
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Strategies for the mitigation of salt precipitation in zero-gap CO2 electrolyzers producing CO
Joey
Disch
 ab,
Luca
Bohn
ab,
Lukas
Metzler
a and
Severin
Vierrath
*ab
ab,
Luca
Bohn
ab,
Lukas
Metzler
a and
Severin
Vierrath
*ab
aElectrochemical Energy Systems, IMTEK – Department of Microsystems Engineering, University of Freiburg, Georges-Koehler-Allee 103, 79110 Freiburg, Germany. E-mail: Severin.Vierrath@imtek.uni-freiburg.de
bUniversity of Freiburg, Institute and FIT – Freiburg Center for Interactive Materials and Bioinspired Technologies, Georges-Köhler-Allee 105, 79110 Freiburg, Germany
First published on 27th February 2023
Abstract
Salt precipitation in the cathode gas diffusion electrodes of zero-gap CO2 electrolyzers producing CO is a major challenge to the stability and durability of this technology. In this review, we examine different strategies proposed in the literature for mitigating salt precipitation in anion-exchange-membrane-based zero-gap cells. We discuss the advantages and limitations of these approaches, with a focus on material developments and systemic approaches. By normalizing all data to the cumulative amount of converted CO2, we provide a quantified comparison and recommendations for future development. Our review shows that material developments, such as the use of hydrophobic or superhydrophobic coatings, can effectively reduce salt precipitation, but may not be suitable for long-term operation due to their limited durability. Systemic approaches, such as adjusting the operating conditions or incorporating salt removal methods, can also be effective in mitigating salt precipitation, but may come with additional cost and complexity. Overall, our results suggest that a combination of these approaches may be the most effective in achieving the long-term stability and durability required for commercialization.
 Joey Disch | Joey Disch received his Bachelor's degree (2017) and his Master's degree (2019) in Environmental Engineering from the University of Stuttgart where he specialized in renewable energy technologies and chemical process engineering. He is currently pursuing his PhD at the University of Freiburg under the supervision of Severin Vierrath. His research focus is on the development and characterization of membrane electrode assemblies for the electrochemical reduction of CO2 and anion-exchange-membrane-based water electrolysis. |
 Luca Bohn | Luca Bohn received his Bachelor's degree (2019) and Master's degree (2022) in Microsystems Engineering from the University of Freiburg. He is currently pursuing his PhD under the supervision of Severin Vierrath at the University of Freiburg. His research focuses on the development and characterization of membrane electrode assemblies and operation conditions for CO2 electrolyzers. |
 Lukas Metzler | Dr. Lukas Metzler is a Senior Researcher in the group “Electrochemical Energy Systems”. His work focuses mainly on the development and characterization of ion exchange membranes and electrode properties. He studied Chemistry at the University of Freiburg (Germany). At the Institute of Macromolecular Chemistry, he developed his research interests in the groups of Dr Michael Sommer and Dr Rolf Mülhaupt as well as in an internship in the lab of Dr Steven Holdcroft (Simon Fraser University, Canada). In 2021, he completed his PhD in the Lab for Chemistry and Physics of Interfaces of Dr Jürgen Rühe. |
 Severin Vierrath | Dr Severin Vierrath has been involved in the field of electrochemical converters since 2013. After studying industrial engineering at RWTH Aachen University, he completed his doctorate at the University of Freiburg on fuel cells. Since 2017, he has been leading the research group “Electrochemical Energy Systems” (EES) at the University of Freiburg and at Hahn-Schickard research institute. The scientific excellence of the group is evidenced by more than 40 publications and several patents. His research focuses on the fundamental understanding of electrochemical energy converters, the integration of new materials – catalysts and polymers – and the engineering of membrane electrode assemblies. |
Introduction
To meet global net-zero CO2 emission goals, not only fossil energy sources but also fossil feedstocks of the chemical industry have to be replaced with sustainable solutions. One pivotal technology to reduce the carbon footprint of the chemical industry is the electrochemical reduction of CO2. This technology allows producing a variety of feedstock chemicals from captured CO2, water and renewable energy. A multitude of reactor designs1 and target products ranging from C1 to C3 products2 have been thoroughly investigated and their advantages and disadvantages are well known.1,3–5 In low-temperature CO2 electrolysis a gas-fed, zero-gap cell architecture comprising an anion exchange membrane (AEM) is considered most promising, due to high CO2 conversion rates and low ohmic losses compared to cells using liquid catholytes.3,6,7 Partial current densities of ∼1 A cm−2 for the production of carbon monoxide (CO) have been demonstrated8 and electrolyzers producing CO are already reaching pilot scale.7 For economic viability in industrial application, operating times of at least 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–80000 hours are considered necessary.5,9 Despite the fast advancement in this field, many groups report grand challenges regarding stability and performance durability on a significantly lower time scale. A decrease in CO2 reduction rate or even cell failure are supposedly linked to electrode flooding and precipitation of carbonate salts on the cathode side.10 Liquids and precipitated salts can block gaseous reactants from reaching active catalyst sites, and hinder products from leaving the catalyst layer. In the worst case precipitates can fully block the gas channels of the cathode flow field, leading to a pressure build-up and consequently to complete cell failure.11–13 The reported time scale for the occurrence of these effects ranges from minutes to days depending on material properties and operating conditions.
000–80000 hours are considered necessary.5,9 Despite the fast advancement in this field, many groups report grand challenges regarding stability and performance durability on a significantly lower time scale. A decrease in CO2 reduction rate or even cell failure are supposedly linked to electrode flooding and precipitation of carbonate salts on the cathode side.10 Liquids and precipitated salts can block gaseous reactants from reaching active catalyst sites, and hinder products from leaving the catalyst layer. In the worst case precipitates can fully block the gas channels of the cathode flow field, leading to a pressure build-up and consequently to complete cell failure.11–13 The reported time scale for the occurrence of these effects ranges from minutes to days depending on material properties and operating conditions.
Fig. 1a displays the key components and reactions occurring in a zero-gap CO2 electrolyzer with a gas-fed cathode producing CO. An anion exchange membrane divides the cathode and the anode compartment and enables the ionic charge transfer between the half-cells. The anode comprises an oxygen evolution catalyst and porous transport layers (see review by Vass et al.14). Usually, the anode is supplied with aqueous electrolyte, while gaseous CO2 is supplied to the cathode catalyst layer from the back side of the gas diffusion electrode. In the cathode, the CO2 is reduced at a catalyst, e.g. silver, under the consumption of water, producing CO and hydroxide ions as illustrated in Fig. 1b. Ideally, the hydroxide ions would migrate to the anode as a charge carrier and form water and oxygen. However, as CO2 is present in the cathode, the produced hydroxide ions tend to form bicarbonate ions (OH− + CO2 → HCO3−) and carbonate ions (HCO3− + OH− → CO32− + H2O) instead (Fig. 1c). As cations, typically K+ from the KOH electrolyte, migrate from the anode side (bi)carbonate salts can form on the cathode side (Fig. 1d). Thus, during operation, larger amounts of salts can accumulate in the cathode gas diffusion electrode (Fig. 1e). Especially at high reaction rates, i.e. high water consumption and high anion production, the solubility limit of the salts can be exceeded easily, which leads to the aforementioned accumulation of precipitates and blockage of the GDE pores.15,16
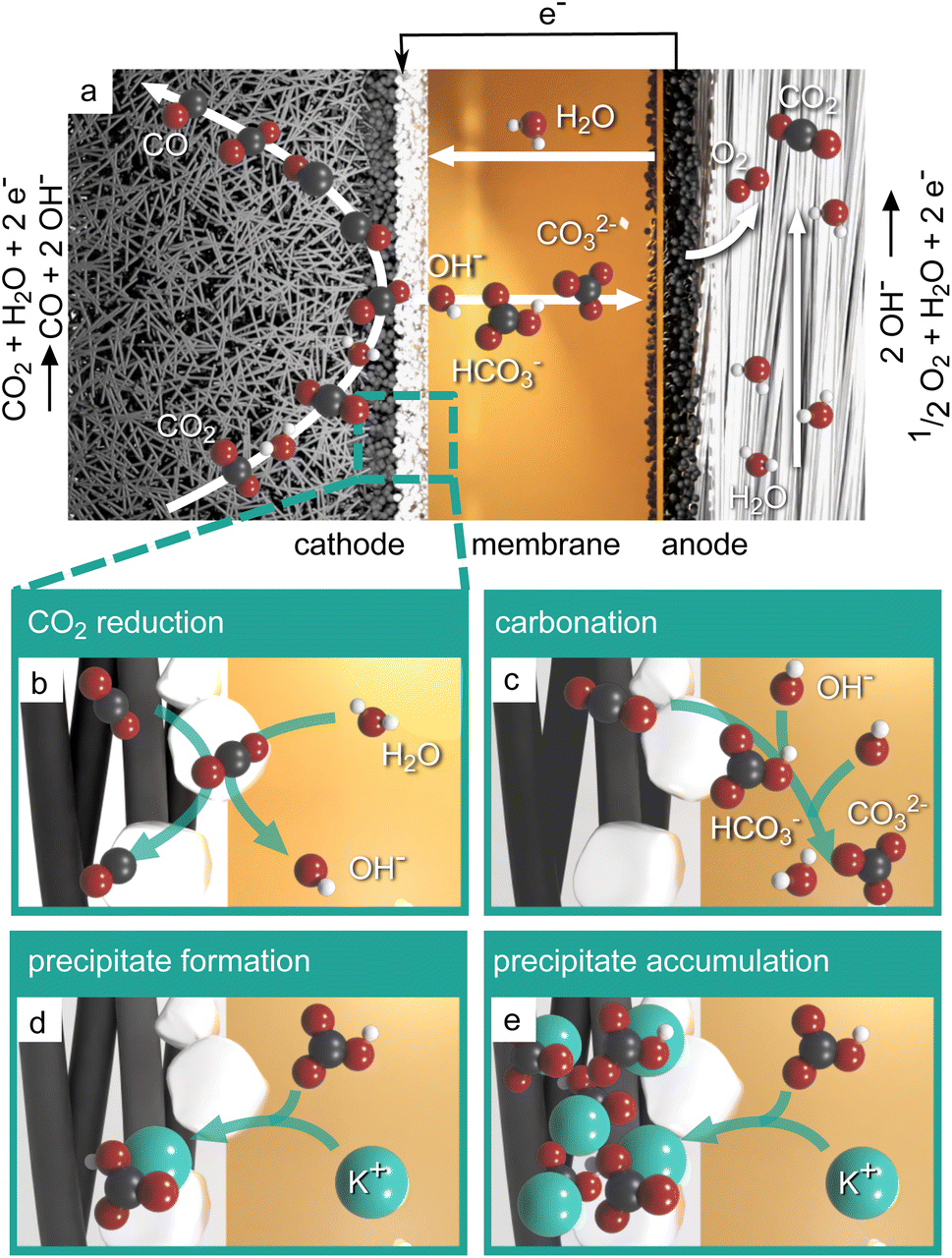 | ||
| Fig. 1 Salt precipitation process in zero-gap CO2 electrolysis. (a) Desired reactions and transport during CO2 reduction. (b) Reduction of CO2 producing CO and OH−. (c) Carbonation reaction: CO2 reacts with the produced hydroxide ions. (d) Precipitation of potassium (bi)carbonate. (e) Precipitates accumulating in the gas diffusion electrode blocking pores. All figures with oxygen (red), carbon (black), hydrogen (white) and potassium (petrol). | ||
(Bi)carbonate salts are generally porous and hydrophilic and thus increase the capillary pressure in the pores of the generally hydrophobic GDE.17 Flooding events and salt precipitation are thereby closely linked and are the major obstacle for constant operation at high current densities.18 As a consequence, strategies for the mitigation of salt precipitation and electrode flooding are urgently needed.12
This work aims at giving a comprehensive overview on current strategies and influence factors to mitigate salt precipitation in zero-gap CO2 electrolyzers with a gas-fed cathode. As depicted in Fig. 2, the strategies can generally be divided into two categories: (i) strategies that try to mitigate the salt precipitation by tuning material properties of the electrolysis cell components. This addresses especially the properties of the membrane and electrodes, but also the properties of the liquid electrolyte. (ii) Strategies that try to mitigate precipitation during cell operation. This entails all operating conditions, electrochemical operation modes as well as strategies to remove the salt precipitates.
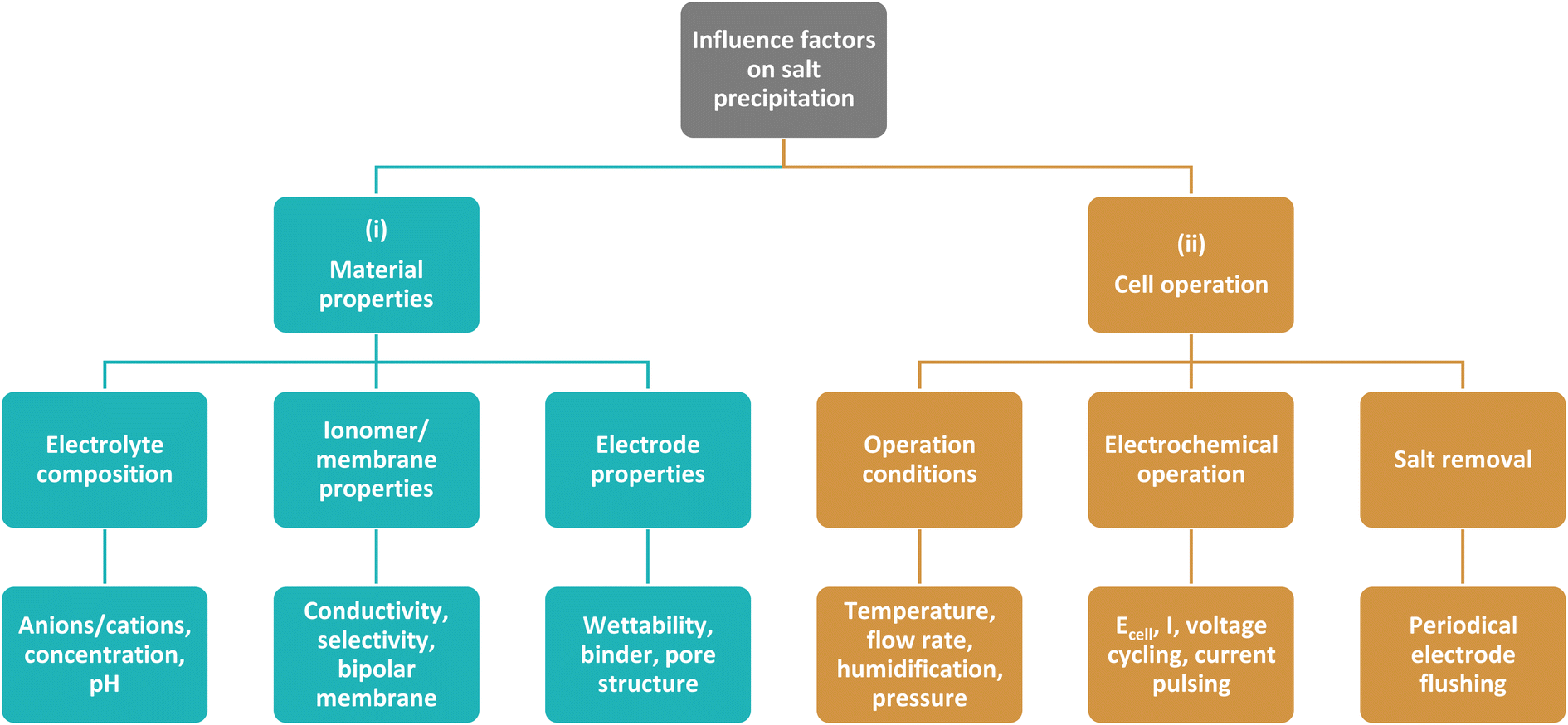 | ||
| Fig. 2 Influence factors and mitigation strategies for salt precipitation in zero-gap CO2 electrolyzers. Group (i) comprises different components and their material properties that have an influence on the salt precipitation behavior. Group (ii) includes all strategies and operating conditions that have an influence during cell operation. | ||
Influence of material properties on salt precipitation
Material properties of the components influence the salt precipitation and electrode flooding behavior in CO2 electrolyzers. The following subsections summarize the effects of electrolyte, membrane and electrode properties observed and discussed in literature.Electrolyte
In gas-fed zero-gap electrolyzers, the anode side is typically supplied with liquid electrolyte. The electrolyte composition defines the environment in the anode, i.e. pH and thus conductivity and catalyst activity. It has therefore significant influence on the energetic efficiency and cannot be modified independently. Furthermore, during operation electrolyte constituents cross the membrane and change the environment on the cathode side. Therefore, the composition of the anolyte can have a significant influence on the CO2 reduction rate and product selectivity.The employed electrolytes are typically aqueous solutions of alkali metal hydroxide, bicarbonate or carbonate salts. In general, salt precipitation occurs more rapidly with increasing electrolyte concentration, as the cation crossover from the anode side is also increased. Using pure water as electrolyte has been investigated.19,20 Although it effectively inhibits precipitation, employing pure water has strong disadvantages for the cathode and anode reactions and membrane conductivity. The neutral environment on the anode side leads to sluggish kinetics for the oxygen evolution reaction and requires scarce and expensive catalysts like iridium.13 Furthermore, the presence of alkali metal cations plays a vital role for the activity of the CO2 reduction reaction on Ag catalysts.21,22
Most works therefore choose the salt concentration either for optimal electrochemical performance or for durability, depending on the scope of the study. Most publications reporting long-term tests (>100 h) use low anolyte salt concentration between 0.01–0.1 M. However, it is reported that utilizing low salt concentrations only delays the formation of precipitates in the cathode and even with anolyte concentrations of 10 mM significant precipitation is observed after a few days, if no further measures are taken.8,23
In addition to electrolyte concentration, the electrolyte composition also influences the precipitation behavior. The most commonly investigated electrolytes consist of either OH−, HCO3− or CO32− as anions and either Li+, Na+, K+, Rb+ or Cs+ as cations. Table 1 lists the solubility and prices of different alkali metal salts. A high solubility can be advantageous, as it allows higher ion concentrations before salt crystallites form. The alkali metal hydroxide salts show comparably high solubility compared to the respective (bi)carbonate form. However, the solubility of the (bi)carbonate salts is more relevant for the formation of salt crystals in the cathode GDE, as the hydroxide ions produced in the cathode reactions react with the present CO2 forming bicarbonate and carbonate ions. (Bi)carbonate salts comprising Cs+ or Rb+ cations show the highest solubility but are rarely used due their high price (4–28 times higher compared to potassium carbonate). The shape and size of the formed salt crystallites can differ for different alkali metal salts. Fig. 3 shows scanning electron micrographs and elemental maps of the cathode catalyst layer after cell operation with different electrolytes reported by Cofell et al.24 They found that Cs+ containing electrolytes form smaller and more evenly distributed precipitates (bottom row) compared to cells using potassium containing electrolytes (top row), resulting in a slower decrease in CO2 reduction performance.24
 | ||
| Fig. 3 Precipitate distribution on a silver catalyst layer SEM-EDX maps of the cathode catalyst layer after operation in different electrolytes. Reprinted with permission from Cofell et al.24 Copyright © 2021 American Chemical Society. | ||
There are more possible electrolyte properties that could influence the salt precipitation behavior, which might not be investigated because other electrolyzer requirements render them impractical. For instance, multivalent cations might appear attractive due to potentially stronger membrane exclusion and thus less migration. On the other hand, they feature lower solubility and significantly reduced CO2 reduction rates compared to monovalent alkali metal cations.25 Furthermore, the utilization of precipitation inhibiting additives known from other applications like water treatment and oil-refining has been proposed.24 However, there are no studies yet showing successful employment.
Membrane properties
The membrane is a key component of CO2 electrolyzers, as it separates the reactant and product streams of anode and cathode while allowing the required ionic charge transfer between the half-cells. Anion exchange membranes (AEMs) are currently favored over cation exchange membranes for zero-gap electrolyzers, as the alkaline to neutral environment is favorable for CO2 reduction selectivity over the hydrogen evolution reaction.26 Generally desired characteristics of AEMs employed in CO2 electrolysis have been extensively discussed by Salvatore and Gabardo et al.27 In this work, the permselectivity, anion conductivity and water uptake are found to be the most important factors for salt precipitation.Endrődi et al. hypothesized that a high conductivity of the membrane is beneficial, as it decreases the ohmic losses due to membrane resistance and lowers the concentration gradient of (bi)carbonates, by enabling fast anion transport from the cathode to the anode.8,21 A study by Mardle et al. confirms this by comparing membranes with different thicknesses and ion exchange capacities and their influence on electrode flooding and salt precipitation (Fig. 4).12 They found that a cell using an Aemion™ membrane with higher IEC, thus higher anion conductivity, shows lower cell voltages and higher CO Faraday efficiencies, than a cell with a low IEC Aemion™ membrane (Fig. 3cvs.3e). Using a thicker membrane increases the membrane resistance and has consequently a similar effect as a lower IEC – higher cell voltage, lower carbonate crossover and faster decrease in CO selectivity.
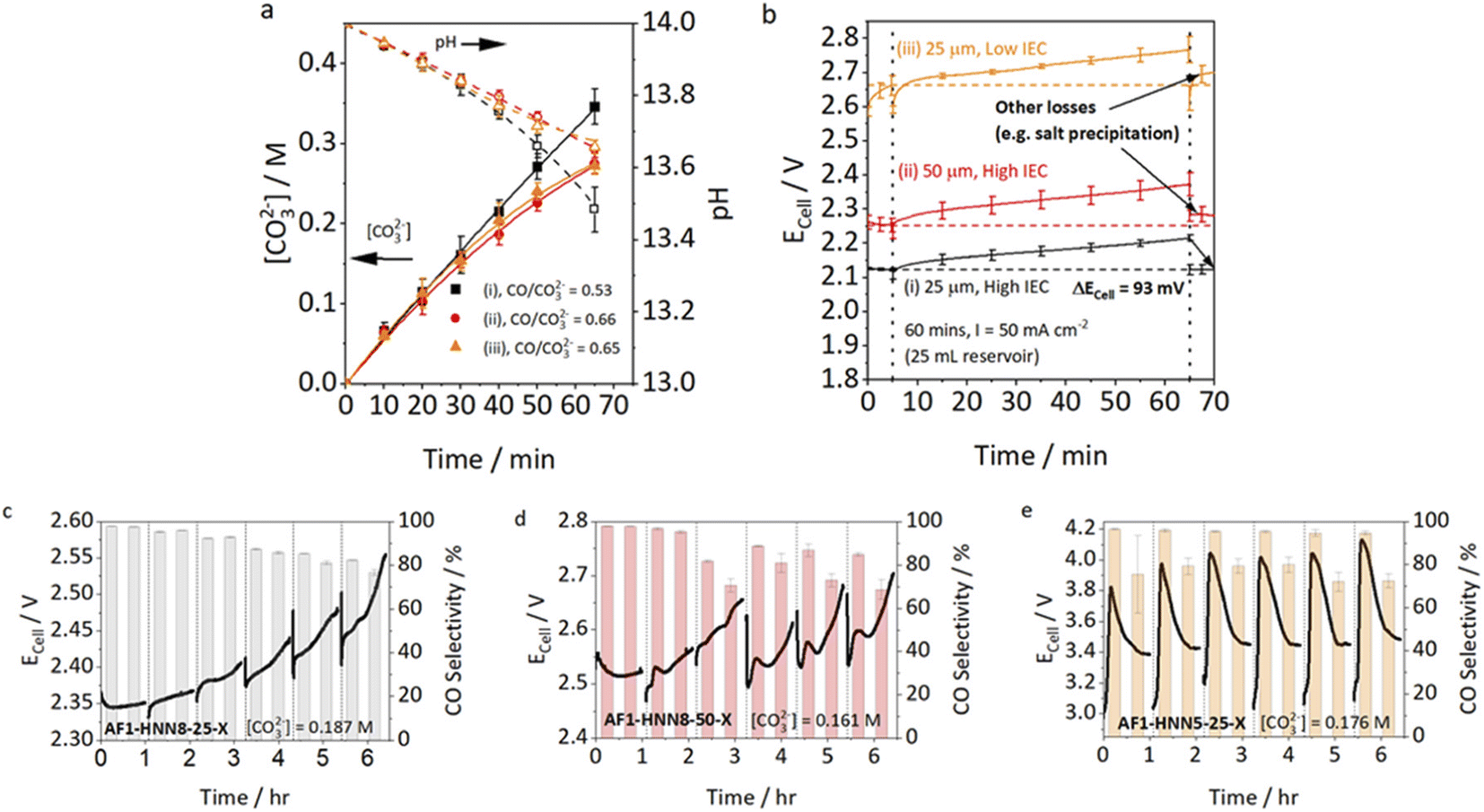 | ||
| Fig. 4 Cell voltage and CO selectivity for membranes with different thickness and IEC from Mardle et al.12 (a) [CO32−] and pH change for the three Aemion AEMS over the course of 65 min (60 min chronopotentiometry + 5 min EIS). AEMS were (i) AF1-HNN8-25-X (25 μm, high IEC), (ii) AF1-HNN8-50-X (50 μm, high IEC), and (iii) AF1-HNN5-25-X (25 μm, low IEC). (b) Average Ecell for the three Aemion membranes at 50 mA cm−2 for 5 min in single pass, 60 min in recirculating anolyte (volume = 25 mL), and 5 min purge in single pass. ΔEcell corresponds to the reversible voltage increase by anolyte carbonation. (c–e) Cell voltage during six 1 h periods of operation at i = 100 mA cm−2, with a water purge (2 mL) conducted every 1 h. For this experiment, a 500 mL reservoir of 1 M KOH was recirculated at 10 mL min−1. Reprinted with permission from Mardle et al.12 Copyright 2021 American Chemical Society. | ||
However, the ideal properties of a membrane remain a tradeoff between several requirements. An increase in ion exchange capacity typically leads to higher water uptake, which reduces the ionic resistance but also lowers permselectivity of the membrane.28 While a lower ionic resistance is desired, a lower permselectivity leads to an increased (unwanted) cation crossover from the anode to the cathode. Furthermore, the carbonate crossover from cathode to anode is generally undesired, as it limits the single-pass efficiency, raises the downstream separation costs and additionally neutralizes the anode electrolyte in case of employment of alkaline electrolytes.26,29 This demonstrates the complexity of opposing optimization interests for components in CO2 electrolysis.
An alternative to AEMs are bipolar membranes, i.e. combining an anion exchange layer (AEL) and a cation exchange layer (CEL). Bipolar membranes promise to solve multiple problems currently discussed in CO2 electrolysis.26 The CEL can mitigate CO2 crossover as carbonate ions are not conducted. Furthermore, a pH gradient can be established, providing ideal reaction environments to the anode and cathode simultaneously. Bipolar membranes can be used in two different operation modes; the so-called reverse bias and forward bias depending on the membrane orientation (see Fig. 5). Both modes show specific advantages and challenges. In reverse bias operation (CEL facing the cathode, Fig. 5a), water is split at the AEL/CEL interface. Hydroxide ions migrate to the anode and protons migrate to the cathode, providing an acidic reaction environment in the cathode.26 The high proton concentration at the cathode, however, promotes the HER and strongly reduces the selectivity towards CO2 reduction products.30 The overpotential for the water dissociation reaction at the AEL/CEL interface additionally increases the cell potential.
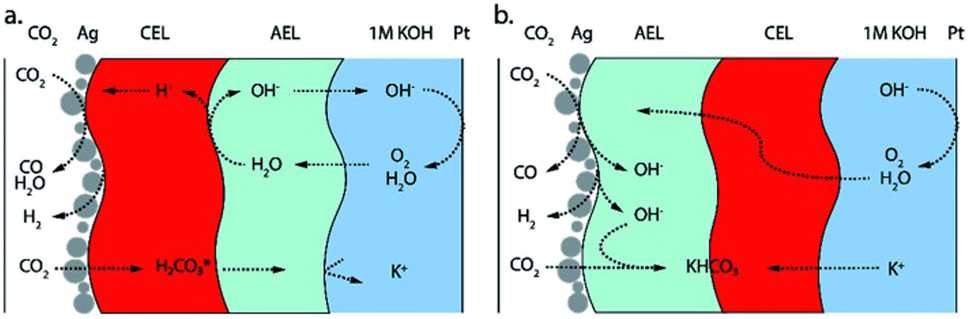 | ||
| Fig. 5 Orientation of bipolar membranes in CO2 electrolysis to CO. (a) Reverse bias orientation and (b) forward bias orientation. Adapted with permission from Blommaert et al.31 with permission from the Royal Society of Chemistry. | ||
In forward bias operation (AEL facing the cathode, Fig. 5b), anions from the cathode and protons from the anode recombine at the AEL/CEL interface and water is produced at the membrane interlayer. With KOH as anolyte the precipitation problem is being shifted to the AEL/CEL interface, as cations and (bi)carbonate ions recombine at the interface.31 Operation with pure water solves this issue by reducing the alkali metal cation concentration, but in turn entails sluggish kinetics and iridium catalysts as discussed above. While studies show that bipolar membranes can mitigate CO2 crossover and salt precipitation, they involve higher cell potentials and lower reaction rates than AEMs and have additional challenges like the delamination of both membrane layers.32 Thus, despite their huge potential, significant advances are needed in bipolar membrane development to pose a serious alternative to AEMs.
Finally, pure cation exchange membranes could solve the salt precipitation and carbonate crossover problem, but would require highly selective CO2 reduction catalysts in acidic environments.33
Electrode properties
In general, the electrode must enable the electric contact of the catalyst and has to facilitate mass transport of reactants and products towards and away from the catalysts active sites. For the application in low-temperature CO2 electrolysis carbon-based GDEs prevail (see recently published reviews about GDEs for CO2 electrolysis10,34,35). The base of the GDE typically consists of a so-called gas diffusion layer (GDL), comprising carbon fibers with diameters in the range of 5–10 μm. A second layer is deposited on top, the so-called micro porous layer (MPL) made from carbon black (spheres of 50–100 nm). The properties considered most relevant for the precipitation of carbonic salts are the electrode wettability and the pore structure of the GDE.17 Most works employ GDEs with a hydrophobic treatment or impregnation to mitigate electrode flooding. However, it was observed that the hydrophobicity of the GDE can decrease during cell operation.11,36 The decay of hydrophobicity is generally attributed to the hygroscopic precipitates or the decomposition of the hydrophobic chemical structure of the GDE in the alkaline environment.11,17Kong et al. compared the carbonate precipitation in various GDEs.36,37 They found that GDEs with cracks in the MPL showed non-uniform carbonate surface coverage but deeper penetration of the GDL. They hypothesize that a crack-free MPL hinders the carbonates from entering deeper areas of the GDL leading to early performance decrease, as (bi)carbonate salts accumulate in the catalyst layer and block the access to the catalysts active sites. This implies that a designed structure of the MPL could improve the removal of precipitates from the catalyst layer. The employed GDLs are typically developed and optimized for the application in fuel cells or water electrolyzers and might not have ideal properties for the application in CO2 electrolysis. Therefore, tailoring the GDL and MPL to the requirements of CO2 electrolyzers could potentially further improve the CO2 reduction performance and electrolyzer durability. Wu et al., for example, proposed to increase the hydrophobicity of the electrode to mitigate electrode flooding by adding a layer of PTFE particles to the back side of the MPL.38 The employment of PTFE-based GDL has also been investigated to mitigated electrode flooding.16
Besides the GDL and MPL, the composition of the cathode catalyst layer also plays an important role for the formation of precipitates. The binder significantly influences the transport properties in the catalyst layer. A hydrophilic anion-conductive binder could mitigate the accumulation of alkali metal cations at the catalyst surface by Donnan exclusion, provide water for the cathode reactions and decrease the (bi)carbonate concentration by enabling fast anion transport away from the catalyst surface. Adding hydrophobic components to the catalyst layer, for example PTFE particles, was also observed to have a positive effect on salt precipitation, potentially by mitigating early flooding of the catalyst layer.39
Furthermore, catalyst engineering or tuning of the cathode catalyst microenvironment could enable efficient pure water operation and thus mitigate the accumulation of alkali metal cations at the cathode. For instance, functional groups of the anion exchange polymers can compensate the co-catalytic effect of the alkali metal cations.40,41 Furthermore, Garg et al. reported improved performance and durability by functionalizing the Ag catalyst layer with urea (200 hours operation at 100 mA cm−2, faradaic efficiency of ∼85%, with 10 mM KHCO3 anolyte).23
Mitigation strategies during cell operation
Almost all operational parameters can potentially influence the salt precipitation and electrode flooding behavior during cell operation. The effect of those parameters, however, might drastically differ for the different experimental setups and cell materials employed (as discussed in the previous section “Influence of material properties on salt precipitation”). The following subchapters summarize the main findings.Gas humidification
The humidity of the CO2 feed can have a critical effect on cell performance, stability and durability.42–44 Cell operation without gas humidification can even lead to membrane dry-out and cell failure.8 Wheeler et al. presented a study on the influence of gas humidification by using a cell fixture with a window allowing to look at the back side of the cathode GDL during cell operation (Fig. 6).45 After 120 min of cell operation at 100 mA cm−2 without humidification, the cathode flow field is visibly flooded with precipitates, while the cell with a humidified CO2 gas stream operated at 150 mA cm−2 shows no precipitates in the flow field after the same time. As the cathode reactions consume water and the produced anions drag water from the cathode to the anode side via electroosmotic drag, a sufficient supply of water to the cathode has to be ensured. They found that a dry cathode feed increases the water flux from the anode to the cathode via diffusion, as water can only be supplied through the membrane. This water flux in turn increases the cation crossover and thus salt formation. | ||
| Fig. 6 Wheeler et al.45 humidification vs. no humidification. (a) Image of the measurement setup, (b) images of the back side of the cathode GDE at with and without humidification and at different current densities. Reprinted with permission from Wheeler et al.45 with permission from the Royal Society of Chemistry. | ||
The water balance, however, further depends on other factors like temperature, flow rate or material properties like membrane water-uptake. Therefore, one has to be careful when generalizing findings from a single study, as the utilized materials and operating conditions might strongly differ from setup to setup. Nonetheless, various studies conclude that cathode gas humidification generally delays the formation of salt crystallites.45,46
While controlled gas humidification can be seen as an important mitigation strategy, one has to consider that most studies use custom-built CO2 electrolysis test setups. Unlike commercial fuel cell test benches, the self-build test setups typically humidify the cathode gas inlet by bubbling the gas feed through a gas washing bottle filled with water. The relative humidity is then controlled by adjusting the temperature of the bubbler. As the gas line between bubbler and electrolysis cell is often not thermally isolated or heated, condensate formation could also play a role in the removal of salt precipitates, when condensate droplets are flushed through the cathode compartment.47
Another strategy to supply water to the cathode without an advanced gas humidifying unit, was investigated by De Mot et al. in an electrolyzer producing formic acid.48 They proposed the continuous injection of small amounts of liquid water (0.1–1 mL min−1) through an interdigitated cathode flow field. In their case, a minimum of 0.2 mL min−1 water injection was necessary to prevent salt crystallization in a potentiostatic measurement for 25 hours at 3 V. By this procedure, the water supply can be decoupled from the gas flow rate. However, it is not clear whether this strategy is applicable for zero-gap electrolyzers producing CO and for scale-up to higher active cell areas.
Gas flow rate
There has been little research examining the effect of CO2 flow rate on salt precipitation. However, some studies suggest that flow rate may have an impact on precipitation behavior. Verma et al., for example, reported that a low flow rate of 17 sccm led to blocked flow field channels, while they achieved to eliminate the issue by increasing the flow rate to 75 sccm.49Considering the effect of gas humidification as discussed above, the flow rate can have a significant influence on the water balance of the cell and thus on salt precipitation. It is to note, that depending on the relative humidity of the gas feed, a higher gas flow rate can either remove water from the cathode or provide more water to the cathode.
However, in industrial application, the flow rate will most likely be chosen with the goal to maximize the single pass efficiency and might therefore be an impractical option to manage the water balance.
Cell temperature
The operating temperature has an influence on many relevant properties like kinetics, ion conductivity, solubility, gas partial pressure, relative humidity and diffusivity. Zero-gap CO2 electrolyzers are often operated at low temperatures (<30 °C), as a reduced selectivity for CO was reported for elevated temperatures.19 Nonetheless, this observation is not universally valid, as high CO partial current densities were demonstrated in cells operating at temperatures of up to 60 °C.8 The CO2 partial pressure at the electrode immersed in a liquid electrolyte decreases with rising temperature, as the solubility of CO2 in water decreases.50 This generally leads to a temperature dependent decrease of CO2 reduction performance in H-cell measurements. The temperature dependence of gas solubility might as well affect the selectivity of gas-fed electrolysis cells that suffer from electrode flooding.Furthermore, the cell temperature has an effect on the formation of salt crystallites as the solubility of salts like KHCO3 and K2CO3 in water increases with rising temperature51 allowing higher ion concentrations before salts precipitate at the cathode. At the same time, the ion conductivity of the membrane increases significantly with rising temperatures. For instance, the HCO3− conductivity of Sustainion membranes approximately doubles from ∼8 mS cm−1 to ∼16 mS cm−1, when the temperature is increased from 30 °C to 60 °C.8 This allows faster anion transport to the anode side, reducing the anion concentration at the cathode.
Finally, there might also be negative effects of elevated temperatures on the salt precipitation. The evaporation of water is enhanced, if the gas feed is not sufficiently humidified, which again increases the water flux from anode to cathode. Despite the potentially drastic effects, there are no publications systematically investigating the effects of temperature on precipitate formation in zero-gap CO2 electrolysis.
Precipitate removal
Besides trying to prevent the formation of precipitates, several groups showed that the periodic removal of precipitates during cell operation can restore the CO2 reduction capacity.12,46,48,49,52 The most common approach is to rinse the cathode GDE by flushing water through the cathode compartment. Fig. 4c–e shows the constant current experiments conducted by Mardle et al. with three different membranes, while flushing the cathode compartment every hour with 2 mL of pure water.12Fig. 4e illustrates how the CO selectivity is partially recovered by the flushing procedure in a cell using a low IEC membrane. In the cell utilizing a membrane with high IEC, the decrease in FE is less significant (see Fig. 4c) and therefore also the influence of the flushing procedure.Leonard et al. found that the hydrophobicity of the employed GDLs, can reduce the efficiency of the flushing procedure, as the water cannot easily penetrate the pores from the back side of the GDL, leading to some carbonates remaining in the catalyst layer.52 They therefore rinsed and vacuum dried the cathode after dissembling the cell, showing higher recovery rates of the CO2 reduction capacity. As periodic disassembly is not applicable beyond lab-scale, one possible way to improve the cleaning procedure is to use other solvents or solvent mixtures with better wettability of the GDLs.21 One has to note, that the choice of solvent is restricted by the ionomer and membrane compatibility. Eventually, although it was shown that it is possible to restore the CO2 reduction capacity of the cathode by rinsing off the carbonates, the practicability of this approach has not yet been proven in long-term experiments of several hundred hours or with high cell areas.
Subramanian et al. investigated different flow field designs in terms of failure resistance due flow channel blockage by salt precipitates and found a serpentine flow channel design superior to parallel or interdigitated flow fields.53 They primarily attribute this to a higher driving pressure in the system, which allows for more dispersed in-plane transport of CO2 through the GDE.53 The flow field design might also be important for the removal of the precipitates by cathode flushing. In multi-channel designs, for example parallel or multi-channel serpentine, the flushing solution might not easily reach blocked regions in the cell, as it can bypass through channels that are not fully blocked.
Influence of electrochemical operating conditions
The employed electrochemical methods and experiment settings for the evaluation of CO2 electrolyzer performance strongly differ, as there are no standardized measurement protocols so far. Nwabara et al. made a proposition for accelerated durability test protocols, similar to standardized testing protocols that are well established for other electrochemical applications.54 Up to now, the stability and durability is most commonly evaluated by conducting potentiostatic or galvanostatic measurements. The duration of the measurements varies significantly between different studies. In some studies the measurement duration seems to be chosen arbitrarily or failure modes are not indicated. Begin of test (BoT) and end of test (EoT) characterization, e.g. electrochemical impedance spectroscopy, and the utilization of suitable figures of merits to characterize the voltage stability (e.g. in V h−1), the degradation rate of the energy efficiency and of the product selectivity (e.g. in FE% h−1) would increase the comparability of different studies.A relation between precipitation and cell potential or applied current has been observed.45,47 At higher currents, problems with salt precipitation generally appear earlier. Leonard et al. uncovered that in their setup the decay of the CO Faraday efficiency correlates to the amount of transferred charge independent from the applied current density.52 This finding is in line with the theoretical understanding that the salt generation rate directly correlates with current density, and thus the accumulation of salt precipitates with electrical charge. Consequently, salt precipitation is dependent of the product of current and operation time.
Cell operation at constant potentials or at constant currents might further not accurately emulate realistic electrolyzer operation using intermittent energy sources,55 nor be the most suitable operation mode in terms of stability and durability. Some studies suggest non-constant electrochemical operation. Fig. 7 illustrates an operation strategy proposed by Xu et al.16 Instead of applying a constant potential they suggest voltage pulsing (see Fig. 7c). In this operation mode, one cycle is split into two phases at different potentials. In the normal operation or CO2 reduction phase at a potential well above the onset potential, the concentration of HCO3−/CO32− at the cathode increases (see Fig. 7b). Before the solubility limit is exceeded and salts can precipitate, the voltage is decreased and the second phase or regeneration phase is initiated. In this phase without reduction of CO2 in the cathode, the anion concentration at the cathode decreases as they migrate to the anode (Fig. 7d). In this work, Xu et al. demonstrated that alternating between 60 s at 3.6 V and 30 s at 2.0 V successfully prevented salt precipitation and the associated decay in Faraday efficiency for 18 h of CO2 to CO reduction. For an electrolyzer setup producing C2-products a ∼5 times longer cell operation compared to potentiostatic cell operation was demonstrated, only increasing the energy demand by less than 1% and fully mitigating the formation of precipitates in the cathode flow field channels.16
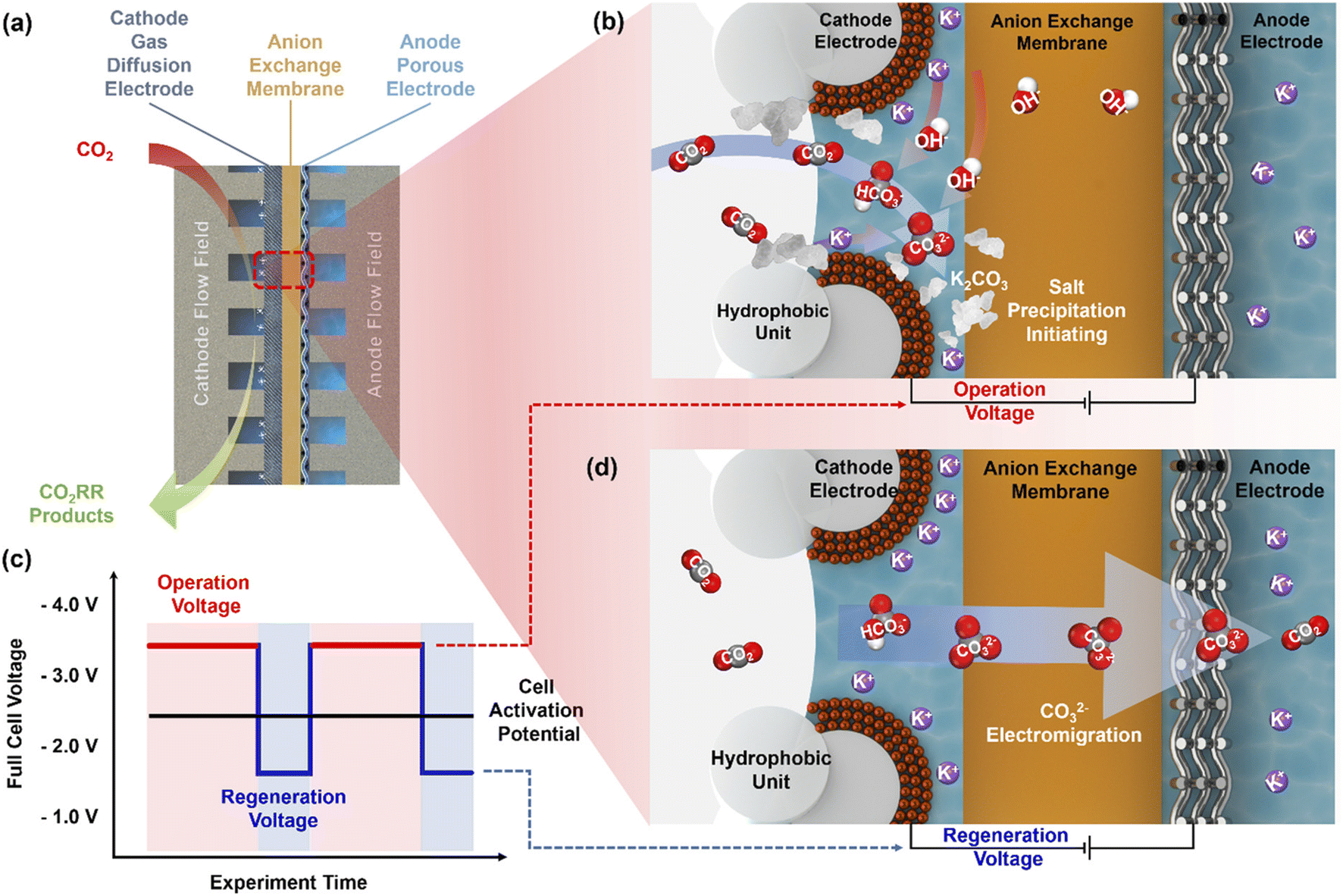 | ||
| Fig. 7 Carbonate formation in MEA CO2 electrolyzers and the self-cleaning CO2 reduction strategy. (a) Schematic of the MEA CO2 electrolyzer. (b) CO2 conversion to bicarbonate and carbonate during regular electrolyzer operation. (c) Strategy to mitigate carbonate formation by cycling between operational and regeneration cell voltages. (d) Carbonate migration during cell operation at the regeneration voltage. Reprinted with permission from Xu et al.16 Copyright 2021 American Chemical Society. | ||
A similar approach could be chosen in galvanostatic cell operation, by alternating between low and high current densities. Samu et al.55 conducted a measurement with an alternating current profile with currents between 312.5 mA cm−2 and 437.5 mA cm−2 to emulate cell operation with a dynamic power load. They demonstrated stable cell performance over the duration of 500 hours.55 Considering this, the operation of zero-gap CO2 electrolyzers coupled with the dynamic power load from intermittent power sources like solar energy could even be beneficial for the electrolyzer operation in terms of salt precipitation and durability.
Discussion and comparison of strategies
In the previous chapters, the individual strategies and factors on salt precipitation have been summarized and discussed. It has to be considered that there are no standardized methods for the characterization of CO2 electrolyzers yet, which make a direct comparison of the measurements impossible. Additionally, many groups do not yet conduct durability tests, probably due to limited test automation, limited testing capacities, different scope or rapid performance decrease. The choice of measurement duration therefore often appears to be arbitrary and in many reports, the measurements are ended before cell failure or without mentioning the failure mode.14,34Despite the lack of comprehensive data, general trends can be derived. Fig. 8 shows the average CO2 partial current density (a), the anolyte concentration (b) and the average cell voltage (c) in relation to the approximate amount of CO2 that has been converted during the experiments of different works5,8,12,16,19–21,23,41,46,54–59 normalized to the active cell area (also see Table 2). The amount of converted CO2 was chosen, as it can be seen as a direct measure for stable performance without too much salt precipitation. Only measurements were included that produced CO in a two-compartment cell with a zero-gap cell architecture, a gas-fed cathode and that cumulatively converted a significant amount of CO2 (>0.001 kgCO2 cm−2).
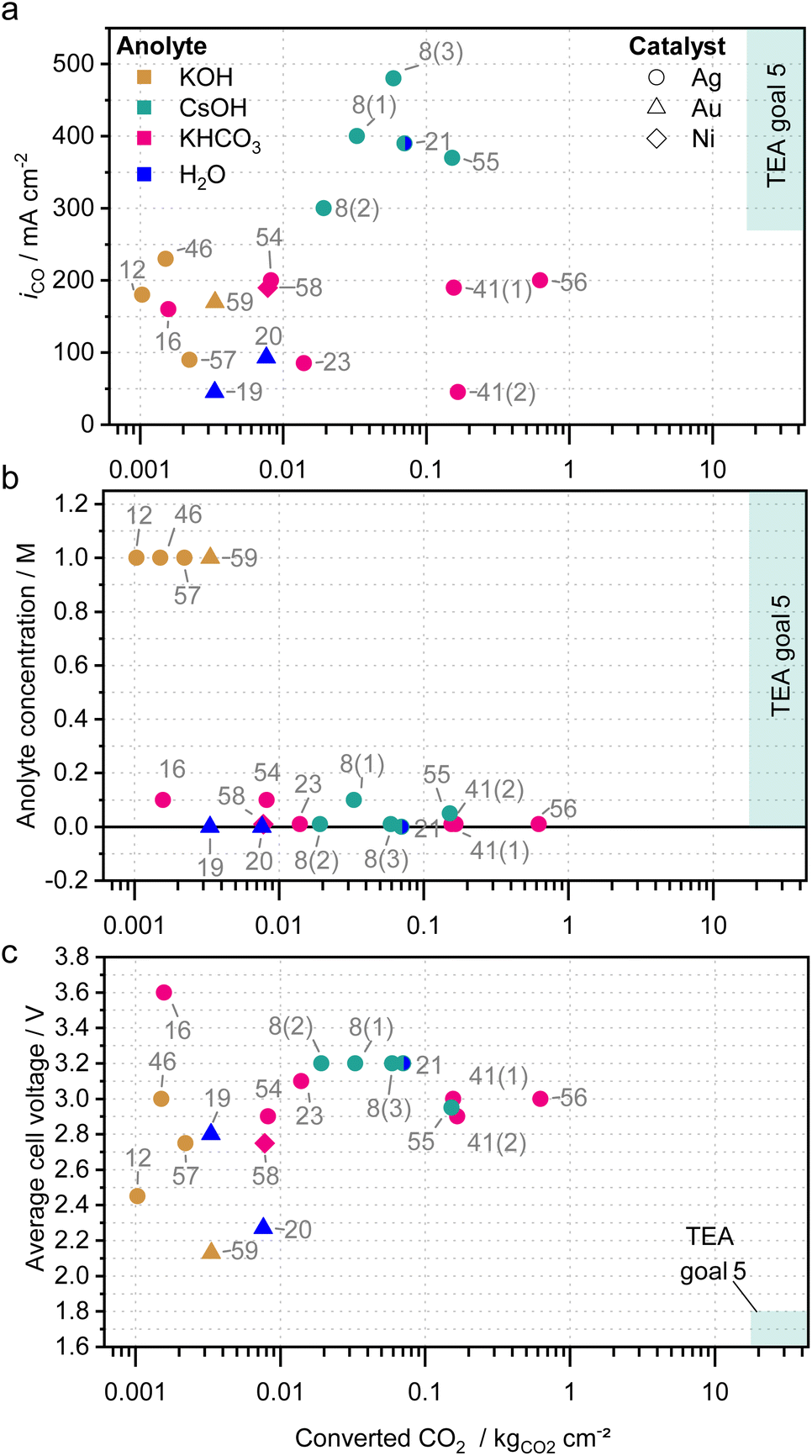 | ||
| Fig. 8 Reported long-term performance tests of zero-gap CO2 electrolyzers producing CO. (a) CO partial current density iCO, (b) anolyte concentration and (c) the average cell voltage plotted against the total amount of converted CO2 normalized to the active cell area. The converted CO2 has been derived by the duration of the measurement and approximate average partial current densities for CO. Symbols indicate the catalysts, colors indicate the anolyte. The annotations represent the literature reference. Different measurement of one publication are numbered in brackets. Only experiments comprising a zero-gap cell architecture and a significant area specific CO2 conversion (>0.001 kgCO2 cm−2) are considered in the graphs. More details to the references can be found in Table 2.5,8,12,16,19–21,23,41,46,54–59 | ||
| Reference | Cathode catalyst | Temp./°C | Electrolyte composition | Average cell voltage/V | Average iCO/mA cm−2 | Measurement duration/h | Converted CO2/kgCO2 cm−2 | Mitigation strategy (comments) |
|---|---|---|---|---|---|---|---|---|
| Sargent5 | — | — | — | 1.8 | 270 | 80000 |
17.7 | Goals from TEA |
| Janáky8 (1) | Ag | 60 | 0.1 M CsOH | 3.2 | 400 | 100 | 0.033 | Electrolyte, membrane (100 cm2 cell) |
| Janáky8 (2) | Ag | 60 | 0.01 CsOH | 3.2 | 300 | 78 | 0.019 | No humidification (8 cm2 cell) |
| Janáky8 (3) | Ag | 60 | 0.01 CsOH | 3.2 | 480 | 150 | 0.059 | With humidification (8 cm2 cell) |
| Holdcroft12 | Ag | 25 | 1 M KOH | 2.45 | 180 | 7 | 0.001 | Rinsing |
| Sargent16 | Ag | 0.1 M KHCO3 | 3.6 | 160 | 12 | 0.002 | Voltage cycling | |
| Wu19 | Au/CN | 25 | H2O | 2.8 | 45 | 90 | 0.003 | Pure water |
| Zhuang20 | Au/C | 50 | H20 | 2.27 | 92.5 | 100 | 0.008 | Pure water |
| Janáky21 | Ag | 60 | H2O/CsOH | 3.2 | 390 | 220 | 0.070 | Electrolyte/operation |
| Rufford23 | Ag | 25 | 0.01 M KHCO3 | 3.1 | 85 | 200 | 0.014 | Electrolyte |
| Masel41 (1) | Ag | 25 | 0.01 M KHCO3 | 3 | 190 | 1000 | 0.156 | Electrolyte |
| Masel41 (2) | Ag | 25 | 0.01 M KHCO3 | 2.9 | 45 | 4500 | 0.166 | Electrolyte |
| Janáky46 | Ag | 60 | 1 M KOH | 3 | 230 | 8 | 0.002 | Rinsing |
| Kenis54 | Ag | 25 | 0.1 M KHCO3 | 2.9 | 200 | 50 | 0.008 | |
| Janáky55 | Ag | 60 | 0.05 M CsHCO3 | 2.95 | 370 | 500 | 0.152 | Electrolyte, current pulsing |
| Masel56 | Ag | 25 | 0.01 M KHCO3 | 3 | 200 | 3800 | 0.624 | Electrolyte |
| Strasser57 | Ag-coral | 25 | 1 M KOH | 2.75 | 90 | 30 | 0.002 | |
| Wu58 | Ni-SAC | 25 | 0.01 M KHCO3 | 2.75 | 190 | 50 | 0.008 | Electrolyte |
| Lee59 | AuAg | 1 M KOH | 2.13 | 170 | 24 | 0.003 |
The majority of the studies employ silver-based catalysts (circle symbol), while only few use Au-catalysts (triangle) or Ni-catalyst (diamond) and meet the criteria for Fig. 8. The highest CO partial current densities during long-term testing have been achieved with CsOH-based electrolytes (green symbols in Fig. 8a), which is in line with the general trend of CO2 reduction activity in the presence of alkali metal cations (Li+ < Na+ < K+ < Rb+ < Cs+).22,60 The CsOH studies are also the only ones to meet the goals derived from a techno-economic analysis (TEA) with CO partial current densities above 270 mA cm−2.5
The highest area specific amount of converted CO2 was, however, demonstrated by Liu et al. (ref. 56) with 10 mM KHCO3 solution as anolyte and a Sustainion-based cell operating at room temperature and 200 mA cm−2. Measurement 41(1) and 41(2), both from the same publication by Kutz et al.,41 show that similarly high cumulative CO2 conversion can be achieved, both at relatively high (200 mA cm−2 for 1000 hours) and low (50 mA cm−2 for 4500 hours) current densities. It can further be concluded that CsOH and KHCO3 appear to be well suited for high conversion amounts, while all KOH-based studies showed only low amounts.
Despite these low amounts, electrolyzer performance is typically evaluated using KOH as anolyte at concentrations well above 0.1 M.14 Although the electrochemical cell performance might be better under those conditions for a short period, the formation of salt precipitates is accelerated. Therefore, the CO2 conversion capacity is limited in those systems and only few studies were found with a cumulative CO2 conversion above 0.001 kgCO2 cm−2 (e.g., ref. 12, 46, 57 and 59). Fig. 8b shows that a low electrolyte concentration is an effective strategy to delay salt precipitation and to enable longer measurements. In fact, all measurements with a total CO2 conversion above 0.003 kgCO2 cm−2 used anolyte concentrations below 0.1 M.
For the efficiency or cell voltage, no direct correlation to the amount of converted CO2 can be derived with cell voltages ranging from 2.1 V to 3.6 V (Fig. 8c). However, it can be observed that cells optimized for high efficiency and low voltage typically only convert low amounts of CO2, while those converting high amounts of CO2 show worse efficiency or higher voltage. As discussed before this is most likely linked to the electrolyte, which is either very conductive, but also prone to salt precipitation or vice versa.
The studies suggesting alternating operation as discussed in section “Influence of electrochemical operating conditions” can also be found in this chart. Samu et al. (ref. 55), who emulated dynamic power load, is among the highest area specific amounts of converted CO2, while also showing twice as high partial current densities. Xu et al. (ref. 16), who showed promising results using a regeneration phase below the onset potential, reaches rather small amounts of converted CO2. To show the effectiveness of this approach longer duration would be very beneficial.
Endrődi et al. (ref. 21) is unique as they combine multiple mitigation strategies at once with precise gas humidification, 60 °C, pure water as anolyte, periodical cathode rinsing, cathode activation with Cs+ cations and a membrane with high carbonate conductivity.21 To make use of the co-catalytic properties of Cs+, despite the utilization of pure water as electrolyte, 5 mL of 1 M CsOH in a mixture of 1:3 isopropanol/water are flushed through the cathode compartment every 12 hours. At the same time, it removes precipitates that might have formed in the GDE or cathode flow field. Consequently, it could be theorized that these measures allowed achieving high efficiency without compromising the amount of converted CO2.
Kibria et al. identified the operation of an electrolyzer for 80000 hours at 300 mA cm−2 and a FE of 90% as techno-economically required.5 That translates to an area specific CO2 conversion of approximately 17.7 kgCO2 cm−2 (ref. 5 in Fig. 8). The highest area specific CO2 conversion in Fig. 8 (ref. 56) counts 0.62 kgCO2 cm−2. This value lies over one order of magnitude below the requirements from the techno-economic analysis, emphasizing the need for improvement.
Table 3 summarizes the advantages and disadvantages of all mitigation strategies, considering the effectiveness, the technological readiness, the potential influences on cell operation and operational expenditures (OpEx), effects on capital expenditures (CapEx) and scalability. Some of these approaches help to reduce or delay the formation of precipitates, but might have to be complemented with other measures. Most of the strategies have only been proven on lab-scale. While the composition and concentration of the liquid anolyte can be easily changed also for larger cells or for cell stacks, the scale up is more challenging for other strategies. Cathode flushing might work well in a single cell with a serpentine flow field. On stack level, however, partially blocked channels can lead to significantly changed flow resistances between individual cells of the stack. This might lead to an imbalance between the individual cells. The freedom to adjust the operating condition to reduce salt precipitation might also be limited on stack level. Adjusting the amount of water provided by the cathode gas feed via variation of the flow rate or gas humidification, can conflict with the optimization of the single-pass efficiency or be limited by the gas supply's ability to hold water. Material-property-based approaches can most probably also be used in lager cells or on stack level, if the fabrication methods of those materials can be scaled. However, the scalability has to be assessed for each material development individually.
| Mitigation strategy | Effectiveness | Readiness | Effect on OpEx | Effect on CapEx | Scalability |
|---|---|---|---|---|---|
| Low concentrated anolyte/pure water | Effective | Ready | Lowers voltage efficiency | Components have to be adapted | Easily scalable |
| Periodic cathode flushing | No mitigation but effective recovery | Ready on single cell level | Down time/potential electrode flooding | Low/none | Effectiveness of flushing might decrease with cell area |
| Voltage/current cycling | Unclear | More research necessary | Partial down time | Requires larger cell area | Scalable |
| Operating conditions | Insufficient/additional strategies needed | Ready | Competing with performance optimization | Low | Scalable/unclear |
| Cell components | Insufficient/additional strategies needed/unclear | Ready/more research necessary | Competing with performance optimization | Unclear | Scalable |
Conclusions
Salt precipitation in alkaline zero-gap CO2 electrolyzers is still a major hurdle to long-term operation and thus for industrial commercialization. Many different factors influence the formation of salt precipitates in zero-gap CO2 electrolyzers, and the comparison of mitigation strategies is challenging, as used materials and measurement conditions vary significantly between the studies. Consequently, no strategy has emerged as a simple standalone solution to this problem up to now. While most strategies try to keep the cation or anion concentration below the nucleation concentration, other strategies, like electrode rinsing, accept the formation of precipitates and try to remove them periodically. The studies with the highest area specific CO2 conversion employed both, material related and operation related strategies to enable long-term operation. However, all studies with a cumulative CO2 conversion above 0.003 kgCO2 cm−2 had in common, that they used low anolyte concentrations (≤0.1 M). Therefore, the anolyte concentration might be considered the most important factor.Advancements in material development still have a great potential to promote new solutions to mitigate salt precipitation. For instance, while the technological maturity of bipolar membranes is substantially lower than of AEMs for CO2 electrolysis, they show high potential to solve several issues like salt precipitation and carbonate crossover at the same time. Furthermore, the development of CO2 reduction catalysts that enable operation under acidic conditions could allow pure water operation and the employment of cation exchange membranes.60 In this case, the lack of alkali metal cations would mitigate salt precipitation. As proton conductivity of state-of-the-art cation exchange membranes is significantly higher than the carbonate conductivity of state-of-the-art AEMs, their employment could significantly lower the overpotentials related to ion transport thereby increase the energy efficiency.
Finally, another key finding is the potential trade-off between optimizing for low salt precipitation and high performance. This trade-off could be solved when techno-economic goals are included, i.e. quantifying the cost of efficiency or durability. Yet, the comparison with a recent techno-economic study shows that the durability of electrolyzers needs to be at least one order of magnitude higher than in current experiments. Therefore, it is essential to further understand the underlying processes and develop further mitigation strategies.
Author contributions
S. V. acquired the funding and supervised this work. J. D. mainly conceptualized and wrote the original draft. L. B., L. M. and S. V. all reviewed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the funding from the Vector foundation (CO2-to-X).References
- S. Liang, N. Altaf, L. Huang, Y. Gao and Q. Wang, Electrolytic cell design for electrochemical CO2 reduction, J. CO2 Util., 2020, 35, 90–105, DOI:10.1016/j.jcou.2019.09.007.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces, Energy Environ. Sci., 2012, 5, 7050, 10.1039/c2ee21234j.
- T. Burdyny and W. A. Smith, CO2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions, Energy Environ. Sci., 2019, 12, 1442–1453, 10.1039/c8ee03134g.
- X. She, Y. Wang, H. Xu, S. C. E. Tsang and S. P. Lau, Challenges and Opportunities of Electrocatalytic CO2 Reduction to Chemicals and Fuels, Angew. Chem., Int. Ed., 2022, 61, e202211396, DOI:10.1002/anie.202211396.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C.-T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Electrochemical CO2 Reduction into Chemical Feedstocks, Adv. Mater., 2019, 31, e1807166, DOI:10.1002/adma.201807166.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Towards membrane-electrode assembly systems for CO2 reduction: a modeling study, Energy Environ. Sci., 2019, 12, 1950–1968, 10.1039/c9ee00909d.
- R. I. Masel, Z. Liu, H. Yang, J. J. Kaczur, D. Carrillo, S. Ren, D. Salvatore and C. P. Berlinguette, An industrial perspective on catalysts for low-temperature CO2 electrolysis, Nat. Nanotechnol., 2021, 16, 118–128, DOI:10.1038/s41565-020-00823-x.
- B. Endrődi, E. Kecsenovity, A. Samu, T. Halmágyi, S. Rojas-Carbonell, L. Wang, Y. Yan and C. Janáky, High carbonate ion conductance of a robust PiperION membrane allows industrial current density and conversion in a zero-gap carbon dioxide electrolyzer cell, Energy Environ. Sci., 2020, 13, 4098–4105, 10.1039/D0EE02589E.
- I. E. L. Stephens, K. Chan, A. Bagger, S. W. Boettcher, J. Bonin, E. Boutin, A. Buckley, R. Buonsanti, E. Cave, X. Chang, S. W. Chee, A. H. M. da Silva, P. de Luna, O. Einsle, B. Endrődi, M. E. Escribano, J. V. Ferreira de Araujo, M. C. Figueiredo, C. Hahn, K. U. Hansen, S. Haussener, S. Hunegnaw, Z. Huo, Y. J. Hwang, C. Janáky, B. S. Jayathilake, F. Jiao, Z. P. Jovanov, P. Karimi, M. T. M. Koper, K. Kuhl, W. H. Lee, Z. Liang, X. Liu, S. Ma, M. Ma, H.-S. Oh, M. Robert, B. R. Cuenya, J. Rossmeisl, C. Roy, M. P. Ryan, E. H. Sargent, P. Sebastián-Pascual, B. Seger, L. Steier, P. Strasser, A. S. Varela, R. E. Vos, X. Wang, B. Xu, H. Yadegari and Y. Zhou, 2022 Roadmap on Low Temperature Electrochemical CO2 Reduction, J. Phys.: Energy, 2022, 4, 42003, DOI:10.1088/2515-7655/ac7823.
- G. Li, T. Yan, X. Chen, H. Liu, S. Zhang and X. Ma, Electrode Engineering for Electrochemical CO2 Reduction, Energy Fuels, 2022, 36, 4234–4249, DOI:10.1021/acs.energyfuels.2c00271.
- M. de Jesus Gálvez-Vázquez, P. Moreno-García, H. Xu, Y. Hou, H. Hu, I. Z. Montiel, A. V. Rudnev, S. Alinejad, V. Grozovski, B. J. Wiley, M. Arenz and P. Broekmann, Environment Matters, ACS Catal., 2020, 10, 13096–13108, DOI:10.1021/acscatal.0c03609.
- P. Mardle, S. Cassegrain, F. Habibzadeh, Z. Shi and S. Holdcroft, Carbonate Ion Crossover in Zero-Gap, KOH Anolyte CO2 Electrolysis, J. Phys. Chem. C, 2021, 125, 25446–25454, DOI:10.1021/acs.jpcc.1c08430.
- L. Giordano, B. Han, M. Risch, W. T. Hong, R. R. Rao, K. A. Stoerzinger and Y. Shao-Horn, pH dependence of OER activity of oxides, Catal. Today, 2016, 262, 2–10, DOI:10.1016/j.cattod.2015.10.006.
- Á. Vass, A. Kormányos, Z. Kószó, B. Endrődi and C. Janáky, Anode Catalysts in CO2 Electrolysis, ACS Catal., 2022, 12, 1037–1051, DOI:10.1021/acscatal.1c04978.
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, Advances and challenges in electrochemical CO2 reduction processes, J. Mater. Chem. A, 2020, 8, 1511–1544, 10.1039/C9TA13298H.
- Y. Xu, J. P. Edwards, S. Liu, R. K. Miao, J. E. Huang, C. M. Gabardo, C. P. O'Brien, J. Li, E. H. Sargent and D. Sinton, Self-Cleaning CO2 Reduction Systems, ACS Energy Lett., 2021, 6, 809–815, DOI:10.1021/acsenergylett.0c02401.
- M. Li, M. N. Idros, Y. Wu, T. Burdyny, S. Garg, X. S. Zhao, G. Wang and T. E. Rufford, The role of electrode wettability in electrochemical reduction of carbon dioxide, J. Mater. Chem. A, 2021, 9, 19369–19409, 10.1039/d1ta03636j.
- S. Park, D. T. Wijaya, J. Na and C. W. Lee, Towards the Large-Scale Electrochemical Reduction of Carbon Dioxide, Catalysts, 2021, 11, 253, DOI:10.3390/catal11020253.
- Y. Gu, J. Wei, L. Wang and X. Wu, Insights into Highly-Efficient CO2 Electroreduction to CO on Supported Gold Nanoparticles in an Alkaline Gas-Phase Electrolyzer, J. Electrochem. Soc., 2022, 169, 54513, DOI:10.1149/1945-7111/ac6c50.
- Z. Yin, H. Peng, X. Wei, H. Zhou, J. Gong, M. Huai, L. Xiao, G. Wang, J. Lu and L. Zhuang, An alkaline polymer electrolyte CO2 electrolyzer operated with pure water, Energy Environ. Sci., 2019, 12, 2455–2462, 10.1039/c9ee01204d.
- B. Endrődi, A. Samu, E. Kecsenovity, T. Halmágyi, D. Sebők and C. Janáky, Operando cathode activation with alkali metal cations for high current density operation of water-fed zero-gap carbon dioxide electrolyzers, Nat. Energy, 2021, 6, 439–448, DOI:10.1038/s41560-021-00813-w.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Understanding cation effects in electrochemical CO2 reduction, Energy Environ. Sci., 2019, 12, 3001–3014, 10.1039/C9EE01341E.
- S. Garg, M. Li, T. Hussain, M. N. Idros, Y. Wu, X. S. Zhao, G. G. X. Wang and T. E. Rufford, Urea-Functionalized Silver Catalyst toward Efficient and Robust CO2 Electrolysis with Relieved Reliance on Alkali Cations, ACS Appl. Mater. Interfaces, 2022, 14, 35504–35512, DOI:10.1021/acsami.2c05918.
- E. R. Cofell, U. O. Nwabara, S. S. Bhargava, D. E. Henckel and P. J. A. Kenis, Investigation of Electrolyte-Dependent Carbonate Formation on Gas Diffusion Electrodes for CO2 Electrolysis, ACS Appl. Mater. Interfaces, 2021, 13, 15132–15142, DOI:10.1021/acsami.0c21997.
- S. S. Bhargava, E. R. Cofell, P. Chumble, D. Azmoodeh, S. Someshwar and P. J. A. Kenis, Exploring multivalent cations-based electrolytes for CO2 electroreduction, Electrochim. Acta, 2021, 394, 139055, DOI:10.1016/j.electacta.2021.139055.
- M. A. Blommaert, S. Subramanian, K. Yang, W. A. Smith and D. A. Vermaas, High Indirect Energy Consumption in AEM-Based CO2 Electrolyzers Demonstrates the Potential of Bipolar Membranes, ACS Appl. Mater. Interfaces, 2022, 14, 557–563, DOI:10.1021/acsami.1c16513.
- D. A. Salvatore, C. M. Gabardo, A. Reyes, C. P. O'Brien, S. Holdcroft, P. Pintauro, B. Bahar, M. Hickner, C. Bae, D. Sinton, E. H. Sargent and C. P. Berlinguette, Designing anion exchange membranes for CO2 electrolysers, Nat. Energy, 2021, 6, 339–348, DOI:10.1038/s41560-020-00761-x.
- G. M. Geise, M. A. Hickner and B. E. Logan, Ionic resistance and permselectivity tradeoffs in anion exchange membranes, ACS Appl. Mater. Interfaces, 2013, 5, 10294–10301, DOI:10.1021/am403207w.
- E. Jeng and F. Jiao, Investigation of CO2 Single-Pass Conversion in a Flow Electrolyzer, React. Chem. Eng., 2020, 5, 1768–1775, 10.1039/D0RE00261E.
- B. Siritanaratkul, M. Forster, F. Greenwell, P. K. Sharma, E. H. Yu and A. J. Cowan, Zero-Gap Bipolar Membrane Electrolyzer for Carbon Dioxide Reduction Using Acid-Tolerant Molecular Electrocatalysts, J. Am. Chem. Soc., 2022, 144, 7551–7556, DOI:10.1021/jacs.1c13024.
- M. A. Blommaert, R. Sharifian, N. U. Shah, N. T. Nesbitt, W. A. Smith and D. A. Vermaas, Orientation of a bipolar membrane determines the dominant ion and carbonic species transport in membrane electrode assemblies for CO2 reduction, J. Mater. Chem. A, 2021, 9, 11179–11186, 10.1039/d0ta12398f.
- B. Pribyl-Kranewitter, A. Beard, T. Schuler, N. Diklić and T. J. Schmidt, Investigation and Optimisation of Operating Conditions for Low-Temperature CO2 Reduction to CO in a Forward-Bias Bipolar-Membrane Electrolyser, J. Electrochem. Soc., 2021, 168, 43506, DOI:10.1149/1945-7111/abf063.
- X. Sheng, W. Ge, H. Jiang and C. Li, Engineering the Ni-N-C Catalyst Microenvironment Enabling CO2 Electroreduction with Nearly 100% CO Selectivity in Acid, Adv. Mater., 2022, 34, 2201295, DOI:10.1002/adma.202201295.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Gas diffusion electrodes, reactor designs and key metrics of low-temperature CO2 electrolysers, Nat. Energy, 2022, 7, 130–143, DOI:10.1038/s41560-021-00973-9.
- J. C. Bui, C. Kim, A. J. King, O. Romiluyi, A. Kusoglu, A. Z. Weber and A. T. Bell, Engineering Catalyst–Electrolyte Microenvironments to Optimize the Activity and Selectivity for the Electrochemical Reduction of CO2 on Cu and Ag, Acc. Chem. Res., 2022, 55, 484–494, DOI:10.1021/acs.accounts.1c00650.
- Y. Kong, H. Hu, M. Liu, Y. Hou, V. Kolivoška, S. Vesztergom and P. Broekmann, Visualisation and quantification of flooding phenomena in gas diffusion electrodes used for electrochemical CO2 reduction, J. Catal., 2022, 408, 1–8, DOI:10.1016/j.jcat.2022.02.014.
- Y. Kong, M. Liu, H. Hu, Y. Hou, S. Vesztergom, M. d. J. Gálvez-Vázquez, I. Zelocualtecatl Montiel, V. Kolivoška and P. Broekmann, Cracks as Efficient Tools to Mitigate Flooding in Gas Diffusion Electrodes Used for the Electrochemical Reduction of Carbon Dioxide, Small Methods, 2022, 6, 2200369, DOI:10.1002/smtd.202200369.
- Y. Wu, L. Charlesworth, I. Maglaya, M. N. Idros, M. Li, T. Burdyny, G. Wang and T. E. Rufford, Mitigating Electrolyte Flooding for Electrochemical CO2 Reduction via Infiltration of Hydrophobic Particles in a Gas Diffusion Layer, ACS Energy Lett., 2022, 7, 2884–2892, DOI:10.1021/acsenergylett.2c01555.
- U. O. Nwabara, A. D. Hernandez, D. A. Henckel, X. Chen, E. R. Cofell, M. P. de-Heer, S. Verma, A. A. Gewirth and P. J. A. Kenis, Binder-Focused Approaches to Improve the Stability of Cathodes for CO2 Electroreduction, ACS Appl. Energy Mater., 2021, 4, 5175–5186, DOI:10.1021/acsaem.1c00715.
- E. W. Lees, B. A. W. Mowbray, F. G. L. Parlane and C. P. Berlinguette, Gas diffusion electrodes and membranes for CO2 reduction electrolysers, Nat. Rev. Mater., 2022, 7, 55–64, DOI:10.1038/s41578-021-00356-2.
- R. B. Kutz, Q. Chen, H. Yang, S. D. Sajjad, Z. Liu and I. R. Masel, Sustainion Imidazolium-Functionalized Polymers for Carbon Dioxide Electrolysis, Energy Technol., 2017, 5, 929–936, DOI:10.1002/ente.201600636.
- R. Wang, H. Haspel, A. Pustovarenko, A. Dikhtiarenko, A. Russkikh, G. Shterk, D. Osadchii, S. Ould-Chikh, M. Ma, W. A. Smith, K. Takanabe, F. Kapteijn and J. Gascon, Maximizing Ag Utilization in High-Rate CO2 Electrochemical Reduction with a Coordination Polymer-Mediated Gas Diffusion Electrode, ACS Energy Lett., 2019, 4, 2024–2031, DOI:10.1021/acsenergylett.9b01509.
- D. Salvatore and C. P. Berlinguette, Voltage Matters When Reducing CO2 in an Electrochemical Flow Cell, ACS Energy Lett., 2019, 5, 215–220, DOI:10.1021/acsenergylett.9b02356.
- D. A. Salvatore, D. M. Weekes, J. He, K. E. Dettelbach, Y. C. Li, T. E. Mallouk and C. P. Berlinguette, Electrolysis of Gaseous CO2 to CO in a Flow Cell with a Bipolar Membrane, ACS Energy Lett., 2017, 3, 149–154, DOI:10.1021/acsenergylett.7b01017.
- D. G. Wheeler, B. A. W. Mowbray, A. Reyes, F. Habibzadeh, J. He and C. P. Berlinguette, Quantification of water transport in a CO2 electrolyzer, Energy Environ. Sci., 2020, 13, 5126–5134, 10.1039/d0ee02219e.
- B. Endrődi, E. Kecsenovity, A. Samu, F. Darvas, R. V. Jones, V. Török, A. Danyi and C. Janáky, Multilayer Electrolyzer Stack Converts Carbon Dioxide to Gas Products at High Pressure with High Efficiency, ACS Energy Lett., 2019, 4, 1770–1777, DOI:10.1021/acsenergylett.9b01142.
- J. Disch, L. Bohn, S. Koch, M. Schulz, Y. Han, A. Tengattini, L. Helfen, M. Breitwieser and S. Vierrath, High-resolution neutron imaging of salt precipitation and water transport in zero-gap CO2 electrolysis, Nat. Commun., 2022, 13, 6099, DOI:10.1038/s41467-022-33694-y.
- B. de Mot, M. Ramdin, J. Hereijgers, T. J. H. Vlugt and T. Breugelmans, Direct Water Injection in Catholyte-Free Zero-Gap Carbon Dioxide Electrolyzers, ChemElectroChem, 2020, 7, 3839–3843, DOI:10.1002/celc.202000961.
- S. Verma, Y. Hamasaki, C. Kim, W. Huang, S. Lu, H.-R. M. Jhong, A. A. Gewirth, T. Fujigaya, N. Nakashima and P. J. A. Kenis, Insights into the Low Overpotential Electroreduction of CO2 to CO on a Supported Gold Catalyst in an Alkaline Flow Electrolyzer, ACS Energy Lett., 2017, 3, 193–198, DOI:10.1021/acsenergylett.7b01096.
- L. W. Diamond and N. N. Akinfiev, Solubility of CO2 in water from −1.5 to 100 °C and from 0.1 to 100 MPa, Fluid Phase Equilib., 2003, 208, 265–290, DOI:10.1016/S0378-3812(03)00041-4.
- Y. Wu, N. R. Mirza, G. Hu, K. H. Smith, G. W. Stevens and K. A. Mumford, Precipitating Characteristics of Potassium Bicarbonate Using Concentrated Potassium Carbonate Solvent for Carbon Dioxide Capture. Part 1. Nucleation, Ind. Eng. Chem. Res., 2017, 56, 6764–6774, DOI:10.1021/acs.iecr.7b00699.
- M. E. Leonard, L. E. Clarke, A. Forner-Cuenca, S. M. Brown and F. R. Brushett, Investigating Electrode Flooding in a Flowing Electrolyte, Gas-Fed Carbon Dioxide Electrolyzer, ChemSusChem, 2020, 13, 400–411, DOI:10.1002/cssc.201902547.
- S. Subramanian, K. Yang, M. Li, M. Sassenburg, M. Abdinejad, E. Irtem, J. Middelkoop and T. Burdyny, Geometric Catalyst Utilization in Zero-Gap CO2 Electrolyzers, ACS Energy Lett., 2022, 8, 222–229, DOI:10.1021/acsenergylett.2c02194.
- U. O. Nwabara, M. P. de Heer, E. R. Cofell, S. Verma, E. Negro and P. J. A. Kenis, Towards accelerated durability testing protocols for CO2 electrolysis, J. Mater. Chem. A, 2020, 8, 22557–22571, 10.1039/D0TA08695A.
- A. A. Samu, A. Kormányos, E. Kecsenovity, N. Szilágyi, B. Endrődi and C. Janáky, Intermittent Operation of CO2 Electrolyzers at Industrially Relevant Current Densities, ACS Energy Lett., 2022, 7, 1859–1861, DOI:10.1021/acsenergylett.2c00923.
- Z. Liu, H. Yang, R. Kutz and R. I. Masel, CO2 Electrolysis to CO and O2 at High Selectivity, Stability and Efficiency Using Sustainion Membranes, J. Electrochem. Soc., 2018, 165, J3371–J3377, DOI:10.1149/2.0501815jes.
- W. H. Lee, Y.-J. Ko, Y. Choi, S. Y. Lee, C. H. Choi, Y. J. Hwang, B. K. Min, P. Strasser and H.-S. Oh, Highly selective and scalable CO2 to CO - Electrolysis using coral-nanostructured Ag catalysts in zero-gap configuration, Nano Energy, 2020, 76, 105030, DOI:10.1016/j.nanoen.2020.105030.
- Q. Fan, P. Gao, S. Ren, Y. Qu, C. Kong, J. Yang and Y. Wu, Total conversion of centimeter-scale nickel foam into single atom electrocatalysts with highly selective CO2 electrocatalytic reduction in neutral electrolyte, Nano Res., 2022, 14, 765, DOI:10.1007/s12274-022-4472-6.
- H. Seong, M. Choi, S. Park, H.-w. Kim, J. Kim, W. Kim, J. S. Yoo and D. Lee, Promoting CO2-to-CO Electroreduction via the Active-Site Engineering of Atomically Precise Silver Nanoclusters, ACS Energy Lett., 2022, 7, 4177–4184, DOI:10.1021/acsenergylett.2c02018.
- J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Modulating electric field distribution by alkali cations for CO2 electroreduction in strongly acidic medium, Nat. Catal., 2022, 5, 268–276, DOI:10.1038/s41929-022-00761-y.
| This journal is © The Royal Society of Chemistry 2023 |