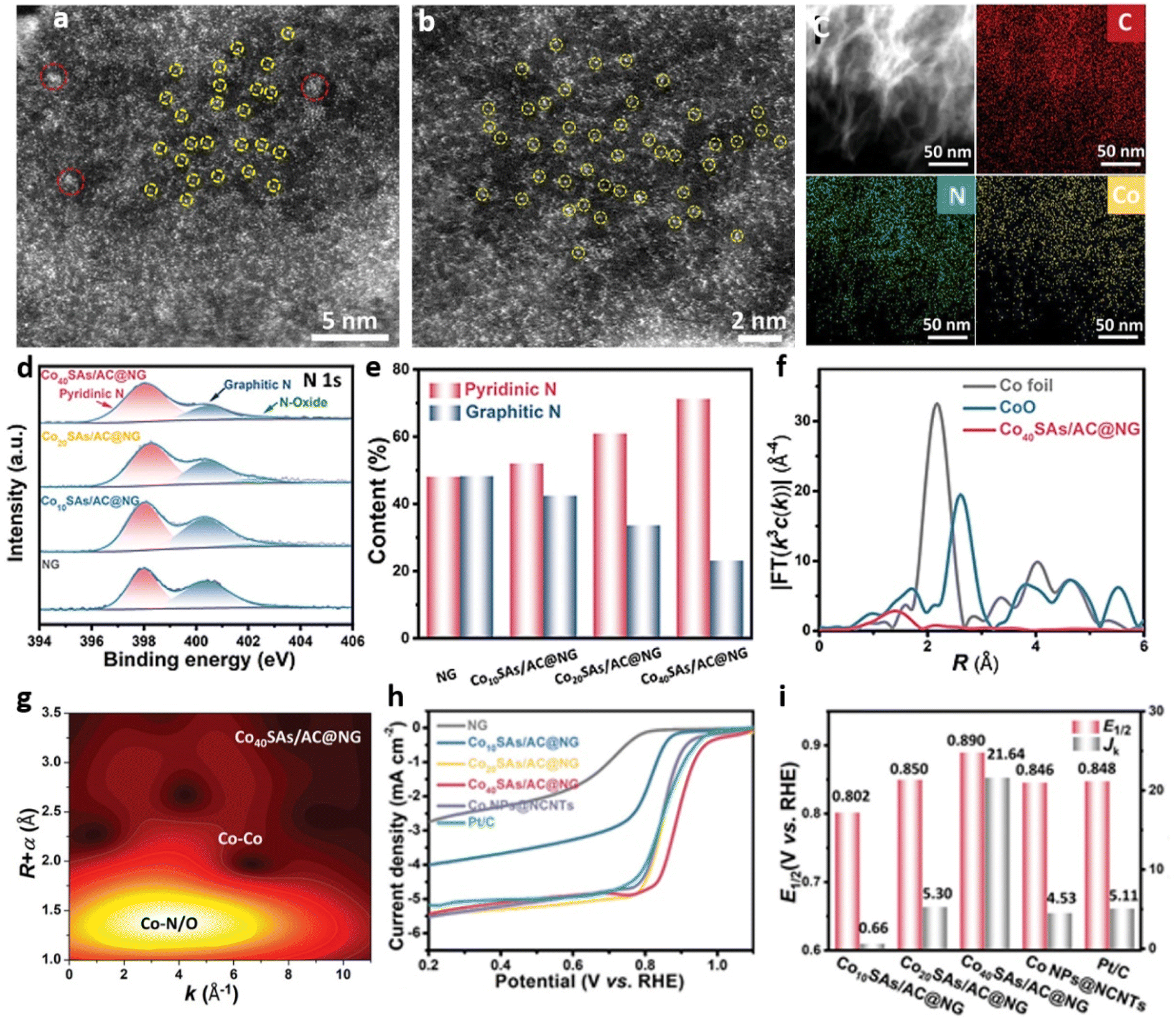 | ||
| Fig. 33 (a and b) HAADF-STEM images of Co40SAs/AC@NG catalyst (aberration-corrected), (c) EDS mappings of Co40SAs/AC@NG by HAADF-STEM; yellow and red circles represent Co single atoms and Co clusters respectively, (d) XPS N 1s spectra of NG and CoSAs/AC@NG, (e) N content of NG and Co SAs/AC@NG, (f and g) EXAFS spectra and WT-EXAFS plots of Co40SAs/AC@NG, (h and i) Comparison of RDE polarisation curves at a scan rate of 5 mV s at 1600 rpm, Jk values at 0.85 V & E1/2 values of the samples for ORR activity in O2-saturated 0.1 M KOH solution. Reproduced with permission from ref. 150. Copyright 2022 John Wiley and Sons. | ||
Table 11 lists the OER and ORR applications of g-C3N4 based SACs.
| System | Single atom & loading | Synthesis method | OER & ORR activity (onset potential, E1/2, current density, mass activity) | Coordination site or structure & oxidation state | Ref. |
|---|---|---|---|---|---|
| Ni–CN | Ni 2.4 at% | Template free direct synthesis | OER: onset potential 1.54 eV | Nitrogen | 142 |
| M-C3N4/CNT | M = Cr, Mn, Fe, Co, Ni, Cu, Zn; Co: 0.2 at% | Template free direct synthesis | Co–C3N4/CNT ORR: onset potential: 0.9 V, current density: 5 mA cm−2, ORR: onset potential: 1.5 V, current density: 10 mA cm−2 at 1.6 V | Co–N2, +2 | 143 |
| Au1Nx/C3N4 | Au 0.7 wt% | Post synthesis-electrostatic adsorption | ORR: Mass activity of 9000 A gAu−1 at E1/2 of 0.76 V, OER: mass activity of 1500 A gAu−1 at overpotential 0.45 V | Au1Nx, +1 | 22 |
| C-FeHZ8 @g-C3N4 | Fe 3.17 wt% | g-C3N4 as a fugitive template | ORR: E1/2:0.18 V in acidic condition 0.845 V in alkaline condition |
Fe–N4, +2 | 144 |
| NCM–CN–Fe | Fe | g-C3N4 as a fugitive template | ORR: Onset potential: 0.0 V current density −6.42 mA cm−2 | Fe–N4.1 | 145 |
| Co–O–C3N4@CS & Co–C3N4@CS | Co 0.13 & 0.09 wt% | Post synthesis | OER: Co–O–C3N4@CS: overpotential 0.23 V current density: 10 mA cm−2 Co–C3N4@CS: overpotential 0.47 V current density: 50 mA cm−2 | Co–O & Co–N | 146 |
| TM/g-C3N4 | TM = Ti - Au | DFT study | Pd/g-C3N4 is predicted to exhibit good ORR activity with an overpotential of 0.46 V | — | 147 |
| Fe-g-C3N4/HPNCPs | Fe 2.15 wt% | g-C3N4 as a fugitive template | ORR: E1/2: 0.902 V | Fe–N2, oxidation state in between 0 & +3 | 148 |
| TM g−1-CN | TM = Ti, V, Cr, Mn, Fe, Co, Ni | DFT study | Co1/g-CN & Ni1/g-CN SACs are predicted to be the most effective OER catalysts with overpotential values of 0.61 V and 0.40 V respectively | Nitrogen | 137 |
| TM/VN-CN | TM = Ti to Au | DFT study | Rh/VNCN is predicted to have optimal activity with overpotential values of 0.32 and 0.43 V for OER & ORR respectively | C/N | 10 |
| Fe/N/C | Fe 1.5 wt% | g-C3N4 as a fugitive template | ORR: onset potential: 0.972 V, E1/2: 0.89 V, mass specific activity: 9.103 mA mg−1 @ 0.9 V | Fe–N4, +2 | 149 |
| CoSAs/AC@NG | Co 14 wt% | g-C3N4 as a fugitive template | ORR: E1/2:0.890 V, kinetic current density (Jk):17.07 mA cm−2, mass activity: 305 A g−1 at 0.85 V |
Co–N4, +2 | 150 |
6 Conclusion and future perspectives
Single atom catalysts have emerged as potential systems with attractive features for a multitude of applications. The use of g-C3N4 as an efficient matrix for the development of SACs has been hitherto established to a fair level of certainty. The ease of anchoring single atoms on the matrix through abundant nitrogen sites in g-C3N4 facilitated the development of a variety of SACs for applications in different domains of catalysis. Advanced analytical tools like EXAFS, XANES, aberration corrected HAADF-STEM and EELS are used to confirm the single atom formation with a positive (partial/full) oxidation state. Computational studies have also been employed to validate the geometrical and coordination structures as well as to elucidate and predict the catalytic mechanisms.Conventional approaches of synthesising g-C3N4 based SACs like wet chemical reduction, high-temperature heat treatment of precursor mix under an inert atmosphere, and supramolecular self-assembly techniques are presumed to be advantageous over hardware intensive atomic layer deposition method (ALD). Even though precise control of single atom dispersion is possible in ALD, metal loading realisable is relatively low. The scale up possibilities in kilogram batches have been recently demonstrated in a multistep high temperature treatment under an inert atmosphere. Single atoms of 15 transition metals in ultra-high density have been developed on g-C3N4 support. The process is thus amenable for large-scale production in appreciable yields. Downsizing metal species to single atoms significantly improves metal utilisation efficiency imparting desirable catalytic properties. However, enhanced metal–support interaction, though beneficial for catalyst stability, often impedes catalytic activity due to unfavourable product desorption. A balance has to be thus established between catalyst stability and activity for which an in-depth analysis of the metal–support interaction and mechanistic reaction pathways are essential. Real-time analysis involving operando techniques has to be explored further for g-C3N4 based SACs.
The use of synchrotron based spectroscopic tools is an essential requirement to elucidate the metal oxidation state and coordination structure in SACs. Apart from this catalyst characterisation, the study of the catalytic mechanism necessitates real-time analysis as mentioned before, the accessibility of which is scarce.
Of the reported systems thus far, g-C3N4 SACs are primarily based on single metal systems predominantly of precious metals. The techno-economic feasibility of such systems for large-scale applications is limited due to cost and availability. The shortcoming can presumably be circumvented by the use of dual or multiple metals as atomic dispersions over g-C3N4. This can be realised by substituting precious metals partly with non-precious ones to balance affordability with catalytic performance. This is particularly true for applications demanding multi-site synergism like CO2 reduction and organic transformations. For example, in photocatalytic CO2 reduction SAC with single sites normally facilitates the reduction of CO2 to C1 products like HCOOH, CO, CH3OH, HCHO, CH4,etc. The product selectivity mainly depends upon the stability of the intermediates formed during the multi-electron–proton transfer. SACs with multiple metal sites can offer better stability to the intermediates compared to single metal sites thereby promoting extended electron proton transfer as well as C–C coupling to produce higher hydrocarbons. Along with dual metal sites, it is also feasible to incorporate single atom alloy catalysts based on g-C3N4 for enhanced activity by tuning electron density distributions around the multi centers.
Researchers have already employed DFT calculations to prove the efficiency of g-C3N4 based SACs for electrocatalytic applications such as CO2 reduction, N2 reduction, etc but the experimental verification of the hypothesis is still required. The poor conductivity of g-C3N4 support may impede its electrocatalytic activity to a larger extent and it may be advantageous to develop hybrid matrices incorporating materials of high electrical conductivity and charge transfer capacity (graphene, N-doped carbon, etc.).
One area of high practical relevance for the use g-C3N4 based SACs is believed to be organic reaction catalysis as a majority of the industrially relevant reactions employ homogeneous catalysts for enhanced kinetics and high yield. Recovery of the catalyst, particularly of precious metal compositions is vital for recyclability and cost effectiveness. g-C3N4 SACs are affordable solutions to bridge this gap and are presumed to be of high practical viability even though most of the works reported still employ Pt, Pd, Au based SACs. However, the recent report on the large-scale synthesis of g-C3N4 Cu SAC and its demonstration for organic transformation opens up myriad possibilities for the industrial utilisation of SACs. Nevertheless, this would necessitate a thorough understanding of the underlying mechanisms leading to the regiospecificity and selectivity of products. This review comprehensively analysed the developments in g-C3N4 based SACs and overviewed the synthesis, characterisation, analysis and applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
One of the authors (SP) acknowledges University Grants Commission for research fellowship.References
- P. Suyana, P. Ganguly, B. N. Nair, S. C. Pillai and U. Hareesh, Chem. Eng. J. Adv., 2021, 8, 100148 CrossRef CAS.
- Z. Li, B. Li, Y. Hu, X. Liao, H. Yu and C. Yu, Small Struct., 2022, 3, 2200041 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 11265–11269 CrossRef PubMed.
- G. Gao, Y. Jiao, E. R. Waclawik and A. Du, J. Am. Chem. Soc., 2016, 138, 6292–6297 CrossRef CAS PubMed.
- Z. Chen, S. Mitchell, E. Vorobyeva, R. K. Leary, R. Hauert, T. Furnival, Q. M. Ramasse, J. M. Thomas, P. A. Midgley, D. Dontsova and J. Pérez-Ramírez, Adv. Funct. Mater., 2017, 27, 1605785 CrossRef.
- T. Tong, B. He, B. Zhu, B. Cheng and L. Zhang, Appl. Surf. Sci., 2018, 459, 385–392 CrossRef CAS.
- P. Zhou, X. Hou, Y. Chao, W. Yang, W. Zhang, Z. Mu, J. Lai, F. Lv, K. Yang, Y. Liu and S. Guo, Chem. Sci., 2019, 10, 5898–5905 RSC.
- S. Ji, Y. Qu, T. Wang, Y. Chen, G. Wang, X. Li, J. Dong, Q. Chen, W. Zhang and Z. Zhang, Angew. Chem., Int. Ed., 2020, 59, 10651–10657 CrossRef CAS PubMed.
- H. Niu, X. Wan, X. Wang, C. Shao, J. Robertson, Z. Zhang and Y. Guo, ACS Sustainable Chem. Eng., 2021, 9, 3590–3599 CrossRef CAS.
- X. Hai, S. Xi, S. Mitchell, K. Harrath, H. Xu, D. F. Akl, D. Kong, J. Li, Z. Li and T. Sun, Nat. Nanotechnol., 2022, 17, 174–181 CrossRef CAS PubMed.
- R. Shibuya, T. Kondo and J. Nakamura, Carbon-Based Metal-Free Catalysts: Design and Applications, 2018, 1, pp. 227–249 Search PubMed.
- C. Gao, J. Low, R. Long, T. Kong, J. Zhu and Y. Xiong, Chem. Rev., 2020, 120, 12175–12216 CrossRef CAS PubMed.
- M. Kathiresan, in Nanoscale Graphitic Carbon Nitride, Elsevier, 2022, pp. 1–16 Search PubMed.
- S. Panneri, P. Ganguly, B. N. Nair, A. A. P. Mohamed, K. G. K. Warrier and U. N. S. Hareesh, Environ. Sci. Pollut. Res., 2017, 24, 8609–8618 CrossRef CAS PubMed.
- P. Chen, B. Lei, X. a. Dong, H. Wang, J. Sheng, W. Cui, J. Li, Y. Sun, Z. Wang and F. Dong, ACS Nano, 2020, 14, 15841–15852 CrossRef CAS PubMed.
- Z. Chen, S. Pronkin, T.-P. Fellinger, K. Kailasam, G. Vilé, D. Albani, F. Krumeich, R. Leary, J. Barnard, J. M. Thomas and D. Dontsova, ACS Nano, 2016, 10, 3166–3175 CrossRef CAS PubMed.
- Y. Li, B. Li, D. Zhang, L. Cheng and Q. Xiang, ACS Nano, 2020, 14, 10552–10561 CrossRef CAS PubMed.
- W. Zhang, Y. Fu, Q. Peng, Q. Yao, X. Wang, A. Yu and Z. Chen, Chem. Eng. J., 2020, 394, 124822 CrossRef CAS.
- C. Chu, D. Huang, Q. Zhu, E. Stavitski, J. A. Spies, Z. Pan, J. Mao, H. L. Xin, C. A. Schmuttenmaer and S. Hu, ACS Catal., 2018, 9, 626–631 CrossRef.
- P. Sharma, S. Kumar, O. Tomanec, M. Petr, J. Zhu Chen, J. T. Miller, R. S. Varma, M. B. Gawande and R. Zbořil, Small, 2021, 17, 2006478 CrossRef CAS PubMed.
- L. Liu, H. Su, F. Tang, X. Zhao and Q. Liu, Nano Energy, 2018, 46, 110–116 CrossRef CAS.
- H. Su, W. Che, F. Tang, W. Cheng, X. Zhao, H. Zhang and Q. Liu, J. Phys. Chem. C, 2018, 122, 21108–21114 CrossRef CAS.
- Y. Cao, S. Chen, Q. Luo, H. Yan, Y. Lin, W. Liu, L. Cao, J. Lu, J. Yang and T. Yao, Angew. Chem., Int. Ed., 2017, 56, 12191–12196 CrossRef CAS PubMed.
- P. Zhou, F. Lv, N. Li, Y. Zhang, Z. Mu, Y. Tang, J. Lai, Y. Chao, M. Luo, F. Lin and S. Guo, Nano Energy, 2019, 56, 127–137 CrossRef CAS.
- Z. Chen, Q. Zhang, W. Chen, J. Dong, H. Yao, X. Zhang, X. Tong, D. Wang, Q. Peng and C. Chen, Adv. Mater., 2018, 30, 1704720 CrossRef PubMed.
- R. Zhang, P. Li, F. Wang, L. Ye, A. Gaur, Z. Huang, Z. Zhao, Y. Bai and Y. Zhou, Appl. Catal., B, 2019, 250, 273–279 CrossRef CAS.
- D. Deng, X. Chen, L. Yu, X. Wu, Q. Liu, Y. Liu, H. Yang, H. Tian, Y. Hu and P. Du, Sci. Adv., 2015, 1, e1500462 CrossRef PubMed.
- K. O'Connell and J. R. Regalbuto, Catal. Lett., 2015, 145, 777–783 CrossRef.
- P. Suyana, P. Ganguly, B. N. Nair, A. P. Mohamed, K. Warrier and U. Hareesh, Environ. Sci.: Nano, 2017, 4, 212–221 RSC.
- M. Varela, A. Lupini, K. Van Benthem, A. Borisevich, M. Chisholm, N. Shibata, E. Abe and S. Pennycook, Annu. Rev. Mater. Res., 2005, 35, 539–569 CrossRef CAS.
- S. Pennycook, A. Lupini, M. Varela, A. Borisevich, Y. Peng, M. Oxley, K. V. Benthem and M. Chisholm, in Scanning Microscopy for Nanotechnology, Springer, 2006, pp. 152–191 Search PubMed.
- X. Xiao, L. Zhang, H. Meng, B. Jiang and H. Fu, Sol. RRL, 2021, 5, 2000609 CrossRef CAS.
- R. Brydson and N. Hondow, Aberration-Corrected Analytical Transmission Electron Microscopy, 2011, pp. 163–210 Search PubMed.
- R. Brydson, Electron Energy Loss Spectroscopy, Garland Science, 2020 Search PubMed.
- T. Zhang, Z. Chen, A. G. Walsh, Y. Li and P. Zhang, Adv. Mater., 2020, 32, 2002910 CrossRef CAS PubMed.
- Y. Wang, J. Mao, X. Meng, L. Yu, D. Deng and X. Bao, Chem. Rev., 2018, 119, 1806–1854 CrossRef PubMed.
- M. Kottwitz, Y. Li, H. Wang, A. I. Frenkel and R. G. Nuzzo, Chem.: Methods, 2021, 1, 278–294 CAS.
- B. Zhang, T. Fan, N. Xie, G. Nie and H. Zhang, Adv. Sci., 2019, 6, 1901787 CrossRef CAS PubMed.
- S. Qiao, Q. He, P. Zhang, Y. Zhou, S. Chen, L. Song and S. Wei, J. Mater. Chem. A, 2022, 10, 5771–5791 RSC.
- S. K. Dutta, S. K. Mehetor and N. Pradhan, J. Phys. Chem. Lett., 2015, 6, 936–944 CrossRef CAS PubMed.
- A. Kubacka, M. Fernandez-Garcia and G. Colon, Chem. Rev., 2012, 112, 1555–1614 CrossRef CAS PubMed.
- X.-S. Zhang, K. Tian, J.-Y. Hu and H. Jiang, Chemosphere, 2015, 141, 127–133 CrossRef CAS PubMed.
- B. Zhu, L. Zhang, B. Cheng and J. Yu, Appl. Catal., B, 2018, 224, 983–999 CrossRef CAS.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- B. Xia, Y. Zhang, J. Ran, M. Jaroniec and S.-Z. Qiao, ACS Cent. Sci., 2021, 7, 39–54 CrossRef CAS PubMed.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- R. Liu, Z. Chen, Y. Yao, Y. Li, W. A. Cheema, D. Wang and S. Zhu, RSC Adv., 2020, 10, 29408–29418 RSC.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- M. Marszewski, S. Cao, J. Yu and M. Jaroniec, Mater. Horiz., 2015, 2, 261–278 RSC.
- A. K. Singh, J. H. Montoya, J. M. Gregoire and K. A. Persson, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- P. Huang, J. Huang, S. A. Pantovich, A. D. Carl, T. G. Fenton, C. A. Caputo, R. L. Grimm, A. I. Frenkel and G. Li, J. Am. Chem. Soc., 2018, 140, 16042–16047 CrossRef CAS PubMed.
- J. Wang, T. Heil, B. Zhu, C.-W. Tung, J. Yu, H. M. Chen, M. Antonietti and S. Cao, ACS Nano, 2020, 14, 8584–8593 CrossRef CAS PubMed.
- L. Cheng, H. Yin, C. Cai, J. Fan and Q. Xiang, Small, 2020, 16, 2002411 CrossRef CAS PubMed.
- Z. Zhao, W. Liu, Y. Shi, H. Zhang, X. Song, W. Shang and C. Hao, Phys. Chem. Chem. Phys., 2021, 23, 4690–4699 RSC.
- B. Singh, V. Sharma, R. P. Gaikwad, P. Fornasiero, R. Zbořil and M. B. Gawande, Small, 2021, 17, 2006473 CrossRef CAS PubMed.
- S. Wang, J. Zhang, B. Li, H. Sun and S. Wang, Energy Fuels, 2021, 35, 6504–6526 CrossRef CAS.
- K. Villa, J. R. Galán-Mascarós, N. López and E. Palomares, Sustainable Energy Fuels, 2021, 5, 4560–4569 RSC.
- A. Mishra, A. Mehta, S. Basu, N. P. Shetti, K. R. Reddy and T. M. Aminabhavi, Carbon, 2019, 149, 693–721 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- X. Li, J. Yu, J. Low, Y. Fang, J. Xiao and X. Chen, J. Mater. Chem. A, 2015, 3, 2485–2534 RSC.
- A. Naseri, M. Samadi, A. Pourjavadi, A. Z. Moshfegh and S. Ramakrishna, J. Mater. Chem. A, 2017, 5, 23406–23433 RSC.
- S. Y. Tee, K. Y. Win, W. S. Teo, L. D. Koh, S. Liu, C. P. Teng and M. Y. Han, Adv. Sci., 2017, 4, 1600337 CrossRef PubMed.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS PubMed.
- W. Liu, L. Cao, W. Cheng, Y. Cao, X. Liu, W. Zhang, X. Mou, L. Jin, X. Zheng, W. Che and S. Wei, Angew. Chem., 2017, 129, 9440–9445 CrossRef.
- M. Ou, S. Wan, Q. Zhong, S. Zhang and Y. Wang, Int. J. Hydrogen Energy, 2017, 42, 27043–27054 CrossRef CAS.
- W. Zhang, Q. Peng, L. Shi, Q. Yao, X. Wang, A. Yu, Z. Chen and Y. Fu, Small, 2019, 15, 1905166 CrossRef CAS PubMed.
- Z. Zeng, Y. Su, X. Quan, W. Choi, G. Zhang, N. Liu, B. Kim, S. Chen, H. Yu and S. Zhang, Nano Energy, 2020, 69, 104409 CrossRef CAS.
- G. Wang, T. Zhang, W. Yu, R. Si, Y. Liu and Z. Zhao, ACS Catal., 2020, 10, 5715–5722 CrossRef CAS.
- M. Ren, X. Zhang, Y. Liu, G. Yang, L. Qin, J. Meng, Y. Guo and Y. Yang, ACS Catal., 2022, 12, 5077–5093 CrossRef CAS.
- B.-B. Xu, X.-B. Fu, X.-M. You, E. Zhao, F.-F. Li, Z. Chen, Y.-X. Li, X. L. Wang and Y.-F. Yao, ACS Catal., 2022, 12, 6958–6967 CrossRef CAS.
- R. Shi, Y. Zhao, G. I. Waterhouse, S. Zhang and T. Zhang, ACS Catal., 2019, 9, 9739–9750 CrossRef CAS.
- S. Hu, X. Qu, J. Bai, P. Li, Q. Li, F. Wang and L. Song, ACS Sustainable Chem. Eng., 2017, 5, 6863–6872 CrossRef CAS.
- C. Ling, X. Niu, Q. Li, A. Du and J. Wang, J. Am. Chem. Soc., 2018, 140, 14161–14168 CrossRef CAS PubMed.
- X.-W. Guo, S.-M. Chen, H.-J. Wang, Z.-M. Zhang, H. Lin, L. Song and T.-B. Lu, J. Mater. Chem. A, 2019, 7, 19831–19837 RSC.
- L. Li, Y. Yu, S. Lin, W. Chu, D. Sun, Q. Su, S. Ma, G. Du and B. Xu, Catal. Commun., 2021, 153, 106294 CrossRef CAS.
- S. Dong, J. Feng, M. Fan, Y. Pi, L. Hu, X. Han, M. Liu, J. Sun and J. Sun, RSC Adv., 2015, 5, 14610–14630 RSC.
- D. S. Bhatkhande, V. G. Pangarkar and A. A. C. M. Beenackers, J. Chem. Technol. Biotechnol., 2002, 77, 102–116 CrossRef CAS.
- M. M. Khan, D. Pradhan and Y. Sohn, Nanocomposites for Visible Light-Induced Photocatalysis, Springer, 2017 Search PubMed.
- D. Chatterjee and S. Dasgupta, J. Photochem. Photobiol., C, 2005, 6, 186–205 CrossRef CAS.
- M. A. Fox and M. T. Dulay, Chem. Rev., 1993, 93, 341–357 CrossRef CAS.
- Y. Wang, X. Zhao, D. Cao, Y. Wang and Y. Zhu, Appl. Catal., B, 2017, 211, 79–88 CrossRef CAS.
- F. Wang, Y. Wang, Y. Li, X. Cui, Q. Zhang, Z. Xie, H. Liu, Y. Feng, W. Lv and G. Liu, Dalton Trans., 2018, 47, 6924–6933 RSC.
- S. An, G. Zhang, T. Wang, W. Zhang, K. Li, C. Song, J. T. Miller, S. Miao, J. Wang and X. Guo, ACS Nano, 2018, 12, 9441–9450 CrossRef CAS PubMed.
- Z. Zhao, W. Zhang, W. Liu, Y. Li, J. Ye, J. Liang and M. Tong, Sci. Total Environ., 2020, 742, 140642 CrossRef CAS PubMed.
- L. Zhang, J. Zhang, M. Qi, X. Guo and X. Feng, Energy Fuels, 2020, 34, 12792–12799 CrossRef CAS.
- Y. Yang, G. Zeng, D. Huang, C. Zhang, D. He, C. Zhou, W. Wang, W. Xiong, B. Song and H. Yi, Small, 2020, 16, 2001634 CrossRef CAS PubMed.
- G. Vilé, P. Sharma, M. Nachtegaal, F. Tollini, D. Moscatelli, A. Sroka-Bartnicka, O. Tomanec, M. Petr, J. Filip, I. S. Pieta and M. B. Gawande, Sol. RRL, 2021, 5, 2100176 CrossRef.
- F. Chen, X. L. Wu, C. Shi, H. Lin, J. Chen, Y. Shi, S. Wang and X. Duan, Adv. Funct. Mater., 2021, 31, 2007877 CrossRef CAS.
- G. Liu, Y. Huang, H. Lv, H. Wang, Y. Zeng, M. Yuan, Q. Meng and C. Wang, Appl. Catal., B, 2021, 284, 119683 CrossRef CAS.
- L. Wang, R. Tang, A. Kheradmand, Y. Jiang, H. Wang, W. Yang, Z. Chen, X. Zhong, S. P. Ringer and X. Liao, Appl. Catal., B, 2021, 284, 119759 CrossRef CAS.
- K. P. Bryliakov, Chem. Rev., 2017, 117, 11406–11459 CrossRef CAS PubMed.
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- C. Dong, Y. Yang, X. Hu, Y. Cho, G. Jang, Y. Ao, L. Wang, J. Shen, J. H. Park and K. Zhang, Nat. Commun., 2022, 13, 1–11 Search PubMed.
- Z. P. Ifkovits, J. M. Evans, M. C. Meier, K. M. Papadantonakis and N. S. Lewis, Energy Environ. Sci., 2021, 14, 4740–4759 RSC.
- C. Wang, X. Zhang and Y. Liu, Appl. Surf. Sci., 2015, 358, 28–45 CrossRef CAS.
- J. Liu, Y. Zou, B. Jin, K. Zhang and J. H. Park, ACS Energy Lett., 2019, 4, 3018–3027 CrossRef CAS.
- X. Zeng, Y. Liu, X. Hu and X. Zhang, Green Chem., 2021, 23, 1466–1494 RSC.
- Z. Chen, D. Yao, C. Chu and S. Mao, Chem. Eng. J., 2022, 138489 Search PubMed.
- Z. Teng, Q. Zhang, H. Yang, K. Kato, W. Yang, Y.-R. Lu, S. Liu, C. Wang, A. Yamakata and C. Su, Nat. Catal., 2021, 4, 374–384 CrossRef CAS.
- Z. Teng, W. Cai, W. Sim, Q. Zhang, C. Wang, C. Su and T. Ohno, Appl. Catal., B, 2021, 282, 119589 CrossRef CAS.
- H. Arnold, F. Dobert, J. Gaube, G. Ertl, H. Knozinger and J. Weitkempl, Handbook of heterogeneous catalysis, ed. G. Ertl, H. Knoezinger and J. Weitkamp, 2008, pp. 2165–2186 Search PubMed.
- J. C. Védrine, Catalysts, 2017, 7, 341 CrossRef.
- X. Liu, L. He, Y.-M. Liu and Y. Cao, Acc. Chem. Res., 2014, 47, 793–804 CrossRef CAS PubMed.
- F. Zhang, Y. Zhu, Q. Lin, L. Zhang, X. Zhang and H. Wang, Energy Environ. Sci., 2021, 14, 2954–3009 RSC.
- C. Bolm and M. Beller, Transition Metals for Organic Synthesis, Wiley-VCH, Weinheim, 2004 Search PubMed.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- J. Li, M. F. Stephanopoulos and Y. Xia, Chem. Rev., 2020, 120, 11699–11702 CrossRef CAS PubMed.
- Z. Chen, E. Vorobyeva, S. Mitchell, E. Fako, M. A. Ortuño, N. López, S. M. Collins, P. A. Midgley, S. Richard and G. Vilé, Nat. Nanotechnol., 2018, 13, 702–707 CrossRef CAS PubMed.
- P. Yang, S. Zuo, F. Zhang, B. Yu, S. Guo, X. Yu, Y. Zhao, J. Zhang and Z. Liu, Ind. Eng. Chem. Res., 2020, 59, 7327–7335 CrossRef CAS.
- F. Hu, L. Leng, M. Zhang, W. Chen, Y. Yu, J. Wang, J. H. Horton and Z. Li, ACS Appl. Mater. Interfaces, 2020, 12, 54146–54154 CrossRef CAS PubMed.
- Z. Chen, Y. Chen, S. Chao, X. Dong, W. Chen, J. Luo, C. Liu, D. Wang, C. Chen and W. Li, ACS Catal., 2020, 10, 1865–1870 CrossRef CAS.
- N. Kong, X. Fan, F. Liu, L. Wang, H. Lin, Y. Li and S.-T. Lee, ACS Nano, 2020, 14, 5772–5779 CrossRef CAS PubMed.
- Y. Xiong, W. Sun, Y. Han, P. Xin, X. Zheng, W. Yan, J. Dong, J. Zhang, D. Wang and Y. Li, Nano Res., 2021, 14, 2418–2423 CrossRef CAS.
- G. Vilé, G. Di Liberto, S. Tosoni, A. Sivo, V. Ruta, M. Nachtegaal, A. H. Clark, S. Agnoli, Y. Zou and A. Savateev, ACS Catal., 2022, 12, 2947–2958 CrossRef.
- K. Eid, M. H. Sliem, M. Al-Ejji, A. M. Abdullah, M. Harfouche and R. S. Varma, ACS Appl. Mater. Interfaces, 2022, 14, 40749–40760 CrossRef CAS PubMed.
- Y. Zhang, X. Cao and Z. Cao, ACS Appl. Mater. Interfaces, 2022, 14, 35844–35853 CrossRef CAS PubMed.
- H. Zhang, W. Cheng, D. Luan and X. W. Lou, Angew. Chem., Int. Ed., 2021, 60, 13177–13196 CrossRef CAS PubMed.
- H. Hu, J. Wang, P. Tao, C. Song, W. Shang, T. Deng and J. Wu, J. Mater. Chem. A, 2022, 10, 5835–5849 RSC.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang and Z. Jin, Adv. Sci., 2018, 5, 1700275 CrossRef PubMed.
- J. Feng, H. Gao, L. Zheng, Z. Chen, S. Zeng, C. Jiang, H. Dong, L. Liu, S. Zhang and X. Zhang, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- S. Fu, X. Liu, J. Ran, Y. Jiao and S.-Z. Qiao, Appl. Surf. Sci., 2021, 540, 148293 CrossRef CAS.
- C. Cometto, A. Ugolotti, E. Grazietti, A. Moretto, G. Bottaro, L. Armelao, C. Di Valentin, L. Calvillo and G. Granozzi, npj 2D Mater. Appl., 2021, 5, 63 CrossRef CAS.
- N. Zhang, M. I. Hussain, M. Xia and C. Ge, Nano, 2021, 16, 2150016 CrossRef CAS.
- X. Wen and J. Guan, Nanoscale, 2020, 12, 8065–8094 RSC.
- G. F. Chen, S. Ren, L. Zhang, H. Cheng, Y. Luo, K. Zhu, L. X. Ding and H. Wang, Small Methods, 2019, 3, 1800337 CrossRef.
- X. Yan, D. Liu, H. Cao, F. Hou, J. Liang and S. X. Dou, Small Methods, 2019, 3, 1800501 CrossRef.
- X. Cui, C. Tang and Q. Zhang, Adv. Energy Mater., 2018, 8, 1800369 CrossRef.
- W. Song, K. Xie, J. Wang, Y. Guo, C. He and L. Fu, Phys. Chem. Chem. Phys., 2021, 23, 10418–10428 RSC.
- Z. Chen, J. Zhao, C. R. Cabrera and Z. Chen, Small Methods, 2019, 3, 1800368 CrossRef.
- C. Ren, Q. Jiang, W. Lin, Y. Zhang, S. Huang and K. Ding, ACS Appl. Nano Mater., 2020, 3, 5149–5159 CrossRef CAS.
- S. Agarwal, R. Kumar, R. Arya and A. K. Singh, J. Phys. Chem. C, 2021, 125, 12585–12593 CrossRef CAS.
- X. Wang, W. Wang, M. Qiao, G. Wu, W. Chen, T. Yuan, Q. Xu, M. Chen, Y. Zhang and X. Wang, Sci. Bull., 2018, 63, 1246–1253 CrossRef CAS PubMed.
- H. Liu, X. Peng and X. Liu, ChemElectroChem, 2018, 5, 2963–2974 CrossRef CAS.
- X. Lv, W. Wei, H. Wang, B. Huang and Y. Dai, Appl. Catal., B, 2020, 264, 118521 CrossRef CAS.
- X. Liu, Y. Deng, L. Zheng, M. R. Kesama, C. Tang and Y. Zhu, ACS Catal., 2022, 12, 5517–5526 CrossRef CAS.
- M. Tahir, L. Pan, F. Idrees, X. Zhang, L. Wang, J.-J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- T. He, S. K. Matta, G. Will and A. Du, Small Methods, 2019, 3, 1800419 CrossRef.
- Z. Xue, X. Zhang, J. Qin and R. Liu, J. Energy Chem., 2021, 55, 437–443 CrossRef CAS.
- S. Ohn, S. Y. Kim, S. K. Mun, J. Oh, Y. J. Sa, S. Park, S. H. Joo, S. J. Kwon and S. Park, Carbon, 2017, 124, 180–187 CrossRef CAS.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336–3339 CrossRef CAS PubMed.
- Y. Deng, B. Chi, X. Tian, Z. Cui, E. Liu, Q. Jia, W. Fan, G. Wang, D. Dang, M. Li and X. Sun, J. Mater. Chem. A, 2019, 7, 5020–5030 RSC.
- C. Wang, H. Zhao, J. Wang, Z. Zhao, M. Cheng, X. Duan, Q. Zhang, J. Wang and J. Wang, J. Mater. Chem. A, 2019, 7, 1451–1458 RSC.
- Q. Song, J. Li, L. Wang, L. Pang and H. Liu, Inorg. Chem., 2019, 58, 10802–10811 CrossRef CAS PubMed.
- H. Niu, X. Wang, C. Shao, Y. Liu, Z. Zhang and Y. Guo, J. Mater. Chem. A, 2020, 8, 6555–6563 RSC.
- T. Sun, P. Zhang, W. Chen, K. Wang, X. Fu, T. Zheng and J. Jiang, Chem. Commun., 2020, 56, 798–801 RSC.
- X.-B. Ding, L. Zhang, Y.-H. Qin, L. Yang, C. Wang and C. Peng, Chem. Commun., 2021, 57, 6935–6938 RSC.
- M. Zhang, H. Li, J. Chen, F. X. Ma, L. Zhen, Z. Wen and C. Y. Xu, Adv. Funct. Mater., 2023, 33, 2209726 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |