
Cruciate DNA probes for amplified multiplexed imaging of microRNAs in living cells†
Zhe
Dong‡
ab,
Xizhu
Xu‡
a,
Jing
Ni
a,
Yuancheng
Li
a,
Kang
An
a,
Ling
Meng
a and
Han
Wu
 *a
*a
aSchool of Public Health, Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan, Shandong 250117, China
bState Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha, Hunan 410082, China
First published on 29th November 2022
Abstract
The real-time imaging of low-abundance tumor-related microRNAs (miRNAs) in living cells holds great potential for early clinical diagnosis of cancers. However, the relatively low detection sensitivity and possible false-positive signals of a probe in complex cellular matrices remain critical challenges for accurate RNA detection. Herein, we developed a novel aptamer-functionalized cruciate DNA probe that enabled amplified multiple miRNA imaging in living cells via catalytic hairpin assembly (CHA). The cross-shaped design of the cruciate DNA probe improved the stability against nucleases and acted as a modular scaffold for CHA circuits for efficient delivery into tumor cells. The cruciate DNA probe allowed self-assembly through thermal annealing and displayed excellent performance for sensitive miRNA detection in vitro. The cruciate DNA probe could be internalized into nucleolin-overexpressed cells specifically via cell-targeting of the AS1411 aptamer, achieving amplified fluorescence imaging and quantitative evaluation of the expression of miRNAs in living cells. Through the simultaneous detection of intracellular multiple miRNAs, the developed cruciate DNA probe could provide more accurate information and reduce the chances of false positive signals for cancer diagnosis. This approach offers a new opportunity for promoting the development of miRNA-related biomedical research and tumor diagnostic applications.
Introduction
MicroRNAs (miRNAs) as a class of endogenous, single-stranded ribonucleic acids that contain 18–25 nt in length are pivotal biomarker molecules for evaluating different clinical stages of diseases.1–3 The abnormalities of tumor-associated miRNA expression are involved in malignant progression of a variety of cancers.4,5 Real-time monitoring of the expression of miRNAs at the cellular level facilitates understanding the regulatory mechanism of miRNAs in cancer pathogenesis and metastasis.6–8 However, a single biomarker is often involved in multiple cellular events, which is likely to result in potential false-positive signals hindering disease diagnosis.9,10 Therefore, monitoring multiple miRNAs simultaneously provides the possibility to improve the diagnostic accuracy of cancers. Currently, there are various appealing strategies exploited for detecting miRNAs using fluorescence molecular probes, including spherical nucleic acids,11–13 molecular beacons,14–16 and light-up RNA aptamers.17,18 However, these strategies are still confronted with many limitations, such as the limited RNA detection sensitivity due to the low abundance of targets and enzyme instability of nucleic acid-based probes.19,20The nucleic acid amplification methods represented by the hybridization chain reaction (HCR) and catalyzed hairpin assembly (CHA) have enabled the programming of DNA molecular cascading hybridization, which generates multiple signal readouts in response to one target molecule.21–24 Among these methods, the catalyzed hairpin assembly, which was pioneered by the Pierce group25 and adapted to analytical applications by the Chen group,26 has been widely applied to intracellular biomarker analysis including RNAs.27 These enzyme-free isothermal amplification strategies offer an attractive option for the highly sensitive detection of low-level miRNAs in living cells. Hence, a facile assembled probe that can maintain stability and effective signal amplification for multiple miRNA imaging is highly needed.
Herein, we developed an aptamer-functionalized cruciate DNA probe via self-assembly for amplified imaging of target miRNAs in living cells. The self-assembled DNA nanostructure via the Watson–Crick base-pairing is a valuable tool for the targeted delivery of nucleic acid probes, holding promising prospects for biomarker sensing and early diagnosis of tumors.28 In recent years, various two- or three-dimensional DNA nanodevices have been developed and successfully realized the sensitive detection of RNAs with high biological stability in living cells or in vivo.29–31 As shown in Scheme 1, the cruciate DNA probe was synthesized via 7 single-stranded DNAs (ssDNAs) of a cross-shaped scaffold and 4 hairpin substrates through the one-pot thermal annealing. MiRNA-21 and miRNA-155, which were overexpressed in various carcinoma cell lines, were selected as the targets for detection.32,33 The fluorescence reporter Cy5 and a quencher moiety BHQ2 were labeled on the stem region of H2 for miRNA-21 detection, and the stem region of H4 was attached to another fluorescein FAM and a quencher DABCYL for miRNA-155 visualization. After incubating the cruciate DNA probe with tumor cells, the AS1411 aptamer of the cruciate DNA probe, which specifically targets nucleolin receptors on the cell membrane, can efficiently enter cells via endocytosis.34 The presence of target miRNAs mediated numerous double-stranded DNA assembly between hairpin probes via the CHA circuits, generating amplified fluorescence responses for the detection. Therefore, the developed multi-color cruciate DNA probe afforded high specificity and sensitivity for multiple low-abundance miRNA imaging in living cells, holding great potential for accurate biomarker detection and early clinical diagnosis of different diseases.
 | ||
| Scheme 1 Schematic diagram for the design of the cruciate DNA probe and amplified imaging of multiplexed miRNAs in living cells under catalytic hairpin assembly. The dye–quencher pairs of Cy5/BHQ2 and FAM/DABCYL are labeled on the stem regions of hairpin H2 and H4, respectively. Under the trigger of miRNA-21 and miRNA-155, the quenched fluorescence signals of Cy5 and FAM are activated via CHA circuits for target detection. | ||
Experimental sections
Reagents and materials
The chemicals and reagents of experiments are listed in the ESI† in detail.Assembly of cruciate DNA probes
The cross-shaped scaffold Cross-A (L1, L2, L3, L4), Cross-B (L5, L6, L7, L4), and Cross-C (L1, L2, L3, L4, L5, L6, L7, L4) were assembled via mixing equal amounts of DNA strands in the TAE–Mg2+ buffer (40 mM Tris buffer, 1 mM EDTA, and 12.5 mM MgCl2, pH 8.0) to the final concentrations of 1 μM. Then, the mixtures of DNA strands were subject to the following thermal annealing protocol: 95 °C for 5 min and cooling from 95 °C to room temperature for more than 8 hours using a PCR instrument. The cruciate DNA probe (CrossC-H1–H2–H3–H4) was constructed by self-assembling L1, L2, L3, L4, L5, L6, L7, hairpin probes H1, H2, H3, and H4 through one-step thermal annealing. The prepared products were stored at 4 °C for future use.Gel electrophoresis analysis
The prepared DNA samples were characterized using the precast 3% native agarose gel in 0.5 × TBE buffer (44.5 mM Tris-HCl, 1 mM EDTA, pH 8.0) at 80 V for 60 min. DNA ladders were used as the molecular weight standards. After electrophoresis, the gel was stained using 4S Red Plus and visualized using the iBright CL1000 imaging system (Thermo Fisher, USA).Stability analysis of cruciate DNA probes
The 1 μM cruciate DNA probe and 1 μM hairpin probe were diluted in the TAE–Mg2+ buffer containing 10% fetal bovine serum (FBS) for 0–12 h incubation at 37 °C. Then, the samples were analyzed by 3% agarose gel electrophoresis. The degradation rates of the cruciate DNA probe and the hairpin probe were measured by “ImageJ” software.In vitro detection of RNA targets
The non-target miRNAs and different concentrations of target miRNAs (miRNA-21 and miRNA-155) were incubated with 100 nM cruciate DNA probe in the TAE–Mg2+ buffer at 37 °C for 3 h. The fluorescence spectra were recorded at room temperature on the Cary Eclipse Fluorescence Spectrometer (Varian, USA). Time-dependent fluorescence monitoring was performed on the F-320 Fluorescence Spectrophotometer from Gangdong Sci.&Tech. Co., Ltd (Tianjin, China). For FAM, the fluorescence spectra were recorded in the range from 500 to 600 nm at 488 nm excitation. For Cy5, the fluorescence spectra were measured between 650 and 750 nm at 635 nm excitation at a PMT voltage of 850 V.Cell culture and confocal fluorescence imaging
MDA-MB-231, A549, and HeLa cells were grown in the Dulbecco's-Modified Eagle Medium (DMEM) supplemented with 10% FBS, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin. L-02 cells were grown in the RPMI 1640 medium with 10% FBS, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin. All cell lines were cultured at 37 °C in a humidified incubator with 5% CO2.The cells were seeded in a 35 mm confocal dish and grown to 80% density in a 2 mL culture medium at 37 °C for 24 h, then the cells were incubated with 1 mL corresponding medium containing 100 nM cruciate DNA probe or control probes at 37 °C for 3 h. Next, the cells were washed three times with cold PBS and incubated with fresh medium containing 10 μg mL−1 Hoechst 33342 for 20 min followed by imaging. To regulate the expressions of intracellular miRNA-21 and miRNA-155, the synthetic antisense miRNA-21 and miRNA-155 (300 nM) were transfected into cells with lipofectamine 3000. After transfection over 24 h, the cells were washed with cold PBS and incubated with 1 mL medium containing 100 nM cruciate DNA probe at 37 °C for 3 h followed by confocal imaging. For the cellular uptake study of the cruciate DNA probe, MDA-MB-231 cells were incubated at 37 °C, 4 °C or pretreated with 0.1% NaN3 for 1 h. Following, 100 nM cruciate DNA probe was added and further incubated for 3 h, then washed with PBS before confocal imaging.
All confocal images were acquired with a 60× oil immersion objective lens using an FV3000 Olympus confocal laser scanning microscope (Olympus, Japan). The fluorescence signal of Cy5 was recorded using a red laser with 640 nm excitation, the fluorescence signal of FAM was recorded using a green laser with 488 nm excitation, and a laser with 405 nm excitation was used for the detection of Hoechst 33342.
Flow cytometry analysis of miRNA expression
MDA-MB-231, A549, HeLa, and L-02 cells were grown in 35 mm dishes with 2 mL culture medium at 37 °C for 24 h (∼105 cells), then the cells were incubated with 1 mL medium containing 100 nM cruciate DNA probe or control probes at 37 °C for 3 h. Then, the cells were washed three times with cold PBS, trypsinized, and suspended in a fresh medium. Later, the cells were centrifuged at 1000 g for 5 min, washed with PBS, and re-suspended in 1 mL PBS for flow cytometry analysis using a BD AccuriTM C6 Plus flow cytometer (BD Biosciences, USA).Results and discussion
Design and synthesis of the cruciate DNA probe
The cross-shaped scaffold was synthesized via self-assembly of seven single-stranded DNAs (L1, L2, L3, L4, L5, L6, L7), each containing a 32-mer region to form the double-stranded cross scaffold and a sticky end for the hybridization of hairpin substrates. To construct a cell-targeting module, L4 was designed with an aptamer sequence of AS1411 for specific cellular delivery. The hairpin probes H1, H2 and H3, H4 were designed and further optimized for CHA circuits according to the sequences of target miRNA-21 and miRNA-155, respectively. For the CHA reaction of H1 and H2 initiated by miRNA-21, the target miRNA hybridizes with the domains 1, 2, and 3 of H1 to open the hairpin. Then, the exposed toehold domains 3* and 4* in the target-H1 intermediate further trigger the hybridization of domains 3-4-3*-2* of H2, followed by displacement of target miRNA for the next cycle of CHA circuit (Fig. 1a). The CHA reaction of H3 and H4 triggered by miRNA-155 is similar as above.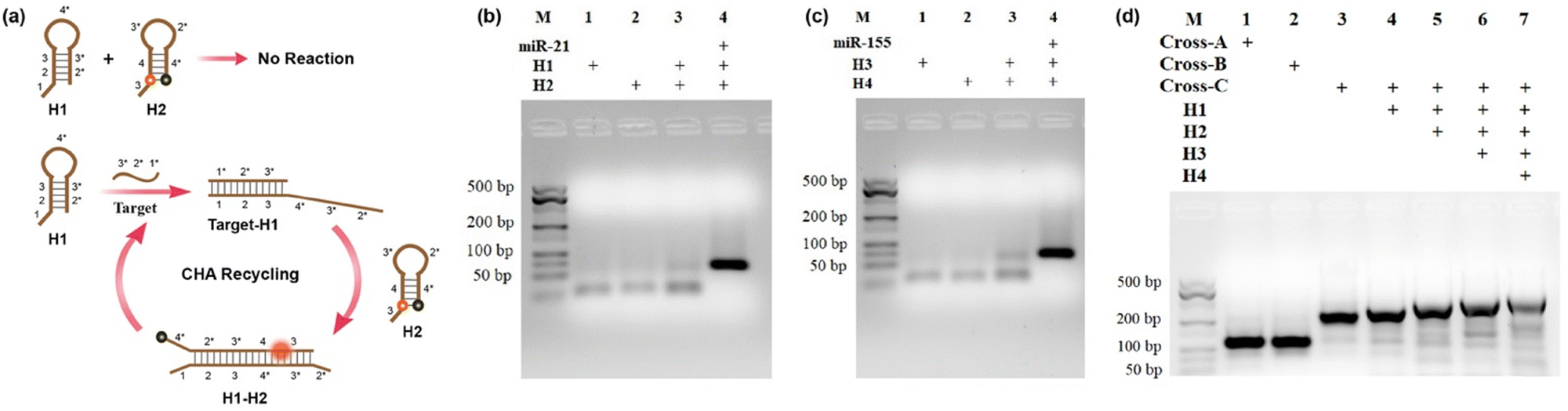 | ||
| Fig. 1 (a) The assembly process for the CHA circuit with hairpin substrates H1 and H2. (b and c) Agarose gel electrophoresis images of CHA circuits triggered by target miRNA-21 and miRNA-155. (d) Agarose gel electrophoresis image for the cruciate DNA probe assembly. The concentrations of Cross-A, Cross-B, Cross-C, H1, H2, H3, and H4 are 1 μM, and the concentrations of target miRNA-21 and miRNA-155 are 100 nM. M denotes the molecular weight marker. | ||
Gel electrophoresis analysis was performed to validate the process of the CHA reaction. In the absence of targets, slight background bands appeared in the gel images (Fig. 1b and c, lane 3), which were caused by the interaction of transiently unpaired hairpins H1 with H2 or H3 with H4.26 Compared to the bands without targets triggered, major bands with larger molecular weights were obtained upon incubation with target miRNAs (Fig. 1b and c, lane 4). The results clearly confirmed that the hairpin substrates were successfully designed to minimize background signals of non-specific initiations and maximize response signals toward targets. As exhibited in Fig. 1d, the successful synthesis of the cruciate DNA probe was verified by gel electrophoresis. Compared with Cross-A (L1, L2, L3, L4) and Cross-B (L5, L6, L7, L4), a lower mobility band corresponding to Cross-C appeared after the incubation of seven ssDNAs (L1, L2, L3, L4, L5, L6, L7), demonstrating the formation of cross-shaped DNA scaffold (Fig. 1d, lane 1–3). With the addition of hairpin substrates, the bands with increasing molecular weights were observed in the gel image (Fig. 1d, lane 4–6). Moreover, a major band with further decreased mobility was shown when L1, L2, L3, L4, L5, L6, L7 and H1, H2, H3, and H4 were self-assembled through one-pot thermal annealing (Fig. 1d, lane 7), indicating the efficient synthesis of a complete cruciate DNA probe.
The nucleic acid probes might be degraded by endogenous nuclease under a complex physiological environment, resulting in high background values or even false positive results. It has been reported that the DNA nanostructure could improve the stability of nucleic acid probes.30,31 We then investigated the serum stability of the cruciate DNA probe in the fresh medium containing 10% FBS (Fig. S1a and b, ESI†). Compared with the hairpin probe, the cruciate DNA probe could retain integrity even after 6 h incubation, indicating that it had good stability to resist nuclease degradation. Moreover, the grayscale statistical analysis showed that the degradation rate of the hairpin probe is relatively higher than that of the cruciate DNA probe (Fig. S1c, ESI†). Additionally, further real-time fluorescence responses of cruciate DNA probes and hairpin probes in cell lysate were performed. As shown in Fig. S2 (ESI†), the cruciate DNA probe displayed little fluorescence increase in the MDA-MB-231 cell lysate, while the hairpin probes displayed fast non-specific fluorescence enhancements in both Cy5 and FAM channels. The results showed that the cruciate DNA probe could reduce false positive signals caused by the hairpin substrate's degradation, holding great potential for RNA imaging applications in a cellular environment.
In vitro evaluation of cruciate DNA probes for miRNA detection
The sensitivity of the cruciate DNA probe for target miRNA detection was verified by monitoring the fluorescence intensities in the presence of varying concentrations of miRNAs in the range from 0 to 10 nM (Fig. 2a and b). The fluorescence curves exhibited dynamic responses with the increasing concentrations of target miRNAs. When the concentrations of miRNAs were 5 pM–6 nM (miRNA-21) and 5 pM–5 nM (miRNA-155), the relationship between fluorescence values and miRNA concentrations displayed a good linear correlation, and the limits of detection (LODs) were estimated to be 3 pM (miRNA-21) and 2 pM (miRNA-155), respectively (Fig. 2c and d), which exhibited comparable or even better performance than other existing strategies for miRNA detection (Table S2, ESI†).35–39 The approximate 16-fold and 18-fold fluorescence enhancement of the cruciate DNA probe were obtained after incubating with 10 nM miRNA-21 and miRNA-155, respectively. Therefore, the cruciate DNA probe had a high signal-noise ratio (SNR) for target detection, which hardly caused false positive signals due to non-specific assembly. | ||
| Fig. 2 Fluorescence curves of 100 nM cruciate DNA probe incubated with miRNA-21 (a) and miRNA-155 (b) in the range from 0 to 10 nM. The plot of the fluorescence peak intensities versus different concentrations (0–10 nM) of miRNA-21 (c) and miRNA-155 (d). Insets exhibited a linear relationship between the fluorescence intensities and miRNA concentrations. The selectivity evaluation of 100 nM cruciate DNA probe for target miRNA-21 (e) and miRNA-155 (f) against other non-target miRNAs. The concentrations of both target and non-target miRNAs are 10 nM. Error bars represent standard deviations from three parallel assays. | ||
Furthermore, the specificity of the cruciate DNA probe for miRNA detection was investigated by introducing target miRNAs and other non-target miRNAs. The miRNA-10b, miRNA-151, and let-7b were used as negative controls, and only faint fluorescence signals were observed. Moreover, three analogues of miRNA-21 and miRNA-155 with single-base (MM1-21, MM1-155), two-base (MM2-21, MM2-155), and three-base mismatch (MM3-21, MM3-155) were designed for selectivity evaluation. As exhibited in Fig. 2e and f, significant differentiations were obtained between the target miRNAs and other mismatched sequences. These results proved that the cruciate DNA probe was capable of discriminating target miRNAs from non-target and base-mismatched miRNAs, displaying excellent specificity for the target detection.
To investigate the reaction kinetics of the developed cruciate DNA probe, real-time fluorescence monitoring of CHA circuits was conducted (Fig. S3, ESI†). The cruciate DNA probe exhibited negligible fluorescence signals in the absence of target miRNAs, while the fluorescence intensities of the cruciate DNA probe increased quickly and reached saturation at about 60 min with the addition of 10 nM target miRNAs. These results exhibited that the cruciate DNA probe could respond to targets with fast kinetics and had the high reaction efficiency of CHA circuits, providing great potential for miRNA detection in living cells.
Detection of target miRNAs in living cells with the cruciate DNA probe
Having demonstrated the excellent performance of the cruciate DNA probe in vitro, then we explored the capability of intracellular miRNA imaging by the aptamer-functionalized cruciate DNA probe. The MDA-MB-231 cell line was employed as the model, which was reported for high expressions of endogenous miRNA-21 and miRNA-155 simultaneously.40 A control cruciate DNA probe (CrossC–H1′–H2–H3′–H4) assembled with mismatched versions of H1’ and H3’ was incubated with MDA-MB-231 cells, and the rare fluorescence intensities were observed in cytosol compared with the cruciate DNA probe (CrossC-H1–H2–H3–H4) incubation (Fig. 3a and c). The results confirmed the efficient and specific response of the cruciate DNA probe to endogenous target miRNAs. Then, the tumor cell-specific delivery of the cruciate DNA probe was verified. The cruciate DNA probes with and without AS1411 aptamer (ssDNA L4′ replaced the aptamer sequence of L4 with poly-T) were incubated with MDA-MB-231 cells for 3 h, respectively. The fluorescence images indicated that the fluorescence signals of FAM and Cy5 were significantly enhanced after MDA-MB-231 cells were treated with cruciate DNA probe with AS1411 aptamer, while the cells incubated with cruciate DNA probe without AS1411 aptamer showed little fluorescence signals (Fig. 3b and c). From the results, we evidenced that the AS1411 aptamer could obviously increase the uptake efficiency of cruciate DNA probes to carcinoma cells. Flow cytometry analysis showed remarkably increased fluorescence signals of MDA-MB-231 cells treated with cruciate DNA probe over control probe and cruciate DNA probe without AS1411 aptamer, which was consistent with confocal fluorescence images (Fig. 3d and e). | ||
| Fig. 3 Fluorescence images of MDA-MB-231 cells incubated with 100 nM CrossC-H1′–H2–H3′–H4 (a), 100 nM cruciate DNA probe without (b) and with (c) AS1411 aptamer sequences. (d and e) Flow cytometry analysis of MDA-MB-231 cells corresponding to FAM and Cy5 channels in panels a–c. Scale bar = 10 μm. | ||
Evaluation of relative miRNA levels in living cells with the cruciate DNA probe
The capability of the cruciate DNA probe to assess relative miRNA levels in living cells was crucial for cellular miRNA imaging. MDA-MB-231 cells were pretreated with the antisense miRNAs which were complementary to the targets for decreasing miRNA expression.41 Compared with untreated cells, the fluorescence images of MDA-MB-231 cells revealed much weaker fluorescence intensities in Cy5 or FAM channel after being treated with antisense miRNA-21 or antisense miRNA-155, respectively (Fig. S4a–e, ESI†). The flow cytometric experiments were also conducted and exhibited consistent results with fluorescence imaging (Fig. S4f and g, ESI†). These findings revealed the strong correlation between the fluorescence intensities and miRNA concentrations, implying the great potential for quantitative detection of multiplexed miRNAs in living cells.Furthermore, MDA-MB-231, A549, HeLa, and L-02 cells were selected for quantitative miRNA imaging via the cruciate DNA probe. It is known that miRNA-21 and miRNA-155 were highly expressed in MDA-MB-231 cells simultaneously, while HeLa and A549 cells showed relatively overexpressed miRNA-21 and miRNA-155, respectively.42,43 L-02 cells displayed low levels of both miRNA-21 and miRNA-155, and served as the negative control for targets fluorescence imaging.44 The images demonstrated increased fluorescence intensities in the FAM or Cy5 channel after incubating the cruciate DNA probe with A549 or HeLa cells, respectively. When MDA-MB-231 cells were incubated with the cruciate DNA probe for 3 h at 37 °C, the activated fluorescence signals of FAM and Cy5 appeared simultaneously in the cytoplasm, while L-02 cells showed rare fluorescence responses due to the lack of targets (Fig. 4a–f). The results of confocal images were in accordance with literature45 and RT-PCR analysis (Fig. S5, ESI†). The flow cytometry assays also exhibited similar results, further indicating the capability of the cruciate DNA probe for quantitative miRNAs detection in different cell lines (Fig. 4g–h).
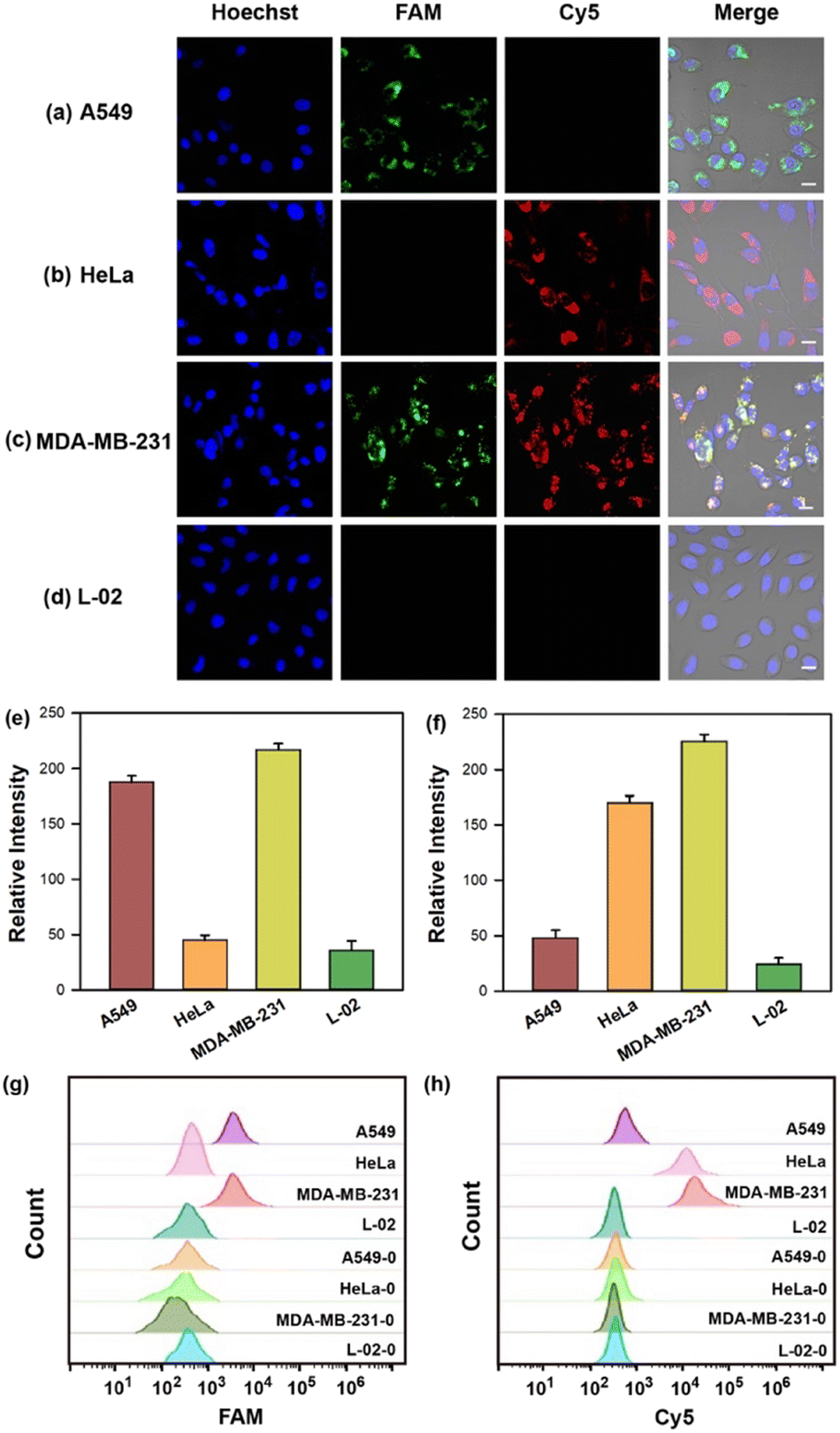 | ||
| Fig. 4 (a–d) Fluorescence images of A549, HeLa, MDA-MB-231, and L-02 cells incubated with 100 nM cruciate DNA probe. Scale bar = 10 μm. The mean fluorescence intensities (e and f) and flow cytometry analysis (g and h) of different cell lines before (MDA-MB-231-0, HeLa-0, A549-0, and L-02-0) and after (MDA-MB-231, HeLa, A549, and L-02) incubation with cruciate DNA probes corresponding to FAM (e and g) and Cy5 (f and h) channels in panels a–d. Error bars represent standard deviations from three parallel assays. | ||
The endocytosis mechanism of cruciate DNA probes
For investigating the endocytosis mechanism of the cruciate DNA probe, MDA-MB-231 cells were treated with NaN3, which is an ATPase inhibitor of energy-dependent uptake processes.46 The rare fluorescence was observed in the cytoplasm of MDA-MB-231 cells after the cruciate DNA probe incubation. Besides, MDA-MB-231 cells incubated with the cruciate DNA probe at 4 °C also exhibited fluorescence with decreased brightness (Fig. S6, ESI†). These results manifested that the cellular internalization of the cruciate DNA probe followed a temperature- and energy-dependent endocytosis mechanism. Furthermore, the z-stacked images clearly implied that the CHA products were localized in the cytoplasm instead of attached to the cell membrane (Fig. S7, ESI†).Conclusions
In summary, we have established a cruciate DNA probe for amplified multiplexed imaging of miRNAs with high specificity and sensitivity in carcinoma cells. The cruciate DNA probe allowed modular self-assembly by one-pot annealing with high yield, and selectively targeting tumor cells with the AS1411 aptamer to improve the cell internalization efficiency. After the uptake of the cruciate DNA probe, the CHA circuits were activated by intracellular multiple miRNAs and produced amplified fluorescence for low-abundance target identification with ideal biocompatibility and stability. Confocal fluorescence imaging manifested that the fluorescence intensities of the cruciate DNA probe were dynamically correlated with target miRNA expression, achieving quantitative assessment of multi-miRNAs in different carcinoma cell lines. Furthermore, due to the programmability of single-stranded DNAs, we anticipate that the cruciate DNA probe can be applied as a universal tool to image more than three intracellular miRNAs with different abundances by simply adjusting the sequences of hairpin substrates. This strategy may extend the applications of functional DNA probes in biomedical research, and the newly developed cruciate DNA probes are expected to be widely utilized for the accurate diagnosis and prognosis of different cancers.Author contributions
Zhe Dong: conceptualization, investigation, methodology, data curation, formal analysis, and writing – original draft. Xizhu Xu: methodology, formal analysis, investigation, and visualization. Jing Ni: resources, validation, and data curation. Yuancheng Li: formal analysis, resources, and supervision. Kang An: validation and data curation. Ling Meng: supervision and visualization. Han Wu: conceptualization, methodology, project administration, supervision, funding acquisition, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22204091), the academic promotion program of Shandong First Medical University (No. 921001012002, 001003100).Notes and references
- B. Ortiz-Quintero, Cell Proliferation, 2016, 49, 281–303 CrossRef CAS.
- J. F. Chen, E. M. Mandel, J. M. Thomson, Q. L. Wu, T. E. Callis, S. M. Hammond, F. L. Conlon and D. Z. Wang, Nat. Genet., 2006, 38, 228–233 CAS.
- G. A. Calin and C. M. Croce, Nat. Rev. Cancer, 2006, 6, 857–866 CrossRef CAS PubMed.
- N. Kosaka, H. Iguchi and T. Ochiya, Cancer Sci., 2010, 101, 2087–2092 CrossRef CAS.
- S. Volinia, G. A. Calin, C. G. Liu, S. Ambs, A. Cimmino, F. Petrocca, R. Visone, M. Iorio, C. Roldo, M. Ferracin, R. L. Prueitt, N. Yanaihara, G. Lanza, A. Scarpa, A. Vecchione, M. Negrini, C. C. Harris and C. M. Croce, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2257–2261 CrossRef CAS.
- H. Dong, J. Lei, L. Ding, Y. Wen, H. Ju and X. Zhang, Chem. Rev., 2013, 113, 6207–6233 CrossRef CAS.
- S. Tyagi, Nat. Methods, 2009, 6, 331–338 CrossRef CAS PubMed.
- Y. Xia, R. Zhang, Z. Wang, J. Tian and X. Chen, Chem. Soc. Rev., 2017, 46, 2824–2843 CAS.
- L. V. Von Voithenberg, A. F. Khartchenko, D. Huber, P. Schraml and G. V. Kaigala, Nucleic Acids Res., 2020, 48, e17 Search PubMed.
- B. Kura, B. Szeiffova Bacova, B. Kalocayova, M. Sykora and J. Slezak, Int. J. Mol. Sci., 2020, 21, 358 CAS.
- D. S. Seferos, D. A. Giljohann, H. D. Hill, A. E. Prigodich and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 15477–15479 CAS.
- J. I. Cutler, E. Auyeung and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 1376–1391 CAS.
- H. Li, B. Zhang, X. Lu, X. Tan, F. Jia, Y. Xiao, Z. Cheng, Y. Li, D. O. Silva, H. S. Schrekker, K. Zhang and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4340–4344 CAS.
- S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303–308 CAS.
- H. Zhang, F. Li, B. Dever, X.-F. Li and X. C. Le, Chem. Rev., 2013, 113, 2812–2841 CAS.
- J. Zheng, R. Yang, M. Shi, C. Wu, X. Fang, Y. Li, J. Li and W. Tan, Chem. Soc. Rev., 2015, 44, 3036–3055 CAS.
- R. L. Strack, M. D. Disney and S. R. Jaffrey, Nat. Methods, 2013, 10, 1219–1224 CAS.
- Z.-M. Ying, Z. Wu, B. Tu, W. Tan and J.-H. Jiang, J. Am. Chem. Soc., 2017, 139, 9779–9782 CAS.
- R. Duan, X. Zuo, S. Wang, X. Quan, D. Chen, Z. Chen, L. Jiang, C. Fan and F. Xia, J. Am. Chem. Soc., 2013, 135, 4604–4607 CAS.
- T. Xue, W. Liang, Y. Li, Y. Sun, Y. Xiang, Y. Zhang, Z. Dai, Y. Duo, L. Wu, K. Qi, B. N. Shiyananju, L. Zhang, X. Cui, H. Zhang and Q. Bao, Nat. Commun., 2019, 10, 28 CAS.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CAS.
- Z. Cheglakov, T. M. Cronin, C. He and Y. Weizmann, J. Am. Chem. Soc., 2015, 137, 6116–6119 CAS.
- S. Bi, S. Yue and S. Zhang, Chem. Soc. Rev., 2017, 46, 4281–4298 CAS.
- L. He, D. Lu, H. Liang, S. Xie, X. Zhang, O. Liu, Q. Yuan and W. Tan, J. Am. Chem. Soc., 2018, 140, 258–263 CAS.
- P. Yin, H. M. T. Choi, C. R. Calvert and N. A. Pierce, Nature, 2008, 451, 318–322 CAS.
- B. Li, A. D. Ellington and X. Chen, Nucleic Acids Res., 2011, 39, e110 CAS.
- C. Wu, S. Cansiz, L. Zhang, I.-T. Teng, L. Qiu, J. Li, Y. Liu, C. Zhou, R. Hu, T. Zhang, C. Cui, L. Cui and W. Tan, J. Am. Chem. Soc., 2015, 137, 4900–4903 CAS.
- D. S. Lee, H. Qian, C. Y. Tay and D. T. Leong, Chem. Soc. Rev., 2016, 45, 4199–4225 CAS.
- W. Ma, B. Chen, R. Jia, H. Sun, J. Huang, H. Cheng, H. Wang, X. He and K. Wang, Anal. Chem., 2021, 93, 10511–10518 CAS.
- H. Wu, T. Chen, X. Wang, Y. Ke and J.-H. Jiang, Chem. Sci., 2020, 11, 62–69 CAS.
- X. Fu, G. Ke, F. Peng, X. Hu, J. Li, Y. Shi, G. Kong, X.-B. Zhang and W. Tan, Nat. Commun., 2020, 11, 1518 CAS.
- F. Liu, Z. Yang, J. Zhou, Y. Chai, R. Yuan and S. Wei, Anal. Chem., 2022, 94, 9026–9032 CAS.
- P. Zhang, Y. Ouyang, Y. S. Sohn, R. Nechushtai, E. Pikarsky, C. Fan and I. Willner, ACS Nano, 2021, 15, 6645–6657 CAS.
- S. Soundararajan, W. Chen, E. K. Spicer, N. Courtenay-Luck and D. J. Fernandes, Cancer Res., 2008, 68, 2358–2365 CAS.
- L. Li, Y. Ren, X. Wen, Q. Guo, J. Wang, S. Li, M. Yang and K. Wang, Anal. Chem., 2021, 93, 13919–13927 CAS.
- L. Mo, D. Liang, W. He, C. Yang and W. Lin, J. Mater. Chem. B, 2021, 9, 8341–8347 CAS.
- C.-P. Liang, P.-Q. Ma, H. Liu, X. Guo, B.-C. Yin and B.-C. Ye, Angew. Chem., Int. Ed., 2017, 56, 9077–9081 CAS.
- M. Liu, R. Shen, H. Li, Y. Jia, P.-I. Mak and R. P. Martins, J. Mater. Chem. C, 2022, 10, 655–664 CAS.
- Y. Yu, L. Li, G. Li, X. Zhou, T. Deng, M. Liang and G. Nie, Chem. Commun., 2021, 57, 3753–3756 CAS.
- W. Zhou, D. Li, C. Xiong, R. Yuan and Y. Xiang, ACS Appl. Mater. Interfaces, 2016, 8, 13303–13308 CAS.
- E. Leone, E. Morelli, M. T. Di Martino, N. Amodio, U. Foresta, A. Gullà, M. Rossi, A. Neri, A. Giordano, N. C. Munshi, K. C. Anderson, P. Tagliaferri and P. Tassone, Clin. Cancer Res., 2013, 19, 2096–2106 CAS.
- J. A. Chan, A. M. Krichevsky and K. S. Kosik, Cancer Res., 2005, 65, 6029–6033 CAS.
- C. J. Cheng, R. Bahal, I. A. Babar, Z. Pincus, F. Barrera, C. Liu, A. Svoronos, D. T. Braddock, P. M. Glazer, D. M. Engelman, W. M. Saltzman and F. J. Slack, Nature, 2015, 518, 107–110 CAS.
- D. Zhu, Y. Wei, T. Sun, C. Zhang, L. Ang, S. Su, X. Mao, Q. Li, C. Fan, X. Zuo, J. Chao and L. Wang, Anal. Chem., 2021, 93, 2226–2234 CAS.
- J. Zhang, H. Fu and X. Chu, Anal. Chem., 2021, 93, 14675–14684 CAS.
- F. Zhao, Y. Zhao, Y. Liu, X. Chang, C. Chen and Y. Zhao, Small, 2011, 7, 1322–1337 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb02027k |
| ‡ Contributed equally to the implementation of the project. |
| This journal is © The Royal Society of Chemistry 2023 |