
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and evaluation of poly(propylene fumarate)-grafted graphene oxide as nanofiller for porous scaffolds†
Andreea M.
Pandele
ab,
Aida
Selaru
c,
Sorina
Dinescu
c,
Marieta
Costache
c,
Eugeniu
Vasile
d,
Constanţa
Dascălu
e,
Matei D.
Raicopol
 *f and
Mircea
Teodorescu
g
*f and
Mircea
Teodorescu
g
aAdvanced Polymer Materials Group, University Politehnica of Bucharest, 1-7 Gheorghe Polizu St., 011061, Bucharest, Romania
bDepartment of Analytical Chemistry and Environmental Engineering, University Politehnica of Bucharest, 1-7 Gheorghe Polizu St., 011061, Bucharest, Romania
cDepartment of Biochemistry and Molecular Biology, University of Bucharest, 91-95 Splaiul Independentei, 050095, Bucharest, Romania
dDepartment of Science and Engineering of Oxide Materials and Nanomaterials, University Politehnica of Bucharest, 1-7 Gheorghe Polizu St., 011061, Bucharest, Romania
eDepartment of Physics, University Politehnica of Bucharest, 313 Splaiul Independenţei, 060042, Bucharest, Romania
f“Costin Nenitzescu” Department of Organic Chemistry, University Politehnica of Bucharest, 1-7 Gheorghe Polizu St., 011061, Bucharest, Romania. E-mail: m_raicopol@chim.upb.ro; Tel: +40214023912
gDepartment of Bioresources and Polymer Science, University Politehnica of Bucharest, 1-7 Gheorghe Polizu St., 011061, Bucharest, Romania
First published on 7th August 2023
Abstract
In an effort to obtain porous scaffolds with improved mechanical properties and biocompatibility, the current study discusses nanocomposite materials based on poly(propylene fumarate)/N-vinyl pyrrolidone(PPF/NVP) networks reinforced with polymer-modified graphene oxide (GO@PPF). The GO@PPF nanofiller was synthesized through a facile and convenient surface esterification reaction, and the successful functionalization was demonstrated by complementary techniques such as FT-IR, XPS, TGA and TEM. The PPF/NVP/GO@PPF porous scaffolds obtained using NaCl as a porogen were further characterized in terms of morphology, mechanical properties, sol fraction, and in vitro degradability. SEM and nanoCT examinations of NaCl-leached samples revealed networks of interconnected pores, fairly uniform in size and shape. We show that the incorporation of GO@PPF in the polymer matrix leads to a significant enhancement in the mechanical properties, which we attribute to the formation of denser and more homogenous networks, as suggested by a decreased sol fraction for the scaffolds containing a higher amount of GO@PPF. Moreover, the surface of mineralized PPF/NVP/GO@PPG scaffolds is uniformly covered in hydroxyapatite-like crystals having a morphology and Ca/P ratio similar to bone tissue. Furthermore, the preliminary biocompatibility assessment revealed a good interaction between PPF/PVP/GO@PPF scaffolds and murine pre-osteoblasts in terms of cell viability and proliferation.
1. Introduction
Recent advances in nanotechnology and polymer chemistry enabled the development of many advanced scaffolds for tissue engineering and regenerative medicine. Therefore, a variety of materials including hydrogels, porous nanocomposites and nanofibers have been investigated as potential candidates for bone repair.1 Several aspects should be considered when designing scaffolds for tissue engineering applications, including mechanical properties, biodegradation rate, and biocompatibility in order to allow cell adhesion, differentiation and proliferation.2 Moreover, a key morphological feature of tissue engineering scaffolds is the network of interconnected pores, necessary for cellular infiltration and transport of nutrients and metabolic waste.3,4 To date, various approaches were considered for obtaining porous scaffolds: 3D printing,5–9 magnetically-guided assembly on 3D-printed poly(ε-caprolactone) containing bioresorbable Fe-doped hydroxyapatite,10 thermally induced phase separation,2 salt leaching11 and gas foaming.4One of the most promising polymers employed in biodegradable scaffolds for guided bone regeneration is poly(propylene fumarate) (PPF),12,13 an amorphous, unsaturated linear polyester which contains double bonds along its backbone that allow covalent crosslinking. PPF is generally considered biocompatible and biodegradable to nontoxic products following non-enzymatic hydrolysis.14
Unfortunately, the poor mechanical properties of neat PPF make it unsuitable for load-bearing orthopedic applications. To address the requirements of bone tissue engineering, various nanofillers can be incorporated into the PPF matrix. For example, graphene oxide (GO) was employed in numerous studies.15 Because a homogeneous dispersion of the nanofiller combined with improved compatibility and adhesion with the polymer matrix are prerequisites for obtaining high performance composites, polymer-grafted GO obtained via both covalent and non-covalent approaches can provide a suitable solution as it allows fine-tuning the properties of the biomaterials. To date, various approaches were used for the covalent functionalization of GO with polymer chains:16 reversible addition–fragmentation chain transfer,17 atom transfer radical polymerization,18 nitroxide-mediated polymerization,19 radical polymerization,20 and ring-opening polymerization.21 However, grafting from methods are unsuitable for surface functionalization with condensation polymers such as PPF.
Our group recently developed a series of PPF-based hybrid materials containing GO covalently modified with 2-hydroxyethyl methacrylate (HEMA), which showed significantly improved mechanical properties compared to neat PPF. Furthermore, the hybrid materials induced mineralization and enhanced the metabolic activity and proliferation rate of pre-osteoblasts without displaying cytotoxicity.22 In the current study we extend our previous results towards the synthesis of porous composite scaffolds whose structure is more appropriate for bone tissue engineering. In contrast with our previous paper, a polymer-grafted graphene oxide nanofiller is employed here, in order to improve its dispersability and increase the number of surface double bonds available for crosslinking with the matrix. Furthermore, a new crosslinking agent, i.e. N-vinyl pyrrolidone, was employed instead of poly(ethylene glycol) dimethacrylate, in an attempt to overcome recent concerns regarding the immunogenicity of poly(ethylene glycol).23,24
2. Materials and methods
Carboxylated graphene oxide (GO–COOH) with 0.7 mmol –COOH equivalents per g was purchased from NanoInnova. Acetone, tetrahydrofuran (THF), anhydrous N,N-dimethylformamide (DMF), 1,1′-carbonyldiimidazole (CDI), 1,8-diazabicyclo[5.4.0]-undec-7-ene (DBU), 1-vinyl-2-pyrrolidone (NVP), benzoyl peroxide (BP), N,N-dimethyl-p-toluidine (DMT) and bromine were purchased from Sigma-Aldrich and used without further purification. Tris(hydroxymethyl)aminomethane (Tris), NaCl, KCl, MgCl2, NaHCO3, CaCl2, KH2PO4, Na2HPO4 and 37% HCl were purchased from Sigma-Aldrich or Merck and used as received.Poly(propylene fumarate) with Mn = 1400 g![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mol−1 and PDI = 1.85 was synthesized following a literature protocol,25 as described in detail in our previous publication.22
mol−1 and PDI = 1.85 was synthesized following a literature protocol,25 as described in detail in our previous publication.22
2.1. Synthesis of GO@PPF
In a 500 mL two-neck round-bottom flask containing 250 mg of GO–COOH were added 250 mL of anhydrous DMF. The flask was flushed with argon and then sonicated in an ultrasonic bathf or 4 hours at 20–25 °C. Next, 1 g of CDI was added and the resulting mixture was sonicated again for 4 hours at 20–25 °C. The suspension was transferred under argon to a stirred pressure filtration cell (Millipore XFUF04701) fitted with a 0.2 μm Teflon membrane. The precipitate was further washed with anhydrous DMF (3 × 25 mL), resuspended in 250 mL of anhydrous DMF and transferred under argon back into the round-bottom flask. The suspension was sonicated at 20–25 °C for 4 hours. Then 3 g of PPF and 1.9 mL of DBU catalyst were added, a magnetic follower was placed in the flask and the reaction mixture was stirred at room temperature. After 24 hours, the suspension was transferred again into the pressure filtration cell and the functionalized graphene was filtered through a 0.2 μm Teflon membrane. The precipitate was washed with 2 × 25 mL DMF followed by 2 × 25 mL acetone and finally dried at room temperature under vacuum (5 mmHg, 24 h).2.2. Fabrication of hybrid porous scaffolds
To 1 g of PPF/NVP monomers in 2.5:1 (w:w) ratio was added the required amount of GO@PPF nanofiller in order to obtain the desired concentration (0, 0.25, 0.5, 1 wt%), followed by 2.5 μL DMT accelerator, and the resulting mixtures were ultrasonicated for 30 min. Then, 4 g of NaCl porogen (200–500 μm crystal size) were added and mixed thoroughly for 10 min, followed by 1 wt% BP radical initiator. The samples were stirred rapidly for 10 s, poured into cylindrical polyethylene vials, and then cured under vacuum (10 mmHg) at 60 °C for 20 hours. After curing, the samples were leached for 3 days in distilled water, which was changed daily. Finally, the scaffolds were dried under vacuum (5 mmHg) for 24 h.
2.3. Gas-phase chemical derivatization for XPS
Samples of ∼2.5 mg GO–COOH and GO@PPF placed in small glass vials were inserted in larger glass tubes fitted with Teflon stoppers. Bromine (∼0.1 mL) was carefully added in the outer tubes. After 24 h at room temperature, the inner vials were taken out and placed under vacuum (5 mmHg) for 24 h.262.4. Characterization
ATR-FTIR spectra were recorded on a VERTEX 70 spectrometer (Bruker) in the 4000–600 cm−1 range, at a resolution of 4 cm−1. Thermogravimetric analysis (TGA) was performed with a Q500 thermogravimetric analyzer (TA Instruments), by heating the samples under a nitrogen atmosphere from room temperature to 900 °C, at a rate of 10 °Cmin−1. Scanning electron microscopy (SEM) investigations were carried out using a Quanta Inspect F (FEI) field emission scanning electron microscope equipped with an energy dispersive X-ray spectroscopy (EDAX) system. Transmission electron micrsocopy (TEM) images were recorded on a Tecnai G2 F30 S-Twin (FEI) high resolution electron microscope. Small pieces of the synthesized materials were embedded in epoxy resin and, after complete curing, thin sections were cut with an ultramicrotome and transferred to copper grids covered with holey carbon films. Nano computed tomography (nanoCT) was performed with a SkyScan 2727 system (Bruker), on cylindrical specimens (∼6 mm diameter and ∼7 mm length). Image acquisition was performed at a resolution (pixel size) of 100 nm, a rotation step of 0.2°, and 7 average frames per capture. The Nrecon 1.7.1.6 software package (Bruker) was used to reconstruct the raw images. The total porosity and structure thickness were quantified using the CTAn analysis software (Bruker). For that, the 3D reconstructed images were subjected to a sequence of steps including the selection of a region of interest, image binarization after thresholding and morphometric 3D analysis. After thresholding, the binarized images lack intermediary grey tones: the scaffold material appears white, and the pores (empty space) appear black. The width of the pore walls is defined by the structure thickness, i.e. the product between the number of white pixels and the scanning resolution (pixel size). X-ray photoelectron spectra were acquired on a K-Alpha XPS spectrometer (Thermo Scientific), fitted with a monochromatic Al Kα source (1486.6 eV). Surface charging was compensated by a flood gun and binding energies were referenced to the C1s peak which was set at 284.6 eV. The pass energy for the survey spectra and high-resolution spectra were 200 eV and 20 eV, respectively. The deconvolution of core-level spectra was performed with mixed Gaussian–Lorentzian functions after subtracting a Shirley background. Compressive mechanical testing was performed in accordance to European Standard EN ISO 604:2003, using a Model 3382 universal testing machine (Instron), at an ambient relative humidity of 45–50%. Cylindrical specimens (∼6 mm diameter and 12 mm length) were compressed along their long axis until failure, at a crosshead speed of 1 mmmin−1. Young's Modulus was calculated from the initial slope of the stress–strain curve and the compressive strength was calculated as the intersection of the stress–strain curve with a line drawn parallel to the initial slope, which begins at 1% strain. Five specimens were tested for each scaffold formulation, and results are reported as average ± standard deviation.
The sol fraction (SF) in toluene was determined gravimetrically, as described in detail in ref. 22 The following equation was used to calculate the sol fraction:
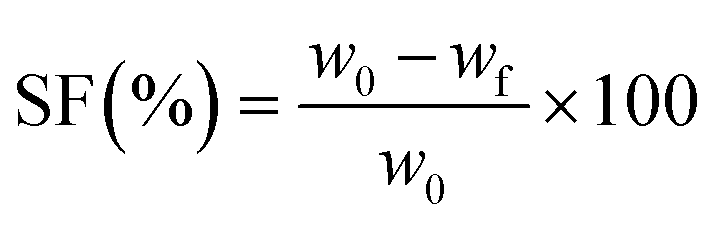
The non-enzymatic degradation of the scaffolds was conducted by keeping the samples for 30 weeks in PBS solution, at 37 °C. Dry samples (∼0.1 g) were weighed and placed into stoppered vials containing 20 mL of PBS (pH 7.4), which was changed daily during the first week and weekly afterwards. After 30 weeks, the samples were taken out and dried in a vacuum oven until constant weight (37 °C, 10 mmHg). The weight loss (WL) was calculated using the equation:
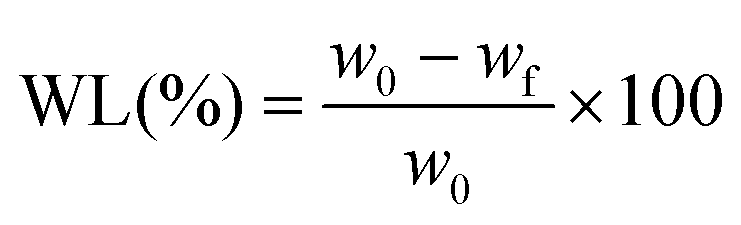
Scaffold mineralization was accomplished using the protocol developed by Taguchi et al.27 Briefly, scaffolds were cut in pieces of about 0.5 g and were soaked alternately for 24 h in 200 mM CaCl2/tris (pH adjusted to 7.4 with 0.5 N HCl) and 120 mM Na2HPO4 solutions maintained at 37 °C. The soaking cycle was repeated twice, and after two days the scaffolds were gently washed with distilled water to remove soluble salts and dried in an oven at 37 °C for 48 h. The mineralized scaffolds were further examined by SEM.
The cell-scaffold systems were obtained by seeding murine pre-osteoblasts from the MC 3T3-E1 cell line (ATCC, CRL-2593), at a density of 2 × 105 cells per cm2, on the surface of scaffolds sterilized in advance by exposure to UV light. This system was maintained for 7 days in Dulbecco's modified Eagle medium (DMEM, Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS, Life Technologies) and 1% antibiotic (Sigma-Aldrich), under standard cell culture conditions (37 °C, 5% CO2 and 95% relative humidity). The MTT assay was performed using a commercial kit (MTT, Sigma-Aldrich), by incubating the cell-scaffold systems for 4 h with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution in the dark, dissolving the formazan crystals in isopropanol and measuring the absorbance of the resulting solution at 550 nm. The cytotoxicity was evaluated using a commercial kit (TOX7, Sigma-Aldrich), the release of lactate dehydrogenase (LDH) being quantified by measuring absorbance at 490 nm. A live/dead kit (Invitrogen, Life Technologies) was employed in order to qualitatively evaluate cell viability. Scaffolds were incubated for 1 hour in the dark and at room temperature, in 500 μL of staining solution prepared according to the manufacturer's instructions. Subsequent microscopic examination was performed using a laser-scanning confocal microscope (A1R, Nikon). All biocompatibility experiments were performed in triplicate (n = 3); results expressed as average ± standard deviation (SD) were assessed using the one-way ANOVA method and Bonferroni post-test, using GraphPad Prism Software 3.0 (GraphPad Software Inc.).
3. Results
3.1. Synthesis and characterization of the polymer-grafted nanofiller (GO@PPF)
For the synthesis of polymer-grafted graphene oxide nanofiller, a facile and convenient surface esterification reaction was employed. Briefly, the –COOH groups on the surface of commercial carboxylated graphene oxide were converted to highly reactive N-acylimidazole groups by treatment with N,N′-carbonyldiimidazole in anhydrous DMF22 (Scheme 1a). In the next step, GO@PPF was synthesized by reacting, under basic catalysis, the activated intermediate with the hydroxyl end groups of PPF. In order to prevent side reactions such as crosslinking between graphene sheets, this step was performed using high dilution conditions and a large excess of polymer. Finally, as-synthesized GO@PPF was thoroughly washed with DMF and acetone in order to remove the excess of PPF. | ||
| Scheme 1 (a) Synthesis of GO@PPF: CDI – N,N′-carbonyldiimidazole; DMF – N,N-dimethylformamide; PPF – poly(propylene fumarate); DBU – 1,8-diazabicyclo[5.4.0]undec-7-ene. (b) Preparation of PPF/PVP/GO@PPF porous scaffolds. | ||
The infrared spectrum of GO@COOH (Fig. 1) displays the following characteristic bands: 1718 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1240 cm−1 (C–O stretching) and 1041 cm−1.28 The spectrum of GO@PPF displays several new bands due to the grafted polymer, which confirm the successful functionalization: 2933 cm−1 (aliphatic C–H stretching), 1725 cm−1 (α,β-unsaturated ester CO stretching), 1646 cm−1 (CC stretching), 1322 cm−1 (ester C–O stretching), 984 cm−1 (alkene C–H bending).29
O stretching), 1240 cm−1 (C–O stretching) and 1041 cm−1.28 The spectrum of GO@PPF displays several new bands due to the grafted polymer, which confirm the successful functionalization: 2933 cm−1 (aliphatic C–H stretching), 1725 cm−1 (α,β-unsaturated ester CO stretching), 1646 cm−1 (CC stretching), 1322 cm−1 (ester C–O stretching), 984 cm−1 (alkene C–H bending).29
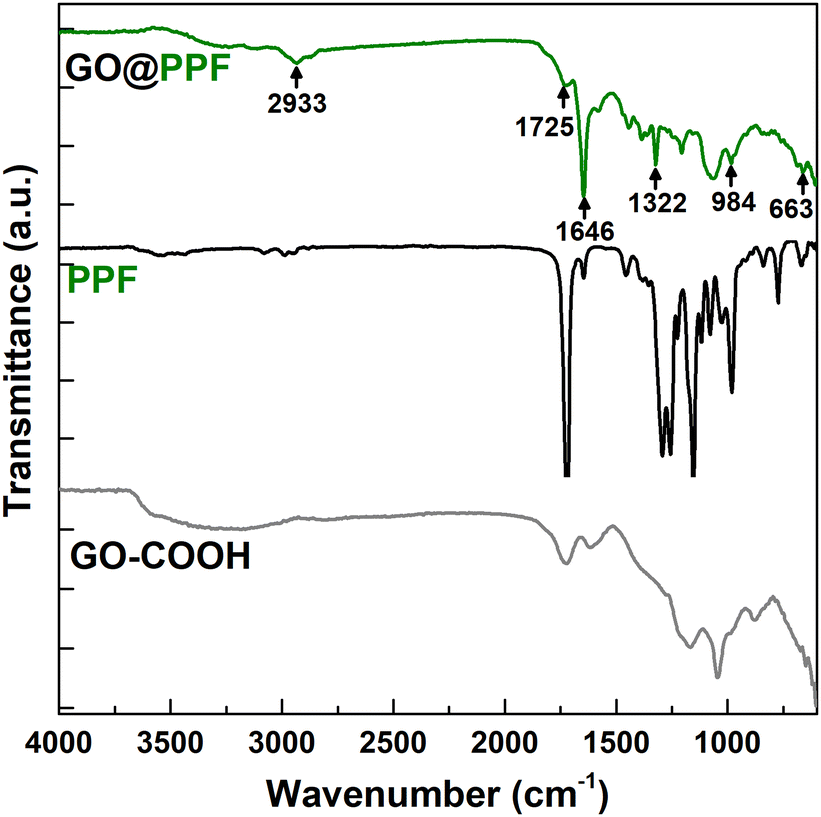 | ||
| Fig. 1 FT-IR spectra of GO@COOH, PPF, and GO@PPF. | ||
The surface composition of the polymer-grafted nanofiller was further investigated using X-ray photoelectron spectroscopy. As expected, the survey spectrum of GO@PPF (Fig. 2a) shows the C1s, O1s and N1s peaks, the presence of nitrogen indicating the presence of unreacted acylimidazole groups.22
 | ||
| Fig. 2 XPS survey spectra of GO–COOH and GO@PPF recorded before (a) and after gas-phase derivatization with bromine (b). | ||
The C1s core-level spectrum of GO@PPF (Fig. S1, ESI†) was deconvoluted into four components centered at 284.5 eV (CC), 285.2 eV (C–C), 286.5 eV (C–O) and 287.9 eV (O-CO). Similarly, two components were used for the deconvoluting the O1s core-level spectrum, at 532.5 (C–O) and 530.7 (CO), respectively.
A highly sensitive XPS method for evidencing surface CC bonds is the gas-phase derivatization with bromine.22 In our case, the atomic C/Br ratio determined from the survey spectrum of GO@PPF is almost 7.5 times lower than for GO–COOH (Fig. 2b), suggesting a significantly larger amount of bromine reacted with CC bonds from the unsaturated polyester backbone, confirming the presence of the surface-grafted polymer.
The surface functionalization was also assessed by thermal analysis. According to Fig. 3, the TGA curves corresponding to GO–COOH and GO@PPF display similar trends, with two main degradation steps: one that starts around 200 °C, assigned to the pyrolysis of labile oxygen-containing functional groups, and the second one above 600 °C, attributed to the removal of stable oxygen-containing functionalities.30 In the case of GO@PPF, a decrease in the onset degradation temperature accompanied by a significantly larger weight loss suggests the presence of surface-grafted PPF chains.31
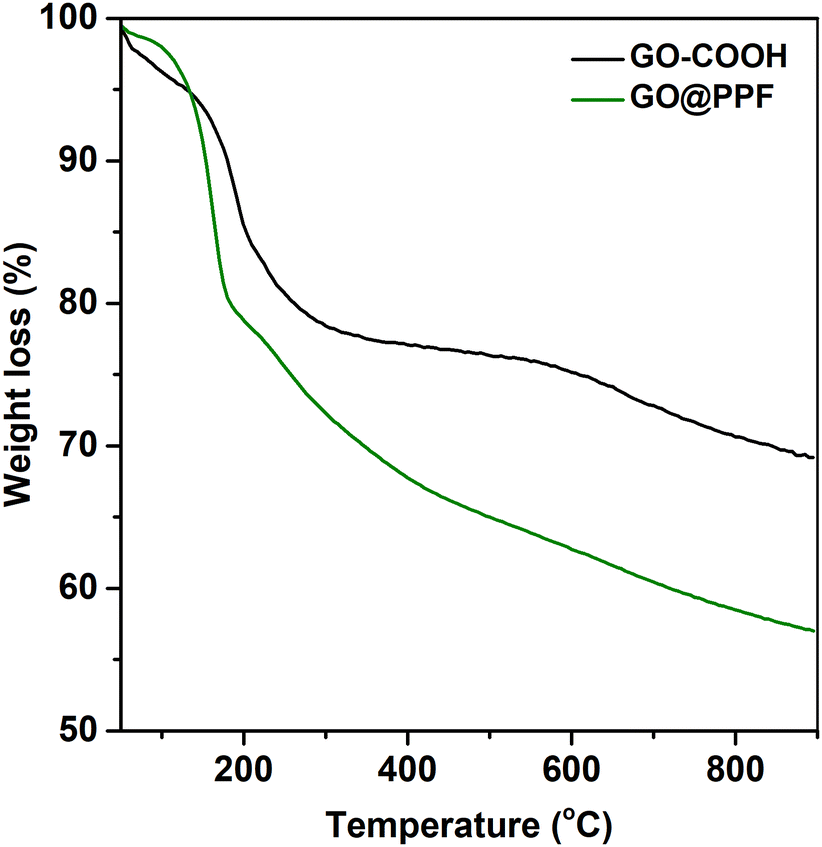 | ||
| Fig. 3 TGA curves of carboxylated GO before and after grafting with PPF. | ||
The morphology of GO@PPF was investigated by TEM, and the corresponding micrograph (Fig. S2, ESI†) exhibits thin, partially folded graphene flakes, suggesting that GO@PFF remains highly exfoliated after functionalization. Also, there is no evidence of crosslinking between GO@PPF sheets, a potential side-reaction during the surface esterification with PPF.
3.2. Fabrication and characterization of PPF/PVP/GO@PPF porous scaffolds
Porous scaffolds were fabricated by incorporating the GO@PPF nanofiller in a PPF matrix crosslinked with N-vinyl pyrrolidone (Scheme 1b).In order to achieve an optimal crosslinking density and mechanical properties of the scaffolds, a PPF:NVP weight ratio of 2.5:1 was employed for all the scaffold formulations, in accordance with results published in the literature.32,33 Based on our previous findings, the highest nanofiller concentration employed here was 1%.22 Moreover, all formulations contained 80 wt% NaCl as porogen, since it was demonstrated that lower concentrations do not lead to fully interconnected pores, and higher amounts have a detrimental effect on the mechanical properties.1,34
Since the microstructure is a key factor in tissue engineering applications, cross-sections of NaCl-leached scaffolds were first examined using SEM. The corresponding micrographs display networks of interconnected pores, fairly uniform in size and shape (Fig. 4a–d). The pore morphology was also assessed using nanoCT, and the 3D reconstruction models of the porous scaffolds also reveal networks of interconnected cubic pores (Fig. 4e–h).
 | ||
| Fig. 4 SEM micrographs (a)–(d) and reconstructed nano-CT images (e)–(h) corresponding to NaCl-leached PPF/PVP (a) and (e) and PPF/PVP/GO@PPF containing 0.25 wt% (b) and (f), 0.5 wt% (c) and (g) and 1 wt% GO@PPF (d) and (h). | ||
Furthermore, the total porosity and pore wall thickness were estimated from the nanoCT data, and the values are reported in Table 1.
| Sample | Total porosity (%) | Pore wall thickness (μm) | Sol fraction (%) | Degradation weight loss (%) |
|---|---|---|---|---|
| PPF/PVP | 72.2 ± 2.7 | 31.5 ± 1.3 | 3.4 ± 0.2 | 11.7 ± 0.5 |
| PPF/PVP/GO@PPF 0.25 wt% | 79.9 ± 0.5 | 14.1 ± 0.6 | 3.5 ± 0.4 | 11.1 ± 0.8 |
| PPF/PVP/GO@PPF 0.5 wt% | 80.3 ± 2.6 | 17.6 ± 0.8 | 0.9 ± 0.1 | 11.7 ± 0.2 |
| PPF/PVP/GO@PPF 1 wt% | 79.2 ± 1.5 | 16.7 ± 0.9 | 1.3 ± 0.2 | 11.2 ± 0.4 |
The total porosity increases slightly with increasing nanofiller concentration, from ∼72% for neat PPF/PVP to ∼80% for scaffolds containing the highest amount of GO@PPF, and concomitently the average pore wall thickness decreases, from ∼30 μm to ∼17 μm for the scaffolds with the highest concentration of GO@PPF. We tentatively attribute this to an increase in crosslinking density of the nanocomposite scaffolds, as discussed below.
The thermal stability of PPF/PVP scaffolds containing GO@PPF was investigated using TGA. As expected, the incorporation of the nanofiller enhances the thermal stability, reflected by an increase in both decomposition temperature (Td5% – temperature at 5% mass loss) and residual mass (Table S1, ESI†).
Next, the compressive mechanical properties of NaCl-leached scaffolds were evaluated. Neat PPF/PVP scaffolds display a compressive modulus of 16 ± 2 MPa and a compressive strength of 0.26 ± 0.02 MPa (Fig. 5). The compressive modulus and yield strength increase gradually with the GO@PPF concentration, being almost three times higher at 1 wt% GO@PPF. The mechanical properties of the porous scaffolds are determined by a complex interplay between the homogenous distribution and adhesion of the nanofiller, its strength and stiffness, the matrix crosslinking density and scaffold porosity. Since the porosity is similar for all the investigated samples, the reinforcing effect of GO@PPF is probably due to the high intrinsic modulus of GO35 and increased crosslinking density of scaffolds containing higher amounts of polymer-grafted nanofiller (see below). Therefore, the addition of GO@PPF nanofiller at concentrations as low as 1 wt% allows an optimal trade-off between mechanical properties and pore interconnectivity.
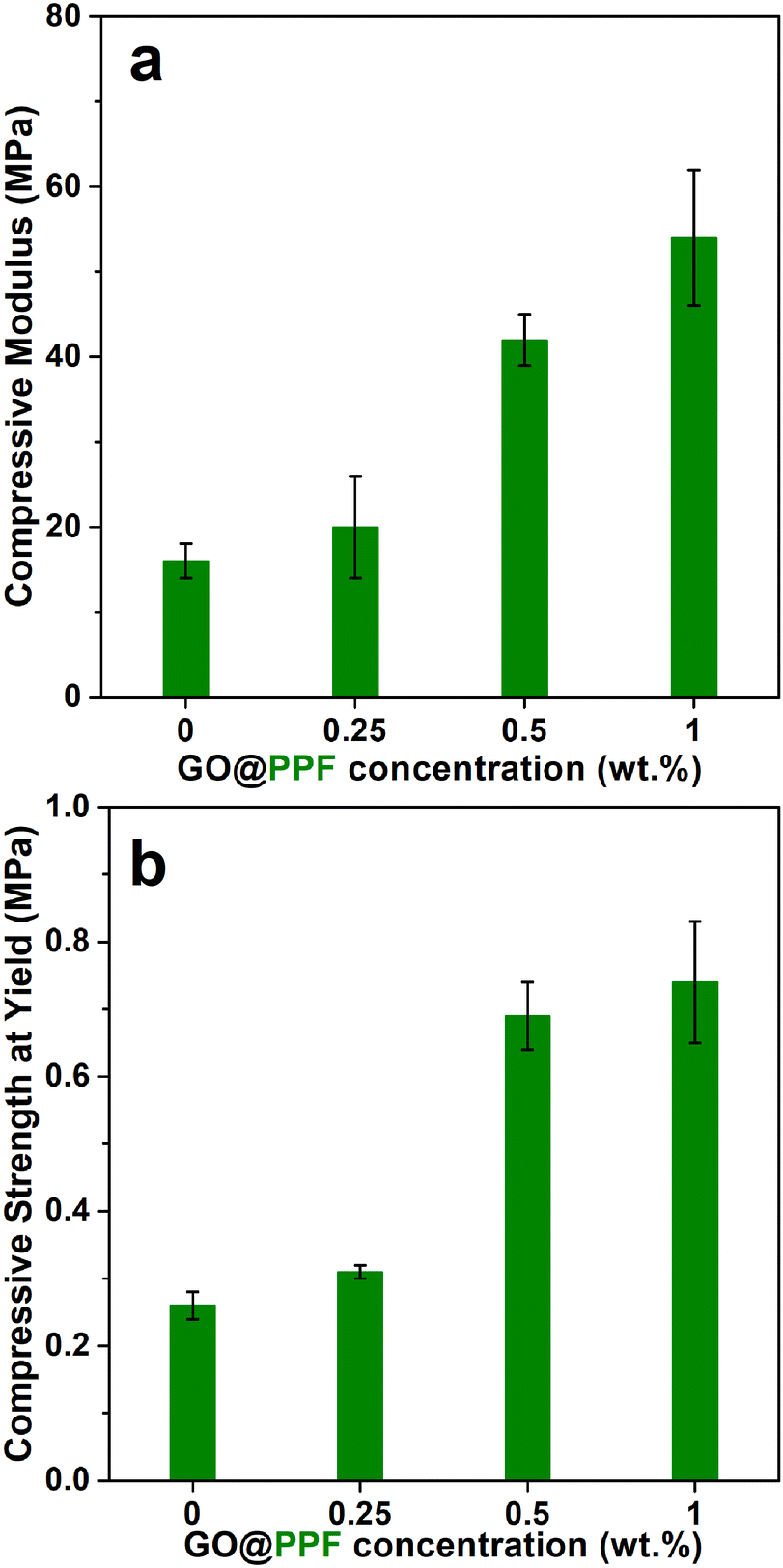 | ||
| Fig. 5 The compressive modulus (a) and compressive strength (b) of PPF/PVP/GO@PPF scaffolds. | ||
Values for the network properties of PPF/PVP scaffolds are also provided in Table 1.
A decrease in sol fraction for the scaffolds containing a higher amount of GO@PPF suggests that relative crosslinking densities increase with increasing nanofiller concentration.
A plausible explanation is that additional of grafted GO within the polymer matrix leads to denser, more homogeneous networks, as every GO@PPF sheet involved in the curing process contains additional grafting points.36
Various studies demonstrated that the degradation rate of PPF-based scaffolds is slow, and depends on both crosslinking density and molecular weight.1,37–39 In our case however, the presence of GO@PPF within the polymer matrix has no significant influence on the in vitro degradation rate, since after 30 weeks in PBS at 37 °C, all the PPF/PVP/GO@PPF scaffolds displayed a similar weight loss of about 11%.
The nucleation and growth of a biomimetic hydroxyapatite (HA) mineral phase on the scaffold surface is crucial for bone tissue engineering applications.
The in vitro mineralization of PPF/PVP/GO@PPF scaffolds was performed using an alternate soaking process,40 and the corresponding SEM images show that all the mineralized samples are uniformly covered with a continuous phase consisting of spherulitic agglomerations of plate-like crystals (Fig. 6).
 | ||
| Fig. 6 SEM micrographs (a)–(d) and EDX spectra (e)–(h) of mineralized PPF/PVP (a) and (e) and PPF/PVP/GO@PPF scaffolds containing 0.25 wt% (b) and (f), 0.5 wt% (c) and (g) and 1 wt% GO@PPF (d) and (h). | ||
The Ca/P ratios determined from EDX for neat PPF/PVP and the composite with 0.25 wt% GO@PPF are 1.32 and 1.28, respectively, indicating that HA is Ca deficient. However, the Ca/P ratio increases at higher nanofiller concentrations, reaching 1.70 at 1 wt% GO@PPF, a value which is very close to stoichiometric HA. Because the mineral phase has a composition and morphology similar to bone tissue, PPF/PVP/GO@PPF scaffolds might interact favorably with bone-forming cells.
Next, the cytotoxicity of the scaffolds was evaluated using the 3T3-E1 pre-osteoblast cell line. An MTT assay revealed that, after 3 days of culture, the cells-maintained viability in contact with PFF/PVP/GO@PPF hybrid porous materials, with no significant differences between samples (Fig. 7).
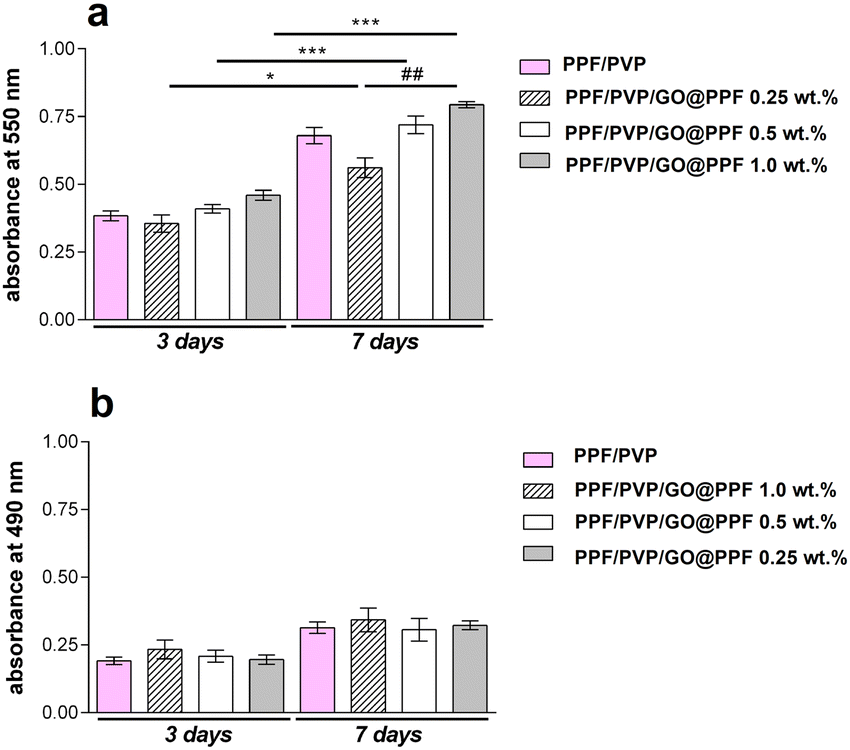 | ||
| Fig. 7 (a) 3T3-E1 viability and proliferation profile in contact with PFF/PVP/GO@PPF materials during one week of culture as revealed by MTT assay (*p < 0.05, ##p < 0.01, ***p < 0.001); (b) PFF/PVP/GO@PPF cytotoxicity evaluation by LDH assay during one week of in vitro cell culture. | ||
Interestingly, at 3 and 7 days of culture, both cell viability and proliferation rate were higher (p < 0.01 and p < 0.001) on PFF/PVP scaffolds containing 0.5 and 1 wt% GO@PPF as compared to 0.25 wt% GO@PPF, where a lower rate (p < 0.05) was registered. Overall, it was concluded that scaffolds containing GO@PPF supported murine pre-osteoblasts’ viability and proliferation during 7 days of culture.
Furthermore, an LDH release assay indicated that during one week of culture these materials had no cytotoxic effects upon the 3T3-E1 pre-osteoblast cell line. Low levels of LDH were found at 3 days post-seeding, indicating that the addition of GO@PPF up to 1 wt% does not induce cell death. Moreover, since only a minor increase in LDH levels was registered at 7 days of culture, GO@PPF doesn’t exhibit toxicity even on a longer period.
The results of MTT and LDH assays were further confirmed by a cytotoxicity assay based on fluorescent dye staining. The micrographs registered at 3 days post-seeding show small groups cells emitting green fluorescence (Fig. 8), suggesting that PFF/PVP/GO@PPF scaffolds offer optimal conditions for cell growth. After 7 days of culture, a substantial number of viable cells was found on all tested scaffolds, indicating that cells proliferated significantly from 3 to 7 days of in vitro cell culture. Moreover, on scaffolds with 0.5 wt% and 1 wt% GO@PPF, the amount of live cells is significantly higher compared to 0.25 wt% GO@PPF. Therefore, all these experiments show that PPF/PVP/GO@PPF scaffolds offer a biocompatible microenvironment for murine pre-osteoblasts.
 | ||
| Fig. 8 Live/dead staining qualitative results: green – cytoplasm of live cells labeled with calcein AM; red – nuclei of dead cells labeled with ethidium bromide. | ||
4. Conclusions
In the current study we report the synthesis and characterization of PPF/PVP-based nanocomposite porous scaffolds employing PPF-grafted GO as reinforcing agent. The GO@PPF nanofiller was obtained through a facile and convenient surface esterification reaction, and the successfull functionalization was demonstrated by complementary techniques such as FT-IR, XPS, TGA and TEM.We show that the incorporation of GO@PPF in a PPF/PVP polymer matrix leads to a significant improvement in mechanical properties, which we attribute to the high intrinsic modulus of GO and increased crosslinking density. Presumably, additional double bonds on the surface of GO@PPF are involved in the curing process, as suggested by a decreased sol fraction for the scaffolds containing a higher amount of nanofiller.
The surface of in vitro mineralized PPF/PVP/GO@PPF nanocomposites caffolds is uniformly covered in hydroxyapatite-like crystals having a morphology and Ca/P ratio similar to bone tissue. Moreover, the preliminary biocompatibility assessment revealed a good interaction between PPF/PVP/GO@PPF scaffolds and murine pre-osteoblasts, as both cell viability and proliferation rate increased, and nocytotoxic effects were evidenced. Therefore, these results suggest that PPF/PVP/GO@PPF scaffolds are promising candidates for bone tissue engineering applications.
Author contributions
A. M. P. and M. D. R. conceptualized the study. A. M. P. secured funding, performed the composite materials synthesis and characterization, data interpretation and developed the manuscript draft. E. V. performed the SEM and TEM analysis. A. S., S. D. and M. C. performed the biocompatibility evaluation and wrote the corresponding part of the manuscript. M. T. and C. D. analyzed some of the results and revised the manuscript. M. D. R. coordinated the team and wrote the final version of the manuscript. All authors read and approved the last version of the manuscript.Conflicts of interest
The authors declare that they have no known competing financialinterests or personal relationships that could have appeared to influencethe work reported in this paper.Acknowledgements
This work was supported by a grant of the Ministry of Research, Innovation and Digitization, CNCS-UEFISCDI, project number PN-III-P1-1.1-TE-2021-1107, within PNCDI III, TE 110/2022-POLYREB. The nanoCT equipment was funded by the European Regional Development Fund through Competitiveness Operational Program 2014–2020, Priority axis 1, Project No. P_36_611, MySMIS code 107066, Innovative Technologies for Materials Quality Assurance in Health, Energy and Environmental – Center for Innovative Manufacturing Solutions of Smart Biomaterials and Biomedical Surfaces – INOVABIOMED.Notes and references
- E. L. Hedberg, C. K. Shih, J. J. Lemoine, M. D. Timmer, M. A. K. Liebschner, J. A. Jansen and A. G. Mikos, In vitro degradation of porous poly(propylene fumarate)/poly(DL-lactic-co-glycolic acid) composite scaffolds, Biomaterials, 2005, 26, 3215–3225 CrossRef CAS PubMed.
- J. Guo, X. Liu, A. L. Miller2nd, B. E. Waletzki, M. J. Yaszmenski and L. Lu, Novel Porous Poly(propylene Fumarate-co-caprolactone) Scaffolds Fabricated by Thermally Induced Phase Separation, J. Biomed. Mater. Res., Part A, 2017, 105, 226–235 CrossRef CAS PubMed.
- R. T. Tran, E. Naseri, A. Kolasnikov, X. Bai and J. Yang, A new generation of sodium chloride porogen for tissue engineering, Biotechnol. Appl. Biochem., 2011, 58, 335–344 CrossRef CAS PubMed.
- E. Behravesh, M. D. Timmer, J. J. Lemoine, M. A. K. Liebschner and A. G. Mikos, Evaluation of the in Vitro Degradation of Macroporous Hydrogels Using Gravimetry, Confined Compression Testing, and Microcomputed Tomography, Biomacromolecules, 2002, 3, 1263 CrossRef CAS PubMed.
- K. W. Lee, S. Wang, L. Lu, E. Jabbari and B. L. Currier,M.J. Yaszemski, Fabrication and characterization of poly(propylene fumarate) scaffolds with controlled pore structures using 3-dimensional printing and injection molding, Tissue Eng., 2006, 12, 2801–2811 CrossRef CAS PubMed.
- K. W. Lee, S. Wang, B. Fox, E. L. Ritman, M. J. Yaszemski and L. Lu, Poly(propylene fumarate) bone tissue engineering scaffold fabrication using stereolithography: effects of resin formulations and laser parameters, Biomacromolecules, 2007, 8, 1077–1084 CrossRef CAS PubMed.
- K. W. Lee, S. Wang, M. Dadsetan, M. J. Yaszemski and L. Lu, Enhanced Cell Ingrowth and Proliferation through Three-Dimensional Nanocomposite Scaffolds with Controlled Pore Structures, Biomacromolecules, 2010, 11, 682–689 CrossRef CAS PubMed.
- K. W. Lee, S. Wang, L. Lu, E. Jabbari, L. B. Currier and M. J. Yaszemski, Fabrication and Characterization of Poly(Propylene Fumarate) Scaffolds with Controlled Pore Structures Using 3-Dimensional Printing and Injection Molding, Tissue Eng., 2006, 12, 2801–2811 CrossRef CAS PubMed.
- D. Ronca, F. Langella, M. Chierchia, U. D’Amora, T. Russo, M. Domingos, A. Gloria, P. Bartolo and L. Ambrosio, Bone Tissue Engineering: 3D PCL-based nanocomposite scaffolds with tailored properties, Procedia CIRP, 2016, 49, 51–54 CrossRef.
- V. Goranov, T. Shelyakova, R. De Santis, Y. Haranava, A. Makhaniok, A. Gloria, A. Tampieri, A. Russo, E. Kon, M. Marcacci, L. Ambrosio and V. A. Dediu, 3D Patterning of cells in Magnetic Scaffolds for Tissue Engineering, Sci. Rep., 2020, 10, 2289 CrossRef CAS PubMed.
- A. S. Mistry, S. H. Cheng, T. Yeh, E. Christenson, J. A. Jansen and A. G. Mikos, Fabrication and in vitro degradation of porous fumarate-based polymer/alumoxane nanocomposite scaffolds for bone tissue engineering, J. Biomed. Mater. Res., Part A, 2009, 89, 68–79 CrossRef PubMed.
- B. Farshid, G. Lalwani, M. S. Mohammadi, J. Simonsen and B. Sitharaman, Boron nitride nanotubes and nanoplatelets as reinforcing agent of polymeric matrices for bone engineering, J. Biomed. Mater. Res., Part B, 2017, 105, 406–419 CrossRef CAS PubMed.
- B. Farshid, G. Lalwani, M. S. Mohammadi, J. S. Sankaran, S. Patel, S. Judex, J. Simonsen and B. Sitharaman, Two-dimensional graphene oxide reinforced porous biodegradable polymeric nanocomposites for bone tissue engineering, J. Biomed. Mater. Res., 2019, A107, 1143–1153 CrossRef PubMed.
- S. He, M. D. Timmer, M. J. Yaszemski, A. W. Yasko, P. S. Engel and A. G. Mikos, Synthesis of biodegradable poly(propylene fumarate) networks with poly(propylene fumarate)-diacrylate macromers as crosslinking agents and characterization of their degradation products, Polymer, 2001, 42, 1251–1260 CrossRef CAS.
- G. Lalwani, A. M. Henslee, B. Farshid, L. Lin, F. K. Kasper, Y. X. Qin, A. G. Mikos and B. Sitharaman, Two-Dimensional Nanostructure-Reinforced Biodegradable Polymeric Nanocomposites for Bone Tissue Engineering, Biomacromolecules, 2013, 14, 900–909 CrossRef CAS PubMed.
- H. J. Salavagione, G. Martinez and G. Ellis, Recent Advances in the Covalent Modification of Graphene With Polymers, Macromol. Rapid Commun., 2011, 32, 1771–1789 CrossRef CAS PubMed.
- L. Yang and W. Zhen, Synthesis of Graphene Oxide-Polystyrene Graft Polymer Based on Reversible Addition Fragmentation Chain Transfer and Its Effect on Properties, Crystallization, and Rheological Behavior of Poly (Lactic Acid), Adv. Polym. Technol., 2020, 9364657 CAS.
- H. Roghani-Mamaqani and V. Haddadi-Asl, In-plane functionalizing graphene nanolayers with polystyrene by atom transfer radical polymerization: Grafting from hydroxyl groups, Polym. Compos., 2014, 35, 386–395 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, New Polymer Synthesis by Nitroxide Mediated Living Radical Polymerizations, Chem. Rev., 2001, 101, 3661–3668 CrossRef CAS PubMed.
- Z. Sekhavat and M. Ghaemy, Polymer grafted graphene oxide: For improved dispersion in epoxy resin and enhancement of mechanical properties of nanocomposite, Compos. Sci. Technol., 2016, 136, 145–157 CrossRef.
- M. Yuan, Y. Chen, M. Yuan, H. Li, X. Xia and C. Xiong, Functionalization of Graphene Oxide with Low Molecular Weight Poly (Lactic Acid), Polymers, 2018, 10, 177 CrossRef PubMed.
- E. Vasile, A. M. Pandele, C. Andronescu, A. Selaru, S. Dinescu, M. Costache, A. Hanganu, M. D. Raicopol and M. Teodorescu, Hema-Functionalized Graphene Oxide: a Versatile Nanofller for Poly(Propylene Fumarate)-Based Hybrid Materials, Sci. Rep., 2019, 9, 18685 CrossRef CAS PubMed.
- M. Teodorescu and M. Bercea, Poly(vinylpyrrolidone) – A Versatile Polymer for Biomedical and Beyond Medical Applications, Polym.-Plast. Technol. Mater., 2015, 54, 923–943 CrossRef CAS.
- N. d’Avanzo, C. Celia, A. Barone, M. Carafa, L. D. Marzio, H. A. Santos and M. Fresta, Immunogenicity of Polyethylene Glycol Based Nanomedicines: Mechanisms, Clinical Implications and Systematic Approach, Adv. Ther., 2020, 3, 1900170 CrossRef.
- K. F. Kasper, K. Tanahashi, J. P. Fisher and A. G. Mikos, Synthesis of poly(propylene fumarate), Nat. Protoc., 2009, 4, 518–525 CrossRef PubMed.
- V. I. Povstugar, A. M. Lyakhovich and A. M. Shakov, Bromination reaction in CC double bond determination by XPS, J. Electron Spectrosc. Relat. Phenom., 1994, 68, 565–568 CrossRef CAS.
- T. Taguchi, A. Kishida and M. Akashi, Hydroxiapatite formation on poly(vinyl alcohol) hydrogel matrices using an alternate soakingprocess, Chem. Lett., 1998, 711–712 CrossRef CAS.
- S. Eigler and A. M. Dimiev, Characterization Techniques, in Graphene Oxide: Fundamentals and Applications, ed. S. Eigler and A. M. Dimiev, John Wiley & Sons, Chichester, 2017, pp. 100–111 Search PubMed.
- A. K. Shung, M. D. Timmer, S. Jo, P. S. Engle and A. G. Mikos, Kinetics of poly(propylene fumarate) synthesis by step polymerization of diethyl fumarate and propylene glycol using zinc chloride as a catalyst, J. Biomater. Sci., Polym. Ed., 2002, 13, 95–108 CrossRef CAS PubMed.
- C. M. Damian, M. I. Necolau, I. Neblea, E. Vasile and H. Iovu, Synergistic effect of graphene oxide functionalized with SiO2 nanostructures in the epoxy nanocomposites, Appl. Surf. Sci., 2020, 507, 145046 CrossRef CAS.
- D. Kadir, C. Mehmet and E. Kaya, A detailed study of thermal degradation of poly(2-hydroxyethyl methacrylate), Polym. Degrad. Stab., 2001, 72, 75–80 CrossRef.
- Z. Cai, T. Zhang, L. Di, D. M. Xu, D. H. Xu and D. A. Yang, Morphological and histological analysis on the in vivo degradation of poly (propylene fumarate)/(calcium sulfate/B-tricalcium phosphate), Biomed. Microdevices, 2011, 13, 623–631 CrossRef CAS PubMed.
- Z. Y. Cai, D. A. Yang, N. Zhang, C. G. Ji, L. Zhu and T. Zhang, Poly(propylenefumarate)/(calcium sulphate/B-tricalcium phosphate) composites: Preparation, characterization and in vitro degradation, Acta Biomater., 2009, 5, 628–635 CrossRef CAS PubMed.
- J. P. Fisher, T. A. Holland, D. Dean, P. S. Engel and A. G. Mikos, Synthesis and properties of photocross-linked poly(propylene fumarate) scaffolds, J. Biomater. Sci., Polym. Ed., 2001, 12, 673–687 CrossRef CAS PubMed.
- J. W. Suk, R. D. Piner, J. An and R. S. Ruoff, Mechanical properties of graphene oxides, ACS Nano, 2014, 4, 6557–6564 CrossRef PubMed.
- M. Teodorescu, B. Cursaru and P. O. Stanescu, Swelling and diffusion characteristics of hydrogels synthesized from diepoxy-terminated poly(ethylene glycol)s and aliphatic polyamines, Soft Mater., 2010, 8, 288–306 CrossRef CAS.
- M. D. Timmer, C. G. Ambrose and A. G. Mikos, In vitro degradation of polymeric networks of poly(propylene fumarate) and the crosslinking macromer poly(propylene fumarate)-diacrylate, Biomaterial, 2003, 24, 571–577 CrossRef CAS PubMed.
- A. M. Henslee, S. R. Shah, M. E. Wong, A. G. Mikos and F. K. Kasper, Degradable, antibiotic releasing poly(propylene fumarate)-based constructs for craniofacial space maintenance applications, J. Biomed. Mater. Res., 2015, A103, 1485–1497 CrossRef PubMed.
- M. D. Timmer, C. Ambrose and A. G. Mikos, Evaluation of thermal and photo-crosslinked biodegradable poly(propylene fumarate)- based networks, J. Biomed. Mater. Res., 2003, 66, 811–818 CrossRef PubMed.
- T. Taguchi, Y. Muraoka, H. Matsuyama, A. Kishada and M. Akashi, Apatite coating on hydrophilic polymer-grafted poly(ethylene) films using an alternate soaking process, Biomaterials, 2001, 22, 53–58 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb01232h |
| This journal is © The Royal Society of Chemistry 2023 |