
Constructing a rhenium complex supported on g-C3N4 for efficient visible-light-driven photoreduction of CO2 to CO via a novel Z-scheme heterojunction†
Phuong Ngoc
Nguyen
 ab,
Trang Thanh
Tran
a,
Quynh Anh
Thi Nguyen
c,
Yoshiyuki
Kawazoe
def,
S. V. Prabhakar
Vattikuti
g,
Long V.
Le
h,
Viet Quoc
Bui
*c,
Tuan Manh
Nguyen
*bi and
Nam Nguyen
Dang
jk
ab,
Trang Thanh
Tran
a,
Quynh Anh
Thi Nguyen
c,
Yoshiyuki
Kawazoe
def,
S. V. Prabhakar
Vattikuti
g,
Long V.
Le
h,
Viet Quoc
Bui
*c,
Tuan Manh
Nguyen
*bi and
Nam Nguyen
Dang
jk
aInstitute of Applied Materials Science, Vietnam Academy of Science and Technology (VAST), 29TL Street, Thanh Loc Ward, District 12, Ho Chi Minh City 700000, Vietnam. E-mail: nguyenmanhtuan@iams.vast.vn; bqviet@ac.udn.vn
bGraduate University of Science and Technology, VAST, 18 Hoang Quoc Viet Street, Cau Giay, Ha Noi 100000, Vietnam
cAdvanced Institute of Science and Technology, The University of Danang, 41 Le Duan, Danang, Vietnam
dNew Industry Creation Hatchery Center, Tohoku University, 6-6-4 Aramaki Aza Aoba, Aoba-ku, Sendai, Miyagi, 980-8579 Japan
eSchool of Physics, Institute of Science, Suranaree University of Technology, 111 University Avenue, Nakhon Ratchasima, 30000 Thailand
fPhysics and Nanotechnoloy, SRM Institute of Science and Technology, Kattankurathur, Tamil Nadu, 603203 India
gSchool of Mechanical Engineering, Yeungnam University, Gyeongsan, Republic of Korea
hInstitute of Materials Science, Vietnam Academy of Science and Technology (VAST), Hanoi 100000, Vietnam
iInstitute of Applied Informatics and Mechanics, Vietnam Academy of Science and Technology (VAST), 291 Dien Bien Phu Street, Ward 7, District 3, Ho Chi Minh City 700000, Vietnam
jFuture Materials & Devices Lab., Institute of Fundamental and Applied Sciences, Duy Tan University, Ho Chi Minh City 700000, Vietnam
kThe Faculty of Environmental and Chemical Engineering, Duy Tan University, Danang 550000, Vietnam
First published on 3rd July 2023
Abstract
Visible-light-driven photocatalytic CO2 reduction is a promising approach to addressing the problem of global warming and the energy crisis. A Z-scheme photocatalyst comprising a Re(I) complex and a polymeric semiconductor (bulk graphitic carbon nitride (g-C3N4)) converted CO2 to CO even under the irradiation of low-intensity visible light. The electronic interaction between Re(I) and g-C3N4 units and the injection of electrons from g-C3N4 to the Re(I) complex improved the photocatalytic efficiency. The lower recombination of electron–hole pairs and the prolonged emission decay (the average emission lifetime τ = 5.8 ms) contributed to the increased overall efficiency of the hybrid system. The maximum turnover number (TON) of CO formation reached 28.56 after 240 min with a high CO selectivity (99.8%). Compared to the homogeneous photocatalyst Re(I), the TON of CO formation was increased by 5.6 times and CO selectivity was higher. Density Functional Theory (DFT) was employed to investigate the impact of the substrate-supported Re complex (Re(bpy-COOH)/g-C3N4) on the CO2 Reduction Reaction (CO2RR) activity. The results indicate that Re(bpy-COOH)/g-C3N4 presents a lower energy barrier for *CO2 to *COOH conversion, promoting *COOH formation more effectively than pure g-C3N4. Moreover, the energy barrier for *CO desorption is lower in Re(bpy-COOH)/g-C3N4 than in Re(bpy-COOH), suggesting a reduction in the strong binding of adsorbed CO intermediates, highlighting the intermediate ensemble effect at the active site. The improved CO2RR activity in Re(bpy-COOH)/g-C3N4 can be attributed to the balance between the enhancement of reaction activity and the binding energy of intermediates at the active site. The study of photocatalytic CO2 reduction (PCO2R) under low-intensity-visible-light irradiation and DFT investigation has reveal an insight into the CO2RR activity for CO2 reduction under various irradiation conditions.
1. Introduction
Carbon dioxide is known as a heat-trapping gas, which causes global heating. Heavy industries and human activities are the main CO2-emission sources that primarily impact global climate change and relate to severe natural events, including wildfires, floods, and droughts. Concerning climate change, several ways have been attempted to reduce CO2 concentration, such as using sustainable energy sources instead of burning fossil fuels and utilizing CO2 as a new chemical feedstock for producing energy-rich carbon compounds. Solar energy conversion of CO2 into carbon compounds has been a promising research field that can solve global warming, shortage of fossil fuels, energy crisis, etc. To this end, numerous investigations have been carried out to develop many different kinds of visible-light-driven photocatalysts. Transition metal complexes (e.g., Ru, Mn and Re) have been known to be potential homogeneous photocatalysts for CO2 reduction with high selectivity and activity.1–3 They possess a long-lived excited state,4,5 tunable structure via changing the coordination ligand to the metal center,6,7 and intense emission properties.8 However, transition metal complexes, which have low quantum efficiency9 and strong absorption in the UV region,4 are high-cost and scarce, and could suffer from progressive structural degradation because of photochemical instability that may severely reduce catalytic performances. For those reasons, various hybrid photocatalytic materials including molecular/semiconductor hybrid materials10–15 and transition metal complex/nanoparticle16 hybrid materials have been introduced to overcome the drawbacks of homogeneous photocatalytic systems. [Re(2,2′-bipyridine-4,4′-bisphosphonic acid) (CO)3(L)]n+ (ReP; L = 3-picoline or bromide) immobilized on TiO2 nanoparticles17 was reported for a visible-light-driven hybrid displaying improvement in CO2 reduction with the turnover number (TON) increasing 24 times in comparison with a ReP homogeneous photocatalyst (the TON of the ReP–TiO2 hybrid was observed to be 48 molCO molRe−1 in DMF). The metal–organic framework (MOFs supported Ru carbonyl complex18 for PCO2R under visible light showed twofold enhanced catalytic activity compared to the pure Ru carbonyl complex.To date, the incorporation of metal complexes with a semiconductor to achieve visible-light-driven photocatalysts19 has attracted attention, and the metal complexes/semiconductor hybrid system15 can utilize the advantages of both metal complexes and semiconductors, as well as reducing the usage of noble metals. To design a metal complexes/semiconductor system, the conduction band of semiconductors and the reduction potential of metal complexes need to be compatible to maximize electron transfer between the two components for efficient CO2 photocatalysis conversion. The rational structure can be achieved by adsorption or by covalent links between metal complexes and semiconductors. Generally, semiconductors play the role of a light absorber and host oxidation reaction sites. Depending on the conduction-band (CB) potential of the semiconductor and the lowest occupied molecular orbital (LUMO) of the metal complexes, the system can be categorized into a hybrid system in which semiconductors function as photosensitizers and metal complexes catalyze CO2 when the CB potential of semiconductors is more negative than the LUMO of metal complexes, and a Z-scheme system when the CB potential of semiconductors is less negative than the LUMO of metal complexes. In the Z-scheme system, both the semiconductor and metal complex undergo photo-excitation together, resulting in electron transfer and enhancement in the driving force of interfacial electron transfer.
Graphitic carbon nitride has been known as a polymeric semiconductor with high chemical stability and a narrow band gap (∼2.7 eV, corresponding to an absorption edge of ≈460 nm), low cost, and ease of preparation.20,21 C3N4 has been applied for photocatalytic reactions, including water reduction/oxidation22 and environmental treatment.23–25 However, g-C3N4 still has common issues during a photocatalytic process such as insufficient visible-light absorption,26 low quantum yield and fast recombination of photogenerated carriers27,28 that restrict the photocatalytic performance. Therefore, heterojunctions based on g-C3N4 with two or three semiconductor materials have been tailored to integrate their own advantages. The g-C3N4 based heterojunctions have been used for PCO2R where the formed products were non-selective and abundant, containing long-chain hydrocarbons (CO, HCOOH, acetone, methanol, and acetaldehyde).29,30 g-C3N4 has a conduction band and valence band at −1.4 eV and 1.34 eV (vs. NHE, pH = 7),31,32 respectively, which are suitable for architecting a hybrid photocatalyst with metal complexes, and have thus been reported for the high selectivity for formed products. In a hybrid CO2-reduction system, bulk g-C3N4 was explored as a semiconductor in conjunction with Ru complexes11,33,34 through carboxylic or phosphonic acid anchoring groups for converting CO2 to formic acid (HCOOH) under visible light (λ > 400 nm). The combination of g-C3N4 and Ru complexes maximized the CO2-reduction efficiency process and improved photocatalytic performance and reached the highest TON (>1000). A manganese (Mn) complex with phosphonic acid anchoring groups in conjunction with bulk g-C3N4 forming a Z-scheme system35 was reported for efficient visible-light photoreduction of CO2 to CO, where the TON of the hybrid system was increased two-fold in comparison with the base Mn complex. Other than Ru and Mn complexes, Ni and Co complexes have been used to combine with g-C3N4 to design hybrid materials for PCO2R.14,36,37 Meanwhile, Re(CO)3(N–N)Cl complexes have not been reported for catalytic CO2 reduction in metal complexes/semiconductor hybrid systems. They are appropriate to incorporate with g-C3N4 to form an efficient visible-light-driven photocatalyst for CO2 conversion, because Re(CO)3(N–N)Cl complexes have efficient CO2-capturing ability, better than that of Mn complexes in the lower CO2 concentration in the presence of TEOA solvent.38 Re(CO)3(bpy-COOH)Cl with a dicarboxylic acid anchor and more negative LUMO potential than the CB of g-C3N4 will form a Z-scheme system, which can enhance efficient CO2 reduction. The adsorption is likely via hydrogen bonding between NH2 groups on g-C3N4 and –COOH anchors. Additionally, the hybrid system can efficiently convert CO2 under low-intensity irradiation which is important for investigating photocatalytic systems working under various illumination.
In this study, we report the straightforward synthetic process of a hybrid material Re(bpy-COOH)/g-C3N4. The characterization of chemical functional groups, chemical states, optical properties, and the morphology was performed by FTIR, XPS, UV-vis DRS, PL, and PL decay spectroscopy, and using SEM and TEM images. PCO2R to CO of this hybrid material was studied in DMF/TEOA mixed solution under low-intensity visible light irradiation (21 mW cm−2) for the first time. Utilizing density functional theory (DFT) calculations, we examined the influence of Re(bpy-COOH)/g-C3N4 on CO2 Reduction Reaction (CO2RR) activities, including factors such as activation energies and charge transfer between intermediates and substrates. Our findings indicate that CO2RR in Re(bpy-COOH)/g-C3N4 is favorable, facilitated by an increase in reaction activity at the active site and a balanced binding energy of both *COOH and *CO intermediates. These results are critical in understanding the photocatalytic activity of this system under sunlight irradiation under various daytime conditions, providing a pathway for further research in this area.
2. Experimental section
Urea (NH2CONH2), pentacarbonylchlororhenium(I) [Re(CO)5Cl], 2,2′-bipyridine-4,4′ dicarboxylic acid (bpy-COOH), and anhydrous N,N-dimethylformamide (DMF) were purchased from Sigma-Aldrich. Triethanolamine (TEOA – (HOCH2CH2)3N), toluene, methanol, acetone, and hexane solvents were of HPLC grade and were obtained from Merck. All solvents were used without further purification. CO2 and argon (Ar) gases used in all experiments were 99.999% purified.Bulk g-C3N4 was prepared by calcining urea (20 g) under air at 550 °C for 2 hours at a temperature ramp rate of 2 °C min−1 in a muffle furnace. The urea was placed in an alumina crucible with a cover. Then, the muffle furnace was cooled to room temperature, and the resulting light yellow powder was collected as bulk g-C3N4, which was ground finely. The Re(CO)3(bpy-COOH)Cl complex was synthesized following a previous report.39 The complex was synthesized by refluxing the mixture of [Re(CO)5Cl] (200 mg, 0.55 mM) and 0.55 mM bpy-COOH ligand in 50 mL toluene for 6 hours under an Ar atmosphere to give a red-orange solution ([Re(CO)3Cl(2,2′-bipyridine-4,4′ dicarboxylic acid)] Re(bpy-COOH)). Re(bpy-COOH) was recrystallized from acetone-hexane. Yield: 70%. νCO/cm−1 (in MeCN): 2041, 1956, 1897, 1H NMR (500 MHz, d6 – acetone, ppm) δ = 9.33 (2H, d, bpy H6, H6′), 9.18 (2H, s, H3, H3’), 8.24–8.26 (2H, m, H5, H5′). ESI-MS (in MeCN): m/z: (M+ for [C15H8N2O7Re]+), 512.9 (M3+ for [C15H6N2O7Re]+. UV/Vis (in MeCN): 270–350 nm (ligand-to-ligand charge transfer (LLCT)), 350–475 nm (metal-to-ligand charge transfer (MLCT)) (ESI-MS and UV/Vis spectra are shown in Fig. S1b, S2, and S3†).
The hybrid photocatalyst Re(bpy-COOH)/g-C3N4 was prepared following a previously reported process.33,35 The synthesized g-C3N4 (50 mg) was added to NaOH (50 mL, 0.1 M), stirring the mixture for 10 minutes. Then, the mixture was ultrasonically cleaned with ionized water (100 mL) and separated by centrifugation. Re(bpy-COOH)/g-C3N4 with a mol ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 was prepared by dispersion of g-C3N4 (46 mg) in methanol (20 mL) and the suspension was ultrasonicated for 1 hour. Then, 2.75 mg of Re(bpy-COOH) was added to g-C3N4 suspension and the mixture was stirred for 24 hours in the dark and under room temperature. Re(bpy-COOH)/g-C3N4 was collected by filtration and washed with methanol.
:100 was prepared by dispersion of g-C3N4 (46 mg) in methanol (20 mL) and the suspension was ultrasonicated for 1 hour. Then, 2.75 mg of Re(bpy-COOH) was added to g-C3N4 suspension and the mixture was stirred for 24 hours in the dark and under room temperature. Re(bpy-COOH)/g-C3N4 was collected by filtration and washed with methanol.
After Re(bpy-COOH)/g-C3N4 was prepared and collected by filtration. The filtrate was kept in a closed vial and its volume was known. The adsorbed amount (Ads_A) of Re(bpy-COOH) on g-C3N4 was calculated based on the UV/Vis absorption spectrum of the filtrate, using the following equation:
 | (1) |
The crystalline structure of the prepared g-C3N4 was confirmed by using powder X-ray diffraction (XRD) patterns using the D8 Advance XRD with Cu Kα radiation (λ = 0.15418 nm), scanning from 10–70°. The chemical structure of the Re(bpy-COOH) complex was determined by using the proton nuclear magnetic resonance (1H NMR) spectrum obtained using a Bruker Avance II (500 MHz) in acetone-D6. Electrospray ionization mass spectrometry (EIS-MS) of Re(bpy-COOH) was performed in acetonitrile (CH3CN) using an X500R QTOF. The chemical functional groups were determined by Fourier transform infrared (FTIR) spectroscopy in KBr pellets using a PerkinElmer MIR/NIR Frontier instrument in the range of 4000–800 cm−1. The chemical states of the materials were tested using a Thermo Scientific X-ray photoelectron spectroscopy (XPS) instrument with Al Kα radiation. The morphology was observed by using a scanning electron microscope (SEM – Hitachi SM- 4800) and transmission electron microscope (TEM- HRTEM, Tecnai G2 F20 S-Twin). The absorption of g-C3N4 and Re(bpy-COOH)/g-C3N4 was measured by UV-vis diffuse reflectance spectroscopy (UV-vis DRS) performed on a UV-2600 (Shimadzu) in the wavelength range of 300–700 nm. The photoluminescence (PL) spectra were identified on a Cary Eclipse Fluorescence Spectrophotometer (Varian, USA) at 370 nm at room temperature. The emission decay profiles of Re(bpy-COOH)/g-C3N4 and g-C3N4 at 450 nm were recorded by using a 0.6 m grating monochromator (Jobin-Yvon HRD1) and detected with a fast photomultiplier (Hamamatsu model H733, with a rise time of 700 ps), using a 355 nm laser diode as the excitation source.
Photoelectrochemical experiments: Photocurrent transient response and electrochemical impedance spectroscopy (EIS) measurements were carried out on a potentiostat Biologic VSP-300 using a three-electrode system cell with a working electrode, a Ni mesh counter electrode, and an Ag/AgCl as the reference electrode in a 0.5 M Na2SO4 electrolyte. The working electrode was prepared by coating the as-prepared samples (g-C3N4 and Re(bpy-COOH)/g-C3N4) on fluorine-doped tin oxide (FTO) glass. The FTO glass with a size of 2.5 × 2.5 cm was washed in the following order with acetone, ethanol, and DI water under sonication, and then dried in a N2 flow. Dispersing the mixture of a certain amount of sample and polyvinyl alcohol (PVA) in distilled water, the obtained slurry was coated on a FTO glass substrate (photoactive area of 1.2 × 1.2 cm) by the spin-coating technique, and dried at 70 °C in 30 min. Photocurrent transient response was recorded under an applied bias of 0.35 V and irradiated with visible light (15 W energy-saving light bulk, center wavelength λ = 408 nm, and light intensity at a distance of 5 cm of 210 W m−2, see Fig. S4†) with light chopping every 30 s. Electrochemical impedance spectroscopy (EIS) plots were obtained over the frequency range from 0.1 to 105 Hz in the dark.
The photocatalytic CO2 reaction was carried out in a reaction solution (0.75 mL) containing a photocatalyst (Re(bpy-COOH) (0.6 mM) and Re(bpy-COOH)/g-C3N4 (1 mg)) in DMF:TEOA mixed solvent (5:1 v/v). The reaction solution was placed in a visible-light-transparent vial (5 mL) and purged with Ar to remove all air, and then CO2 gas was purged for 10 min. Then, the reaction vial was irradiated with a light-source system (15 W energy-saving light bulk, λ = 408 nm, and light intensity at a distance of 5 cm of 210 W m−2, see Fig. S4†) in the photoreaction box. During irradiation, the solution was stirred slowly with a magnetic bar and the temperature of the solution was controlled at room temperature by using a constant temperature system. The gaseous reaction products were identified by using a gas chromatograph (GC Clarus 680- PerkinElmer), which was equipped with a TCD detector, molecular sieve column (L × I.D.: 30 2 m × 0.32 mm), and Ar carrier gas. The turnover number TON of CO and H2, and the selectivity of CO were calculated by using eqn (2)–(4), respectively.
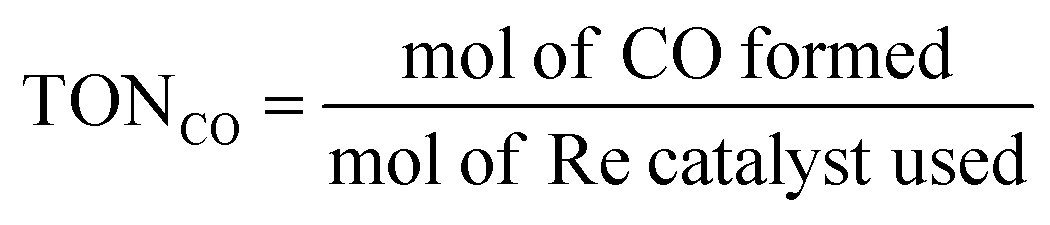 | (2) |
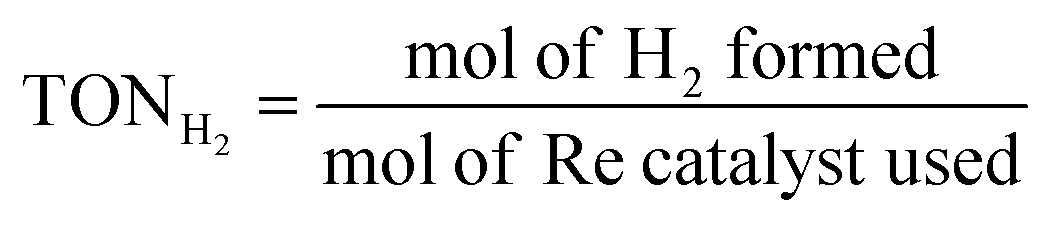 | (3) |
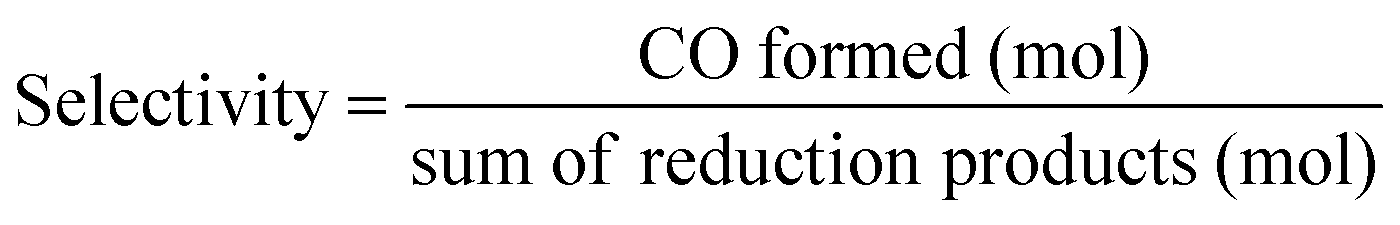 | (4) |
Spin-polarized density functional theory (DFT) calculations were performed using the Vienna ab initio simulation package (VASP).40,41 The generalized gradient approximation (GGA) with parameterization by Perdew, Becke, and Ernzerhof (PBE)42 was employed to describe the exchange–correlation interactions. The interaction between core electrons and valence electrons was treated with the projector-augmented wave (PAW)43 method. The DFT-D3 correction method in Grimme's scheme44 was used to describe the long-range van der Waal interactions between atoms. A cutoff energy of 400 eV was chosen for wave function expansion. For Brillouin zone integration, only a Γ k-point with a Gaussian broadening of 0.05 eV was used. Crystal orbital Hamilton population (COHP) and charge transfer analyses were performed using the Lobster program.45 To investigate the thermodynamics of CO2RR, the Gibbs free energy (ΔG) was calculated using the equation proposed by Nørskov's group:46
| ΔG = ΔE + ΔEZPE − TΔS + ΔGpH + ΔGU | (5) |
In our simulation model, the Re(bpy-COOH) molecule was placed in a cubic unit cell with a = 20 Å, while g-C3N4 and Re(bpy-COOH)/g-C3N4 were constructed using a 2 × 2 hexagonal unit cell with an optimized lattice constant a = 13.98 Å. A vacuum spacing of 15 Å was selected to minimize the artificial interaction between the neighboring slabs. All atomic coordinates are fully relaxed with a force criterion of 1 meV Å−1.
3. Results and discussion
The 1H NMR and ESI-MS confirmed the high purity and the well-structured molecule of the Re(bpy-COOH) complex, as presented in Fig. S1 and S2.† The XRD pattern showed the characteristic diffraction peaks in the crystalline structure of g-C3N4 (Fig. S1(a)†). The two diffraction peaks centered at 13.10 and 27.32°ᵒ correspond to the (100) and (002) crystal planes of graphite phase carbon nitride, respectively, which were in accordance with the planar structure stacking peak of aromatic systems and inter-layer structural packing.47The hybrid material Re(bpy-COOH)/g-C3N4 was optically characterized by FTIR, UV-vis DRS, PL spectroscopy, and TRPL measurement, as shown in Fig. 1. As seen in Fig. 1a, the FTIR spectra showed that the characteristic peaks of g-C3N4 in the range of 1650–800 cm−1 did not change after combining with Re(bpy-COOH). The appearance of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching modes in the FTIR spectrum of the hybrid material, ranging from 2100 to 1800 cm−1, confirmed the successful loading of the Re(bpy-COOH) complex on g-C3N4.
O stretching modes in the FTIR spectrum of the hybrid material, ranging from 2100 to 1800 cm−1, confirmed the successful loading of the Re(bpy-COOH) complex on g-C3N4.
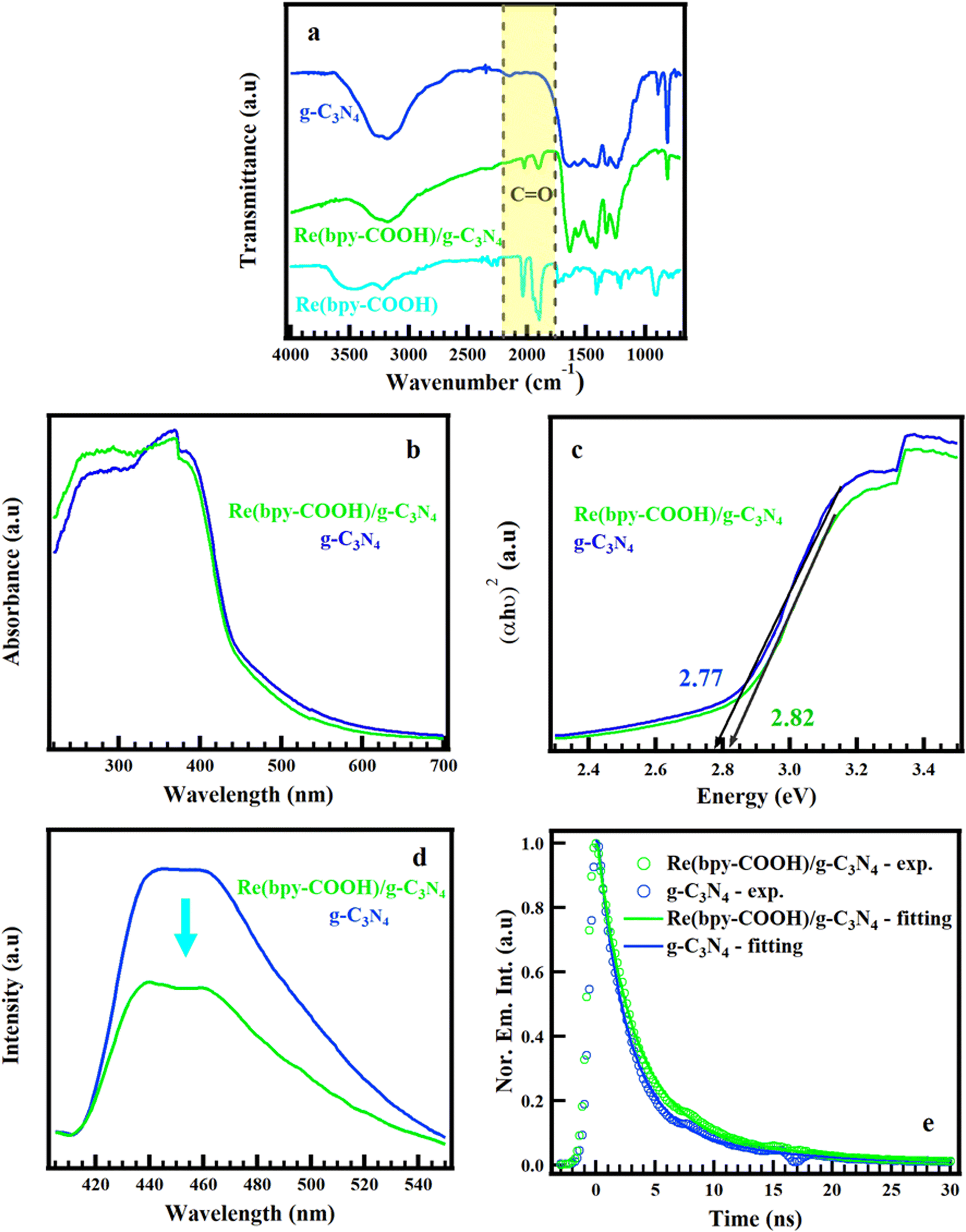 | ||
| Fig. 1 Fourier transform infrared spectra (a), UV-vis diffuse reflectance spectra (b), band gap energy of the corresponding UV-vis DRS determined from the Tauc plot (c), photoluminescence spectra under 365 nm of irradiation (d), and emission decay profiles of Re(bpy-COOH)/g-C3N4 and g-C3N4 (e) at λex = 355 nm and λem = 450 nm without the quenching factor. | ||
The UV-vis DRS spectra and the calculated optical band gaps of the hybrid Re(bpy-COOH)/g-C3N4 and g-C3N4 are presented in Fig. 1(b and c). The g-C3N4 showed broad band absorption in the visible region with a bandgap edge of approximately 2.77 eV which was assigned to the π–π* transition of conjugated heptazine units. This result was consistent with previous reports.48 The absorption edge of the hybrid material Re(bpy-COOH)/g-C3N4 exhibited an insignificant blue-shift from 2.77 to 2.83 eV (∼447–4.38 nm), and the increase of the absorption from 250–315 nm could account for the strongest absorption of Re(bpy-COOH) in the UV region from 250–350 nm (Fig. S3†). Both of the hybrid Re(bpy-COOH)/g-C3N4 and g-C3N4 had the tailing absorption extended to 550 nm, which was ascribed to n–π* transitions involving lone pairs on the edge nitrogen atoms of the heptazine rings.49
Fig. 1c presents the PL of the hybrid Re(bpy-COOH)/g-C3N4 and g-C3N4, which had strong luminescence emission in the range of 420–550 nm. Anchoring Re(bpy-COOH) on g-C3N4, the PL emission peak of the hybrid material was slightly narrow and showed two separated emission peaks at 435 and 455 nm. The PL intensity of Re(bpy-COOH)/g-C3N4 was lower than that of g-C3N4, which indicated that Re(bpy-COOH)/g-C3N4 underwent less recombination of photo-induced electron–hole pairs than g-C3N4 did. The lower recombination rate would lead to the higher photocatalytic activity of Re(bpy-COOH)/g-C3N4. The emission decay profiles of Re(bpy-COOH)/g-C3N4 and g-C3N4 under excitation of 355 nm, and emission of 450 nm without an electron donor source (TEOA), and the fitting curve are shown in Fig. 1e. The emission decay of (bpy-COOH)/g-C3N4 and g-C3N4 had the timescale of nanoseconds, which agreed with a previous report from Zhidong Wei.49,50 The complex [Re(CO)3(N–N)Cl] (N–N: polypyridine) displayed its emission lifetime on the nanosecond timescale (as shown in Fig. S7 in the ESI†).51 The emission lifetime of the hybrid and g-C3N4 was fitted to a triple exponential function and the fitting data are summarized in Table S1.† The lifetime of the hybrid was prolonged (average emission lifetime τ = 4.34 ns) compared to g-C3N4 (τ = 3.47 ns). Under 355 nm excitation, g-C3N4 and Re(bpy-COOH) underwent dual-photoexcitation, resulting in interfacial electron transfer from the conduction band of g-C3N4 to the HOMO of Re(bpy-COOH), minimizing recombination with holes in the valence band. It is noted that the COOH anchor was reported as intrinsically superb in terms of electron transfer due to the greater degree of nonadiabatic coupling,12 which was important for the overall efficiency of the hybrid system. These migrated electrons localized on the HOMO and prevented the combination of photo-induced electrons of Re(bpy-COOH) with holes. That could account for the prolong decay time and the decrease of emission intensity of the PL spectrum. The longer emission lifetime was favorable for promoting PCO2R.
The morphological surface of the materials was observed by using SEM and TEM images, as shown in Fig. 2. The SEM images of g-C3N4 showed the planar structure stacking while Re(bpy-COOH) exhibited a crystal-like morphology with sizes varied from a few dozen to a hundred nanometers, as presented in Fig. 2(a) and (b). The TEM image (Fig. 2(c) and (e)) showed that Re(bpy-COOH) had a particle shape with the size of a few hundred nm and g-C3N4 possessed a sheet-like morphology. Fig. 2(d) and (f) show the morphological images of the hybrid with Re(bpy-COOH) successfully loaded on the surface of g-C3N4.
 | ||
| Fig. 2 SEM images of g-C3N4 (a) and Re(bpy-COOH) (b), and TEM images of g-C3N4 (c), Re(bpy-COOH)/g-C3N4 (d and f), and Re(bpy-COOH) (e). | ||
XPS spectra determined the surface chemical state of g-C3N4, Re(bpy-COOH), and Re(bpy-COOH)/g-C3N4 and the interaction or bonding between Re(bpy-COOH) and g-C3N4 (Fig. 3). Fig. 3a shows the full scan of g-C3N4, Re(bpy-COOH), and Re(bpy-COOH)/g-C3N4. The XPS survey spectrum of g-C3N4 showed the typical structure of graphitic carbon nitride which showed the existence of the elements C, N, and O on the surface. Element O can be the impurity species absorbed on the surface of g-C3N4. The full scan of Re(bpy-COOH) consisted of the peaks of Re (Re 4f and Re 4d), C, N, Cl and O, clearly from the structure of the Re complex, and the obtained spectrum reached an agreement with a previous report of this complex.52 The surface state of Re(bpy-COOH)/g-C3N4 was composed of peaks of Re (Re 4f and Re 4d), C, N, and O. The high-resolution scan of the Re 4f spectrum of the hybrid material Re(bpy-COOH)/g-C3N4 included Re 4f5/2 and 4f7/2 peaking at 44.78 eV and 47.18 eV, respectively; meanwhile the Re 4f spectrum of Re(bpy-COOH) showed those 4f peaks at 45.23 eV and 48.05 eV (Fig. S5(a)†). The peak displacement of Re 4f5/2 and 4f7/2 indicated the interaction between Re with the surrounding elements.
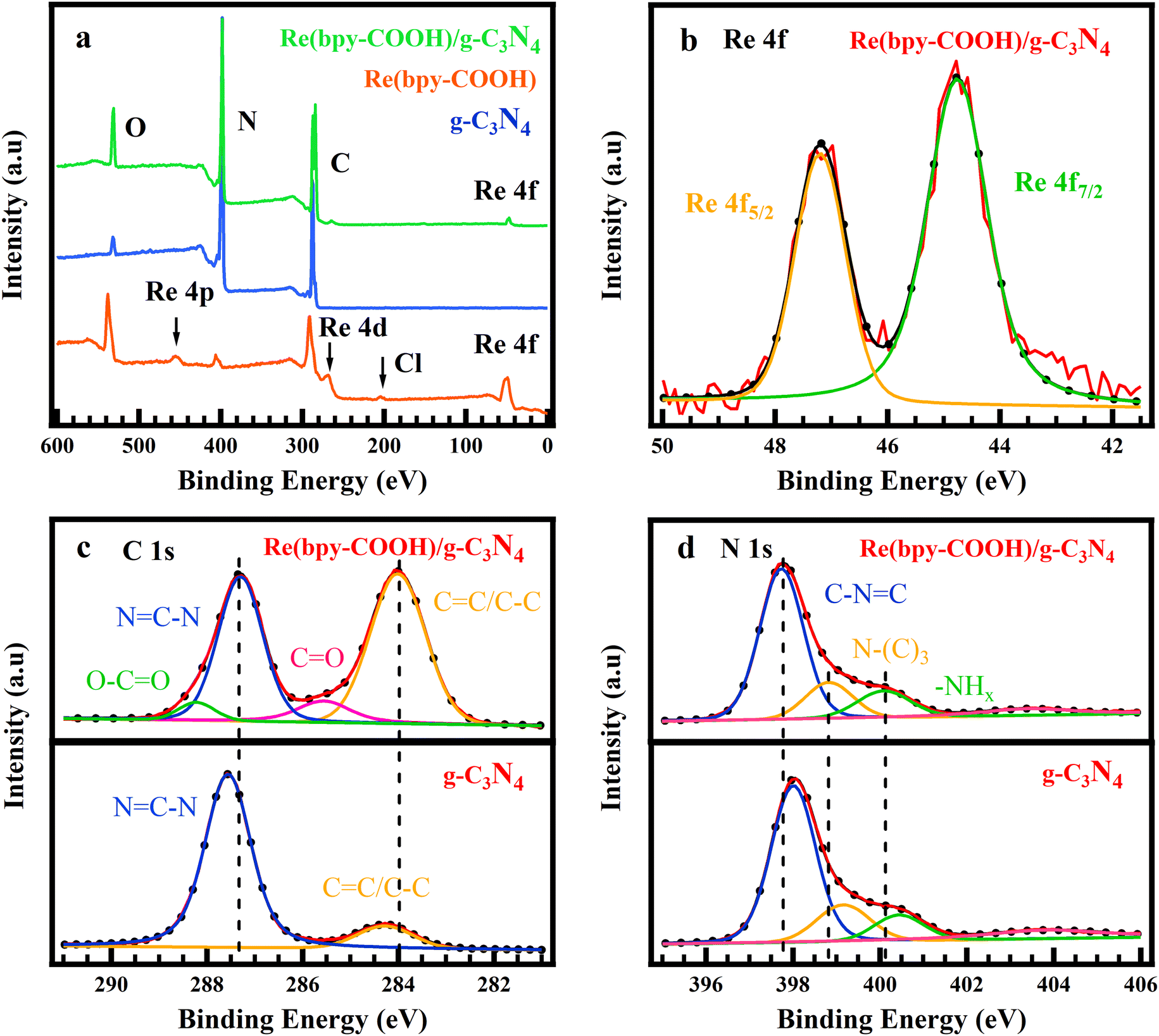 | ||
| Fig. 3 XPS survey scan of g-C3N4, Re(bpy-COOH), and Re(bpy-COOH)/g-C3N4 (a), high resolution scan of Re 4f of Re(bpy-COOH)/g-C3N4 (b), C 1s (c), and N 1s (d) of Re(bpy-COOH)/g-C3N4 (top) and g-C3N4 (bottom). | ||
Fig. 3c presents the high-resolution XPS spectra for C 1s of Re(bpy-COOH)/g-C3N4 and g-C3N4. The C 1s spectrum of Re(bpy-COOH)/g-C3N4 showed the peak changes in the position and intensity, and new peaks appeared at binding energies 285.6 and 288.2 eV assigned to CO and O–CO, respectively. CO and O–CO were the featured groups characterized for Re(bpy-COOH), and hence their appearance on the C 1s spectrum of Re(bpy-COOH)/g-C3N4 indicated the successful linking between Re(bpy-COOH) and g-C3N4. The blue-shift of O–CO at 285.6 eV in Re(bpy-COOH)/g-C3N4 from 286.0 to 285.6 eV (compared to O–CO in Re(bpy-COOH), see Fig. S5(b)†) determined the chemical bonding of the anchor group –OOC of Re(bpy-COOH) on g-C3N4. The binding energy at 287.6 and 284.3 eV corresponded to typical sp2 bonded-carbon NC–N and CC/C–C of g-C3N4, respectively, which reached agreement with previous reports.53
The N 1s spectra were deconvoluted into three peaks at 398.0, 399.2, and 400.5 eV, which were assigned to sp2-hybridized pyridine N (C–NC), tertiary pyrrolic N (N-(C)3), and graphitic N, respectively54,55 (Fig. 3d). The binding energy of the corresponding peaks obtained in N 1s of Re(bpy-COOH)/g-C3N4 (397.8, 398.9, and 400.1 eV) were shifted by 0.2, 0.3, and 0.4 eV, respectively. The peak shifts revealed the interfacial interaction between Re(bpy-COOH) and g-C3N4.
Fig. 4 represents the (EIS) results in the form of Nyquist, Bode, and transient photocurrent response of g-C3N4 and Re(bpy-COOH)/g-C3N4. The reduction of the arc diameter as shown in Fig. 4a suggests a decrease of film and charge transfer resistance, indicating an enhanced charge transfer of g-C3N4 by Re(bpy-COOH) addition. Fig. 4b indicates the local surface defects that are detected at the high-frequency data, the pore and substrate/film interface can be detected at the medium and low-frequency data, respectively. It also indicates a significant reduction in the impedance aperture and phase angles when Re(bpy-COOH) was doped in g-C3N4. The data indicate two phase constants in an equivalent circuit inserted in Fig. 4a. This equivalent circuit was combined with the ZSimpWin program to determine the optimized charge transfer resistance values of the investigated specimens. The fitting results indicated that the charge transfer resistance values reached 1.645 × 105 and 1.182 × 105 for g-C3N4 and Re(bpy-COOH)/g-C3N4 specimens, respectively. The significant reduction in charge transfer resistance values suggested that Re(bpy-COOH) doping increases the resistivity of g-C3N4. The current response upon light irradiation of g-C3N4 and Re(bpy-COOH) was undetermined, whereas that of Re(bpy-COOH)/g-C3N4 showed a large (approximately 1 (μA cm−2)) and fast rising of the current (Fig. 4c). The slightly attenuation of the current density was observed after the second cycle. As expected, linking Re(bpy-COOH) and g-C3N4 enhanced electronic communication and reduced the recombination of photo-induced carriers.
 | ||
| Fig. 4 Electrochemical impedance spectroscopy (EIS) results in the form of Nyquist (insert equivalent circuit) (a) and Bode plots (b), and photocurrent under visible light in Na2SO4 of g-C3N4 and Re(bpy-COOH)/g-C3N4 (c). | ||
PCO2R of g-C3N4, Re(bpy-COOH) and Re(bpy-COOH)/g-C3N4 was conducted in DMF:TEOA mixed solution under low-intensity visible irradiation. Generally, photocatalytic activity was tested under standard test conditions of AM 1.5G, 100 mW cm−2.56 This investigation studied the photocatalytic CO2 conversion and the selectivity of the hybrid system under low-light intensity. DMF solvent was the best choice for the highest selectivity of CO formation. TEOA was used as an effective sacrificial electron donor that scavenges holes generated in the valence band of g-C3N4. As shown in Fig. 5a and b, for the photocatalytic system with g-C3N4 as the photocatalyst, there was no CO and H2 formation found, which met agreement with a previous report.57 The CO generation of the hybrid system reached the highest TON = 28.56 at 240 min and then reached saturation, while the highest TON of Re(bpy-COOH) was 5.6 at 120 min before saturation, as shown in Fig. 5a. Ono et al. reported the total amount of CO production of Re(bpy-COOH) reached TONCO = 6.59 for 2 h.58 In our experiment, the TONCO of Re(bpy-COOH) obtained was lower than that in the Ono report, which can be explained by the low-intensity irradiation used. The amount of CO formation increased significantly when Re(bpy-COOH) was anchored to g-C3N4 (increased by 5.6 times at 240 min of irradiation), indicating that Re(bpy-COOH) and g-C3N4 combination enhanced the efficiency of the PCO2R.
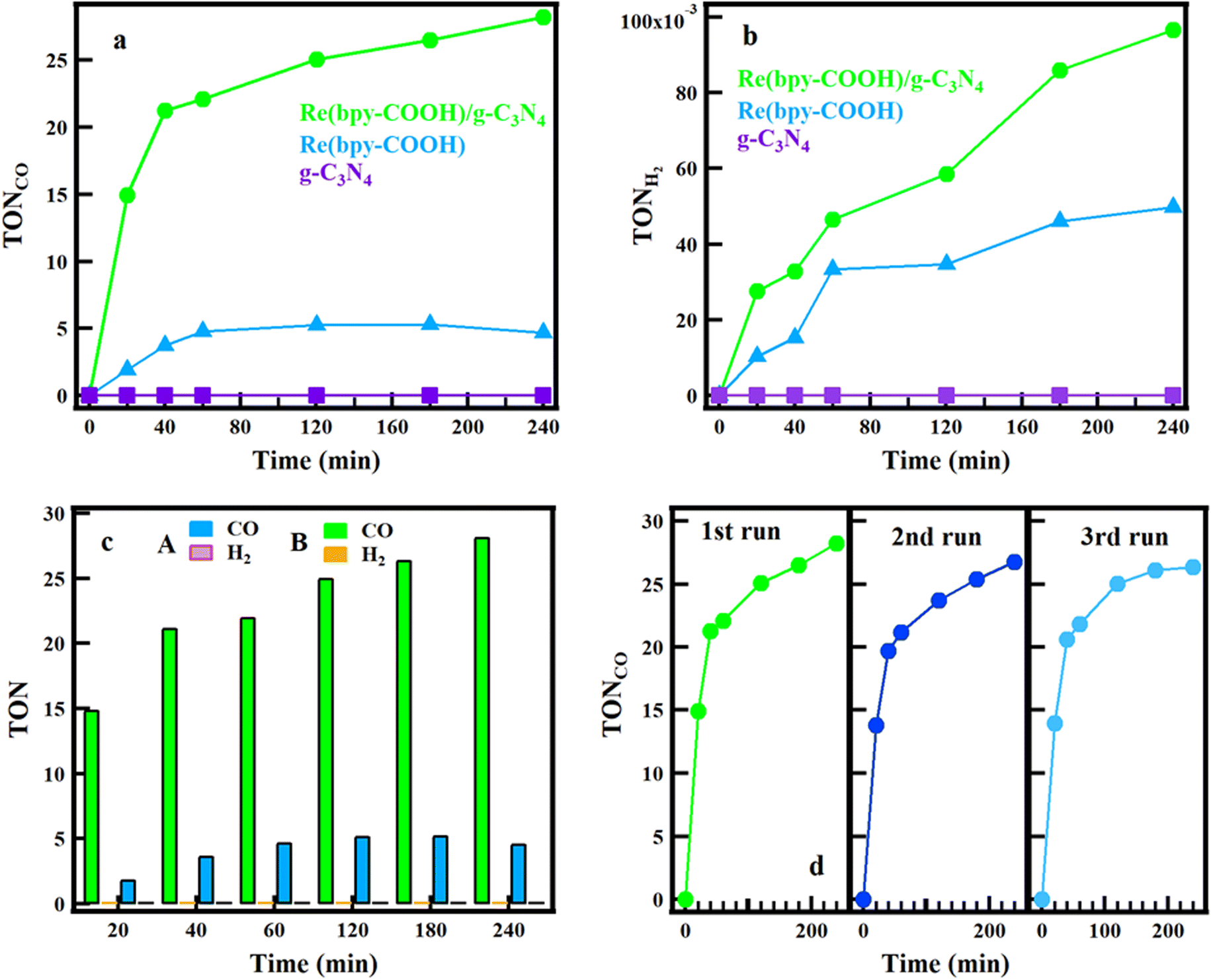 | ||
| Fig. 5 The formation of Co (a) and H2 (b) during photocatalytic reduction by time of g-C3N4, Re(bpy-COOH), and Re(bpy-COOH)/g-C3N4, the comparison of Co and H2 formation of Re(bpy-COOH) (a – Co: turquoise blue and H2: pink), and Re(bpy-COOH)/g-C3N4 (Co: green and H2: orange) (c), and three consecutive 240 min irradiation cycles using the same hybrid material (d). | ||
Hydrogen formation of the three materials is presented in Fig. 5b. Namely, the TON of H2 formation reached 0.1 and 0.05 for Re(bpy-COOH)/g-C3N4 and Re(bpy-COOH) at 240 min, respectively. The comparison of CO and H2 formation of Re(bpy-COOH)/g-C3N4 and Re(bpy-COOH) is shown in Fig. 5c, in which the amount of CO was dominant. Table 1 shows the TON of CO and H2 formation, and the selectivity of CO of Re(bpy-COOH) and Re(bpy-COOH)/g-C3N4 at 60 min. The selectively of CO formation was determined to be 99.4 and 99.8% for Re(bpy-COOH) and Re(bpy-COOH)/g-C3N4, respectively (as shown in Table 1). [Re(CO)3(N–N)Cl] complexes presented high CO selectivity (greater than 90%), and the CO selectivity of Re(bpy-COOH) under low-intensity irradiation was evaluated to be 99.4% that agreed with previous studies.58 The CO selectivity of the hybrid system was greater than that of the homogeneous catalyst, and thus combining Re(bpy-COOH) with g-C3N4 improved the photocatalytic performance and the selectivity of CO.
| Photocatalyst | TONCO | TONH2 | CO selectivity (%) |
|---|---|---|---|
| g-C3N4 | — | — | — |
| Re(bpy-COOH) | 5.14 | 0.03 | 99.4 |
| Re(bpy-COOH)/g-C3N4 | 22.44 | 0.05 | 99.8 |
To study the stability, the PCO2R of the same hybrid material was re-employed for three successive cycles. After each cycle, the hybrid material was washed with acetonitrile and then dried at 70 °C. After that, the hybrid material was dispersed into DMF:TEOA solution as the preparation process for PCO2R measurement in the experimental section and then started the new cycle. The results (Fig. 5d) showed the stability of CO production in the three cycles. The CO formation rate and generation remained stable in three cycles; however, in the second and third cycles, a slightly insignificant reduction of CO yield was observed. That could be explained by the photo-degradation of Re(bpy-COOH) under irradiation and the loss of the catalyst due to collecting and washing the catalyst after each cycle run.
This hybrid system and a similar hybrid system Mn(I)/g-C3N4 (Mn(I):Mn(bipyridineCOOH)(CO)3Br) possessed similar photocatalytic properties such as photoreduction CO2 products (PCO2P) (CO and H2) and photocatalytic structure (Z-scheme model and g-C3N4 surface with the –COOH anchor). The comparison of the two systems showed that the enhancement of PCO2P of the Re(bpy-COOH)/g-C3N4 hybrid was lower than that of the Mn(I)/g-C3N4 system.57 Namely, TONCO was increased approximately 10 times, whereas TONCO in this study was increased 5.6 times. It should be noted that Re(bpy-COOH)/g-C3N4 worked under the low-intensity visible light, and meanwhile Mn(I)/g-C3N4 was studied under a 300 W Xe lamp (100 mW cm−2). The different light-irradiation intensities in the two research conditions might affect the CO generation. Referring to the PCO2P, the hybrid was evaluated to be more dominant in the number of products formed. PCO2P of Ru complex/C3N4 included HCOOH (80% selectively), CO, and H2,11 whereas that of Re(bpy-COOH)/g-C3N4 generated only two products CO (more than 90% selectively) and H2.
To clarify the mechanism of PCO2R of the hybrid system, the reduction potential of Re(bpy-COOH) and the energy level of g-C3N4 need to be mentioned. Re(bpy-COOH) is a good electrocatalyst for CO2 reduction, with LUMO potential39 at −1.94 vs. NHE (or one-electron reduction potential, Ered = −1.69 V vs. Ag/AgNO3) and the HOMO potential for absorbing visible light is approximately 0.75 eV vs. NHE (MLCT band, see the UV/Vis absorbance spectrum of Re(bpy-COOH) in Fig. S3†). Bulk g-C3N4 has a conduction band and valence band at −1.4 and 1.34 eV (vs. NHE, pH = 7),31,32 respectively, which is sufficiently negative for the injection of one electron from the CB to the HOMO of Re(bpy-COOH). The energy structure of Re(bpy-COOH) and g-C3N4 forms a Z-scheme hybrid system59–61 as illustrated in Scheme 1.
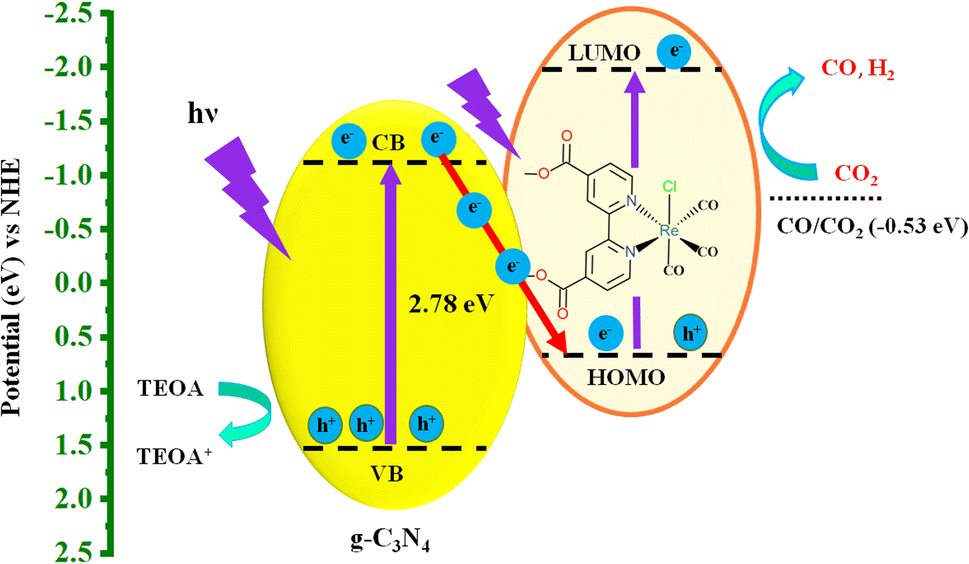 | ||
| Scheme 1 The illustration of the photocatalytic CO2 reduction process. | ||
Under visible-light irradiation, both g-C3N4 and Re(bpy-COOH) undergo photoexcitation that generates electron–hole pairs. Due to the high potential difference between the CB of g-C3N4 and the HOMO of Re(bpy-COOH) interfacial photo-induced electron transfer occurred, which reduces electron–hole pair recombination on Re(bpy-COOH). The photo-induced electrons generated on the LUMO on the catalyst side reduce CO2 molecules while the holes in the VB of g-C3N4 are scavenged by TEOA. The efficient interfacial electron transfer and the decrease in electron–hole pair recombination manifest via photoluminescent decay and PL spectra sufficiently boost the photocatalytic performance of the system.
To enhance our understanding of the ensemble effect within the Re(bpy-COOH)/g-C3N4 catalyst and its implications for the critical intermediate adsorption in CO2RR, we performed Density Functional Theory (DFT) calculations. Fig. 6 depicts the computed Gibbs free energy (ΔG) progression for CO2RR across g-C3N4, Re(bpy-COOH), and Re(bpy-COOH)/g-C3N4. This includes a focus on four fundamental steps: (1) CO2 adsorption onto the N atom of g-C3N4 or the Re atom of Re(bpy-COOH); (2) formation of COOH through hydrogenation; (3) creation of CO via disproportionation; (4) CO desorption. Our computations reveal that the ΔG of both Re(bpy-COOH) and Re(bpy-COOH)/g-C3N4 is less than that of g-C3N4, suggesting a more favorable environment for CO2RR processes in the former materials. Remarkably, in Re(bpy-COOH) and Re(bpy-COOH)/g-C3N4, the conversion of *CO2 to *COOH via hydrogenation overcomes the Gibbs energy barrier with just 0.26 and 0.61 eV, respectively, a significant reduction compared to the 1.53 eV needed in g-C3N4. Additionally, *CO formation is energetically exothermic at −0.55 eV for Re(bpy-COOH) and −0.62 eV for Re(bpy-COOH)/g-C3N4, compared to −0.42 eV for g-C3N4. Interestingly, the Gibbs energy barrier for CO desorption is only 0.64 eV in the hybrid material, lower than the 0.92 eV required in the Re(bpy-COOH) system. Our findings suggest that, despite a slightly higher Gibbs energy barrier for *COOH, the CO2RR processes favoring CO formation in the Re(bpy-COOH)/g-C3N4 system are more likely, aligning with our experimental results.
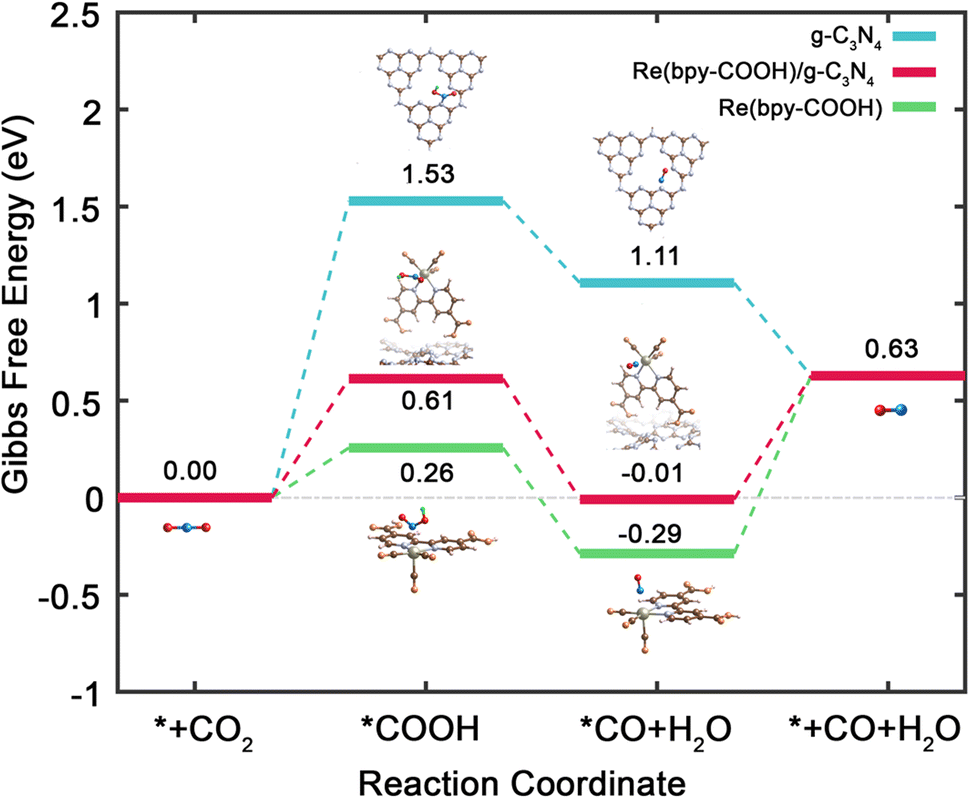 | ||
| Fig. 6 Calculated Gibbs free energy diagram of CO2RR processes with the corresponding intermediates for g-C3N4 (blue), Re(bpy-COOH) (green), and Re(bpy-COOH)/g-C3N4 (red). The asterisk (*) denotes the adsorption site. | ||
For a deeper understanding of the CO2RR catalytic activity, we delved into the charge transfer analysis between *COOH, *CO, and g-C3N4, Re(bpy-COOH), and Re(bpy-COOH)/g-C3N4, respectively. The Mulliken charge and corresponding charge transfer for each atom involved in the *COOH and *CO intermediates are detailed in Table S2 (see the ESI†). Positive and negative charge transfer values denote electron loss and gain, respectively. Remarkably, the charge transfers in both Re(bpy-COOH) and Re(bpy-COOH)/g-C3N4 scenarios register negative values (−0.51 and −0.48 for *COOH, and −0.15 and −0.13 for *CO, respectively). This contrasts with the negligible charge transfers observed in the case of g-C3N4. These findings suggest that the chemical interactions between CO and COOH with Re(bpy-COOH) and Re(bpy-COOH)/g-C3N4 are more potent compared to those with g-C3N4. It's noteworthy that the charge transfer of Re(bpy-COOH)/g-C3N4 is slightly less than that of Re(bpy-COOH) alone, indicating that g-C3N4 reduces the charge transfer between Re(bpy-COOH) and the intermediates, thus moderating their bond strength. This observation provides an explanation for the reduced Gibbs energy barrier for CO desorption observed in Re(bpy-COOH)/g-C3N4, as depicted in Fig. 6.
Additionally, we employed COHP analysis to unravel the interactions between the orbital pairs of intermediates and substrates. This strategy facilitates an understanding of the bonding and antibonding states emanating from each atomic orbital. In Fig. S6 (in the ESI†), we show the energy-resolved COHP between the 2p-orbitals of adsorbed C and a surface N 2p-orbital of g-C3N4, or a Re d-orbital of Re(bpy-COOH) and Re(bpy-COOH)/g-C3N4 for *COOH and *CO intermediates, respectively. Our analysis indicates weak C–N interactions in g-C3N4, as nearly zero COHP is observed around the Fermi level (E_F). In contrast, the C–Re interactions display a peak of the bonding state at E_F in the case of Re(bpy-COOH) and Re(bpy-COOH)/g-C3N4, signifying substantial interaction. These strong chemical interactions between C–Re atoms contribute to the favorable CO2RR observed in Re(bpy-COOH) and Re(bpy-COOH)/g-C3N4. Our DFT calculations affirm that Re(bpy-COOH)/g-C3N4 acts as a more efficient catalyst for CO2RR to CO conversion compared to Re(bpy-COOH) and g-C3N4. In line with this, our composite catalysts achieve a peak TON (Turnover Number) of CO formation and CO faradaic efficiency of 22.44 and 99.8%, respectively.
4. Conclusions
The hybrid photocatalytic material comprising the molecular catalyst Re(bpy-COOH) and the graphitic carbon nitride g-C3N4 is among the first examples of PCO2R under low-intensity visible-light irradiation. Re(bpy-COOH)/g-C3N4 is highly selective for CO production and has a high TON of CO formation. The combination of Re(bpy-COOH) and g-C3N4 formed a Z-scheme structure that improves the quantum efficiency and interfacial electron transfer, and decreases electron–hole pair recombination, hence boosting the photocatalytic efficiency. Utilizing DFT calculations, it was found that the Re(bpy-COOH)/g-C3N4 hybrid system exhibits efficient charge transfer and demonstrates lower energy barriers for *CO2 to *COOH conversion, as well as an increased propensity for *COOH formation compared to pure g-C3N4. Furthermore, the desorption activation energy of the *CO intermediate is also lower in the Re(bpy-COOH)/g-C3N4 system than in pure Re(bpy-COOH), indicating that the hybrid system achieves a balance between two rate-limiting steps for CO2RR. The unique combination of the two components contributes to the development and tailoring of highly active photocatalysts for low-intensity visible-light irradiation.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors appreciate the financial support from Vietnam Academy of Science and Technology under Grant Number CSCL19.03/23-24. The authors also gratefully acknowledge the Center for Computational Materials Science, Institute for Materials Research, Tohoku University, Japan, for the use of MASAMUNE-IMR, Cray XC50-LC supercomputer facility.References
- H. Takeda and O. Ishitani, Development of efficient photocatalytic systems for CO2 reduction using mononuclear and multinuclear metal complexes based on mechanistic studies, Coord. Chem. Rev., 2010, 254, 346–354 CrossRef CAS.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Photocatalytic CO2 reduction with high turnover frequency and selectivity of formic acid formation using Ru(II) multinuclear complexes, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673–15678 CrossRef CAS PubMed.
- H. Takeda, H. Koizumi, K. Okamoto and O. Ishitani, Photocatalytic CO2 reduction using a Mn complex as a catalyst, Chem. Commun., 2014, 50, 1491–1493 RSC.
- A. E. Nahhas, C. Consani, A. M. Blanco-Rodríguez, K. M. Lancaster, O. Braem, A. Cannizzo, M. Towrie, T. P. Clark, S. Záliš, M. Chergui and A. Vlček Jr, Ultrafast excited-state dynamics of rhenium(I) photosensitizers [Re(Cl)(CO)3(N,N)] and [Re(imidazole)(CO)3(N,N)]+: diimine effects, Inorg. Chem., 2011, 50, 2932–2943 CrossRef PubMed.
- S. Sato, Y. Matubara, K. Koike, M. Falkenstrom, T. Katayama, Y. Ishibashi, H. Miyasaka, S. Taniguchi, H. Chosrowjan, N. Mataga, N. Fukazawa and S. Koshihara, Photochemistry of fac-[Re(bpy)(CO)3Cl], Chem.–Eur. J., 2012, 18, 15722–15734 CrossRef CAS PubMed.
- A. Vlček and M. Busby, Ultrafast ligand-to-ligand electron and energy transfer in the complexes fac-[ReI(L)(CO)3(bpy)]n+, Coord. Chem. Rev., 2006, 250, 1755–1762 CrossRef.
- C. Gourlaouen, J. Eng, M. Otsuka, E. Gindensperger and C. Daniel, Quantum chemical interpretation of ultrafast luminescence decay and intersystem crossings in rhenium(I) carbonyl bipyridine complexes, J. Chem. Theory Comput., 2015, 11, 99–110 CrossRef CAS PubMed.
- R. C. Evans, P. Douglas and C. J. Winscom, Coordination complexes exhibiting room-temperature phosphorescence: evaluation of their suitability as triplet emitters in organic light emitting diodes, Coord. Chem. Rev., 2006, 250, 2093–2126 CrossRef CAS.
- J. N. Demas and G. A. Crosby, Quantum efficiencies of transition-metal complexes. I. d–d luminescence, J. Am. Chem. Soc., 1970, 92, 7262–7270 CrossRef.
- A. Nakada, T. Nakashima, K. Sekizawa, K. Maeda and O. Ishitani, Visible-light-driven CO2 reduction on a hybrid photocatalyst consisting of a Ru(II) binuclear complex and a Ag-loaded TaON in aqueous solutions, Chem. Sci., 2016, 7, 4364–4371 RSC.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Visible-light-driven CO2 reduction with carbon nitride: enhancing the activity of ruthenium catalysts, Angew. Chem., Int. Ed., 2015, 54, 2406–2409 CrossRef CAS PubMed.
- K. Maeda, Metal-complex/semiconductor hybrid photocatalysts and photoelectrodes for CO2 reduction driven by visible light, Adv. Mater., 2019, 31, 1808205 CrossRef.
- R. Kuriki and K. Maeda, Development of hybrid photocatalysts constructed with a metal complex and graphitic carbon nitride for visible-light-driven CO2 reduction, Phys. Chem. Chem. Phys., 2017, 19, 4938–4950 RSC.
- H. Kasap, C. A. Caputo, B. C. M. Martindate, R. Godin, V. W. H. Lau, B. V. Lotsch, J. R. Durrant and E. Reisner, Solar-driven reduction of aqueous protons coupled to selective alcohol oxidation with a carbon nitride–molecular Ni catalyst system, J. Am. Chem. Soc., 2016, 138, 9183–9192 CrossRef CAS PubMed.
- A. Nakada, H. Kumagai, M. Robert, O. Ishitani and K. Maeda, Molecule/semiconductor hybrid materials for visiblelight CO2 reduction: design principles and interfacial engineering, Acc. Mater. Res., 2021, 2, 458–470 CrossRef CAS.
- C. Quintana, M. P. Cifuentes and M. G. Humphrey, Transition metal complex/gold nanoparticle hybrid materials, Chem. Soc. Rev., 2020, 49, 2316–2341 RSC.
- C. D. Windle, E. Pastor, A. Reynal, A. C. Whitwood, Y. Vaynzof, J. R. Durrant, R. N. Perutz and E. Reisner, Improving the photocatalytic reduction of CO2 to CO through immobilisation of a molecular Re catalyst on TiO2, Chem.–Eur. J., 2015, 21, 3746–3754 CrossRef CAS PubMed.
- D. Sun, Y. Gao, J. Fu, X. Zeng, Z. Chen and Z. Li, Construction of supported Ru complex on bifunctional MOF-253 for photocatalytic CO2 reduction under visible light, Chem. Commun., 2015, 51, 2645–2648 RSC.
- B. Shan, S. Vanka, T. T. Li, L. Troian-Gautier, M. K. Brennaman, Z. Mi and T. J. Meyer, Binary molecular-semiconductor p–n junctions for photoelectrocatalytic CO2 reduction, Nat. Energy, 2019, 4, 290–299 CrossRef CAS.
- F. Paquin, J. Rivnay, A. Salleo, N. Stingelin and C. Silva, Multi-phase semicrystalline microstructures drive exciton dissociation in neat plastic semiconductors, J. Mater. Chem. C, 2015, 3, 10715–10722 RSC.
- Q. Gu, Y. Liao, J. Long, X. Wang and C. Xue, Template-free synthesis of porous graphitic carbon nitride microspheres for enhanced photocatalytic hydrogen generation with high stability, Appl. Catal. B Environ., 2015, 165, 503–510 CrossRef CAS.
- X. Zhang, Q. Wu, Z. Du, Y. Zheng and Q. Li, Green synthesis of g-C3N4 -Pt catalyst and application to photocatalytic hydrogen evolution from water splitting, Fullerenes, Nanotub. Carbon Nanostruct., 2018, 26, 688–695 CrossRef CAS.
- W. Zhang, L. Zhou and H. Deng, Ag modified g-C3N4 composites with enhanced visible-light photocatalytic activity for diclofenac degradation, J. Mol. Catal. A Chem., 2016, 423, 270–276 CrossRef CAS.
- K. Qi, Y. Li, Y. Xie, S. Y. Lui, K. Zheng, Z. Chen and R. Wang, Ag loading enhanced photocatalytic activity of g-C3N4 porous nanosheets for decomposition of organic pollutants, Front. Chem., 2019, 7, 1–9 CrossRef PubMed.
- D. Hao, J. Ren, Y. Wang, H. Arandian, M. Garbrecht, X. Bai, H. K. Shin, W. Wei and B. J. Ni, A green synthesis of Ru modified g-C3N4 nanosheets for enhanced photocatalytic ammonia synthesis, Energy Mater. Adv., 2021, 2021, 1–12 Search PubMed.
- G. Mamba and A. K. Mishra, Graphitic carbon nitride (g-C3N4) nanocomposites: a new and exciting generation of visible light driven photocatalysts for environmental pollution remediation, Appl. Catal. B Environ., 2016, 198, 347–377 CrossRef CAS.
- S. Patnaik, S. Martha and K. M. Parida, An overview of the structural, textural and morphological modulations of g-C3N4 towards photocatalytic hydrogen production, RSC Adv., 2016, 6, 46929–46951 RSC.
- J. Fu, J. Yu, C. Jiang and B. Cheng, g-C3N4-based heterostructured photocatalysts, Adv. Energy Mater., 2018, 8, 1–31 CAS.
- Z. Sun, H. Wang, Z. Wu and L. Wang, g-C3N4 based composite photocatalysts for photocatalytic CO2 reduction, Catal. Today, 2018, 300, 160–172 CrossRef CAS.
- N. Q. Thang, A. Sabbah, L. C. Chen, K. H. Chen, L. V. Hai, C. M. Thi and P. V. Viet, Localized surface plasmonic resonance role of silver nanoparticles in the enhancement of long-chain hydrocarbons of the CO2 reduction over Ag-gC3N4/ZnO nanorods photocatalysts, Chem. Eng. Sci., 2021, 229, 116049 CrossRef CAS.
- A. Alaghmandfard and K. Ghandi, A comprehensive review of graphitic carbon nitride (g-C3N4)–metal oxide-based nanocomposites: potential for photocatalysis and sensing, Nanomaterials, 2022, 12, 294 CrossRef CAS.
- X. Ma, Y. Lv, J. Xu, Y. Liu, R. Zhang and Y. Zhu, A strategy of enhancing the photoactivity of g-C3N4 via doping of nonmetal elements: a first-principles study, J. Phys. Chem. C, 2012, 116, 23485–23493 CrossRef CAS.
- K. Maeda, R. Kuriki and O. Ishitani, Photocatalytic activity of carbon nitride modified with a ruthenium(II) complex having carboxylic- or phosphonic acid anchoring groups for visible-light CO2 reduction, Chem. Lett., 2016, 45, 182–184 CrossRef CAS.
- K. Maeda, D. An, R. Kuriki, D. Lu and O. Ishitani, Graphitic carbon nitride prepared from urea as a photocatalyst for visible-light carbon dioxide reduction with the aid of a mononuclear ruthenium(II) complex, Beilstein J. Org. Chem., 2018, 14, 1806–1812 CrossRef CAS PubMed.
- X. Ma, C. Hu and Z. Bian, Hybrid photocatalytic systems comprising a manganese complex anchored on g-C3N4 for efficient visible-light photoreduction of CO2, Inorg. Chem. Commun., 2020, 117, 107951 CrossRef CAS.
- J. J. Walsh, C. Jiang, J. Tang and A. J. Cowan, Photochemical CO2 reduction using structurally controlled g-C3N4, Phys. Chem. Chem. Phys., 2016, 18, 24825–24829 RSC.
- B. Ma, G. Chen, C. Fave, L. Chen, R. Kuriki, K. Maeda, O. Ishitani, T. C. Lau, J. Bonin and M. Robert, Efficient visible-light-driven CO2 reduction by a cobalt molecular catalyst covalently linked to mesoporous carbon nitride, J. Am. Chem. Soc., 2020, 142, 6188–6195 CrossRef CAS PubMed.
- H. Koizumi, H. Chiba, A. Sugihara, M. Iwamura, K. Nozaki and O. Ishitani, CO2 capture by Mn(i) and Re(i) complexes with a deprotonated triethanolamine ligand, Chem. Sci., 2019, 10, 3080–3088 RSC.
- P. N. Nguyen, T. B. N. Dao, T. T. Tran, T. A. Nguyen, T. D. L. Phan, L. P. Nguyen, V. Q. Dang, T. M. Nguyen and N. Nguyen Dang, Electrocatalytic CO2 reduction by [Re(CO)3Cl(3-(pyridin-2-yl)-5-phenyl-1,2,4-triazole)] and [Re(CO)3Cl(3-(2-pyridyl)-1,2,4-triazole)], ACS Omega, 2022, 7, 34089–34097 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, Crystal orbital Hamilton population (COHP) analysis as projected from plane-wave basis sets, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef CAS PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanebe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, A metal-free polymeric photocatalyst for hydrogen production from water under visible light, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- J. Liu, T. Zhang, Z. Wang, G. Dawson and W. Chen, Simple pyrolysis of urea into graphitic carbon nitride with recyclable adsorption and photocatalytic activity, J. Mater. Chem., 2011, 21, 14398–14401 RSC.
- H. Zhang and A. Yu, Photophysics and photocatalysis of carbon nitride synthesized at different temperatures, J. Phys. Chem. C, 2014, 118, 11628–11635 CrossRef CAS.
- Z. Wei, J. Liu, W. Fang, M. Xu, Z. Qin, Z. Jiang and W. Shangguan, Photocatalytic hydrogen evolution with simultaneous antibiotic wastewater degradation via the visible-light-responsive bismuth spheres-g-C3N4 nanohybrid: waste to energy insight, Chem. Eng. J., 2019, 358, 944–954 CrossRef CAS.
- A. E. Nahhas, A. Cannizzo, F. V. Mourik, A. M. Blanco-Rodriguez, A. Vlcek Jr. and M. Chergui, Ultrafast excited-state dynamics of [Re(L)(CO)3(bpy)] n complexes: involvement of the solvent, J. Phys. Chem. A, 2010, 114, 6361–6369 CrossRef PubMed.
- A. Zhanaidarova, S. C. Jones, E. Despagnet-Ayoub, B. R. Pimentel and C. P. Kubiak, Re(tBu-bpy)(CO)3Cl supported on multi-walled carbon nanotubes selectively reduces CO2 in water, J. Am. Chem. Soc., 2019, 141, 17270–17277 CrossRef CAS PubMed.
- L. Tan, J. Xu, X. Zhang, Z. Hang, Y. Lia and S. Wang, Synthesis of g-C3N4/CeO2 nanocomposites with improved catalytic activity on the thermal decomposition of ammonium perchlorate, Appl. Surf. Sci., 2015, 356, 447–453 CrossRef CAS.
- Z. Huang, Q. Sun, K. Lv, Z. Zhang, M. Li and B. Li, Effect of contact interface between TiO2 and g-C3N4 on the photoreactivity of g-C3N4/TiO2 photocatalyst: (001) vs (101) facets of TiO2, Appl. Catal. B Environ., 2015, 164, 420–427 CrossRef CAS.
- S. W. Cao, Y. P. Yuan, J. Barber, S. C. J. Loo and C. Xue, Noble-metal-free g-C3N4/Ni(dmgH)2 composite for efficientphotocatalytic hydrogen evolution under visible light irradiation, Appl. Surf. Sci., 2014, 319, 344–349 CrossRef CAS.
- S. Roy and E. Reisner, Visible-light-driven CO2 reduction by mesoporous carbon nitride modified with polymeric cobalt phthalocyanine, Angew. Chem., 2019, 131, 12308–12312 CrossRef.
- X. Ma, L. Zheng and Z. Bian, Visible-light-driven CO2 reduction with g-C3N4-based composite: enhancing the activity of manganese catalysts, Chem. Eng. Sci., 2021, 229, 116042 CrossRef CAS.
- Y. Ono, J. Nakamura, M. Hayashi and K. I. Takahashi, Effect of substituent groups in rhenium bipyridine complexes on photocatalytic CO2 reduction, Am. J. Appl. Chem., 2014, 2, 74 CrossRef CAS.
- R. Guo, Z. Bi, Z. Lin, X. Hua, J. Wang, X. Chen and W. Pan, Carbon quantum dots-modified Z-scheme Bi12O17Cl2/NiAl-LDH for significantly boosting photocatalytic CO2 reduction, J. Colloid Interface Sci., 2022, 627, 343–354 CrossRef CAS PubMed.
- X. Ji, R. T. Guo, Y. Miao, Z. Lin, L. Hong, Y. Yuan, Z. Li and W. Pan, Construction of full solar-spectrum-driven Cu2-xS/Ni-Al-LDH heterostructures for efficient photocatalytic CO2 reduction, ACS Appl. Energy Mater., 2022, 5, 2862–2872 CrossRef CAS.
- Y. Miao, R. Guo, J. Gu, Y. Liu, G. Wu, C. Duan and W. Pan, Z-Scheme Bi/Bi2O2CO3/layered double-hydroxide nanosheet heterojunctions for photocatalytic CO2 reduction under visible light, ACS Appl. Nano Mater., 2021, 4, 4902–4911 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01502e |
| This journal is © The Royal Society of Chemistry 2023 |