
Random double-cable conjugated polymers with controlled acceptor contents for single-component organic solar cells†
Baiqiao
Liu‡
ab,
Shijie
Liang‡
a,
Safakath
Karuthedath‡
 cg,
Chengyi
Xiao
*a,
Jing
Wang
d,
Wen Liang
Tan
e,
Ruonan
Li
b,
Hao
Li
f,
Jianhui
Hou
f,
Zheng
Tang
d,
Frédéric
Laquai
c,
Christopher R.
McNeill
e,
Yunhua
Xu
*b and
Weiwei
Li
*a
cg,
Chengyi
Xiao
*a,
Jing
Wang
d,
Wen Liang
Tan
e,
Ruonan
Li
b,
Hao
Li
f,
Jianhui
Hou
f,
Zheng
Tang
d,
Frédéric
Laquai
c,
Christopher R.
McNeill
e,
Yunhua
Xu
*b and
Weiwei
Li
*a
aBeijing Advanced Innovation Center for Soft Matter Science and Engineering, State Key Laboratory of Organic-Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: xiaocy@mail.buct.edu.cn; liweiwei@iccas.ac.cn
bSchool of Physical Science and Engineering, Beijing Jiaotong University, Beijing 100044, P. R. China. E-mail: yhxu@bjtu.edu.cn
cKing Abdullah University of Science and Technology (KAUST), KAUST Solar Center (KSC), Physical Sciences and Engineering Division (PSE), Material Science and Engineering Program (MSE), Thuwal 23955-6900, Kingdom of Saudi Arabia
dCenter for Advanced Low-dimension Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, P. R. China
eDepartment of Materials Science and Engineering, Monash University, Wellington Road, Clayton, Victoria 3800, Australia
fState Key Laboratory of Polymer Physics and Chemistry, Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China
gInstitute of Materials Research, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen, 518055, China
First published on 16th May 2023
Abstract
Double-cable conjugated polymers contain electron-donating (D) backbones and electron-accepting (A) side units, in which the nanophase separation of the donor and acceptor segments is a crucial factor to determine the photovoltaic performance of single-component organic solar cells (SCOSCs). In this work, three random double-cable conjugated polymers (denoted as P1–P3 with enhanced acceptor contents) have been designed to tailor the nanophase separation of D/A to realize high-performance SCOSCs. These new random double-cable conjugated polymers contain identical polymer backbones with varied contents of near-infrared acceptor side units. It is observed that the acceptor contents could effectively tune the aggregation degree of the backbone and acceptor (shown in the absorption spectra and grazing-incidence wide-angle X-ray scattering measurement) and further influence the construction of charge-transporting pathways. Therefore, a moderate content of acceptor side units provides balanced D/A aggregation and optimal nanophase separation, resulting in a high efficiency of 9.4% in SCOSCs. These results demonstrate that random double-cable conjugated polymers are an excellent model for studying the impact of their aggregation/crystallinity so as to realize high-performance SCOSCs.
1. Introduction
Single-component organic solar cells (SCOSCs) are recognized as a simplified version of bulk-heterojunction organic solar cells (BHJOSCs), representing an exciting target with fundamental and technological implications.1 Double-cable conjugated polymers, as one class of single-component materials used in SCOSCs, have attracted enormous attention driven by recent material innovation.2 Double-cable conjugated polymers, with pendant aromatic side units as the electron acceptor (A) and conjugated backbones as the electron donor (D), simultaneously ensure hole and electron percolation pathways.3 In the beginning, most double-cable conjugated polymers were fabricated with a polythiophene backbone and fullerene side units; however, their narrow absorption spectrum severely limited the efficiency of SCOSCs.4 Therefore, Li and coworkers have been working on developing double-cable conjugated polymers with novel structures through a “functionalization-polymerization” method since 2017.3,5–15 Impressively, double-cable polymers containing rylene diimides as acceptor units have propelled the performance of SCOSCs, boosting the power conversion efficiency (PCE) to over 8%.16 However, rylene diimides do not absorb light in the near-infrared (NIR) spectral region, thus hampering further PCE enhancement. In BHJOSCs, A–D–A type push–pull structured non-fullerene electron acceptors have been successfully incorporated to boost their PCEs to over 19%, due to intramolecular charge transfer and extended NIR absorption.17–19 Obviously, incorporating A–D–A NIR acceptor blocks into double-cable polymers is a promising strategy for next generation SCOSCs.20Furthermore, well-ordered nanophase separation in double-cable polymers is another pathway to obtaining high-efficiency SCOSCs. However, double-cable polymers with donor and acceptor segments being covalently linked in one molecule exhibit a more complex crystalline behavior owing to their mutual restraint, resulting in inferior PCEs compared to that of state-of-the-art BHJOSCs.21–24 Therefore, various strategies have been developed to control the nanophase separation of the donor backbone and acceptor units consisting of rylene diimide-based double-cable polymers, for instance, thermal annealing,6 tuning the miscibility of donors and acceptors,7 changing the linker length between the backbone and side unit,25,26etc.3,16 However, there are very few reports focusing on double-cable conjugated polymers with pendant NIR electron acceptors, in which the acceptors exhibit a 3D stacking network.27–30 In our previous work, a molecular symmetry strategy was used to obtain optimized morphology in these polymers.20 The asymmetric design led to an enhanced PCE of above 10% due to appropriate nanophase separation, while the symmetric polymer exhibited only <4% PCE with a low fill factor (FF) of 0.39 as a result of strongly aggregated acceptor side units. This result showed the importance of phase-separation control between the donor backbone and acceptor side units in controlling the nanophase separation of NIR-type double-cable polymers.
In this work, a random copolymer design was used to optimize the nanophase separation of double-cable polymers via tuning the molar ratio of acceptors. Three random double-cable conjugated copolymers (named P1, P2, and P3) with different feed ratios of TPDIC acceptors (Scheme 1) have been prepared.20 When lowering acceptor contents, the ordered/crystallized backbone would cause the acceptor units to adopt the periodicity of the backbone. At this circumstance, the low acceptor content can not form a continuous electron pathway in thin films, resulting in low PCEs in SCOSCs. When double-cable polymers contain excessive acceptor contents, the crystallized/aggregated acceptor units may cause the backbone locally to be disordered, thus lowering the hole-transporting ability. Consequently, the double-cable conjugated copolymer P2 with optimal acceptor contents exhibited a more balanced degree of backbone and acceptor aggregation and more balanced charge transport, delivering an excellent PCE of 9.4%. The results demonstrate that random copolymer design is an effective method to tune the D/A phase separation of double-cable polymers toward efficient SCOSCs.
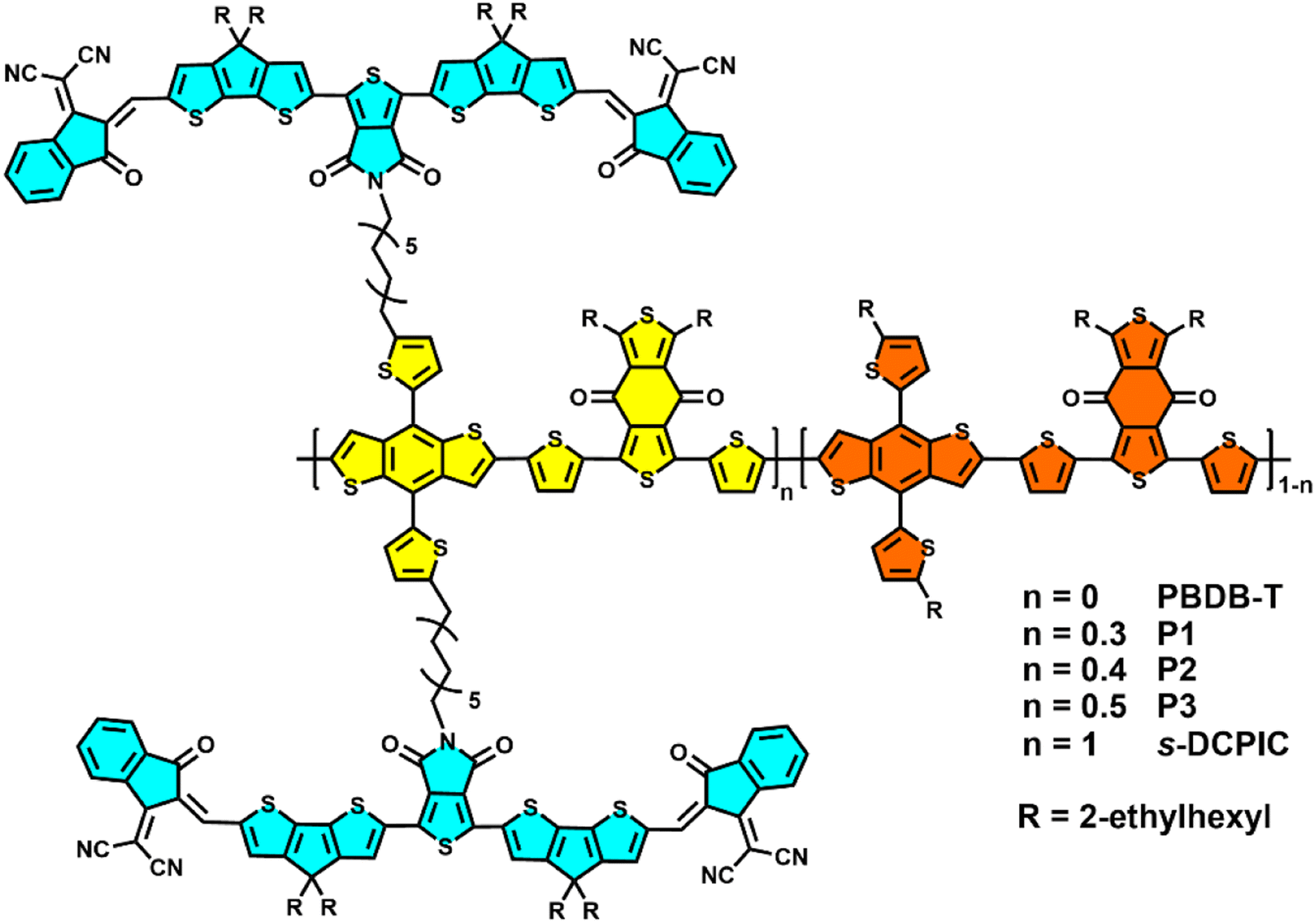 | ||
| Scheme 1 Chemical structures of the double-cable copolymers in this work. | ||
2. Results and discussion
The polymers used in this work are shown in Scheme 1, and the detailed preparation procedures are summarized in Scheme S1.† All the random double-cable polymers were synthesized using the “functionalization-polymerization” method.2 They exhibit good solubility in chlorobenzene (CB) and ortho-dichlorobenzene (o-DCB). Gel permeation chromatography measurement (GPC) was used to determine the molecular weight of the double-cable polymers with 1,2,4-trichlorobenzene as the eluent at 150 °C. The number-average molecular weights (Mn) of P1, P2, and P3 are 41.4, 67.9, and 47.4 kDa (Fig. S1–S3† and Table 1), respectively. The double-cable polymers show excellent thermal stability with 5% weight loss above 300 °C via thermal gravimetric analysis (TGA) (Fig. S4†). Different scanning calorimetry measurements (DSC) show no phase transition peaks in the measured temperature range (Fig. S5†). The donor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acceptor mass ratios of P1, P2, and P3 are 1:0.73, 1:1, and 1:1.3, respectively, which are calculated from nuclear magnetic resonance (NMR) spectra, as shown in Fig. S19–S21.†
:acceptor mass ratios of P1, P2, and P3 are 1:0.73, 1:1, and 1:1.3, respectively, which are calculated from nuclear magnetic resonance (NMR) spectra, as shown in Fig. S19–S21.†
| M n (kDa) | M w (kDa) | Đ M | λ sol,onset (nm) | λ film,onset (nm) | E filmg , (eV) | E HOMO (eV) | E LUMO (eV) | |
|---|---|---|---|---|---|---|---|---|
|
a Absorption onsets in o-DCB:3% DIO solution.
b Absorption onsets in a thin-film state.
c Optical bandgap was calculated through the onset of the thin-film absorption edge.
d Calculated from EHOMO = −4.80 eV − Eox.
e
E
LUMO = −Ered − 4.80 eV.
|
||||||||
| P1 | 41.4 | 125.6 | 3.04 | 777 | 837 | 1.48 | −5.54 | −3.75 |
| P2 | 67.9 | 149.2 | 2.20 | 777 | 846 | 1.47 | −5.54 | −3.75 |
| P3 | 47.4 | 123.1 | 2.60 | 777 | 846 | 1.47 | −5.54 | −3.75 |
The absorption spectra of these double-cable polymers are shown in Fig. 1 and the data are summarized in Table 1. All the polymers exhibited similar absorption spectra both in solutions and thin films, in which the intensity contribution from the acceptor was enhanced from P1 to P3 due to the enhanced ratios of TPDIC (650 nm ∼750 nm). Furthermore, the absorption spectra of the polymers showed large red shifts of ∼60 nm when going from solutions to thin films, indicating pronounced aggregation of acceptor segments in thin films. We selected the intensity of the 0–0 transition to the 0–1 transition as an indication of the relative degree of aggregation, which can be done separately for the donor backbone (marked as D: 0–0 and D: 0–1) and for the TPDIC acceptor (marked as A: 0–0 and A: 0–1) according to the previous report,20 as shown in Fig. 1b and Table S1.† We found that the D: 0–0/D: 0–1 ratio increased going from P1 to P2 and decreased from P2 to P3, indicating the aggregation degree of the donor backbone. Also, the A: 0–0/A: 0–1 ratio increased from P1 to P2 and decreased from P2 to P3, showing the same trend in the aggregation of the acceptor segments. These changes illustrate that the aggregation/crystallinity degrees of both the donor backbone and acceptor units are in the order of P1 < P3 < P2. Electrochemical cyclic voltammetry (CV) measurements were performed to determine the frontier energy levels of these random double-cable polymers, as shown in Fig. S6† and Table 1. The double-cable polymers show identical highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies because of the same donor backbone and acceptor segments used in these polymers.
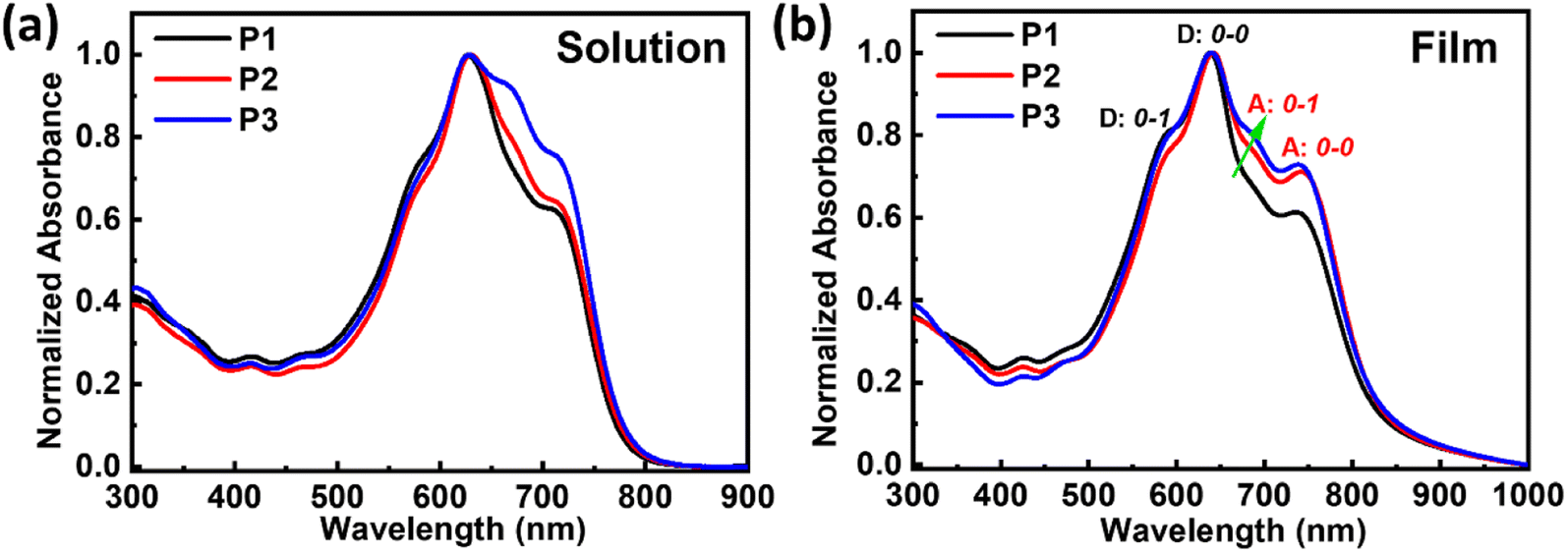 | ||
| Fig. 1 Optical absorption spectra of the double-cable polymers in (a) o-DCB:3%DIO solutions and (b) thin films annealed at 150 °C for 10 min. | ||
SCOSCs based on P1, P2, or P3 were fabricated with the inverted configuration of ITO/ZnO/active layers/MoO3/Ag, and the detailed preparation conditions are summarized in the ESI.†20 The J–V characteristics are shown in Fig. 2a, and the photovoltaic parameters are summarized in Table 2 and Tables S2–S4.† P2-based SCOSCs provided the highest PCE of 9.45%, while devices based on P1 and P3 showed PCEs of ∼6%. The SCOSCs based on P1 and P2 have the same VOCs (open-circuit voltages) of ∼0.738 V, while P3-based devices show a reduced VOC of ∼0.717 V. The high performance of SCOSCs based on P2 is due to the enhanced JSC (short-circuit current density) of 20.17 mA cm−2 and FF (fill factor) of 0.63. The improved JSC in P2-based SCOSCs is also reflected in its high EQEs, as shown in Fig. 2b. It should be noted that the JSCs and FFs increased significantly with enhanced acceptor contents from P1 to P2, while they decreased from P2 to P3 when further enhancing the acceptor contents. Moreover, the BHJOSCs based on PBDB-T:s-DCPIC were fabricated to demonstrate the necessity of the copolymer design (Fig. S7 & Table S5†), in which s-DCPIC is the double-cable polymer prepared in our previous work.20 A weight ratio of PBDB-T:s-DCPIC = 1:2.26 (w/w) was applied with respect to the copolymer P2. BHJ-type OSCs exhibited a champion PCE of 6.92% with a high JSC of 16.54 mA cm−2 and FF of 0.53 but a low VOC of 0.79 V, which is higher than that of the reported symmetrical double-cable polymer s-DCPIC20 but lower than that of P2-based SCOSCs.
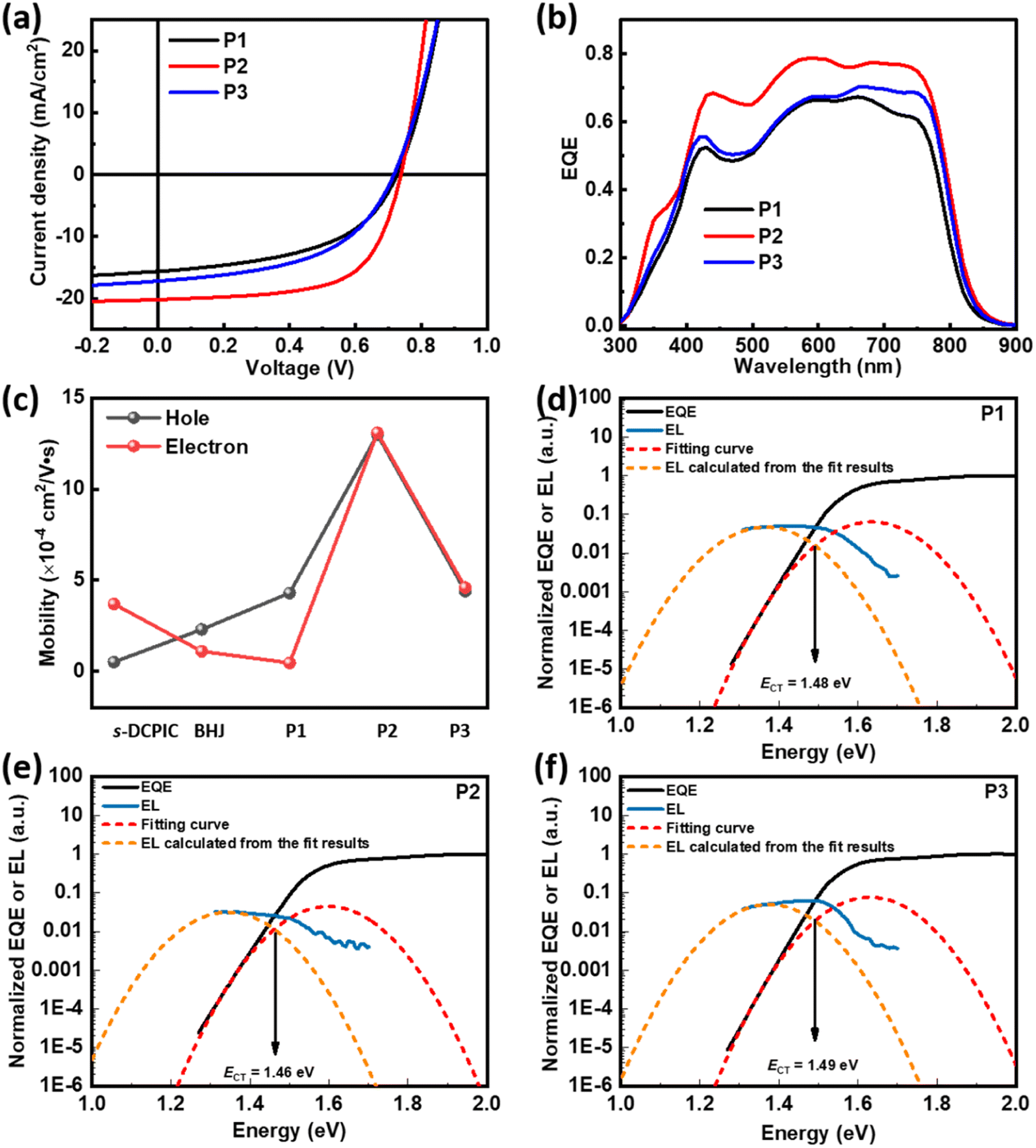 | ||
| Fig. 2 (a) J–V characteristics of OSCs based on P1–P3. (b) The corresponding EQE spectra. (c) The calculated μhs and μes of s-DCPIC, BHJ, and P1–P3. EQE and EL spectra of the OSCs based on (d) P1 (e) P2, and (f) P3. | ||
| Polymers | V OC (V) | J SC (mA cm−2) | FFa | PCEa (%) | μ h (cm2 V−1 s−1) | μ e (cm2 V−1 s−1) |
|---|---|---|---|---|---|---|
| a The average parameters are obtained from 6 parallel devices. b The parameters are from our previous work.31 | ||||||
| s-DCPICb | 0.85 (0.845 ± 0.002) | 11.94 (11.98 ± 0.24) | 0.39 (0.38 ± 0.01) | 3.98 (3.87 ± 0.08) | 5.1 × 10−5 | 1.1 × 10−4 |
| BHJ | 0.792 (0.795 ± 0.002) | 16.54 (16.58 ± 0.72) | 0.53 (0.50 ± 0.02) | 6.92 (6.57 ± 0.15) | 2.3 × 10−4 | 3.7 × 10−4 |
| P1 | 0.737 (0.732 ± 0.007) | 15.55 (15.95 ± 0.45) | 0.52 (0.50 ± 0.02) | 6.00 (5.79 ± 0.15) | 4.3 × 10−4 | 4.6 × 10−5 |
| P2 | 0.738 (0.734 ± 0.003) | 20.17 (20.44 ± 0.59) | 0.63 (0.62 ± 0.01) | 9.45 (9.32 ± 0.09) | 1.3 × 10−3 | 1.1 × 10−3 |
| P3 | 0.717 (0.714 ± 0.001) | 17.18 (17.07 ± 0.26) | 0.51 (0.50 ± 0.01) | 6.26 (6.10 ± 0.11) | 4.4 × 10−4 | 4.6 × 10−4 |
We also investigated the thermal stability of the devices, as shown in Fig. S8.† The VOCs of all the devices have little decay during the long heat treatment. It is noted that the JSCs of the devices decrease and FFs increase during the initial time, which is attributed to the degradation of the test light source. All the devices have similar PCE degradation curves and maintain over 80% of the initial efficiency after more than 350 hours. The results indicate that the double-cable conjugated polymers are advantageous for achieving stable organic solar cells.
To reveal the origin of the distinct performance, we used the space charge-limited current (SCLC) method to extract the hole/electron mobilities of these cells, as shown in Fig. 2c and S9.† Devices based on PBDB-T:s-DCPIC showed an enhanced hole mobility (μh) of 2.3 × 10−4 cm2 V−1 s−1 compared to that of s-DCPIC with μh of 5.1 × 10−5 cm2 V−1 s−1, caused by the additional hole transport path of PBDB-T.20 The copolymer P1 with low content of TPDIC gave a low electron mobility (μe) of 4.6 × 10−5 cm2 V−1 s−1 and μh of 5.1 × 10−4 cm2 V−1 s−1. Surprisingly, P2 showed an enhanced μh of 1.3 × 10−3 cm2 V−1 s−1 and μe of 1.1 × 10−3 cm2 V−1 s−1. However, copolymer P3 with improved acceptor content exhibited a decreased μh of 4.4 × 10−4 cm2 V−1 s−1 and μe of 4.6 × 10−4 cm2 V−1 s−1. The changes in the charge transport properties are responsible for the difference in JSCs and FF values. Considering the same PBDB-T/TPDIC ratios of the PBDB-T:s-DCPIC blend and P2, the higher μe and μh of P2 indicate a well-ordered molecular packing motif in thin films. We also measured the mobilities of the PBDB-T:TPDIC blend film with different mass ratios, as shown in Fig. S10 and Table S6.† The hole mobilities of the blend film are in the order of BHJ-2 > BHJ-3 > BHJ-1, which is similar to that of the random double-cable conjugated polymer. However, the electron mobilities of the blend film increase continuously with enhanced acceptor content, which is different from that of the double-cable conjugated polymer. This difference may be attributed to the unique microstructure of the random double-cable conjugated polymer.
We further explore the difference in exciton dissociation between SCOSCs and BHJOSCs, using P2 and PBDB-T:s-DCPIC as active layers due to their similar D/A mass ratio. Photoluminescence (PL) was used to study the quenching efficiencies in P2 and BHJ films. The exciton quenching efficiencies in the donor phase of BHJ and P2 films are almost 100% (as shown in Fig. S11a†), so the exciton dissociation of the two devices is mainly determined by the quenching efficiencies of excitons in the acceptor phase. As shown in Fig. S11b,† compared to the 50% quenching efficiency of the excitons in the s-DCPIC phase of the BHJ film, the excitons in the s-DCPIC phase of the P2 film have a 65% quenching efficiency, which should be related to the more ordered molecular stacking in the P2 film.
We noticed that P1- and P2-based SCOSCs exhibited similar VOCs of ∼0.738 V, while P3-based cells showed a lower VOC of ∼0.717 V, which may be related to different voltage losses (Vloss). Therefore, we performed electroluminescence (EL) and highly sensitive EQE (sEQE) measurements to determine the energy of the charge transfer state (ECT) of the solar cells by using Marcus theory,32 and the statistics are summarized in Table 3. The detailed procedure is provided in the ESI.† Devices studied in this work showed different ECTs, as 1.49 eV for PBDB-T:s-DCPIC, 1.48 eV for P1, 1.46 eV for P2, and 1.49 eV for P3, respectively (Fig. 2d–f and S12†). These ECT values are higher than those reported for s-DCPIC-based devices which showed an ECT value of 1.44 eV, so they have a larger Vloss of 0.70–0.77 eV (the difference between ECT/q and Voc, where q is the elementary charge). Interestingly, the EQEEL (sensitive EQE spectra calculated from the EL spectra, Fig. S13†) of the devices is more than 10 times higher than that of devices based on s-DCPIC, indicating a lower non-radiative recombination loss (ΔVnr). Therefore, a higher radiative recombination loss (ΔVr) with values of 0.50–0.55 eV was observed in the devices, which is consistent with NDI-based devices.7 Moreover, these three SCOSCs exhibit different ECT and ΔVr values, resulting in different voltage losses. As mentioned above, compared with P1 and P3 films, P2 films should have more ordered molecular packing motifs, and the more ordered the interfacial order is, the smaller the CT state energy will be;33 therefore, P2-based devices should have smaller ECT. From the ECT results of the three SCOSCs, as shown in Table 3, the P2-based device indeed has the smallest ECT value. Since Vloss = ECT − Voc, among the three SCOSC devices, the P2-based device with the smallest ECT has the lowest Vloss. Another noteworthy point is that disordered stacking leads to substantial broadening of the density of electronic states (DOS) of the photoactive layer;34–36 therefore, the highly ordered P2-based active layer should have narrower DOS, resulting in smaller ΔVr for the device.37 However, the relative disorder in P1- and P3-based active layers leads to larger ΔVr and Vloss.
Atomic force microscopy (AFM) was performed to study these polymers' surface morphology. Fig. S14† shows a smooth surface in the PBDB-T:s-DCPIC blended films with a root-mean-square (RMS) roughness value of 0.846 nm. Thin films of these double-cable polymers show a relatively rough surface with RMS values of 1.01–1.16 nm and fiber-like phase separation (Fig. S15†). We then used two-dimensional wide-angle grazing-incidence X-ray scattering (GIWAXS) measurement to further study the phase separation of these copolymers (Fig. 3) and the crystallographic parameters are summarized in Table 4 & S7.† As shown in Fig. 3a and g, the donor backbone PBDB-T exhibited a preferential face-on orientation as indicated by in-plane (IP) lamellar (h00) peaks, (002) backbone repeat peaks, and out-of-plane (OOP) π–π (010) peak. The TPDIC acceptor has a highly crystalline phase with three-dimensional structural order as indicated by a large number of sharp peaks with many of these peaks located off-axis. The appearance of the IP peak at q ∼ 1.65 Å−1 matches with the characteristic position of the π–π stacking peak, indicating the edge-on orientation of TPDIC (Fig. 3b and g). The plentiful, sharp multiple peaks both in the IP and OOP directions have suggested a complicated unit cell and highly crystalline nature of the TPDIC film, as reported in many single-crystal structures of non-fullerene acceptors.28,29 However, physically mixed PBDB-T and s-DCPIC simultaneously disrupt the packing of both the donor backbone and acceptor unit (Fig. 3c and g), which can help to explain the low performance in PBDB-T:s-DCPIC-based OSCs.
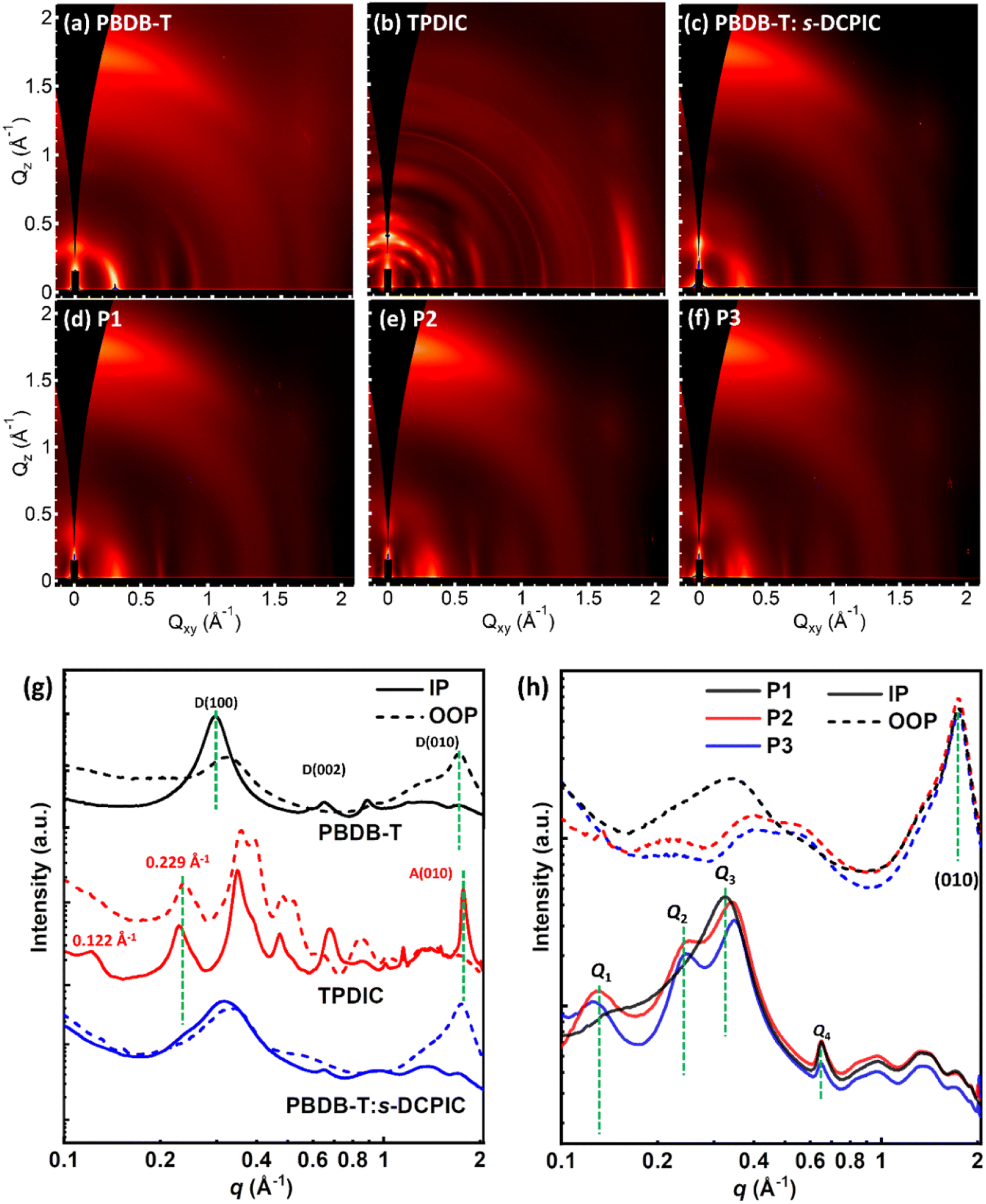 | ||
| Fig. 3 GIWAXS profiles on the Si substrates of (a) PBDB-T, (b) TPDIC, (c) PBDB-T:s-DCPIC, and (d and f) P1–P3 thin films. (g and h) In-plane and out-of-plane plots of the corresponding GIWAXS images. | ||
All three copolymers exhibited a preferential “face-on” orientation with IP lamellar (h00) peaks, (002) backbone repeat peaks, and OOP (010) peaks, as shown in Fig. 3d–f. The corresponding IP and OOP line cuts of these patterns are shown in Fig. 3h. For the OOP direction, all three polymers showed the (010) peak at q = 1.74 Å−1, which coincides with the location of the π–π stacking peaks of both the PBDB-T polymer and TPDIC acceptor in Fig. 3a–b. The CL values of the (010) diffraction peaks are 3.11 nm, 3.13 nm, and 2.99 nm for double-cable copolymers P1, P2, and P3, respectively (Table 4), indicating different ordering in the (010) direction. For the IP direction, we denoted the peak at q ∼ 0.13 Å−1, 0.24 Å−1, 0.34 Å−1 and 0.65 Å−1 as Q1–Q4, respectively. The peaks Q1–Q3 are related to the lamellar packing behavior for the double-cable molecular structure and peak Q4 is identified as a backbone stacking peak matching that found in the donor polymer PBDB-T. According to the GIWAXS profiles of PBDB-T and TPDIC in Fig. 3g, we speculate that there are at least two modes of packing in the film state: one form (phase I) where the acceptor units are highly organized (the existence of Q1 and Q2) at the expense of backbone planarity (reduced intensity of Q4); other regions (phase II) where the backbone is highly ordered (strong Q4 peak) but the acceptor units and side-chain ordering are more disordered (broader Q3 peak that appears at lower q). For these copolymers, phases I & II existed in all copolymers with different relative contents. Peak Q1 was enhanced from P1 to P2 and P3, and meanwhile the Q2 peak only appeared in P2 and P3. This is reasonable since P2 and P3 have more TPDIC contents than P1, which may enhance the phase I contents and electron-transporting ability. Moreover, we summarized the calculated crystallographic parameters of Q2–Q4 and the (010) peak in Tables 4 and S6.† We infer that, in thin films, there is competition within the backbone ordering and acceptor units ordering, resulting in different phase I and phase II contents as well as charge-transporting properties. This result emphasizes the importance of the donor/acceptor segment ratio in constructing effective charge transport pathways.
To determine the photo-generated charge carrier dynamics, we performed transient absorption (TA) spectroscopy over a wide temporal (picosecond to microseconds) and spectral energy (2.2 eV–0.7 eV) range using our home-built pump–probe spectroscopy setup.38 Positive ΔT/T signals represent ground state bleaching (GSB), while negative ΔT/T signals indicate regions of photo-induced absorption (PA) regions. Fig. 4a–c show the picosecond–nanosecond (ps–ns) TA spectra of P1, P2, and P3 films, respectively, measured under a dynamic vacuum after photoexciting at 650 nm with 2 μJ cm−2. In all three cases, a PA band at 1.15 eV was present immediately after excitation, which we assigned to singlet exciton-induced absorption of PBDB-T molecules (see Fig. S17† for TA spectra of neat films). Within a few ps, the TA spectra evolved into broader and distinct PA bands, which we assigned to charge carrier-induced absorption. The charge generation continued until ∼50 ps. The shaded area under the spectra at 1–2 ns (blue line) indicates the yield of charge carriers at 1–2 ns time. All three films showed very similar charge generation rates and decay transients in the ps-ns timescale (see Fig. 4d). Although all films showed similar charge carrier dynamics, P1 and P3 showed the presence of triplets at ∼0.85–0.9 eV. A small rise in the kinetics after 500 ps is visible for P1 and P3, which is absent in P2 (Fig. 4e, circled with a grey shade). However, we note that the triplet formation is comparably small and does not explain the significant PCE differences between the devices. The charge carrier recombination is not completed at 2 ns; thus, we performed TA experiments on the nanosecond-microsecond (ns–μs) time scale. Fig. 4f–h show the charge carrier decay dynamics (ns–μs time range) at various excitation fluences after photoexciting with 532 nm laser pulses. The corresponding ns–μs TA spectra are provided in the ESI in Fig. S18.† The decay of the charge-induced absorption is fluence independent at early times, indicating geminate recombination of charge-transfer states, and fluence dependent, indicating non-geminate recombination of free charges, at later times.38,39 We parameterized the decay dynamics using a well-established two-pool charge carrier recombination model to quantify the recombination rates.40 Details of this model can be found in the ESI† and our previous studies.39,41 Here, we assumed the charge carrier cross-section to be 1 × 10−16 cm−2, similar to values typically determined for similar material systems.42 The fit parameters obtained from the global fit to the carrier dynamics are listed in Table 5.
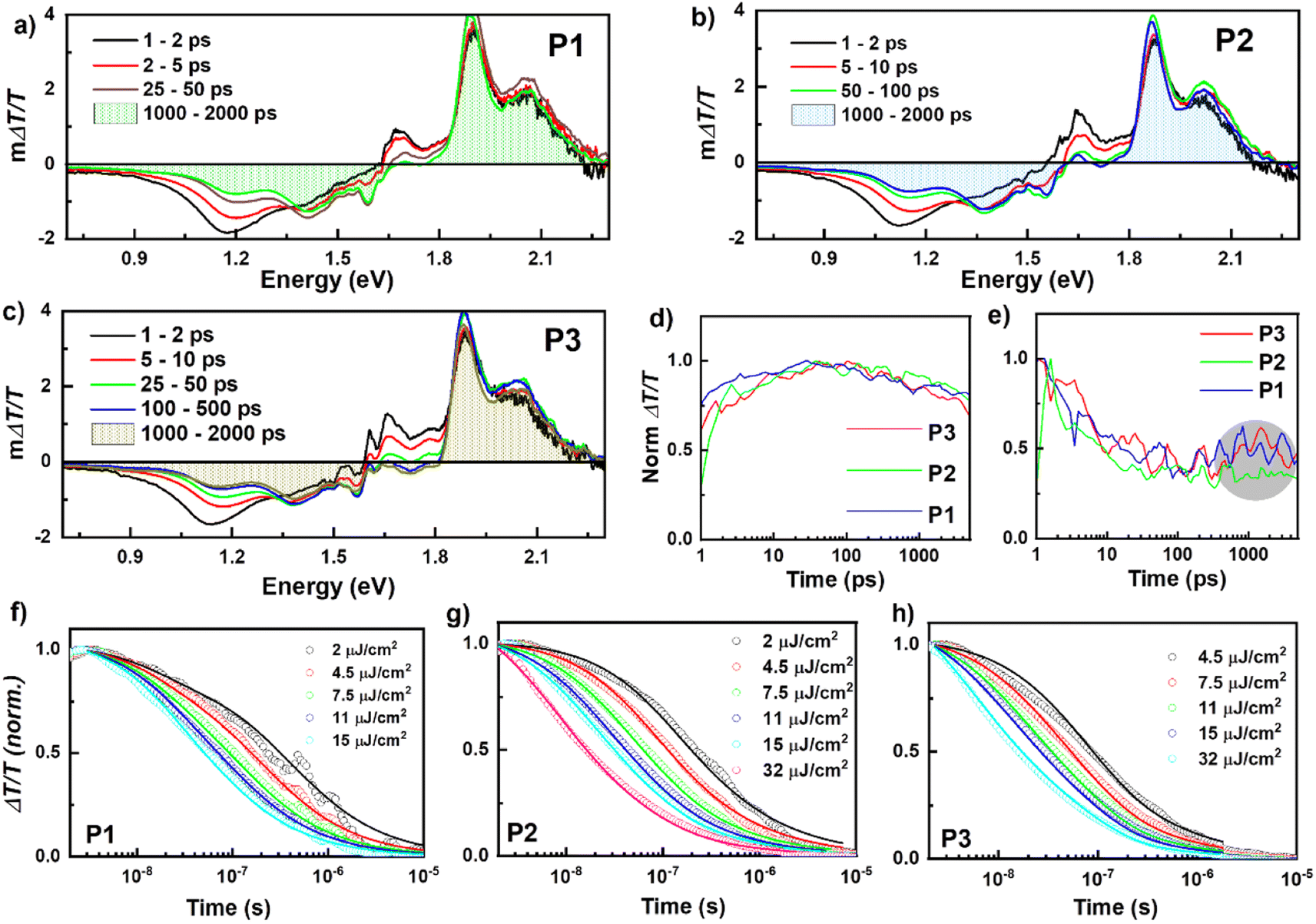 | ||
| Fig. 4 ps–ns TA spectra of (a) P1, (b) P2, and (c) P3 after exciting at 650 nm. (d) Integrated ps–ns TA kinetics of 1.35–1.45 eV and (e) 0.85–0.9 eV spectral regions at 2 μJ cm−2. The region around 1.5 eV in the x-axis of ps–ns TA spectra is affected by the scattering of the 800 nm pump used for creating white light. The ns–μs TA kinetics (open symbols) of the 1.35–1.45 eV region (charges) of (f) P1, (g) P2, and (h) P3 for a wide range of fluences as indicated in the legend. The solid line shows the global fit to the experimental data. | ||
| Parameter | P1 | P2 | P3 |
|---|---|---|---|
| f | 0.86 | 0.96 | 0.83 |
| 1 − f | 0.14 | 0.04 | 0.17 |
| λ + 1 | 2.25 | 2.51 | 2.55 |
| γ | 1 × 10−16 | 1.8 × 10−21 | 1 × 10−21 |
| β | 9 × 10−13 | 2.6 × 10−13 | 7.9 × 10−13 |
The fit to the two-pool model revealed that in P2, ∼96% of the charge carrier population are initially spatially-separated charges, which can be extracted as photocurrent. In P1 and P3 films, the percentage of free charge carriers is lower (86% and 82%, respectively), indicating more geminate recombination, which is directly related to the device performance differences. Besides a reduced fraction of geminate recombination, P2 showed also a reduced non-geminate recombination coefficient (β), indicating that free charge carriers are very long-lived and thus can be extracted effectively. In contrast, the P1 and P3 exhibited rather high β values, indicating fast non-geminate recombination of free charge carriers in addition to the larger losses due to geminate recombination. The reduced recombination in P2 is due to the formation of favorable morphology with good balance in backbone ordering and acceptor organization. This reduction in the recombination is well reflected in the improvement of FF of P2.41 Overall, the TA results revealed the mechanism and efficiency of charge carrier recombination channels, and they indicate that carrier recombination (both geminate and non-geminate) is the bottleneck in P1 and P3 films.
3. Conclusion
Random double-cable conjugated polymers with different acceptor contents were applied to SCOSCs in this work. The double-cable polymer with a medium content acceptor, coded P2, gave the best PCE of 9.45%, while the polymers with lower and higher content of acceptors, coded P1 and P3, showed comparably smaller PCEs of ∼6%. Further in-depth morphological and photophysical studies revealed that P2 has a good balance in backbone ordering and acceptor organization, which is beneficial for creating efficient charge transport pathways, thus promoting the photovoltaic performance of SCOSCs. Transient absorption spectroscopy demonstrated that polymers P1 and P3 exhibit more geminate and faster non-geminate charge carrier recombination than polymer P2. Our results demonstrate that the copolymerization strategy is an important method to control nanophase separation and construct efficient charge-transfer channels for high-performance double-cable polymer based SCOSCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was jointly supported by MOST (2018YFA0208504), the Beijing Natural Science Foundation (JQ21006) and NSFC (92163128, 52073016, 52163018, and 21905018) of China. This work was further supported by the Fundamental Research Funds for the Central Universities (buctrc201828, XK1802-2), and the Opening Foundation of State Key Laboratory of Organic-Inorganic Composites of Beijing University of Chemical Technology (oic-202201006). This work was performed in part at the SAXS/WAXS beamline at the Australian Synchrotron, part of ANSTO. The research reported in this publication was supported by funding from the King Abdullah University of Science and Technology (KAUST).References
- J. Roncali and I. Grosu, Adv. Sci., 2019, 6, 1801026 CrossRef PubMed.
- S. Liang, X. Jiang, C. Xiao, C. Li, Q. Chen and W. Li, Acc. Chem. Res., 2021, 54, 2227–2237 CrossRef CAS PubMed.
- G. Feng, J. Li, F. J. M. Colberts, M. Li, J. Zhang, F. Yang, Y. Jin, F. Zhang, R. A. J. Janssen, C. Li and W. Li, J. Am. Chem. Soc., 2017, 139, 18647–18656 CrossRef CAS PubMed.
- S. Miyanishi, Y. Zhang, K. Hashimoto and K. Tajima, Macromolecules, 2012, 45, 6424–6437 CrossRef CAS.
- B.-Q. Liu, Y.-H. Xu, F. Liu, C.-C. Xie, S.-J. Liang, Q.-M. Chen and W.-W. Li, Chin. J. Polym. Sci., 2022, 40, 898–904 CrossRef CAS.
- G. T. Feng, J. Y. Li, Y. K. He, W. Y. Zheng, J. Wang, C. Li, Z. Tang, A. Osvet, N. Li, C. J. Brabec, Y. P. Yi, H. Yan and W. W. Li, Joule, 2019, 3, 1765–1781 CrossRef CAS.
- X. Jiang, J. Yang, S. Karuthedath, J. Li, W. Lai, C. Li, C. Xiao, L. Ye, Z. Ma, Z. Tang, F. Laquai and W. Li, Angew. Chem., Int. Ed., 2020, 59, 21683–21692 CrossRef CAS PubMed.
- S. Liang, J. Wang, Y. Ouyang, W. L. Tan, C. R. McNeill, Q. Chen, Z. Tang and W. Li, Macromolecules, 2022, 55, 2517–2523 CrossRef CAS.
- W. Lai, C. Li, J. Zhang, F. Yang, F. J. M. Colberts, B. Guo, Q. M. Wang, M. Li, A. Zhang, R. A. J. Janssen, M. Zhang and W. Li, Chem. Mater., 2017, 29, 7073–7077 CrossRef CAS.
- F. Yang, J. Y. Li, C. Li and W. W. Li, Macromolecules, 2019, 52, 3689–3696 CrossRef CAS.
- Z. F. Yang, S. J. Liang, B. Q. Liu, J. Wang, F. Yang, Q. M. Chen, C. Y. Xiao, Z. Tang and W. W. Li, Polym. Chem., 2021, 12, 6865–6872 RSC.
- P. Yu, G. Feng, J. Li, C. Li, Y. Xu, C. Xiao and W. Li, J. Mater. Chem. A, 2020, 8, 2790–2797 CAS.
- Z. Hu, C. Xiao, W. L. Tan, B. Liu, S. Liang, X. Jiang, C. R. McNeil and W. Li, Macromolecules, 2022, 55, 5188–5196 CrossRef CAS.
- S. J. Liang, Y. H. Xu, C. Li, J. Y. Li, D. Wang and W. W. Li, Polym. Chem., 2019, 10, 4584–4592 RSC.
- D. Wang, Z. F. Yang, F. Liu, C. Y. Xiao, Y. G. Wu and W. W. Li, Chin. Chem. Lett., 2022, 33, 466–469 CrossRef CAS.
- C. Li, X. Wu, X. Sui, H. Wu, C. Wang, G. Feng, Y. Wu, F. Liu, X. Liu, Z. Tang and W. Li, Angew. Chem., Int. Ed., 2019, 58, 15532–15540 CrossRef CAS PubMed.
- Y. Z. Lin, J. Y. Wang, Z.-G. Zhang, H. T. Bai, Y. F. Li, D. B. Zhu and X. W. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C. C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- S. Liang, B. Liu, S. Karuthedath, J. Wang, Y. He, W. L. Tan, H. Li, Y. Xu, N. Li, J. Hou, Z. Tang, F. Laquai, C. R. McNeill, C. J. Brabec and W. Li, Angew. Chem., Int. Ed., 2022, 61, 202209316 Search PubMed.
- J. Wang and X. Zhan, Acc. Chem. Res., 2020, 54, 132–143 CrossRef PubMed.
- H. Yao, J. Wang, Y. Xu, S. Zhang and J. Hou, Acc. Chem. Res., 2020, 53, 822–832 CrossRef CAS PubMed.
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed.
- K. Yang, Z. Chen, Y. Wang and X. Guo, Acc. Mater. Res., 2023, 4, 237–250 CrossRef CAS.
- G. Feng, W. Tan, S. Karuthedath, C. Li, X. Jiao, A. C. Y. Liu, H. Venugopal, Z. Tang, L. Ye, F. Laquai, C. R. McNeill and W. Li, Angew. Chem., Int. Ed., 2021, 60, 25499–25507 CrossRef CAS PubMed.
- J. Wang, X. D. Jiang, H. B. Wu, G. T. Feng, H. Y. Wu, J. Y. Li, Y. P. Yi, X. D. Feng, Z. F. Ma, W. W. Li, K. Vandewal and Z. Tang, Nat. Commun., 2021, 12, 6679 CrossRef CAS PubMed.
- H. Lai, H. Chen, J. Zhou, J. Qu, P. Chao, T. Liu, X. Chang, N. Zheng, Z. Xie and F. He, iScience, 2019, 17, 302–314 CrossRef CAS PubMed.
- C. Y. Xiao, C. Li, F. Liu, L. Zhang and W. W. Li, J. Mater. Chem. C, 2020, 8, 5370–5374 RSC.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- D. Meng, J. L. Yang, C. Xiao, R. Wang, X. Xing, O. Kocak, G. Aydin, I. Yavuz, S. Nuryyeva, L. Zhang, G. Liu, Z. Li, S. Yuan, Z. K. Wang, W. Wei, Z. Wang, K. N. Houk and Y. Yang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 20397–20403 CrossRef CAS PubMed.
- S. Liang, B. Liu, S. Karuthedath, J. Wang, Y. He, W. L. Tan, H. Li, Y. Xu, N. Li, J. Hou, Z. Tang, F. Laquai, C. R. McNeill, C. J. Brabec and W. Li, Angew. Chem., Int. Ed., 2022, 61, e202209316 CAS.
- K. Vandewal, K. Tvingstedt, A. Gadisa, O. Inganäs and J. V. Manca, Phys. Rev. B, 2010, 81, 125204 CrossRef.
- C. Kästner, K. Vandewal, D. A. M. Egbe and H. Hoppe, Adv. Sci., 2017, 4, 1600331 CrossRef PubMed.
- S. D. Collins, C. M. Proctor, N. A. Ran and T.-Q. Nguyen, Adv. Energy Mater., 2016, 6, 1501721 CrossRef.
- G. Garcia-Belmonte, P. P. Boix, J. Bisquert, M. Lenes, H. J. Bolink, A. La Rosa, S. Filippone and N. Martín, J. Phys. Chem. Lett., 2010, 1, 2566–2571 CrossRef CAS.
- N. I. Craciun, J. Wildeman and P. W. M. Blom, Phys. Rev. Lett., 2008, 100, 056601 CrossRef CAS PubMed.
- J. C. Blakesley and D. Neher, Phys. Rev. B, 2011, 84, 075210 CrossRef.
- S. Karuthedath, Y. Firdaus, R. Z. Liang, J. Gorenflot, P. M. Beaujuge, T. D. Anthopoulos and F. Laquai, Adv. Energy Mater., 2019, 9, 1901443 CrossRef.
- S. Karuthedath, J. Gorenflot, A. Melianas, Z. Kan, M. Kemerink and F. Laquai, J. Phys. Chem. Lett., 2020, 11, 2838–2845 CrossRef CAS PubMed.
- I. A. Howard, R. Mauer, M. Meister and F. Laquai, J. Am. Chem. Soc., 2010, 132, 14866–14876 CrossRef CAS PubMed.
- S. Karuthedath, A. Melianas, Z. Kan, V. Pranculis, M. Wohlfahrt, J. I. Khan, J. Gorenflot, Y. Xia, O. Inganäs, V. Gulbinas, M. Kemerink and F. Laquai, J. Mater. Chem. A, 2018, 6, 7428–7438 RSC.
- S. Karuthedath, Y. Firdaus, A. D. Scaccabarozzi, M. I. Nugraha, S. Alam, T. D. Anthopoulos and F. Laquai, Small Struct., 2022, 3, 2100199 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and measurements, synthesis, GPC, TGA, DSC, CV, OSCs, SCLC, EQEEL, AFM, GIWAXS, transient absorption spectroscopy, and NMR spectra. See DOI: https://doi.org/10.1039/d3ta01501g |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |