
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConjugation of gold nanoparticles with multidentate surfactants for enhanced stability and biological properties†
Rany
Rotem‡
,
Marco
Giustra‡
,
Federica
Arrigoni
,
Jessica A.
Bertolini
,
Stefania
Garbujo
,
Maria A.
Rizzuto
,
Lucia
Salvioni
,
Linda
Barbieri
 ,
Luca
Bertini
,
Luca
De Gioia
,
Miriam
Colombo
* and
Davide
Prosperi
*
,
Luca
Bertini
,
Luca
De Gioia
,
Miriam
Colombo
* and
Davide
Prosperi
*
Department of Biotechnology and Bioscience, University of Milano-Bicocca, Piazza della Scienza 2, 20126, Milano, Italy. E-mail: davide.prosperi@unimib.it
First published on 7th November 2022
Abstract
This work originated from the need to functionalize surfactant-coated inorganic nanoparticles for biomedical applications, a process that is limited by excess unbound surfactant. These limitations are connected to the bioconjugation of targeting molecules that are often in equilibrium between the free aliquot in solution and that which binds the surface of the nanoparticles. The excess in solution can play a role in the biocompatability in vitro and in vivo of the final nanoparticles stock. For this purpose, we tested the ability of common surfactants – monothiolated polyethylene glycol and amphiphilic polymers – to colloidally stabilize nanoparticles as excess surfactant is removed and compared them to newly appearing multidentate surfactants endowed with high avidity for inorganic nanoparticles. Our results showed that monothiolated polyethylene glycol or amphiphilic polymers have an insufficient affinity to the nanoparticles and as the excess surfactant is removed the colloidal stability is lost, while multidentate high-avidity surfactants excel in the same regard, possibly allowing improvement in an array of nanoparticle applications, especially in those stated.
1. Introduction
Inorganic colloidal nanoparticles (NPs) are a promising tool in the field of biomedical applications. However, the research community is divided over the current state and direction necessary to fulfil that promise,1 with some calling for reevaluation of long standing drug delivery paradigms. Here we go back to take a closer look at surfactant coated NP and their functionalization.Most often NPs are covered by a surfactant to prevent aggregation and allow conjugation to other ligands e.g., for active targeting.2–10 The conjugation can be limited by presence of excess unbound surfactant. Hypothetically it can lead to a mixture of ligand-surfactant-NP and ligand-unbound surfactant, or equivalent scenarios when binding NPs to surfaces (e.g., electrode) or to each other (e.g., for sensing). Removal of excess surfactant however might destabilize the NPs. This might also impact in in vivo applications where NPs and surfactant are naturally separated due to different clearance times and routes.11–13 Furthermore, excess surfactant may modulate the electrocatalytic properties of NPs promoting many reactions useful in industrial applications (i.e. hydrogenation of unsaturated molecules, CO2 and O2 reduction).14,15 This is a critical point to consider for in vitro and in vivo administration.
The coverage and passivation of the NP surface by a surfactant can be explained using the Langmuir adsorption equation, utilizing a single equilibrium constant. However, the surface of different NPs might not behave so ideally.16 Thus, here we examine practically the ability of a surfactant to colloidally stabilize NPs as excess surfactant is removed.
Most of currently used surfactants can be divided to monodentate surfactants and amphiphilic polymers.6 Monodentate ligands interact directly with the surface through functionalities with high affinity for the inorganic surface (e.g., poly(ethylene glycol)-thio or PEG-thio2,17), (Fig. 1A); amphiphilic polymers are ligands capable of intercalating by hydrophobic interactions (Fig. 1B).
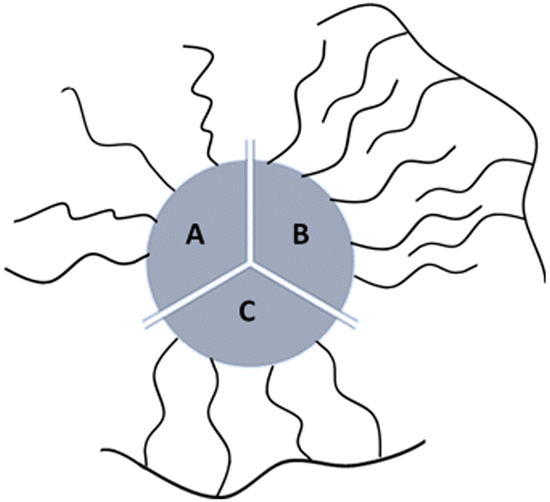 | ||
| Fig. 1 Cartoon of the three types of synthetic coatings that can bind NPs surface: (A) monodentate ligands; (B) intercalating surfactant; (C) multidentate surfactant. | ||
A newly emerging approach is the preparation of multidentate polymers by grafting with ligands able to interact with the surface. In this last case the chelating effect predominates (Fig. 1C). These can be prepared separately,18–22 or by polymerization directly on the NP.6,22–26 In the first case, polymers as poly(isobutylene-alt-maleic anhydride) or poly(acrylic acid) can be used as precursors to produce a library of multi-branching polymers containing primary amines. In fact, by exploiting succinic anhydrides or carboxylic acids in the monomeric units, molecules as dopamine, PEG (polyethylene glycol), and synthetic ones (i.e. lipoic acid and biotin modified with ethylenediamine) can be conjugated as the literature shows. In the second case, small molecules on the NPs can be used as initiators to obtain polymer coatings: i.e. photo-polymerization on diacetylene as henicosa-10,12-diyn-1-yl disulfide are used to coat AuNPs; use of coupling agents such as DCC (Dicyclohexylcarbodiimide) in presence of DPA (diaminopimelic acid) allows amidation with mercaptoundecanoic acid coated NPs otherwise, i.e. Nichel NPs can be use as catalyst to produce polystyrene starting from olefins.
We present here a new multidentate surfactant and quantitatively compare its ability to stabilize NPs as a function of the removal of its excess with two common surfactants. Pyridilthio-cysteamine-methoxypolyoxyethyleneamine-grafted-poly(isobutylene-alt-maleic-anhydride) (PCP) is the resulting polymer of the nucleophilic addition of pyridilthiocysteamine and short amino PEG (750 Da). These two ligands are selected given that: (1) the presence of sulfur-group has high affinity to specific inorganic surfaces such as gold; (2) the presence of short PEG improves the solubility of the surfactant in organic and aqueous solvents. The well-known Dopa-PIMA-PEG (PDP) has been used to coat Iron Oxide NPs in comparison to PCP. Our results highlight limitations in traditional surfactants and show a major improvement using multidentate surfactants. In the present work, the capability of our new multi-branching polymer in stabilizing colloidal nanoparticles and some ensuing benefits are shown. We provide the opportunity to produce a wide library of surfactant selecting suitable ligands that coat NPs properly, according to surface chemistry. In particular, the employment of poly(isobutylene-alt-maleic anhydride) allows facile and fast synthesis and purification exploiting basic knowledges of organic chemistry (i.e. not using expensive catalysts). Moreover, the possibility to obtain the NPs stability removing the surfactant excess from the solution will make the use of these polymers family possible and promising in the medical field.
2. Experimental
2.1. Chemicals and materials
Hydrogen tetrachloroaurate hydrate, 99.999%, (trace metal basis) Acros Organics™ was purchased from FischerScientific. Sodium citrate dihydrate (99%), dodecylamine (99%), tetra-n-octylammonium bromide (98%) (TOAB), NaOH, L-ascorbic acid (98.0%), sodium borohydride (≥99%), Toluene, MeOH, HCl, NaCl, CHCl3, pentane, dimethylformamide, tris(2-carboxyethyl)phosphine hydrochloride, tetrahydrofuran, triethylamine (Et3N), trifluoroacetic acid (TFA), poly(isobutylene-alt-maleic anhydride) Mw ∼ 6000, dopamine hydrochloride, carboxy-X-rhodamine N-succimidyl ester, fluorescamine, DMSO, Boric acid, n-hexane, N,N′-diisopropylcarbodiimide (DIC), albumin from bovin serum (BSA), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysulfosuccinimide sodium salt (sulfo NHS), were purchased from Sigma-Aldrich; Poly(ethylene glycol) or PEGs: Meo-PEG-SH (2 kDa), Carboxyl-PEG-SH (2 kDa), and Meo-PEG-NH2 (750 Da) were purchased from RappPolymere. (S)-2-Pyridylthio cysteamine HCl was purchased from CombiBlocks; Phosphate buffered saline (PBS), Dulbecco's Modified Eagle Medium (DMEM), Fetal Bovine Serum (FBS), PenStrep were purchased from EuroClone; Alexa Fluor 647 – Cadaverine was purchased from ThermoFischer Scientific. Water used is purified by passing through MilliQ Millipore system.2.2. Synthesis of spherical gold nanoparticles
The synthesis of spherical gold nanoparticles (5 nm) was made in organic solvent using Brust's protocol.27 For 300 mg of HAuCl4 dissolved in 25 mL of MilliQ water, 2.17 g of Tetra-n-octylammonium bromide (TOAB) were dissolved in 80 mL of toluene. These solutions were added to a separating funnel, shaked gently for 5 min and the aqueous phase was discarded. 334 mg of NaBH4 were dissolved in 25 mL of MilliQ water and added dropwise throughout 1 min. The solution was kept under stirring for one hour. In a separating funnel, the solution was mixed with 25 mL of HCl 10 mM and the aqueous phase was discarded. This was repeated with 25 mL of NaOH 10 mM and four times with 25 mL of MilliQ water. The solution was then kept under stirring and overnight. 9.6 mL of pure dodecylamine were added for a final concentration 0.4 M and the solution was left for three hours at 65 °C. The sample was centrifugated at 2000 RCF for 10 min and the precipitate was discarded. 250 mL of MeOH was added and the solution was centrifugated at 2000 RCF discarding the supernatant. The pellet was dried and dissolve in CHCl3. The suspension was later centrifugated again to remove aggregates.The synthesis of spherical gold nanoparticles (15 nm) was made in MilliQ water. 1.2 mL of HAuCl4 25 mM were added into boiling, 150 mL of a tri-sodium Citrate solution 1.32 mM. After 15 min the red solution was characterized using UV-Vis and Dynamic light scattering. To move the NPs to CHCl3 the NPs were left with MeO-PEG-SH (2 kDa) in a molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30000 overnight and then stirred with 50 mL of Dodecylamine 0.4 M in CHCl3 until the phase transfer was completed. The solution was characterized using UV-Vis, dynamic light scattering and transmission electron microscopy (TEM) and Infrared Spectroscopy (IR). 5 μL of NPs suspension were dried on a carbon film-coated copper grid. TEM images were collected using Jeol JEM 2100Plus (Jeol, Tokyo, Japan) electron microscope.
:30000 overnight and then stirred with 50 mL of Dodecylamine 0.4 M in CHCl3 until the phase transfer was completed. The solution was characterized using UV-Vis, dynamic light scattering and transmission electron microscopy (TEM) and Infrared Spectroscopy (IR). 5 μL of NPs suspension were dried on a carbon film-coated copper grid. TEM images were collected using Jeol JEM 2100Plus (Jeol, Tokyo, Japan) electron microscope.
2.3. Synthesis of PMDA
For the synthesis of PMDA, 1 g of poly(isobutene-alt-maleic anhydride) (PMA) and 0.898 g of dodecylamine were added to 30 mL THF anhydrous and sonicated until well dispersed. The temperature was increased to 60 °C for three hours. The volume of THF was reduced under vacuum to 10 mL and the sample was left overnight at 60 °C. The solvent was evaporated and the product dissolved in CHCl3.2.4. Synthesis of multidentate polymers
The concentrate was recovered, lyophilized, and redissolved in CHCl3. The reaction yield could be roughly estimated by UV absorption.
2.5. Labelling of surfactants using AlexaFluor 647 Cadaverine
:1. Given the poor solubility of the fluorescent dye in CHCl3, the unconjugated fraction was removed by dissolving PMDA in this solvent.
:3. The reaction was stirred at RT for 16 hours in the dark. The solvent was evaporated, and the polymer was dissolved in sodium borate buffer (SBB) 0.5 M pH12. The presence of DMSO, by-products and the unconjugated dye were removed by Amicon Ultra centrifugal filter (30 kDa). Trifluoroacetic acid (TFA) was added to the solution to protonate the carboxylates. The solvent was evaporated, and the polymer was dissolved in CHCl3.
PDP. 5.3 μL of AlexaFluor 647 Cadaverine 1 mg × mL−1 in DMSO and 11.6 μL of pure DIC were added to 1 mg of PDP dissolved in 500 μL of THF. The reaction was stirred at RT for 16 hours in the dark. The THF was evaporated, and the polymer was precipitated from DMSO leftovers by adding 3 mL 9
:1 hexane/diethylether mixture. The polymer precipitate was then dissolved in CHCl3. Given the poor solubility of the fluorescent dye in CHCl3, the unconjugated fraction was removed at that step.
PCP. 15 μL of AlexaFluor 647 Cadaverine 1 mg × mL−1 in DMSO and 390 μL of EDC 3.3 M in MilliQ water were added to 1 mg of surfactant dissolved in 20 mM borate buffer pH 9. The reaction was stirred at RT for 16 hours in the dark. The presence of DMSO, by-products, buffer, and the unconjugated dye were removed by Amicon Ultra centrifugal filter (10 kDa). The solvent was evaporated, and the polymer was dissolved in CHCl3.
2.6. Preparation of polymer coated gold and iron oxide NPs.
Gold NPs (5 nm). 546 μL NPs 2.71 μM and 84.6 μL of AlexaFluor 647-PEG-SH 0.07 g × L−1 were added to 0.5 mL of CHCl3 and left to react for 2 hours in the dark at 40 °C. Then 580 μL of MeO-PEG-SH 50 g × L−1 were added and left to react for another 4 hours in the dark at 40 °C.
Gold NPs (15 nm). 300 μL NPs 58.4 nM and 100 μL of AlexaFluor 647-PEG-SH 0.07 g × L−1 were added to 0.5 mL of CHCl3 and left to react for 2 hours in the dark at 40 °C. Then 149.3 μL of MeO-PEG-SH 2.3 g × L−1 were added and left to react for another 4 hours in dark at 40 °C.
To remove DDA, the NPs were diluted 1/20 with pentane and precipitated at 1500 RCF for 5 min. The supernatant was discarded, and the pellet was dispersed in CHCl3. This was repeated twice, and the last pellet was dried and dispersed in MilliQ water.
Gold NPs (5 nm). 546 μL NPs 2.71 μM and 550 μL of PMDA 0.1 M were added into 1 mL of CHCl3.
Gold NPs (15 nm). 300 μL NPs 58.4 nM and 535 μL of PMDA 0.1 M were added into 100 μL of CHCl3.
Iron oxide NPs. Iron oxide suspended in organic solvent were synthesized by solvothermal decomposition according to the protocol published by Park and co-workers.28 70 μL NPs 1.43 mM and 288 μL of PMDA 0.1 M were added into 1 mL of CHCl3.
The CHCl3 was evaporated under vacuum at 50 °C, and the NPs dispersed in aqueous SBB 100 mM pH 12 with sonication. The buffer was then replaced to SBB 10 mM pH9 using Amicon Ultra centrifugal filter (10 kDa).
Gold NPs (5 nm). 256 μL NPs 2.71 μM and 30.8 μL of PCP 26 mM were added into 40 μL of CHCl3.
Gold NPs (15 nm). 300 μL NPs 58.4 nM and 183 μL of PCP 26 mM were added into 1100 μL of CHCl3.
Iron oxide NPs. 70 μL NPs 1.43 mM and 1.77 mL of PDP 49 mM were added into 100 μL of CHCl3.
The solutions were left in the dark at 50 °C overnight. To remove DDA, the NPs were diluted 1/20 with pentane and precipitated at 1500 × g for 5 min. The supernatant was discarded, and the pellet was dispersed in CHCl3. This was repeated other 2 times, and the last pellet was dried and dispersed in SBB pH9 10 mM.
2.7. Fluorescamine assay to detect primary amine
To quantify the dodecylamine removed from the NPs, the pentane supernatant was evaporated and solid leftovers were dissolved in SBB pH8.3 20 mM. 200 μL of this solution were moved to a 96 well plate and added 10 μL Fluorescamine in DMSO 3 mg × mL−1. The resulting emission was quantified at 485 nm.2.8. Removal and quantification of surfactant excess
PMDA. The stock solution was split into six 10 mL conical centrifuge tubes (7.5 mL each). NPs were precipitated at 200
000 RCF (swing bucket), 5 hours. After each precipitation, the supernatants were collected, and the pellets were dispersed in fresh buffer (or in its own supernatant) to reach the desired dilution factor.
PEG and PCP. Each stock solution was split into six Amicon Ultra centrifugal filter (100 kDa). After each centrifugation, the filtrate was collected, and the concentrate was diluted in fresh buffer (or in its own filtrate) to reach the desired dilution factor.
Gold NPs (15 nm). Each stock solution was split into six 15 mL centrifuge tubes (3 mL each). NPs were precipitated at 5000 RCF (swing buckets) for 4 hours. After each precipitation, the supernatants were collected, and the pellets were dispersed in fresh buffer (or in its own supernatant) to reach the desired dilution factor.
Iron oxide NPs. Each stock solution was split into six microcentrifuge tubes. NPs were precipitated at 10
000 RCF (fixed angle rotor), for 2 hours. After each precipitation, the supernatants were collected, and the pellets were dispersed in fresh buffer (or in its own supernatant) to reach the desired dilution factor.
Quantification of the surfactant in the supernatant and in the pellet was done by fluorescence emission.
2.9. Stability test and characterization after removal of surfactant excess
After the serial removal of the surfactant, the NPs were diluted in PBS pH7.2 20 mM and left at 27 °C for 4 days before characterization by UV-Vis absorption and dynamic light scattering.2.10. Serum stability of PCP coated 15 nm Gold NPs
20 μL NPs 17.6 nM (with 99% of initial surfactant removed) were added to 980 μL of DMEM supplemented with 10% FBS (fetal bovine serum) and 1% penicillin-streptomycin and L-glutamine, and incubated at 37 °C. Before measuring the size distribution using DLS, the NPs were diluted to 12 mL in PBS 20 mM pH 7.2 and precipitated at 5000 RCF (swing buckets), for 4 hours. After discarding the supernatant, the pellets (∼200 μL) were dispersed in 12 mL of PBS pH 7.2 20 mM and the procedure was repeated twice more before the DLS measurement.2.11. Conjugation of bovine serum albumin (BSA) to PCP coated 15 nm Gold NPs
10 mL NPs in a 15 mL centrifuge tube were precipitated at 3000 RCF overnight and 9.9 mL of the supernatant was discarded to remove 99% of the free surfactant. To 40 μL NPs 7 nM in 10 mM SBB pH 9 added 2 μL EDC 6 mM and 2 μL sulfoNHS 2 mM. The solution was incubated at RT for 20 min, diluted to 4.5 mL in MilliQ water and reconcentrated to 100 μL using a 30 kDa centrifugal filter. To the concentrate added 25 μL 24 g × L−1x-rhodamine labelled BSA and the solution was incubated at 4 °C overnight. The conjugates were then mounted on top a continuous sucrose gradient (900 μL 5% sucrose, 1 μM (PMA monomer) PCP, 10 mM SBB pH 9, on top of 500 μL 20% sucrose, 1 μM (PMA monomer) PCP, 10 mM SBB pH 9. Tilted rotation to form a continuous gradient) and centrifugated at 4500 RCF for 4 hours. The pellet (∼100 μL) was diluted to 10 mL in SBB pH 9 with 1 μM (PMA monomer) PCP and centrifugated at 5000 RCF for 8 hours. BSA-rhodamine emission was then quantified in the pellet and supernatant and NPs were quantified using UV-Vis absorption.2.12. Cell viability assay
HeLa cells were cultured with DMEM supplemented with 10% FBS (fetal bovine serum) and 1% penicillin-streptomycin and L-glutamine, and seeded (5000 cells per well) on 96 wells plates. After 24 h from seeding, cells were treated with the different formulation and concentration of 15 nm gold NPs specified in the paper and incubated at 37 °C; untreated cells were used as control. After 24–48–72 h of incubation, cells were washed with PBS and tested with CellTiter 96® Non-Radioactive Cell Proliferation Assay (MTT; Promega Corporation), according to the manufacturer's instructions. Absorbance was read immediately in a plate reader (EnSightTM multimode plate reader, PerkinElmer), using a testing wavelength at 570 nm and a reference wavelength at 630 nm. The results were normalized on viability of untreated cells (set at 100%) and expressed as means ± standard deviation of three independent experiments in quadruplicate each.2.13. Density functional theory calculations
All geometry optimizations have been carried out with the TURBOMOLE 7.2.1 suite of programs,29 using the pure PBE exchange-correlation (XC) functional.30,31 A triple-ζ basis set with polarization functions for all atoms (def2-TZVP) has been used.32 For gold atoms, a relativistic effective core potential (ECP) was used for core electrons, in order to include (scalar) relativistic corrections.33 The resolution-of-identity (RI) technique34 has been used to speed up calculations, by reducing all four-center two-electron Coulomb integrals to precomputed three- and two-center integrals. Solvent effect has been implicitly evaluated according to the COnductor-like Screening MOdel (COSMO), setting ε = 80 (water).35,36 Grimme's corrections37 for dispersion forces have been added to the computational protocol. Additionally, to further validate our computational results, we also carried out geometry optimizations with an alternative GGA functional that implicitly accounts for vdW contacts, namely B97-D.38 A consensus for binding energies has been found between the two theoretical schemes (see ESI† for details). Binding energies have been computed and reported as ΔEbinding = EAu–ligand − (EAu + Eligand) were EAu–ligand indicates the energy of the bound state, EAu and Eligand the ones of the gold cluster alone and of the free ligand, respectively. According to previous investigations, Au nanoparticles can be modelled considering the Au(100), Au(111) and, to a lesser extent, Au(110) facets derived from bulk gold cleavage, along the respective planes.39 Thus, in the first instance, we used published xyz coordinates40 of three-layers Au(100) and Au(111) finite-sized nanoclusters, derived from bulk gold, of 110 and 85 Au atoms, respectively. Then, we built larger models for both Au(100) and Au(111) facets (referred to as AuNP(100) and AuNP(111)), cut from a 5 nm diameter nanoparticle whose atomistic structure was created using the Nanoparticle Builder Module of the OpenMD software.41 Both models comprise three Au layers, for a total of 149 and 160 Au atoms for AuNP(100) and AuNP(111), respectively. The nearest Au–Au distance is 2.884 Å, which is fully consistent with the lattice constant for bulk gold. Calculations for models mimicking Au(100) or Au(111) containing an even number of Au atoms were performed setting a singlet state (S = 0), while for the ones with a odd number of Au atoms a S = 1 spin state was imposed, according to the unrestricted formalism. EAu values have been calculated keeping Au models at their fixed geometry while structural perturbation was allowed only to the Au atom(s) directly involved in ligand binding, for the calculation of EAu–ligand. Ligands were instead fully relaxed, in both bound and unbound states.3. Results and discussion
Our aim in this work was to accurately quantify both free and NP-bound surfactant and estimate the NP's colloidal stability as the excess unbound surfactant is removed. For this purpose, we investigated two of the most broadly utilized surfactants, representing the two synthetic approaches – the ligand exchange with high-affinity ligands and the hydrophobic interaction-based coating with amphiphilic surfactants – and compared them with a new class of multidentate ligands with enhanced affinity to the NP surface.Each surfactant was fluorescently labelled in advance. This fluorescent label can be conceptually thought of as any other molecule to be conjugated to the NPs (e.g., drug, targeting ligand, reporter probe). Lack of colloidal stability would mean that the NPs were either aggregated or precipitated out of the solution; for this reason, dynamic light scattering (DLS) and UV absorption measurements were used to track any change in colloidal stability.
The first model NPs explored was 15 nm gold NPs stabilized in chloroform using dodecylamine hydrophobic ligands (Fig. 2, for generalization to other NPs see ESI†). As a model for surfactants using a single high affinity anchoring group to the NP we chose 2 kDa PEG-SH, a very common surfactant used for both metallic and semiconductor NPs. In this method, we used a mix of methoxy-PEG-SH and carboxy-PEG-SH, where fluorescent dye was bound through an amide bond (Fig. 2). For obtaining fluorescent PEGylated NPs, a common procedure was used and is made of two steps: first, labeled PEG was added to the NPs; later, unlabeled methoxy-PEG-SH was added to saturate the residual unreacted surface and reach the necessary amount for successful transfer to water. This two-step process was later shown to have little effect on preferential dye binding to the NPs (Fig. 2). The NPs were characterized by TEM (Table 1) and IR (Fig. 3).
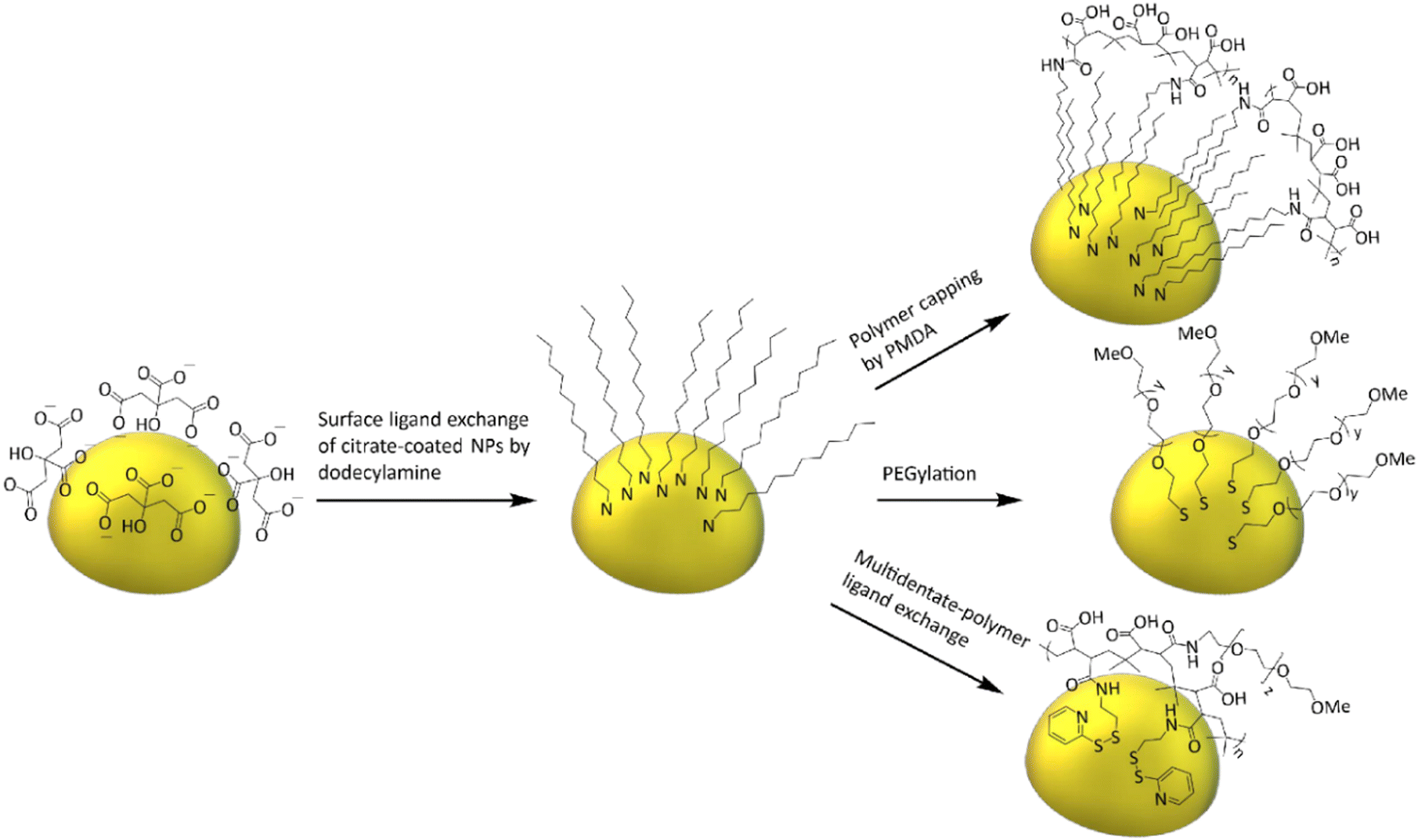 | ||
| Fig. 2 General scheme of model NPs preparation. As-synthesized 15 nm gold-citrate NPs were moved to chloroform and capped with dodecylamine, followed by transferring the NPs to water using one of the three representative surfactants. | ||
| Coating of 15 nm gold Nps | TEM image | Particle size distribution |
|---|---|---|
| Citrate |

|
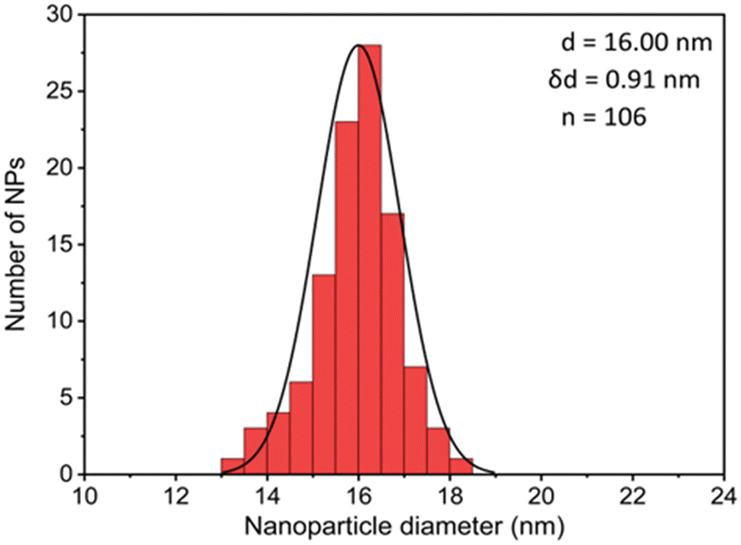
|
| PEG |

|
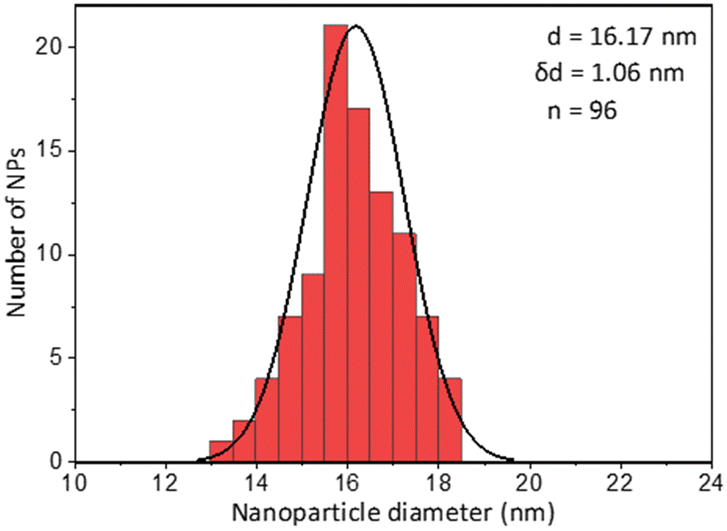
|
| PMDA |

|
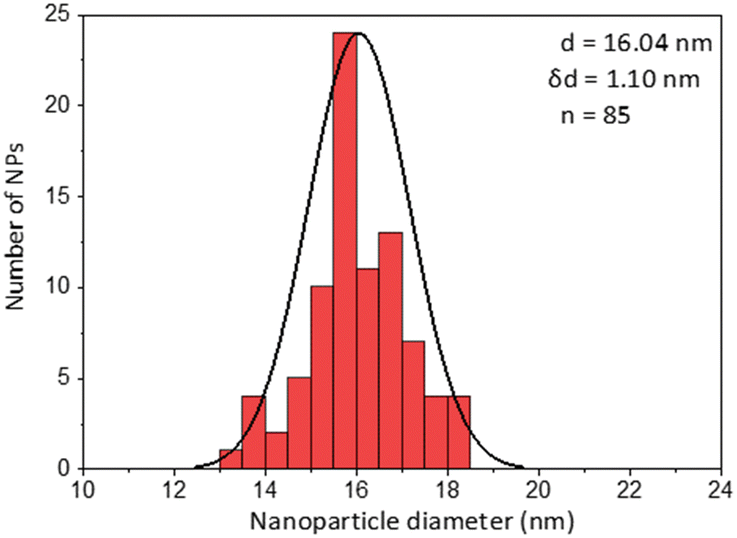
|
| PCP |

|

|
 | ||
Fig. 3 IR comparison of coatings onto 15 nm Gold NPs using gold colloidal solution as blank. PEG is recognizable by typical δ(C–O ether) around 1100 cm−1. PMDA in water presents ν(O–H) around 3500 cm−1 and ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at 1730 cm−1 of carboxylic acid. PCP shows δ(C–H substituted aromatic ring) at 750 cm−1 demonstrating the presence of 2-pyridiylthio cysteamine. O) at 1730 cm−1 of carboxylic acid. PCP shows δ(C–H substituted aromatic ring) at 750 cm−1 demonstrating the presence of 2-pyridiylthio cysteamine. | ||
Secondly, as a model for amphiphilic polymer surfactants relying on hydrophobic interactions with the NP, we chose poly[isobutene-alt-maleic anhydride] grafted with 75% dodecylamine (PMDA).42 The polymer was pre-labeled through an amide bond and added to the NPs in chloroform to allow transfer to water phase according to established protocols (Fig. 2). The NPs were characterized by TEM (Table 1) and IR spectroscopy (Fig. 3).
Lastly a new polymer, poly[isobutene-alt-maleic anhydride] grafted with both 2-pyridylthio cysteamine and 750 Da PEG (PCP), models multidentate polymers having multiple high affinity groups. The polymer was made using a facile, one pot reaction. The polymer was fluorescently labelled and added to the NPs in chloroform to allow phase transfer to water (Fig. 2). The NPs were characterized by TEM (Table 1) and IR spectroscopy (Fig. 3).
Immediately after transfer to water, the NPs were divided into six centrifuge tubes and repeatedly sedimented/resuspended by centrifugation 5 times (Fig. S1A, ESI†). The centrifugation parameters were optimized (4 h, 5000 RCF for 15 nm gold NPs), as longer times or higher centrifugation speeds destabilized all the NP samples, probably as a result of the excessive drag force applied to the NP's surface. The NPs were resuspended to give an accurate dilution factor of the unbound surfactant (1/30 dilution each time). To isolate the effect of unbound surfactant removal (rather than the sedimentation process) the first tube was used as a control, where NPs were sedimented and resuspended each time, without removing and diluting the supernatant (SUP). The second tube was diluted just in the first sedimentation, and sedimented-resuspended four times more without dilution, etc. In tube number six the supernatant was sequentially diluted five times. After the centrifugations, the NP suspensions were diluted into pH 7.2 phosphate buffered saline (PBS) and incubated at 27 °C for three days before measuring UV absorption, fluorescence emission and DLS. Emission allowed quantification of the surfactant in both the precipitate and the supernatant (to give insight to bound vs. free surfactant), while absorption and DLS were meant to detect any NP instability, agglomeration, or sedimentation.
As preliminary insight, we expected the free surfactant in the supernatant to be progressively removed following the dilution factor. Assuming the bound surfactant was irreversibly attached to the NP surface, the surfactant in the precipitate fraction (bottom ∼3% of the solution) is expected to diminish while most of it was initially unbound, and to reach a plateau once all unbound surfactant was eventually removed (Fig. S1B, ESI†). In contrast, a mixed behaviour (i.e., no plateau) would indicate a dynamic equilibrium between bound and free surfactant, and loss of “bound” surfactant as free surfactant was removed, leading to NP destabilization.
Fig. 4 shows the results for 15 nm gold NPs stabilized with 2 kDa PEG-SH. In the emission data (Fig. 4A) we observed that (1) even though the labelled PEG reacted with the NPs first – initially most of it was not bound to the NP, probably due to dynamic exchange of ligands between the NP surface and the solution; (2) three rounds of dilutions (1/27 k) were necessary to obtain a sample in which most of the PEG in solution was indeed bound to the NPs; (3) the fluorescent signal from the NPs actually never reached a plateau; (4) at the maximal dilution, the surfactant's bound/free ratio was 2.4.
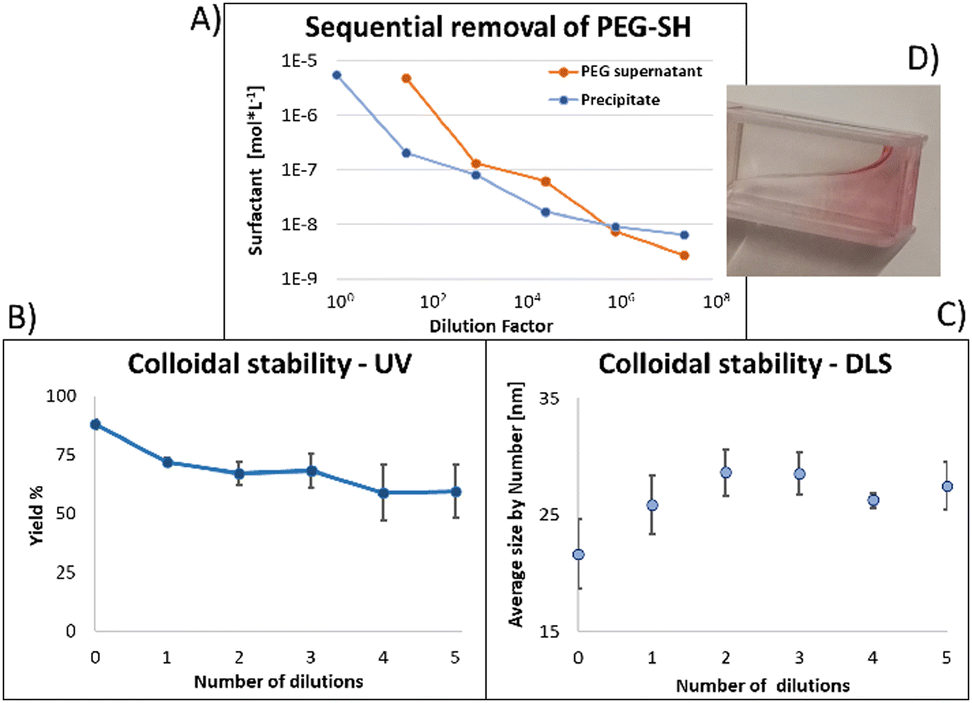 | ||
| Fig. 4 (A) Progressive removal of surfactant. Initially most surfactant was free in solution and appeared to be in equilibrium with the bound surfactant. (B) Absorption and (C) DLS data shows progressive instability of NPs as excess PEG-SH was removed, highlighting limitations for the use of monodentate ligands in stabilization and conjugation to NPs. Error bars represent standard deviations from 3 independent experiments. (D) Showing NPs attaching to plastic cuvette walls. | ||
Next, addressing the NP's stability, Fig. 4B showed progressive elimination of the NPs from solution as more surfactant was removed. Specifically, we found these NPs attaching to the plastic tubes, as shown in Fig. 4D. The absorption measurement was performed after sonication, thus re-dispersing some NPs from the plastic. Likewise, Fig. 4C shows progressive increase in hydrodynamic size possibly due to aggregation as surfactant was removed. Referring to the fourth sample, in which most of the surfactant in solution was estimated to be attached to the NPs, 22% of the plasmon absorption of the NPs was lost and the average NP size had increased 30%.
This observation of fast dynamic equilibrium between bound and unbound monothiolated ligands is in agreement with experiments of self-assembled 2D monolayers on gold surfaces and NPs,43–45 and highlights underestimated limitations in the use of PEG-SH for conjugation of materials to NPs. In fact, PEG is also a non-biodegradable polymer,46–48 alters the fate of NPs (known as “PEG dilemma”) and proved to be immunogenic.48,49 Furthermore, the existence of such equilibrium suggests limited reliability in applications in which the surfactant is naturally eliminated at a fast rate (e.g., in in vivo experiments), where the elimination half-life of PEG is much faster than that of NPs (in the order of min compared to days).12,13
Similar observations apply to PMDA, as shown in Fig. 5: this time, 2 dilutions were necessary to obtain a sample in which most surfactant was attached to the NPs (A); once again at that point, due to loss of surfactant from the NP's surface, 53% of the NPs absorption was lost, and the average size increased by 70% ((B) and (C), respectively). At maximal dilution, the surfactant bound/free ratio was 12.
 | ||
| Fig. 5 The surfactant PMDA seems to be in equilibrium between NP bound and free in solution. As free PMDA is removed (A) the NPs lose their colloidal stability as evident by UV absorption (B) and DLS (C). This will limit the use of such surfactants for conjugation of chemicals to NPs. Error bars represents standard deviation from three independent experiments. | ||
Fig. 6 shows the same experiment done with NPs coated with PCP. Looking at the fluorescence emission data (A), the most striking difference compared to the two previous surfactants was the plateau in the NPs emission intensity. Another compelling indication of improved avidity of PCP is the final ratio of bound/free surfactant: 137 compared to 12 for PMDA and 2.4 for PEG-SH. More importantly, this value was achieved without losing the colloidal stability of the NPs (B and C), contrary to PMDA and PEG-SH.
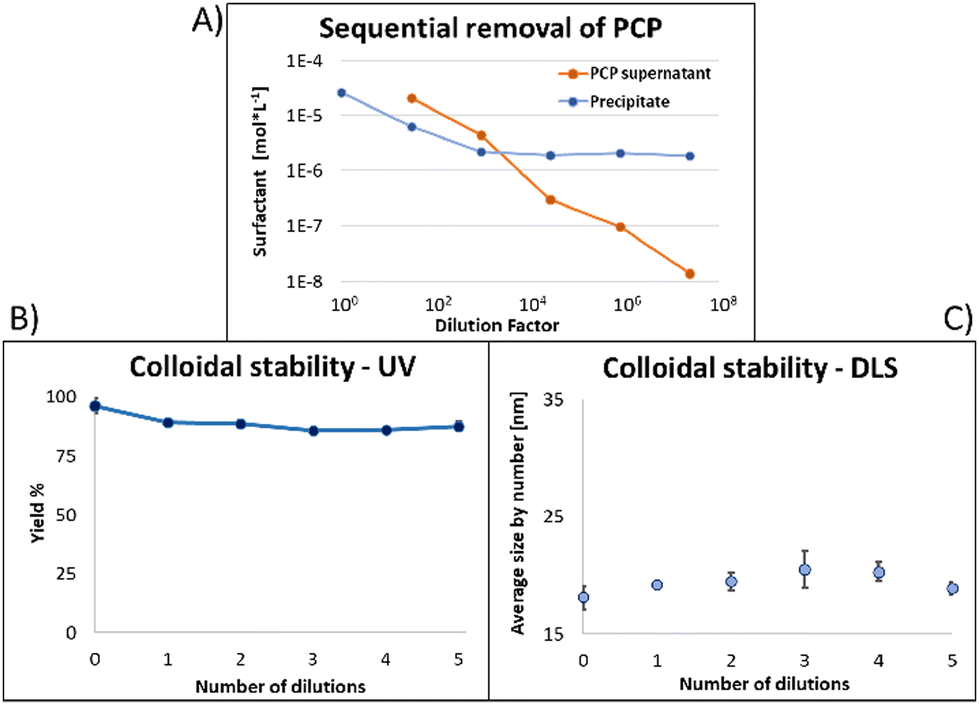 | ||
| Fig. 6 (A) As free surfactant is removed the signal from the NPs reaches a plateau and the ratio between free and bound surfactant exceeds 100. (B) 87% of NPs are recovered after 5 dilutions with no trend of instability. (C) No significant change in size due to removal of free surfactant. | ||
Using PCP as surfactant, at the maximal dilution only 10% of the NPs absorption was lost, with no significant size increase. Such improved performance is expected due to the chelate effect, or denticity of this polymer and implies a revolutionary advantage of such multidentate surfactants.
We further show that this result holds for NPs of different size (5 nm gold, Fig. S2–S4, ESI†) and chemistry (iron oxide NPs stabilized by poly[isobutene-alt-maleic anhydride] grafted with both dopamine and 750 Da PEG,18,19 Fig. S5 and S6, ESI†).
To further validate the potential of PCP specifically, we demonstrated a few practical applications. The cytotoxicity of 15 nm gold NPs covered by each surfactant was tested using the MTT cell viability assay showing PCP to have the least toxic effect at each dosage (Fig. 7). 15 nm gold NPs@PMDA clearly confirm the toxicity of surfactant excess especially with this type of polymer. While 15 nm gold NPs@PEG-SH show a toxicity quite similar to PCP coated one, immunogenicity effects and renal clearance in according to molecular weight of the surfactant have to be considered for in vivo studies.50–52
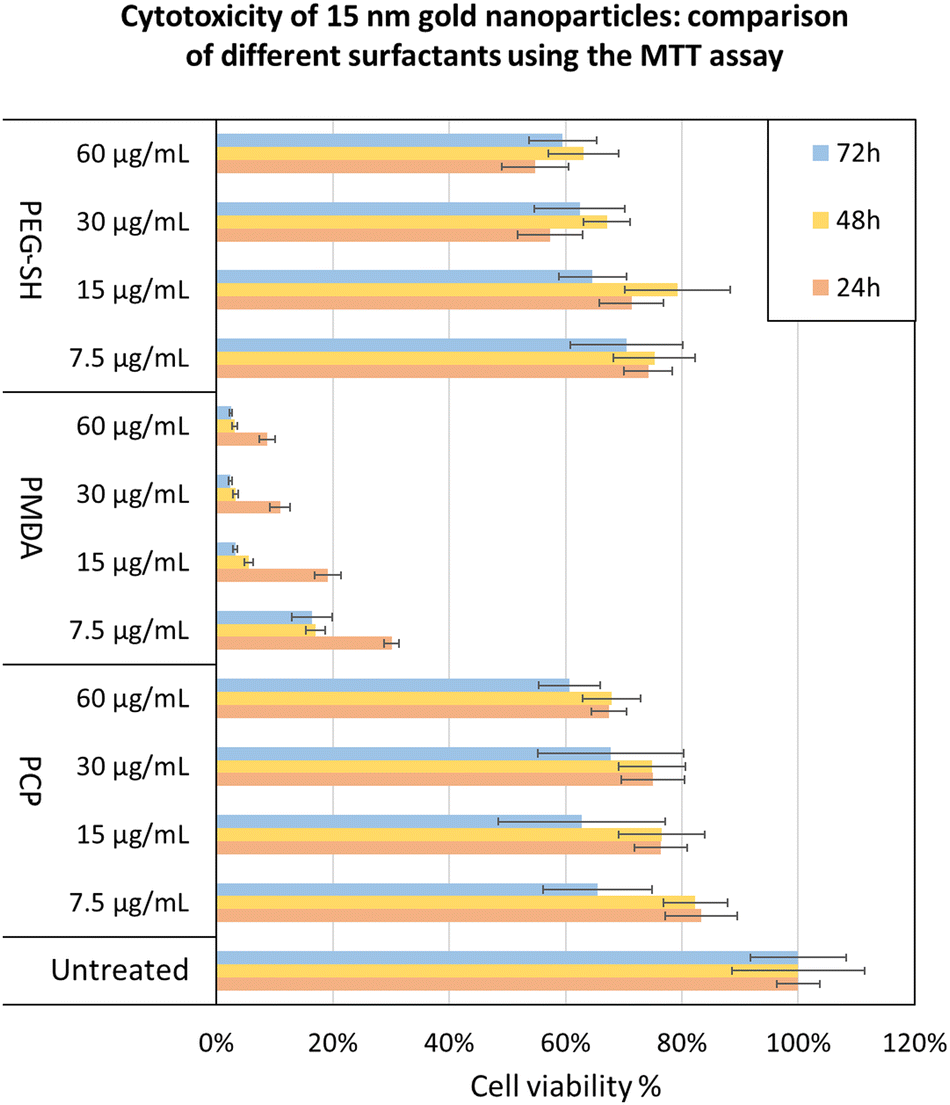 | ||
| Fig. 7 MTT assay to compare the toxicity of gold NPs stabilized by different surfactant – PMDA and PEG-SH with polymer excess, and PCP without polymer excess. We choose this comparison due to our conclusion that it is not practical to use PMDA and PEG-SH without excess surfactant. Test was done using HeLa cells. The values are the mean of three different experiments in quadruplicates each. All samples are relative to the untreated cells, which are set as 100%. Error bars represents standard deviation. | ||
We demonstrated the conjugation and purification of a protein (bovine serum albumin (BSA)) to PCP covered NPs, such that most of the protein in the mixture was actually bound to the NPs (Fig. S10, ESI†).
In addition, PCP covered NPs were shown to be colloidally stable in 10% blood plasma, through lyophilization and re-solvation, and able to transfer the NPs back and forth between water and chloroform depending on the pH (Fig. S8 and S9, ESI†).
3.1. Insight for applying multidentate ligands to different NPs: not all multidentate ligands perform equally
 | ||
| Fig. 8 (A) a cartoon of the polymer unfolding and re-organization following NP binding. (B) Calculated binding energies (PBE-D3/TZVP/COSMO) of each single ligand, supporting (together with published experimental data) the idea multidentate polymers need to unfold and reorganize to orient ligands to the NP's surface, and that excessively high binding energy and/or low surface mobility might limit this process, resulting in NP crosslinking. (C) Representative image from DFT calculation of covalent binding energy of aminothiophenol to gold (for detailed explanation of the calculation see ESI†). | ||
Alternatively, 4-aminothiophenol have higher affinity to gold - theoretically confirmed by calculations (Fig. 8B) and experimentally supported by previous studies.54,55 However, polymers of 4-aminothiophenol caused immediate aggregation even at low degrees of functionalization (3 ligands on average). Again, we could rationalize this effect by the same process of polymer unfolding and reorganization on the NP's surface. Having higher affinity and lower surface mobility (characteristic of covalent binding compared to physical adsorption) could limit this process, resulting in dangling ligands and aggregation. Overall, these results indicate that ligands having intermediate affinity and high surface mobility would be ideal for multidentate polymers.
4. Conclusions
In this work, we showed that currently common surfactants have a fundamental limitation in the context of conjugation and in vivo applications: the presence of excess surfactant in solution and the loss of colloidal stability once that excess is removed. We believe that this underestimated problem represents one of the big issues behind current failure of colloidal nanoparticles in reaching the expected outcome in nanomedicine.where θ is the surface coverage fraction, C is the surfactant concentration and K is a constant, we can anticipate that as the total surface area, or the NP concentration decreases, a higher ratio of free/bound surfactant will be necessary to maintain the NP stability. Unfortunately, it is generally the case, especially for biological applications, that NPs are used in nanomolar concentrations or lower. The data presented here and in ESI†cover many orders of magnitude of NP concentration (about 0.5 nM for 15 nm gold prepared by the citrate method, 25 nM for 5 nm gold prepared by the Brust–Schriffin protocol, and 0.4 mM for iron oxide), yet the trend of free/bound surfactant ratio and its implications on NP stability remain the same.

Searching for a possible solution, here we proposed a new multidentate polymer and showed its improved performance, possibly enabling improved performance in the same applications. To demonstrate this in practice, a fundamental application in nanobiotechnology, the nanoparticle bioconjugation, was successfully demonstrated. A second application, i.e., improved stability in vivo compared to monothiolated polyethylene glycol is left to be demonstrated in follow-up studies.
Finally, showing the feasibility of the same strategy while changing different parameters, including size and material composition (e.g., iron oxide) of the nanoparticles, we tried to generalize these results to indicate a general solution (multidentate ligands) not limited to a specific nanoparticle, and to provide general guidelines for successful application of such surfactants.
We hope this work will have a significant impact on nanoparticle assisted drug delivery and bioimaging.17,56,57 This result can be generalized to nanoparticle functionalization in different fields of materials science and technology well beyond nanobiotechnology, ranging from nanoparticle sensitized solar cells58,59 to sensing,60 catalysis,61 and others.62
Author contributions
R. R. designed the research, executed the experiments and analyzed the data; M. G. executed the experiments, R. R. and F. A. wrote the first draft; F. A. performed the theoretical DFT calculations; J. A. B. performed cytotoxicity tests; M. G. and S. G. synthesized the NPs; M. A. R., L. S. and L. B. discussed the data; L. B., L. D. G., M. C. and D. P. designed the research, discussed the results and gave critical comments; D. P. and M. C. provided funding, wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Tiziano Catelani for TEM images, and Emanuele Patriarca for NMR analyses. This project was partly supported by the Regione Lombardia, the Italian Ministry of University and Research (MIUR) through grant ‘Dipartimenti di Eccellenza-2017’, and Inthena s.r.l. This work has received funding from the European Union's HORIZON 2020 Program for research, technological development and demonstration under grant agreement no. 642028 H2020-MSCA-ITN-2014.References
- The two directions of cancer nanomedicine, Nat. Nanotechnol., 2019, 14, 1083 DOI:10.1038/s41565-019-0597-5.
- A. J. Clark and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12486–12491 CrossRef CAS.
- M. Colombo, L. Fiandra, G. Alessio, S. Mazzucchelli, M. Nebuloni, C. De Palma, K. Kantner, B. Pelaz, R. Rotem, F. Corsi, W. J. Parak and D. Prosperi, Nat. Commun., 2016, 7.1, 1–14 Search PubMed.
- L. Fiandra, S. Mazzucchelli, C. De Palma, M. Colombo, R. Allevi, S. Sommaruga, E. Clementi, M. Bellini, D. Prosperi and F. Corsi, ACS Nano, 2013, 7, 6092–6102 CrossRef CAS PubMed.
- E. E. Lees, T. L. Nguyen, A. H. A. Clayton and P. Mulvaney, ACS Nano, 2009, 3, 1121–1128 CrossRef CAS PubMed.
- R. A. Sperling and W. J. Parak, Philos. Trans. R. Soc., A, 2010, 368, 1333–1383 CrossRef CAS.
- S. Halivni, A. Sitt, I. Hadar and U. Banin, ACS Nano, 2012, 6, 2758–2765 CrossRef CAS PubMed.
- P. Khashayar, G. Amoabediny, B. Larijani, M. Hosseini and J. Vanfleteren, Sci. Rep., 2017, 7.1, 1–8 Search PubMed.
- S. Mourdikoudis, M. Menelaou, N. Fiuza, G. Zheng, S. Wei, J. Perez-Juste, L. Polavarapu and Z. Sofer, Nanoscale Horiz., 2022, 7, 941–1015 RSC.
- C. Iriarte-Mesa, Y. C. López, Y. Matos-Peralta, K. de la Vega-Hernández and M. Antuch, Top. Curr. Chem., 2020, 378, 1–40 CrossRef.
- H. Soo Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. Itty Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef.
- C. H. J. Choi, J. E. Zuckerman, P. Webster and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6656–6661 CrossRef CAS PubMed.
- T. Yamaoka, Y. Tabata and Y. Ikada, J. Pharm. Sci., 1994, 83, 601–606 CrossRef CAS PubMed.
- M. D. Khan, M. Opallo and N. Revaprasadu, Dalton Trans., 2021, 50(33), 11347–11359 RSC.
- L. Lu, S. Zou and B. Fang, ACS Catal., 2021, 11(10), 6020–6058 CrossRef CAS.
- C. Vericat, G. A. Benitez, D. E. Grumelli, M. E. Vela and R. C. Salvarezza, J. Phys.: Condens. Matter, 2008, 20, 18 CrossRef.
- B. Ouyang, W. Poon, Y. N. Zhang, Z. P. Lin, B. R. Kingston, A. J. Tavares, Y. Zhang, J. Chen, M. S. Valic, A. M. Syed, J. Couture-Senécal, G. Zheng and W. C. Chan, Nat. Mater., 2020, 1–10 Search PubMed.
- W. Wang, G. Palui, X. Ji, F. Aldeek and H. Mattoussi, Colloidal Nanoparticles for Biomedical Applications IX, SPIE, 2014, vol. 8955, pp. 37–44 Search PubMed.
- W. Wang, X. Ji, H. B. Na, M. Safi, A. Smith, G. Palui, J. Manuel Perz and H. Mattoussi, Langmuir, 2014, 30, 6197–6208 CrossRef CAS.
- W. Wang, A. Kapur, X. Ji, B. Zeng, D. Mishra and H. Mattoussi, Bioconjug. Chem., 2016, 27, 2024–2036 CrossRef CAS PubMed.
- E. Giovanelli, E. Muro, G. Sitbon, M. Hanafi, T. Pons, B. Dubertret and N. Lequeux, Langmuir, 2012, 28, 15177–15184 CrossRef CAS.
- G. Palui, F. Aldeek, W. Wang and H. Mattoussi, Chem. Soc. Rev., 2015, 44, 193–227 RSC; W. Jiang, S. Mardyani, H. Fischer and W. C. Chan, Chem. Mater., 2006, 18(4), 872–878 CrossRef CAS.
- W. Jiang, S. Mardyani, H. Fischer and W. C. Chan, Chem. Mater., 2006, 18(4), 872–878 CrossRef CAS.
- T. K. Mandal, M. S. Fleming and D. R. Walt, Nano Lett., 2002, 2(1), 3–7 CrossRef.
- M. Alloisio, A. Demartini, C. Cuniberti, M. Muniz-Miranda, E. Giorgetti, A. Giusti and G. Dellepiane, Phys. Chem. Chem. Phys., 2008, 10(16), 2214–2220 RSC.
- W. H. Le Roux, A. J. van Reenen and R. Malgas-Enus, Appl. Catal., A, 2022, 633, 118526 CrossRef CAS . 4 193–227 (2015).
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- J. Park, K. An, Y. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. Park, N. Hwang and T. Hyeon, Nat. Mater., 2004, 3(12), 891–895 CrossRef CAS PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS , Erratum: (Physical Review Letters (1996) 77 (3865)).
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- A. Klamt, J. Phys. Chem., 1996, 100, 3349–3353 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- N. E. Singh-Miller and N. Marzari, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 235407 CrossRef.
- H. Al-Johani, E. Abou-Hamad, A. Jedidi, C. M. Widdifield, J. Viger-Gravel, S. S. Sangaru, D. Gajan, D. H. Anjum, S. Ould-Chikh, M. Nejib Hedhili, A. Gurinov, M. J. Kelly, M. El Eter, L. Cavallo, L. Emsley and J. M. Basset, Nat. Chem., 2017, 9, 890–895 CrossRef CAS.
- J. D. Gezelter, S. Kuang, J. Marr, K. Stocker, C. Li, C. Vardeman and M. Meineke A OpenMD, an open source engine for molecular dynamics. openmd.net.
- J. Hühn, C. Carrillo-Carrion, M. G. Soliman, C. Pfeiffer, D. Valdeperez, A. Masood, I. Chakraborty, L. Zhu, M. Gallego, Z. Yue, M. Carril, N. Feliu, A. Escudero, A. M. Alkilany, B. Pelaz, P. Del Pino and W. J. Parak, Chem. Mater., 2017, 29, 399–461 CrossRef.
- T. Bürgi, Nanoscale, 2015, 7, 15553–15567 RSC.
- M. Yang, C. M. Hellas, A. Yau and H. L. Chan, Langmuir, 1998, 14, 6121–6129 CrossRef CAS.
- S. H. Chen and C. W. Frank, Langmuir, 1989, 5, 978–987 CrossRef CAS.
- J. Ulbricht, R. Jordan and R. Luxenhofer, Biomaterials, 2014, 35(17), 4848–4861 CrossRef CAS.
- M. J. Ernsting, M. Murakami, A. Roy and S. D. Li, J. Controlled Release, 2013, 172(3), 782–794 CrossRef CAS PubMed.
- F. S. Mozar and E. H. Chowdhury, Curr. Pharm. Des., 2018, 24(28), 3283–3296 CrossRef CAS PubMed.
- S. Dufort, L. Sancey and J. L. Coll, Adv. Drug Delivery Rev., 2012, 64(2), 179–189 CrossRef CAS PubMed.
- J. D. Friedl, V. Nele, G. De Rosa and A. Bernkop-Schnürch, Adv. Funct. Mater., 2021, 31(34), 2103347 CrossRef CAS.
- D. Stanicki, L. Larbanoix, S. Boutry, T. Vangijzegem, I. Ternad, S. Garifo and S. Laurent, J. Mater. Chem. B, 2021, 9(25), 5055–5068 RSC.
- B. M. Chen, T. L. Cheng and S. R. Roffler, ACS Nano, 2021, 15(9), 14022–14048 CrossRef CAS.
- T. Weidner, N. Ballav, U. Siemeling, D. Troegel, T. Walter, R. Tacke, D. G. Catner and M. Zharnikov, J. Phys. Chem. C, 2009, 113, 19609–19617 CrossRef CAS.
- M. S. Inkpen, Z. F. Liu, H. Li, L. M. Campos, J. B. Neaton and L. Venkataraman, Nat. Chem., 2019, 11, 351–358 CrossRef CAS PubMed.
- N. Garg, E. Carrasquillo-Molina and T. R. Lee, Langmuir, 2002, 18, 2717–2726 CrossRef CAS.
- Z. Wang, R. Qiao, N. Tang, Z. Lu, H. Wang, Z. Zhang, X. Xue, Z. Huang, S. Zhang, G. Zhang and Y. Li, Biomaterials, 2017, 127, 25–35 CrossRef CAS.
- L. Y. T. Chou, K. Zagorovsky and W. C. W. Chan, Nat. Nanotechnol., 2014, 9, 148–155 CrossRef CAS PubMed.
- K. Yao, K. Yao, S. Leng, Z. Liu, L. Fei, Y. Chen, S. Li, N. Xhou, J. Zhang, Y. Xu, L. Zhou, H. Huang and A. K. Y. Jen, Joule, 2019, 3, 417–431 CrossRef CAS.
- I. Mora-Seró, Adv. Energy Mater., 2020, 10.33, 2001774 CrossRef.
- L. Qin, G. Zeng, C. Lai, D. Huang, P. Xu, C. Zhang, M. Cheng, X. Liu, S. Liu, B. Liu and H. Yi, Coord. Chem. Rev., 2018, 359, 1–31 CrossRef CAS.
- Z. B. Shifrina, V. G. Matveeva and L. M. Bronstein, Chem. Rev., 2020, 120, 1350–1396 CrossRef CAS PubMed.
- H. Heinz, C. Pramanik, O. Heinz, Y. Ding, R. K. Mishra, D. Marchon, M. J. Flatt, I. Estrela-Lopis, J. Llop, S. Moya and R. F. Ziolo, Surf. Sci. Rep., 2017, 72, 1–58 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb01528e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |