
Intrinsically robust cubic MnCoOx solid solution: achieving high activity for sustainable acidic water oxidation†
Jingjing
Zhang‡
ab,
Ali
Raza‡
 c,
Yang
Zhao‡
a,
Song
Guo
*a,
Zaheer Ud Din
Babar
cd,
Liangliang
Xu
*f,
Changhai
Cao
ae and
Gao
Li
*ab
c,
Yang
Zhao‡
a,
Song
Guo
*a,
Zaheer Ud Din
Babar
cd,
Liangliang
Xu
*f,
Changhai
Cao
ae and
Gao
Li
*ab
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: gaoli@dicp.ac.cn; Fax: +86-411-82463017; Tel: +86-411-82463009
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cDepartment of Physics “Ettore Pancini”, University of Naples Federico II, Piazzale Tecchio, 80, 80125 Naples, Italy
dScuola Superiore Meridionale (SSM), University of Naples Federico II, Largo S. Marcellino, 10, 80138, Italy
eKey Laboratory of Biofuels and Biochemical Engineering, SINOPEC Dalian Research Institute of Petroleum and Petrochemicals Co., Ltd, Dalian 116045, China
fDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-Ro, Yuseong-Gu, Daejeon 34141, Republic of Korea
First published on 9th October 2023
Abstract
This article reveals the promising potential of MnCoOx solid solutions as an effective catalyst for the oxygen evolution reaction (OER). The solid oxide solution was synthesized by utilizing redox deposition of KMnO4 and Co(NO3)2 on a carbon paper (CP) support with varying Co/Mn ratios. XRD and TEM analyses confirmed the homogeneous distribution of Co and Mn species in MnCoOx. According to OER tests in acid electrolyte, the catalytic efficiency of Mn1Co5Ox surpassed that of Co3O4, MnOx, and IrO2, demonstrating overpotentials of 275 and 569 mV at a current density of 10 and 100 mA cm−2, respectively. The investigation emphasizes the crucial roles of Co3+ and Mn4+ cations as well as oxygen vacancies (Ov) sites in facilitating the OER mechanism. These essential characteristics were duly observed in a Mn1Co5Ox solid solution catalyst. Furthermore, the introduction of a Mn dopant contributed to the durability of Mn1Co5Ox in acidic OER, thereby exhibiting outstanding catalytic stability for nearly 300 hours at 100 mA cm−2 current density. In addition, DFT studies provided insight into how Mn1Co5Ox enhances the catalytic performance of the OER. In conclusion, this study offers valuable insight into designing efficient OER catalysts using MnCoOx solid solutions as a promising candidate.
Introduction
Using renewable energy sources for water electrolysis presents a reliable and sustainable approach to produce clean hydrogen (H2) fuel.1–4 In conventional electrolysis systems, the slow kinetics of their proton-coupled electron transfer electrochemical reaction and poor stability impede the progress of overall water splitting.5–8 Thus, for optimizing the efficiency to meet such systems, the oxygen evolution reaction (OER) plays a decisive role.9–11 The water electrolysis system featuring a proton exchange membrane (PEM) is attracting global interest due to its various benefits, for instance, elevated current densities, exceptionally pure gases, minimal ohmic losses, and superior compactness.12,13 However, this technique requires electrocatalysts that resist corrosion and can function effectively in highly acidic environments. Due to their impressive durability and exceptional effectiveness, only Ir-based catalysts, especially IrO2, are considered practical electrocatalysts for the OER in PEM devices.14–16 Nevertheless, limited availability and high cost restrict their widespread applications. Moreover compared to Ir, ruthenium is approximately ten times less expensive, making it a desirable substitute for acidic OER.17–19 RuO2 has satisfactory OER activity in an acidic environment. However the OER long-term stability of Ru-based catalysts in acids or PEM reactors remains a significant issue.20–24 As a result, it's an enormous task to develop OER catalysts that are both inexpensive and effective for applications in an acidic medium.Several 3d transition metals, particularly cobalt-based catalysts, have been extensively studied as potential OER catalysts, such as Co2TiO4,25 Co3O4,26–28 CoFePbOx,29 and hetero-N-coordinated Co single atom catalysts.30 Regardless, they are subjected to severe degradation under strong acidic and oxidative states. This results in a limited selection of OER catalysts with high abundance on earth that exhibit high activity and stability under acidic conditions.31–34 An essential approach involves generating catalysts from transition metal oxides with enhanced resistance to acid-induced corrosion. For example, MnOx has shown promise in acidic OER investigations by a self-regeneration mechanism under acidic conditions demonstrated by Huynh et al.35 Meanwhile, Zhou et al.36 discovered that the rutile alloy Mn–Sb–O systems can achieve 50 mA cm−2 at an overpotential of 580 mV for acidic OER, but the stability of Mn–Sb oxides was not better than that of the artwork of IrO2. Additionally, the correlation between cobalt oxides' active sites and the mechanism in acidic media is rarely studied. It is also reported that the intrinsic oxygen evolution reaction activity of Co3O4 can be enhanced by modifying its redox properties.37 As a result, investigating the connection between redox properties and catalytic OER performance is intended to encourage mechanistic understanding.
In our previous investigation, we have reported the preparation of clusters based on cobalt through the bonding of ligands.38,39 These clusters demonstrated exceptional electrochemical efficiency in the OER and oxygen reduction reaction (ORR). Thenceforth, we have been exploring the use of the potassium permanganate redox method to print cobalt–manganese oxide materials onto carbon paper (CP) surfaces. The Mn1Co5Ox prepared in our study shows notable distinctions from doped compounds. In this process, we employ the oxidizing properties of potassium permanganate to transform low-valent cobalt into a high-valent state, leading to the creation of a homogeneous solid solution of Mn1Co5Ox (which can be viewed as a Mn-rich doped Co3O4). A remarkable characteristic of the solid solution crystal structure is its ability to retain the crystal structure of the original solvent, thus maintaining identical composition, structure, and properties.40 This solid solution belongs to a single phase and displays exceptional catalytic performance for the OER. The Mn1Co5Ox catalyst exhibited superior performance in an acid electrolyte, with overpotentials of 275 mV@10 mA cm−2 and 569 mV@100 mA cm−2. Moreover, the catalyst displayed remarkable catalytic stability for 300 hours at a current density of 100 mA cm−2, which highlights the potential of MnCoOx as a promising catalyst for electrocatalytic applications. Scheme 1 illustrates the utilization of MnCoOx as a highly effective catalyst for OER mechanisms in acidic media. Furthermore, it highlights the novel aspects of this material in terms of material design and exceptional performance features in OER applications.
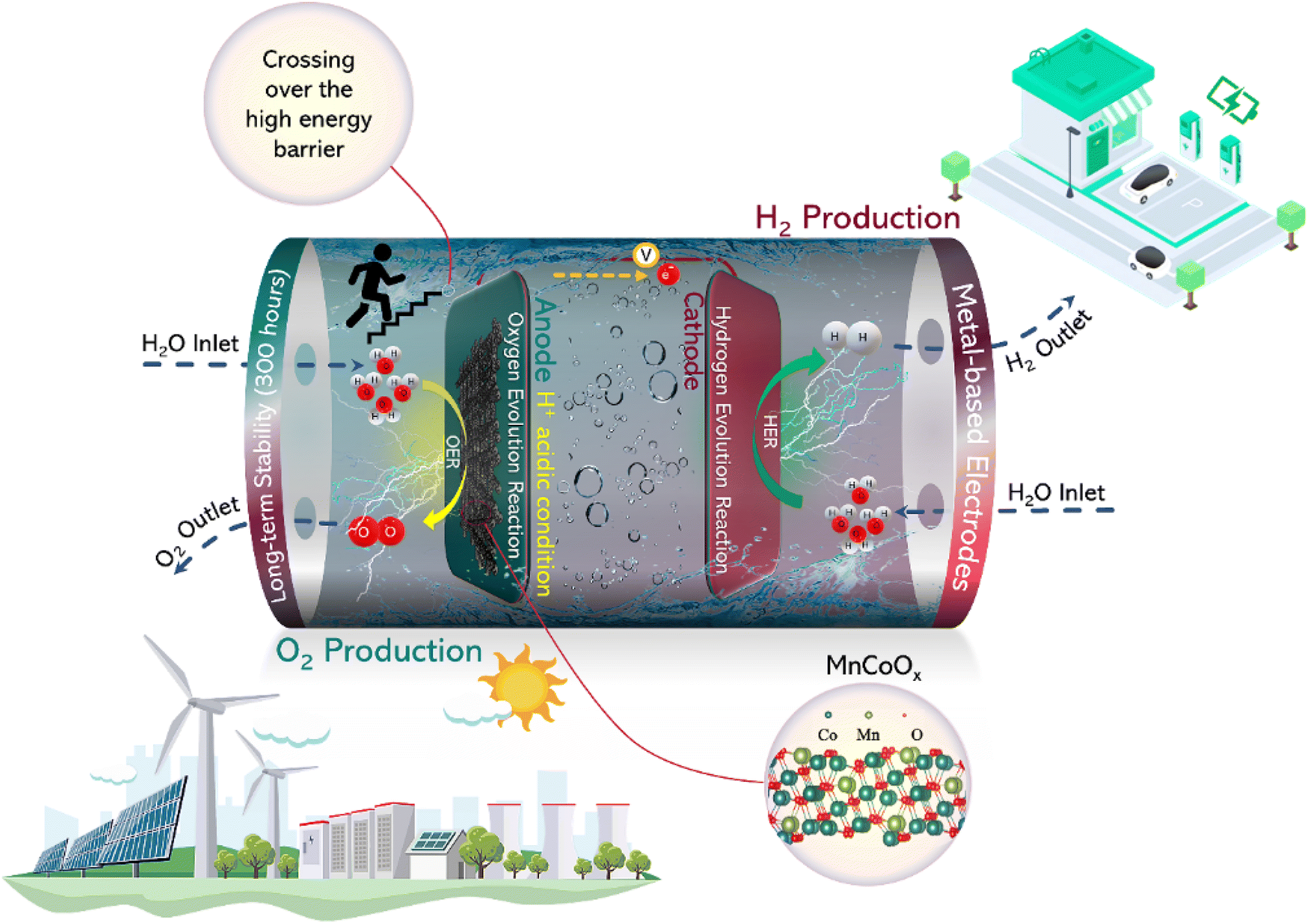 | ||
| Scheme 1 Pictorial demonstration of the OER mechanism using MnCoOx catalysts under acidic conditions and highlighting the paramount aspects of this study. | ||
Experimental section
Electrocatalyst preparation
A 2 × 4 cm2 CP substrate was cleaned using ultrasonication in ethanol and water, followed by treatment with HNO3. MnCoOx catalysts were synthesized using cobalt nitrate and potassium permanganate dissolved in water. The resulting solution was drop-cast onto CP and subsequently dried in an oven at 80 °C for 30 min. After rinsing, the CP was subjected to sonication for 10 seconds to remove the loosely attached particles. The drop-casting, drying, and cleaning steps were repeated to ensure complete and consistent coverage. The electrode precursor was then calcined at 300 °C for 2 hours, resulting in a catalyst loading of ∼2 mg on the CP substrate with a geometric area of 0.25 cm2.Characterization
The powder X-ray diffraction (XRD) patterns were obtained on an X-ray powder diffractometer (SmartLab, Rigaku Corporation) with Cu Kα radiation at 40 kV and 30 mA from 10° to 70° at a scan rate of 10° min−1 to identify the phase structure of these electrodes. X-ray photoelectron spectroscopy (XPS) analyses were conducted on a Theta Probe system (base pressure: ∼10−8 Pa), equipped with a Phoibos 100 hemispherical analyzer and XR 50 X-ray source (SPECS GmbH) operated in the constant pass energy mode at 50 eV. Using CasaXPS, all spectra were corrected by using the C 1s signal (284.8 eV). Subsequently, peaks were fitted after a Shirley-type background subtraction utilizing a mixed Gauss–Lorentz method, with peak positions and full-width half maxima left unconstrained. The concentration of the surface Co3+ (Co3+% = ACo3+/(ACo3+ + ACo2+)), Mn4+ (Mn4+% = AMn4+/(AMn4+ + AMn3+)), and surface oxygen vacancies (Ov% = AOv/(AOv + AOl + AOa)) was calculated based on the area under the peaks of Co 2p, Mn 2p, and O 1s, respectively. Note that Ai represents the peak area of the i species. Inductively coupled plasma-optical emission spectroscopy (ICP-OES) was carried out on an ICP-8100 (Shimadzu). The samples of MnCoOx were dissolved in an aqua regia solution. The actual ratios of Co/Mn in the electrocatalyst were determined to be 1.53 (for Mn1Co1.5Ox), 2.46 (Mn1Co2.5Ox), 5.12 (Mn1Co5Ox), and 8.13 (Mn1Co8Ox) by ICP-OES. Transmission electron microscopy (TEM) and high-angle annular dark-field scanning TEM (HAADF-STEM) were carried out on a Thermo Fisher FEI Talos F200x (Thermo Fisher Scientific). X-ray absorption spectroscopy (XAS) measurements were performed on a laboratory device (easyXAFS300, easyXAFS LLC), which is based on Rowland circle geometries with spherically bent crystal analyzers (SBCA) and a silicon drift detector. Si (5,5,1) SBCA was used for Mn measurement at 35 kV, and Si (5,3,3) SBCA was used for Co measurement at 30 kV. The powder samples were thoroughly ground and mixed with polyethylene glycol using an agate mortar and pestle and pressed into Ø = 13 mm pellets. The pressed pellets were then sandwiched using Kapton tape.Electrochemical measurements
All the electrochemical measurements were carried out on an electrochemical workstation CHI 760E using a standard three-electrode system with a platinum electrode (Tianjin Aida Hengsheng Technology Development Co., Ltd.) as the counter electrode and Hg/Hg2Cl2 electrode (saturated KCl solution) as the reference electrode. The potential of the Hg/Hg2Cl2 reference electrode was corrected using a reversible hydrogen electrode (RHE) after the experiments. The working electrode was cycled for 20 seconds at 100 mV s−1 in 0.5 M H2SO4 (pH 0) to activate the electrode and then linear sweep voltammetry (LSV) was recorded at a scan rate of 5 mV s−1. The current density was calculated based on the geometric area (0.25 cm2) of the electrode exposed to the electrolyte solutions. All the potentials were referred to the reversible hydrogen electrode by calibrating the reference electrodes using Pt sheets (purchased from Aldrich) as both the working and counter electrode in corresponding solutions. The iR compensation was performed manually after the measurement for 80% of the resistance.DFT
In this work, all the calculations were conducted using the Viena ab initio simulation package (VASP),41,42 and the structure optimization was based on density functional theory (DFT). The modeling of exchange–correlation energy was performed with the implemented generalized gradient approximation (GGA), proposed by Perdew–Burke–Ernzerhof (PBE).43,44 The valence electronic states were expanded with the plane waves; and the core–valence interactions were described using the projector augmented wave (PAW) approach,45 with the corresponding cutoff kinetic energies set to 400 eV. The DFT+U method with U = 2.0 eV and U = 3.2 eV was used to describe the 3d orbital of Co and Mn, respectively.46,47 The Brillouin zone was set with a Gamma-centered k-point grid of 1 × 1 × 1 (ref. 48), and to further improve the convergence of the simulated systems, a Gaussian functionalized Fermi surface, with a broadening magnitude of 0.1 eV, was used. The structures were first relaxed with the implemented conjugate-gradient algorithm, until the convergence tolerance of Hellmann–Feynman forces and energy on each atom was less than 0.05 eV Å−1 and 10−4 eV per atom, respectively. Moreover, due to the periodic boundary conditions (PBC), there exist non-negligible interactions among periodical images; to reasonably overcome this, a separate vacuum layer with a thickness of 15 Å along the z-direction was applied. Also, the van der Waals (vdW) interactions were described by using the Grimme's DFT-D3 scheme with the application of dispersion correction.49,50 In addition, for all the calculations, spin polarization was considered.The Gibbs free energy was calculated via the following equation:
| ΔG = EDFT + ZPE-TS | (1) |
The OER pathways are depicted as follows:
| * + H2O → OH* + H+ + e− | (2) |
| OH* + OH− → O* + H2O + e− | (3) |
| O* + H2O → OOH* + H+ + e− | (4) |
| OOH* + H2O → * + H+ + e | (5) |
The adsorption Gibbs free energy was calculated as follows:
| ΔG(O*) = E(O*) − E(*) − E(H2O) + E(H2) | (6) |
| ΔG(OH*) = E(OH*) − E(*) − E(H2O) + 1/2E(H2) | (7) |
| ΔG(OOH*) = E(OOH*) − E(*) − 2E(H2O) + 3/2E(H2) | (8) |
Results and discussion
Synthesis and structural and morphological characterization of MnCoOx solid solution
The synthetic route of MnCoOx solid solutions with varying Co/Mn ratios is illustrated in Fig. 1a; they were synthesized directly on CP by a redox reaction of Co2+ and MnO4−, followed by annealing in air. XRD analysis was performed to determine the crystalline structure of MnCoOx (Fig. 1b). Six strong diffraction lines were observed at 2θ ∼19.0°, 31.3°, 38.5°, 44.8°, 59.3°, and 65.2°, which correspond to the planes (111), (220), (311), (400), (511) and (440) of cubic Co3O4 in the Fd-3m (227) space group (JCPDS #42-1467).52,53 Furthermore, due to the significant dispersion of manganese ions throughout the cobalt framework, no reflections corresponding to MnOx species (e.g. MnO, Mn2O3, MnO2, etc.) were found in the XRD patterns of these MnCoOx samples even at a relatively high concentration of MnOx (∼40 mol%). By examining the XRD patterns of MnCoOx catalysts with varying doping levels, a slight shift towards lower angles of around 36.8° for (311) was observed with the increasing amount of Mn dopants (Fig. S1†), indicating that the Mn cations were well doped into the lattice of Co3O4 to form MnCoOx solid solutions, and we also obtained the XRD pattern of Mn1Co5Ox annealed at different temperatures. The results showed that the crystalline phase of the Co3O4 structure was formed at 200 °C. And the crystallinity was improved when the sample was calcined at high temperatures. Of note, no peaks for Mn oxides were observed even after the annealing at 500 °C, indicating the thermal stability of Mn1Co5Ox composites (Fig. S2†).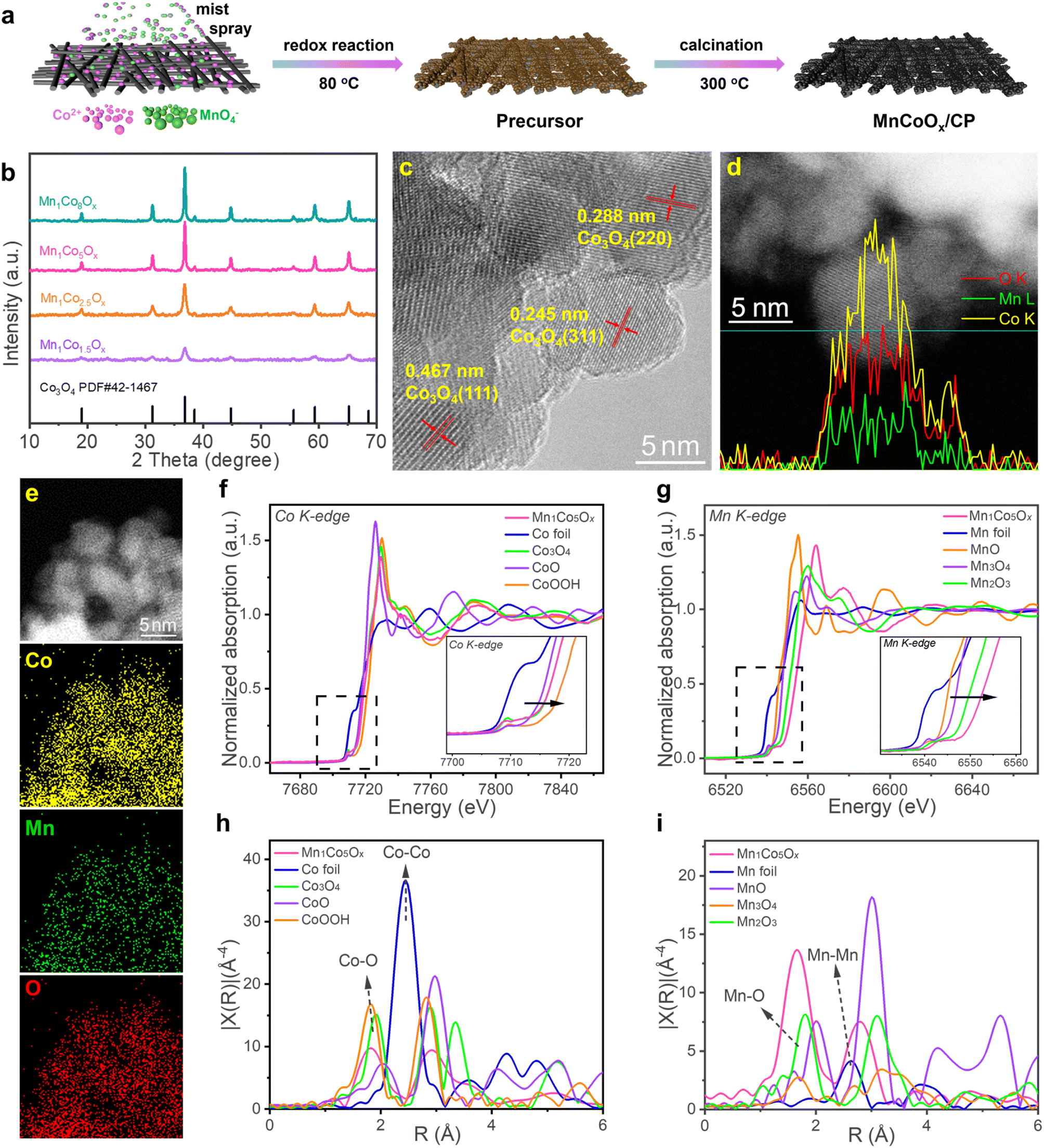 | ||
| Fig. 1 Structural and morphological characterization of MnCoOx electrocatalysts: (a) schematic illustration of the synthesis process, (b) XRD patterns, (c) TEM image of the Mn1Co5Ox catalyst, (d) linear-scanning profiles of the cross-section of the Mn1Co5Ox catalyst, and (e) HAADF-STEM image with EDS elemental mapping (cobalt, green; manganese, purple; oxygen, pink). XANES spectra of (f) Co K-edge and (g) Mn K-edge. Corresponding Fourier transforms of (h) Co K-edge and (i) Mn K-edge EXAFS spectra for Mn1Co5Ox and the standard references. | ||
Subsequently, the MnCoOx solid solutions were analyzed by TEM to determine their elemental composition and morphology. TEM images revealed that the majority of oxide particles ranged in size from 6 to 8 nm (as depicted in Fig. S3†). The lattice fringes observed in Fig. 1c exhibit an interplanar distance of 0.288 nm, 0.245 nm, and 0.467 nm, which correspond to the (220), (311), and (111) planes of cubic Co3O4, respectively.54–56 Intriguingly, no lattice fringes for MnOx species were observed, indicating that Mn was successfully doped within the lattice of Co3O4. Furthermore, the linear-scanning profiles depicted in Fig. 1d also confirmed that Mn atoms were successfully distributed within the lattice of Co3O4. Uniform lattice fringes can be seen in the line sweep area, and the distribution of elements obtained in the line sweep area also show that Co and Mn exist simultaneously with the content of Co more than that of Mn. The elemental mapping, illustrated in Fig. 1e, provides more evidence that Co and Mn species are homogeneously distributed within the MnCoOx solid solutions.
X-ray absorption near edge structure (XANES) spectra, obtained at the Co and Mn K-edge, were utilized to examine the electronic structure and local coordination of Mn1Co5Ox.39 As displayed in Fig. 1f, the peak intensity of Mn1Co5Ox is similar to that of Co3O4 and different from that of Co-foil. Meanwhile, it can also be observed that the pre-line of Mn1Co5Ox completely coincides with that of Co3O4. This confirms that Co2+ and Co3+ co-exist as the major valence states of Co species in Mn1Co5Ox. In contrast, the XANES spectra at the Mn K-edge revealed that the peak intensity of the white-line for Mn1Co5Ox is significantly higher than that of the reference Mn3O4 and Mn2O3 (Fig. 1g). Moreover, the pre-line for Mn1Co5Ox shifted toward higher binding energies compared to that of Mn2O3, indicating that the average valence of Mn species in Mn1Co5Ox should be higher than +3. This also suggests a state of electron deficiency near the Mn site, which is related to the use of potassium permanganate oxidation. To further investigate the coordination environment of the Mn1Co5Ox catalyst, R-space extended X-ray absorption fine structure (EXAFS) fitting analysis was performed at Co and Mn K-edges, as shown in Fig. 1h and i. Mn1Co5Ox exhibited an expansion of the Co–O bond distance from 1.81 Å to 1.83 Å compared to the control group of CoOOH. In contrast Mn1Co5Ox showed a reduction of the Co–O bond distance from 1.91 Å to 1.83 Å when compared to that of Co3O4.57 Furthermore, Mn1Co5Ox exhibited a reduced Mn–O bonding distance from 1.80 Å to 1.67 Å relative to that of Mn2O3, plausibly due to Mn's valence state being higher than +3. Specifically, the Co–O bonds in Mn1Co5Ox had an electron density similar to that of Co3O4, which is slightly larger than CoOOH and relatively less than Co3O4 in length. The Mn–O bonds in Mn1Co5Ox with low electron density is smaller than that of Mn2O3, which facilitated the formation of a solid-solution structure with an approximately uniform M–O bond length and accelerated electron transfer between Mn and Co atoms, promoting the catalytic activity in the OER (vide infra).
Electrocatalytic OER in acidic solution
To assess the electrochemical activity of MnCoOx samples towards electrocatalytic water splitting, OER tests of the as-synthesized nanocomposites deposited on CP were performed in a solution of 0.5 M H2SO4 (pH 0) employing a conventional three-electrode setup. Before the measurements, the reference electrode was calibrated relative to the reversible hydrogen electrode (RHE) (Fig. S4†).58 Electrocatalytic measurements suggested that Mn1Co5Ox exhibited superior OER response and outperformed its MnCoyOx (y: 1.5, 2.5, and 8) counterparts (Fig. 2a). Apparently, the Mn1Co5Ox catalyst demonstrated the highest current density and lowest Tafel slope (Fig. S5†). We further investigated the effect of annealing temperatures in the range of 200 to 500 °C and discovered that Mn1Co5Ox displays optimal OER activity after annealing at 300 °C for 2 h (Fig. S6†). Moreover, the OER polarization curves demonstrated that the Mn1Co5Ox composite exhibited superior catalytic efficiency compared to Co3O4, MnOx,, a CP support and IrO2, indicating that the OER occurred over the Mn1Co5Ox surface (Fig. 2b).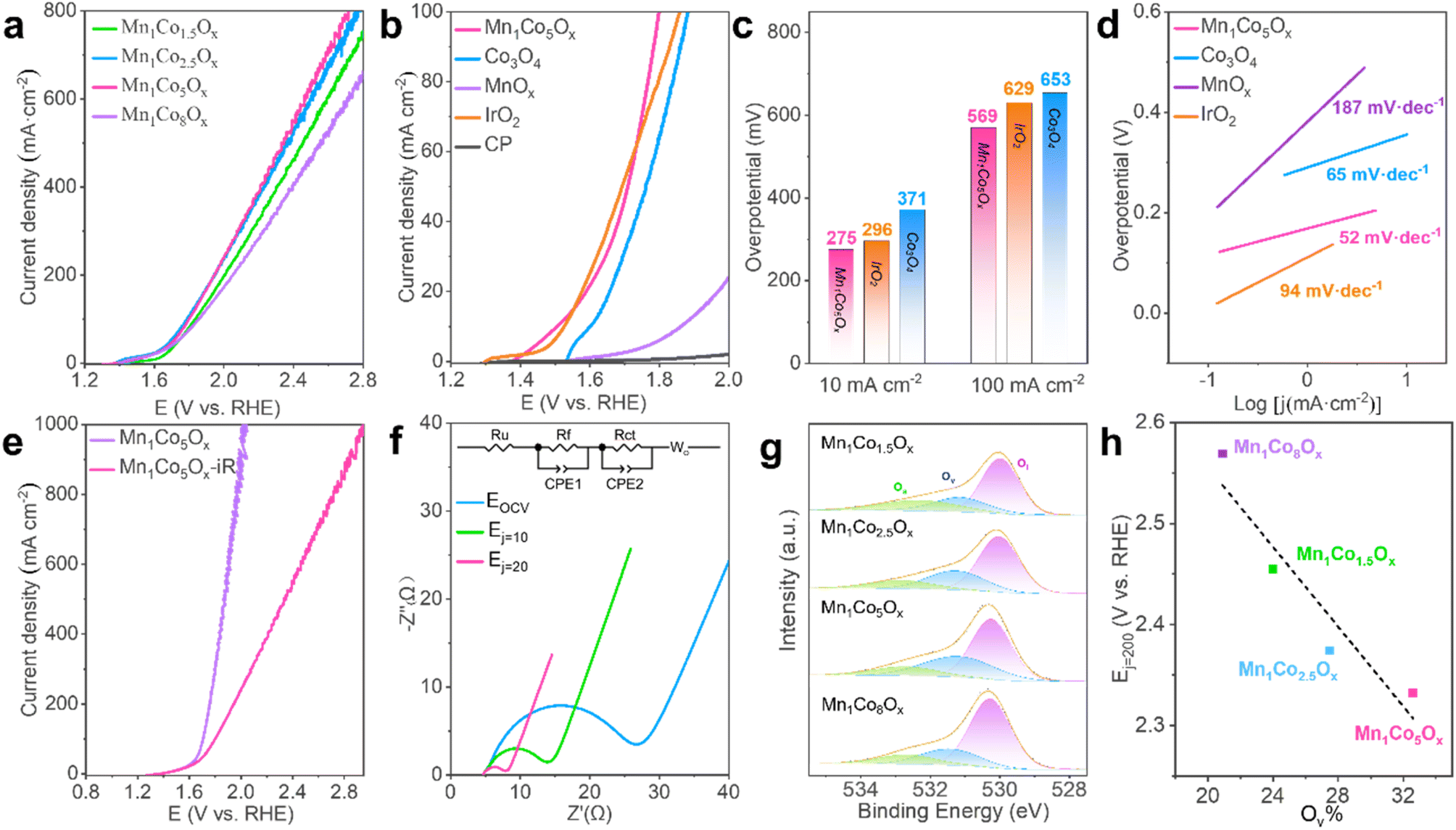 | ||
| Fig. 2 Catalytic OER performance. (a) OER polarization curves with different Mn/Co ratios of MnCoOx. (b) OER polarization curves of Mn1Co5Ox, Co3O4, MnOx, IrO2, and CP in a 0.5 M H2SO4 solution. (c) Overpotentials of Mn1Co5Ox (pink), IrO2 (orange) and Co3O4 (blue) at a current density of 10 and 100 mA cm−2. (d) Tafel plots derived from (a). (e) OER polarization curves of Mn1Co5Ox before and after iR correction. (f) Nyquist plots of Mn1Co5Ox at EOCV, Ej=10, and Ej=20. (g) O 1s XPS spectra of MnCoOx catalysts. (h) The behavior of E (V vs. RHE) at 200 mA cm−2 in different Ov populations. | ||
Specifically, the Mn1Co5Ox showed overpotentials of 275 and 569 mV at a current density of 10 and 100 mA cm−2, respectively (Fig. 2c). These values are in good agreement with those of IrO2 (296 mV@10 mA cm−2 and 629 mV@100 mA cm−2) and are much lower than those of Co3O4 (371 mV@10 mA cm−2 and 653 mV@100 mA cm−2). Such improved catalytic OER activity of Mn1Co5Ox can be ascribed to the cooperation between the Mn and Co components. Additionally, the surface of Mn1Co5Ox effectively interacts with the intermediates through an oxygen atom, and the bonding interactions (–Mn–O–Co–O–) in the intermediates appear to be essential in determining the OER activity as a whole.59–63 The superior OER activity over the Mn1Co5Ox surface stems from its smallest Tafel slopes of 52 mV dec−1, which is considerably lower than those of Co3O4 (65 mV dec−1), IrO2 (94 mV dec−1) and MnOx (187 mV dec−1), hence suggesting the favorable OER kinetics of Mn1Co5Ox (Fig. 2d). Furthermore, after iR compensation, the maximum current density of the Mn1Co5Ox catalyst can reach up to 1 A cm−2 at 2.0 V (vs. RHE), as shown in Fig. 2e. It is worth noting that similar results were also obtained from the test on the PtTi substrate, indicating that the current originated from the activity of Mn1Co5Ox (Fig. S7†).
To order to enhance our understanding of the electrode kinetics during the OER, electrochemical impedance spectroscopy (EIS) was conducted at open circuit voltage (OCV), Ej=10, and Ej=20 with frequencies ranging from 106 Hz to 10 mHz (Fig. 2f). The equivalent circuit used to model the data is shown in the inset of Fig. 2f. The electrical circuit consists of the solution resistance (Ru), film resistance (Rf), charge transfer resistance (Rct), and diffusion resistance (Wo). The capacitive elements used, CPE1 and CPE2, represent the constant phase element of the film and the electrical double layer, respectively. The Rct of Mn1Co5Ox was measured to be 18.11 Ω, 7.56 Ω, and 2.72 Ω at OCV, Ej=10, and Ej=20, respectively (here, “j” stands for current density). The smaller Rct means better electron-transfer kinetics during the OER. Moreover, we examined the intrinsic activity of the catalyst, as characterized by its electrochemical active surface area (ECSA), which is linearly proportional to the double-layer capacitance (Cdl) derived from cyclic voltammetry (CV) curves with various sweep rates in the non-faradic region (Fig. S8 and S9†). As shown in Fig. S10,† the Cdl value of Mn1Co5Ox was significantly larger than that of Co3O4, indicating an increase in active sites available for the OER.
Oxygen vacancies (Ov) exist widely in transition metal oxides due to their low formation energy. The formation energy of Ov increases with the energy difference between the upper edge of the O 2p band and the Fermi level. Thus, only a certain amount of Ov can be created in oxides and they have a direct influence on the M–O bonds near the Ov. They can promote the adsorption of water molecules on the catalyst surface, thereby improving the reactivity of the active site and OER performance. Therefore, we obtained XPS spectra to analyze the Ov in MnCoOx; the binding energy peaks at 530.2 eV, 531.2 eV and 532.7 eV belong to the lattice oxygen (Ol), oxygen vacancy (Ov) and adsorbed oxygen (Oa) respectively (Fig. 2g). The Mn1Co5Ox sample had the highest Ov proportion of 32.6%, followed by Mn1Co2.5Ox at 27.5%, Mn1Co1.5Ox at 24.0%, and Mn1Co8Ox at 20.9%, as shown in Table S1.† Correspondingly, the reaction potentials at 200 mA cm−2 decreased with the increase in oxygen vacancy content, with E (Mn1Co5Ox) < E (Mn1Co2.5Ox) < E (Mn1Co1.5Ox) < E (Mn1Co8Ox). Next, we correlated the Ov concentration with the corresponding OER activity. As shown in Fig. 2h, the OER activity over these MnCoOx composites increases proportionally to the surface Ov proportion, i.e., Mn1Co5Ox with the highest Ov proportion showed the best activity (lowest Ej=20 value). These results distinctly indicate that the presence of surface Ov in these MnCoOx composites plays a crucial role in promoting OER activity, which is further substantiated by the DFT studies, vide infra.
The ability of the MnCoOx catalyst to withstand acidic electrolytes is a crucial aspect for practical applications. The chronopotentiometry (CP) measurements were carried out to evaluate its long-term stability at varied constant current densities of 100, 200, 500, and 1000 mA cm−2 in 0.5 M H2SO4 (pH 0), as shown in Fig. S11.† As evidenced by the potential remaining at around 1.97 V@100 mA cm−2 (for ∼300 hours) and 2.01 V@200 mA cm−2 (∼110 hours), as shown in Fig. 3b, our results demonstrate a remarkable durability of the Mn1Co5Ox catalyst, compared to various other reported catalysts (Fig. 3a and Table S2†). These experimental outcomes substantiate that Mn1Co5Ox electrocatalysts exhibited promising electrochemical activity and superior durability for the OER.
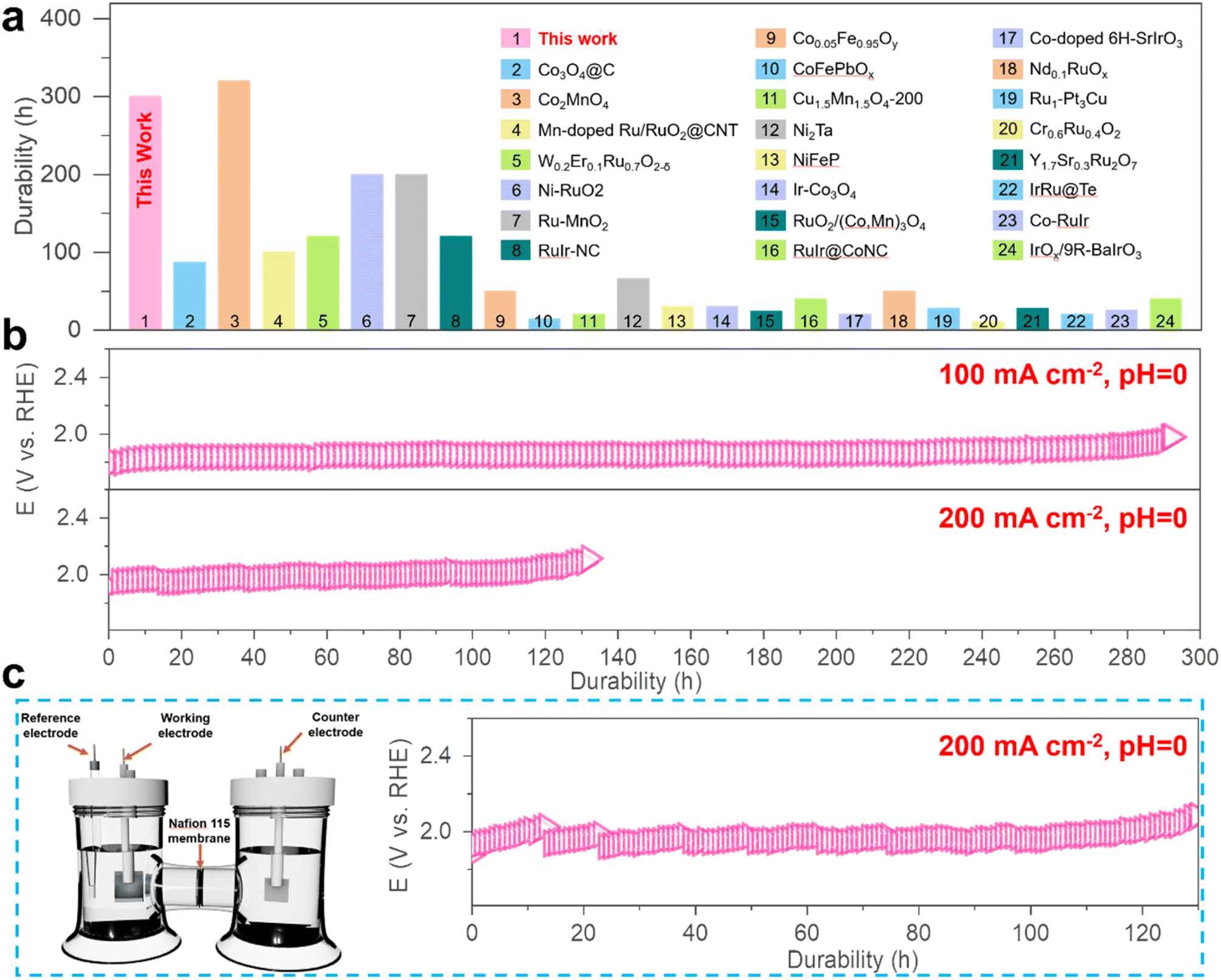 | ||
| Fig. 3 Stability of MnCoOx. (a) Comparison of the durability of Mn1Co5Ox catalysts in acidic solution with that of other reported catalysts. Of note, samples 1–5 were measured at 100 mA cm−2, and the others, at 10 mA cm−2. (b) Potential required to reach 100 and 200 mA cm−2 in 0.5 M H2SO4 for Mn1Co5Ox measured using chronopotentiometry method. (c) Chronopotentiometric measurement of the Mn1Co5Ox catalyst at a constant OER current density of 200 mA cm−2 recorded in an H-type water electrolysis cell in 0.5 M H2SO4 with the anode and cathode separated by a Nafion 115 membrane at room temperature. | ||
Furthermore, the Mn1Co5Ox catalyst demonstrated a ∼15-fold increase, which is a significantly longer lifetime compared to that of Co3O4 at 100 mA cm−2 (Fig. S12†), thus indicating that the Mn dopant substantially enhances the stability of MnCoOx during the acidic OER. For further investigations, chronopotentiometry measurements of the Mn1Co5Ox catalyst were performed while maintaining a constant OER current density of 200 mA cm−2 using a water electrolysis cell designed in an H-type configuration, featuring a Nafion 115 membrane to separate the anode and cathode (Fig. 3c). The results showed a monotonic increase in the Ej=200 in the case of the Mn1Co5Ox catalyst from 666 mV to 825 mV over 100 hours of continuous operation.
Study of the utilized MnCoOx catalyst
This study investigated the activity loss of MnCoOx/CP electrocatalysts during the electrochemical process by comparing the pristine and utilized catalysts. The XRD analysis was used for comparison, which shows that the peak intensities at 2θ = 31.3° (for Co3O4 (220) facet), 44.8° (Co3O4 (400)), and 59.3° (Co3O4 (551)) were less intense in the utilized Mn1Co5Ox/CP catalyst (Fig. 4a), hence indicating the deformation of the MnCoOx oxide structure, significant to the deactivation of the OER activity. In contrast, the CP support can be identified at 2θ = 26.5°. Furthermore, the TEM image of the utilized Mn1Co5Ox catalyst showed an amorphous layer of about 1 nm thickness, as shown in Fig. 4b and S13,† indicating the formation of Co(OH)x hydroxide species. Furthermore, XPS was applied to investigate and compare the surface composition and oxidation state of the pristine and utilized MnCoOx catalysts. In the high-resolution spectra of Co 2p, the deconvoluted binding energies (B.E.) at 781.72 eV and 780.40 eV can be assigned to Co2+ 2p3/2 and Co3+ 2p3/2 species, respectively (Fig. 4c).52,64,65 Also, for the Mn 2p spectra shown in Fig. 4d, the B.E. peaks at ∼642.4 eV and 653.7 eV can be associated with Mn3+ species.64,66 In comparison, other peaks at 644.0 and 655.3 eV are affiliated with Mn4+.66 As listed in Table S1,† the Co3+ and Mn4+ species were reduced to Co2+ and Mn3+ during the OER process. The O 1s B.E. peaks of fresh Mn1Co5Ox can be identified by two components centered at 530.1 eV and 531.8 eV (Fig. S15†), depicting the Ol and Ov species.67,68 Interestingly, the hydroxyl species (OH) was found at ∼533.1 eV in the utilized Mn1Co5Ox catalyst, signifying the surface amorphous structure of the utilized oxide electrocatalyst, which is well consistent with the observations in the XRD and STEM analyses. These results imply that the Co3+ and Mn4+ species and Ov sites in the MnCoOx solid solution catalysts play an important role in the OER, which is consistent with the previous literature studies.67,69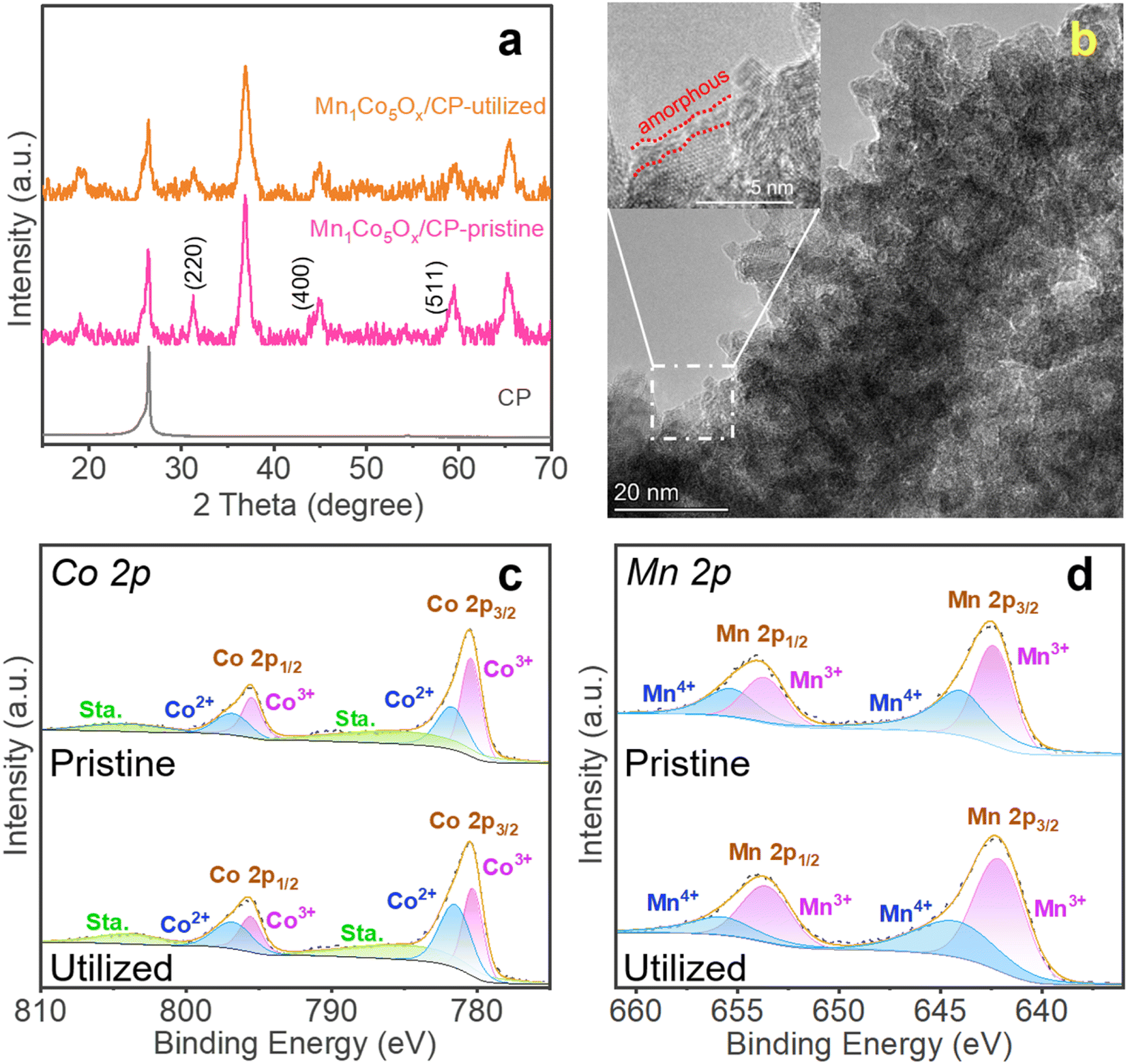 | ||
| Fig. 4 (a) XRD pattern of the pristine and utilized Mn1Co5Ox/CP electrocatalysts. (b) STEM image of utilized Mn1Co5Ox. XPS spectra of the pristine and utilized Mn1Co5Ox catalysts: (c) Co 2p and (d) Mn 2p. Electrolysis conditions: a 0.5 M H2SO4 solution for 10 h at 100 mA cm−2 at 25 °C. | ||
The stability of Mn1Co5Ox primarily arises from the incorporation of Mn into the crystal structure of Co3O4. The results of XAS and XPS suggested that the addition of a small amount of Mn enhances the conductivity of Co3O4 by increasing the number of 3d orbital electrons. Simultaneously, the presence of Mn reduces the number of outer orbital electrons in the local vicinity, leading to a decreased local lattice constant and improved stability of the local structure. This, in turn, enhances the overall stability of the crystal structure. TEM images of the spent MnCoOx sample after a 10 hour reaction test reveals the presence of a small amorphous layer on the catalyst surface, while the remaining regions retain the crystal structure characteristics. XRD analysis of the employed catalyst confirms that MnCoOx still maintains an overall crystal structure, as depicted in Fig. 4a. The results from TEM and XRD indicate that the sustained catalytic durability originates from the doped crystal structure.70
To investigate the ion release levels of catalysts during stability testing, Co3O4 and Mn1Co5Ox catalysts were subjected to testing at a current density of 100 mA cm−2. The final electrolyte was collected and prepared for inductively coupled plasma (ICP) analysis to determine the concentrations of Co and Mn ions in the solution. Upon comparative analysis of the results presented in Table S3,† it is worth noting that the Co3O4 catalyst exhibited a concentration of 89 ppm Co cations in the solution following a 14 hour stability test. In contrast, the Mn1Co5Ox catalyst showed low concentrations of 38 ppm Co and 6 ppm Mn cations in the solution after a 25 hour stability testing period. These outcomes revealed that the addition of Mn enhances the structural durability of the catalyst, retarding the release of Co cations in the solution and thus extending the lifespan of the catalyst.
DFT calculations
Density functional theory (DFT) calculations were performed to elucidate further the fundamental mechanism behind the enhanced OER activity of Mn1Co5Ox. We considered the plausibility of Mn doping sites to locate their low energy configurations and calculated the configurations of 10 random bulk Mn1Co5Ox molecules, as shown in Fig. S16,† with the isomers arranged from low to high relative energies (high to low stability). This proves that the chosen lowest energy configuration remains relatively thermodynamically stable. The band structure of bulk Co3O4 was calculated, and the results indicate a band gap of 1.32 eV (Fig. S17†), demonstrating semiconductor behavior. Furthermore, we also calculated the OER activity of bare Co3O4(311), MnCoOx(311) and Ov_MnCoOx(311), respectively, and the configurations of the calculated OER are shown in Fig. S18 and S19.† The free energy diagrams of the OER at 0 V and 1.23 V are shown in Fig. 5a for Co3O4(311), MnCoOx(311) and Ov_MnCoOx(311). In contrast, the formation of the intermediate O* has the highest activation energy, which is thermodynamically unfavorable. However, after Mn doping,71 the overpotential of the OER is sharply reduced from 2.79 eV (Co3O4(311)) to 0.82 eV (MnCoOx(311)).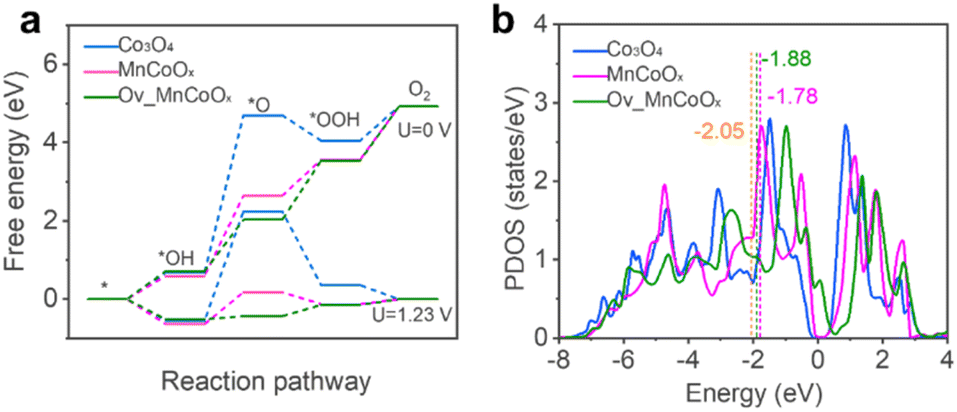 | ||
| Fig. 5 (a) Reaction-free energies of the OER pathways at 0 V and 1.23 V. (b) The d-band center of Co active sites. | ||
Furthermore, the overpotential of the OER decreases to 0.29 eV upon the introduction of Ov. The d-band center is typically used as a descriptor to explain the direct interaction of the catalyst with the adsorbate, and the OER activity is regulated by modulation of the d-band center energy level (Ed). Therefore, we also calculated the projected density of states (PDOS) of the active site Co for Co3O4(311) and MnCoOx(311), respectively. The results are shown in Fig. 5b; the incorporation of Mn can significantly elevate the d-band center position of Co3O4 (Ed = −2.05 eV) to near the Fermi level (Ed = −1.78 eV). Interestingly, after the introduction of Ov, the position of the d-band center at the active site is equilibrated (Ed = −1.88 eV). During the OER process, a moderate adsorption strength of intermediate species can lower the energy barrier of the OER. Therefore, the incorporation of Mn and the introduction of Ov can stabilize reaction intermediates, thus reducing the energy barrier of the OER and enhancing catalytic activity, as shown in Fig. S20 and S21.† (ref. 72) The results of the theoretical calculations also agree with the experimental phenomena.
Conclusions
The study highlights the promising potential of MnCoOx solid solutions as an efficient catalyst for the OER. The solid solutions were synthesized by redox deposition of KMnO4 and Co(NO3)2 on a CP support under varying Co/Mn ratios. The crystalline nature of MnCoOx was evaluated by XRD analysis, and TEM analysis verifies the homogeneous distribution of Co and Mn species. OER tests of MnCoOx solid solutions deposited on CP revealed that Mn1Co5Ox was the most efficient catalyst, outperforming Co3O4, MnOx, the CP support, and IrO2, suggesting its practical application in electrocatalysis. Moreover, Mn1Co5Ox exhibited excellent durability and increased active sites, making it a promising candidate for OER applications. Following the XPS analysis, this study showed that Co3+, Mn4+, and Ov sites in the MnCoOx solid solution catalyst play an essential role in the OER, with the Mn dopant significantly enhancing the catalyst's stability during acidic OER. Corroborative DFT computations validated the empirical observations and provided insight into how Mn1Co5Ox improves OER catalytic performance. This study offers valuable guidance for designing efficient OER catalysts, highlighting MnCoOx solid solutions as a promising candidate.Author contributions
G. L. and S. G. designed the concept of this work. J. J. Z. and Y. Zhao performed major experiments. J. J. Z. and analyzed the experimental data. J. J. Z. and S. G. wrote the first manuscript. A. R., Z. U. D. B., and G. L. wrote the final manuscript. L. X. performed the computational study. Y. C. performed TEM. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (22172167).Notes and references
- X. Cao, T. Wang and L. Jiao, Adv. Fiber Mater., 2021, 3, 210–228 CrossRef CAS
.
- J. Z. Hassan, A. Zaheer, A. Raza and G. Li, Sustainable Mater. Technol., 2023, 36, e00609 CrossRef CAS
- L. Li, P. Wang, Q. Shao and X. Huang, Chem. Soc. Rev., 2020, 49, 3072–3106 RSC
- A. Raza, A. A. Rafi, J. Z. Hassan, A. Rafiq and G. Li, Appl. Surf. Sci. Adv., 2023, 15, 100402 CrossRef
- P. P. Lopes, D. Y. Chung, X. Rui, H. Zheng, H. He, P. Farinazzo Bergamo Dias Martins, D. Strmcnik, V. R. Stamenkovic, P. Zapol, J. F. Mitchell, R. F. Klie and N. M. Markovic, J. Am. Chem. Soc., 2021, 143, 2741–2750 CrossRef CAS PubMed
- S. Li, Y. Gao, N. Li, L. Ge, X. Bu and P. Feng, Energy Environ. Sci., 2021, 14, 1897–1927 RSC
- G. Shen, R. Zhang, L. Pan, F. Hou, Y. Zhao, Z. Shen, W. Mi, C. Shi, Q. Wang, X. Zhang and J.-J. Zou, Angew. Chem., Int. Ed., 2020, 59, 2313–2317 CrossRef CAS PubMed
- S. B. Scott, R. R. Rao, C. Moon, J. E. Sørensen, J. Kibsgaard, Y. Shao-Horn and I. Chorkendorff, Energy Environ. Sci., 2022, 15, 1977–1987 RSC
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433 CrossRef
- K. A. Lewinski, D. van der Vliet and S. M. Luopa, ECS Trans., 2015, 69, 893–917 CrossRef CAS
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed
- L. A. King, M. A. Hubert, C. Capuano, J. Manco, N. Danilovic, E. Valle, T. R. Hellstern, K. Ayers and T. F. Jaramillo, Nat. Nanotechnol., 2019, 14, 1071–1074 CrossRef CAS PubMed
- A. Li, H. Ooka, N. Bonnet, T. Hayashi, Y. Sun, Q. Jiang, C. Li, H. Han and R. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 5054–5058 CrossRef CAS PubMed
- D. A. Kuznetsov, M. A. Naeem, P. V. Kumar, P. M. Abdala, A. Fedorov and C. R. Müller, J. Am. Chem. Soc., 2020, 142, 7883–7888 CrossRef CAS PubMed
- N. Li, L. Cai, C. Wang, Y. Lin, J. Huang, H. Sheng, H. Pan, W. Zhang, Q. Ji, H. Duan, W. Hu, W. Zhang, F. Hu, H. Tan, Z. Sun, B. Song, S. Jin and W. Yan, J. Am. Chem. Soc., 2021, 143, 18001–18009 CrossRef CAS
- C. W. Song, J. Lim, H. B. Bae and S.-Y. Chung, Energy Environ. Sci., 2020, 13, 4178–4188 RSC
- Y. Lin, Z. Tian, L. Zhang, J. Ma, Z. Jiang, B. J. Deibert, R. Ge and L. Chen, Nat. Commun., 2019, 10, 162 CrossRef PubMed
- L. Cao, Q. Luo, J. Chen, L. Wang, Y. Lin, H. Wang, X. Liu, X. Shen, W. Zhang, W. Liu, Z. Qi, Z. Jiang, J. Yang and T. Yao, Nat. Commun., 2019, 10, 4849 CrossRef PubMed
- S. Hao, M. Liu, J. Pan, X. Liu, X. Tan, N. Xu, Y. He, L. Lei and X. Zhang, Nat. Commun., 2020, 11, 5368 CrossRef CAS
- Z. L. Zhao, Q. Wang, X. Huang, Q. Feng, S. Gu, Z. Zhang, H. Xu, L. Zeng, M. Gu and H. Li, Energy Environ. Sci., 2020, 13, 5143–5151 RSC
- Q. Feng, J. Zou, Y. Wang, Z. Zhao, M. C. Williams, H. Li and H. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 4520–4530 CrossRef CAS
- N. Hodnik, P. Jovanovič, A. Pavlišič, B. Jozinović, M. Zorko, M. Bele, V. S. Šelih, M. Šala, S. Hočevar and M. Gaberšček, J. Phys. Chem. C, 2015, 119, 10140–10147 CrossRef CAS
- S. Cherevko, A. R. Zeradjanin, A. A. Topalov, N. Kulyk, I. Katsounaros and K. J. J. Mayrhofer, ChemCatChem, 2014, 6, 2219–2223 CrossRef CAS
- K. Wang, Y. Wang, B. Yang, Z. Li, X. Qin, Q. Zhang, L. Lei, M. Qiu, G. Wu and Y. Hou, Energy Environ. Sci., 2022, 15, 2356–2365 RSC
- S. Anantharaj, K. Karthick and S. Kundu, Inorg. Chem., 2019, 58, 8570–8576 CrossRef CAS PubMed
- J. S. Mondschein, J. F. Callejas, C. G. Read, J. Y. C. Chen, C. F. Holder, C. K. Badding and R. E. Schaak, Chem. Mater., 2017, 29, 950–957 CrossRef CAS
- X. Yang, H. Li, A.-Y. Lu, S. Min, Z. Idriss, M. N. Hedhili, K.-W. Huang, H. Idriss and L.-J. Li, Nano Energy, 2016, 25, 42–50 CrossRef CAS
- K.-L. Yan, J.-F. Qin, J.-H. Lin, B. Dong, J.-Q. Chi, Z.-Z. Liu, F.-N. Dai, Y.-M. Chai and C.-G. Liu, J. Mater. Chem. A, 2018, 6, 5678–5686 RSC
- M. Chatti, J. L. Gardiner, M. Fournier, B. Johannessen, T. Williams, T. R. Gengenbach, N. Pai, C. Nguyen, D. R. MacFarlane, R. K. Hocking and A. N. Simonov, Nat. Catal., 2019, 2, 457–465 CrossRef CAS
- J. Zhang, Y. Xie, Q. Jiang, S. Guo, J. Huang, L. Xu, Y. Wang and G. Li, J. Mater. Chem. A, 2022, 10, 16920–16927 RSC
- L. An, C. Wei, M. Lu, H. Liu, Y. Chen, G. G. Scherer, A. C. Fisher, P. Xi, Z. J. Xu and C. H. Yan, Adv. Mater., 2021, 33, 2006328 CrossRef CAS
- C. Spöri, J. T. H. Kwan, A. Bonakdarpour, D. P. Wilkinson and P. Strasser, Angew. Chem., Int. Ed., 2017, 56, 5994–6021 CrossRef PubMed
- F.-Y. Chen, Z.-Y. Wu, Z. Adler and H. Wang, Joule, 2021, 5, 1704–1731 CrossRef CAS
- S. Geiger, O. Kasian, M. Ledendecker, E. Pizzutilo, A. M. Mingers, W. T. Fu, O. Diaz-Morales, Z. Li, T. Oellers, L. Fruchter, A. Ludwig, K. J. J. Mayrhofer, M. T. M. Koper and S. Cherevko, Nat. Catal., 2018, 1, 508–515 CrossRef CAS
- M. Huynh, D. K. Bediako and D. G. Nocera, J. Am. Chem. Soc., 2014, 136, 6002–6010 CrossRef CAS
- L. Zhou, A. Shinde, J. H. Montoya, A. Singh, S. Gul, J. Yano, Y. Ye, E. J. Crumlin, M. H. Richter, J. K. Cooper, H. S. Stein, J. A. Haber, K. A. Persson and J. M. Gregoire, ACS Catal., 2018, 8, 10938–10948 CrossRef CAS
- J. Huang, H. Sheng, R. D. Ross, J. Han, X. Wang, B. Song and S. Jin, Nat. Commun., 2021, 12, 3036 CrossRef CAS
- J. Zhao, X. Li, M. Zhang, Z. Xu, X. Qin, Y. Liu, L. Han and G. Li, Nanoscale, 2023, 15, 4612–4619 RSC
- Y. Cao, S. Guo, C. Yu, J. Zhang, X. Pan and G. Li, J. Mater. Chem. A, 2022, 8, 15767–15773 RSC
- Y. Wang, Q. Jiang, L. Xu, Z. Han, S. Guo, G. Li and A. Baiker, ACS Appl. Mater. Interfaces, 2021, 13, 61078–61087 CrossRef CAS PubMed
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 12947–12954 CrossRef
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed
- D.-E. Jiang and S. Dai, Phys. Chem. Chem. Phys., 2011, 13, 978–984 RSC
- Y. Wang, Y.-Z. Zhang, Y.-Q. Gao, G. Sheng and J. E. Elshof, Nano Energy, 2020, 68, 104306 CrossRef CAS
- D. J. Chadi, Phys. Rev. B: Condens. Matter Mater. Phys., 1977, 16, 1746–1747 CrossRef
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed
- J. Zhou, Z. Han, X. Wang, H. Gai, Z. Chen, T. Guo, X. Hou, L. Xu, X. Hu, M. Huang, S. V. Levchenko and H. Jiang, Adv. Funct. Mater., 2021, 31, 2102066 CrossRef CAS
- X. Wei, S. Barkaoui, J. Chen, Q. Fang, G. Cao, Z. Wu, F. Wang and G. Li, Nanoscale Adv., 2021, 3, 1741–1746 RSC
- Q. Shi, Z. Li, C. Cao, G. Li and S. Barkaouib, Nanoscale Adv., 2023, 5, 5385–5389 RSC
- S. Niu, X.-P. Kong, S. Li, Y. Zhang, J. Wu, W. Zhao and P. Xu, Appl. Catal., B, 2021, 297, 120442 CrossRef CAS
- Y. Sun, Q. Wang and Z. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 43508–43516 CrossRef CAS PubMed
- F. Kollhoff, J. Schneider, G. Li, B. Sami, W. Shen, T. Berger, O. Diwald and J. Libuda, Phys. Chem. Chem. Phys., 2018, 20, 24858–24868 RSC
- Y. Cao, Y. Su, L. Xu, X. Yang, Z. Han, R. Cao and G. Li, J. Energy Chem., 2022, 71, 167–173 CrossRef CAS
- S. Niu, S. Li, Y. Du, X. Han and P. Xu, ACS Energy Lett., 2020, 5, 1083–1087 CrossRef CAS
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed
- M. García-Mota, M. Bajdich, V. Viswanathan, A. Vojvodic, A. T. Bell and J. K. Nørskov, J. Phys. Chem. C, 2012, 116, 21077–21082 CrossRef
- K. Zhu, T. Wu, M. Li, R. Lu, X. Zhu and W. Yang, J. Mater. Chem. A, 2017, 5, 19836–19845 RSC
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS
- L. Zhang, L. Shi, L. Huang, J. Zhang, R. Gao and D. Zhang, ACS Catal., 2014, 4, 1753–1763 CrossRef CAS
- Y. Liu, H. Zhang, H. Wan, W. Zhang, N. Jiang, G. Huang, Z. Wang, S. Luo and H. Sun, J. Alloys Compd., 2019, 787, 720–727 CrossRef CAS
- Q. Shi, Y. Zhang, Z. Li, Z. Han, L. Xu, A. Baiker and G. Li, Nano Res., 2023, 16, 6951–6959 CrossRef CAS
- Q. Shi, Z. Qin, A. Waheed, Y. Gao, H. Xu, H. Abroshan and G. Li, Nano Res., 2020, 13, 939–946 CrossRef CAS
- H. Zhang, T. Ling and X.-W. Du, Chem. Mater., 2014, 27, 352–357 CrossRef
- Y. Chen, Y. Li, W. Chen, W. W. Xu, Z. Han, A. Waheed, Z. Ye, G. Li and A. Baiker, Nano Res., 2022, 15, 1366–1374 CrossRef CAS
- L. An, H. Zhang, J. Zhu, S. Xi, B. Huang, M. Sun, Y. Peng, P. Xi and C.-H. Yan, Angew. Chem., Int. Ed., 2023, 62, e202214600 CrossRef CAS PubMed
- A. Li, S. Kong, C. Guo, H. Ooka, K. Adachi, D. Hashizume, Q. Jiang, H. Han, J. Xiao and R. Nakamura, Nat. Catal., 2022, 5, 109–118 CrossRef CAS
- C. Li, J. Zhao, L. Xie, J. Wu, Q. Ren, Y. Wang and G. Li, Angew. Chem., Int. Ed., 2021, 60, 18129 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05233h |
| ‡ J. J. Z., A. R. and Y. Z. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |