
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDivergent approach to nanoscale glycomicelles and photo-responsive supramolecular glycogels. Implications for drug delivery and photoswitching lectin affinity†
Elena
Romero-Ben‡
 a,
M Carmen
Castillejos‡
a,
Cristian
Rosales-Barrios
a,
María
Expósito
a,
Pilar
Ruda
a,
Paula M.
Castillo
a,
Stefania
Nardecchia
b,
Juan
de Vicente
b and
Noureddine
Khiar
*a
a,
M Carmen
Castillejos‡
a,
Cristian
Rosales-Barrios
a,
María
Expósito
a,
Pilar
Ruda
a,
Paula M.
Castillo
a,
Stefania
Nardecchia
b,
Juan
de Vicente
b and
Noureddine
Khiar
*a
aAsymmetric Synthesis and Functional Nanosystems Group, Instituto de Investigaciones Químicas (IIQ), CSIC-Universidad de Sevilla, Avda. Américo Vespucio 49, 41092, Seville, Spain. E-mail: khiar@iiq.csic.es
bDepartment of Applied Physics and Excellence Research Unit ‘Modeling Nature’ (MNat), Faculty of Sciences, University of Granada, C/Fuentenueva s/n, 18071 – Granada, Spain
First published on 9th October 2023
Abstract
The field of stimuli-responsive supramolecular biomaterials has rapidly advanced in recent years, with potential applications in diverse areas such as cancer theranostics, tissue engineering, and catalysis. However, designing molecular materials that exhibit predetermined hierarchical self-assembly to control the size, morphology, surface chemistry, and responsiveness of the final nanostructures remains a significant challenge. In this study, we present a divergent synthetic approach for the fabrication of spherical micelles and functional 1D-glyconanotube-based photoresponsive gels from structurally related diazobenzene/diacetylene glycolipids. The resulting nanostructures were characterized using NMR, TEM, and SEM, confirming the formation of spherical and tubular nanostructures in both the gel and solution states. Upon UV irradiation, a reversible gel–sol transition was observed, resulting from the photoswitching of the azobenzene unit from the stretched trans form to the compact, metastable cis form. Our gels were shown to enable spatio-temporal control of the adhesion and release of the lectin Concanavalin A, demonstrating potential use as regenerable biomaterials to fight against infections with toxins and pathogens. Additionally, our micelles and gels were evaluated as nanocontainers for loading and controlled release of hydrophobic dyes and antitumoural agents, suggesting their possible use as smart theranostic drug delivery systems.
Introduction
The development of complex nanoscale systems able to receive and execute commands from external stimuli has received increased attention in recent years due to their potential use in drug delivery, tissue engineering, biomineralization, molecular electronics, and catalysis.1–7 Of relevance for the design of these systems as cancer theranostics, structure–activity relationship studies have shown that topology and size are key factors for their cellular uptake, circulation time and interaction with specific receptors.8–12 Indeed, while spherical nanomaterials are well suited to processes involving rapid cellular uptake,13 1D-rod structures are more appropriate for those requiring a longer circulation time.14 On the other hand, it has been shown that spherical micelles are potent inhibitors of globular receptors, while 1D-nanofibers as well as 3D-gels are more suitable for adhesion and inhibition of bacterial motility.15–18 Therefore, developing divergent and cost-effective synthetic approaches allowing the synthesis of smart functionalized organic materials and modulating their sizes, topology and functionality from well-designed molecular monomers is highly desirable, although it remains an outstanding challenge.19 One family of molecules suitable for the creation of supramolecular diversity are the amphiphiles, whose self-organization can lead to the formation of species with a wide range of sizes and whose functionality can range from ordinary soap to extracellular matrix mimics.,20,21A paradigmatic example are supramolecular gels,22,23 obtained from low molecular weight organogelators (LMOGS),24–26 whose hierarchical self-organization lead to flexible 1D-nanofibers with a high aspect ratio having sizes ranging from submicron to hundreds of micrometers which by entanglement, form the macroscopic supramolecular gels of millimeter size.27,28 This hierarchy provides an excellent opportunity to design intelligent supramolecular systems with functional responses by incorporating motifs responsive to external stimuli into the molecular structures of amphiphiles.29–31
Among the different stimuli, using light offers the advantage of being easily turned on and off, employing adjustable wavelength and intensity, and offering high spatial and temporal control.29,30,32–36 Moreover, an interesting class of amphiphiles are glycolipids whose hierarchical supramolecular organization leads to complex systems endowed with important biological activities due to the involvement of carbohydrates in vital processes such as pathogen adhesion, transplant rejection, fertilization, and cell proliferation/differentiation.29,37–40 Based on these premises and within our interest in the development of functional soft glyconanomaterials,41–44 we report, herein, a new modular synthetic approach of self-associative multifunctional neoglycolipids 1–3 for the divergent nanofabrication of functional spherical micelles, 1D-lipid glyconanotubes, and photoresponsive 3D-gels, Fig. 1.
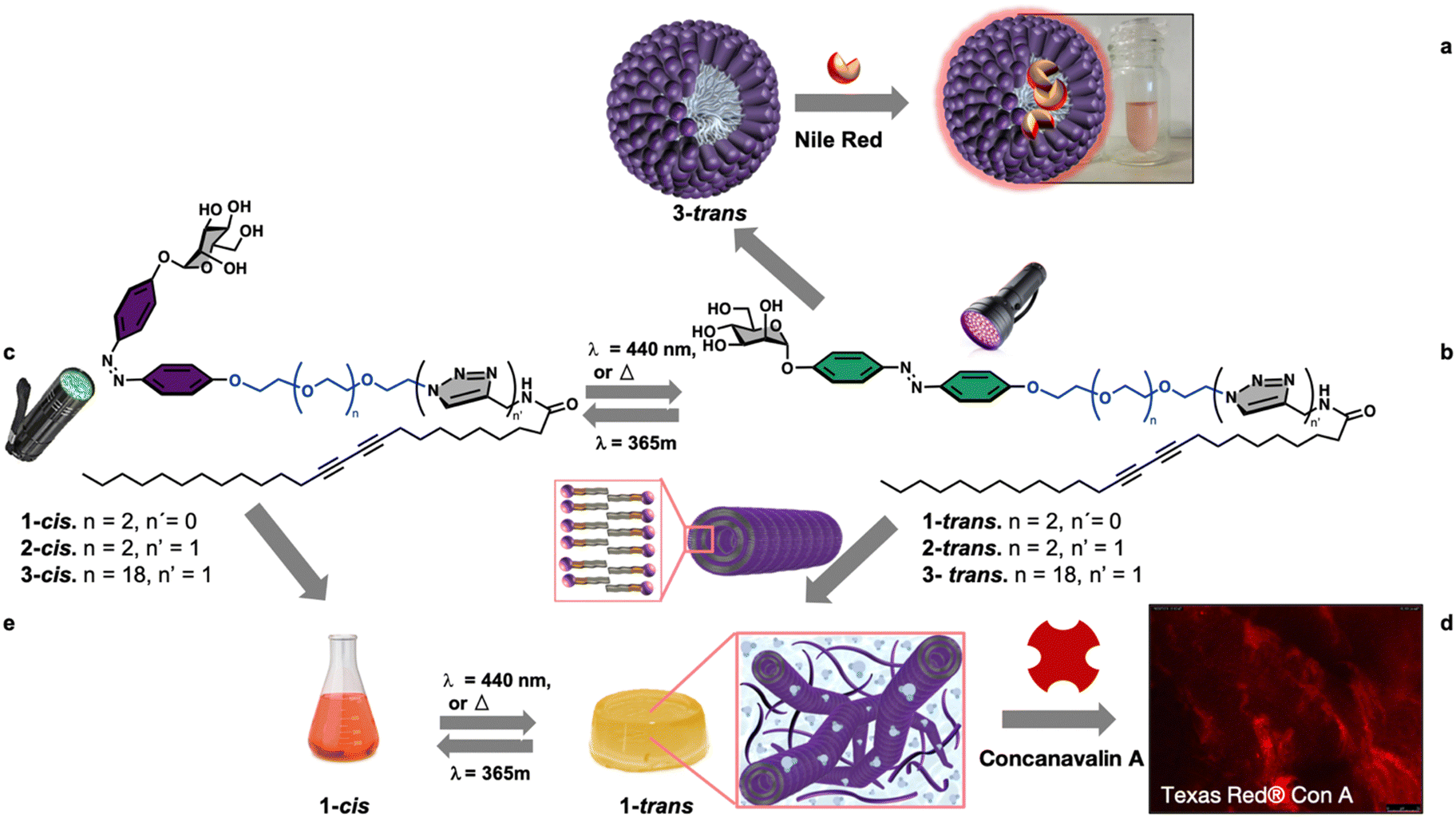 | ||
| Fig. 1 Divergent synthesis of mannose-coated micelles (a) and photoresponsive mannose-coated gel (b) through supramolecular self-organization of sugar-coated diazobenzene/diacetylene amphiphiles. Amhiphile 3 with a large PEG20 chain self-organize into spherical micelle able to host and solubilize the hydrophobic dye Nile red (a), while amphiphile 1 with a short tetraethyleneglycol chain self-organize into 1D-nanotubes (b) which evolve to tridimensional gels, able to establish selective interactions with mannose-specific lectin Texas red® concanavalin A (d), and to perform a reversible gel–sol transition in response to light irradiation (c and e). | ||
Results and discussion
Synthetic design
In the design of the self-associative monomers, diazobenzene, linked directly to the biorecognition element mannose, was used as a light-responsive group.29,34 The design also includes a variable oligoethylene glycol to fine-tune the hydrophobic–hydrophilic balance of the neoglycolipid. Furthermore, as hydrophobic tail a 25 carbon chain bearing a photo-polymerizable diacetylenic function, known for its ability to form nanomaterials with interesting chromatic properties was used.44–49 The design was validated by synthesizing functional spherical micelles (Fig. 1a), and 1D-lipid glyconanotubes (Fig. 1b), that entangle to form light responsive 3D-gels (Fig. 1c).Synthesis of the monomers
The synthesis of the three compounds (1, 2 and 3), use tetra-O-acetyl-α-D-mannopyranosyl trichloroacetimidate 4, obtained in two steps from mannose pentaacetate, as starting material, Scheme 1. Monoglycosylation of 4,4′ -dihydroxyazobenzene 5 acceptor,50 using donor 4 and boron trifluoride as an activator gives the desires compound 6 with 86% yield. For the modulation of the hydrophilic–hydrophobic balance, as well as for unravelling the effect of the connecting group between the polar head group and the apolar tail, bifunctional spacers 7–9, derived from tetra-ethyleneglycol (7, 8) or eicosaetyleneglycol (9), were used, Scheme 1. The condensation of alcohol 6 with tosylated spacer 7 in the presence of K2CO3, LiCl in acetonitrile gives the corresponding derivative 10 in 60% yield, Scheme 1a. Next, the deprotection of the NHBoc derivative 10 using trifluoroacetic acid in methylene chloride gives the free amine 11. Amidation with pentacosadiinoic acid (PCDA) in the presence of TBTU and DIPEA in DMF gives the corresponding amide 12 with 60% yield and, finally, Zemplen deacetylation gives the desired neoglycolipid 1, Scheme 1a. For the synthesis of the neoglycolipids 2 and 3, Scheme 1b, condensation of potassium alcoholate of 6 with the tosylated azido spacers derived from tetraethyleneglycol 8 or eicosaethyleneglycol spacer 9, in acetonitrile gives the ether derivatives 13 and 14 in 67% and 70% yields respectively. Cu(I)-catalyzed azide alkyne 1,3-dipolar cycloadditions (CuAAC) between azides 13 or 14 and the N-(2-propynyl)pentacosa-10,12-diynamide alkyne 15, itself obtained in one step from PCDA and propargylamine, in the presence of sodium ascorbate and in a methylene chloride/water mixture, afforded regioselectively the 1,2,3-triazole derivatives 16 and 17 with 60 and 80% yield respectively. Finally, Zemplen deacetylation gives the desired compounds 2 and 3 with 97 and 99% chemical yield. The structures of the 3 monomers were confirmed by monodimensional and bidimensional NMR as well as by mass spectroscopy (see experimental part and ESI†).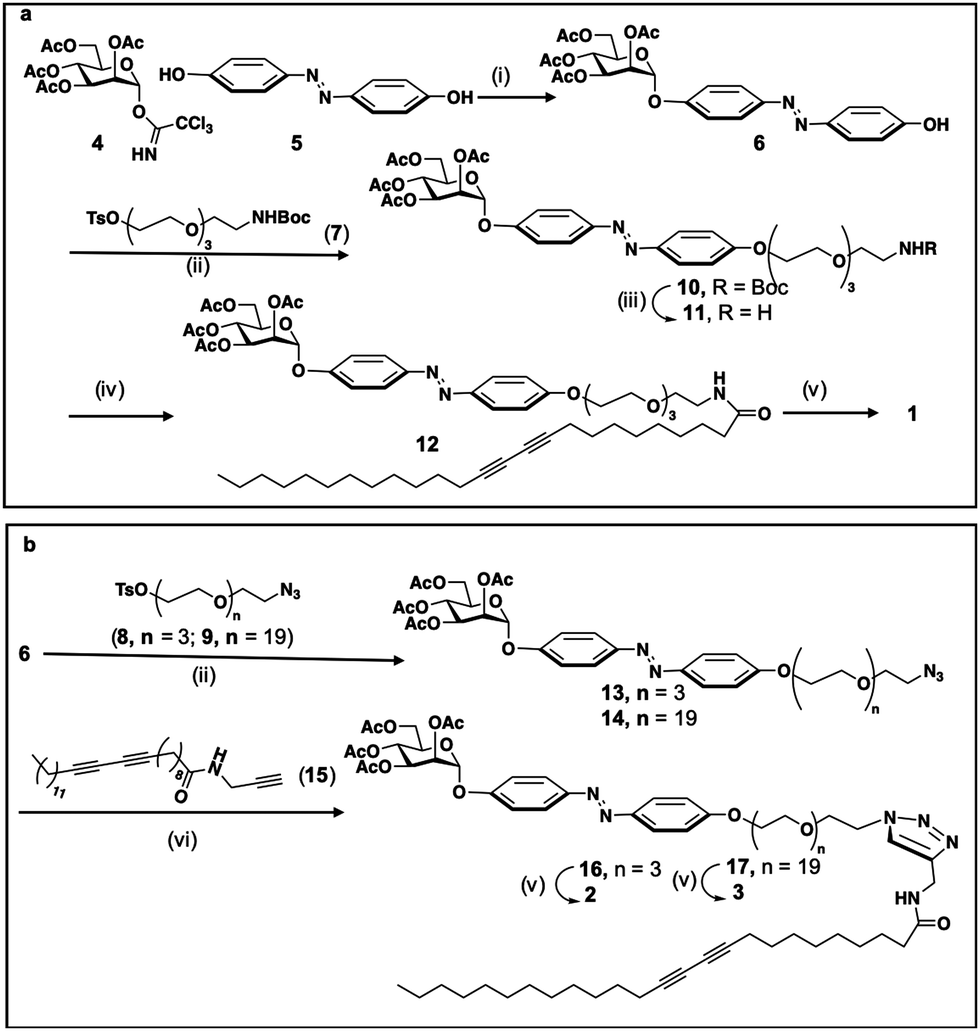 | ||
| Scheme 1 Synthesis of self-associatives neoglycolipid 1 (a), 2 and 3 (b). (i) BF3·Et2O, CH3CN, 0 °C; (ii) K2CO3, LiCl, CH3CN; (iii) TFA, CH2Cl2; (iv) TBTU, DIPEA, DMF; (v) MeONa, MeOH; (vi) CuSO4, AsCNa, CH2Cl2/H2O. | ||
Photoisomerization study of the monomers
Azobenzene compounds are well-known for their reversible photoisomerization between the trans and cis forms under alternating UV and visible light irradiation.51 The light-responsive behavior of three neoglycolipids 1–3, Fig. 1, was studied by 1H NMR (Fig. 2b and c), UV-Vis (Fig. 2d and e), and HPLC-MS spectroscopies (vide infra). The most significant protons for studying the isomerization kinetics of neoglycolipids 1–3 by 1H NMR are the diazobenzene protons, the proton 5 of the triazole and the anomeric proton of sugar. In the stable trans (E) isomers the signals of protons 3, 5 and 3′, 5′ of the diazobenzene moiety appear as two doublets at 6.90 ppm and 7.10 ppm respectively, while protons 2.6 and 2′.6′ appear together at a lower field (about 7.75 ppm). The triazole proton is observed as a singlet at 7.90 ppm, and the anomeric proton appears either as a broad singlet (in the case of 1-trans and 2-trans) or as a doublet with a small coupling constant (1.34 Hz) in the case of 3-trans, at approximately 5.60 ppm. In the cis (Z) isomers [1-cis, 2-cis (see ESI†), and 3-cis], the signals of the 3, 5 and 3′, 5′ protons undergo a slight shift at 7.00 and 6.90 ppm, while the 2, 6 and 2′, 6′ protons alpha to the diazo function undergo a pronounced shielding effect, from 8.00 ppm to 6.80 ppm. Although the anomeric proton undergoes a slight chemical shift change (from 5.60 ppm to 5.50 ppm), its location in a position where there are no other signals allows it to be used as an additional control to monitor and quantify the trans–cis isomers. To gain further structural insights, we conducted selective 1D-NOESY and 1H/1H EASY ROESY 2D experiments (see ESI†). In the case of the 2-trans derivative, our NOESY 1D experiments facilitated more precise signal assignments for both aromatic rings (see Fig. S53–S56 in the ESI†). Additionally, 2D 1H/1H EASY-ROESY experiments, carried out on a predominantly 2-cis configuration, revealed contact NOEs between the H3 and H5 protons of the aromatic ring linked to the sugar and a methylene protons of the PEG chain (see Fig. S60 in the ESI†).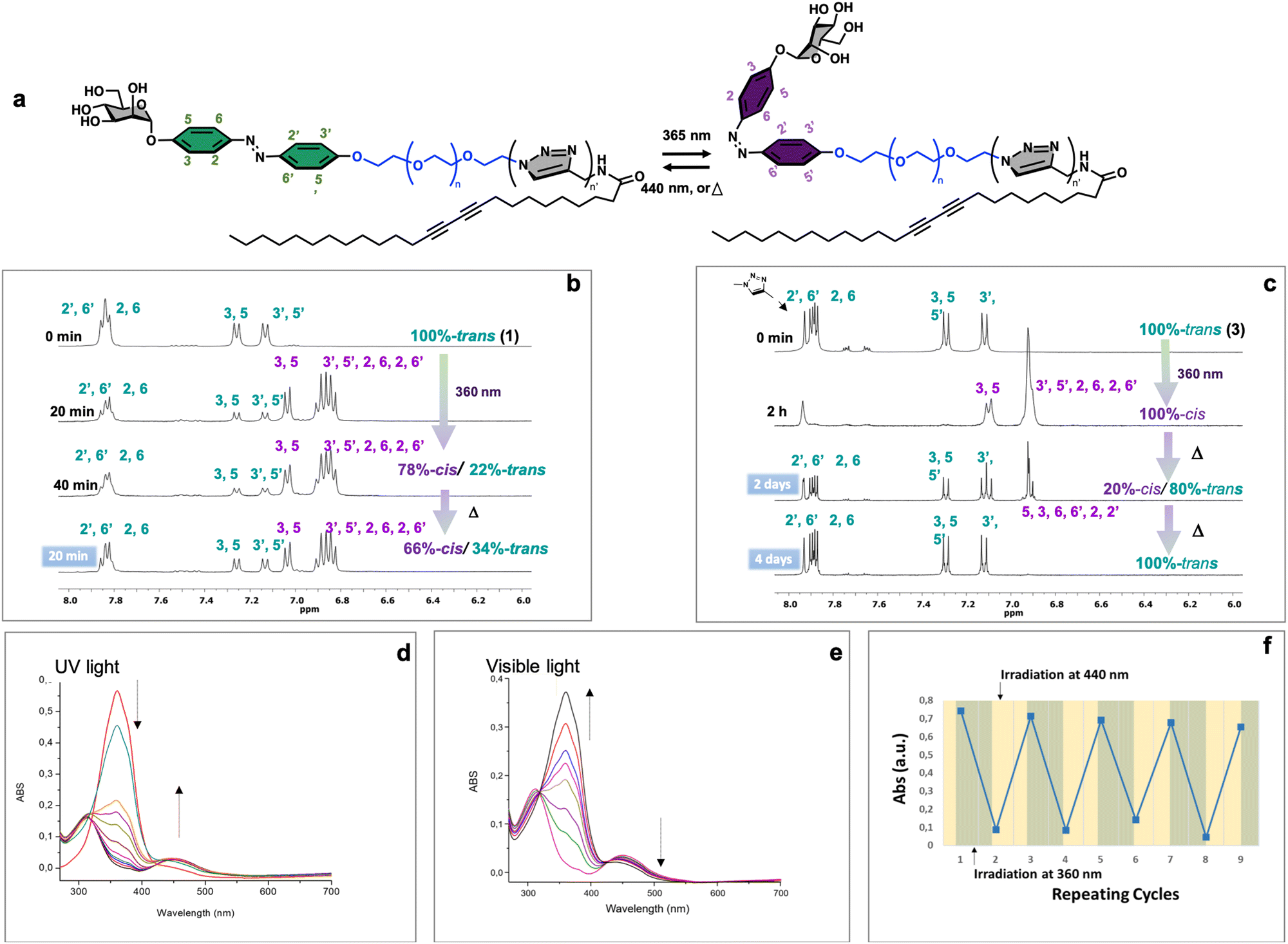 | ||
| Fig. 2 Schematic representation of the photoisomerization of neoglycolipids 1–3 (a). 1HNMR studies of trans–cis and cis–trans photo and thermal isomerization of the neoglycolipid 1 in DMSOd6 (b), and 3 in MeOD (c). UV-Vis studies of trans–cis (d) photoisomerization and cis–trans recovery under blue light irradiation (e) of neoglycolipid 1 (0,005 wt/v% in DMSO). Cycle of absorption after alternate irradiation with UV (360 nm, green bar) and visible light (460 nm, yellow bar) (f). | ||
Our study allowed us to quantify the photostationary (Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E) state of the three neoglycolipids in DMSO. Neoglycolipid 1 exhibited a photostationary state of 75% cis (Z) and 25% trans (E) isomers (Fig. 2b), while neoglycolipid 2 displayed a photostationary state of 82% cis (Z) and 18% trans (E) isomers (see ESI†). Remarkably, and contrary to most reported diazobenzene glycolipids, neoglycolipid 3 showed a unique behavior as its photoswitching was independent of the solvent and complete even in methanol, with a photostationary state of 100% cis (Z) isomer (Fig. 2c). The complete isomeric conversion of neoglycolipid 3 is of great interest in studying the influence of carbohydrate orientation on lectin–carbohydrate interactions. The slow thermal return from pure 3-cis to pure 3-trans isomer, occurring over a period of 96 hours in the dark, further adds to the significance of neoglycolipid 3. Moreover, under blue light irradiation (460 nm), the return from the 3-cis to 3-trans isomer in neoglycolipid 3 was significantly accelerated, taking only 2 hours (Fig. 2c).
:E) state of the three neoglycolipids in DMSO. Neoglycolipid 1 exhibited a photostationary state of 75% cis (Z) and 25% trans (E) isomers (Fig. 2b), while neoglycolipid 2 displayed a photostationary state of 82% cis (Z) and 18% trans (E) isomers (see ESI†). Remarkably, and contrary to most reported diazobenzene glycolipids, neoglycolipid 3 showed a unique behavior as its photoswitching was independent of the solvent and complete even in methanol, with a photostationary state of 100% cis (Z) isomer (Fig. 2c). The complete isomeric conversion of neoglycolipid 3 is of great interest in studying the influence of carbohydrate orientation on lectin–carbohydrate interactions. The slow thermal return from pure 3-cis to pure 3-trans isomer, occurring over a period of 96 hours in the dark, further adds to the significance of neoglycolipid 3. Moreover, under blue light irradiation (460 nm), the return from the 3-cis to 3-trans isomer in neoglycolipid 3 was significantly accelerated, taking only 2 hours (Fig. 2c).
UV/Vis spectroscopic studies were conducted using DMSO as the solvent and under gelation conditions (see details below). Glycolipids exhibit a broad absorption peak around 320–350 nm, primarily attributed to the trans form of the azobenzene group (Fig. 2d and e). Upon irradiation, the photo-induced trans–cis isomerization was observed to be reversible, as the cis form of the azo-surfactants relaxed back to the trans form after exposure to visible light or slowly in the absence of light (Fig. 2e). Furthermore, a series of successive “on–off” switching experiments on the absorption bands of glycolipid 1 was carried out. The results of these experiments demonstrated that the process was reversible for at least nine cycles, indicating that sample 1 exhibits good stability (Fig. 2f).
Supramolecular self-assembly of amphiphile 1 and characterization of the photoresponsive-glycogel
In order to determine the supramolecular self-association capacities of the monomers, we determined first their water and organic solvents solubilities. Although monomers 1 and 2 are insoluble in most solvents, the more hydrophilic monomer 3 is soluble in various solvents including water (vide infra). The use of mixture of solvents reveals water–ethanol as the best mixture. Indeed, in 1:1 or 1:2 water–ethanol mixture, monomer 1 showed a great ability to form gels. In the same conditions monomer 2 was insoluble while monomer 3 gave a clear yellowish solution.
Interestingly, while in the 1:2 water ethanol mixture, the gelation of 1 takes place at a concentration of 1% (w/v), in 50% aqueous ethanol solution it takes place at a concentration as low as 0.1%. Additionally, in the latter case the compound gelificate instantaneously, Fig. 3, at room temperature with no need of heating–cooling or sonication. The structure of the as-formed gel as well as the xerogel formed from monomer 1 at 1% was characterized by transmission electron microscopy (TEM) and field emission scanning electron microscopy (FESEM) (Fig. 3). Interlocked agglomerated tubules with a uniform diameter of 31 nm, a thickness of 9 nm, and micron-scale lengths were regularly found in the glycogel-1 under TEM analysis (Fig. 3a). The observation of xerogel-1 under SEM at the tens of micrometer scale showed interesting intertwined and twisted structures in a bush-form (see Fig. S32, ESI†). Moreover, FESEM analysis allowed observing the detailed fibrillar aggregates forming the bundles shown in the standard SEM images (Fig. 3b).
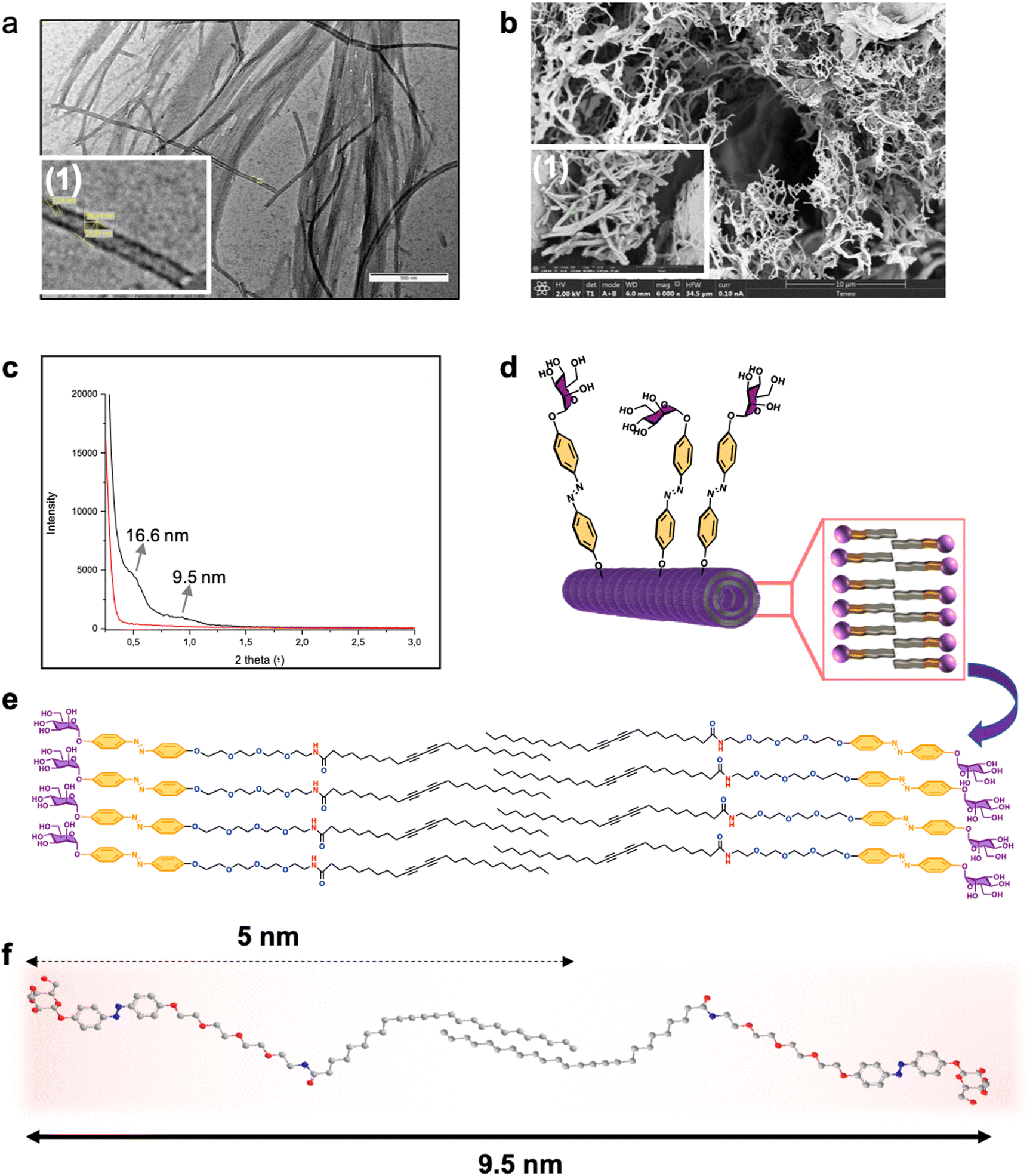 | ||
| Fig. 3 Characterization of the glycogel formed by supramolecular self-organization of neoglycolipid 1. Large scale high (a) and enlarged (1) TEM images. (1). Large-scale height (b) and enlarge micrograph of xerogel. Small angle X-ray scattering (c). Model of the molecular bilayer nanotube derived from SAXS study (d). Schematic illustration of the molecular packing in the self-assembled state (e). Chem 3D schematic illustration of the size of self-assembled amphiphile 1. | ||
Small-angle X-ray scattering (SAXS) studies allowed us to advance in the exact structural determination of the fibrillar glycogel-1 (Fig. 3c). Maximum peaks corresponding to averaged repeating distances of about 9.5 nm were obtained (Fig. 3c). It should be noted that the starting monomer is approximately 5 nm in length when extended, as determined by Chem3D analysis (Fig. 3f). This suggests that a single bilayer would extend for a maximum of 10 nm, indicating that the tube walls are likely formed by a single amphiphilic bilayer with some interpenetration of the lipid tail (as shown in Fig. 3d–f). A second peak in the SAXS spectrum at 2θ = 0.5°, corresponding to a separation of 16.6 nm, can be assigned to the inner diameter of the lipid nanotube, which is likely influenced by the packing of the amphiphilic bilayer. Under such supramolecular self-organization, the tube in the gel would expose the mannose residue usually used for the adhesion of pathogens to the external phase, whose specific receptor is involved in several biological processes of interest.51,52 On the other hand, the internal cavity of the tube, larger than 10 nm, could accommodate molecules of interest such as cytotoxic compounds, dyes or could also serve as a confined medium for organic reactions.53
The mechanical properties of gel-1 were characterized through rheological strain sweep and frequency sweep experiments, revealing its high low-shear viscosity and shear-thinning behavior. The gel exhibited a hysteresis loop in the flow curve, indicating thixotropic behavior. Dynamic oscillatory shear tests showed that the gel behaved as a solid-like material with a linear viscoelastic range, followed by non-linear behavior and yielding at higher strain amplitudes. The full study with detailed results can be found in the ESI.†
Diacetylenic amphiphiles (DA) possess photoresponsive properties as they can undergo polymerization through a 1,4-addition reaction of diradicals generated by UV or γ irradiation. This process leads to the formation of smart polydiacetylene (PDA)-nanomaterials, exhibiting intriguing chromatic properties. Prior to investigating the functionality of gel 1, we examined the ability of the self-associated DA monomer 1 to photopolymerize in the gel state using Raman spectroscopy. The results of these studies revealed that photopolymerization occurs only in the dry state and not in the gel state (for detailed information, see the ESI†).
Studies on the UV-induced reversible gel–sol transition
To investigate the light-responsive behavior of glycogel 1, we, first, conducted UV/Vis spectroscopic studies using dilution solutions in ethanol/water (Fig. 4). The results revealed two distinct absorption peaks at 365 nm and 445 nm, which correspond to the π–π* and an n–π* transitions, respectively. Upon exposure to UV light, a noticeable decrease in the π–π* transition band was observed, accompanied by a slight increase in the n–π* transition, indicating the occurrence of trans–cis photoisomerization of monomer 1 upon UV irradiation (Fig. 4a). After 20 minutes of exposure, the absorption bands stabilized, indicating that the photostationary state had been reached. Furthermore, we conducted High-Performance Liquid Chromatography (HPLC) to separate the two isomers, and the results showed that 1-cis eluted at 11.3 minutes, while 1-trans eluted at 11.7 minutes (Fig. 4c and d). This separation allowed us to quantify the proportions of both isomers at different time points. The analysis revealed that neoglycolipid 1 exhibited a photostationary state with 88% in the cis (Z) isomer and 12% in the trans (E) isomer (Fig. 4c). An important observation for practical applications is that this photo-induced isomerization is reversible. When the glycolipid is in the cis form, it relaxes back to the trans form after exposure to visible light (Fig. 4d) or gradually in the dark (see ESI† for more details). Photoexcitation of glycogel 1 with UV light at 365 nm causes a gel–sol transition accompanied by a color change from yellow to orange, (Fig. 4e). Remarkably, the gel–sol transition takes place faster (after a few minutes, see video in ESI†), in the case of gel formed in a 33% compared to that formed in 50% aqueous ethanol mixture (10–12 h). The resulting solution was analysed by transmission electron microscopy, which revealed the presence dispersed lipid nanotubes (Fig. 4f). Interestingly, the diameter of the tubes (about 39 nm) was found to be slightly larger than that of the tube in the gel state. It is believed that the increase in diameter is mainly due to the widening of the inner cavity of the tube, as the walls remained the same size. The precise cause of this change is challenging to elucidate, but it is likely attributed to alterations in the curvature of the lipid layer resulting from the isomerization of the glycogel-1 molecules. The analysis of cis-glycogel-1 through SAXS studies (see ESI†) indicates that the size of the tube walls, formed by a single amphiphilic bilayer, remained unchanged. However, the inner diameter of the lipid nanotubes exhibited an increase and showed non-uniformity. Specifically, we observed three distinct peaks corresponding to averaged repeating distances of approximately 9.17 nm, 17.86 nm, and 20.18 nm. These findings suggest that the diameter increment primarily arises from the widening of the inner cavity of the tube, rather than the widening of the tube walls. The gel is also capable to maintain several cycles of formation and disintegration promoted by light stimuli with no noticeable degradation (data not shown).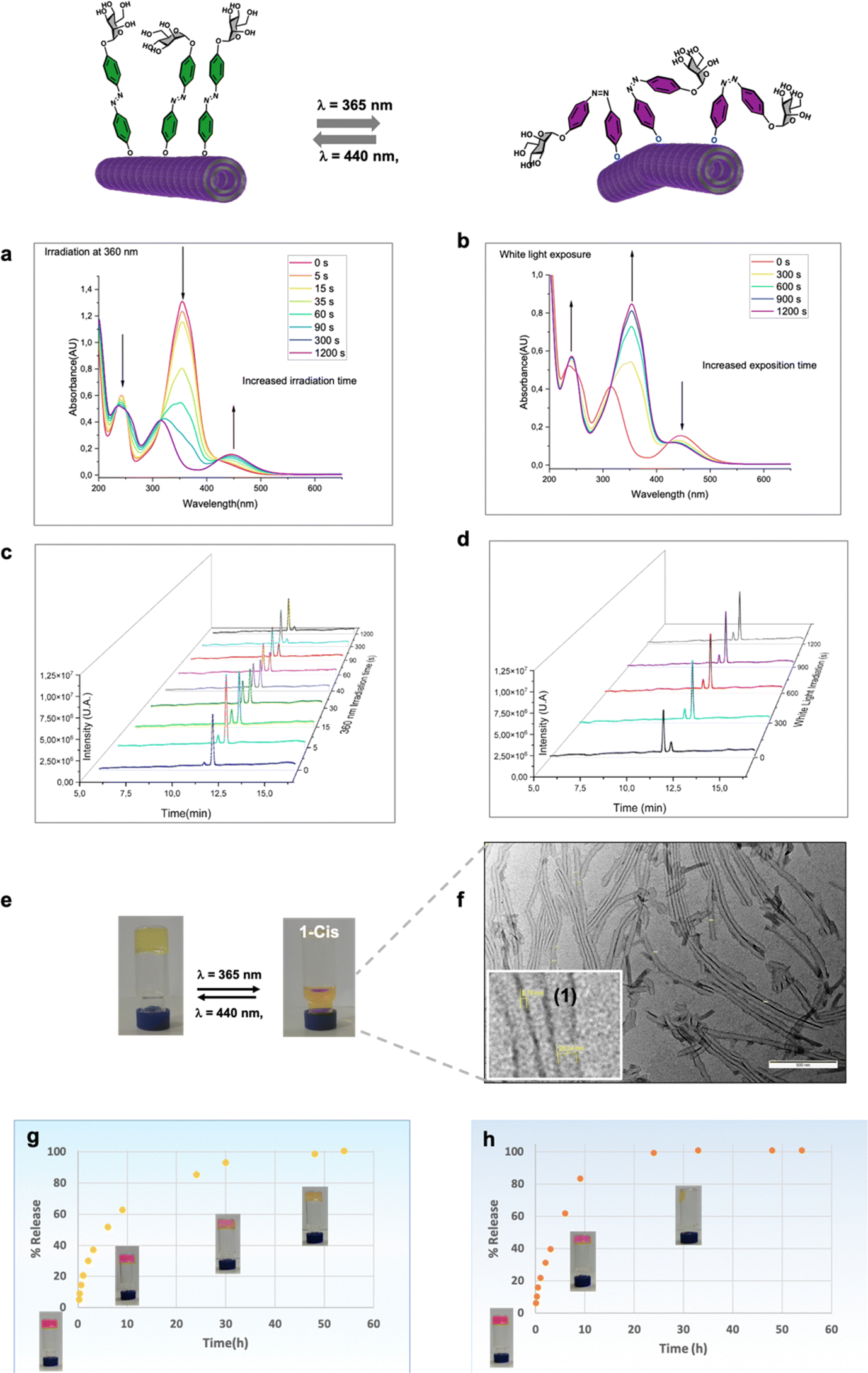 | ||
| Fig. 4 Reversible gel–sol transition and rhodamine release studies. UV-Vis studies of trans–cis (a) photoisomerization and cis–trans recovery under blue light irradiation (b) of 1 (0,005% in EtOH/H2O). HPLC chromatograms after exposure of 1 to UV light (c) and to white light from 0 to 20 minutes (d). Photographs of vials showing the gel–sol transition and color change under UV irradiation (e). Large-scale height (f) and enlarge phase (1) TEM images of spherical nanotubes in the solution. Rhodamine release in the dark (g) and under UV irradiation (h). | ||
Study of rhodamine storage and controlled release from glycogel-1
Next, in order to investigate the capability of glycogel-1 for small molecules storage, as well as passive and light-controlled release we measured the diffusion of rhodamine B (a common biological dye) from gel-1 formed at 0.5% in the dark and upon UV light irradiation (Fig. 4g and h). For this, freshly prepared Rho/gel-1 was covered with the same hydroalcoholic solution used for its preparation and, at different time intervals, the entire supernatant was removed, replaced, and the absorptions at 550 nm recorded. As deduced from sigmoid curve of Fig. 5d and the reach of a plateau, a total rhodamine release from Rho/gel-1 was attained at 50 h (Fig. 4g) in the dark. In contrast, irradiation at 365 nm led to the release of 100% of the drug within 15 h, Fig. 4h, indicating that the diffusion of rhodamine from the gel-1 in the darkness is at least 3-fold slower than its release under UV light irradiation. | ||
| Fig. 5 Fluorescence study of the selective interaction of mannose-coated glycogel-1 with lectins. The images for the detection of the green fluorescence were acquired upon excitation at 480/40 nm with a band pass filter at 527/30 nm. The images for the detection red fluorescence were acquired after excitation at 560/40 nm with a band filter 645/75. Images of the glycogel-1 in the trans form alone (a) and (b), after incubation with PNA-FITC (c), after interaction with ConA-Texas Red (d), after incubation with reversely labelled lectins ConA-FITC (e), and with PNA-Alexa Fluor™ 594 (f). | ||
Spatiotemporal control of adhesion and release of the lectin concanavalin A
Once demonstrated the ability of the gel to entrap and release molecules of interest in a controlled manner, we focused on the additional functionality conferred by spatial orientation of the mannose residue. It is pertinent to recall at this point that spectroscopic and microscopic studies (vide supra), support the proposition of a model according to which glycogel-1 derives from the interconnection of nanotubes whose walls are formed by a single bilayer of the amphiphile. If this model is correct, the external face of the nanotubes will expose mannose residues, providing an excellent opportunity to study the presentation and orientation of carbohydrate moieties in their multivalent interactions with specific receptors for the first time in the gel state. The interaction of the plant lectin concanavalin A (ConA), obtained from jack bean (Concavalia ensiformis), with multivalent mannosylated materials is well studied and provides an excellent tool to validate our model.54–56 ConA is known to selectively recognizes α-mannopyranoside, α-glucopyranoside and to a lesser extent α-N-acetylglucosamine.57–59 As a lectin control we used the Peanut agglutinin lectin (PNA) from Arachis hypogaea, known to selectively recognize β-galactose but not α-mannose epitopes. The studies were conducted by fluorescence spectroscopy using Texas Red®-labelled ConA and FITC-labelled PNA. Considering that PDA derivatives can exhibit autofluorescence, we conducted a lectin-free control assay (Fig. 5a and b), which showed that glycogel-1 lacks both green (Fig. 5a) and red fluorescence (Fig. 5b).However, an image showing a gel having a red fluorescence was observed upon treatment of glycogel-1 with ConA-Texas Red® (Fig. 5d), indicating an effective interaction between lectin and mannose on the surface of the fibers. Conversely, no fluorescence was observed when glycogel-1 was treated with PNA-FITC (Fig. 5c), indicating that glycogel-1 is able to establish selective interactions and is resistant to non-specific interactions. As an additional control, the same assay was performed using reverse labelled lectins (ConA-FITC and PNA-AlexaFluor™). In this case (Fig. 5e and f), and as expected, glycogel-1 exhibited fluorescence only in green. It is also remarkable that this specific recognition takes place in a very short time (30 s), comparable to the best systems reported in the literature.60 This efficiency can be explained by the so-called glycoside cluster effect,61–63 consequence of mannose multivalent exposition on the 1D-glyconanotubes in glycogel-1. In this sense we recently reported that mannose-coated single-walled carbon nanotubes are among the most efficient binder to ConA.31
Taking advantage of the possibility of visualizing sugar-lectin interactions by fluorescence microscopy, we conducted a study on the effect of the spatiotemporal arrangement of mannose on these interactions (Fig. 6) in the gel state.
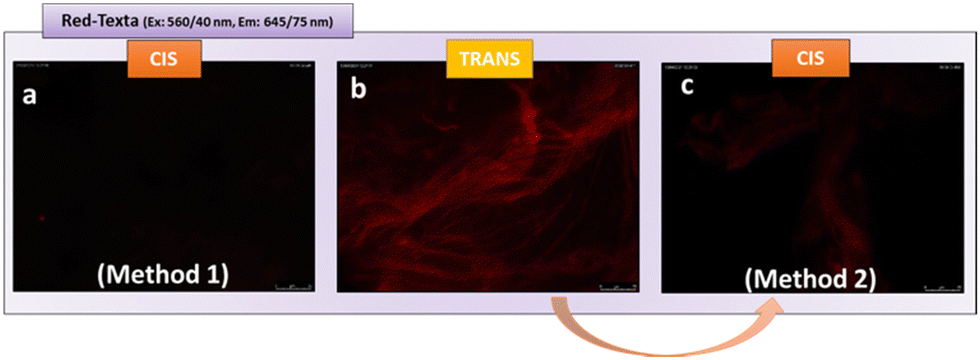 | ||
| Fig. 6 Fluorescence study of the effect of the spatiotemporal arrangement of mannose on gel-1-lectin interactions. Method 1: Image of the cisgel-1 after incubation with ConA-Texas Red (a). Method 2: Image of the transgel-1 after incubation with ConA-Texas Red (b), image of the transgel-1-ConA Texas Red after photoisomerization (c) | ||
For this, we carried out two different tests. The initial one consisted of isomerizing the gel from the stable trans form to the cis form, followed by incubation with lectin. After carrying out successive washes, we analysed the fluorescence of the aggregates obtained (method 1, Fig. 6). No fluorescence was detected in this case (Fig. 6a), highlighting the absence of ConA-mannose interaction. This result indicates for the first time, that after isomerization, the mannose within gel-1 in the cis form is no longer accessible and therefore cannot establish effective interactions with ConA. This observation is in line with previous research conducted by the Jayaraman group,64 the Hartmann group,65 and the Lindhorst group.66,67 These studies have highlighted the critical role of carbohydrate ligand orientation in lectin recognition, mainly in solution. The Jayaraman group pioneered the development of photoswitchable multivalent carbohydrate ligands, showcasing photoinduced variations in binding affinities.64 Similarly, the Hartmann group reported changes in inhibitory concentrations upon photoswitching, underscoring the importance of ligand structure and orientation.65 Additionally, the Lindhorst group demonstrated light-dependent differences in bacterial adhesion on immobilized glycosylated azo-benzene derivatives, both on microtiter plates66 and human cell membranes.67
Interestingly, in the second test, once the glycogel-1-ConA Texas Red aggregate was obtained (Fig. 6b), UV irradiation (360 nm, 30 min), followed by successive washes, led to the loss of the initial red fluorescence (method 2, Fig. 6c). This test indicates that glycogel-1 is able to release the adherent lectin by changing from the trans form to the cis form. The ability of glycogel-1 to capture and release soluble lectins highlights its potential for the development of regenerable biomaterials to fight against toxins and pathogen infections.
Supramolecular self-assembly of the more hydrophilic monomer 3
Finally, we focused on the supramolecular self-organization of monomer 3, which unlike the other two neoglycolipids is water soluble. We were especially interested in knowing if this monomer is capable of forming micelles or liposomes due to their importance as smart drug delivery systems. The amphiphilic neoglycolipid 3 showed a critical micellar concentration (CMC) in water of 22 μM as determined by DLS technique using a Zetasizer Nano ZS system. Formation of the nanomicellar system ManMic-3 was carried out by a simple dispersion of the neoglycolipid 3 in water at a concentration above the CMC. The photo-polymerization of the diacetylene function upon ultraviolet irradiation (254 nm) afforded a partially conjugated polydiacetylene backbone of alternating enyne groups (Fig. 7a). The characteristic size and morphology of the formed nanosystems were determined by DLS and TEM. The TEM micrograph shows the formation of several entities, being the spherical micelles ManMic-3 with sizes of 15 nm the most abundant (Fig. 7b). TEM analysis also shows the presence of liposome with approximately 150 nm in size (Fig. 7c). DLS analysis confirms the presence of vesicle having an average diameter of 184 nm, uniform in shape and monodisperse as indicated by the PDI of 0.05 (Fig. 7d). However, the micelles detected by TEM, could not be detected by DLS, since the signal is dominated by the powerful scattering of the liposome thus hiding the micelles present. Indeed, being micelles more than one order of magnitude smaller, scatter more than three orders of magnitude less for similar volume phase. As for the gel, we studied the photopolymerization of the starting DA monomers in the micelle by Raman spectroscopy (Fig. 7g).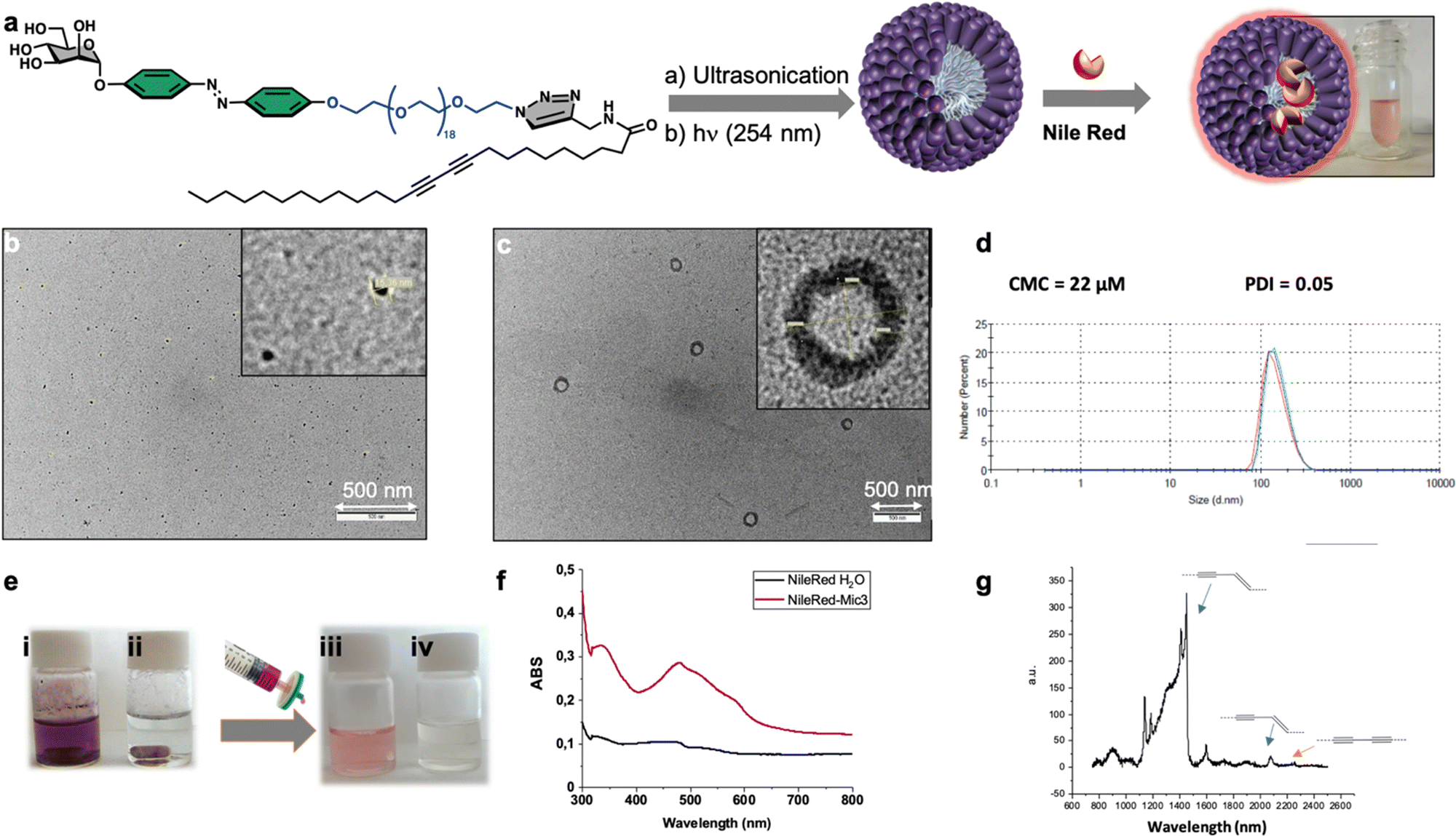 | ||
| Fig. 7 Synthesis and use as nanocontainer of static mannose-coated nanomicelles ManMic-3 through sonication-promoted supramolecular self-assembly followed by intermolecular photo-polymerization of neoglycolipid 3 in water (a). TEM images (b) and (c) of the formed micelles and liposomes. CMC and DLS size determination of MicMan-3 (d). Nile Red encapsulation study (e). Absorption spectra of Nile Red in water (black line) and in ManMic-3 solution (purple line) (f). Raman spectrum of the polymerized ManMic-3 (g). | ||
In this case the Raman spectrum shows the presence of the peak at 2268 cm−1 corresponding to the triple bond in the monomer together with the peaks at 2084 cm−1 and 1454 cm−1 corresponding to the conjugated ene-yne system, indicating an incomplete photopolymerization of the DA monomers in the micelles (Fig. 7g). The obtained micelles ManMic-3 present a sheltered hydrophobic inner cavity formed by the hydrocarbon tails, a hydrophilic PEG chain surrounded by a diazobenzene and mannose moieties. Interestingly, both the internal hydrophobic area and the diazobenzene corona of these micelles can, host hydrophobic guest molecules such as cytotoxic or image enhancing agents.30 As a preliminary test to show the encapsulation performance of ManMic-3 micelles, Nile Red was chosen as a model of hydrophobic drug and dye. Nile Red is insoluble in water (solubility <1 μg mL−1), Fig. 7e(i), the fluorescence of which increases substantially in hydrophobic environments. The micelles formulated with Nile Red were prepared simply mixing the previously formed ManMic3 micelles with Nile Red (Fig. 7e). After stirring and heating at 50 °C for 24 hours, filtration for elimination of the non-encapsulated dye, a pink solution without visible precipitation was obtained (Fig. 7e(iii)). In comparison, applying the same procedure in water without micelles resulted in a clear colorless solution (Fig. 7e(iv)), which also did not show the characteristic band absorbance for Nile Red (Fig. 7f).
Subsequently, we investigated the potential of MicMan3 as a nanocarrier for hydrophobic drugs, with the aim of improving their water solubility and protecting them within the hydrophobic core. Docetaxel (DTX), a drug widely used in clinical practice but with very low aqueous solubility (solubility 0.3 μg mL−1 at 37 °C), presented a significant challenge. In response to this challenge, we endeavored to create inclusion complexes using a solvent-free method involving stirring and heating. This approach eliminated the need for organic solvents. The quantity of incorporated DTX was determined by measuring the difference in mass between the loaded and empty micelles, complemented by HPLC analysis. The drug loading content (DLC) and encapsulation efficiency (DLE) were calculated using eqn (1) and (2) as follows:
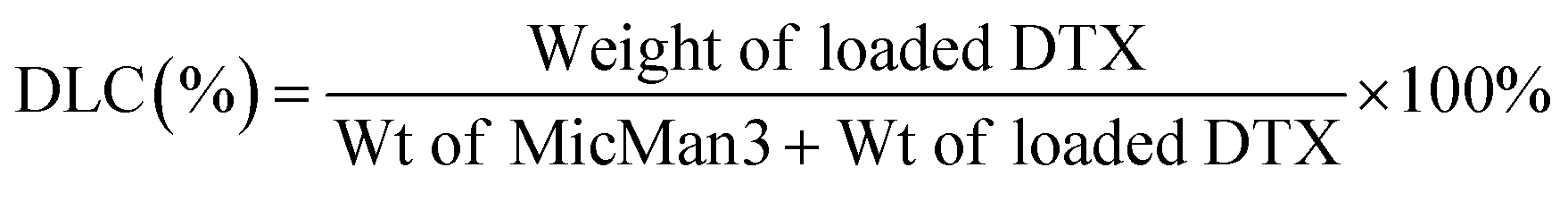 | (1) |
 | (2) |
The drug loading content and encapsulation efficiency of MicMan3 for DTX were 2.8% and 11.2%. These preliminary findings underscore the capability of MicMan3 to enhance the water solubility of highly insoluble organic molecules, which is of utmost significance for their potential utility as effective drug delivery systems.
Conclusions
In conclusion, we have developed a versatile design for the synthesis of multifunctional diazobenzene/diacetylene glycoamphiphiles. By adjusting the spacer length and the presence of a triazole ring, we can control the hierarchical self-organization of these molecules. Neoglycolipid 3 forms spherical micelles (MicMan-3) with a sheltered inner core, capable of solubilizing hydrophobic molecules like Nile Red and docetaxel. On the other hand, monomer 1 forms a hydro-alcoholic gel without the need for additional processing, consisting of entangled 1D-glyconanotubes with a nanometric inner cavity. This gel exhibits photosensitivity, undergoing gel–sol transition upon UV irradiation and regenerating gels through thermal or visible light stimuli. The gel shows controlled release properties for hydrophilic and hydrophobic dyes, making it a potential smart drug delivery system. Furthermore, the gel selectively interacts with lectins such as Texas red labeled Concanavalin A, thanks to the exposed mannose residues on the entangled nanotubes. Additionally, the gel demonstrates the ability to capture and release soluble lectins, offering potential applications in regenerable biomaterials for combating toxin and pathogen infections.Experimental section
General methods
Chemicals employed all over this work were purchased from Sigma Aldrich Chemical Co. Dry solvents were purchased from SDS in HPLCs grade and in addition dried in a solvent purification system (Pure Solv MD5, Innovative Technology). The monitoring of the reactions was carried out by TLC, employing aluminum sheets coated with silica gel 60 F254 (normal phase) purchased from Merck, with detection by charring with phosphomolybdic acid/EtOH and sulfuric acid/EtOH. For flash chromatography, silica Gel (Merck 230–400 mesh) was used. The organic extracts were dried over anhydrous sodium sulfate and concentrated under vacuum. Columns were eluted with positive air pressure. Chromatographic eluents are given as volume to volume ratios (v/v). NMR spectra were recorded with a BRUKER AC-500 apparatus. Deuterated solvents are indicated in brackets. Chemical shift values (δ) are referred to tetramethylsilane (TMS), utilized as internal reference; then, the spectral signals were calibrated according to the non-deuterated residual peak of the solvent. Optical rotations [α]20D were determined with a PerkinElmer 341 polarimeter using a sodium lamp (λ = 589 nm) with a 10 cm cell length. UV/Vis spectra were recorded on a UV/vis PerkinElmer Lambda 12, using quartz cuvettes. HR-MS were recorded on a Kratos MS-80RFA 241-MC apparatus. Transmission Electron Microscopy (TEM) images were taken by Philips CM 10 or CM 200 apparatuses with an accelerating voltage of 80 kV or 200 kV, respectively. Typically, a very small volume of the aqueous solutions (20 μL) was deposited over carbon-coated copper grids and uranyl acetate 2% as the negative stain. High resolution transmission electron microscopy (HRTEM) images were taken by a JEOL JEM-2200FS microscope, equipped with a field emission gun working at an accelerating voltage of 200 kV, a CEOS spherical aberration corrector and an Omega filter. Scanning electron microscopy (SEM) images were obtained on a JEOL JSM-5400 apparatus. Samples were prepared by depositing 15 μl of the suspension onto grids, allowing the grids to absorb for 2 minutes. Small angle X-ray scattering (SAXS) was performed on a PANalytical X'Pert PRO:1)), affording the tetraacetylated mannose (lactol) as a white syrup in 88% yield. Subsequently, to a solution of the lactol (314 mg 0.90 mmol) in dry CH2Cl2 (6.30 mL) at −5 °C, DBU solution was added dropwise for 1 hour. Once the reaction finished, the crude was filtered over a pad of Celite® and the organic extracts were washed with CH2Cl2 and evaporated under reduced pressure. Finally, after purifying by flash column chromatography (EtOAc/Hexanes (3:7), 4 was obtained as a yellow syrup with a yield of 70%. Rf: 0.41 EtOAc/Hexanes (2:3). [α]20D: +23.5(c 1.4, CHCl3). 1H NMR (400 MHz, CDCl3): δ 8.81 (s,1H), 6.30 (d, 1H, J = 1.92 Hz), 5.50 (t, 1H, J = 2.47 Hz), 5.44–5.42 (m, 2H), 4.31 (dd, 1H, J = 11.5 Hz, J = 4.55 Hz), 4.22–4.17 (m, 2H), 2.22 (s,3H), 2.11 (s, 3H), 2.09 (s, 3H), 2.03 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 170.6, 169.8, 169.7, 169.6, 159.8, 94.5, 90.5, 71.2, 68.8, 67.9, 65.4, 62.0, 20.7, 20.3. HRMS: calcd. for C16H20O10NCl3Na 514.0045 [M + Na]+, found 514.0043.
:2)), affording 5 as a red solid in 30% yield. Rf: 0.387 EtOAc/Hexanes (1:2). 1H NMR (400 MHz, MeOD): δ 7.77 (d, 4H, J = 8.85 Hz), 6.91 (d, 4H, J = 8.79 Hz), 3.33 (s, 2H). 13C NMR (100 MHz, CDCl3): δ 159.9, 146.2, 123.9, 115.5. HRMS: calcd. for C12H11N2O2 215.0815 [M + H]+, found 215.0813.
:3)), affording 6 as a yellow solid in 89% yield. Rf: 0.373 EtOAc/Hexanes (2:3). [α]20D: +9.8 (c 0.5, CHCl3). 1H NMR (400 MHz, MeOD): δ 7.86 (dd, 4H, J = 8.6 Hz, J = 29.2 Hz), 7.32 (d, 2H, J = 9.17 Hz), 6.92 (d, 2H, J = 8.02 Hz), 5.75 (s, 1H), 5.54–5.48 (m, 2H), 5.35 (t, 1H, J 10.31 Hz), 4.27 (dd, 2H, J= 5.16, J = 12.03Hz), 4.16–4.07 (m, 2H), 2.21 (s, 3H), 2.08 (s, 3H), 2.03 (s, 3H), 1.98 (s, 3H). 13C NMR (100 MHz, MeOD): δ 170.8, 170.2, 160.5, 157.1, 148.4, 146.1, 124.3, 123.6, 116.7, 115.3, 95.7, 69.4, 69.1, 68.9, 65.6, 61.9, 19.2. HRMS: calcd. for C26H28N2O11Na 567.1585 [M + Na]+, found 567.1578.
:1)), affording the protected monoamine spacer as a yellow syrup in 91% yield. Next, a solution of p-toluenesulfonyl choride (1.1 g, 5.79 mmol) in THF (1.6 mL) is added dropwise on a solution of the Boc protected spacer (1.7 g, 5.79 mmol) and Et3N (0.49 mL) in THF (1.6 mL) at 0 °C, and the reaction mixture was stirred overnight at room temperature. The reaction product was filtered over a pad of Celite®, washed with ethyl acetate and purified by column chromatography (EtOAc/Hexanes (2:1)), affording the spacer 7 as a yellow syrup in 42% yield. Rf: 0.343 EtOAc/Hexanes (1:1). 1H NMR (400 MHz, CDCl3): δ 7.77 (d, 2H, J = 8.25 Hz), 7.32 (d, 2H, J = 8.12 Hz), 5.28 (s, 1H), 4.14 (t, 2H, J = 4.75 Hz), 3.67 (t, 2H, J = 4.92 Hz), 3.61–3.56 (m, 8H), 3.50 (t, 2H, J = 5.08 Hz), 3.27 (t, 2H, J = 4.98 Hz), 2,42 (s, 3H), 1.41 (s, 9H). 13H NMR (100 MHz, CDCl3): δ 155.9, 144.8, 132.9, 129.8, 127.9, 79.1, 70.7, 68.6, 40.5, 28.4, 21.6. HRMS: calcd. for C20H33O8SNNa 470.1819 [M + Na] +, found 470.1815.
:1)), affording compound 8 as a yellow syrup in 40% yield. Rf: 0.447 EtOAc/Hexanes (1:1). 1H NMR (400MHz, CDCl3): δ 7.81 (d, 2H, J = 7.67 Hz), 7.36 (d, 2H, J = 7.29 Hz), 4.18 (t, 2H, J = 4.55 Hz), 3.75–3.55 (m, 12H), 3.39 (t, 2H, J = 4.7Hz), 2.46 (s, 3H). 13C NMR (100MHz, CDCl3): δ 144.6, 132.9, 129.8, 127.9, 70.6, 69.3, 68.6, 50.6, 21.6. HRMS: calcd. for C15H23O6N3SNa 396.1200 [M + Na]+, found 396.1193.
:1)), affording the desired compound 9 as a white solid in 67% yield. Rf: 0.54 CH2Cl2/MEOH (20:1). 1H NMR (400MHz, CDCl3): δ 7.81 (d, 2H, J = 7.95Hz), 7.36 (d, 2H, J = 7.95Hz), 4.18 (t, 2H, J = 3.67Hz), 3.75–3.45 (m, 76H, 38), 3.41 (t, 2H, J = 4.70 Hz), 2.65 (s, 3H). 13C NMR (100MHz, CDCl3): δ 144.8, 133.0, 129.9, 128.0, 70.6, 70.1, 69.3, 68.2, 50.7, 21.7. HRMS: calcd. for C47H87O22N3NaS a 1100.5394 [M + Na]+, found 1100.5394.
:3)), to give the pure compound 10 as a yellow syrup in 70% yield. Rf: 0.3 Et2O/Hexanes (6:1). [α]20D: +3.05 (c 1, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.89 (d, 4H, J = 8.56 Hz), 7.23 (d, 2H, J = 8.45 Hz), 7.04 (d, 2H, J = 8.26 Hz), 5.64 (d, 1H, J = 1.42 Hz) 5.61 (dd, 1H, J = 3.33 Hz, J = 10.02 Hz) 5.50 (dd, 1H, J = 1.42 Hz, J = 3.33 Hz), 5.40 (t, 1H, J= 10.13 Hz), 4.32 (dd, 1H, J = 12.19 Hz, J = 5.51 Hz), 4.27–4.21 (t, 2H, J = 3.54 Hz), 4.16–4.07 (m, 2H), 3.95–3.89 (t, 2H, J = 4.90 Hz), 3.80–3.62 (m, 8H), 3.56 (t, 2H, J = 4.44 Hz) 2.23 (s, 3H), 2.08 (s, 3H), 2.07 (s, 3H), 2.05 (s, 3H), 1.46 (s, 9H). 13C NMR (100 MHz, CDCl3): δ 170.5, 170.0, 169.9, 169.9, 161.1, 157.2, 148.5, 147.0, 124.4, 124.2, 116.7, 114.8, 95.7, 79.2, 70.2, 69.6, 69.3, 68.8, 67.8, 65.9, 62.0, 40.4, 28.4, 20.9. HRMS: calcd. for C39H53N3O16Na 842.3329 [M + Na]+, found 842.3308.
:1)), affording compound 11 as a yellow syrup in 97% yield. Rf: 0.481 CH2Cl2/MeOH (5:1). [α]20D: +33.42 (c 0.9, CHCl3). 1H NMR (400 MHz, MeOD): δ 7.90 (dd, 4H, J = 8.89 Hz, J = 1.85 Hz), 7.31 (d, 2H, J = 8.88 Hz), 7.11 (d, 2H, J = 8.06 Hz), 5.76 (br, 1H), 5.54–5-49 (m, 2H), 5.36 (t, 1H, J = 10.22 Hz), 4.30–4.24 (m, 3H), 4.16–4.07 (m, 2H), 3.91 (t, 2H, J = 4.5 Hz), 3.79–3.74 (m, 2H), 3.73–3.66 (m, 10H), 3.12 (t, 2H, J = 4.86 Hz) 2.21 (s, 3H), 2.08 (s, 3H), 2.03 (s, 3H), 1.98 (s, 3H). 13C NMR (100 MHz, MeOD): δ 170.8, 170.2, 170.1, 170.0, 161.3, 157.4, 148.4, 146.9, 124.2, 124.2, 116.8, 114.6, 95.7, 70.2, 70.0, 69.4, 69.3, 68.9, 67.6, 66.4, 61.9, 39.2, 19.2. HRMS: calcd. for C34H46N3O14 720.2974 [M + H]+, found a 720.2966.
:1)) affording compound 12 as a yellowish solid in 60% yield. Rf: 0.451 CH2Cl2/MeOH (40:1). [α]20D: +23.68 (c 0.5, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.90 (dd, 4H, J = 8.65 Hz, J = 3.34 Hz), 7.23 (d, 2H, J = 7.23 Hz), 7.05 (d, 2H, J = 8.9 Hz), 6.14 (s, 1H), 5.64 (d, 1H, J = 1.09 Hz), 5.61 (dd, 1H, J = 3.41 Hz, J = 10.07 Hz), 5.51 (dd, 1H, J = 3.5 Hz, J = 1.85 Hz), 5.41 (t, 1H, J =10.11 Hz), 4.33 (dd, 1H, J = 12.20 Hz, J = 5.81 Hz), 4.25 (t, 2H, J = 4.73 Hz), 4.16–4.09 (m, 1H), 3.93 (t, J = 4.6 Hz), 3.81–3.63 (8H), 3.58 (t, 2H, J = 4.59 Hz) 3.50–3.45 (m, 2H), 2.29–2.21 (m, 7H), 2.18 (t, 2H, J = 7.56 Hz) 2.09 (s, 3H), 2.08(s, 3H), 2.06 (s, 3H), 1.69–1.59 (m, 2H) 1.55–1.50 (dd, 2H, J =7.41Hz, J = 7.48 Hz), 1.43–1.25 (m, 26H), 0.90 (t, 3H, J = 6.9 Hz). 13C NMR (100 MHz, CDCl3): δ 173.2, 170.5, 170.0, 169.8, 161.0,157.2, 148.5, 147.1, 124.6, 124.2, 116.7, 114.8, 95.7, 70.9, 70.0, 69.6, 69.4, 69.3, 68.8, 67.7, 65.9, 65.3, 65.2, 62.1, 39.1, 36.7, 33.4, 28.3, 25.7, 22.7, 20.9, 19.1, 14.1. HRMS: calcd. for C59H85N3O15Na 1098.5873 [M + Na]+, found 1198.5858.
:3). [α]20D: +39.7 (c 0.7, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.90 (dd, 4H, J = 8.74 Hz, J = 3.66 Hz), 7.32 (d, 2H, J = 9.00 Hz), 7.05 (d, 2H, J = 9.02 Hz), 5.63 (d, 1H, J = 1.54 Hz) 5.60 (dd, 1H, J = 10.06 Hz, J = 3.56 Hz), 5.51–5.50 (m, 1H), 5.41 (t, 1H, J = 10.10 Hz), 4.32 (dd, 1H, J = 12.19 Hz, J = 5.51 Hz, 4.26–4.23 (m, 2H), 4.14–4.09 (m, 2H), 3.94–3.91 (t, 2H, J = 4.90), 3.79–3.75 (m, 2H) 3.62 (m, 8H), (m, 2H), 2.23 (s, 3H), 2.09 (s, 3H), 2.07 (s, 3H), 2.06 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 170.5, 170.0, 169.9, 169, 161.2, 157.2, 148.4, 146.9 124.6, 124.3 116.7, 114.9, 95.7, 70.9, 70.7–70.7 (m), 70.1, 69.6, 69.3, 68.8, 67.8, 65.9, 62.1, 50.7, 20.7. HRMS: calcd. for C34H43N5O14Na 768.2699 [M + Na]+, found 768.2692.
:1). [α]20D: +10.2 (c 0.35, CHCl3). 1H NMR (400 MHz, MeOH): δ 7.93–7.89 (dd, 4H, J = 4.67 Hz, J = 9.75 Hz), 7.347.31 (d, 2H, J = 9.00 Hz), 7.14–7.12 (d, 2H, J = 9.63 Hz), 5.78 (d, 1H, J = 0.93 Hz), 5.54–5.51 (m, 2H), 5.36 (t, 1H, J = 9.99 Hz), 4.30–4.24 (m, 3H), 4.16–4.08 (m, 2H), 3.91 (t, 2H, J = 3.9 Hz), 3.76–3.72 (m, 2H), 3.71–3.65 (m, 72H), 3.39 (t, 2H, J = 5.27 Hz, J = 9.61 Hz), 2.22 (s, 3H), 2.09 (s, 3H), 2.04 (s, 3H), 1.99 (s, 3H). 13C NMR (100 MHz, MeOH): δ 170.8, 170.2–170.1 (3C), 161.5, 157.3, 148.4, 146.9, 124.2, 123.8, 116.8, 114.7, 95.7, 70.2–70.1 (m), 69.7, 69.5, 69.1, 68.9, 67.7, 65.6, 61.9, 50.0, 19.2. HRMS: calcd. for C66H107O30N5Na a 1450.6879 [M + Na]+, found 1472.6893.
:3)), affording compound 15 as a white solid in 83% yield. Rf: 0.50 EtOAc/Hexanes (1:3). 1H NMR (500 MHz, CDCl3): δ 5.50 (br. s, 1H), 4.06–4.04 (m, 2H), 2.23 (t, 4H, J = 6.9 Hz), 2.18 (t, 2H, J = 7.6 Hz, COC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif)
![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) CH2), 1.64–1.60 (m, 2H, COCH2C), 1.52–1.47 (m, 4H), 1.40–1.25 (m, 26H), 0.88 (t, 3H, J = 6.5 Hz). 13C NMR (125 MHz, CDCl3): δ 172.6, 79.7, 77.6, 77.4, 71.6, 65.3, 65.2, 36.5, 31.9, 29.6, 29.5, 29.3, 29.2, 29.1, 28.9, 28.7, 28.4, 28.3, 25.5, 22.6, 19.2, 19.1, 14.1. HRMS: calcd. for C28H45ONNa a 434.3398 [M + Na]+, found 434.3394.
:1)) to give the compound 16 as a yellow solid in 60% yield. Rf: 0.283 EtOAc/Hexanes (20:1). [α]20D: +16.7 (c 0.4, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.89 (d, 4H, J = 8.67 Hz), 7.73 (s, 1H), 7.23 (d, 2H, J = 8.92 Hz), 7.03 (d, 2H, J = 8.98 Hz), 6.29 (t, 1H, J = 4.8 Hz), 5.63 (d, 1H, J = 1.2 Hz), 5.61 (dd, 1 H, J = 10.08 Hz, J = 3.38 Hz), 5.51–5.50 (m, 1H) 5.43–5.38 (t, 1H, J = 10.29 Hz), 4.55–4.50 (m, 4H) 4.34–4.60 (dd, 1H, J = 5.37 Hz, J = 12.45 Hz), 4.24–4.22 (m, 1H), 4.15–4.09 (m, 2H, H-5), 3.92–3.86 (m, 4H), 3.77–3.62 (m, 8 H), 2.27–2.24 (m, 4 H), 2.21–2.17 (m, 2H), 2.11 (s, 3H), 2.08 (s, 3H), 2.07 (s, 3H), 2.06 (s, 3H), 1.64–1.49 (m, 2H), 1.40–1.35 (m, 4H), 1.33–1.27 (s, 26 H), 0.91–0.88 (t, 3H, J = 6.34 Hz). 13C NMR (100 MHz, CDCl3): δ 173.1, 170.5, 169.9, 169.7, 161.1, 157.2, 148.5, 147.0, 144.4, 124.5–124.2, 123.2, 116.7, 114.8, 95.7, 70.9, 70.5, 69.4, 69.3, 68.9, 67.7, 65.9, 65.23, 62.2, 50.3, 36.5, 34.8, 31.1–28.9 (m), 28.4, 25.6, 22.8, 20.9, 20.7, 19.2,14.1. HRMS: calcd. for C62H88N6O15 1179.6200 [M + Na]+, found 1179.6190.
:1)), affording compound 17 as a yellow solid within 80% yield. Rf: 0.18 CH2Cl2/MeOH (15:1). [α]20D: +16.8 (c 0.73, CHCl3). 1H NMR (400 MHz, MeOD): δ 7.98 (s, 1H), 7.91 (d, 4H, J = 4.07 Hz, J = 9.69 Hz), 7.32 (d, 2H, J = 8.50 Hz), 7.14–7.11 (d, 2H, J = 8.43 Hz), 5.78 (br. s, 1H), 5.56–5.48 (m, 2H), 5.37 (t, 1H, J = 9.19 Hz), 4.58 (t, 2H) 4.45 (t, 2H) 4.31–4.22 (m, 3H), 4.17–4.07 (m, 2H, H-5), 3.92–3.86 (m, 4H), 3.77–3.54 (m, 72H), 2.27–2.19 (m, 9 H), 2.09 (s, 3H), 2.04 (s, 3H), 1.99 (s, 3H), 1.68–1.59 (m, 2H), 1.57–1.47 (m, 4H), 1.45–1.24 (s, 26H), 0.92 (t, 3H, J = 6.34 Hz). 13C NMR (100 MHz, MeOD): δ 170.8, 170.2–170.0, 161.5, 157.3, 148.4, 146.9, 145.7, 124.3, 123.8, 116.9, 114.8, 95.7, 76.6, 70.7–69.8 (m), 69.5, 69.0, 68.9, 67.7, 65.6, 65.1, 61.9, 50.2, 35.7, 34.1, 31.5, 29.4–28.4 (m), 28.1, 25.5, 22.4, 19.2, 18.3, 13.1. HRMS: calcd. for C94H152O31N6Na a 1884.0394 [M + Na]+, found 1884.0330.
:1). [α]20D: −7.93 (c 0.2, CHCl3). 1H NMR (400 MHz, DMSO d6): δ 7.84 (t, 5H, J = 7.80 Hz), 7.26 (d, 2H, J = 8.56 Hz), 7.13 (d, 2H, J = 8.85 Hz), 5.52 (brs, 1H), 4.21 (br, 2H), 3.87 (s, 1H), 3.79 (br, 2H), 3.72 (dd, 1H, J = 3.32 Hz, J = 6.02 Hz), 3.66–3.48 (m, 13H,), 3.38 (m, 1H), 3.19 (m), 2.27 (m, 4H), 2.04 (t, J = 7.5 Hz), 1.47–1.39 (m, 6H), 1.31–1.23 (m, 26H), 1.31–1.23 (m, 26H), 0.86 (t, 3H, J = 6.88 Hz). 13C NMR (100 MHz, DMSO d6): δ 172.7, 161.3, 158.91, 147.4, 146.7, 124.7, 124.4, 117.5, 115.5, 99.2, 78.5, 75.7, 71.2, 70.6–70.0 (m), 69.7, 68.0, 67.1, 65.8, 61.5, 39.0, 35.9, 31.9–28.7 (m) 28.2, 25.9, 22.6, 18.8, 14.4. HRMS: calcd. for C51H77N3O11Na 930.5450 [M + Na]+, found 930.5441.
:1). [α]20D: 35.37 (c 0.3, CHCl3). 1H NMR (400 MHz, DMSO d6): δ 8.33–8.24 (br, 1H), 7.87–7.82 (m, 5H), 7.27–7.25 (d, 2H, J = 8.36 Hz), 7.14–7.12 (d, 2H, J = 8.34 Hz), 5.52 (brs, 1H), 4.49 (br, 2H), 4.28 (d, 2H, J = 5.25 Hz), 4.20 (s, 2H), 3.88 (s, 1H), 3.83–3.76 (m, 4H) 3.71 (d, 1H, J = 8.5 Hz) 3.64–3.12 (m, 12H), 2.26 (m, 4H), 2.08 (t, 2H, J = 6.6 Hz), 1.53–1.37 (m, 6H) 1.36–1.18 (m, 26H), 0.86 (t, 3H, J = 6.37 Hz). 13C NMR (100 MHz, DMSO d6): δ 172.6, 161.3, 158.9, 147.4, 146.7, 145.4, 124.7, 124.4, 123.6, 117.6, 115.5, 99.2, 78.5, 75.6, 71.1, 70.6–69.7 (m), 69.3, 68.7, 67.1 (C4), 65.8 (2C), 60.7, 49.7, 35.9, 34.5, 31.9–28.7 (m), 28.2, 25.6, 22.5, 18.8, 14.4. HRMS: calcd. for C54H80N6O11:1011.5777 [M + Na]+, found: 1011.5769.
:1). [α]20D: +15.3 (c 0.55, CHCl3). 1H NMR (400 MHz, MeOD): δ 7.92 (s, 1H), 7.89 (dd, 4H, J = 5.21 Hz, J = 8.63 Hz), 7.29 (d, 2H, J = 8.81 Hz), 7.13–7.11 (d, 2H, J = 8.81 Hz), 5.62 (d, 1H, J1,2 = 1.34 Hz), 4.59–4.57 (t, 2H, J = 5.05 Hz, J = 9.77 Hz), 4.45 (s, 2H), 4.27–4.25 (m, 2H), 4.06 (br, 1H), 3.97–3.86 (m, 5H), 3.83–3.72 (m, 6H), 3.72–3.57 (m, 70H), 2.28–2.19 (m, 6H), 1.63 (t, 2H, J = 7.8 Hz), 1.56–1.47 (m, 4H), 1.46–1.26 (m, 26H), 0.92 (t, 3H, J = 7.15 Hz). 13C NMR (100 MHz, MeOD): δ. 174.8, 161.3, 158.5, 147.9, 146.9, 145.1, 124.1, 123.8, 116.6, 114.6, 98.8, 76.6, 74.4, 70.1–70.0, 69.4, 69.0, 67.7, 66.9, 65.1, 61.2, 50.1, 35.6, 34.2, 31.7, 29.4–28.4, 28.2, 25.5, 22.4, 18.3, 13.1 (CH3). HRMS: calcd. for C86H144O27N6Na a 1715.9972 [M + Na]+,
:1 or 1:2), then the suspension was heated to 50 °C during 5 min. Cooling the obtained clear solution to room temperature, induced the gelation of the mixture. The as obtained gel and the lyophilized gel (Xerogel) were characterized by scanning electron microscopy and SAXS as described below.
CH2), 1.64–1.60 (m, 2H, COCH2C), 1.52–1.47 (m, 4H), 1.40–1.25 (m, 26H), 0.88 (t, 3H, J = 6.5 Hz). 13C NMR (125 MHz, CDCl3): δ 172.6, 79.7, 77.6, 77.4, 71.6, 65.3, 65.2, 36.5, 31.9, 29.6, 29.5, 29.3, 29.2, 29.1, 28.9, 28.7, 28.4, 28.3, 25.5, 22.6, 19.2, 19.1, 14.1. HRMS: calcd. for C28H45ONNa a 434.3398 [M + Na]+, found 434.3394.
:1)) to give the compound 16 as a yellow solid in 60% yield. Rf: 0.283 EtOAc/Hexanes (20:1). [α]20D: +16.7 (c 0.4, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.89 (d, 4H, J = 8.67 Hz), 7.73 (s, 1H), 7.23 (d, 2H, J = 8.92 Hz), 7.03 (d, 2H, J = 8.98 Hz), 6.29 (t, 1H, J = 4.8 Hz), 5.63 (d, 1H, J = 1.2 Hz), 5.61 (dd, 1 H, J = 10.08 Hz, J = 3.38 Hz), 5.51–5.50 (m, 1H) 5.43–5.38 (t, 1H, J = 10.29 Hz), 4.55–4.50 (m, 4H) 4.34–4.60 (dd, 1H, J = 5.37 Hz, J = 12.45 Hz), 4.24–4.22 (m, 1H), 4.15–4.09 (m, 2H, H-5), 3.92–3.86 (m, 4H), 3.77–3.62 (m, 8 H), 2.27–2.24 (m, 4 H), 2.21–2.17 (m, 2H), 2.11 (s, 3H), 2.08 (s, 3H), 2.07 (s, 3H), 2.06 (s, 3H), 1.64–1.49 (m, 2H), 1.40–1.35 (m, 4H), 1.33–1.27 (s, 26 H), 0.91–0.88 (t, 3H, J = 6.34 Hz). 13C NMR (100 MHz, CDCl3): δ 173.1, 170.5, 169.9, 169.7, 161.1, 157.2, 148.5, 147.0, 144.4, 124.5–124.2, 123.2, 116.7, 114.8, 95.7, 70.9, 70.5, 69.4, 69.3, 68.9, 67.7, 65.9, 65.23, 62.2, 50.3, 36.5, 34.8, 31.1–28.9 (m), 28.4, 25.6, 22.8, 20.9, 20.7, 19.2,14.1. HRMS: calcd. for C62H88N6O15 1179.6200 [M + Na]+, found 1179.6190.
:1)), affording compound 17 as a yellow solid within 80% yield. Rf: 0.18 CH2Cl2/MeOH (15:1). [α]20D: +16.8 (c 0.73, CHCl3). 1H NMR (400 MHz, MeOD): δ 7.98 (s, 1H), 7.91 (d, 4H, J = 4.07 Hz, J = 9.69 Hz), 7.32 (d, 2H, J = 8.50 Hz), 7.14–7.11 (d, 2H, J = 8.43 Hz), 5.78 (br. s, 1H), 5.56–5.48 (m, 2H), 5.37 (t, 1H, J = 9.19 Hz), 4.58 (t, 2H) 4.45 (t, 2H) 4.31–4.22 (m, 3H), 4.17–4.07 (m, 2H, H-5), 3.92–3.86 (m, 4H), 3.77–3.54 (m, 72H), 2.27–2.19 (m, 9 H), 2.09 (s, 3H), 2.04 (s, 3H), 1.99 (s, 3H), 1.68–1.59 (m, 2H), 1.57–1.47 (m, 4H), 1.45–1.24 (s, 26H), 0.92 (t, 3H, J = 6.34 Hz). 13C NMR (100 MHz, MeOD): δ 170.8, 170.2–170.0, 161.5, 157.3, 148.4, 146.9, 145.7, 124.3, 123.8, 116.9, 114.8, 95.7, 76.6, 70.7–69.8 (m), 69.5, 69.0, 68.9, 67.7, 65.6, 65.1, 61.9, 50.2, 35.7, 34.1, 31.5, 29.4–28.4 (m), 28.1, 25.5, 22.4, 19.2, 18.3, 13.1. HRMS: calcd. for C94H152O31N6Na a 1884.0394 [M + Na]+, found 1884.0330.
:1). [α]20D: −7.93 (c 0.2, CHCl3). 1H NMR (400 MHz, DMSO d6): δ 7.84 (t, 5H, J = 7.80 Hz), 7.26 (d, 2H, J = 8.56 Hz), 7.13 (d, 2H, J = 8.85 Hz), 5.52 (brs, 1H), 4.21 (br, 2H), 3.87 (s, 1H), 3.79 (br, 2H), 3.72 (dd, 1H, J = 3.32 Hz, J = 6.02 Hz), 3.66–3.48 (m, 13H,), 3.38 (m, 1H), 3.19 (m), 2.27 (m, 4H), 2.04 (t, J = 7.5 Hz), 1.47–1.39 (m, 6H), 1.31–1.23 (m, 26H), 1.31–1.23 (m, 26H), 0.86 (t, 3H, J = 6.88 Hz). 13C NMR (100 MHz, DMSO d6): δ 172.7, 161.3, 158.91, 147.4, 146.7, 124.7, 124.4, 117.5, 115.5, 99.2, 78.5, 75.7, 71.2, 70.6–70.0 (m), 69.7, 68.0, 67.1, 65.8, 61.5, 39.0, 35.9, 31.9–28.7 (m) 28.2, 25.9, 22.6, 18.8, 14.4. HRMS: calcd. for C51H77N3O11Na 930.5450 [M + Na]+, found 930.5441.
:1). [α]20D: 35.37 (c 0.3, CHCl3). 1H NMR (400 MHz, DMSO d6): δ 8.33–8.24 (br, 1H), 7.87–7.82 (m, 5H), 7.27–7.25 (d, 2H, J = 8.36 Hz), 7.14–7.12 (d, 2H, J = 8.34 Hz), 5.52 (brs, 1H), 4.49 (br, 2H), 4.28 (d, 2H, J = 5.25 Hz), 4.20 (s, 2H), 3.88 (s, 1H), 3.83–3.76 (m, 4H) 3.71 (d, 1H, J = 8.5 Hz) 3.64–3.12 (m, 12H), 2.26 (m, 4H), 2.08 (t, 2H, J = 6.6 Hz), 1.53–1.37 (m, 6H) 1.36–1.18 (m, 26H), 0.86 (t, 3H, J = 6.37 Hz). 13C NMR (100 MHz, DMSO d6): δ 172.6, 161.3, 158.9, 147.4, 146.7, 145.4, 124.7, 124.4, 123.6, 117.6, 115.5, 99.2, 78.5, 75.6, 71.1, 70.6–69.7 (m), 69.3, 68.7, 67.1 (C4), 65.8 (2C), 60.7, 49.7, 35.9, 34.5, 31.9–28.7 (m), 28.2, 25.6, 22.5, 18.8, 14.4. HRMS: calcd. for C54H80N6O11:1011.5777 [M + Na]+, found: 1011.5769.
:1). [α]20D: +15.3 (c 0.55, CHCl3). 1H NMR (400 MHz, MeOD): δ 7.92 (s, 1H), 7.89 (dd, 4H, J = 5.21 Hz, J = 8.63 Hz), 7.29 (d, 2H, J = 8.81 Hz), 7.13–7.11 (d, 2H, J = 8.81 Hz), 5.62 (d, 1H, J1,2 = 1.34 Hz), 4.59–4.57 (t, 2H, J = 5.05 Hz, J = 9.77 Hz), 4.45 (s, 2H), 4.27–4.25 (m, 2H), 4.06 (br, 1H), 3.97–3.86 (m, 5H), 3.83–3.72 (m, 6H), 3.72–3.57 (m, 70H), 2.28–2.19 (m, 6H), 1.63 (t, 2H, J = 7.8 Hz), 1.56–1.47 (m, 4H), 1.46–1.26 (m, 26H), 0.92 (t, 3H, J = 7.15 Hz). 13C NMR (100 MHz, MeOD): δ. 174.8, 161.3, 158.5, 147.9, 146.9, 145.1, 124.1, 123.8, 116.6, 114.6, 98.8, 76.6, 74.4, 70.1–70.0, 69.4, 69.0, 67.7, 66.9, 65.1, 61.2, 50.1, 35.6, 34.2, 31.7, 29.4–28.4, 28.2, 25.5, 22.4, 18.3, 13.1 (CH3). HRMS: calcd. for C86H144O27N6Na a 1715.9972 [M + Na]+,
:1 or 1:2), then the suspension was heated to 50 °C during 5 min. Cooling the obtained clear solution to room temperature, induced the gelation of the mixture. The as obtained gel and the lyophilized gel (Xerogel) were characterized by scanning electron microscopy and SAXS as described below.
Studies on the release of RhoB from gel-1/RhoB complex
:1) was performed in the maximum absorbance peak wavelength of 550 nm in UV/Vis PerkinElmer Lambda 12 (Fig. S43, ESI†).
Gel-1/RhoB complex was covered by 1 mL of the same solvent mixture used to form of gels, and at a given time (5 min, 15 min, 30 min, 1 h, 2 h, 3 h, 6 h, 10 h, 24 h, 33 h and 48 h), the covered layer was removed for analysis, and replaced by the same volume (1 mL) of solvent mixture. The RhoB present in the removed layer was quantified by measuring its absorbance at 550 nm; the experiments were performed in triplicate.
Studies of DTX encapsulation in Nanomicelles ManMic-3
To determine the drug-loading content (DLC) and encapsulation efficiency (DLE); DTX was added to a 1 mM solution of MicMan3 in Milli-Q water and left stirring at 54 °C for 24 h. The solution obtained was centrifuged twice at 2000 rpm for 20 minutes, the supernatant (corresponding to the drug-loaded micelle solution) was separated from the precipitate (non-incorporated DTX) and the samples were freeze-dried (Ilshin BioBase, Freeze Dryer) at −76 °C. The incorporated drug amount was determined by HPLC (Waters Alliance 2695 with a UV/Vis detector Waters 996 Photodiode Array Detector set at a wavelength of 254 nm). A reverse-phase C18 column (5 μm, 4.6 × 150 mm) was used. The mobile phase consisted of acetonitrile (0.1% TFA): Milli-Q water (0.05% TFA) (50:50) at a flow rate of 1 mL/min.
The drug-loading (DLC) content and drug loading efficiency (DLE) were calculated by the following equations:
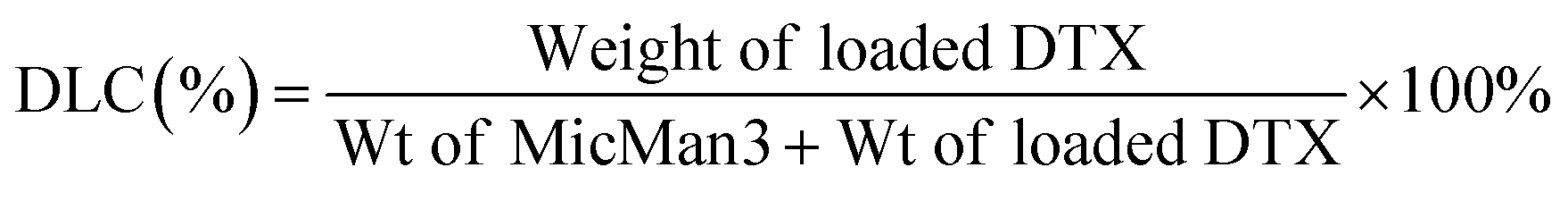
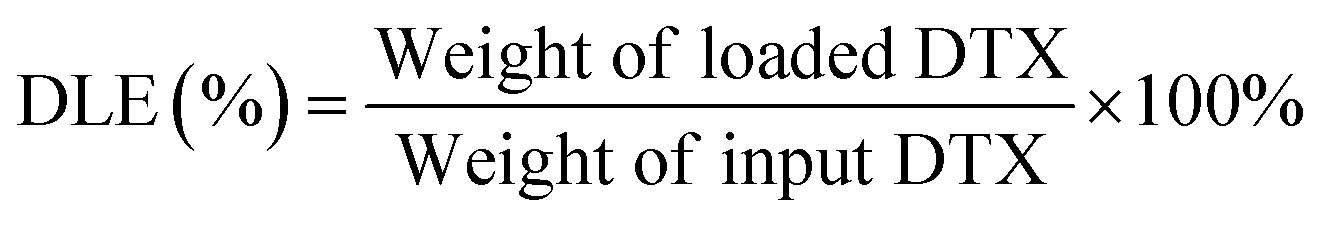
Author contributions
NK conceived the project, and wrote the manuscript. ERB help in writing the manuscript. ERB, MCC, ME and PMC, conducted the synthesis and characterization of the nanomaterials and release studies. CR, ME, MCC and PR conducted the synthesis and characterization of the self-associative monomers. JV and SN conducted the reological studies. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the Spanish Ministry of Science and Innovation (Ref: PID2020-119949RB-I00/AEI/10.13039/501100011033), the Andalusian Ministry of Economy, Science and Innovation cofinanced by the European Regional Development Fund (ERDF) from FEDER and the European Social Fund (ESF) (PY20_00882 and CV20-04221), the CSIC (CSIC-COV19-047) the PAIDI Program from the Andalusian Government (FQM-313). The COST action CA-18132 “Functional Glyconanomaterials for the Development of Diagnostic and Targeted Therapeutic Probe” is also acknowledged. ERB, was supported by FPU predoctoral fellowship (FPU15/04267), from Spanish Ministry of Education, Culture and Sports. We also thank the Centre of Research Technology and Innovation of the University of Seville (CITIUS) for the use of TEM, SEM, FESEM, and NMR facilities.Notes and references
- D. Roy, J. N. Cambre and B. S. Sumerlin, Future Perspectives and Recent Advances in Stimuli-Responsive Materials, Prog. Polym. Sci., 2010, 35(1–2), 278–301, DOI:10.1016/j.progpolymsci.2009.10.008.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Emerging Applications of Stimuli-Responsive Polymer Materials, Nat. Mater., 2010, 9(2), 101–113, DOI:10.1038/nmat2614.
- A. B. Shodeinde, A. C. Murphy, H. F. Oldenkamp, A. S. Potdar, C. M. Ludolph and N. A. Peppas, Recent Advances in Smart Biomaterials for the Detection and Treatment of Autoimmune Diseases, Adv. Funct. Mater., 2020, 30(37), 1–22, DOI:10.1002/adfm.201909556.
- S. Municoy, M. I. Álvarez Echazú, P. E. Antezana, J. M. Galdopórpora, C. Olivetti, A. M. Mebert, M. L. Foglia, M. V. Tuttolomondo, G. S. Alvarez, J. G. Hardy and M. F. Desimone, Stimuli-Responsive Materials for Tissue Engineering and Drug Delivery, Int. J. Mol. Sci., 2020, 21(13), 1–39, DOI:10.3390/ijms21134724.
- M. Vázquez-González and I. Willner, Stimuli-Responsive Biomolecule-Based Hydrogels and Their Applications, Angew. Chem., Int. Ed., 2020, 59(36), 15342–15377, DOI:10.1002/anie.201907670.
- M. Ding, L. Jing, H. Yang, C. E. Machnicki, X. Fu, K. Li, I. Y. Wong and P. Y. Chen, Multifunctional Soft Machines Based on Stimuli-Responsive Hydrogels: From Freestanding Hydrogels to Smart Integrated Systems, Mater. Today Adv., 2020, 8, 100088 CrossRef.
- N. Annabi, A. Tamayol, J. A. Uquillas, M. Akbari, L. E. Bertassoni, C. Cha, G. Camci-Unal, M. R. Dokmeci, N. A. Peppas and A. Khademhosseini, 25th Anniversary Article: Rational Design and Applications of Hydrogels in Regenerative Medicine, Adv. Mater., 2014, 26(1), 85–124, DOI:10.1002/adma.201303233.
- M. Massignani, C. Lopresti, A. Blanazs, J. Madsen, S. P. Armes, A. L. Lewis and G. Battaglia, Controlling Cellular Uptake by Surface Chemistry, Size, and Surface Topology at the Nanoscale, Small, 2009, 5(21), 2424–2432, DOI:10.1002/smll.200900578.
- V. Mailänder and K. Landfester, Interaction of Nanoparticles with Cells, Biomacromolecules, 2009, 10(9), 2379–2400, DOI:10.1021/bm900266r.
- A. Albanese, P. S. Tang and W. C. W. Chan, The Effect of Nanoparticle Size, Shape, and Surface Chemistry on Biological Systems, Annu. Rev. Biomed. Eng., 2012, 14, 1–16, DOI:10.1146/annurev-bioeng-071811-150124.
- P. Decuzzi, B. Godin, T. Tanaka, S.-Y. Lee, C. Chiappini, X. Liu and M. Ferrari, Size and Shape Effects in the Biodistribution of Intravascularly Injected Particles, J. Controlled Release, 2010, 141(3), 320–327, DOI:10.1016/j.jconrel.2009.10.014.
- J. Wang, Q. Li, J. Xue, W. Chen, R. Zhang and D. Xing, Shape Matters: Morphologically Biomimetic Particles for Improved Drug Delivery, Chem. Eng. J., 2021, 410, 127849 CrossRef.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Accumulation of Sub-100 Nm Polymeric Micelles in Poorly Permeable Tumours Depends on Size, Nat. Nanotechnol., 2011, 6(12), 815–823, DOI:10.1038/nnano.2011.166.
- X. Huang, X. Teng, D. Chen, F. Tang and J. He, The Effect of the Shape of Mesoporous Silica Nanoparticles on Cellular Uptake and Cell Function, Biomaterials, 2010, 31(3), 438–448, DOI:10.1016/j.biomaterials.2009.09.060.
- Y. B. Lim, S. Park, E. Lee, J. H. Ryu, Y. R. Yoon, T. H. Kim and M. Lee, Tunable Bacterial Agglutination and Motility Inhibition by Self-Assembled Glyco-Nanoribbons, Chem. – Asian J., 2007, 2(11), 1363–1369, DOI:10.1002/asia.200700163.
- J. H. Ryu, E. Lee, Y. B. Lim and M. Lee, Carbohydrate-Coated Supramolecular Structures: Transformation of Nanofibers into Spherical Micelles Triggered by Guest Encapsulation, J. Am. Chem. Soc., 2007, 129(15), 4808–4814, DOI:10.1021/ja070173p.
- E. Romero-Ben, J. J. Cid, M. Assali, E. Fernández-García, R. E. Wellinger, N. Khiar, V. Valdivia, E. M. Sánchez-Fernández, J. M. Garcia Fernández, R. E. Wellinger, I. Fernández, N. Khiar, E. Romero-Ben, T. Mena Barragán, E. García De Dionisio, E. M. Sánchez-Fernández, J. M. Garcia Fernández, E. Guillén-Mancina, M. López-Lázaro and N. Khiar, Surface Modulation of Single-Walled Carbon Nanotubes for Selective Bacterial Cell Agglutination, J. Mater. Chem. B, 2019, 4(39), 2028–2037, 10.1039/c9tb01218d.
- J. J. Cid Martín, M. Assali, E. Fernández-García, V. Valdivia, E. M. Sánchez-Fernández, J. M. Garcia Fernández, R. E. Wellinger, I. Fernández and N. Khiar, Tuning of Glyconanomaterial Shape and Size for Selective Bacterial Cell Agglutination, J. Mater. Chem. B, 2016, 4(11), 2028–2037 RSC.
- L. Li, R. Sun and R. Zheng, Tunable Morphology and Functionality of Multicomponent Self-Assembly: A Review, Mater. Des., 2021, 197, 109209, DOI:10.1016/j.matdes.2020.109209.
- F. della Sala, W. Verbeet, S. Silvestrini, I. Fortunati, C. Ferrante and L. J. Prins, Stepwise Hierarchical Self-Assembly of Supramolecular Amphiphiles into Higher-Order Three-Dimensional Nanostructures, ChemNanoMat, 2018, 4(8), 821–830, DOI:10.1002/cnma.201800097.
- S. Chen, R. Costil, F. K. C. Leung and B. L. Feringa, Self-Assembly of Photoresponsive Molecular Amphiphiles in Aqueous Media, Angew. Chem., Int. Ed., 2021, 60(21), 11604–11627, DOI:10.1002/anie.202007693.
- G. Yu, X. Yan, C. Han, F. Huang, X. Du, J. Zhou, J. Shi and B. Xu, Supramolecular Hydrogelators and Hydrogels: From Soft Matter to Molecular Biomaterials, Chem. Soc. Rev., 2013, 42(16), 13165–13307, DOI:10.1021/acs.chemrev.5b00299.
- Z. L. Pianowski, J. Karcher and K. Schneider, Photoresponsive Self-Healing Supramolecular Hydrogels for Light-Induced Release of DNA and Doxorubicin, Chem. Commun., 2016, 52(15), 3143–3146, 10.1039/c5cc09633b.
- J. Hoque, N. Sangaj and S. Varghese, Stimuli-Responsive Supramolecular Hydrogels and Their Applications in Regenerative Medicine, Macromol. Biosci., 2019, 19(1), 1–16, DOI:10.1002/mabi.201800259.
- R. Dong, Y. Pang, Y. Su and X. Zhu, Supramolecular Hydrogels: Synthesis, Properties and Their Biomedical Applications, Biomater. Sci., 2015, 3(7), 937–954, 10.1039/c4bm00448e.
- J. Y. C. Lim, Q. Lin, K. Xue and X. J. Loh, Recent Advances in Supramolecular Hydrogels for Biomedical Applications, Mater. Today Adv., 2019, 3, 100021 CrossRef.
- Q. Zhang, D. H. Qu, H. Tian and B. L. Feringa, Bottom-Up: Can Supramolecular Tools Deliver Responsiveness from Molecular Motors to Macroscopic Materials?, Matter, 2020, 3(2), 355–370, DOI:10.1016/j.matt.2020.05.014.
- A. Barnard and D. K. Smith, Self-Assembled Multivalency: Dynamic Ligand Arrays for High-Affinity Binding, Angew. Chem., Int. Ed., 2012, 6572–6581, DOI:10.1002/anie.201200076.
- M. J. Clemente, R. M. Tejedor, P. Romero, J. Fitremann and L. Oriol, Photoresponsive Supramolecular Gels Based on Amphiphiles with Azobenzene and Maltose or Polyethyleneglycol Polar Head, New J. Chem., 2015, 39(5), 4009–4019, 10.1039/c4nj02012j.
- I. N. Lee, O. Dobre, D. Richards, C. Ballestrem, J. M. Curran, J. A. Hunt, S. M. Richardson, J. Swift and L. S. Wong, Photoresponsive Hydrogels with Photoswitchable Mechanical Properties Allow Time-Resolved Analysis of Cellular Responses to Matrix Stiffening, ACS Appl. Mater. Interfaces, 2018, 10(9), 7765–7776, DOI:10.1021/acsami.7b18302.
- L. Li, J. M. Scheiger and P. A. Levkin, Design and Applications of Photoresponsive Hydrogels, Adv. Mater., 2019, 31(26), 1807333 CrossRef PubMed.
- M. Baroncini, J. Groppi, S. Corra, S. Silvi and A. Credi, Light-Responsive (Supra)Molecular Architectures: Recent Advances, Adv. Opt. Mater., 2019, 7(16), 1900392 CrossRef.
- J. Zhang, Q. Zou and H. Tian, Photochromic Materials: More than Meets the Eye, Adv. Mater., 2013, 25(3), 378–399, DOI:10.1002/adma.201201521.
- W. Feng, W. Luo and Y. Feng, Photo-Responsive Carbon Nanomaterials Functionalized by Azobenzene Moieties: Structures, Properties and Application, Nanoscale, 2012, 4(20), 6118–6134, 10.1039/c2nr31505j.
- D. Bléger and S. Hecht, Visible-Light-Activated Molecular Switches, Angew. Chem., Int. Ed., 2015, 54(39), 11338–11349, DOI:10.1002/anie.201500628.
- E. Merino, Synthesis of Azobenzenes: The Coloured Pieces of Molecular Materials, Chem. Soc. Rev., 2011, 40(7), 3835–3853, 10.1039/c0cs00183j.
- V. Chandrasekaran, E. Johannes, H. Kobarg, F. D. Sönnichsen and T. K. Lindhorst, Synthesis and Photochromic Properties of Configurationally Varied Azobenzene Glycosides, ChemistryOpen, 2014, 3(3), 99–108, DOI:10.1002/open.201402010.
- L. Möckl, A. Müller, C. Bräuchle and T. K. Lindhorst, Switching First Contact: Photocontrol of E. Coli Adhesion to Human Cells, Chem. Commun., 2016, 52(6), 1254–1257, 10.1039/c5cc08884d.
- M. J. Clemente, P. Romero, J. L. Serrano, J. Fitremann and L. Oriol, Supramolecular Hydrogels Based on Glycoamphiphiles: Effect of the Disaccharide Polar Head, Chem. Mater., 2012, 24(20), 3847–3858, DOI:10.1021/cm301509v.
- Y. Ogawa, C. Yoshiyama, T. Kitaoka, M. Bhattacharya, M. M. Malinen, P. Lauren, Y. R. Lou, S. W. Kuisma, L. Kanninen, M. Lille, A. Corlu, C. Guguen-Guillouzo, O. Ikkala, A. Laukkanen, A. Urtti and M. Yliperttula, Nanofibrillar Cellulose Hydrogel Promotes Three-Dimensional Liver Cell Culture, Langmuir, 2012, 28(9), 291–298, DOI:10.1016/j.jconrel.2012.06.039.
- J. J. Cid, M. Assali, E. Fernández-García, V. Valdivia, E. M. Sánchez-Fernández, J. M. Garcia Fernández, R. E. Wellinger, I. Fernández and N. Khiar, Tuning of Glyconanomaterial Shape and Size for Selective Bacterial Cell Agglutination, J. Mater. Chem. B, 2016, 4(11), 2028–2037, 10.1039/c5tb02488a.
- M. Assali, J.-J. Cid, M. Pernía-Leal, M. Muñoz-Bravo, I. Fernández, R. E. Wellinger and N. Khiar, Glyconanosomes: Disk-Shaped Nanomaterials for the Water Solubilization and Delivery of Hydrophobic Molecules, ACS Nano, 2013, 7(3), 2145–2153 CrossRef PubMed.
- E. Romero-Ben, T. Mena Barragán, E. García De Dionisio, E. M. Sánchez-Fernández, J. M. Garcia Fernández, E. Guillén-Mancina, M. López-Lázaro and N. Khiar, Mannose-Coated Polydiacetylene (PDA)-Based Nanomicelles: Synthesis, Interaction with Concanavalin A and Application in the Water Solubilization and Delivery of Hydrophobic Molecules, J. Mater. Chem. B, 2019, 7(39), 5930–5946, 10.1039/c9tb01218d.
- M. Assali, J. J. Cid, I. Fernández and N. Khiar, Supramolecular Diversity through Click Chemistry: Switching from Nanomicelles to 1D-Nanotubes and Tridimensional Hydrogels, Chem. Mater., 2013, 25(21), 4250–4261, DOI:10.1021/cm4022613.
- R. Jelinek and M. Ritenberg, Polydiacetylenes-Recent Molecular Advances and Applications, RSC Adv., 2013, 3(44), 21192–21201, 10.1039/c3ra42639d.
- X. Quian and B. Städler, Recent Developments in Polydiacetylene-Based Sensors, Chem. Mater., 2019, 31, 1196–1222 CrossRef.
- M. Gou, X. Qu, W. Zhu, M. Xiang, J. Yang, K. Zhang, Y. Wei and S. Chen, Bio-Inspired Detoxification Using 3d-Printed Hydrogel Nanocomposites, Nat. Commun., 2014, 5(May), 1–9, DOI:10.1038/ncomms4774.
- M. P. Leal, M. Assali, I. Fernández and N. Khiar, Copper-Catalyzed Azide-Alkyne Cycloaddition in the Synthesis of Polydiacetylene: “Click Glycoliposome” as Biosensors for the Specific Detection of Lectins, Chem. – Eur. J., 2011, 17(6), 1828–1836 CrossRef PubMed.
- I. Theodorou, P. Anilkumar, B. Lelandais, D. Clarisse, A. Doerflinger, E. Gravel, F. Ducongé and E. Doris, Stable and Compact Zwitterionic Polydiacetylene Micelles with Tumor-Targeting Properties, Chem. Commun., 2015, 51(80), 14937–14940, 10.1039/c5cc05333a.
- M. Z. Alam, A. Shibahara, T. Ogata and S. Kurihara, Synthesis of Azobenzene-Functionalized Star Polymers via RAFT and Their Photoresponsive Properties, Polymer, 2011, 52(17), 3696–3703, DOI:10.1016/j.polymer.2011.06.035.
- E. Dalle Vedove, G. Costabile and O. M. Merkel, Mannose and Mannose-6-Phosphate Receptor–Targeted Drug Delivery Systems and Their Application in Cancer Therapy, Adv. Healthcare Mater., 2018, 7(14), 1–19, DOI:10.1002/adhm.201701398.
- T. S. Patil and A. S. Deshpande, Mannosylated Nanocarriers Mediated Site-Specific Drug Delivery for the Treatment of Cancer and Other Infectious Diseases: A State of the Art Review, J. Controlled Release, 2020, 320(January), 239–252, DOI:10.1016/j.jconrel.2020.01.046.
- T. Shimizu, W. Ding and N. Kameta, Soft-Matter Nanotubes: A Platform for Diverse Functions and Applications, Chem. Rev., 2020, 120(4), 2347–2407, DOI:10.1021/acs.chemrev.9b00509.
- N. Yu. Kostina, D. Söder, T. Haraszti, Q. Xiao, K. Rahimi, B. E. Partridge, M. L. Klein, V. Percec and C. Rodriguez-Emmenegger, Enhanced Concanavalin A Binding to Preorganized Mannose Nanoarrays in Glycodendrimersomes Revealed Multivalent Interactions, Angew. Chem., Int. Ed., 2021, 60(15), 8352–8360, DOI:10.1002/anie.202100400.
- T. Kim, H. Lee, Y. Kim, J. M. Nam and M. Lee, Protein-Coated Nanofibers for Promotion of T Cell Activity, Chem. Commun., 2013, 49(38), 3949–3951, 10.1039/c3cc41215f.
- J. E. Gestwicki, C. W. Cairo, L. E. Strong, K. A. Oetjen and L. L. Kiessling, Influencing Receptor-Ligand Binding Mechanisms with Multivalent Ligand Architecture, J. Am. Chem. Soc., 2002, 124(50), 14922–14933, DOI:10.1021/ja027184x.
- G. N. Patel, R. R. Chance, E. A. Turi and Y. P. Khanna, Energetics and Mechanism of the Solid-State Polymerization of Diacetylenes, J. Am. Chem. Soc., 1978, 100(21), 6644–6649 CrossRef.
- W. Neumann and H. Sixl, The Mechanism of the Low Temperature Polymerization Reaction in Diacetylene Crystals, Chem. Phys., 1981, 58(3), 303–312, DOI:10.1016/0301-0104(81)80066-3.
- M. A. Jermyn, Increasing the Sensitivity of the Anthrone Method for Carbohydrate, Anal. Biochem., 1975, 68(1), 332–335, DOI:10.1016/0003-2697(75)90713-7.
- Y. Ogawa, C. Yoshiyama and T. Kitaoka, Helical Assembly of Azobenzene-Conjugated Carbohydrate Hydrogelators with Specific Affinity for Lectins, Langmuir, 2012, 28(9), 4404–4412, DOI:10.1021/la300098q.
- C. R. Becer, The Glycopolymer Code: Synthesis of Glycopolymers and Multivalent Carbohydrate-Lectin Interactions, Macromol. Rapid Commun., 2012, 33(9), 742–752, DOI:10.1002/marc.201200055.
- D. Deniaud, K. Julienne and S. G. Gouin, Insights in the Rational Design of Synthetic Multivalent Glycoconjugates as Lectin Ligands, Org. Biomol. Chem., 2011, 9(4), 966–979, 10.1039/c0ob00389a.
- J. J. Lundquist and E. J. Toone, The Cluster Glycoside Effect, Chem. Rev., 2002, 102(2), 555–578, DOI:10.1021/cr000418f.
- O. Srinivas, N. Mitra, A. Surolia and N. Jayaraman, Photoswitchable Multivalent Sugar Ligands: Synthesis, Isomerization, and Lectin Binding Studies of Azobenzene-Glycopyranoside Derivatives, J. Am. Chem. Soc., 2002, 124(10), 2124–2125, DOI:10.1021/ja0173066.
- U. Osswald, J. Boneberg and V. Wittmann, Photoswitching Affinity and Mechanism of Multivalent Lectin Ligands, Chem. – Eur. J., 2022, 28(27), e202200267 CrossRef PubMed.
- T. Weber, V. Chrasekaran, I. Stamer, M. B. Thygesen, A. Terfort and T. K. Lindhorst, Switching of Bacterial Adhesion to a Glycosylated Surface by Reversible Reorientation of the Carbohydrate Ligand, Angew. Chem., Int. Ed., 2014, 53(52), 14583–14586, DOI:10.1002/anie.201409808.
- G. Despras, L. Möckl, A. Heitmann, I. Stamer, C. Bräuchle and T. K. Lindhorst, A Photoswitchable Trivalent Cluster Mannoside to Probe the Effects of Ligand Orientation in Bacterial Adhesion, ChemBioChem, 2019, 20(18), 2373–2382, DOI:10.1002/cbic.201900269.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb01713c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |