
Rational design and combinatorial chemistry of ionizable lipids for RNA delivery
Yue
Xu†
 a,
Alex
Golubovic†
a,
Shufen
Xu†
a,
Anni
Pan
a and
Bowen
Li
*ab
a,
Alex
Golubovic†
a,
Shufen
Xu†
a,
Anni
Pan
a and
Bowen
Li
*ab
aDepartment of Pharmaceutical Sciences, Leslie Dan Faculty of Pharmacy, University of Toronto, Toronto, Ontario, Canada. E-mail: bw.li@utoronto.ca
bInstitute of Biomedical Engineering, University of Toronto, Toronto, Ontario, Canada
First published on 5th June 2023
Abstract
In 2018, LNPs enabled the first FDA approval of a siRNA drug (Onpattro); two years later, two SARS-CoV-2 vaccines (Comirnaty, Spikevax) based on LNPs containing mRNA also arrived at the clinic, saving millions of lives during the COVID-19 pandemic. Notably, each of the three FDA-approved LNP formulations uses a unique ionizable lipid while the other three components, i.e., cholesterol, helper lipid, and PEGylated lipid, are almost identical. Therefore, ionizable lipids are critical to the delivery efficiency of mRNA. This review covers recent advances in ionizable lipids used in RNA delivery over the past several decades. We will discuss chemical structures, synthetic routes, and structure–activity relationships of ionizable lipids.
 Bowen Li | Dr Bowen Li is an Assistant Professor in Leslie Dan Faculty of Pharmacy at the University of Toronto, where his laboratory focuses on harnessing biomaterials combined with artificial intelligence and automated robotic platforms to develop delivery systems for engineered, bespoke RNA cargos including mRNA, circRNA, and gene editing tools. Dr Li received his PhD in Bioengineering from the University of Washington in Seattle, followed by postdoctoral training under the supervision of Prof. Daniel Anderson and Robert Langer at MIT. Dr Li currently holds the Canada Research Chair in RNA Vaccines and Therapeutics. |
1. Introduction
The discovery of messenger RNA (mRNA) has opened new avenues for treating diseases caused by loss-of-function mutations, cancer, and infectious diseases.1–4 mRNA-based drugs have the potential to replace disabled genes, express CRISPR-associated RNA or DNA nucleases, and produce antigens that induce protective immunity.5,6 However, delivering these large, polyanionic molecules to the cytoplasm so that they can be translated remains a significant challenge.7–12 Moreover, mRNA is highly unstable and prone to degradation by ubiquitous nucleases.13–18 To address these issues, scientists have developed various non-viral synthetic nanoparticles for mRNA delivery, among which lipid nanoparticles (LNPs) are the most clinically viable.19–21The rapid development of RNA therapy has been greatly promoted in recent years, thanks to a deeper understanding of LNPs. This understanding has enabled researchers to make significant progress in the delivery of RNA-based drugs. As far back as 1990, Gilead Sciences was already developing AmBisome as a treatment for fungal infections and Leishmaniasis by using liposomes to encapsulate the drug Amphotericin B.22 The approval of Patisiran (Brand name Onpattro) by the US Food and Drug Administration (FDA) in August 2018 marked a significant milestone in the development of nucleic acid-based drugs.23 Patisiran is an LNP-encapsulated siRNA preparation that effectively inhibits the synthesis of transthyretin protein (TTR) in the liver, thereby treating polyneuropathy caused by the genetic disease transthyretin-mediated amyloidosis (hATTR).24,25 The approval of Patisiran by the FDA represented a breakthrough in the treatment of this debilitating disease and paves the way for the development of new drugs based on therapeutic nucleic acids.26,27
The successful clinical application of COVID-19 (Coronavirus Disease 2019) vaccines, such as those developed by Pfizer-BioNTech and Moderna using mRNA-LNPs, has accelerated the emergence of LNP-based gene therapy.28–30 These lipid nanoparticles have shown efficacy in delivering both small siRNA and larger mRNA cargo, indicating that LNP delivery technology has the potential to facilitate most forms of nucleic acid-based therapy.31–35
The standard structure of LNPs comprises four key components: ionizable lipids, cholesterol, helper lipids, and PEGylated lipids.36,37 The ionizable lipid plays a paramount role in LNPs as it is necessary for the encapsulation of mRNA into LNPs and its subsequent escape from endosomes into the cytoplasm.31,38–42 Ionizable lipids carry a positive charge under acidic conditions, facilitating RNA loading into LNPs, but remain neutral at body pH to prevent the harmful effects linked to cationic lipids. During the process of endocytosis, these ionizable lipids become protonated in the endosome's acidic milieu and engage with phospholipids, destabilizing the endosomal membrane and enabling mRNA to escape the endosome.43 At their core, ionizable lipids consist of an ionizable head group linked to lipid tails. In this article, we provide a comprehensive review of the cutting-edge developments of ionizable lipids, emphasizing their fundamental importance in RNA delivery. We delve into the characteristics and mechanisms of these lipids, presenting an insight into their logical design and synthetic pathways. Furthermore, we also underscore the potential of incorporating combinatorial chemistry techniques into the design and synthesis of new ionizable lipids.
2. Rational design of ionizable lipids
The chemical composition of ionizable lipids generally comprises three components: headgroup, linker, and tail (Fig. 1). The head group is typically small and ionizable due to the presence of amine groups. Additionally, the linker is ideally stable but biodegradable to maintain integrity during circulation. Finally, the tail is commonly 8 to 18 carbons in length, possessing variable unsaturation and symmetry. These components collectively form a corona-like structure that serves to shield negatively charged RNA within the core of LNP formulations.44 To maintain stability and avoid adverse reactions, it is imperative that LNPs retain electrical neutrality in the bloodstream, as it has been shown that cationic lipids promote cytotoxicity.45 Subsequently, LNPs approach the vicinity of the cell membrane and interact with membrane-associated proteins. This interaction can be influenced by various extracellular proteins, including apolipoprotein E (ApoE), which plays a crucial role in the plasma circulation time of LNPs and is involved in the intracellular transport of lipids.46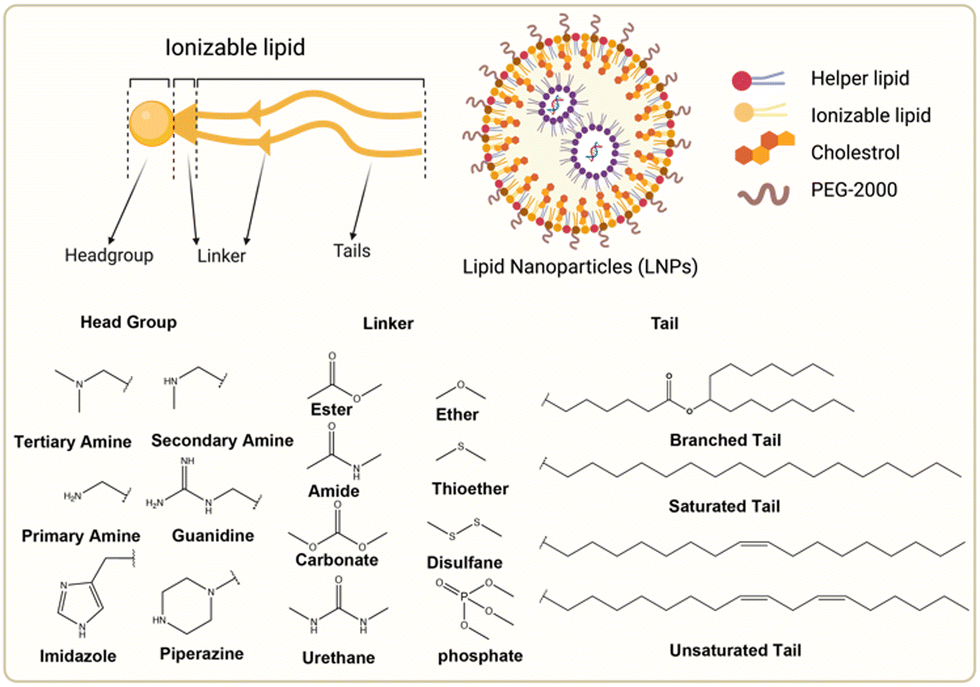 | ||
| Fig. 1 Chemical structures of the ionizable lipid and three segments of lipids. Created with BioRender.com. | ||
LNPs are susceptible to adsorption with these proteins, thus enabling the formation of a biomolecular corona in the vicinity of the interface. This surface coating can be constituted of hundreds of biomolecules, including lipoproteins, immunoglobulins, and clotting factors.47 Some of these biomolecules may bind almost irreversibly to the LNP surface in their natural or denatured form.48 The presence of a biomolecular corona on the surface of LNPs modifies their surface properties and physiochemical attributes and plays a significant role in the nanoparticle's biodistribution and phagocytosis. ApoE is one of the components of the biomolecular corona that influences the delivery of nucleic acids by LNPs. ApoE is primarily synthesized in hepatocytes and is abundant in the liver, contributing to the preferential uptake of intravenously administered LNPs in the liver. ApoE is inherently involved in cholesterol metabolism, forming lipid complexes, and facilitating their translocation to cells expressing the low-density lipoprotein receptor (LDLR), thereby promoting LDLR-mediated endocytosis. The use of polyethylene glycol (PEG)-lipids to attenuate protein adsorption and prolong the circulation of LNPs in vivo is commonplace. Furthermore, research on how altering the structure of individual LNP components and the collective LNP formulation affects protein corona adsorption and LNP tropism is actively ongoing. Despite extensive research, the full potential of the biomolecular corona remains to be fully understood. The biomolecular corona governs LNP recognition, plasma circulation time and biodistribution by the immune system and further exploration into methods for controlling the composition of the biomolecular corona could enable cell-, tissue-, or organ-specific uptake.49
The principle of extrahepatic delivery of LNP has also been extensively studied. Siegwart and colleagues developed a system called selective organ targeting (SORT) LNPs by adding a fifth component to conventional LNPs.50 By incorporating cationic lipids permanently, they achieved selective lung transfection after intravenous injection, overcoming the typical liver tropism associated with conventional LNPs. The lung-selective SORT LNPs displayed a relatively high apparent pKa and had a distinct composition of the biomolecular corona, which refers to the proteins that adsorb onto the surface of the nanoparticles. The biomolecular corona of SORT LNPs was found to be enriched in coagulation-related serum proteins with low isoelectric points, while the concentrations of complement and immune proteins decreased. This suggests that the organ-selectivity of SORT LNPs may be influenced by the charge states or functions of the serum proteins that make up the biomolecular corona. Interestingly, the distribution of the LNPs did not correspond to the site of transfection, indicating that other factors may influence the efficiency of cellular entry or endosomal escape of the particles. Despite having neutral zeta-potentials, it was necessary for PEG desorption to occur following administration in order to expose the charged SORT lipids, which then associate with serum proteins. In the case of lung-targeting SORT LNPs, vitronectin was the most enriched protein, present at a concentration 108-fold higher than its plasma concentration. This enrichment of vitronectin facilitated receptor-mediated endocytosis via αvβ3 integrin, contributing to lung-specific targeting. Overall, this study demonstrates that incorporating cationic lipids permanently in SORT LNPs enables selective lung transfection and overcomes the liver tropism associated with conventional LNPs.51 The distinct composition of the biomolecular corona and the enrichment of vitronectin suggest that the charge states and functions of serum proteins play a role in the organ-selectivity of SORT LNPs. Further research is needed to optimize the design and understand the underlying mechanisms of SORT LNPs for targeted drug delivery.
Active transport proteins help LNPs penetrate the cell membrane and enter the cell interior, where they remain stable due to the neutral cytoplasm environment. However, once in the endosome, the pH drops to nearly 4.5 before lysosomal fusion; this protonates LNPs and is thought to create ion pairs of positively charged ionizable lipids and anionic endosome phospholipids, destabilizing the endosomal membrane and releasing the RNA payload.44,52 The endosomal escape mechanism for LNPs is still unclear, and however, the proton sponge effect is also considered a possibility in this phase.53 In general, to effectively transport and express nucleic acids using LNPs, LNPs must rapidly escape from the endosomes through membrane fusion or other mechanisms to release the delivered nucleic acids. If LNPs are unable to escape from the endosomes, they will be degraded by hydrolases in the lysosomes.
Therefore, ionizable lipids need to be able not only to bind mRNA well but also to ionize rapidly to release mRNA after reaching the appropriate site. This requires a higher pKa value for ionizable lipids, which means easier binding to RNA. In contrast, a lower pKa value is more likely to release RNA. Harashima et al. concluded that a surface pKa value of LNP between 6.5 and 8.0 is optimal for RNA delivery by TNS fluorometry.54 Michael et al. concluded that the optimal value of pKa for the delivery of RNA is between 6.2 and 6.5.55,56
Additionally, it is commonly held that ionizable lipids’ molecular shape affects delivery effectiveness. A cone-shaped lipid is considered ideal as its small head and wider tail disrupt the lipid bilayer and destabilize the endosome, releasing the nucleic acid payload.57 Therefore, when designing ionizable lipids, a smaller headgroup and a longer tail became the consensus. The ionizable lipids SM-102 (Moderna) and ALC-0315 (BioNTech/Pfizer), which are approved for use in mRNA-LNP vaccines, also share this feature.58
2.1 Headgroup
The head group of ionizable lipids plays a vital role in their biological processes. The head group determines the charge and polarity of the lipid, which in turn affects its solubility, membrane interactions, and physiological effects.59 Based on chemical composition, the most prominent polar head categories are amines (primary, secondary, and tertiary amines), guanidine, and heterocyclic compounds and their combinations.60,61Francis et al. conducted a study to investigate the influence of hydrophobic mobility, lipid net charge, and lipid pKa on the transfection efficiency of lysine-based lipids as non-viral vectors.62 The authors synthesized several lysine-based lipids and analyzed the effect of changing pH on the cationicity of the ionizable lipids. In vitro results showed that cell membrane lysis, which is driven by electrostatic forces, positively impacts transfection efficiency. The synthesis route is illustrated in Fig. 2, the synthesis of the selected lipids involved several steps, including the selection of the primary amine lipid tail, condensation with fatty acids to form an amide, reduction to a dialkylamine precursor using lithium aluminum hydride, Michael addition reaction to form intermediate 6, removal of the ester using trifluoroacetic acid, and finally, deprotection to obtain the final product LOA-LysC2. The synthesis of the remaining three ionizable lipids did not differ significantly.62
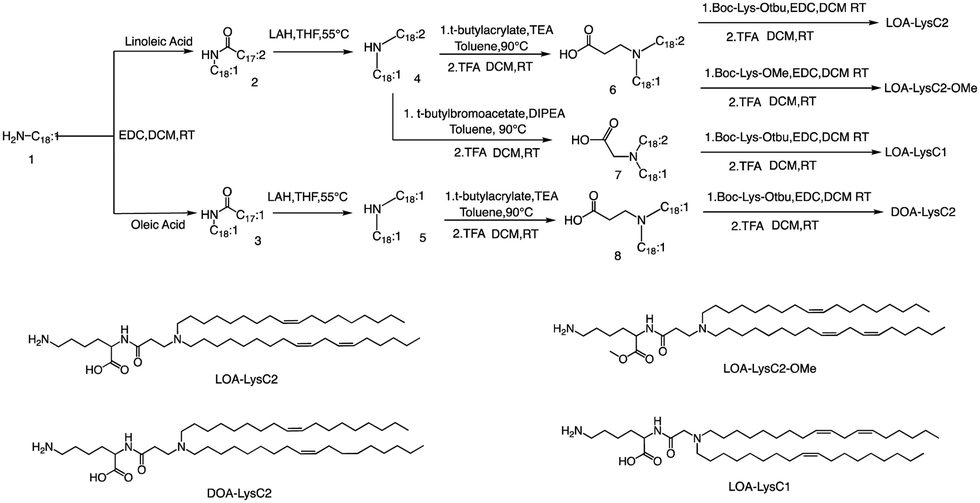 | ||
| Fig. 2 Structure and synthetic routes of lysine-based lipids. Abbreviations: lithium aluminium hydride (LAH), tetrahydrofuran (THF), triethylamine (TEA), dichloromethane (DCM), trifluoroacetic acid (TFA). | ||
A recent publication by Moderna Inc highlights the crucial role of hydrogen bonding between the headgroup and mRNA in enhancing the in vivo expression of LNPs.63 Through their research, the authors discovered a new type of squaramide lipid (lipid 29) in Fig. 3. Molecular dynamics simulations and biophysical evidence demonstrate that squaramide can interact specifically with mRNA to create an LNP with a more stable and efficient structure. In their experiment, they showed that lipid 29's headgroup can form pi stacking and bridging hydrogen bonds with RNA backbone sugars by crossing between nucleobases. This finding highlights the critical role of achieving a balance between optimal hydrogen bonding with sugars and pi packing with nucleobases in determining the significant expression and properties of lipid 29. In addition, the synthesis process of lipid 29 is depicted in Fig. 3, where the intermediate bromoalkyl ester tail was formed via a condensation reaction. Subsequently, the nucleophilic addition of bromoalkyl and amino groups yields mono-substituted secondary amines. The process was repeated with another bromoalkyl lipid tail, forming tertiary amines through nucleophilic substitution with secondary amines. Subsequently, trifluoroacetic acid was utilized to remove the amino protecting group, and the resulting exposed amino group reacted with the methyl ester to form secondary amines, resulting in the final product lipid 29.
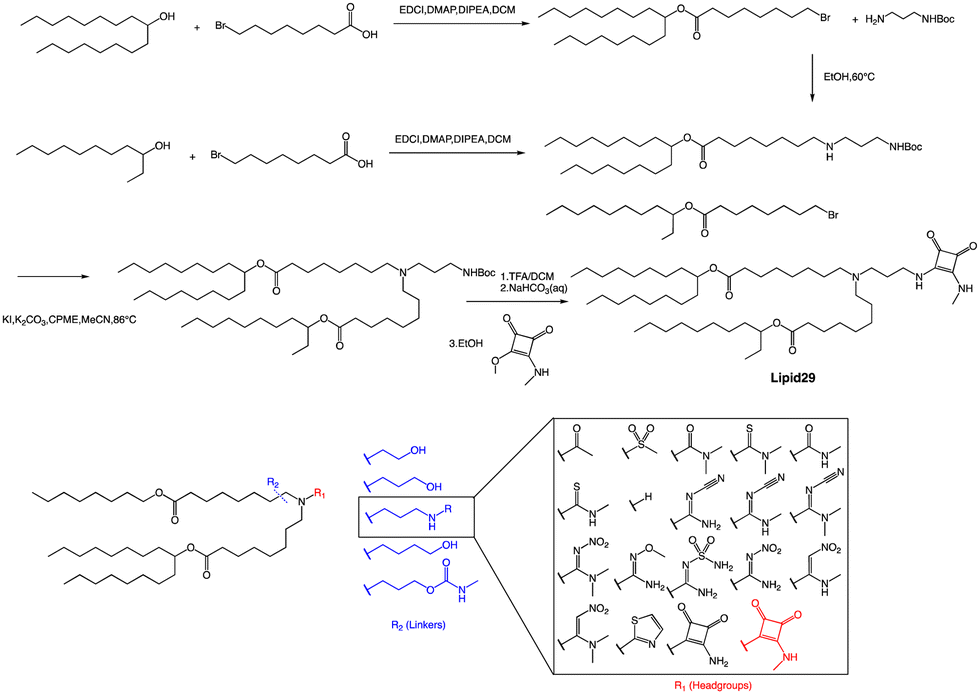 | ||
| Fig. 3 Library of lipids and representative synthetic scheme of squaramide lipid 29. Abbreviations: 1-ethyl-3-(3-dimethyl aminopropyl) carbodiimide (EDCI). 4-Dimethylamino pyridine (DMAP) N,N-diisopropylethylamine (DIPEA). | ||
Overall, LNPs formulated with lipid 29 have a range of desirable biophysical properties that may be attributed to squaramide/mRNA interactions. Once LNPs are formed, they can exhibit pH insensitivity, meaning they remain intact and stable across a range of pH values. Insensitivity to buffer pH indicates that ion interaction is not the only force that binds ionizable lipids and mRNA together, although hydrogen bonds alone are not enough to affect LNP assembly because these particles will not form at neutral pH, which indicates that a certain amount of charge complexation is required. However, the relative importance of hydrogen bonding versus electrostatics may change at different pH values, and there may be a different microenvironment within the LNP compared to the overall environment. The robustness of the lipid 29 formulation offers several advantages, such as improved nanoparticle quality and consistency through reduced heterogeneity and the ability to tune intermolecular interactions to facilitate greater size control. This superior biophysical control translates into increased formulation and process robustness, which in turn enhances in vivo expression.
In a separate study, Harashima et al. conducted a systematic derivatization of the hydrophilic headgroup and hydrophobic tail of the pH-sensitive cationic lipid YSK12-C4 (Fig. 4). They found that the hydrophilic head had a significant impact on the apparent pKa of the final product, which is a crucial factor in determining the intrahepatic distribution and endosomal escape. Specifically, they discovered that the c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP value of the hydrophilic headgroup was strongly correlated with the apparent pKa of the product. In contrast, the structure of the hydrophobic tail had a strong influence on intrahepatic distribution independently of the apparent pKa.64 Interestingly, when Harashima et al. studied pH-sensitive cationic lipids in LNPs, they found that when the lipids were in proximity, this led to lower pKa values due to electrostatic repulsion. Additionally, the hydrophobic interface of the LNP reduced the effectiveness of protonation of pH-sensitive cationic lipids, resulting in a lower apparent pKa. In addition to these findings, the synthesis route of YSK12-C4 is also noteworthy, with γ-Valero lactone (GVL) playing a decisive role in the lipid's synthesis. The synthesis culminated in a six-step reaction to obtain the final lipid.
logP value of the hydrophilic headgroup was strongly correlated with the apparent pKa of the product. In contrast, the structure of the hydrophobic tail had a strong influence on intrahepatic distribution independently of the apparent pKa.64 Interestingly, when Harashima et al. studied pH-sensitive cationic lipids in LNPs, they found that when the lipids were in proximity, this led to lower pKa values due to electrostatic repulsion. Additionally, the hydrophobic interface of the LNP reduced the effectiveness of protonation of pH-sensitive cationic lipids, resulting in a lower apparent pKa. In addition to these findings, the synthesis route of YSK12-C4 is also noteworthy, with γ-Valero lactone (GVL) playing a decisive role in the lipid's synthesis. The synthesis culminated in a six-step reaction to obtain the final lipid.
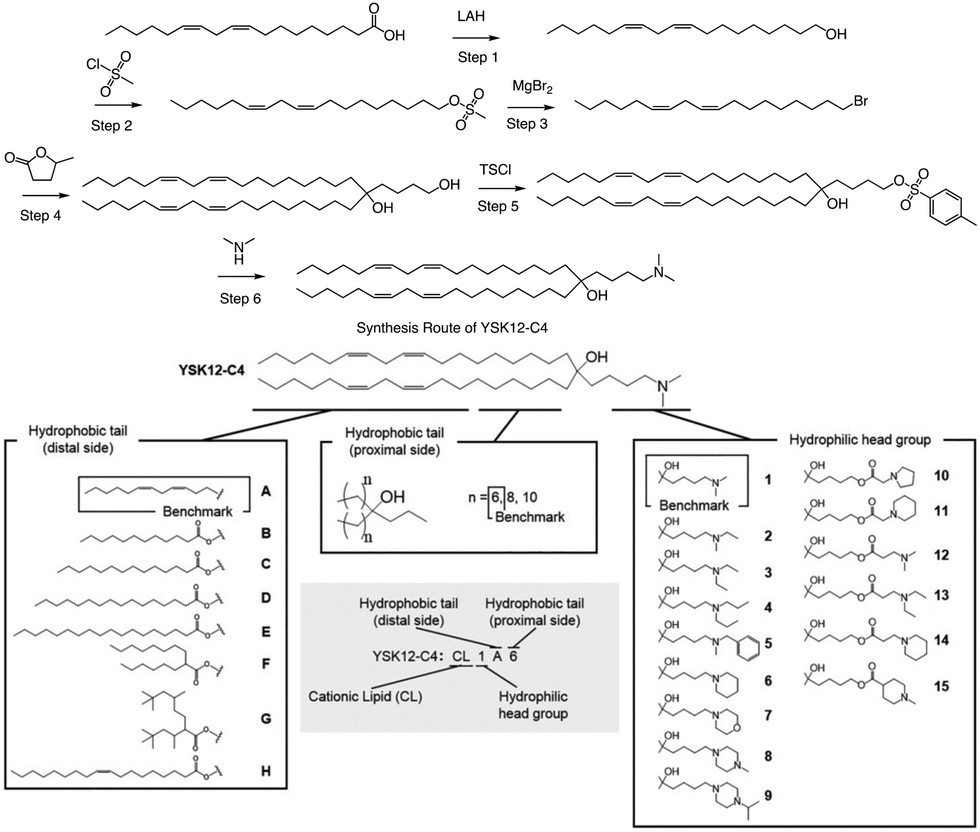 | ||
| Fig. 4 The synthesis route and structure of pH-sensitive cationic lipid YSK12-C4. Magnesium bromide (MgBr2), 4-toluenesulfonyl chloride (TsCl). | ||
Overall, the head group of ionizable lipids is essential for modulating membrane properties, participating in cell signalling, facilitating membrane dynamics, regulating ion transport, and enabling various biomedical applications.
2.2 Linker
To be an optimal component of an LNP product intended for therapeutic use, a lipid should possess specific characteristics. First, it should have a fast in vivo metabolism rate while maintaining excellent chemical stability to ensure a sufficient shelf-life. Second, the resulting metabolites should be non-toxic, easily eliminated, or further metabolized. Lastly, it can be particularly advantageous for the lipid to be rapidly broken down into hydrophilic, water-soluble products that have reduced affinity for biological membranes.Although the linkage groups constitute only a small part of the ionizable lipid structure, they are essential to achieve the objectives mentioned previously. They usually appear between the headgroups and the tail chains, and sometimes in the middle of the tail chains. On one hand, linking groups can regulate or enhance the pH value of the entire structure, enabling ionizable lipids to wrap nucleic acids better, stabilize LNP, promote cell membrane interaction, and facilitate endosomal escape, among other processes. On the other hand, linking groups are crucial for the biodegradability of ionizable lipids and can significantly reduce their cytotoxicity. Therefore, biodegradable groups are the preferred choice for chemists designing ionizable lipids.65–68 Currently, almost all ionizable lipids approved by the FDA for clinical use are biodegradable linking groups. For example, SM102, ALC-0315, and Dlin-MC3-DMA69 all use ester bonds as linking groups, and research has shown that ester bonds can substantially improve the biodegradation rate of ionizable lipids.70–72
Ester bonds are often used as linking groups in the design of ionizable lipids because they are biodegradable and can be easily cleaved by esterase, which is ubiquitous in the body. This results in the efficient removal of lipid nanoparticles from the body, reducing the potential for toxicity and increasing their safety for clinical use. In addition, ester bonds are also chemically stable and can easily be synthesized using standard organic chemistry techniques. They can also be functionalized to introduce additional properties, such as pH sensitivity or increased biocompatibility.
In related research, Akinc and colleagues demonstrated that breaking the lactone bond in the hydrocarbon chain will significantly increase the hydrophilicity of the product, resulting in the release of carboxylic acids and alcohols.55 In addition, they added an ester bond between the headgroup and the hydrocarbon in L319, which would yield a dimethylamino-dicarboxylic acid fragment and two hydrophilic primary alcohols upon hydrolysis (Fig. 5). This allows negatively charged carboxylates to neutralize or even reverse any positive charge associated with the amino headgroup, supporting the dissociation of lipid-siRNA complexes and the release of siRNA from the vector. The resulting hydrolysate may also be a substrate for further metabolism through β-oxidation, which is the natural mechanism of free fatty acid metabolism, making it easier to eliminate. When considering the position of the ester group, they noticed that the presence of sp2-carbon in the ester introduces a kink similar to a double bond in the alkyl chain, thereby maintaining the spacing between the alkyl chains, which has been shown to be important for efficacy. Therefore, they believe that an ester group can replace one of the double bonds of the oil-based chain.
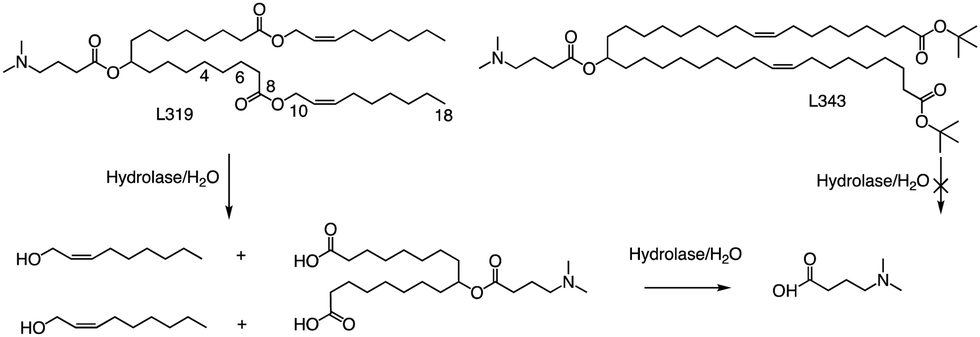 | ||
| Fig. 5 Hydrolysis routes of biodegradable lipid L319 and structure of non-degradable lipid L343. | ||
In L319, it was also shown that the degradable function can be placed in the middle of the hydrocarbon chain by replacing the 9,10-cis double bond with an ester bond. This leads to hydrolysis products that are expected to be more hydrophilic than the parent lipid. To further investigate the impact of the ester bond position, Akinc and colleagues prepared and analyzed additional lipids containing esters at various positions along the alkyl chains (C4, C6, C10, and C18).55 For another example, they also created L343, which contains sterically hindered tert-butyl esters, to serve as a “non-degradable” control due to its higher stability against enzymatic degradation. Experimental results indicate that, following a single intravenous administration of lipid nanoparticles in mice, the metabolic rate of L319 in plasma and liver is significantly higher than that of L343. Therefore, placing the ester bond in the center of the hydrocarbon chain may improve the degradation ability of lipids.
In addition to the common ester bonds, disulphide bonds as linking groups can also promote lipid degradation. Akita and colleagues developed a self-degradable ionizable lipid that utilized disulphide bonds as linking groups between two alkyl chains (Fig. 6).73 This lipid exhibited superior mRNA transfection activity compared to non-biodegradable polymers and did not cause any notable cytotoxicity. The researchers concluded that to optimize the translation efficiency of exogenous mRNA, the carrier must quickly and actively collapse in the cytoplasm. Thioester-containing molecules primarily exist in the form of S-acylated proteins during cellular processes, and acyl protein thioesterases (APT) mediate the deacylation of such proteins. Due to the broad substrate range that APTs can hydrolyze, it is likely that the inverted head compound would also undergo enzymatic degradation in cells. Additionally, as the thioester, an intermediate product of the disulphide degradation, is a relatively unstable structure, nonenzymatic hydrolysis would result in the inverted head compound degrading into a mixture of oleic acid and headgroup.
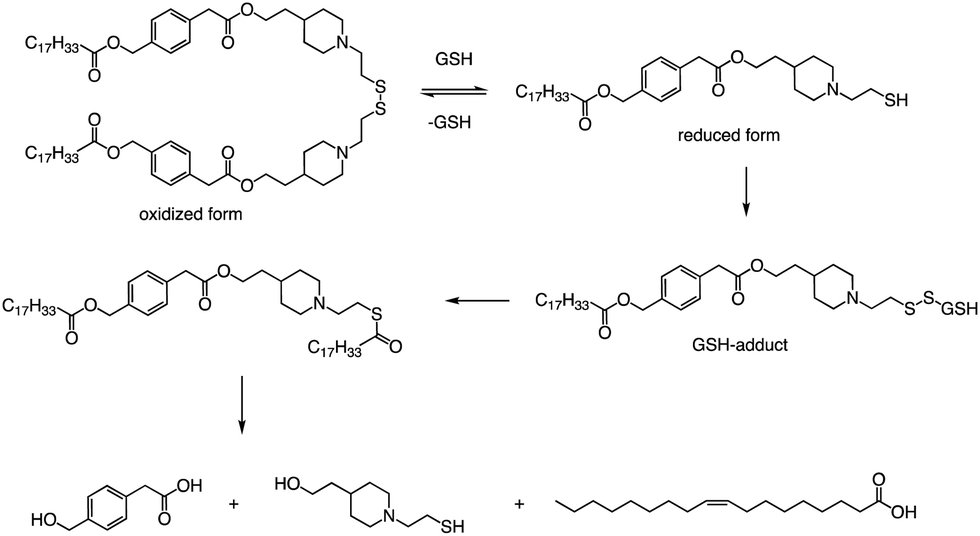 | ||
| Fig. 6 The disulfide bond of lipid is cleaved by reducing agents (GSH; glutathione). Further degradation of the compounds results in the generation of the final products of degradation; headgroup, oleic acid, and oxidized form of glutathione (GSSG). | ||
Apart from a neutral pH, the reducing environment found in the cytoplasm is also a crucial factor that triggers intracellular degradation. Disulfide bonds are commonly utilized to induce cleavage in response to the reducing environment, as the concentration of glutathione (GSH) in the cytoplasm is up to 1000 times greater than that in the extracellular environment. Consequently, the spatial selectivity and high reactivity of disulfide bonds make them an ideal choice for the cytoplasmic release of RNA. The lipids reported by Akita et al. were degraded into hydrophilic fragments under the action of glutathione and fragments were also verified by LCMS (Fig. 6).
In addition to ester bonds and disulphide bonds, amide bonds are also common in the design process of ionizable lipids. Dong's Lab reported a series of ionizable lipids using amide bonds as linking groups as vehicles for mRNA delivery (Fig. 7).74 They used benzene-1,3,5-tricarbonyl trichloride as a core with various biodegradable tails to build the lipid. The study's findings indicated that LNPs containing FTT lipids with branched ester tails, specifically FTT6, had a higher mRNA delivery efficiency to the liver and spleen compared to FTT lipids with linear ester tails like FTT10. FTT5 was found to be the most effective lipid for delivering mRNA to the liver out of all the FTT compounds tested. Cryo-TEM images of FTT5 LNPs revealed their spherical shape. Injection of FTT5 LNPs loaded with FVIII mRNA resulted in robust expression of hFVIII protein in both wild-type mice and hemophilia A mice. Furthermore, FTT5 LNPs induced substantial base editing of PCSK9 in mice at a dosage of 0.125 mg kg−1.74
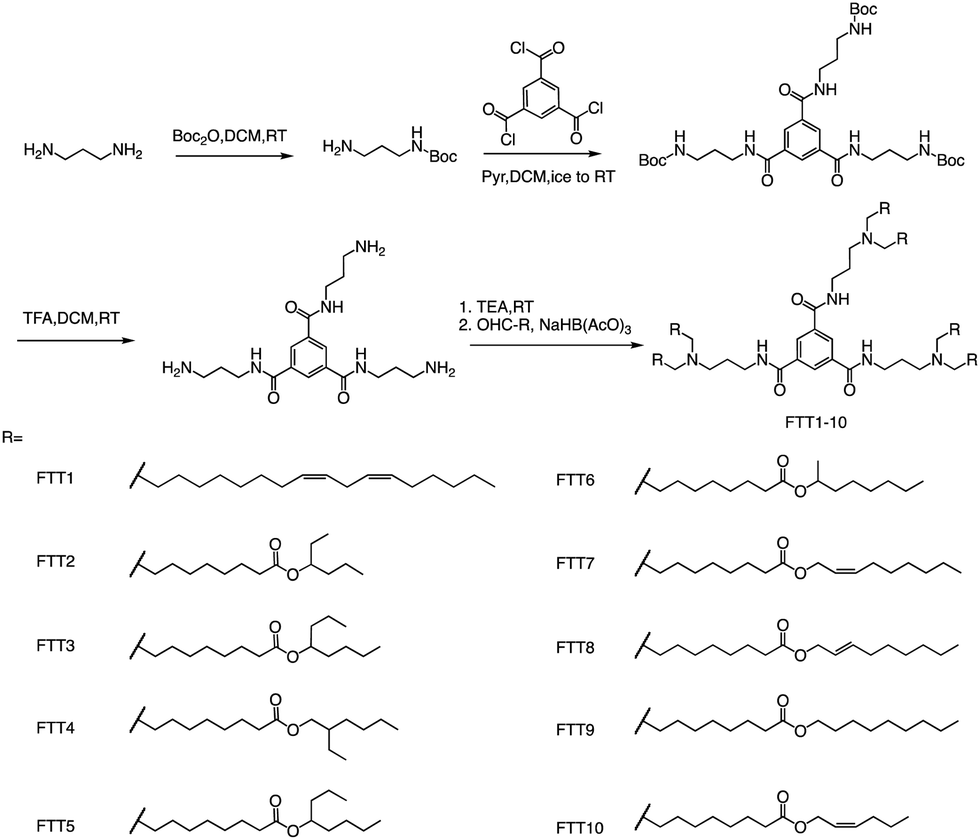 | ||
| Fig. 7 The general method for synthesizing FTT1-10. Abbreviations: di-tert-butyl decarbonate (Boc2O), pyridine (Pyr), and sodium triacetoxyborohydride is NaBH(OAc)3. | ||
2.3 Tail
The tail of an ionizable lipid typically contains 1–4 hydrophobic alkyl chains of about 8–20 carbon atoms in length. These hydrophobic tails not only affect the overall physical properties of the ionizable lipids such as pKa, logP, but also affect the transfection efficiency, biodegradation rate and cytotoxicity of LNPs, which play a pivotal role in the physical properties and transfection efficiency of LNPs. Currently, common tails typically include single/branched chains, saturated/unsaturated chains, and biodegradable/non-biodegradable chains. Many studies have proved that the two tail chains can form a crown structure, which can promote membrane instability and intracellular release of nucleic acids in vivo.
The FDA approved two LNP-based Covid-19 vaccines containing mRNA encoding the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) spike protein, the first mRNA vaccine approved for use in humans.75,76 Brito et al. observed minimal rank agreement between the LNPs in terms of IM and IV expression, indicating that the formulation behavior may vary when administered locally versus systemically. The absence of correlation between IM and IV performance may be attributed to potential differences in optimal physical or chemical properties required for each administration route.77 They compared the ability of each of the precursor LNP formulations to drive the secretion of IgG antibodies in mice's muscles after injection, including MC3 and SM102 (Fig. 8). Except for lipid Q, the IgG serum concentration produced by the other four lipids (Lipid H, Lipid M, Lipid P and Lipid N) was higher than that of MC3 (p < 0.05). Compared with MC3, the tissue histology and tolerability of lipid SM102 shown here are consistent with improved tissue pathology and reduced innate immune stimulation. Inflammatory cell infiltration, muscle fiber damage, and decreased systemic cytokines support the hypothesis that mRNA vaccines may not require a strong adjuvant response to generate an effective immune response.78 Therefore, it is generally believed that the biodegradability and transfection efficiency of SM102 with ester bonds are better than MC3. Another important reason is that the branching chain can greatly improve the transfection efficiency of nucleic acid because SM102 can form a crown structure more easily, which is conducive to the rapid release of nucleic acid in the cytoplasm.
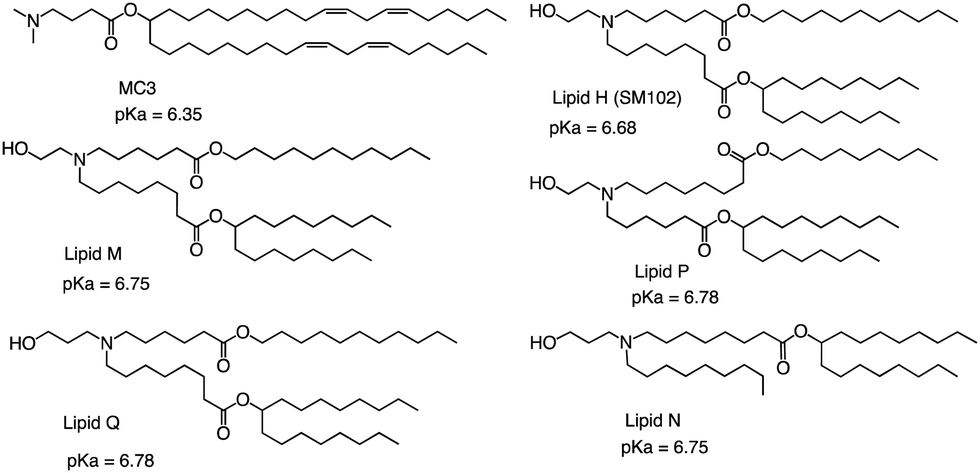 | ||
| Fig. 8 Chemical structures and pKa of MC3 and novel lipids. | ||
In addition to the common two alkyl chains of ionizable lipids, Fenton et al. also synthesized four alkenyl α-amino alcohols (AAA) type analogs of cKK-E12 in 2016 by conjugating alkenyl epoxides derived from biologically relevant fatty acids with the dilysine-derived diketopiperazine core of cKK-E12 (Fig. 9).79 In the following study, they also introduced unsaturated tail chains based on cKK-E12. LNPs of OF-02 (Fig. 10) were used to load EPO mRNA.80 The concentration of EPO was found to be more than twice that of the control cKK-E12 by intravenous administration in mice. This result proves that the delivery efficiency of mRNA is greatly improved by the unsaturated tail chain based on ionizable lipids constructed by the cKK core. Although cKK-E12 and OF-02 exhibited robust transfection efficiency, they displayed high biotoxicity in cells and the liver. To address this issue, the researchers designed a biodegradable tail chain version based on cKK to enhance biological tolerance. So, they designed biodegradable ionizable lipids called Deg-Lin (Fig. 10) to encapsulate the mRNA in another study, which led to the production of functional proteins in mouse B lymphocytes. They subsequently increased the distance between the ester bond and the linker group from two to four carbons and found that OF-C4-Deg-Lin delivered anti-FLuc siRNA and FLuc mRNA more efficiently in HeLa cells than OF-Deg-Lin. In mice, intravenous administration of LNP of OF-C4-Deg-Lin encapsulated firefly luciferase mRNA resulted in its accumulation primarily in the spleen, whereas OF-Deg-Lin was mainly concentrated in the liver. This example serves as a convincing illustration that tail modifications can enhance the biodegradation rate of LNPs and minimize cellular as well as liver toxicity compared to unbiodegradable lipids. It highlights the importance of carefully designing the tail structure when developing ionizable lipids.
 | ||
| Fig. 9 Synthesis route of cKK-A12, cKK-O12 and cKK-E12. | ||
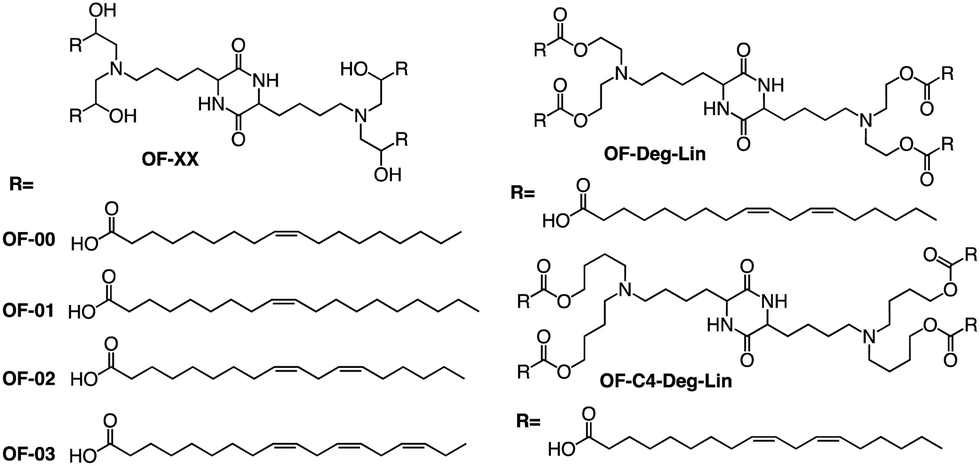 | ||
| Fig. 10 Structure of OF-XX, OF-Deg-Lin and OF-C4-Deg-Lin. | ||
3. Combinatorial chemistry for ionizable lipids
Given the time-consuming, labour-intensive, and high-risk nature of ionizable lipid synthesis, many researchers aim to rapidly generate large quantities of these lipids through combinatorial chemistry for in vitro screening. These libraries are often called lipidoids. Compared to traditional synthetic approaches, combinatorial chemistry offers distinct advantages, including reduced time, lower costs, and the ability to perform high-throughput screening of ionizable lipids. Combinatorial chemistry is usually accompanied by several types of chemical reactions such as Michael addition, ring-opening reactions, condensation reactions, multi-component reactions and enzyme-assisted chemical reactions.3.1 Michael addition
Whitehead et al. used Michael addition chemistry to stoichiometrically bind amine 306 to 11 saturated alkyl acrylate tails ranging in length from 6 to 18 (Fig. 11).81 In this lipid pool, they found that the Oi10 material is more ionized and has a greater ability to transfer mRNA than the lipids containing linear tails. Oi10 has only one additional carbon in the branch compared to the normal linear-tailed lipids. This slight structural difference increases the efficacy of 306O i10 by a factor of ten compared to its similar O10 structure. They believe that the branches of the Oi10 tail act as wedges, increasing the distance between ionizable lipid molecules within the LNP bilayer.82,83 This extra space can facilitate the protonation of the amine due to reduced repulsion between adjacent cations.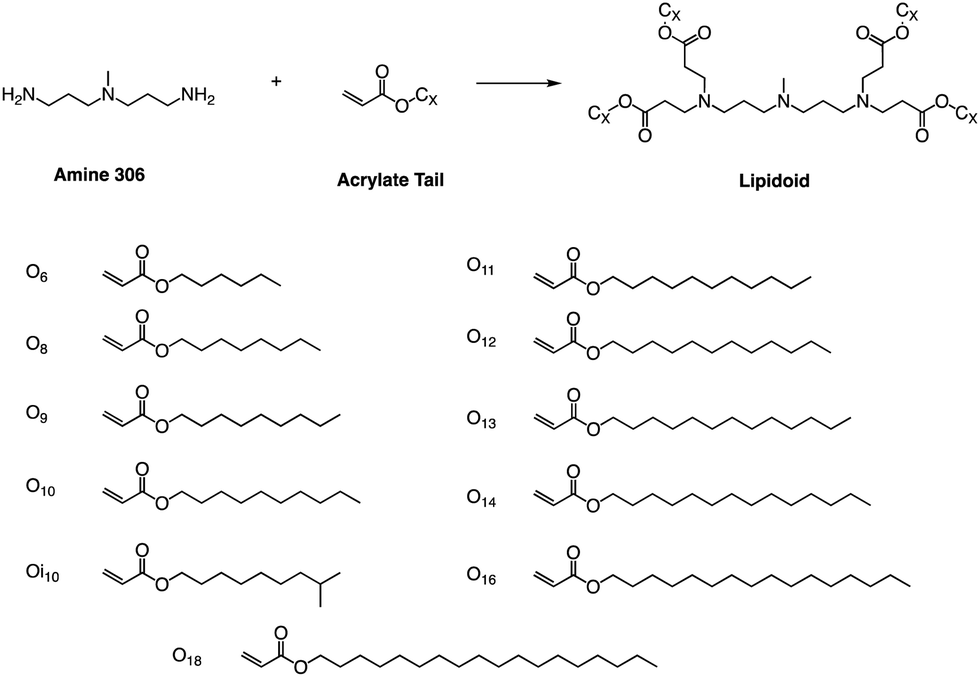 | ||
| Fig. 11 Lipidoids were synthesized using Michael addition chemistry by reacting the alkyl-amine 306 (3,3′-diamino-N-methyldipropylamine) combinatorially with an alkyl-acrylate tail. | ||
Other than that, Yang and colleagues conducted a study on the synthesis of ionizable lipids using Michael addition and investigated the impact of different amine-based headgroups (Fig. 12) on mRNA transfection efficiency.84 Specifically, they assessed the ability of these ionizable lipids to deliver mLuc and hEPO mRNA in vivo. Their results demonstrated that Spermine (structure #1 in Fig. 12) was the most efficient headgroup for mRNA delivery in their library, indicating that it was able to facilitate the successful delivery of the mRNA molecules in vivo. Moreover, the researchers observed a positive correlation between the positive charge density of the ionizable lipid and its transfection efficiency, suggesting that increasing the charge density of the lipid could further enhance mRNA delivery. Overall, this study sheds light on the potential of ionizable lipids as a promising platform for mRNA delivery and highlights the importance of optimizing the headgroup structure to achieve optimal transfection efficiency. Further research in this area could lead to the development of more effective mRNA delivery vehicles for therapeutic applications.
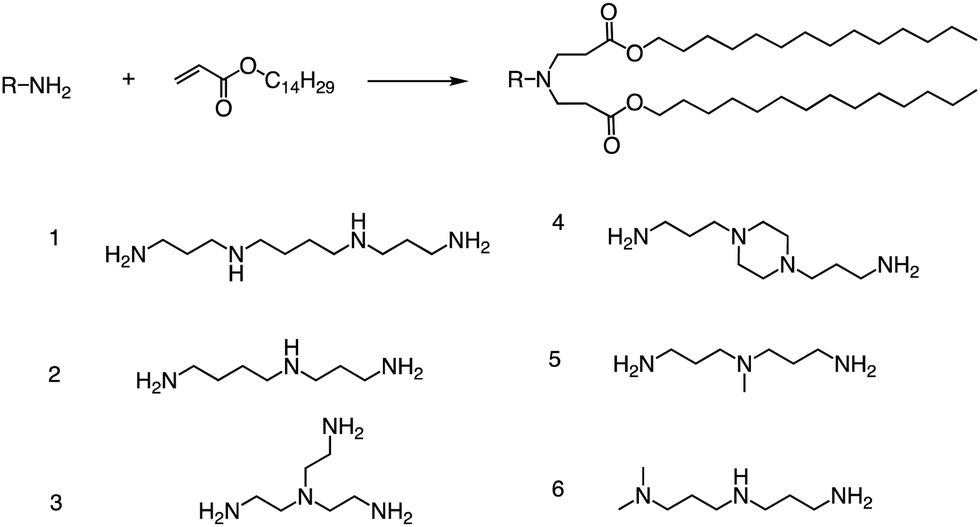 | ||
| Fig. 12 Synthesis route of different lipid materials via the Michael addition of an alkyl-acrylate to amine-head molecules. | ||
In a recent study, Li et al. successfully leveraged Michael addition chemistry to establish a combinatorial library of biodegradable ionizable lipids, the aim being to identify the LNPs that can safely and effectively enable pulmonary mRNA delivery, thereby enabling powerful gene editing within the lung.85 As part of this library, over 700 biodegradable ionizable lipids were synthesized and examined (Fig. 13). This three-component reaction (3-CR) system outperforms the conventional two-component reaction, which directly connects amine headgroups with lipid tails. The 3-CR system streamlines the synthesis process, bolsters lipid structural diversity, and rapidly generates a broad range of biodegradable ionizable lipids for inclusion in the combinatorial library.
 | ||
| Fig. 13 Structures of amine headgroups (72) and alcohol tails (10) in a combinatorial library of 720 biodegradable ionizable lipids. | ||
Although Michael addition reactions are becoming more and more common for lipid synthesis, the reaction conditions and catalysts for such reactions also need to be carefully considered. The lipid synthesis method based on the Michael addition reaction was optimized by Puri et al., who proposed that using Lewis bases can enhance the reaction's rate and yield.86 When allylic boronic esters are activated under photoredox reaction conditions, they produce allylic radicals that readily react with Michael acceptors (Fig. 14). This reaction yields functionalized alkene products that can be transformed into other useful synthetic structures. For example, ionizable lipids have been synthesized using this method for RNA delivery.86 These organoboronic reactions are robust, enabling the branched lipid structures to be derivatized, where amino groups and aliphatic tails can be tuned to optimize these ionizable excipients.
 | ||
| Fig. 14 The library of lipids was developed to expedite the Michael addition reaction by Lewis bases. | ||
3.2 Ring-opening epoxides with amines
In addition to the Michael addition reaction, ring-opening reactions of epoxides by the one-pot method are also frequently used to synthesize new ionizable lipids. Mitchell et al. conducted research to create a specific ionizable lipid for engineering Human CAR T cells.87 The study found that using LNPs containing this lipid for mRNA delivery was less damaging to T cells than electroporation techniques. The investigation tested 24 LNP libraries, and C14-4 (Fig. 15) was found to be the most effective. The results showed that C14-4LNP delivered CAR mRNA to primary T cells and generated functional CAR-T cells. The findings suggest that C14-4 LNP has the potential to be optimized for various mRNA-based T-cell engineering applications. To synthesize the ionizable lipids, Michael addition chemistry was used as the basis of the reaction. This involved the reaction of an excess of alkyl chains with polyamine cores. C14-4 was chosen as a representative reaction in this study (Fig. 15). | ||
| Fig. 15 Structures of the alkyl chains (A) and polyamine cores (B) were used to generate the ionizable lipid library. (C) C14-4 is used here as a representative reaction. | ||
In a recent study, Levkin and colleagues showcased a rapid and straightforward method for synthesizing a diverse set of 288 lipid-like molecules through thiolactone ring-opening reactions (Fig. 16).88 This approach enables the creation of lipids with hydrophobic tails containing both unsaturated bonds and reducible disulphide moieties, expanding the range of available lipids for drug delivery and gene therapy applications. The entire synthesis and purification process is efficient and speedy, taking only a few hours to complete. HEK293T cell-based screening of the generated lipids without auxiliary lipids resulted in the identification of highly stable liposomes with an impressive transfection efficiency of around 95% and minimal toxicity. The developed lipid-like molecules have the potential to serve as promising candidates for the efficient delivery of therapeutic molecules, such as mRNA and DNA, to target cells. This simple and rapid one-pot synthesis of structurally diverse ionizable lipids offers a promising platform for the development of new and improved liposome-based drug delivery systems.
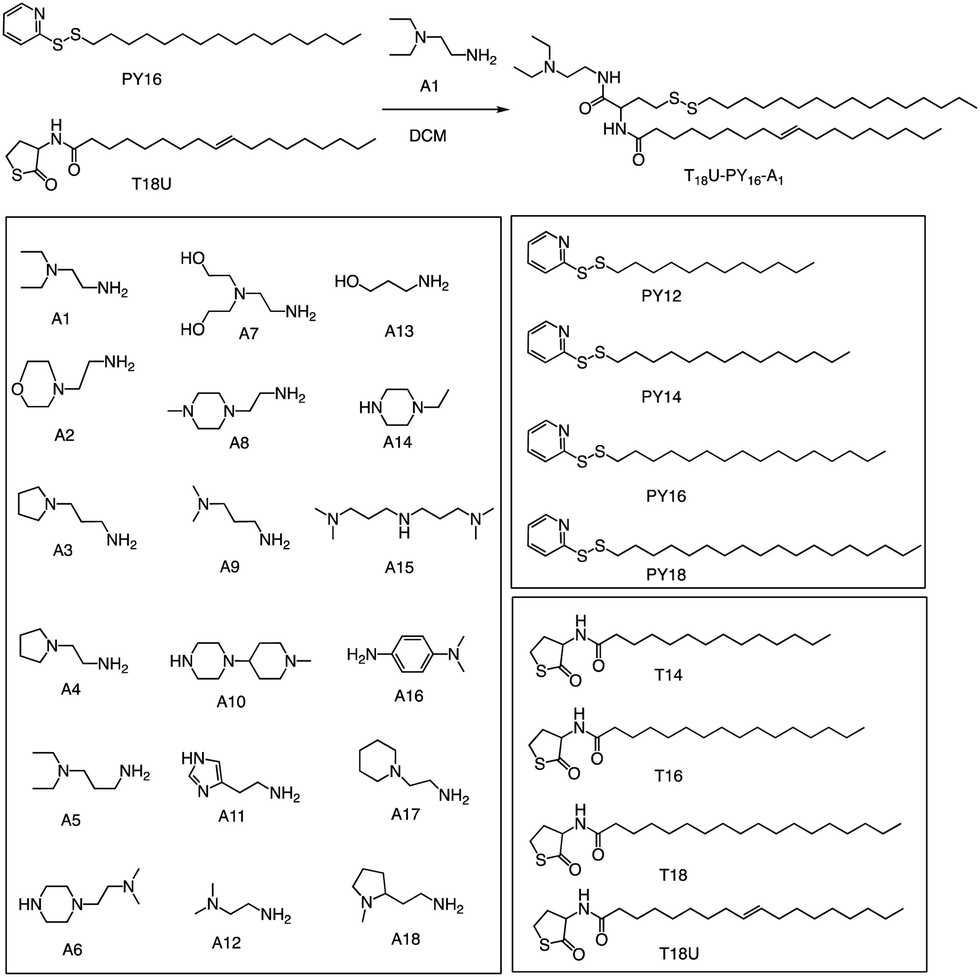 | ||
| Fig. 16 Three-component, one-pot synthesis of lipidoids using nucleophilic ring opening of thiolactones followed by the disulphide exchange reaction. Representative reaction scheme and structures of amines; pyridyl disulphide derivatives; thiolactone derivatives. | ||
Alongside thiol ring-opening reactions, the use of ring-opening epoxides is a prevalent method in the synthesis of lipid pools. Xia's group89 has been screening a range of cationic lipidoids materials using a ring-opening reaction of epoxide, ultimately developing a minimalist nano vaccine with LNP of tail C12 that efficiently delivers mRNA in antigen-presenting cells, while simultaneously activating Toll-like receptor 4 (TLR4) and inducing robust T cell activation (Fig. 17). This nano vaccine enters cells through phagocytosis, resulting in efficient mRNA-encoded antigen expression and presentation. Additionally, the lipids with tail C12 nanoparticle itself trigger the expression of inflammatory cytokines such as IL-12 by stimulating TLR4 signal pathways in dendritic cells. Excitingly, the C1 mRNA nano vaccine demonstrates significant antitumor efficacy in both tumor prevention and therapeutic vaccine settings. In summary, this research introduces an LNP-based mRNA cancer nano vaccine that effectively promotes antigen expression and has self-adjuvant properties. This breakthrough provides a promising platform for developing cancer immunotherapy that could potentially benefit a broad range of tumor types.
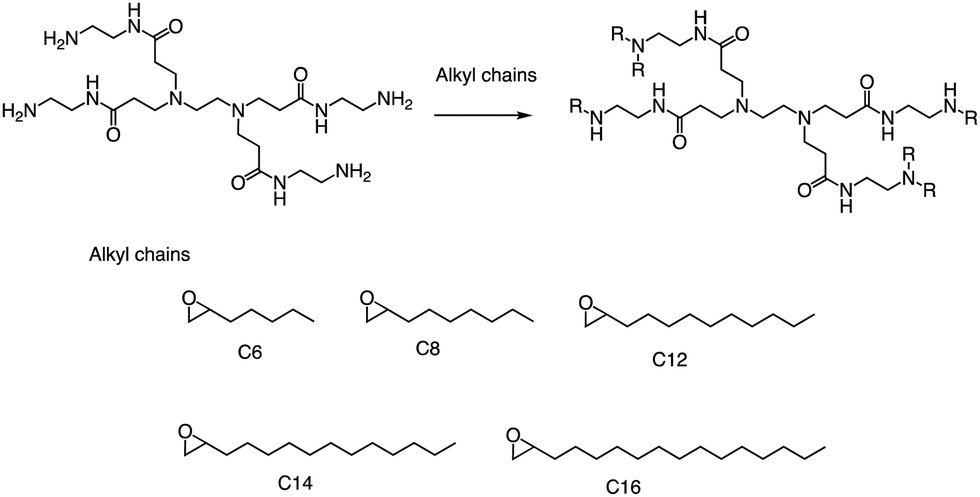 | ||
| Fig. 17 Synthesis of 15 lipidoids through efficient ring-open epoxides by generation 0 of poly(amidoamine) dendrimers for mRNA delivery. | ||
3.3 Reductive amination of aldehydes
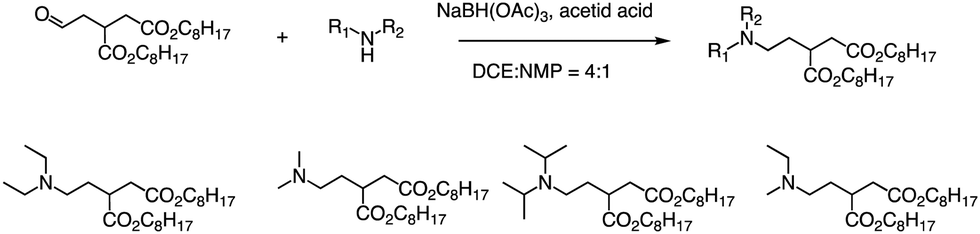
Formation of the amide bond typically requires reductive amination of the aldehyde. Dong et al. drew inspiration from the structure of lipid proteins to develop a set of lipid nanomaterials that were synthesized using amino acids and aldehyde tail chains (Fig. 18).79 Through a reductive amination process, the natural amino acids amino group reacted with the aldehyde tail to create tertiary amines. These nanomaterials were then utilized to deliver siRNA nanoparticles to hepatocytes, resulting in high selectivity towards hepatocytes compared to non-target cell types. The FVII-silenced ED50 was found to be approximately 0.002 mg kg−1, indicating the effectiveness of these materials in the targeted delivery of siRNA nanoparticles to hepatocytes.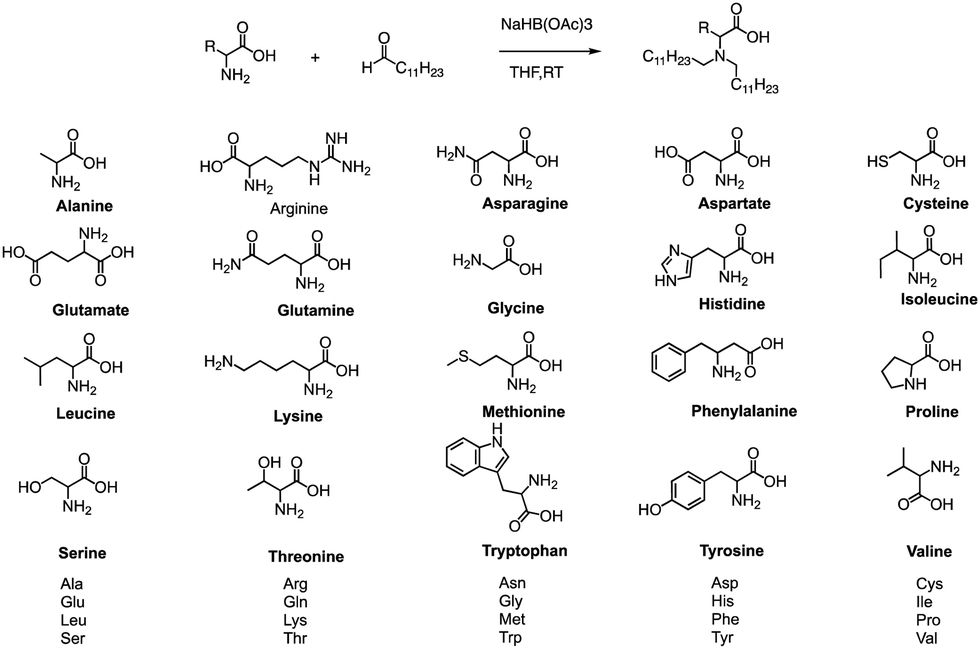 | ||
| Fig. 18 Synthetic routes of lipid amino acid derivatives. | ||
At the same time, Dong's group also used reductive amination of aldehydes to build a lipidoid library for mRNA delivery in vivo (Fig. 19).90 By incorporating the lipid TT3, their best LNP candidate restored human factor IX (hFIX) levels to normal physiological values in FIX knockout mice. Overall, this one-pot type reaction is very useful for building a lipidoid library.
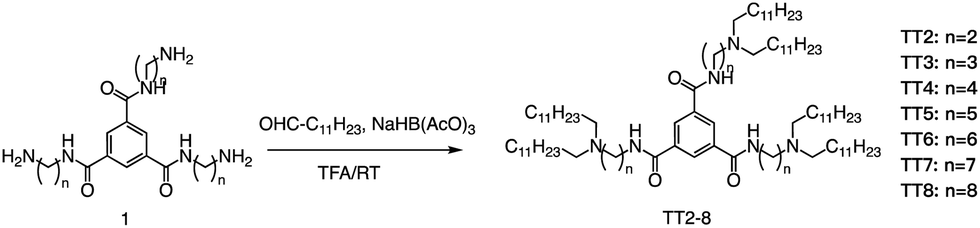 | ||
| Fig. 19 Synthetic route to N1,N3,N5-tris(2-aminoethyl)benzene-1,3,5-tricarboxamide derivatives (TT2–TT8). Compound 1 underwent a reductive amination to afford desired products TT2–TT8. | ||
3.4 Multicomponent reactions
Anderson and his team created a combinatorial library of ionizable lipids to identify mRNA delivery vehicles that could efficiently deliver mRNA in vivo and promote potent and specific immune activation.91 The library consisted of over 1000 lipid formulations that were synthesized and evaluated using a three-dimensional multi-component reaction (MCR) system (Fig. 20). This reaction type was the modified Ugi reaction, and it was the first reported use of MCR in lipidoids synthesis.92–99 Notably, the ethanol used in this reaction as well as the absence of catalysts are of great help in the high-throughput screening of ionizable lipids. The top-performing lipid formulations shared a common structure, which included an unsaturated lipid tail, cyclic amine headgroups, and a dihydroimidazole linker. These formulations proved to be highly effective in inducing a robust immune response and were able to inhibit tumor growth and extend the lifespan of melanoma and human papillomavirus E7 in vivo tumor models. Unlike traditional Toll-like receptor-based vaccines, these top-performing lipid formulations activate antigen-presenting cells through the intracellular stimulator of the interferon genes (STING) pathway. This results in limited systemic cytokine expression while enhancing the anti-tumor efficacy of the vaccine. Overall, the development of the MCR for screening these lipidoids represents an important advancement in the field of mRNA delivery.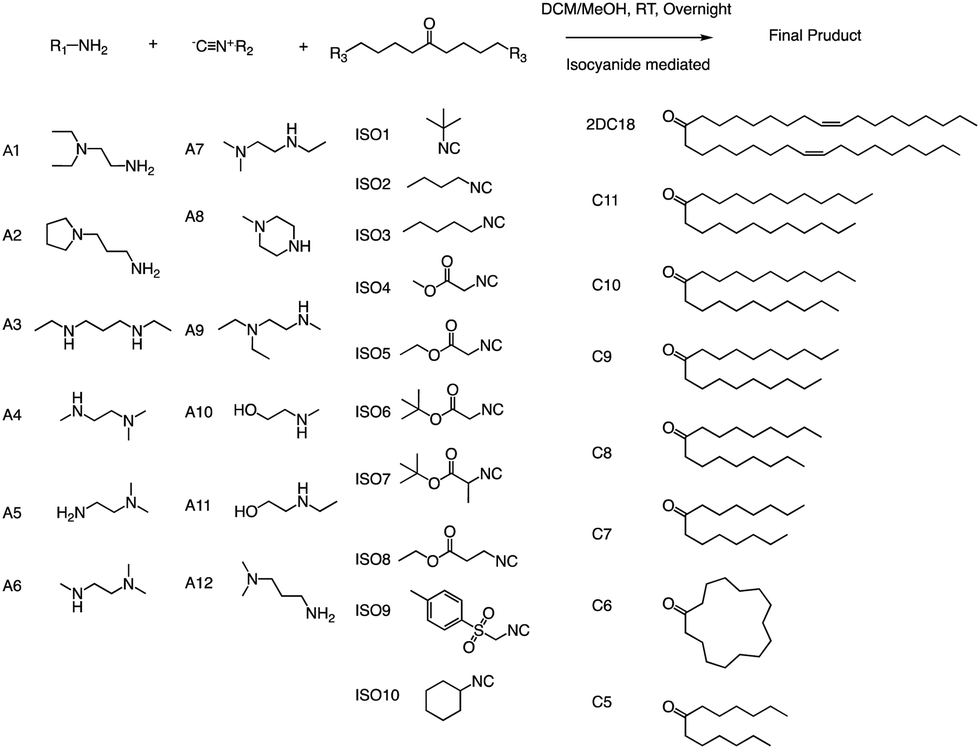 | ||
| Fig. 20 Structures of the three components of the lipidoid (amine, isocyanide and ketone) used in the synthesis library are shown. | ||
3.5 Enzyme-assisted chemical reactions
In addition to traditional organic synthesis methods, biochemical synthesis methods involving enzymes have also been used for the synthesis of lipids. In a recent study, Xu et al. developed a chemical library of ionizable lipids through a one-step chemical-biological enzyme-catalyzed (Candida antarctica Lipase B) esterification method.100 These lipids were utilized to create LNPs for mRNA delivery, and the top-performing AA3-DLin (Fig. 21) LNPs exhibited outstanding mRNA delivery efficacy and long-term storage capability. The researchers encapsulated SARS-CoV-2 spike mRNAs in the AA3-DLin LNPs, and in a BALB/c mouse model, the resulting AA3-DLin LNP COVID-19 vaccines successfully induced strong immunogenicity, as evidenced by antibody titers, virus challenge, and T cell immune response studies. The results of the study demonstrate that the AA3-DLin LNPs are an excellent mRNA delivery platform, with the potential for further applications in vaccine development and gene therapy. The development of this chemical library and the subsequent LNP platform could have significant implications for the delivery of therapeutic molecules, including mRNA-based vaccines, to treat various diseases. Thus, the integration of chemical biology could be a potentially effective strategy to expedite lipid screening, thereby circumventing the need for intricate chemical synthesis.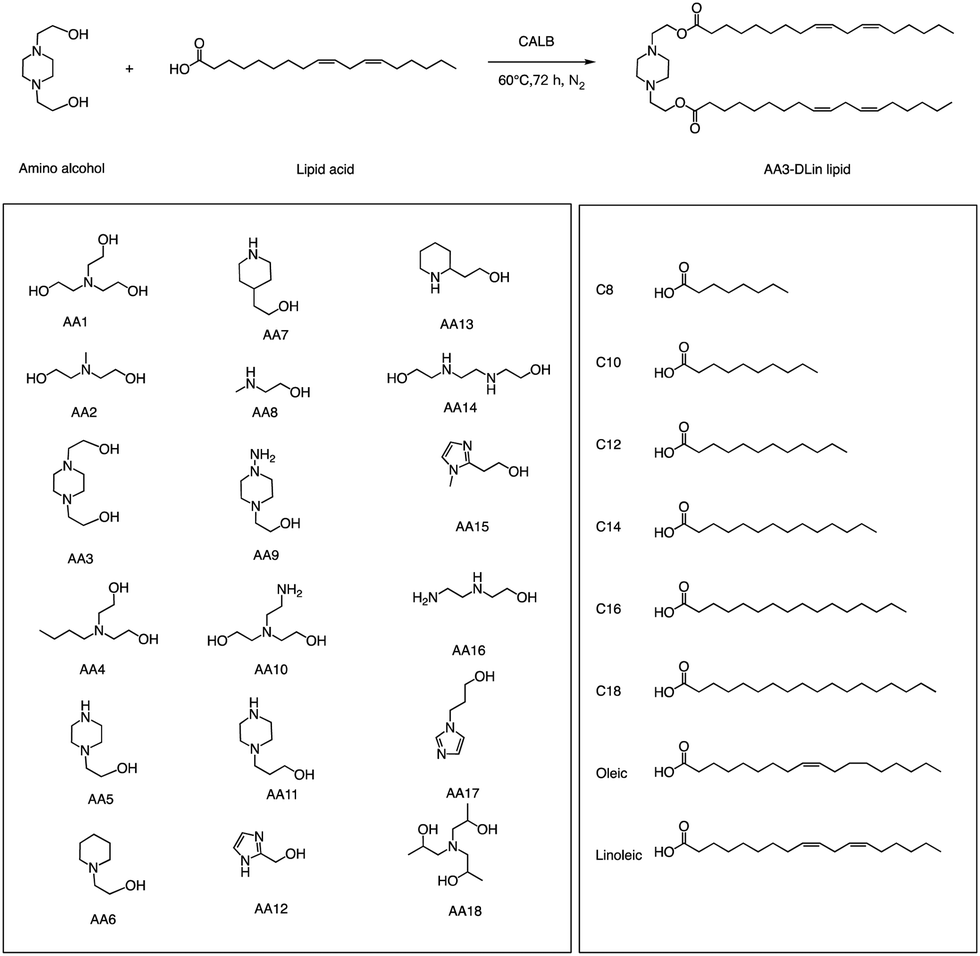 | ||
| Fig. 21 The enzyme-catalyzed synthesis of ionizable lipids library. | ||
4. Conclusions
Synthetic chemists invest a significant amount of time and effort into designing and developing ionizable lipids suitable for various RNA delivery applications. However, with the advent of increasingly advanced ionizable lipids, researchers are gaining valuable insights and knowledge on how to design, synthesize, and screen new lipids for this purpose. The design of ionizable lipids for RNA delivery remains a complex endeavour, with numerous challenges that require further research and discussion. For instance, it is unclear whether modifying ionizable lipids can alter the immune response type, such as the B or T cell response. Some mRNA vaccines have been shown to elicit T cell-mediated protection but lack the ability to induce neutralizing antibodies. Moreover, it is still uncertain whether different ionizable lipids can be used for all RNA delivery types, including siRNA,101 mRNA,69 long noncoding RNA (lncRNA),102 circular RNA (circRNA),103,104 transfer RNA (tRNA)105 and others.106 While biodegradable ionizable lipids can lower the risk of potential adverse reactions, they may also affect the stability of LNPs, leading to issues with biodistribution and pharmacokinetics.107–109 Despite these challenges, the increasing diversity of ionizable lipids holds promise for treating RNA-based diseases, particularly non-treatable targets such as genetic diseases and neurodegenerative disorders.110,111 Simultaneously, the rapid synthesis of ionizable lipids has given rise to a plethora of combinatorial chemistry techniques, which offer distinctive perspectives for conducting high-throughput screening of such lipids.Author contributions
Y. X., A. G., S. X., A. P. and B. L. conceptualized the article, wrote the original draft, and reviewed/edited the article. B. L. served as a project administrator.Conflicts of interest
The authors declare that they have no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the PRiME-UHN Clinical Catalyst Program (no. PRMUHN2022-005), the Leslie Dan Faculty of Pharmacy startup fund, the Connaught Fund (no. 514681), the J. P. Bickell Foundation (grant no. 515159), the Canada Research Chairs Program (no. CRC-2022-00575), Canadian Institutes of Health Research (no. PJH-185722), and the Canada Foundation for Innovation John R. Evans Leaders Fund (no. 43711).Notes and references
- H. Zogg, R. Singh and S. Ro, Int. J. Mol. Sci., 2022, 23, 2736 CrossRef CAS PubMed.
- N. J. Abreu and M. A. Waldrop, Pediatr. Pulmonol., 2021, 56, 710–720 CrossRef PubMed.
- K. Horodecka and M. Düchler, Int. J. Mol. Sci., 2021, 22, 6072 CrossRef CAS PubMed.
- H. Li, Y. Yang, W. Hong, M. Huang, M. Wu and X. Zhao, Signal Transduction Targeted Ther., 2020, 5, 1 CrossRef PubMed.
- Y. Weng, C. Li, T. Yang, B. Hu, M. Zhang, S. Guo, H. Xiao, X.-J. Liang and Y. Huang, Biotechnol. Adv., 2020, 40, 107534 CrossRef CAS PubMed.
- N. Sayed, P. Allawadhi, A. Khurana, V. Singh, U. Navik, S. K. Pasumarthi, I. Khurana, A. K. Banothu, R. Weiskirchen and K. K. Bharani, Life Sci., 2022, 120375 CrossRef CAS PubMed.
- T. C. Roberts, R. Langer and M. J. Wood, Nat. Rev. Drug Discovery, 2020, 19, 673–694 CrossRef CAS PubMed.
- A. Wadhwa, A. Aljabbari, A. Lokras, C. Foged and A. Thakur, Pharmaceutics, 2020, 12, 102 CrossRef CAS PubMed.
- W. Ho, M. Gao, F. Li, Z. Li, X. Q. Zhang and X. Xu, Adv. Healthcare Mater., 2021, 10, 2001812 CrossRef CAS PubMed.
- A. Lacroix and H. F. Sleiman, ACS Nano, 2021, 15, 3631–3645 CrossRef CAS PubMed.
- G. Kara, G. A. Calin and B. Ozpolat, Adv. Drug Delivery Rev., 2022, 114113 CrossRef CAS PubMed.
- A. Golubovic, S. Tsai and B. Li, ACS Bio Med Chem Au, 2023, 3(2), 114–136 CrossRef CAS PubMed.
- S. Qin, X. Tang, Y. Chen, K. Chen, N. Fan, W. Xiao, Q. Zheng, G. Li, Y. Teng and M. Wu, Signal Transduction Targeted Ther., 2022, 7, 166 CrossRef CAS PubMed.
- M. Mehta, D. Tewari, G. Gupta, R. Awasthi, H. Singh, P. Pandey, D. K. Chellappan, R. Wadhwa, T. Collet and P. M. Hansbro, Chem.-Biol. Interact., 2019, 308, 206–215 CrossRef CAS PubMed.
- M. I. Sajid, M. Moazzam, S. Kato, K. Yeseom Cho and R. K. Tiwari, Pharmaceuticals, 2020, 13, 294 CrossRef CAS PubMed.
- L. Nair, H. Chung and U. Basu, Nat. Rev. Mol. Cell Biol., 2020, 21, 123–136 CrossRef CAS PubMed.
- T. Nojima and N. J. Proudfoot, Nat. Rev. Mol. Cell Biol., 2022, 23, 389–406 CrossRef CAS PubMed.
- L. A. Passmore and J. Coller, Nat. Rev. Mol. Cell Biol., 2022, 23, 93–106 CrossRef CAS PubMed.
- E. H. Pilkington, E. J. Suys, N. L. Trevaskis, A. K. Wheatley, D. Zukancic, A. Algarni, H. Al-Wassiti, T. P. Davis, C. W. Pouton and S. J. Kent, Acta Biomater., 2021, 131, 16–40 CrossRef CAS PubMed.
- K. Paunovska, D. Loughrey and J. E. Dahlman, Nat. Rev. Genet., 2022, 23, 265–280 CrossRef CAS PubMed.
- Y. Li, Z. Ye, H. Yang and Q. Xu, Acta Pharm. Sin. B, 2022, 12(6), 2624–2639 CrossRef CAS PubMed.
- A. Akinc, M. A. Maier, M. Manoharan, K. Fitzgerald, M. Jayaraman, S. Barros, S. Ansell, X. Du, M. J. Hope, T. D. Madden, B. L. Mui, S. C. Semple, Y. K. Tam, M. Ciufolini, D. Witzigmann, J. A. Kulkarni, R. van der Meel and P. R. Cullis, Nat. Nanotechnol., 2019, 14, 1084–1087 CrossRef CAS PubMed.
- A. Akinc, M. A. Maier, M. Manoharan, K. Fitzgerald, M. Jayaraman, S. Barros, S. Ansell, X. Du, M. J. Hope and T. D. Madden, Nat. Nanotechnol., 2019, 14, 1084–1087 CrossRef CAS PubMed.
- A.-M. Yu and M.-J. Tu, Pharmacol. Ther., 2022, 230, 107967 CrossRef CAS PubMed.
- M. A. Subhan and V. Torchilin, Transl. Res., 2019, 214, 62–91 CrossRef CAS PubMed.
- Y. Xiao, Z. Tang, X. Huang, W. Chen, J. Zhou, H. Liu, C. Liu, N. Kong and W. Tao, Chem. Soc. Rev., 2022, 51, 3828–3845 RSC.
- M. S. Bami, M. A. R. Estabragh, P. Khazaeli, M. Ohadi and G. Dehghannoudeh, J. Drug Delivery Sci. Technol., 2022, 70, 102987 CrossRef CAS.
- S. Hatziantoniou, H. C. Maltezou, A. Tsakris, G. A. Poland and C. Anastassopoulou, Vaccine, 2021, 39, 2605 CrossRef CAS PubMed.
- M. Papi, D. Pozzi, V. Palmieri and G. Caracciolo, Nano Today, 2022, 43, 101403 CrossRef CAS PubMed.
- A. J. Barbier, A. Y. Jiang, P. Zhang, R. Wooster and D. G. Anderson, Nat. Biotechnol., 2022, 40, 840–854 CrossRef CAS PubMed.
- E. Samaridou, J. Heyes and P. Lutwyche, Adv. Drug Delivery Rev., 2020, 154, 37–63 CrossRef PubMed.
- M. Qiu, Z. Glass, J. Chen, M. Haas, X. Jin, X. Zhao, X. Rui, Z. Ye, Y. Li and F. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2020401118 CrossRef CAS PubMed.
- X. Xu, C. Liu, Y. Wang, O. Koivisto, J. Zhou, Y. Shu and H. Zhang, Adv. Drug Delivery Rev., 2021, 176, 113891 CrossRef CAS PubMed.
- R. Tenchov, R. Bird, A. E. Curtze and Q. Zhou, ACS Nano, 2021, 15, 16982–17015 CrossRef CAS PubMed.
- Y. Yan, X.-Y. Liu, A. Lu, X.-Y. Wang, L.-X. Jiang and J.-C. Wang, J. Controlled Release, 2022, 342, 241–279 CrossRef CAS PubMed.
- L. Schoenmaker, D. Witzigmann, J. A. Kulkarni, R. Verbeke, G. Kersten, W. Jiskoot and D. J. Crommelin, Int. J. Pharm., 2021, 601, 120586 CrossRef CAS PubMed.
- M. Karam and G. Daoud, Asian J. Pharm. Sci., 2022, 17, 491–522 CrossRef PubMed.
- M. Schlich, R. Palomba, G. Costabile, S. Mizrahy, M. Pannuzzo, D. Peer and P. Decuzzi, Bioeng. Transl. Med., 2021, 6, e10213 CAS.
- L. Xu, X. Wang, Y. Liu, G. Yang, R. J. Falconer and C.-X. Zhao, Adv. NanoBiomed Res., 2022, 2, 2100109 CrossRef CAS.
- K. Hashiba, Y. Sato, M. Taguchi, S. Sakamoto, A. Otsu, Y. Maeda, T. Shishido, M. Murakawa, A. Okazaki and H. Harashima, Small Sci., 2023, 3, 2200071 CrossRef CAS.
- R. Verbeke, M. J. Hogan, K. Loré and N. Pardi, Immunity, 2022, 55, 1993–2005 CrossRef CAS PubMed.
- E. Xu, W. M. Saltzman and A. S. Piotrowski-Daspit, J. Controlled Release, 2021, 335, 465–480 CrossRef CAS PubMed.
- L. Zhang, L. Yang, J. Huang, S. Chen, C. Huang, Y. Lin, A. Shen, Z. Zheng, W. Zheng and S. Tang, Asian J. Pharm. Sci., 2022, 17, 666–678 CrossRef PubMed.
- Y. Zhang, C. Sun, C. Wang, K. E. Jankovic and Y. Dong, Chem. Rev., 2021, 121, 12181–12277 CrossRef CAS PubMed.
- S. Cui, Y. Wang, Y. Gong, X. Lin, Y. Zhao, D. Zhi, Q. Zhou and S. Zhang, Toxicol. Res., 2018, 7, 473–479 CrossRef CAS PubMed.
- F. Sebastiani, M. Yanez Arteta, M. Lerche, L. Porcar, C. Lang, R. A. Bragg, C. S. Elmore, V. R. Krishnamurthy, R. A. Russell, T. Darwish, H. Pichler, S. Waldie, M. Moulin, M. Haertlein, V. T. Forsyth, L. Lindfors and M. Cárdenas, ACS Nano, 2021, 15, 6709–6722 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, M. Foy, T. Berggård, S. C. Donnelly, G. Cagney, S. Linse and K. A. Dawson, Angew. Chem., Int. Ed., 2007, 46, 5754–5756 CrossRef CAS PubMed.
- D. Chen, S. Ganesh, W. Wang and M. Amiji, Nanoscale, 2019, 11, 8760–8775 RSC.
- V. Francia, R. M. Schiffelers, P. R. Cullis and D. Witzigmann, Bioconjugate Chem., 2020, 31, 2046–2059 CrossRef CAS PubMed.
- Q. Cheng, T. Wei, L. Farbiak, L. T. Johnson, S. A. Dilliard and D. J. Siegwart, Nat. Nanotechnol., 2020, 15, 313–320 CrossRef CAS PubMed.
- S. A. Dilliard, Q. Cheng and D. J. Siegwart, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2109256118 CrossRef CAS PubMed.
- I. Koltover, T. Salditt, J. O. Rädler and C. R. Safinya, Science, 1998, 281, 78–81 CrossRef CAS PubMed.
- Z. u Rehman, D. Hoekstra and I. S. Zuhorn, ACS Nano, 2013, 7, 3767–3777 CrossRef PubMed.
- N. Shobaki, Y. Sato and H. Harashima, Int. J. Nanomed., 2018, 13, 8395–8410 CrossRef CAS PubMed.
- M. A. Maier, M. Jayaraman, S. Matsuda, J. Liu, S. Barros, W. Querbes, Y. K. Tam, S. M. Ansell, V. Kumar, J. Qin, X. Zhang, Q. Wang, S. Panesar, R. Hutabarat, M. Carioto, J. Hettinger, P. Kandasamy, D. Butler, K. G. Rajeev, B. Pang, K. Charisse, K. Fitzgerald, B. L. Mui, X. Du, P. Cullis, T. D. Madden, M. J. Hope, M. Manoharan and A. Akinc, Mol. Ther., 2013, 21, 1570–1578 CrossRef CAS PubMed.
- M. Jayaraman, S. M. Ansell, B. L. Mui, Y. K. Tam, J. Chen, X. Du, D. Butler, L. Eltepu, S. Matsuda and J. K. Narayanannair, Angew. Chem., Int. Ed., 2012, 124, 8657–8661 CrossRef.
- M. J. Carrasco, S. Alishetty, M.-G. Alameh, H. Said, L. Wright, M. Paige, O. Soliman, D. Weissman, T. E. Cleveland, A. Grishaev and M. D. Buschmann, Commun. Biol., 2021, 4, 956 CrossRef CAS PubMed.
- R. Verbeke, I. Lentacker, S. C. De Smedt and H. Dewitte, J. Controlled Release, 2021, 333, 511–520 CrossRef CAS PubMed.
- M. D. Buschmann, M. J. Carrasco, S. Alishetty, M. Paige, M. G. Alameh and D. Weissman, Vaccines, 2021, 9, 65 CrossRef CAS PubMed.
- D. Zhi, S. Zhang, S. Cui, Y. Zhao, Y. Wang and D. Zhao, Bioconjugate Chem., 2013, 24, 487–519 CrossRef CAS PubMed.
- Y. Eygeris, M. Gupta, J. Kim and G. Sahay, Acc. Chem. Res., 2022, 55, 2–12 CrossRef CAS PubMed.
- C. L. Walsh, J. Nguyen, M. R. Tiffany and F. C. Szoka, Bioconjugate Chem., 2013, 24, 36–43 CrossRef CAS PubMed.
- M. Cornebise, E. Narayanan, Y. Xia, E. Acosta, L. Ci, H. Koch, J. Milton, S. Sabnis, T. Salerno and K. E. Benenato, Adv. Funct. Mater., 2022, 32, 2106727 CrossRef CAS.
- Y. Sato, K. Hashiba, K. Sasaki, M. Maeki, M. Tokeshi and H. Harashima, J. Controlled Release, 2019, 295, 140–152 CrossRef CAS PubMed.
- Y. Dong, D. J. Siegwart and D. G. Anderson, Adv. Drug Delivery Rev., 2019, 144, 133–147 CrossRef CAS PubMed.
- P. T. Bremer and K. D. Janda, Pharmacol. Rev., 2017, 69, 298–315 CrossRef CAS PubMed.
- G. R. Rettig and M. A. Behlke, Mol. Ther., 2012, 20, 483–512 CrossRef CAS PubMed.
- M. Wang, J. A. Zuris, F. Meng, H. Rees, S. Sun, P. Deng, Y. Han, X. Gao, D. Pouli, Q. Wu, I. Georgakoudi, D. R. Liu and Q. Xu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2868–2873 CrossRef CAS PubMed.
- X. Hou, T. Zaks, R. Langer and Y. Dong, Nat. Rev. Mater., 2021, 6, 1078–1094 CrossRef CAS PubMed.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef CAS PubMed.
- O. S. Thomas and W. Weber, Front. Bioeng. Biotechnol., 2019, 7, 415 CrossRef PubMed.
- S. C. Semple, A. Akinc, J. Chen, A. P. Sandhu, B. L. Mui, C. K. Cho, D. W. Y. Sah, D. Stebbing, E. J. Crosley, E. Yaworski, I. M. Hafez, J. R. Dorkin, J. Qin, K. Lam, K. G. Rajeev, K. F. Wong, L. B. Jeffs, L. Nechev, M. L. Eisenhardt, M. Jayaraman, M. Kazem, M. A. Maier, M. Srinivasulu, M. J. Weinstein, Q. Chen, R. Alvarez, S. A. Barros, S. De, S. K. Klimuk, T. Borland, V. Kosovrasti, W. L. Cantley, Y. K. Tam, M. Manoharan, M. A. Ciufolini, M. A. Tracy, A. de Fougerolles, I. MacLachlan, P. R. Cullis, T. D. Madden and M. J. Hope, Nat. Biotechnol., 2010, 28, 172–176 CrossRef CAS PubMed.
- H. Tanaka, T. Takahashi, M. Konishi, N. Takata, M. Gomi, D. Shirane, R. Miyama, S. Hagiwara, Y. Yamasaki, Y. Sakurai, K. Ueda, K. Higashi, K. Moribe, E. Shinsho, R. Nishida, K. Fukuzawa, E. Yonemochi, K. Okuwaki, Y. Mochizuki, Y. Nakai, K. Tange, H. Yoshioka, S. Tamagawa and H. Akita, Adv. Funct. Mater., 2020, 30, 1910575 CrossRef CAS.
- X. Zhang, W. Zhao, G. N. Nguyen, C. Zhang, C. Zeng, J. Yan, S. Du, X. Hou, W. Li, J. Jiang, B. Deng, D. W. McComb, R. Dorkin, A. Shah, L. Barrera, F. Gregoire, M. Singh, D. Chen, D. E. Sabatino and Y. Dong, Sci. Adv., 2020, 6, eabc2315 CrossRef CAS PubMed.
- X. Huang, E. Kon, X. Han, X. Zhang, N. Kong, M. J. Mitchell, D. Peer and W. Tao, Nat. Nanotechnol., 2022, 17, 1027–1037 CrossRef CAS PubMed.
- S. M. Moghimi, Mol. Ther., 2021, 29, 898–900 CrossRef CAS PubMed.
- K. J. Hassett, K. E. Benenato, E. Jacquinet, A. Lee, A. Woods, O. Yuzhakov, S. Himansu, J. Deterling, B. M. Geilich, T. Ketova, C. Mihai, A. Lynn, I. McFadyen, M. J. Moore, J. J. Senn, M. G. Stanton, Ö. Almarsson, G. Ciaramella and L. A. Brito, Mol. Ther.-Nucleic Acids, 2019, 15, 1–11 CrossRef CAS PubMed.
- F. Liang, G. Lindgren, A. Lin, E. A. Thompson, S. Ols, J. Röhss, S. John, K. Hassett, O. Yuzhakov and K. Bahl, Mol. Ther., 2017, 25, 2635–2647 CrossRef CAS PubMed.
- Y. Dong, K. T. Love, J. R. Dorkin, S. Sirirungruang, Y. Zhang, D. Chen, R. L. Bogorad, H. Yin, Y. Chen, A. J. Vegas, C. A. Alabi, G. Sahay, K. T. Olejnik, W. Wang, A. Schroeder, A. K. R. Lytton-Jean, D. J. Siegwart, A. Akinc, C. Barnes, S. A. Barros, M. Carioto, K. Fitzgerald, J. Hettinger, V. Kumar, T. I. Novobrantseva, J. Qin, W. Querbes, V. Koteliansky, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3955–3960 CrossRef CAS PubMed.
- O. S. Fenton, K. J. Kauffman, R. L. McClellan, J. C. Kaczmarek, M. D. Zeng, J. L. Andresen, L. H. Rhym, M. W. Heartlein, F. DeRosa and D. G. Anderson, Angew. Chem., Int. Ed., 2018, 57, 13582–13586 CrossRef CAS PubMed.
- K. A. Hajj, R. L. Ball, S. B. Deluty, S. R. Singh, D. Strelkova, C. M. Knapp and K. A. Whitehead, Small, 2019, 15, 1805097 CrossRef PubMed.
- M. F. W. Trollmann and R. A. Böckmann, Biophys. J., 2022, 121, 3927–3939 CrossRef CAS PubMed.
- C. Hald Albertsen, J. A. Kulkarni, D. Witzigmann, M. Lind, K. Petersson and J. B. Simonsen, Adv. Drug Delivery Rev., 2022, 188, 114416 CrossRef CAS PubMed.
- F. Ding, H. Zhang, J. Cui, Q. Li and C. Yang, Biomater. Sci., 2021, 9, 7534–7546 RSC.
- B. Li, R. S. Manan, S.-Q. Liang, A. Gordon, A. Jiang, A. Varley, G. Gao, R. Langer, W. Xue and D. Anderson, Nat. Biotechnol., 2023 DOI:10.1038/s41587-023-01679-x.
- L. Yan, D. Ulkoski and T. Puri, Tetrahedron Lett., 2022, 105, 154057 CrossRef CAS.
- M. M. Billingsley, N. Singh, P. Ravikumar, R. Zhang, C. H. June and M. J. Mitchell, Nano Lett., 2020, 20, 1578–1589 CrossRef CAS PubMed.
- M. R. Molla, A. Böser, A. Rana, K. Schwarz and P. A. Levkin, Bioconjugate Chem., 2018, 29, 992–999 CrossRef CAS PubMed.
- H. Zhang, X. You, X. Wang, L. Cui, Z. Wang, F. Xu, M. Li, Z. Yang, J. Liu, P. Huang, Y. Kang, J. Wu and X. Xia, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2005191118 CrossRef CAS PubMed.
- B. Li, X. Luo, B. Deng, J. Wang, D. W. McComb, Y. Shi, K. M. L. Gaensler, X. Tan, A. L. Dunn, B. A. Kerlin and Y. Dong, Nano Lett., 2015, 15, 8099–8107 CrossRef CAS PubMed.
- L. Miao, L. Li, Y. Huang, D. Delcassian, J. Chahal, J. Han, Y. Shi, K. Sadtler, W. Gao, J. Lin, J. C. Doloff, R. Langer and D. G. Anderson, Nat. Biotechnol., 2019, 37, 1174–1185 CrossRef CAS PubMed.
- J. C. Flores-Reyes, A. Islas-Jácome and E. González-Zamora, Org. Chem. Front., 2021, 8, 5460–5515 RSC.
- N. P. Tripolitsiotis, M. Thomaidi and C. G. Neochoritis, Eur. J. Org. Chem., 2020, 6525–6554 CrossRef.
- Y.-M. Yan, Y. Rao and M.-W. Ding, J. Org. Chem., 2017, 82, 2772–2776 CrossRef CAS PubMed.
- C. Foley, A. Shaw and C. Hulme, Org. Lett., 2018, 20, 1275–1278 CrossRef CAS PubMed.
- P. Stiernet, P. Lecomte, J. De Winter and A. Debuigne, ACS Macro Lett., 2019, 8, 427–434 CrossRef CAS PubMed.
- R. Butera, A. Shrinidhi, K. Kurpiewska, J. Kalinowska-Tłuścik and A. Dömling, Chem. Commun., 2020, 56, 10662–10665 RSC.
- C. Russo, G. Graziani, R. Cannalire, G. C. Tron and M. Giustiniano, Green Chem., 2022, 24, 3993–4003 RSC.
- P. Brandão, C. Marques, A. J. Burke and M. Pineiro, Eur. J. Med. Chem., 2021, 211, 113102 CrossRef PubMed.
- Z. Li, X.-Q. Zhang, W. Ho, F. Li, M. Gao, X. Bai and X. Xu, ACS Nano, 2022, 16, 18936–18950 CrossRef CAS PubMed.
- X. Han, H. Zhang, K. Butowska, K. L. Swingle, M. G. Alameh, D. Weissman and M. J. Mitchell, Nat. Commun., 2021, 12, 7233 CrossRef CAS PubMed.
- A. M. Reichmuth, M. A. Oberli, A. Jaklenec, R. Langer and D. Blankschtein, Ther. Delivery, 2016, 7, 319–334 CrossRef CAS PubMed.
- A. T. He, J. Liu, F. Li and B. B. Yang, Signal Transduction Targeted Ther., 2021, 6, 185 CrossRef CAS PubMed.
- T. Seimiya, M. Otsuka, T. Iwata, C. Shibata, E. Tanaka, T. Suzuki and K. Koike, Front. Cell Dev. Biol., 2020, 8, 568366 CrossRef PubMed.
- Y. Xiao and J. Shi, Chem. Rev., 2021, 121, 12109–12111 CrossRef CAS PubMed.
- R. L. Ball, K. A. Hajj, J. Vizelman, P. Bajaj and K. A. Whitehead, Nano Lett., 2018, 18, 3814–3822 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS.
- R. L. Setten, J. J. Rossi and S.-P. Han, Nat. Rev. Drug Discovery, 2019, 18, 421–446 CrossRef CAS PubMed.
- T. S. Zatsepin, Y. V. Kotelevtsev and V. Koteliansky, Int. J. Nanomed., 2016, 11, 3077 CrossRef CAS PubMed.
- W. Chen, Y. Hu and D. Ju, Acta Pharm. Sin. B, 2020, 10, 1347–1359 CrossRef CAS.
- S. Ingusci, G. Verlengia, M. Soukupova, S. Zucchini and M. Simonato, Front. Pharmacol., 2019, 10, 724 CrossRef CAS.
Footnote |
| † Yue Xu, Alex Golubovic, Shufen Xu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |