
Aggregation effects on the one- and two-photon excited fluorescence performance of regioisomeric anthraquinone-substituted perylenediimide†
Liang
Xu
 *a,
Xueting
Long
a,
Jiaxin
He
a,
Lingxiu
Liu
b,
Fangyuan
Kang
c,
Ziqi
Deng
d,
Jieyu
Wu
e,
Xiao-Fang
Jiang
e,
Jianguo
Wang
*b and
Qichun
Zhang
*cf
*a,
Xueting
Long
a,
Jiaxin
He
a,
Lingxiu
Liu
b,
Fangyuan
Kang
c,
Ziqi
Deng
d,
Jieyu
Wu
e,
Xiao-Fang
Jiang
e,
Jianguo
Wang
*b and
Qichun
Zhang
*cf
aDepartment of Chemistry and Key Laboratory for Preparation and Application of Ordered Structural Materials of Guangdong Province, Shantou University, Guangdong 515063, China. E-mail: xuliang@stu.edu.cn
bCollege of Chemistry and Chemical Engineering, Inner Mongolia Key Laboratory of Fine Organic Synthesis, Inner Mongolia University, Hohhot 010021, P. R. China. E-mail: wangjg@iccas.ac.cn
cDepartment of Materials Science and Engineering, City University of Hong Kong, Kowloon, Hong Kong SAR, China. E-mail: qiczhang@cityu.edu.hk
dDepartment of Chemistry, University of Hong Kong, Hong Kong, China
eLaboratory of Quantum Engineering and Quantum Material, School of Physics and Telecommunication Engineering, South China Normal University, Guangzhou 510006, China
fCenter of Super-Diamond and Advanced Films (COSDAF), City University of Hong Kong, Hong Kong SAR, 999077, P. R. China
First published on 11th October 2022
Abstract
Regioisomerism is an efficient strategy for finely tuning the structures of molecules in the isolated or the aggregated state. Herein, two regioisomers of perylene diimide (PDI) derivatives have been designed and synthesized. The positions of the substituent anthraquinone groups display distinct influences on their molecular conformation and aggregate structures, resulting in different fluorescence properties upon one- and two-photon excitation. In addition, owing to the bright emission, cellular imaging tests based on their nanoparticles were also conducted.
Introduction
Organic luminescent materials have drawn considerable attention owing to many attractive advantages, such as the diversity of molecular design, high compatibility with organisms, and potential to prepare flexible devices.1 Therefore, they have great potential for applications in biological imaging,2–5 organic nonlinear optical (NLO) materials,6–10etc. The performance of these materials depends on not only the properties of the well-designed molecules with specific functional groups but also their aggregate states.11–13 In 2001, Tang and co-workers first coined the concept of aggregation-induced emission (AIE).14 In 2018, Li and co-workers first coined the concept of molecular uniting set identified characteristic (MUSIC).15 In 2020, Tang and co-workers put forward the concept of “aggregate science”.16 These concepts all highlight the key role of molecular aggregates in the performance of organic luminescent materials.There are mainly two approaches used to adjust the molecular aggregates. One is external force, including the characteristics of solvent (polarity, viscosity, hydrophilia or hydrophobicity), mechanical force, light, and electric field.17 For instance, polymorphs,18,19 mechanoluminescence (ML),20–23 photoinduced room temperature phosphorescence (RTP),24–28 and the optimized nonlinear optical (NLO) properties were achieved after electric poling.29–35 The other is internal force including π–π stacking, anion–π interactions, dispersion forces, electrostatic interactions and hydrogen bonding.36,37 For instance, alkyl chains can suppress molecular motions and favour the RTP properties through the formation of multiple C–H⋯π and C–H⋯X (X presents heteroatoms such as N, O, S, etc.) interactions with surrounding aromatic rings or heteroatoms.38,39 Moreover, the different lengths of alkyl chains can subtly tune intermolecular interactions, resulting in a unique oddeven effect in photoluminescence.40,41 Hydrogen bonding induced by hydroxy, carboxylic acid and amino moieties can promote RTP performance by suppressing the molecular motions in the aggregated states.42 Overall, predesign or modification of the molecular aggregates at the stage of molecular design would be ideal, despite its great difficulty and challenges.
As one kind of molecular design method, regioisomerism is a very promising strategy for adjusting the molecular aggregates. In this way, a simple structural modification may drastically influence the luminescence properties of the molecular aggregates.43 In recent three years, several groups have reported the use of regioisomerism to finely tune the structures of the molecular aggregates, leading to distinct luminescence properties. In 2019, Li and co-workers reported a series of halogen-substituted triphenylamines. Among these, ortho-substituted products are all ML-inactive, while meta-substituted products are all ML-active.44 In 2020, Wang, Zhang and co-workers reported a phenyl-substituted effect in tetraphenylpyrazine. They found that the ortho-phenyl substitution stimulates the AIE characteristics, but substituting at either meta or para position results in non-AIE active compounds.45 In 2020, Tang, Lu, Qian and co-workers reported two triphenylamine-substituted dithienobenzophenazine and found that the end-substituted product is dominated by the ACQ properties while the bay-substituted product is AIE-active.46 In 2020, Promarak and co-workers reported two n-hexylthiophene-substituted naphthothiadiazole with AIE properties. Due to a large steric repulsion between naphthothiadiazole and nearby thiophenes, the molecular conformation of the product substituted at the 3-position of thiophene is more twisted than that of the product substituted at the 4-position of thiophene. As a result, their fluorescence emission peak and quantum yields are totally different in solution and aggregate.47 In 2020, Iyer and co-workers reported two pyridine-substituted triphenylethylene. When the pyridine group is substituted at the ortho position of the phenyl ring, the product is non-AIE active. When the pyridine group is substituted at the para position of the phenyl ring, the product is AIE active.48 In 2021, Wang, Zhi and co-workers reported three aldehyde-substituted tetraaryl ethylenes. They are all AIE-active; however, the AIE amplification factors are totally different. The biggest one is 176.4 for the para-substituted product, while the smallest one is only 9.3 for the meta-substituted product.49
Although perylene diimides (PDIs) are one class of the most explored organic fluorescent materials, the report of the regioisomerism effect on the aggregate emission properties of PDIs is still rare. Tang, Sun and co-workers reported two tetraphenyl ethene-substituted PDIs with AIE properties.50 The AIE amplification factor of the 1,7-substituted product is larger than that of the 1,6-substituted product. In 2021 and 2022, our group reported bay- and ortho-substituted PDIs with the benzanthrone or anthracene groups as the electron-donating moieties.51,52 Very interesting phenomena are observed. With the substitution position of benzanthrone changing from bay to ortho, ACQ-to-AIE conversion is achieved. With the substitution position of anthracene changing from bay to ortho, the AIE process is accelerated and the AIE amplification factor is also enhanced.51,52 However, it is still unknown what will happen if changing the substitution position of electron-withdrawing groups from bay to ortho on PDI.
Inspired by the above points, this work aims to explore the aggregate emission of PDIs with electron-withdrawing anthraquinone groups as the bay- and ortho-substituents. It is anticipated that by changing the anthraquinone group from the ortho position to the bay position of PDI, the resultant steric hindrance may alter the π-conjugated core and affect the molecular aggregates, which may make possible altering the aggregate fluorescence emission properties. Herein, we present the syntheses, UV-Vis absorption and fluorescence emission properties, femtosecond and nanosecond transient absorption properties, two-photon absorption properties, and theoretical calculations in detail. Finally, given the high fluorescence quantum yields of the products, their nanoparticles are also applied for cellular imaging.
Results and discussion
UV-Vis absorption and fluorescence emission spectra
As shown in Fig. 1, the UV-Vis absorption spectrum of PDI in THF shows three main peaks centered at 522, 486 and 455 nm, respectively. The ortho-substituted product O-PDI-ANQ in THF also exhibits structured absorption peaks centered at 524, 488 and 457 nm, respectively. The bay-substituted product B-PDI-ANQ in THF exhibits absorption peaks centered at 530 and 495 nm, respectively. Upon excitation at 525 nm, PDI shows vibronically resolved emission peaks centered at 530, 571 and 620 nm. O-PDI-ANQ exhibits slightly red-shifted emission peaks centered at 535, 576 and 625 nm. B-PDI-ANQ exhibits a broad emission band centered at 567 nm. As a result, the Stokes shifts for PDI, O-PDI-ANQ and B-PDI-ANQ are 8, 11 and 37 nm, respectively. It is worth noting that both the absorption and emission spectra of B-PDI-ANQ appear to be less structured in comparison to those of O-PDI-ANQ. For O-PDI-ANQ, the structured emission spectrum and small Stokes shift reflect its molecular rigidity. For B-PDI-ANQ, the extremely broad emission spectrum and the large Stokes shift suggest a drastic rearrangement of this flexible molecule in the excited state.53 The optimized molecular structures in vacuum indeed reveal that the dihedral between the ANQ group and PDI core changes by 12° from the ground state to the excited state for B-PDI-ANQ, while that is only 6° for O-PDI-ANQ (Fig. S5 and S6, ESI†). Using rhodamine B (ΦF = 97% in ethanol) as the calibration standard, the fluorescence quantum yields (ΦF) of PDI, O-PDI-ANQ and B-PDI-ANQ are 70, 70, and 54%, respectively. Partial reduction in the ΦF value of B-PDI-ANQ when compared with PDI and O-PDI-ANQ could be attributed to the non-radiative decay pathways arising from the bay substitution of the PDI core.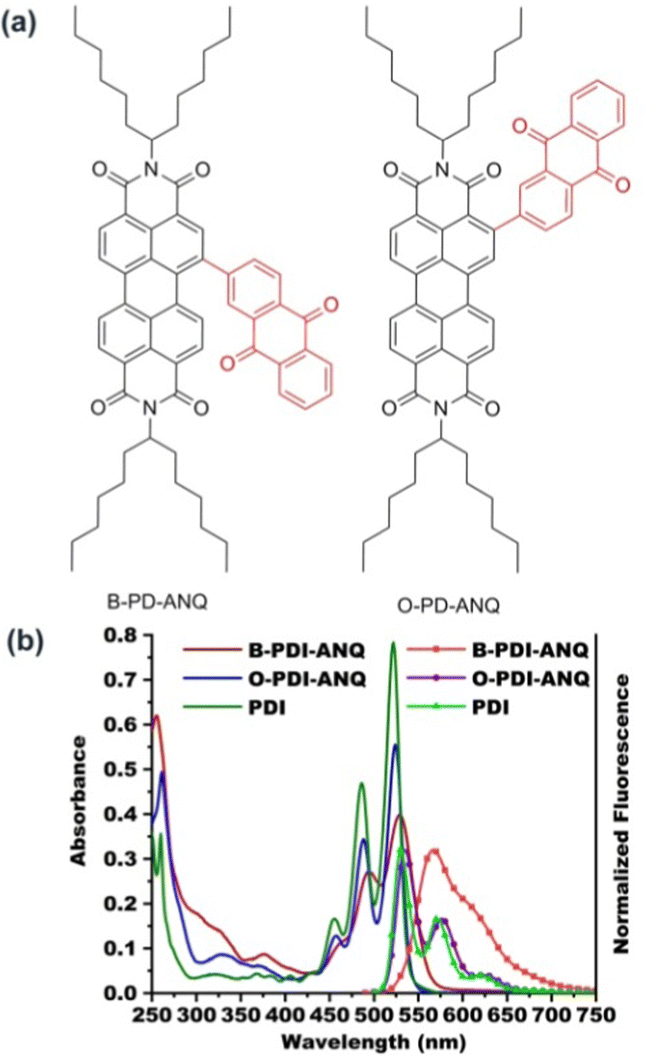 | ||
| Fig. 1 (a) The molecular structures of B-PDI-ANQ and O-PDI-ANQ and (b) UV-Vis absorption and normalized fluorescence emission spectra of PDI, B-PDI-ANQ and O-PDI-ANQ in THF. | ||
Femtosecond and nanosecond transient absorption of B-PDI-ANQ and O-PDI-ANQ in THF
Further insights into the excited state deactivation in bay- and ortho-substituted PDI derivatives come from femtosecond and nanosecond transient absorption measurements. Fig. 2a–d show four distinct processes for B-PDI-ANQ after photoexcitation. First, B-PDI-ANQ exhibits two negative peaks centered at 534 and 493 nm corresponding to ground-state bleaching (GSB),54 where one negative peak centered at 566 corresponds to stimulated emission (SE),55 and two positive peaks centered at 345 and 710 nm come from excited-state absorption (ESA).56 Second, the decrease of GSB signals at 534 nm suggests the occurrence of intramolecular charge transfer (ICT),57 and no significant spectral shift is observed. It indicates that this singlet excited state with ICT character is formed very fast. Third, the transient absorption peaks do not show any change. This is probably due to the solvent rearrangement (SR).58 Fourth, GSB and ESA signals gradually fall down to the baseline and remain at a 534 nm peak, which do not disappear even after reaching the maximum delay time of the fs-TA instrument (7.5 ns). This reveals that an intersystem crossing (ISC)59 may take place to generate a triplet state. Compared to the fs-TA spectrum of B-PDI-ANQ, that of O-PDI-ANQ displays apparently different features. As shown in Fig. 2f, O-PDI-ANQ shows GSB at 532, 487, and 456 nm, SE at 576 nm and ESA at 345 and 706 nm. The SE feature of O-PDI-ANQ is more prominent than that of B-PDI-ANQ, which is consistent with the larger ΦF value of O-PDI-ANQ. As shown in Fig. 2g, the SR feature persists in the transient spectra, but there is no ICT feature observed for O-PDI-ANQ. Therefore, there are two non-radiative decay processes (ICT and ISC) for the excited state of B-PDI-ANQ. In contrast, there is one non-radiative decay process (ISC) for the excited state of O-PDI-ANQ. This is the reason that the ΦF value of O-PDI-ANQ is higher than that of B-PDI-ANQ. To obtain the exact rate constants of the excited state dynamics, the kinetic fittings of 534 nm for B-PDI-ANQ and 532 nm for O-PDI-ANQ are extracted from the data of fs-TA. As shown in Fig. 2e, for B-PDI-ANQ, the 534 nm transient absorption should be fitted by the four exponential functions, resulting in four lifetimes for them. The four lifetimes are τ1 = 0.45 ps (16.2%), τ2 = 6.5 ps (13.1%), τ3 = 231 ps (12%), and τ4 = 7.32 ns (51.5%), respectively. As shown in Fig. 2i, for O-PDI-ANQ, the 532 nm transient absorption should be fitted by the three exponential functions, resulting in τ1 = 0.36 ps (5.7%), τ2 = 254 ps (12.6%) and τ3 = 4.07 ns (77.3%), respectively. To further confirm the generation of triplet states for B-PDI-ANQ and O-PDI-ANQ, purged nitrogen and oxygen experiments for nanosecond transient absorption (ns-TA) were performed. The results and discussion are available in the ESI.†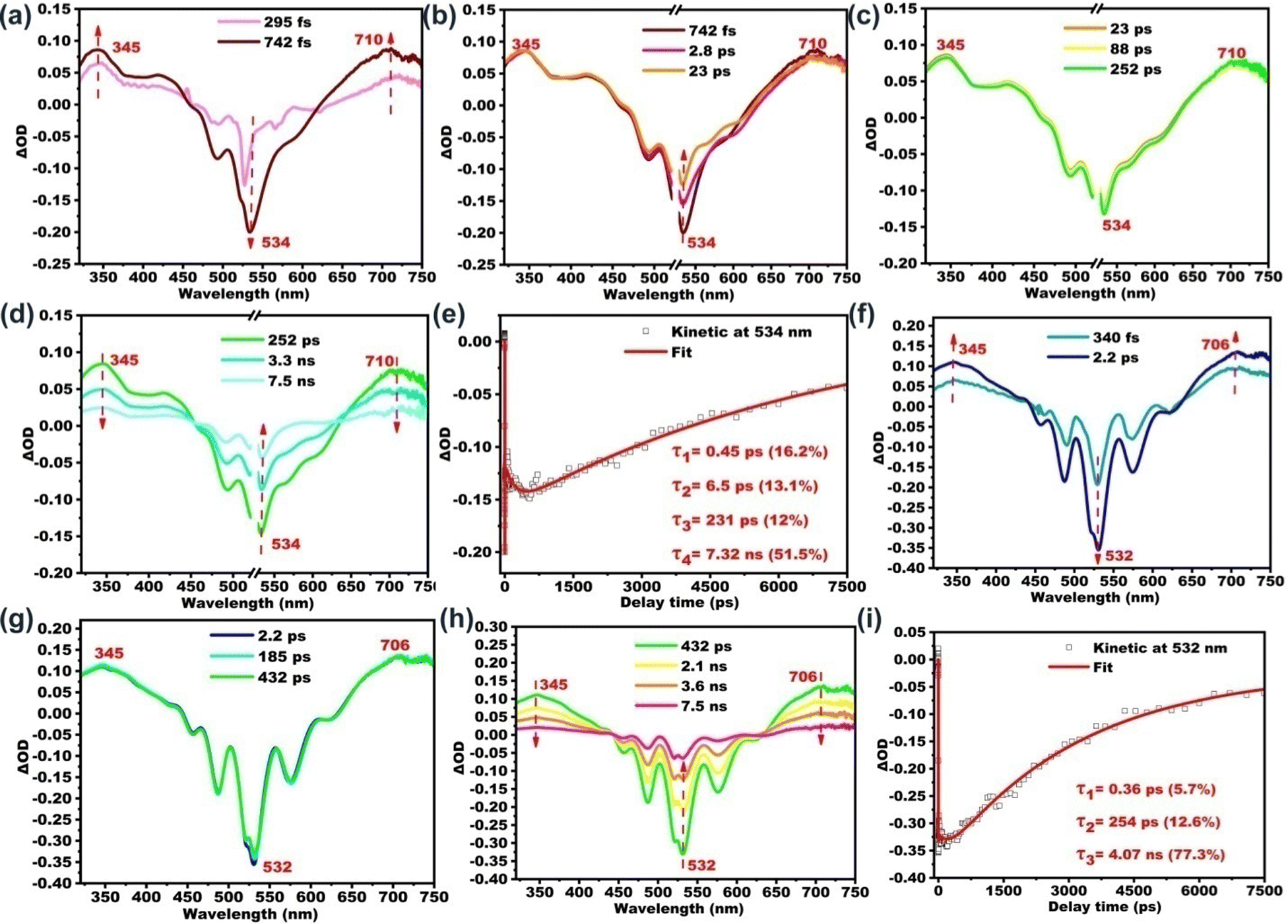 | ||
| Fig. 2 Fs-TA spectra and kinetics of (a–e) B-PDI-ANQ and (f–i) O-PDI-ANQ in THF. | ||
One-photon excited fluorescence properties
In order to investigate the fluorescence properties of B-PDI-ANQ and O-PDI-ANQ in their aggregated state, the fluorescence spectra of these compounds are measured in THF containing various water volume fractions (fw). As shown in Fig. 3, the fluorescence intensities of B-PDI-ANQ are dramatically decreased at the initial stage from fw = 0 to 30%, indicating a grave quenching effect. Unlike B-PDI-ANQ, the fluorescence intensities of O-PDI-ANQ have a very small decrease when fw is below 60%, manifesting a slight quenching effect. When water is progressively added, an evident decrease in the fluorescence intensity of O-PDI-ANQ is observed at fw = 70%.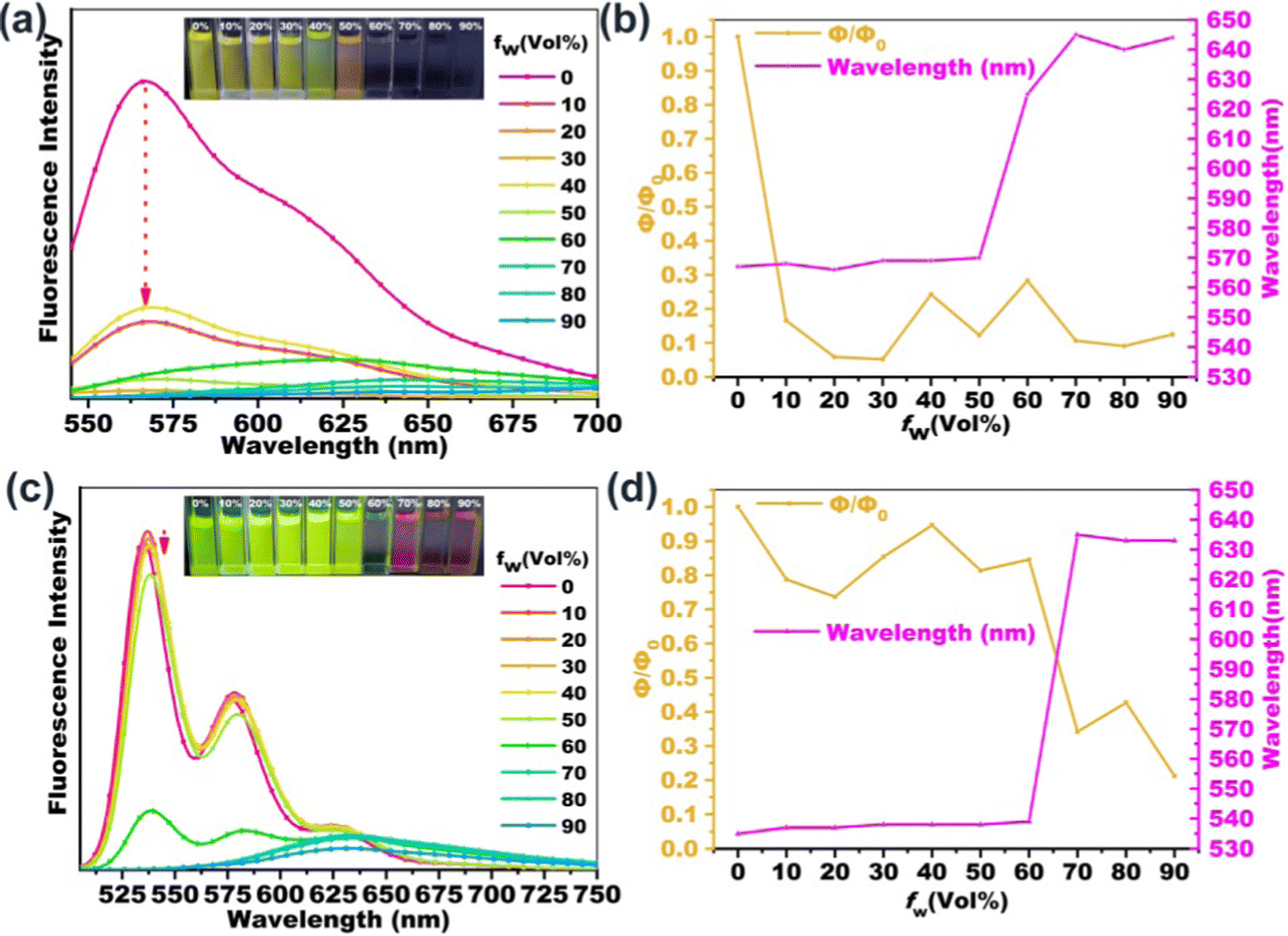 | ||
| Fig. 3 The fluorescence emission spectra of (a) B-PDI-ANQ and (c) O-PDI-ANQ in the THF/water mixture with different water fractions (fw). Variations of the fluorescence quantum yield and the maximum fluorescence emission wavelength for (b) B-PDI-ANQ and (d) O-PDI-ANQ in the THF/water mixture with different fw. The insets of (a and c) show the photographs of FL images (taken under 365 nm UV light) for the corresponding mixtures. | ||
In order to further clarify the quenching characteristics of B-PDI-ANQ and O-PDI-ANQ with the increase in the water content in the solution, the changes in the ΦF values and the maximum emission wavelengths are plotted against the water fraction in the THF solutions. The quenching factor Φq is calculated by using the equation of Φq = (Φsol − Φagg)/Φsol, where Φsol and Φagg are the fluorescence emission quantum efficiency of the dye in dilute solution and in the aggregated state, respectively.60 When fw increases from 0 to 30%, ΦF of B-PDI-ANQ drops from 54 to 2.8%. Φq is calculated to be as high as 95%. Under the same condition, ΦF of O-PDI-ANQ decreases slowly from 70 to 60% and Φq is calculated to be 14%. Only at a very high fw of 70%, ΦF of O-PDI-ANQ decrease to 24% and Φq is calculated to be 66%. It is noted that the emission peak of B-PDI-ANQ gradually red-shifted from 570 to 645 nm when fw is increased from 50 to 70%. This result indicates that the aggregation of B-PDI-ANQ involves intermolecularπ–π interactions, which would increase the coplanarity of the π-conjugated molecules and should result in a red-shift of the emission wavelength due to excimer formation.61 Interestingly, the emission peak of B-PDI-ANQ shows a small blue-shift by approximately 5 nm at fw = 80% as compared with that at fw = 70%. The abnormal phenomenon of spectral shift is related to the degree of aggregation. When the degree of aggregation exceeds a certain range (fw > 70%), the intermolecular π–π interactions are weakened rather than strengthened, leading to a blue-shift of the emission wavelength. A similar behavior of emission wavelength is also observed for O-PDI-ANQ.
Femtosecond transient absorption of B-PDI-ANQ and O-PDI-ANQ in THF/H2O systems
According to the literature, on one hand, molecular aggregation could enhance the ISC rate and thereby suppressing fluorescence. This mechanism is termed “aggregation-induced intersystem crossing” (AI-ISC).62,63 On the other hand, core-twist leads to an enhancement in triplet excited state generation, which is proposed as twisted π-conjugation-induced ISC.64–68 The present study focuses on the impact of aggregation on the fluorescence properties of PDI derivatives in the core-twisted vs. planar conformation. The influence of the aggregation degree on the excited state is also tracked with the femtosecond transient absorption study. We choose aggregated B-PDI-ANQ in the THF/water mixture (with fw of 30%) and O-PDI-ANQ in the THF/water mixture (with fw of 30% and 70%, respectively) as the test samples. As shown in Fig. 4, GSB and ESA bands show an apparent redshift upon aggregation. Four or three components are required for the satisfactory fitting of the data for B-PDI-ANQ and O-PDI-ANQ, respectively. At fw = 30%, the ISC rate or the triplet rise time of aggregated B-PDI-ANQ is 3.70 ns, which is much shorter than that of non-aggregated B-PDI-ANQ (τISC = 7.32 ns), while the ISC takes 3.50 ns in aggregated O-PDI-ANQ, which is only slightly shorter than that in non-aggregated O-PDI-ANQ (τISC = 4.07 ns). Thus, aggregated B-PDI-ANQ displays a large fluorescence quenching at fw = 30%, but aggregated O-PDI-ANQ only exhibits a small one. At fw = 70%, the ISC takes 716 ps in aggregated O-PDI-ANQ, which is much shorter than that in non-aggregated O-PDI-ANQ. Therefore, the fluorescence of aggregated O-PDI-ANQ begins to quench largely then.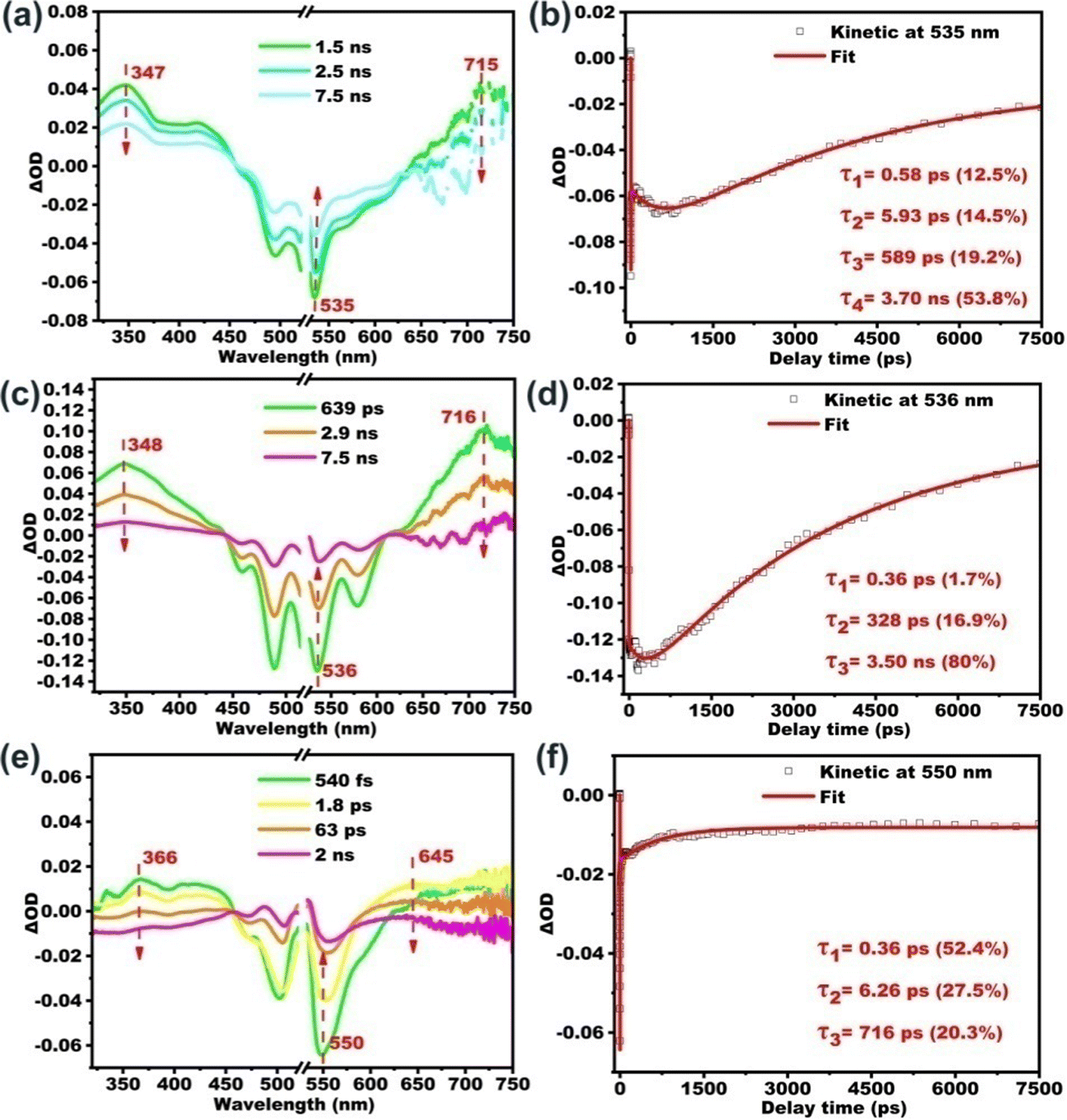 | ||
| Fig. 4 Fs-TA spectra of (a) B-PDI-ANQ in THF/H2O (fw = 30%) and (c and e) O-PDI-ANQ in THF/H2O (fw = 30% and 70%) and (b, d and f) kinetics fitting at the corresponding wavelengths. | ||
Two-photon excited fluorescence properties
The influence of aggregation on the two-photon excited fluorescence properties of PDI derivatives has also been investigated. As shown in Fig. 5, the two-photon excited fluorescence emission spectra of B-PDI-ANQ and O-PDI-ANQ in THF/H2Omixtures with different water fractions are recorded. Interestingly, B-PDI-ANQ and O-PDI-ANQ all show fluorescence quenching with the increasing water fractions; however, there is some variation in the extent of the decline. To quantitatively describe their fluorescence quenching process, the changes in the I/I0 values are plotted against the water fraction in the THF solutions. I0 is the fluorescence intensity in THF. I is the fluorescence intensity in THF/H2O mixtures. The tendencies are very clear: the fluorescence intensity of B-PDI-ANQ decreases relatively fast, with only 23% and 15% of its initial emission at fw = 20% and fw = 40%, respectively. The fluorescence intensity of O-PDI-ANQ shows a relatively slow but apparent decrease since it keeps 37% and 20% of its initial emission at fw = 20% and fw = 40%, respectively.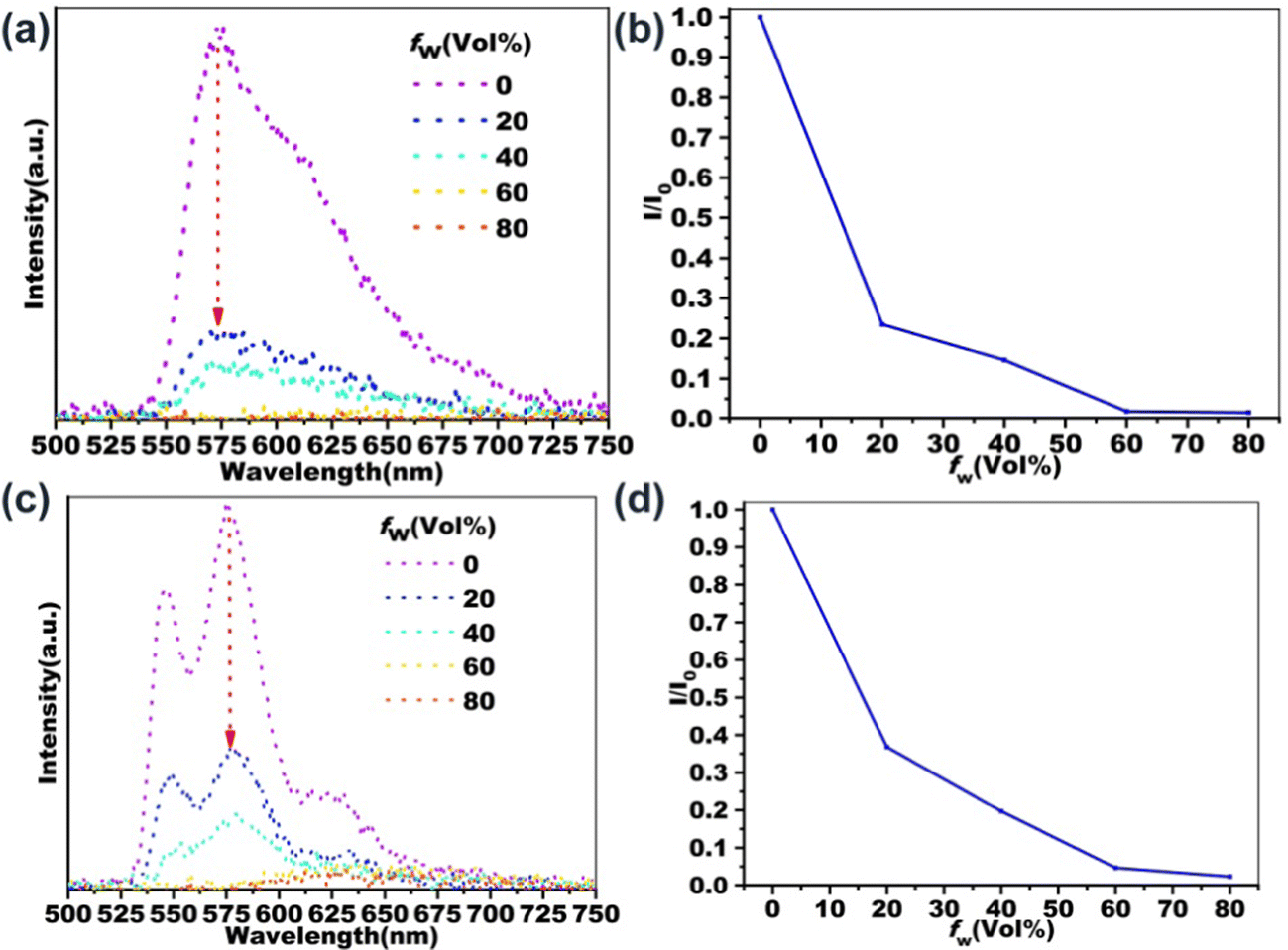 | ||
| Fig. 5 Two-photon excited FL spectra of (a) B-PDI-ANQ and (c) O-PDI-ANQ in the THF/water mixture with different water fraction (fw). Variation of 2PEF intensity for (b) B-PDI-ANQ and (d) O-PDI-ANQ in THF/water mixture with different fw. | ||
Cellular imaging
Banking on their bright emission, cellular imaging ability of B-PDI-ANQ and O-PDI-ANQ were assessed. Before this study, the cytotoxicity of the two PDI derivatives toward HeLa cells was tested. Results in Fig. 6a and b demonstrate that both B-PDI-ANQ and O-PDI-ANQ display negligible influence on the viability of HeLa cells, indicating a good biocompatibility of these two PDI derivatives. Then, cells are incubated with B-PDI-ANQ or O-PDI-ANQ for 8 h. A commercially available probe Lyso-tracker blue (LTB) with lysosome targetability is used for indicating cellular lysosomes. As displayed in Fig. 6c, both B-PDI-ANQ and O-PDI-ANQ exhibit bright orange emission inside cells and the bright emission matches well with the blue emission of probe Lyso-tracker blue, verifying the lysosome-targeting capability of B-PDI-ANQ and O-PDI-ANQ. These data confirm the potential application of B-PDI-ANQ and O-PDI-ANQ in fluorescence imaging of live cells and further visualization of biological procedures that occur inside lysosomes.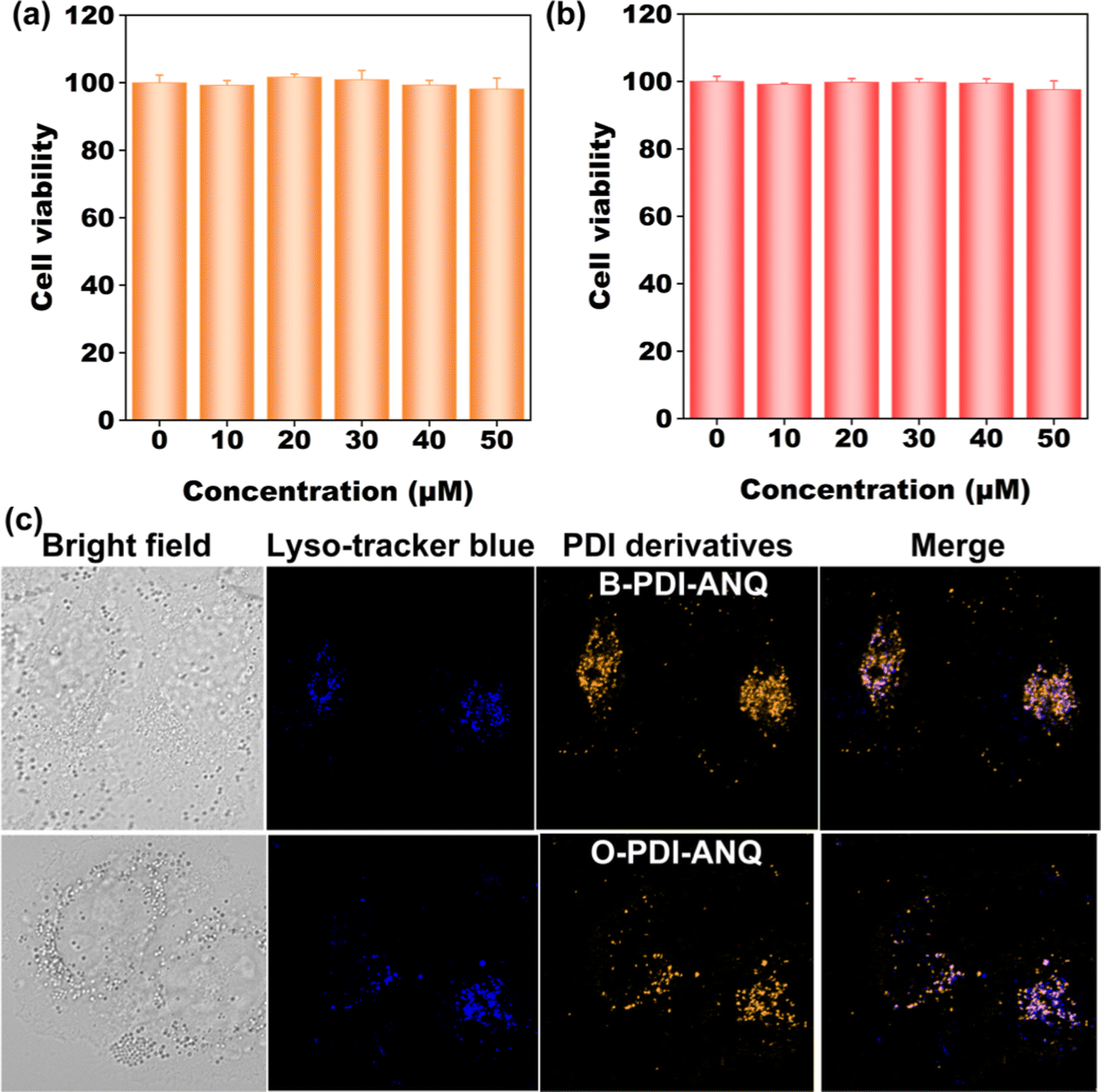 | ||
| Fig. 6 (a and b) Cell viability of HeLa cells incubated with different concentrations of B-PDI-ANQ and O-PDI-ANQ for 8 h. (c) Confocal images of HeLa cells co-stained with B-PDI-ANQ and O-PDI-ANQ (25 μM) and LTB (25 μM). | ||
Conclusions
In summary, the regioisomerism strategy is employed in exploring the structure–property relationship of aggregated PDI derivatives. The experimental and theoretical results reveal that changing the substitution position of the anthraquinone group on the PDI from the bay position to the ortho position can improve the planarity of the PDI core and enhance the structural rigidity of the product. Femtosecond transient absorption studies on their isolated and aggregated state confirm that the regioisomerism from the bay to the ortho position not only blocks the nonradiative decay channels in the non-aggregated state but also decelerates nonradiative excited-state decay in the aggregated state, thus resulting in enhanced fluorescence emission of the isolated molecules and a suppressed fluorescence quenching effect of the aggregated molecules. Two-photon excited fluorescence studies on their aggregated state also show a suppressed fluorescence quenching effect of the ortho-substituted product. Cellular imaging studies reveal their potential application in fluorescence imaging of live cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52073167, 21702132) and the Guangdong Basic and Applied Basic Research Foundation (2021A1515011879). Q. Z. thanks the funding support from the City University of Hong Kong (9380117, 7005620 and 7020040) and Hong Kong Institute for Advanced Study, City University of Hong Kong, Hong Kong, P.R. China.Notes and references
- M. Fang, J. Yang and Z. Li, Prog. Mater. Sci., 2022, 125, 100914 CrossRef CAS
.
-
(a) D. Ding, D. Mao, K. Li, X. Wang, W. Qin, R. Liu, D. S. Chiam, N. Tomczak, Z. Yang, B. Z. Tang, D. Kong and B. Liu, ACS Nano, 2014, 8, 12620–12631 CrossRef CAS PubMed
- J. Qi, C. Sun, D. Li, H. Zhang, W. Yu, A. Zebibula, J. W. Y. Lam, W. Xi, L. Zhu, F. Cai, P. Wei, C. Zhu, R. T. K. Kwok, L. L. Streich, R. Prevedel, J. Qian and B. Z. Tang, ACS Nano, 2018, 12, 7936–7945 CrossRef CAS PubMed
- L. Zong, H. Zhang, Y. Li, Y. Gong, D. Li, J. Wang, Z. Wang, Y. Xie, M. Han, Q. Peng, X. Li, J. Dong, J. Qian, Q. Li and Z. Li, ACS Nano, 2018, 12, 9532–9540 CrossRef CAS PubMed
- J. Wang, X. Gu, P. Zhang, X. Huang, X. Zheng, M. Chen, H. Feng, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16974–16979 CrossRef CAS PubMed
- R. Tang and Z. Li, Chem. Rec., 2017, 17, 71–89 CrossRef CAS PubMed
- L. Xu, H. Zhu, G. Long, J. Zhao, D. Li, R. Ganguly, Y. Li, Q.-H. Xu and Q. Zhang, J. Mater. Chem. C, 2015, 3, 9191–9196 RSC
- L. Xu and Q. Zhang, Sci. China Mater., 2017, 60, 1093 CrossRef CAS
- L. Xu, J. Zhang, L. Yin, X. Long, W. Zhang and Q. Zhang, J. Mater. Chem. C, 2020, 8, 6342–6349 RSC
- L. Xu, W. Lin, B. Huang, J. Zhang, X. Long, W. Zhang and Q. Zhang, J. Mater. Chem. C, 2021, 9, 1520–1536 RSC
- A. Huang, Q. Li and Z. Li, Chin. J. Chem., 2022, 40, 2356–2370 CrossRef CAS
- J. Yang, M. Fang and Z. Li, Aggregate, 2020, 1, 6–18 CrossRef
- Q. Li and Z. Li, Sci. China Mater., 2020, 63, 177–184 CrossRef
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed
- H. Zhang, Z. Zhao, A. T. Turley, L. Wang, P. R. McGonigal, Y. Tu, Y. Li, Z. Wang, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Adv. Mater., 2020, 32, 2001457 CrossRef CAS
- Q. Li and Z. Li, Acc. Chem. Res., 2020, 53, 962–973 CrossRef CAS PubMed
- C. Wang, B. Xu, M. Li, Z. Chi, Y. Xie, Q. Li and Z. Li, Mater. Horiz., 2016, 3, 220–225 RSC
- J. Wang, Z. Chai, J. Wang, C. Wang, M. Han, Q. Liao, A. Huang, P. Lin, C. Li, Q. Li and Z. Li, Angew. Chem., Int. Ed., 2019, 58, 17297–17302 CrossRef CAS PubMed
-
(a) B. Xu, J. He, Y. Mu, Q. Zhu, S. Wu, Y. Wang, Y. Zhang, C. Jin, C. Lo, Z. Chi, A. Lien, S. Liu and J. Xu, Chem. Sci., 2015, 6, 3236–3241 RSC
- M. Fang, J. Yang, Q. Liao, Y. Gong, Z. Xie, Z. Chi, Q. Peng, Q. Li and Z. Li, J. Mater. Chem. C, 2017, 5, 9879–9885 RSC
- F. Liu, J. Tu, X. Wang, J. Wang, Y. Gong, M. Han, X. Dang, Q. Liao, Q. Peng, Q. Li and Z. Li, Chem. Commun., 2018, 54, 5598–5601 RSC
- Y. Yu, Y. Fan, C. Wang, Y. Wei, Q. Liao, Q. Li and Z. Li, J. Mater. Chem. C, 2019, 7, 13759–13763 RSC
- X. Jia, C. Shao, X. Bai, Q. Zhou, B. Wu, L. Wang, B. Yue, H. Zhu and L. Zhu, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4816–4821 CrossRef CAS
- M. Louis, H. Thomas, M. Gmelch, A. Haft, F. Fries and S. Reineke, Adv. Mater., 2019, 31, 1807887 CrossRef
- L. Gu, H. Shi, M. Gu, K. Ling, H. Ma, S. Cai, L. Song, C. Ma, H. Li, G. Xing, X. Hang, J. Li, Y. Gao, W. Yao, Z. Shuai, Z. An, X. Liu and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 8425–8431 CrossRef CAS PubMed
- Q. Dang, L. Hu, J. Wang, Q. Zhang, M. Han, S. Luo, Y. Gong, C. Wang, Q. Li and Z. Li, Chem. – Eur. J., 2019, 25, 7031–7037 CrossRef CAS
- Y. Wang, J. Yang, M. Fang, Y. Gong, J. Ren, L. Tu, B. Z. Tang and Z. Li, Adv. Funct. Mater., 2021, 31, 2101719 CrossRef CAS
- Z. A. Li, Z. Li, C. A. Di, Z. Zhu, Q. Li, Q. Zeng, K. Zhang, Y. Liu, C. Ye and J. Qin, Macromolecules, 2006, 39, 6951–6961 CrossRef CAS
- Z. a Li, Q. Zeng, Z. Li, S. Dong, Z. Zhu, Q. Li, C. Ye, C. a Di, Y. Liu and J. Qin, Macromolecules, 2006, 39, 8544–8546 CrossRef CAS
- Z. a Li, W. Wu, G. Yu, Y. Liu, C. Ye, J. Qin and Z. Li, ACS Appl. Mater. Interfaces, 2009, 1, 856–863 CrossRef CAS PubMed
- W. Wu, Q. Huang, G. Qiu, C. Ye, J. Qin and Z. Li, J. Mater. Chem., 2012, 22, 18486–18495 RSC
- W. Wu, C. Wang, R. Tang, Y. Fu, C. Ye, J. Qin and Z. Li, J. Mater. Chem. C, 2013, 1, 717–728 RSC
- W. Wu, C. Ye, J. Qin and Z. Li, ACS Appl. Mater. Interfaces, 2013, 5, 7033–7041 CrossRef CAS
- R. Tang, S. Zhou, Z. Cheng, G. Yu, Q. Peng, H. Zeng, G. Guo, Q. Li and Z. Li, Chem. Sci., 2017, 8, 340–347 RSC
- Q. Li and Z. Li, Adv. Sci., 2017, 4, 1600484 CrossRef PubMed
- G. Jiang, R. Hu, C. Li, J. Gong, J. Wang, J. W. Y. Lam, A. Qin and B. Zhong Tang, Chem. – Eur. J., 2022 DOI:10.1002/chem.202202388
- Y. Zhou, W. Qin, C. Du, H. Gao, F. Zhu and G. Liang, Angew. Chem., Int. Ed., 2019, 58, 12102–12106 CrossRef CAS PubMed
- Y. Fan, Q. Li and Z. Li, Mater. Chem. Front., 2021, 5, 1525–1540 RSC
- J. Yang, H. Gao, Y. Wang, Y. Yu, Y. Gong, M. Fang, D. Ding, W. Hu, B. Z. Tang and Z. Li, Mater. Chem. Front., 2019, 3, 1391–1397 RSC
- Y. Yu, Y. Fan, C. Wang, Y. Wei, Q. Liao, Q. Li and Z. Li, Mater. Chem. Front., 2021, 5, 817–824 RSC
- M. Fang, J. Yang, X. Xiang, Y. Xie, Y. Dong, Q. Peng, Q. Li and Z. Li, Mater. Chem. Front., 2018, 2, 2124–2129 RSC
- S. Sasaki, K. Igawa and G.-I. Konishi, J. Mater. Chem. C, 2015, 3, 5940–5950 RSC
- Y. Yu, C. Wang, Y. Wei, Y. Fan, J. Yang, J. Wang, M. Han, Q. Li and Z. Li, Adv. Opt. Mater., 2019, 7, 1900505 CrossRef
- H. Wu, X. Song, B. Zhang, Z. Wang, T. Zhang, A. Qin and B. Z. Tang, Mater. Chem. Front., 2020, 4, 1706–1713 RSC
- Y. Li, S. Liu, H. Ni, H. Zhang, H. Zhang, C. Chuah, C. Ma, K. S. Wong, J. W. Y. Lam, R. T. K. Kwok, J. Qian, X. Lu and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 12822–12826 CrossRef CAS PubMed
- P. Funchien, P. Chasing, T. Sudyoadsuk and V. Promarak, Chem. Commun., 2020, 56, 6305–6308 RSC
- L. R. Adil and P. K. Iyer, Chem. Commun., 2020, 56, 7633–7636 RSC
- H.-X. Yu, J. Zhi and J.-L. Wang, J. Mater. Chem. C, 2021, 9, 3882–3891 RSC
- Q. Zhao, S. Zhang, Y. Liu, J. Mei, S. Chen, P. Lu, A. Qin, Y. Ma, J. Z. Sun and B. Z. Tang, J. Mater. Chem., 2012, 22, 7387–7394 RSC
- X. Long, J. Wu, S. Yang, Z. Deng, Y. Zheng, W. Zhang, X.-F. Jiang, F. Lu, M.-D. Li and L. Xu, J. Mater. Chem. C, 2021, 9, 11679–11689 RSC
- L. Xu, X. Long, F. Kang, Z. Deng, J. He, S. Yang, J. Wu, L. Yin, X.-F. Jiang, F. Lu, M.-D. Li and Q. Zhang, J. Mater. Chem. C, 2022, 10, 7039–7048 RSC
- B. Carlotti, I. K. Madu, H. Kim, Z. Cai, H. Jiang, A. K. Muthike, L. Yu, P. M. Zimmerman and T. Goodson, Chem. Sci., 2020, 11, 8757–8770 RSC
- H. Zhu, Y. Yang, K. Wu and T. Lian, Annu. Rev. Phys. Chem., 2016, 67, 259–281 CrossRef CAS PubMed
- S. Yamaguchi and H.-O. Hamaguchi, J. Chem. Phys., 1998, 109, 1397–1408 CrossRef CAS
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed
- S. J. Formosinho and L. G. Arnaut, J. Photochem. Photobiol., A, 1993, 75, 21–48 CrossRef CAS
- J. H. Lee, M. Wulff, S. Bratos, J. Petersen, L. Guerin, J.-C. Leicknam, M. Cammarata, Q. Kong, J. Kim, K. B. Møller and H. Ihee, J. Am. Chem. Soc., 2013, 135, 3255–3261 CrossRef CAS PubMed
- P. Foggi, L. Bussotti and F. V. R. Neuwahl, Int. J. Photoenergy, 2001, 3, 164849 CrossRef
- Y. J. Wang, Z. Li, J. Tong, X. Y. Shen, A. Qin, J. Z. Sun and B. Z. Tang, J. Mater. Chem. C, 2015, 3, 3559–3568 RSC
- Y. Liu, Y. Zhang, X. Wu, Q. Lan, C. Chen, S. Liu, Z. Chi, L. Jiang, X. Chen and J. Xu, J. Mater. Chem. C, 2014, 2, 1068–1075 RSC
- L. Yang, X. Wang, G. Zhang, X. Chen, G. Zhang and J. Jiang, Nanoscale, 2016, 8, 17422–17426 RSC
- J. Zhang, E. Sharman, L. Yang, J. Jiang and G. Zhang, J. Phys. Chem. C, 2018, 122, 25796–25803 CrossRef CAS
- K. Nagarajan, A. R. Mallia, V. S. Reddy and M. Hariharan, J. Phys. Chem. C, 2016, 120, 8443–8450 CrossRef CAS
- K. Nagarajan, A. R. Mallia, K. Muraleedharan and M. Hariharan, Chem. Sci., 2017, 8, 1776–1782 RSC
- Z. Mahmood, A. A. Sukhanov, N. Rehmat, M. Hu, A. Elmali, Y. Xiao, J. Zhao, A. Karatay, B. Dick and V. K. Voronkova, J. Phys. Chem. B, 2021, 125, 9317–9332 CrossRef CAS PubMed
- Y. Dong, B. Dick and J. Zhao, Org. Lett., 2020, 22, 5535–5539 CrossRef CAS PubMed
- Z. Wang, L. Huang, Y. Yan, A. M. El-Zohry, A. Toffoletti, J. Zhao, A. Barbon, B. Dick, O. F. Mohammed and G. Han, Angew. Chem., Int. Ed., 2020, 59, 16114–16121 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc03928a |
| This journal is © The Royal Society of Chemistry 2023 |