
Metal–organic framework for high-performance catalyst layers in proton-exchange membrane fuel cells†
Inyoung
Choi‡
 a,
Jinhyuk
Lim‡
b,
Roberto dos
Reis
cde,
Eunji
Kim
a,
Soo Yeon
Lim
f,
Vinayak P.
Dravid
cde,
Heejin
Kim
*g,
Keun-Hwan
Oh
*bh and
Kwan Woo
Nam
*a
a,
Jinhyuk
Lim‡
b,
Roberto dos
Reis
cde,
Eunji
Kim
a,
Soo Yeon
Lim
f,
Vinayak P.
Dravid
cde,
Heejin
Kim
*g,
Keun-Hwan
Oh
*bh and
Kwan Woo
Nam
*a
aDepartment of Chemical Engineering and Materials Science, and Graduate Program in System Health Science and Engineering, Ewha Womans University, Seoul 03760, Republic of Korea. E-mail: kwanwoo@ewha.ac.kr
bKorea Automotive Technology Institute, 303 Pungse-ro, Chungnam 31214, Republic of Korea
cDepartment of Materials Science and Engineering, Northwestern University, Evanston, IL 60208-3113, USA
dNorthwestern University Atomic and Nanoscale Characterization Experimental Center, Northwestern University, Evanston, IL 60208-3113, USA
eInternational Institute of Nanotechnology, Northwestern University, Evanston, IL 60208-3113, USA
fSamsung SDI, Gongse-ro, Giheung-gu, Yongin-si, Gyeonggi-do 17084, Republic of Korea
gDivision of Analytical Science, Korea Basic Science Institute, 169-148 Gwahak-ro, Yuseong-gu, Daejeon 34133, Republic of Korea. E-mail: heejinkim@kbsi.re.kr
hEnergy Materials Research Center, Korea Research Institute of Chemical Technology (KRICT), Daejeon 34114, Republic of Korea. E-mail: khoh@krict.re.kr
First published on 8th September 2023
Abstract
Proton (H+)-exchange membrane fuel cells (PEMFCs) are considered effective power sources for fuel cell electric vehicles because of their clean emission, air-purification capability, and excellent durability. However, the performance degradation at low relative humidity (RH) remained to be addressed. Water back-diffusion, which refers to the diffusion of water from the cathode to the membrane by a concentration gradient, could lead to a remarkable improvement in the H+ conductivity of membrane/electrode assemblies (MEAs) at low RH. This effect can be achieved by introducing a functional material with a high water content into the cathode catalyst layer. Herein, we propose a Zr-based metal–organic framework (MOF; UiO-66(Zr)-(COOH)2) as a water-rich additive material in the MEA and demonstrate that it increases the power performance of PEMFCs at low RH by promoting water back-diffusion. The high compositional and structural tunability of MOFs will expand the pool of materials to adjust the humidity level of PEMFCs.
Introduction
The global interest in the development of next-generation fuel cell electric vehicles (FCEVs) has increased because of their potential to resolve carbon emission issues.1 Research on developing proton (H+)-exchange membrane fuel cells (PEMFCs), which are regarded as effective power sources for FCEVs, is well underway.2 PEMFCs in commercialized FCEVs typically contain perfluorosulfonic acid (PFSA)-ionomer-based polymeric membranes characterized by high H+ conductivity and excellent mechanical and chemical stability. However, the applications of PFSA-based membranes are limited owing to their high production cost and performance degradation under low relative humidity (RH) conditions.3–6 Low RH conditions not only impair the power performance of membrane/electrode assemblies (MEAs) but also limit the range of their operating conditions.7–10Several approaches have been proposed to address these limitations of PFSA based MEAs under low RH conditions. For example, composite membranes containing hydrophilic inorganic particles or organic materials have been investigated.11–15 Also, controlling the regularity of cracks within membranes at the nanoscale level has been suggested16 in order to increase the humidity level of MEAs. However, the uneven dispersion of hydrophilic particles can increase the ohmic resistance of the cell by limiting the mobility of polymer chains. Furthermore, locally accumulated particles, when they are electrically conductive, can degrade the insulating function of the membrane. Therefore, the development of a new conceptual approach to maintaining the H+ conductivity of the membrane under low RH conditions without directly modifying the membrane is a worthwhile endeavour.
Research17,18 on CL design has led to a better understanding of water back-diffusion, which refers to the movement of water from CL with a high water concentration to a membrane with a low water concentration. The operation of PEMFCs begins with the oxidation of the H2 gas supplied to the anode (Fig. 1a). The generated H+ ions pass through the polymeric membrane towards the cathode, followed by interacting with the O2 gas supplied to the cathode to produce water. The produced water is ejected from the catalyst surface to the flow-field channel by capillary-pressure differences. Despite this water generation, the amount of water back-diffusion in a conventional PEMFC is not usually high because the generated water is discharged outside (Fig. 1b). To realize the water back-diffusion, boehmite (γ-AlOOH) bearing high water content was introduced to the cathode CL and improved the performance of the PEMFC at low RH.18 However, its application to the PEMFC was limited because it exists in an unstable phase and has low thermal stability.
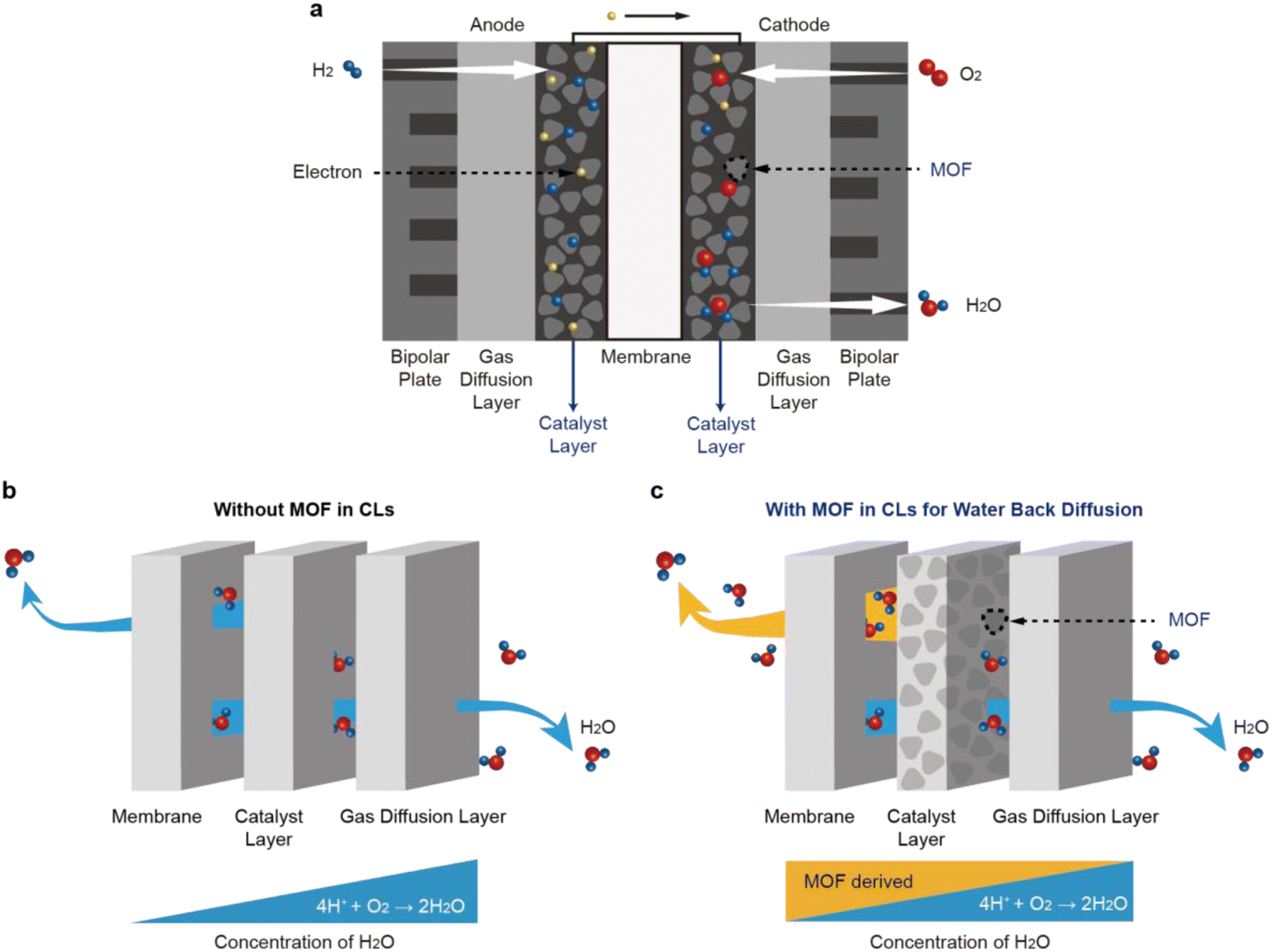 | ||
| Fig. 1 Influence of a metal–organic framework (MOF) on water back-diffusion in the cathode catalyst layer of a PEMFC. (a) Schematic illustrating the principle of PEMFC. (b) Pathways of water molecules in a catalyst layer (CL) without the MOF. (c) Water back-diffusion caused by the presence of MOF, which functions as a water reservoir in the catalyst layer. | ||
Metal–organic frameworks (MOFs) have been studied19,20 actively by chemists as sorbent materials for atmospheric water harvesting in an effort to resolve the global water shortage crisis. They have excellent water-storage capacity and exhibit stable performance on account of their porous structures and large surface areas.19–23 In addition, MOFs are thermally and chemically stable owing to their strong covalent metal–ligand bonds and can withstand high-temperature operation. Also, MOFs have recently been employed to improve the H+ conductivity of the membrane.24,25 We hypothesized, therefore, that MOFs could be ideal materials for realizing water back-diffusion when introduced to the cathode CL of MEAs. Besides, the carboxylic acid group can promote proton transfer by forming an effective proton transport channel, although the ion exchange capacity is lower. For that reason, various studies have been conducted on blending inorganic nanoparticles and polymers functionalized carboxylic and sulfonic acids.26–28
In this investigation, we introduce a water-stable Zr-based UiO-66-type MOF containing benzene-1,2,4,5-tetracarboxylate ligands – hereafter referred to as UiO-66(Zr)-(COOH)2 – as a functional water-storage material for the cathode CL of MEAs (Fig. 1c). UiO-66(Zr)-(COOH)2 is thermally and electrochemically stable, thus imparting robustness and durability during PEMFC operation. The MOF can be readily synthesized in an aqueous solution on the gram scale,29 indicating its production is not only suitable for industrialization but also cost effective.30,31 These water-containing and proton-conducting properties of UiO-66(Zr)-(COOH)2 can assist a PEMFC to retain the power performance, regardless of the RH conditions, when UiO-66(Zr)-(COOH)2 is incorporated into the cathode CL of MEAs. Our approach of grafting MOFs onto MEA presents a promising direction toward the development of novel humidification systems for PEMFCs since the composition and structure of the MOF can be tailored easily to maximize water back-diffusion.
Results and discussion
Synthesis and characterization of UiO-66(Zr)-(COOH)2
UiO-66(Zr)-(COOH)2 was synthesized according to a previously reported procedure29 and then applied to the cathode CL of a PEMFC. The highly porous UiO-66(Zr)-(COOH)2 contains water molecules within its pores because it is prepared under the aqueous condition. Based on powder X-ray diffraction (PXRD) analysis (Fig. 2a), the as-synthesized material could be indexed to a cubic unit cell with the Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. The lattice parameters of the MOF were calculated to be a = b = c = 20.6 Å by Rietveld refinement (Rp = 3.16, Rwp = 3.91, χ2 = 1.88).
m space group. The lattice parameters of the MOF were calculated to be a = b = c = 20.6 Å by Rietveld refinement (Rp = 3.16, Rwp = 3.91, χ2 = 1.88).
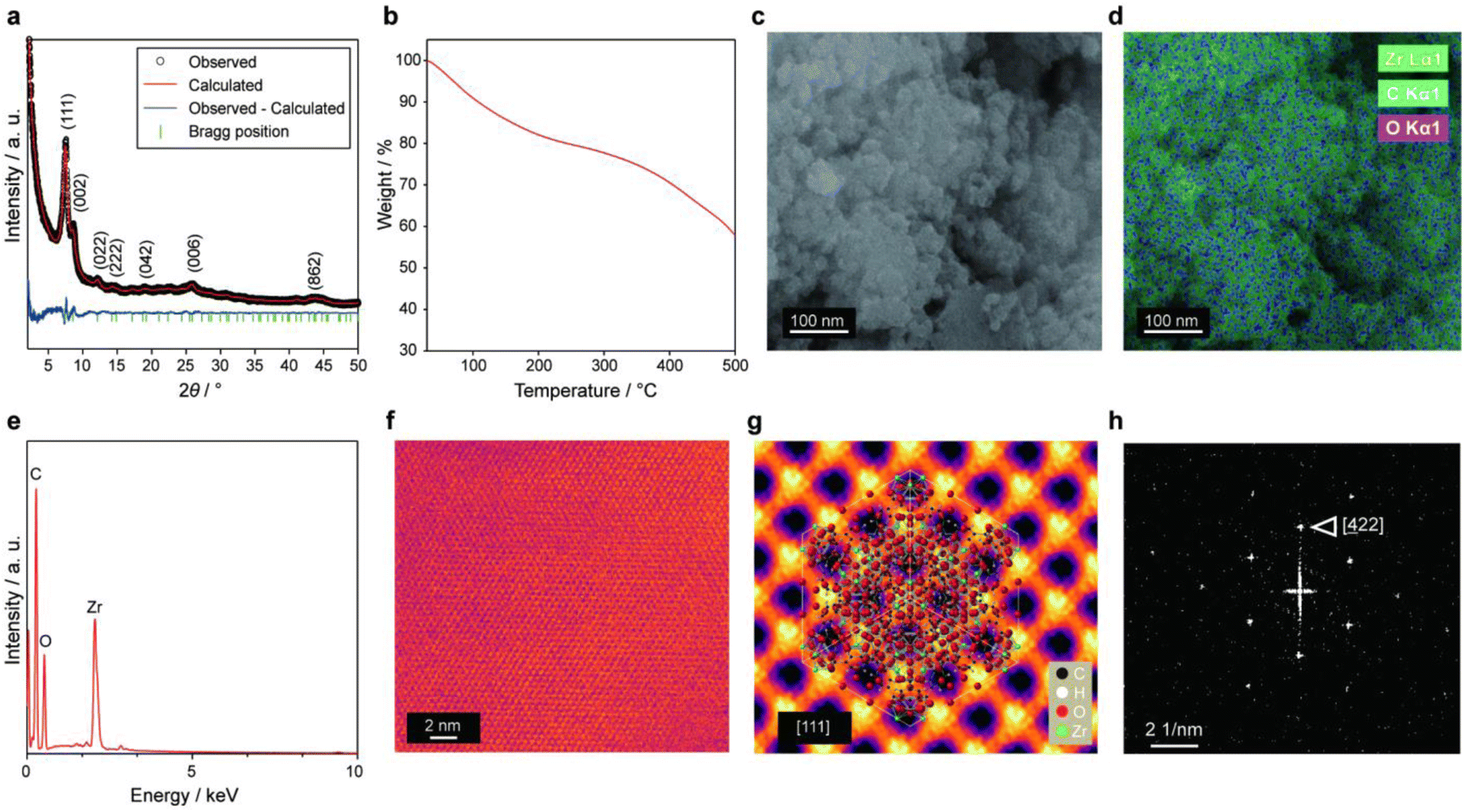 | ||
| Fig. 2 (a) Rietveld refinement of the powder X-ray diffraction pattern of UiO-66(Zr)-(COOH)2. (b) Thermogravimetric profile of UiO-66(Zr)-(COOH)2. (c) Scanning electron microscopy (SEM) and (d) EDS elemental mapping images of pristine UiO-66(Zr)-(COOH)2 for Zr, C, and O. (e) EDS profile of the whole area in (c). (f) Bright-field STEM image of the UiO-66(Zr)-(COOH)2 along the [111] direction. (g) Averaged lattice obtained from (f). (h) Fast Fourier transform of the whole area in (f). | ||
We performed thermal gravimetric analysis (TGA) on the pristine UiO-66(Zr)-(COOH)2 powder in order to investigate its water content. The TGA profile of the MOF shows (Fig. 2b) three distinct weight-loss regions as in the previous studies.29,32–35 The first weight-loss region (11.3 wt%) occurs between 25 and 120 °C and is caused by the evaporation of physisorbed water molecules.33–35 The second weight-loss region (18.1 wt%) takes place between 120 and 400 °C and is attributed to the dehydroxylation of Zr cluster35 and free acid29 remaining inside the MOF pores. These fractions of weight loss were in line with the previous report,29 where the number of water molecules per formula unit of UiO-66(Zr)-(COOH)2 was 16. The third weight-loss occurs above 400 °C and corresponds to the collapse of the framework to produce ZrO2.
The morphology of UiO-66(Zr)-(COOH)2 was examined by field-emission scanning electron microscopy (FE-SEM). The images shown in Fig. 2c and Fig. S1 in ESI† confirm that the MOF consists of secondary particles measuring several micrometres in size; these particles are formed by the aggregation of spherical primary particles having a size of approximately 20–50 nm. Scanning electron microscopy-energy-dispersive X-ray spectroscopy (SEM-EDS) confirms (Fig. 2d and e) that UiO-66(Zr)-(COOH)2 contains Zr, C, and O. A bright-field scanning transmission electron microscopy (BF-STEM) image (Fig. 2f) also reveals the highly porous structure of UiO-66(Zr)-(COOH)2. In addition, the BF-STEM image of the averaged MOF lattice (Fig. 2g) shows large pores with diameters of ∼0.4 nm viewed along the [111] direction. Fast Fourier transform (FFT) (Fig. 2h) confirms that the UiO-66(Zr)-(COOH)2 has a well-developed cubic crystal structure.
Electrochemical performance of MEAs
All MEAs were fabricated using the NR211 membrane and catalyst layer consisting of Nafion ionomer and carbon supported Pt (10F50E), as described in the Experimental section. In order to investigate the effect of UiO-66(Zr)-(COOH)2 on the cathode CL, we measured the current density–voltage (j–V) polarization curves (Fig. 3) of MEAs prepared with and without UiO-66(Zr)-(COOH)2 under various RH states. The performance of the MEA without the MOF (bare-MEA) decreased drastically from 1.09 to 0.90 and 0.63 A cm−2 at 0.6 V (filled green marks in Fig. 3a) as the RH decreased from 100 to 50 and 30%, respectively. This rapid performance degradation of PEMFC under low RH conditions is generally attributed to the reduced H+ conductivity. To verify the H+ conduction in MEAs, we measured the bulk resistance of the entire assemblies (Rohm) by electrochemical impedance spectroscopy (EIS) at a current density of 200 mA cm−2 (Fig. 3b). In the PEMFC case, Rohm generally reflects the extent of H+ conduction across the MEA and can be obtained from the high-frequency intercept of the real axis of the Nyquist plot. The Rohm of the bare-MEA increases sequentially from 68.9 to 93.4 and 142.6 mΩ cm2 (Fig. 3b) as the RH decreases from 100 to 50 and 30%, respectively. As shown in the inset of Fig. 3b, the current densities in Fig. 3a decrease linearly with increasing Rohm. This result indicates that the limiting factor determining the power performance of the cell is H+ conduction in MEAs. Meanwhile, the charge transfer resistance (Rct), which can be obtained from the diameter of the semicircle, was also increased from 409.1 to 453.0 Ω cm2 when the RH decreased from 100 to 50%. The Rct was not comparable at 30% RH due to the incomplete semicircle originating from the non-homogeneous distribution of ion resistance in the CL.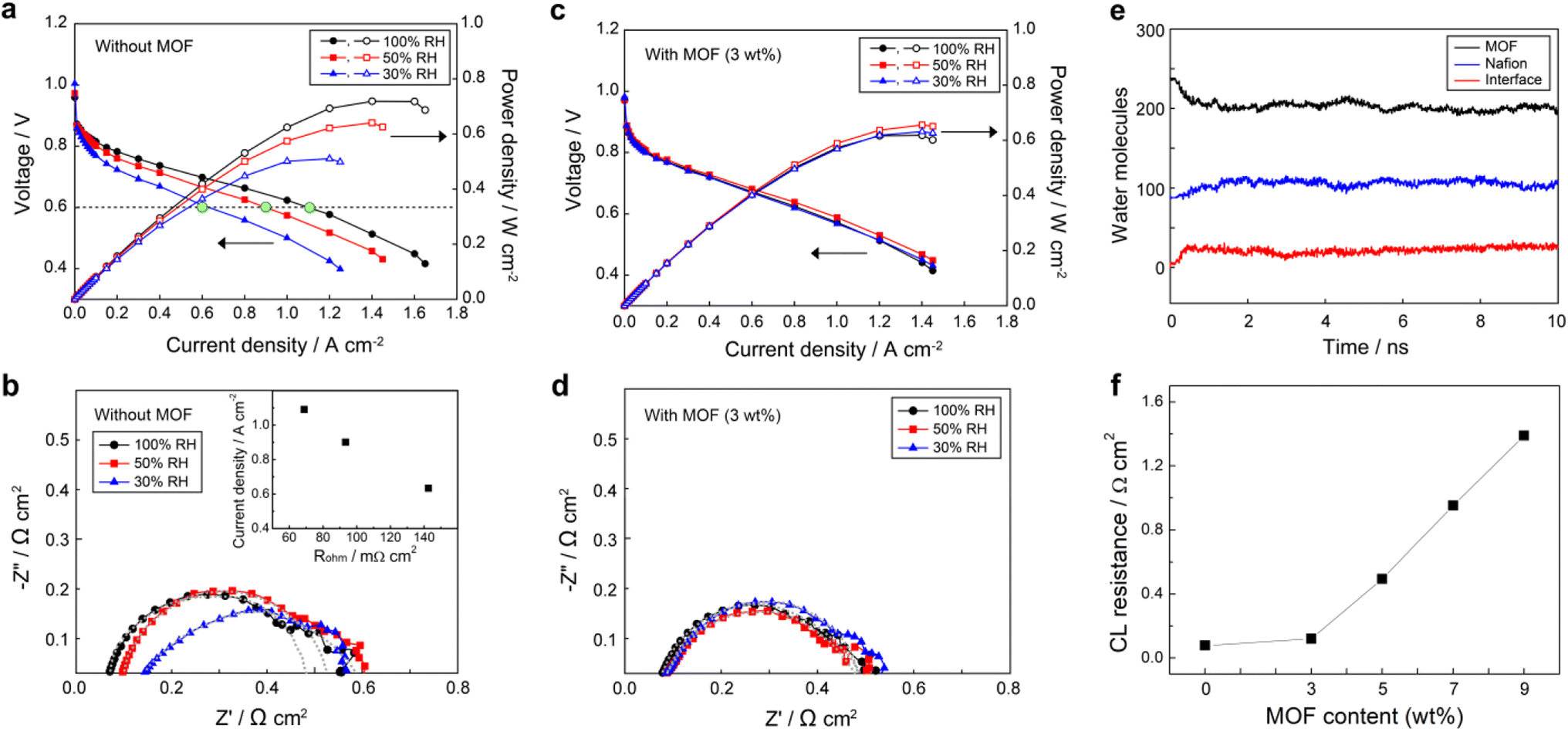 | ||
| Fig. 3 Evaluation of the electrochemical properties and proton conductivities of membrane/electrode assemblies (MEAs). Galvanostatic polarization curves of MEAs prepared (a) without and (c) with UiO-66(Zr)-(COOH)2 in the cathode catalyst layer. Nyquist plots of MEAs prepared (b) without and (d) with UiO-66(Zr)-(COOH)2 in the cathode catalyst layer. (e) Change of the water content in UiO-66(Zr)-(COOH)2 and Nafion when they are in contact (interface of 1 Å). (f) CL resistance at 30% RH condition depending on the MOF content. | ||
In contrast to the pristine membrane cases, the power performance of the MEA with the MOF in its cathode CL (mof-MEA, Fig. 3c) remains relatively constant with a current density of greater than 0.88 A cm−2 at 0.6 V under all humidity conditions. It is noteworthy that when comparing Fig. 3a and c, after adding the MOF in CL, the current density was improved at 30% RH and somewhat decreased at 100% RH while it was maintained at 50% RH condition, resulting in similar power performance between the cells under different humidity conditions. The reduced efficiency of mof-MEA at 100% RH compared to bare-MEA can be attributed to the flooding caused by the MOF that is saturated with water (Fig. S2 in ESI†). However, since FCEVs typically operate at 50% RH or lower, using the water reservoir is practically advantageous. To figure out the result, H+ conduction was compared between the samples. The mof-MEA exhibited relatively similar Rohm of 76.3, 84.3, and 85.3 mΩ cm2 (Fig. 3d) at 100%, 50%, and 30% RH conditions, respectively. The Rct values also became similar (397.0–393.0 mΩ cm2) with decreasing RH from 100% to 30%. These equalized resistances, i.e., H+ conduction, between the samples by the MOF explains the comparable power performances shown in Fig. 3c.
Noticeable performance improvement at low RH conditions suggests that the MOF may have increased the water content of the membrane by promoting the diffusion of water from the cathode to the membrane, so-called water back-diffusion. Fig. 3e shows the change of water contents in UiO-66(Zr)-(COOH)2 and Nafion at 30% RH (assuming λ = 3, where λ is water to SO3− ratio), obtained from molecular dynamics (MD) simulations, when the MOF and Nafion are in contact. Water molecules diffuse from the MOF to Nafion, indicating that water molecules have a preference for Nafion over the MOF at such a low RH. This phenomenon suggests that water can be supplied from the MOF to low RH membranes by direct contact or through the ionomers in the cathode. Such water back-diffusion is known as a major factor determining the water content of the membrane and is caused by the concentration gradient.36 Since the incorporated MOF particles have high water content, the concentration gradient of water between the cathode and the membrane becomes steeper and thus water back-diffusion can be encouraged.
Although the MOF invigorated the low RH conditions, the power performance and H+ conduction of the mof-MEA with respect to the bare-MEA at 100% RH condition were decreased by 14.4% and 10.7%, respectively, of which the numbers are greater than the weight fraction (3%) of the MOF. These results imply that when the H+ conductivity of the membrane is sufficient, other factors limit the power performance. We expect that the additional performance decrease is attributed to the deterioration of CL, such as a reduction of active surface area and particle agglomeration. To investigate the effect of MOF on the deterioration of CL, we prepared standalone CLs with different MOF contents from 3 wt% to 9 wt%. Followed by EIS measurements were conducted under an H2/N2 atmosphere at 30% RH to evaluate the H+ conduction resistance of CLs (Rcl). The Rcl is typically calculated as three times the length between the high-frequency intercept and the intercept of the asymptotic line extending from the slightly inclined low-frequency line on the real axis (Fig. S3, ESI†).37,38Fig. 3f shows Rcl values at 30% RH condition with varying the MOF content. The Rcl was 75.6 mΩ cm2 in the absence of the MOF, and it increased to 120.0 mΩ cm2 when 3 wt% MOF was integrated. With further increasing MOF content, the Rcl increased more rapidly to 494.4, 951.6, and 1390.0 mΩ cm2 for CLs with 5, 7, and 9 wt% MOF, respectively. The reason for this increase of Rcl, i.e., decrease of the H+ conductivity within the CL, can be inferred from the SEM images (Fig. S4, ESI†). The CLs prepared with 0 and 3 wt% MOF exhibited well-dispersed particles; however, the particles were agglomerated in the CL containing 7 wt% MOF. We interpret that excess of the MOF in the CL causes particle agglomeration, leading to a disruption of the H+ conduction pathways in the CL. The agglomeration implies attractive interactions between the MOF and other component particles, such as Pt/C catalysts and PFSA ionomers. Such an attractive interaction between particles negatively affects the uniform dispersibility in the CL, however, it can positively influence long-term stability. Also, while the addition of 3 wt% MOF slightly increased Rcl, its beneficial effect on the resistance of the entire MEA (Rohm) was predominant, improving the overall performance. We also expect that because MOFs are mixed with other components in a particle-to-particle manner, unlike the carbon coating, their direct effect on CL is less significant at low content such as 3 wt%.
The Rohm is a sum of the resistances of charge carriers in the membrane (Rm), CL (Rcl), and their interface (Rint). According to the previous investigations,37–40 the Rint is less influenced by RH conditions and its effect is marginal (3.5–8 mΩ cm2). Meanwhile, we showed that the Rcl increased by 44.4 mΩ cm2 (from 75.6 to 120) after adding the 3 wt% MOF, but Rohm was rather decreased by 57.3 mΩ cm2 (from 142.6 to 85.3) at 30% RH condition. Therefore, one can conclude that the Rm is a major factor determining the Rohm at low RH conditions, and the additive MOF enhanced the power performance of the MEA by improving the membrane by water back-diffusion.
Water adsorption characterization of UiO-66(Zr)-(COOH)2 and effect on the MEA performance
The water molecules inside the membrane can be classified into two types of water: free water, which behaves like bulk water; the other is bound water with a lower freezing temperature due to its limited mobility. Since free water molecules are easily influenced by the environment, such as temperature, increasing the content of bound water is advantageous in securing the performance. In the PFSA membrane, bound water molecules place at the first solvation shell of the sulfonyl group. Their heat of sorption is a maximum of 65 kJ mol−1 when λ = 1–2 and decreases gradually to ∼45 kJ mol−1 with increasing the water content to λ = 4–5.41,42 At higher water contents, free water molecules appear and the binding enthalpy slowly converges to the heat of water condensation (∼40 kJ mol−1). In the UiO-66(Zr), meanwhile, the heat of water adsorption ranges from 40 to 55 kJ mol−1 depending on the adsorbed water content,43 functional modification,44 and defect concentration.45 Although it is difficult to strictly apply these enthalpy values to our MEA system, they still provide some notable points. First, there is a thermodynamic driving force for the migration of water molecules from the MOF to PFSAs under low RH conditions. Since the competing materials for water absorption in the system are MOF and PFSA polymer chains, their difference in water-absorption enthalpy can encourage the water back-diffusion along with the water concentration gradient. Second, at high RH conditions, such an energetic driving force is minimized or disappeared. In this condition, the added MOF may play a high-concentration water reservoir rather than energetically drives water back-diffusion. In other words, similar water absorption enthalpies between PFSAs and the MOF can make balance the water content of the MEA against varying RH. This comparable water absorption between different materials is natural because all their interactions are hydrogen bonds. Thus, we expect that a variety of materials containing water molecules via hydrogen bonds can be used for additive materials of MEAs. Third, the adsorption enthalpy of water molecules in UiO-66(Zr) MOFs is close to that of the bound water. It implies that the addition of the MOF not only increases the total amount of water but also increases the amount of bound water. The increase in the total amount of water at 30% RH was 19.2–32.2% according to the TGA results (Table S1 and Section B, ESI†). To estimate the amount of bound water, we conducted differential scanning calorimetry (DSC) measurements for Nafion–MOF (3 wt%) composite that has been stabilized at 24 °C and 100% RH. The amount of free water was calculated based on the area of the melting peak (Fig. S5, ESI†), and the quantity of bound water was determined by subtracting the free water content from the total (Table S2, ESI†). The Nafion–MOF composite (11.3%) showed a higher amount of bound water compared to Nafion alone (7.6%), confirming that the water molecules in the MOF are bound. This increased bound water can improve the low-temperature performance; also, the importance of the bound water in the MOF is not limited to the temperature conditions. At low RH conditions, where most free water molecules are removed from the MEA, the water transport deteriorates.36 It implies that although water molecules are generated on the CL, their back-diffusion to the membrane is kinetically hindered on a level of CL. When the MOF is integrated, on the other hand, it improves the water back-diffusion at low RH by providing robust transport pathways due to the bound water, and increases the humidity level of the membrane successfully.Long-term performance of MEAs
To see the long-term stability of the UiO-66(Zr)-(COOH)2, we carried out galvanostatic polarization measurements for 450 h Fig. 4a shows the voltage change of the 3 wt% mof-MEA at a current density of 1 A cm−2 under 65 °C and 30% RH. The initial voltage decreased gradually during the operation, and the operation was stopped for every 50–150 h to investigate the reversible and irreversible decay rates separately. Between each step, nitrogen purging procedure was carried out for 30 min for the recovery. Despite the 30% RH condition, the mof-MEA exhibited 185 μV h−1 of irreversible decay rate (ΔVirrev), which is a comparable value to other long-term tests at 100% RH (Table S3, ESI†).46–58 The ΔVirrev can be caused by membrane damage, loss of ionomer and catalyst, and structural change of CL.59 Meanwhile, the MEA showed similar reversible voltage drop (ΔVrev) during the consecutive start-up/shut-down procedures: the ΔVrev values were 0.45, 0.37, and 0.49 V, for 2nd, 3rd, and 4th step, respectively, exhibiting high reversibility. The ΔVrev is generally attributed to the surface oxidation of Pt catalyst during the cell operation. It leads to an increase in charge transport resistance, resulting in a decrease in the overall performance and efficiency of the fuel cell. During the shut-down and rest period, however, the residual hydrogen gas deoxidates the Pt surface by crossover, leading to the recovery of the cell voltage to close to its initial value.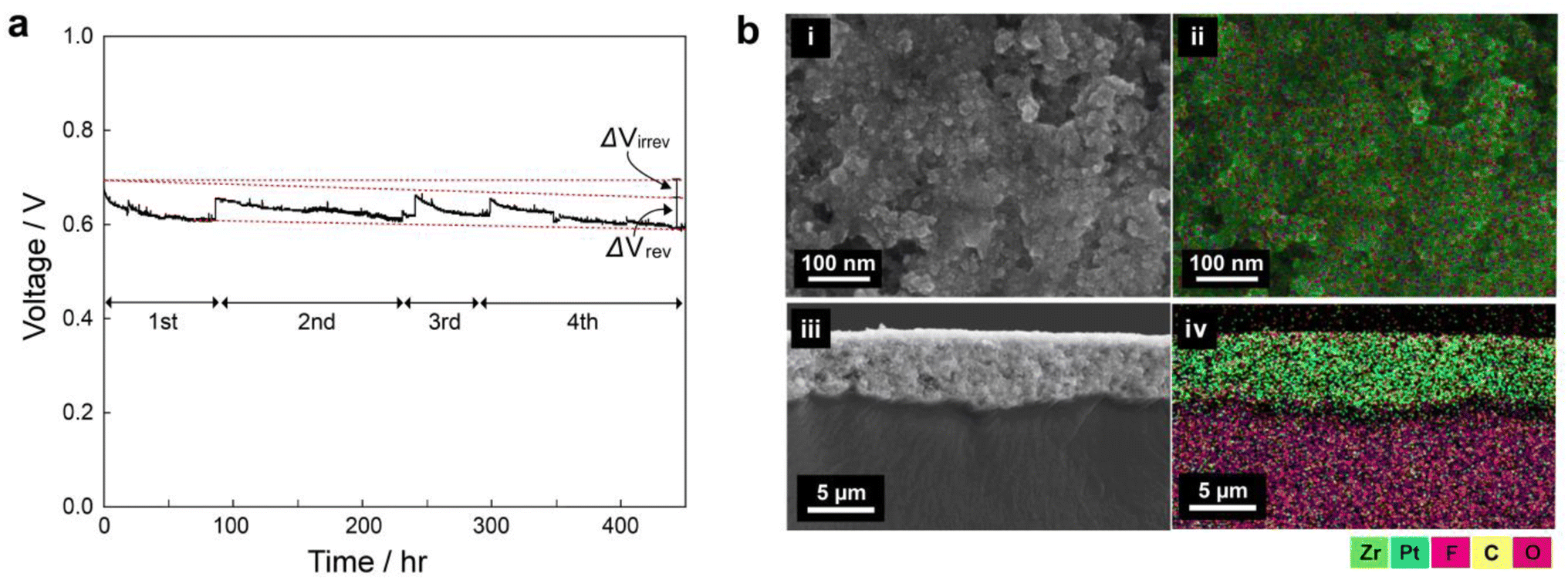 | ||
| Fig. 4 (a) Constant current density operation (1 A cm−2) of 3 wt% MOF membrane/electrode assembly for 450 hours. (b) SEM-EDS maps of surface (i and ii) and cross-section of CL (iii and iv). | ||
To further confirm the stability of MOFs in the CL, SEM-EDS analyses were performed for the disassembled MEA after 450 h of constant current operation (Fig. 4b). Both the surface and cross-section maps indicate that the zirconium (Zr) element from the MOF is evenly distributed throughout the entire CL after the long-term operation, likewise the pristine CL (Fig. S6, ESI†). This suggests that the MOF materials are stably preserved in the CL without significant degradation during long-term operation. This long-term stability indicates the strong binding between the MOF and other particles and is consistent with the agglomeration phenomenon observed in the previous section. The drain water from the cathode outlet was collected at the activation operation and after 450 h of operation for elemental analysis (Table S4, ESI†). The Zr concentration in the drain water was 1.311 ± 0.885 ppb in the initial collection and less than 0.004 ppb after 450 h. This also confirms the stability of the MOF in the CL during long-term operation.
Conclusions
In this article, we demonstrated how UiO-66(Zr)-(COOH)2 can be utilized in the cathode CL of a PEMFC to improve H+ conductivity under low RH conditions. After integrating 3 wt% UiO-66(Zr)-(COOH)2 in the cathode CL of the MEA, the power performance of the cell was improved by around 40% (from 0.63 to 0.88 A cm−2 at 0.6 V) under 30% RH condition. We showed that this enhancement is attributed to the improved H+ conductivity, especially in the membrane, using EIS measurements. Since UiO-66(Zr)-(COOH)2 has a high water content, its integration to CL increases the total water content of the cathode, promoting the concentration-driven diffusion of water from the cathode to the membrane, so-called water back-diffusion. In addition, we found that the absorbed water molecules in UiO-66(Zr)-(COOH)2 are a kind of bound water, which can help the water back-diffusion under low RH conditions. Also, we showed that the integration of UiO-66(Zr)-(COOH)2 is robust, and thus the MEA exhibited excellent long-term stability at low RH conditions. Hence, this investigation established the potential of MOFs as a new class of materials that can be used as water reservoirs in the cathode CL of a PEMFC, without direct modification of the membrane materials. Considering the synthetic diversity of MOFs, increasing their water content or enhancing their H+ conductivity may help improve the power performance of MEAs in PEMFCs under low humidity conditions.Experimental
Materials
All reagents, 1-propanol, and Nafion ionomer (D2021, 5% dispersion) were purchased from Sigma Aldrich and used as received without further purification. Deionized water was purified using a Milli-DI® water purification system (resistivity, 18.2 MΩ cm).Fabrication of MEAs
A total of 1 g of Pt/C (46% Pt, 10F50E, TKK, Japan), deionized water, 1-propanol, Nafion ionomer, and a UiO-66(Zr)-(COOH)2 dispersion were evenly mixed and tip-sonicated for 10 min to prepare the cathode CL. The slurry was cast onto a polyimide substrate using a doctor blade with a gap of 200 μm. The slurry-coated substrate was dried first of all at 35 °C for 30 min and then at 80 °C for 12 h to obtain the cathode CLs. Anode CLs were prepared by the same process but without the addition of the MOF. The mass ratio of the Nafion ionomer to carbon was 1, and the Pt loading of the cathode and anode CLs was controlled within 0.25 ± 0.02 mg cm−2. The active surface area of the CLs was 5 cm × 5 cm.The MEAs were fabricated by transferring the anode and cathode CLs formed on the polyimide substrate to each side of an NR211 membrane at 10 MPa and 130 °C for 3 min. The polyimide film was peeled off from the laminates after cooling to room temperature. A single cell was fabricated by assembling the MEA, a pair of gas diffusion media (JNT20-A3, JNTG, Korea), a pair of glass fiber-reinforced gaskets, and a pair of graphite blocks with four serpentine flow channels for the reactants. The flow fields of the anode and cathode reactants were arranged symmetrically on a single plane.
Characterization of UiO-66(Zr)-(COOH)2
The PXRD patterns of the MOF were collected in transmission geometry mode employing monochromic Cu-Kα 1 radiation for crystal structure analysis using STOE STADI-P (STOE & Cie GmbH, Germany). Scanning was conducted in the 2θ range of 2°–50° with a step size of 0.015° and at an accelerating voltage and current of 40 kV and 40 mA, respectively. The water content of pristine UiO-66(Zr)-(COOH)2 powders was investigated using a thermogravimetric analyzer (Mettler Toledo); the temperature was increased from room temperature to 500 °C at a ramp rate of 2 °C min−1 under an N2 flow. The morphology and elemental composition of the UiO-66(Zr)-(COOH)2 particles and cathodes containing 3 wt% UiO-66(Zr)-(COOH)2 were determined on an FE-SEM instrument (Hitachi S-4800) equipped with an Energy Dispersive X-ray Spectrometer (EDS, Oxford Aztec X-max 80 SDD EDS detector). Scanning transmission electron microscopy (STEM) was performed using a JEOL ARM 200CF Cs-corrected instrument (JEOL, Japan) operated at 200 kV. The probe was set to 27.1 mrad using a condenser aperture of 40 μm and the current to 5 pA. Bright-field STEM was collected using the detection range of 0–11 mrad. The UiO-66(Zr)-(COOH)2 MOF TEM specimens were prepared by first of all dispersing the MOF in EtOH and drop-casting it on lacey carbon Mo-based TEM grids. The data were collected using a Gatan K3-IS direct electron detector. The images were collected at dose rates below 20 e− per pixel per s and cumulative doses in the range of 15–20 e− A−2 in order to prevent the structural degradation of the MOF on a result by exposure to the electron beams.60 Selected area diffraction (SAD) was conducted with the electron beam spread out, and low-magnification data was acquired to prevent sample damage. The SAD patterns were collected using a Gatan OneView camera. EDX data were collected using an SDD EDX detector.Electrochemical tests
The j–V polarization curves of each cell were measured at 65 °C without back pressure using a fuel cell test station (CNL, Korea). H2 gas was fed at the anode side at a flow rate of 300 mL s−1. Air was fed at the cathode side at a flow rate of 1200 mL s−1. The amounts of H2 and air corresponded to an H2/air stoichiometry of 8.6/14.5 at 0.2 A cm−2. Cyclic voltammetry (CV) was conducted in the voltage range of 0.6–0.4 V for over 100 cycles to achieve an equilibrium state, after which the j–V polarization tests were performed. EIS was conducted using an alternating current (AC) impedance analyzer (Bio-Logics, HCP-803). The AC amplitude was 10% of the applied current over the range of 0.1–10,000 Hz. Polarization curves were measured at 65 °C under various RH conditions without back pressure using the galvanostatic method to confirm the effect of the UiO-66(Zr)-(COOH)2 nanoparticles on humidity functions. The physical state of the water molecules in the Nafion/MOF was investigated with differential scanning calorimetry (DSC). Sample was first cooled from 25 to −50 °C and then heated to 50 °C at a rate of 1 °C min−1. Long-term test of mof-MEA was measured at 1 A cm−2 under 65 °C and 30% RH for a duration of 450 h. The reversible and irreversible decay rate were determined by calculating the initial and final cell voltage at the beginning and end of experiment, respectively.Computational methods
The molecular dynamics (MD) simulations were performed using the Dreiding force field61 for Nafion chain and the UiO-66(Zr)-(COOH)2. The parameters for Zr were taken from the universal force field.62 The F3C force field63 was employed for water molecules. The partial charge of UiO-66(Zr)-(COOH)2 was taken from the literature,29 and that of the Nafion chain (Fig. S7, ESI†) was obtained from the electrostatic potential on a grid (ChElPG) method using the B3LYP functional with 6-31+g* basis set. The monomeric molecule (EW = 1032 g mol−1) was used for the atomic charge evaluation. To make a Nafion chain, 10 monomers were connected, resulting in a molecular weight of ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1.64 To construct the interface between the MOF and Nafion chains, each part was generated and relaxed before contact (Fig. S8 in ESI†). The integrated interface model was heated up to the operation temperature (338.15 K) using NVT ensemble with a step size of 10 K. Followed by, the NVT simulation was performed for 10 ns with Nose–Hoover thermostat and a time step of 1 fs. The position of Zr was fixed to prevent the drift of the entire structure.
000 g mol−1.64 To construct the interface between the MOF and Nafion chains, each part was generated and relaxed before contact (Fig. S8 in ESI†). The integrated interface model was heated up to the operation temperature (338.15 K) using NVT ensemble with a step size of 10 K. Followed by, the NVT simulation was performed for 10 ns with Nose–Hoover thermostat and a time step of 1 fs. The position of Zr was fixed to prevent the drift of the entire structure.
Author contributions
I. C., J. L., H. K., K.-H. O., and K. W. N. designed the research. I. C., E. K., and K. W. N. carried out the synthesis and analysis of the MOF materials. K.-H. O. and J. L. worked on the electrochemical measurements and analysis. H. K. conducted MD simulations. R. dR. performed the TEM measurements and analysis. S. L. analyzed the crystal structure of MOF. I. C., J. L., R. dR., E. K., S. L., V. P. D., H. K., K.-H. O. and K. W. N. co-wrote the manuscript. H. K., K.-H. O. and K. W. N. supervised the research. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. W. N. acknowledges support from the National Research Foundation (NRF) of Korea through the Excellent Young Scientist Program (NRF-2022R1C1C1007133). This research was supported by the BK21 FOUR (Fostering Outstanding Universities for Research) funded by the Ministry of Education (MOE, Korea) and NRF (NRF-5199990614253, Education Research Center for 41R-Based Health Care). K.-H. O. acknowledges support from the Technology Innovation Program (1415179321/20012133) funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea) and the Korea Evaluation Institute of Industrial Technology (KEIT), and the Korea Research Institute of Chemical Technology (KRICT) Core Research Program (KS2322-10). H. K. acknowledges support from Korea Basic Science Institute (KBSI, C330110). This research made use of the EPIC facility of Northwestern University's NUANCE Center, which receives support from the SHyNE Resource (NSF ECCS-2025633), the International Institute for Nanotechnology (IIN), and Northwestern's MRSEC program (NSF DMR-1720139).Notes and references
- K. Jiao, J. Xuan, Q. Du, Z. Bao, B. Xie, B. Wang, Y. Zhao, L. Fan, H. Wang, Z. Hou, S. Huo, N. P. Brandon, Y. Yin and M. D. Guiver, Nature, 2021, 595, 361–369 CrossRef CAS PubMed.
- Y. Sun, S. Polani, F. Luo, S. Ott, P. Strasser and F. Dionigi, Nat. Commun., 2021, 12, 5984 CrossRef CAS PubMed.
- J. Fan, M. Chen, Z. Zhao, Z. Zhang, S. Ye, S. Xu, H. Wang and H. Li, Nat. Energy, 2021, 6, 475–486 CrossRef CAS.
- L. Cao, H. Wu, Y. Cao, C. Fan, R. Zhao, X. He, P. Yang, B. Shi, X. You and Z. Jiang, Adv. Mater., 2020, 32, e2005565 CrossRef PubMed.
- F. Yang, G. Xu, Y. Dou, B. Wang, H. Zhang, H. Wu, W. Zhou, J.-R. Li and B. Chen, Nat. Energy, 2017, 2, 877–883 CrossRef CAS.
- C. H. Park, C. H. Lee, M. D. Guiver and Y. M. Lee, Prog. Polym. Sci., 2011, 36, 1443–1498 CrossRef CAS.
- W. Zheng, L. Wang, F. Deng, S. A. Giles, A. K. Prasad, S. G. Advani, Y. Yan and D. G. Vlachos, Nat. Commun., 2017, 8, 418 CrossRef PubMed.
- X. Liu, J. Zhang, C. Zheng, J. Xue, T. Huang, Y. Yin, Y. Qin, K. Jiao, Q. Du and M. D. Guiver, Energy Environ. Sci., 2020, 13, 297–309 RSC.
- K. Miyatake, T. Tombe, Y. Chikashige, H. Uchida and M. Watanabe, Angew. Chem., Int. Ed., 2007, 119, 6766–6769 CrossRef.
- Y. Wang, K. S. Chen, J. Mishler, S. C. Cho and X. C. Adroher, Appl. Energy, 2011, 88, 981–1007 CrossRef CAS.
- S. R. Mokhtaruddin, A. B. Mohamad, L. K. Shyuan, A. A. H. Kadhum and M. Akhmad, Adv. Mater. Res., 2011, 239–242, 263–268 CAS.
- J. Wang, X. Yue, Z. Zhang, Z. Yang, Y. Li, H. Zhang, X. Yang, H. Wu and Z. Jiang, Adv. Funct. Mater., 2012, 22, 4539–4546 CrossRef CAS.
- R. P. Pandey, A. K. Thakur and V. K. Shahi, ACS Appl. Mater. Interfaces, 2014, 6, 16993–17002 CrossRef CAS PubMed.
- J. J. Smith and I. Zharov, Chem. Mater., 2009, 21, 2013–2019 CrossRef CAS.
- Y.-H. Liu, B. Yi, Z.-G. Shao, L. Wang, D. Xing and H. Zhang, J. Power Sources, 2007, 163, 807–813 CrossRef CAS.
- C. H. Park, S. Y. Lee, D. S. Hwang, D. W. Shin, D. H. Cho, K. H. Lee, T. W. Kim, T. W. Kim, M. Lee, D. S. Kim, C. M. Doherty, A. W. Thornton, A. J. Hill, M. D. Guiver and Y. M. Lee, Nature, 2016, 532, 480–483 CrossRef PubMed.
- U. H. Jung, K. T. Park, E. H. Park and S. H. Kim, J. Power Sources, 2006, 159, 529–532 CrossRef CAS.
- H. S. Kang, K.-W. Nam, S. So and K.-H. Oh, J. Ind. Eng. Chem., 2019, 78, 455–460 CrossRef CAS.
- H. Kim, S. Yang, S. R. Rao, S. Narayanan, E. A. Kapustin, H. Furukawa, A. S. Umans, O. M. Yaghi and E. N. Wang, Science, 2017, 356, 430–434 CrossRef CAS PubMed.
- N. Hanikel, M. S. Prevot and O. M. Yaghi, Nat. Nanotechnol., 2020, 15, 348–355 CrossRef CAS PubMed.
- T. A. Goetjen, J. Liu, Y. Wu, J. Sui, X. Zhang, J. T. Hupp and O. K. Farha, Chem. Commun., 2020, 56, 10409–10418 RSC.
- W. Xu and O. M. Yaghi, ACS Cent. Sci., 2020, 6, 1348–1354 CrossRef CAS PubMed.
- Y. Ye, L. Gong, S. Xiang, Z. Zhang and B. Chen, Adv. Mater., 2020, 32, e1907090 CrossRef PubMed.
- P. Wang, J. Lin, Y. Wu and L. Wang, J. Power Sources, 2023, 560, 232665 CrossRef CAS.
- G. Wei, Y. Liu, A. Wu, Y. Min, Z. Liao, R. Zhu, Y. Liang and L. Wang, Mater. Today Chem., 2023, 27, 101276 CrossRef CAS.
- J. Wang, S. Jiang, H. Zhang, W. Lv, X. Yang and Z. Jiang, J. Membr. Sci., 2010, 364, 253–262 CrossRef CAS.
- C. Wang, Z. Feng, Y. Zhao, X. Li, W. Li, X. Xie, S. Wang and H. Hou, Int. J. Hydrogen Energy, 2017, 42, 29988–29994 CrossRef CAS.
- X. Meng, S.-Y. Song, X.-Z. Song, M. Zhu, S.-N. Zhao, L.-L. Wu and H.-J. Zhang, Chem. Commun., 2015, 51, 8150–8152 RSC.
- Q. Yang, S. Vaesen, F. Ragon, A. D. Wiersum, D. Wu, A. Lago, T. Devic, C. Martineau, F. Taulelle, P. L. Llewellyn, H. Jobic, C. Zhong, C. Serre, G. De Weireld and G. Maurin, Angew. Chem., Int. Ed., 2013, 52, 10316–10320 CrossRef CAS PubMed.
- M. J. Katz, Z. J. Brown, Y. J. Colon, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- H. Jiang, Y. Zhao, J. Lin, Y. Chen and S. Wang, J. Porous Mater., 2021, 28, 1737–1747 CrossRef CAS.
- X. Sun, D. Hu, L. Yang, N. Wang, Y. Wang and X. Ouyang, J. Sol-Gel Sci. Technol., 2019, 91, 353–363 CrossRef CAS.
- Q. Yang, H. Zhang, L. Wang, Y. Zhang and J. Zhao, ACS Omega, 2018, 3, 4199–4212 CrossRef CAS PubMed.
- S. M. Chavan, G. C. Shearer, S. Svelle, U. Olsbye, F. Bonino, J. Ethiraj, K. P. Lillerud and S. Bordiga, Inorg. Chem., 2014, 53, 9509–9515 CrossRef CAS PubMed.
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS PubMed.
- D. Malevich, J. G. Pharoah, B. A. Peppley and K. Karan, J. Electrochem. Soc., 2011, MA2011-02, 1080 Search PubMed.
- C. Gabrielli, O. HAAS and H. Takanouti, J. Appl. Electrochem., 1987, 17, 82–90 CrossRef CAS.
- R. Jiang, C. K. Mittelsteadt and C. S. Gittleman, J. Electrochem. Soc., 2009, 156(12), B1440–B1446 CrossRef CAS.
- B. S. Pivovar and Y. S. Kim, J. Electrochem. Soc., 2007, 154(8), B739–B744 CrossRef CAS.
- D. J. Burnett, A. R. Garcia and F. Thielmann, J. Power Sources, 2006, 160, 426–430 CrossRef CAS.
- K. D. Kreuer, Solid State Ionics, 2013, 252, 93–101 CrossRef CAS.
- G. Fu, P. Wu, J. Yang, S. Zhang and X. Huai, Microporous Mesoporous Mater., 2022, 331, 111642 CrossRef CAS.
- G. jajko, P. Gryta, P. Kozira, M. Szufla, D. Matoga, D. Majda and W. Makowski, J. Phys. Chem. C, 2022, 126, 9185–9194 CrossRef CAS.
- G. Jajko, J. J. G. Sevillano, A. Sławek, M. Szufla, P. Kozyra, D. Matoga, W. Makowski and S. Calero, Microporous Mesoporous Mater., 2022, 330, 111555 CrossRef CAS.
- D. Liu and S. Case, J. Power Sources, 2006, 162, 521–531 CrossRef CAS.
- M. Prasanna, E. A. Cho, T.-H. Lim and I.-H. Oh, Electrochim. Acta, 2008, 53, 5434–5441 CrossRef CAS.
- R. Lin, B. Li, Y. P. Hou and J. M. Ma, Int. J. Hydrogen Energy, 2009, 34, 2369–2376 CrossRef CAS.
- K. Panha, M. Fowler, X.-Z. Yuan and H. Wang, Appl. Energy, 2012, 93, 90–97 CrossRef CAS.
- P. Gazdzick, J. Mitzel, D. G. Sanchez, M. Schulze and K. A. Friedrich, J. Power Sources, 2016, 327, 86–95 CrossRef CAS.
- D. Yao, T. C. Jao, W. Zhang, L. Xu, L. Xing, Q. Ma, Q. Xu, H. Li, S. Pasupathi and H. Su, Int. J. Hydrogen Energy, 2018, 43, 21006–21016 CrossRef CAS.
- K. Talukdar, P. Gazdzicki and K. A. Friedrich, J. Power Sources, 2019, 439, 227078 CrossRef CAS.
- J. O. Leader, Y. Yue, M. R. Walluk and T. A. Trabold, Int. J. Hydrogen Energy, 2022, 47, 18820–18830 CrossRef CAS.
- J. Bai, S. Ke, J. Song, K. Wang, C. Sun, J. Zhang and M. Dou, ACS Appl. Mater. Interfaces, 2022, 14, 5287–5297 CrossRef CAS PubMed.
- V. D. Thuc, V. D. C. Tinh and D. Kim, J. Membr. Sci., 2022, 642, 119990 CrossRef CAS.
- K.-H. Oh, Y. Goo, M. Kim, J. Park, K. W. Nam and H. Kim, J. Alloys Compd., 2022, 928, 167227 CrossRef CAS.
- Y. Duan, Y. Pang, B. Liu, L. Wu, X. Hu, Q. Li and C. Zhao, ACS Sustainable Chem. Eng., 2023, 11, 5270–5283 CrossRef CAS.
- X. Shen, X. Liang, Y. Xu, W. Yu, Q. Li, X. Ge, L. Wu and T. Xu, J. Membr. Sci., 2023, 675, 121556 CrossRef CAS.
- Q. Zhang, M. Schulze, P. Gazdzicki and K. A. Friedrich, Appl. Energy, 2021, 302, 117490 CrossRef CAS.
- D. Zhang, Y. Zhu, L. Liu, X. Ying, C.-E. Hsiung, R. Sougrat, K. Li and Y. Han, Science, 2018, 359, 675–679 CrossRef CAS PubMed.
- S. L. Mayo, B. D. Olafson and W. A. Goddard III, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard III and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- M. Levitt, M. Hirshberg, R. Sharon, K. E. Laidig and V. Daggett, J. Phys. Chem. B, 1997, 101, 5051–5061 CrossRef CAS.
- H. Kang, S. H. Kwon, R. Lawler, J. H. Lee, G. Doo, H.-T. Kim, S.-D. Yim, S. S. Jang and S. G. Lee, J. Phys. Chem. C, 2020, 124, 21386–21395 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: SEM and EDS images, DSC and ICP results, atomic charge. See DOI: https://doi.org/10.1039/d3ta04377k |
| ‡ Inyoung Choi and Jinhyuk Lim contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |