
Modulation of the photothermal degradation of epoxy upon ultraviolet irradiation by polyhedral oligomeric silsesquioxane: a multiscale simulation study†
Youngoh
Kim
ab and
Joonmyung
Choi
 *ab
*ab
aDepartment of Mechanical Design Engineering, Hanyang University, 222 Wangsimni-ro, Seongdong-gu, Seoul, 04763, Korea. E-mail: joonchoi@hanyang.ac.kr
bDepartment of Mechanical Engineering, BK21 FOUR ERICA-ACE Center, Hanyang University, 55 Hanyangdaehak-ro, Sangnok-gu, Ansan, 15588, Korea
First published on 10th January 2023
Abstract
Polyhedral oligomeric silsesquioxane (POSS) nanocomposites that improve the ultraviolet (UV) resistance without affecting the transmittance and mechanical properties of the crosslinked epoxy are in the spotlight. In this study, the structural role of POSS nanofillers in the UV resistance of epoxy was theoretically examined using multiscale analysis that integrates classical molecular dynamics and density functional theory. Under deep-UV irradiation, POSS contributes to the energy dissipation of photothermally excited vibrations rather than its role as a photochemical UV absorber. The intrachain energy transfer modulated by POSS suppresses network chain scission and reduces the rate of fragmentation events. In particular, the unique molecular network structure constructed by POSS inhibits fragment aggregation and prevents the morphological collapse of nanocomposites, such as pore growth and phase separation. The proposed mechanism elucidates the fundamental principle of UV resistance improvement owing to POSS insertion in terms of photothermal reaction-induced thermodynamics.
Introduction
Epoxy resins have been regarded as fascinating materials in a wide variety of advanced technological applications because of their outstanding adhesion to different substrates, excellent thermal resistance, chemical stability, and mechanical properties.1–4 However, the poor ultraviolet (UV) stability of epoxy resins is a major drawback to applying it in various outdoor environments.5,6 In particular, UV radiation with deep-UV wavelengths is considered a serious threat to the use of epoxy resins in shielding materials for UV lamps,7,8 electronic devices,9 and space shuttles,10 because they easily degrade polymer chains even with a small number of photons.Recently, attempts to insert polyhedral oligomeric silsesquioxanes (POSSs) as nanofillers to improve the UV stability of epoxy resins have received considerable attention.11 POSS is a hybrid material with an inorganic nanocage as the core structure and organic constituents attached to it as functional groups.12 POSS nanofillers improve the thermal stability of the polymer matrix within the composites and provide excellent durability against atomic oxygen attack owing to their chemical stability.13,14 Moreover, the large transition energy inherent in inorganic cages provides high UV stability.15 In particular, POSS is excellent for retaining its mechanical properties during UV exposure. The tensile strength of the neat polybenzoxazine (PBZ)/epoxy system was reduced by 24% (74 → 56 MPa), whereas that of PBZ/epoxy/POSS was reduced by only 4% (96 → 92 MPa).16 Deng's experiment showed that the high POSS content of monodispersed hybrid microparticles based on POSS significantly inhibited structural collapse after UV exposure.17 These advantages make POSS very promising compared to other inorganic UV absorbers such as ZnO and TiO2, which affect the transmittance and mechanical properties of nanocomposites.18–20
Despite these abundant experimental findings, the theoretical principle by which POSS improves the UV stability of the entire system within polymer nanocomposites has not been fully resolved. Thus far, the properties of conventional UV-absorbing materials have been explained by the presence of p-orbitals formed by parts of inorganic or organic materials that absorb photons.21–23 Although POSS cannot be classified as a UV absorber, it improves the UV resistance of the matrix over its photochemical stability in polymer nanocomposites.11,15,24,25 In other words, the mechanism by which POSS improves the UV resistance of polymer nanocomposites in the absence of direct photochemical contributions remains an unresolved problem. Therefore, a theoretical approach to photothermal reaction-induced thermodynamics in POSS nanocomposites is required.
In this study, the photochemistry and photothermal reaction-induced thermodynamics of POSS nanocomposites were investigated using a multiscale simulation approach. The photochemistry of the epoxy matrix and POSS was explored using time-dependent density functional theory (TD-DFT), and the photothermal induced-thermodynamics of the POSS nanocomposites were analysed by integrating classical molecular dynamics (MD) simulations and density functional theory (DFT) calculations. The deterioration by the vibrationally excited chain under UV irradiation was evaluated, and the morphological changes in the microstructure owing to the growth of pores and by-product aggregation were quantified numerically. The mechanism by which POSS modulates the photothermal reaction-induced thermodynamics of the entire microstructure has been intensively described in terms of the dissipation of vibrational energy. The effect of the unique pore structure composed of inorganic cages on the inhibition of fragment aggregation was also discussed.
Results and discussion
Structural mechanics of POSS
All-atom MD simulations were performed to understand the structural mechanics of neat epoxy and epoxy/POSS nanocomposites. Each molecular component constituting the microstructure was inserted into the simulation box in an uncrosslinked state, and the new covalent bond formation and equilibration processes were repeated until the epoxy resin reached a crosslinking rate of 70% (Fig. 1a). Subsequently, the mechanical properties of the models were evaluated by performing uniaxial tensile loading simulations in the elastic region, with a total strain of 0.3%. The details of the uniaxial tensile loading conditions are provided in Appendix A and Fig. S1 in the ESI.† The improvement in the mechanical properties of the microstructures by POSS insertion was observed from the comparison of the elastic modulus results (Fig. 1b). This suggests that the load transfer between the reinforcement material and the matrix occurred directly because all functional groups of the inorganic cage participated in crosslinking to form a macro network structure. This enhancement of the modulus of elasticity is consistent with the experimental observations in the study by Thitsartarn et al.26 In addition, the mechanical properties of neat epoxy (4.6 GPa) predicted in this MD simulation were quantitatively consistent with the results of other studies (2.3–6.0 GPa),27–29 validating the model preparation process and results. | ||
| Fig. 1 Structural mechanics of crosslinked epoxy and epoxy/POSS nanocomposites. (a) Configurations of the neat epoxy (left) and epoxy/POSS nanocomposite (right) models. The crosslinking reaction simulation scheme of the epoxy resin is also shown. (b) Stress–strain curves of neat epoxy and epoxy/POSS nanocomposites obtained from uniaxial tensile loading simulations. (c) Changes in non-bond and bond potential energy of each component during the deformation. The distributions shown on the left are for the neat epoxy and those shown on the right are for the epoxy/POSS nanocomposites. Each dot represents the energy of atoms constituting the major constituents of the molecule (aromatic ring, backbone chain, epoxy ring, hydroxyl group, and POSS). (d) Mean potential energy change for the aromatic ring, hydroxyl group, and POSS constituents after deformation. The graphs on the left and right represent the bond stretching and non-bond interactions, respectively. (e) The virial stress applied to the aromatic ring, hydroxyl group, and POSS constituents, respectively. (f) Inhomogeneous distribution of interaction energy around the POSS network. The energy conversion is quantified as the absolute value of the product of the change in bond and non-bond potential energy applied to each atom. | ||
The mechanical role of POSS in the nanocomposites was further investigated based on the potential energy variation of each component constituting the microstructures under mechanical loading (Fig. 1c). The aromatic and epoxy ring directly participate in the load transfer within the crosslinked network; therefore, their potential energy changes due to the covalent bonds are particularly pronounced. The hydroxyl group is not contributed by covalent bonding but imparts high adhesion between the chains of the epoxy resin through polarity. In contrast, backbone chains composed of C–C Sigma bonds exhibit little change in potential energy during mechanical loading because of their high deformability.30 One interesting finding was that the POSS of the epoxy/POSS nanocomposites exhibited significant potential energy changes in both covalent and non-covalent bonds. The high dispersion of the potential energy distribution can be explained by the unique geometric properties of POSS at the nanoscale level. Exploring the results specifically from a structural point of view, POSS constructs numerous nanopores on the cage surface, while providing a robust inorganic framework through functional groups at the corners. Therefore, the change in the covalent bond energy due to the elongation of the functional group and the change in the non-covalent bond energy due to the transformation of the cubic structure was simultaneously observed.
The steric deformation of the inorganic cage structure occupied the highest energy share among the components of the load transfer process (Fig. 1d). Because the cubic structure of POSS does not collapse, the applied mechanical load preferentially collapses the nanopores of the epoxy matrix through the deformation of the steric network. As the nanopore area around the POSS decreased, the non-covalent repulsive force increased rapidly, weakening the interaction between POSS and the epoxy matrix. The reduction in the non-bond potential energy applied to the inorganic cage quantitatively explains its behavioural properties. The structural disruption of nanopores also weakens the interactions between polymer chains, thus reducing load transfer via hydroxyl groups. As a result, the mechanical stress to the external load was concentrated toward the POSS, and the effective stress of the neighbouring polymer network also increased under the same strain (Fig. 1e and f). In conclusion, the robust structure of the POSS cage acts as a transporter that converts the non-covalent energy inside the network into a stretching form of covalent bonds on the functional groups.
Multiscale analysis of the photochemical process of epoxy upon UV irradiation
The dominant processes of electronic excitation and vibronic relaxation under deep-UV light were identified to comprehensively understand the photothermal disintegration of polymeric matter under a wide range of outdoor conditions (from the ground state to the low-earth orbit environment). For the molecular structures of diethyltoluenediamine (DETDA), bisphenol F diglycidyl ether (DGEBF), and POSS derivatives (Fig. 2a), the dominant photochemical process was predicted using TD-DFT calculations. The upper threshold of the photon energy was set at 5 eV (≅250 nm), which is the valid wavelength range of solar UV radiation. The limiting of the photon energy range was based on the fact that the irradiance spectrum obtained from the International Space Station during the EXPOSE-R2 mission showed a significant increase in intensity in the wavelength range above 250 nm (Fig. 2b).31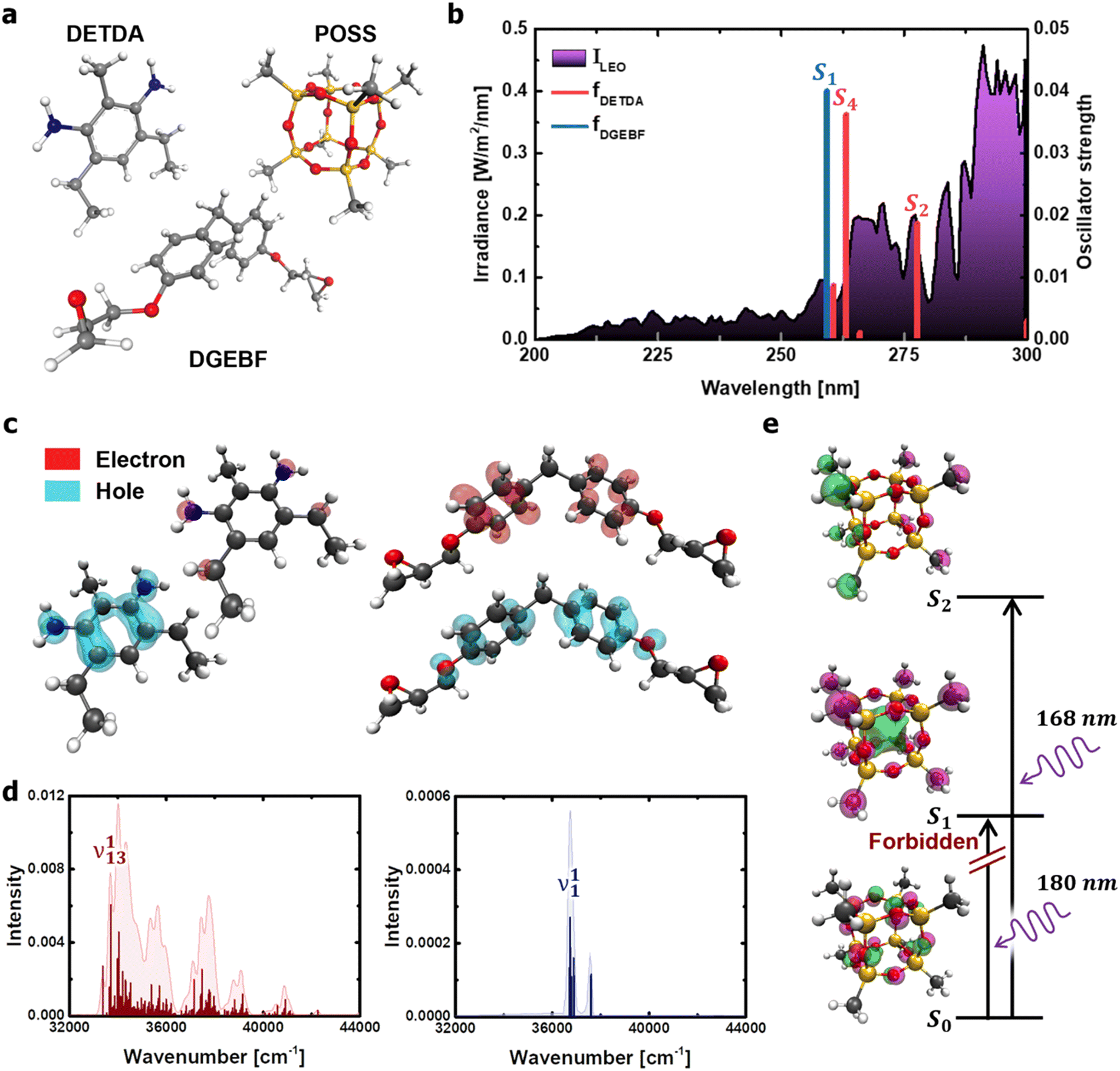 | ||
| Fig. 2 Calculation of photochemical properties of the DETDA, DGEBF, and POSS molecules. (a) Molecular structures considered in the TD-DFT. (b) Transition energies of DETDA and DGEBF. UV irradiance measured in low-earth orbit condition31 is shown (in purple) as a background curve for comparison. (c) Hole and electron distributions of the S2 state of DETDA (left), and the ones of the S4 state of DGEBF (right). (d) Vibronic spectrum of the S2 state of DETDA (left), and the one of the S4 state of DGEBF (right) obtained from the Franck–Condon transition. (e) Molecular orbitals of excited states of the POSS molecule. UV wavelengths required for the transition of the methyl group of the POSS constituent are also shown. | ||
Six excited electronic states were clearly distinguished in the wavelength range in which the irradiance increased significantly. One of the excited states is observed at 260 nm, a well-known peak for aromatic chromophores.32 This transition process originates from the DGEBF molecule, and the remaining five excited states originate from DETDA. Nevertheless, the oscillator strengths of DETDA are smaller than S1 of DGEBF, and even the S4 state of DETDA remains at a similar level of transition energy as S1 of DGEBF. These results suggest that DGEBF in the S1 state and DETDA in the S4 state are competitively allocated excitation energies for a limited number of photons of specific wavelengths. It is also noteworthy that S2 of DETDA has a fairly large oscillator strength within the epoxy system but has no other excited electronic states in the surrounding wavelength region. In other words, all photons with a wavelength of about 280 nm are only consumed to achieve the S2 state of DETDA. The S0 → S2 excitation occurs very frequently due to the abundance of photons, large oscillator strengths, and lone-occupied transition energy levels. In addition, the excitation path makes the S4 state a double excited state that requires more energy than singly excited states. In conclusion, the S2 state in the DETDA molecule is considered to be more potent for photochemical degradation than the S4 state. Among the major excited states, the S2 state of DETDA contains (n, σ*) of the amine group, and thus a large change in electron density around nitrogen is observed during the transition (Fig. 2c). The local change in the electron density gives momentum to the nitrogen of the amine during the transition process and activates the vibrational excitation state of v113 (Fig. 2d and Fig. S2, ESI†). In this case, the correlation between the electron density change and excited vibrational mode follows the Franck–Condon principle,33–36 as it represents the conversion of the photochemical process to thermodynamics by energetic photon irradiation. The vibrational energy in the conversion process, defined as 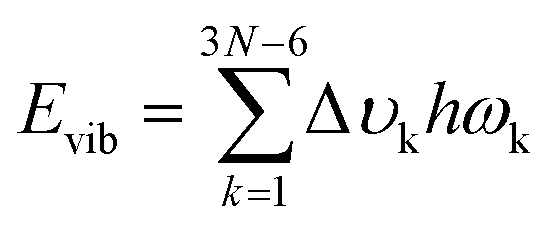 , was calculated to be 0.961 kcal mol−1 for the v113 state. The vibrational energy was clearly driven by the motion of the amine group (Fig. S2, ESI†), which was dissipated in the form of the kinetic energy of DETDA during the relaxation process. However, the S1 state of the DGEBF involves excitation from the π orbital to π* orbital, so the excitation accompanies changes in a wide range of electron densities. The occupation of π* orbital causes the expansion of aromatic rings in DGEBF, while the v11 state with rotation of the aromatic ring is activated during the π → π* transition. Accordingly, the aromatic rings of DGEBF rotate collectively due to the attractive Coulomb interaction between the unhybridized p orbital and counterpart hydrogen. The vibration energy obtained from DGEBF is 0.073 kcal mol−1, which is unambiguously lower than the vibrational excited energy level of the v113 state. The difference in vibrational energy between the v113 and v11 states is attributed to the kinematic deformation of the structure. The v113 state includes N–C–C scissoring and N–H stretching, whereas the v11 state is achieved by the torsional motion of C–O–C–C. In other words, the weak vibrational energy is caused by the v11 state associated with the higher deformation mode, that is, torsion of the C–O–C–C bond.
, was calculated to be 0.961 kcal mol−1 for the v113 state. The vibrational energy was clearly driven by the motion of the amine group (Fig. S2, ESI†), which was dissipated in the form of the kinetic energy of DETDA during the relaxation process. However, the S1 state of the DGEBF involves excitation from the π orbital to π* orbital, so the excitation accompanies changes in a wide range of electron densities. The occupation of π* orbital causes the expansion of aromatic rings in DGEBF, while the v11 state with rotation of the aromatic ring is activated during the π → π* transition. Accordingly, the aromatic rings of DGEBF rotate collectively due to the attractive Coulomb interaction between the unhybridized p orbital and counterpart hydrogen. The vibration energy obtained from DGEBF is 0.073 kcal mol−1, which is unambiguously lower than the vibrational excited energy level of the v113 state. The difference in vibrational energy between the v113 and v11 states is attributed to the kinematic deformation of the structure. The v113 state includes N–C–C scissoring and N–H stretching, whereas the v11 state is achieved by the torsional motion of C–O–C–C. In other words, the weak vibrational energy is caused by the v11 state associated with the higher deformation mode, that is, torsion of the C–O–C–C bond.
To investigate the electron states of an inorganic cage, a POSS molecule in which each corner was terminated using a methyl group was considered (Fig. 2e). The results show that photons with a wavelength less than 180 nm are required to achieve excitation of the POSS. Moreover, the S0 → S1 excitation is strictly forbidden due to the identical angular momentum of S0 and S1 states that are dominantly involved in a lone pair composed of sp3 oxygen hybridized orbitals. In other words, the inorganic cage of POSS is photochemically stable in the deep UV regions with wavelengths greater than 200 nm.
Modulation of photothermal reaction-induced thermodynamics by POSS insertion
The kinetics of the polymer chains during UV irradiation were analysed to understand the role of inorganic cages in nanocomposites. The photothermal reaction-induced thermodynamics of the polymer networks was investigated using MD simulations. A key aspect is that, following the Franck–Condon principle,33–36 high-energy photons vertically excite the electronic state while simultaneously activating vibration modes that depend on the excited-state potential energy surface. Therefore, in the MD simulations covered in this study, photothermal reaction-induced thermodynamics were implemented by providing kinetic energy to the atoms involving the activated molecular vibrational modes. In addition, covalent bonds that exceeded the bond dissociation energy were cleaved during time integration. The description of the UV irradiation process in the MD framework is provided in Appendix B of the ESI.† The polymer networks damaged by UV irradiation were partly decomposed, and the number of independent molecules constituting the system increased accordingly (Fig. 3a). The degradation occurred mainly in the form of protonation and chain scission (Fig. 3b and c). Protonation is well known as the early stage of fatal deterioration of epoxy,37,38 but it was revealed to be an independent factor from UV resistance due to POSS insertion (Fig. 3b). One point that can be drawn from the results is that protonation occurs in the hydroxyl and amine groups by the intensification of the photothermal-induced vibration of the epoxy network (Fig. 3d). Breakage of the polar functional group due to protonation weakens the attractive hydrogen bonds and dipole–dipole interactions between the epoxy chains and cleaves the main chain with Norrish reactions (via beta scission reaction).37 Furthermore, the hydroperoxyl formed by oxygen in the atmosphere during protonation oxidises epoxy networks to cause the aromatic ring opening.13,39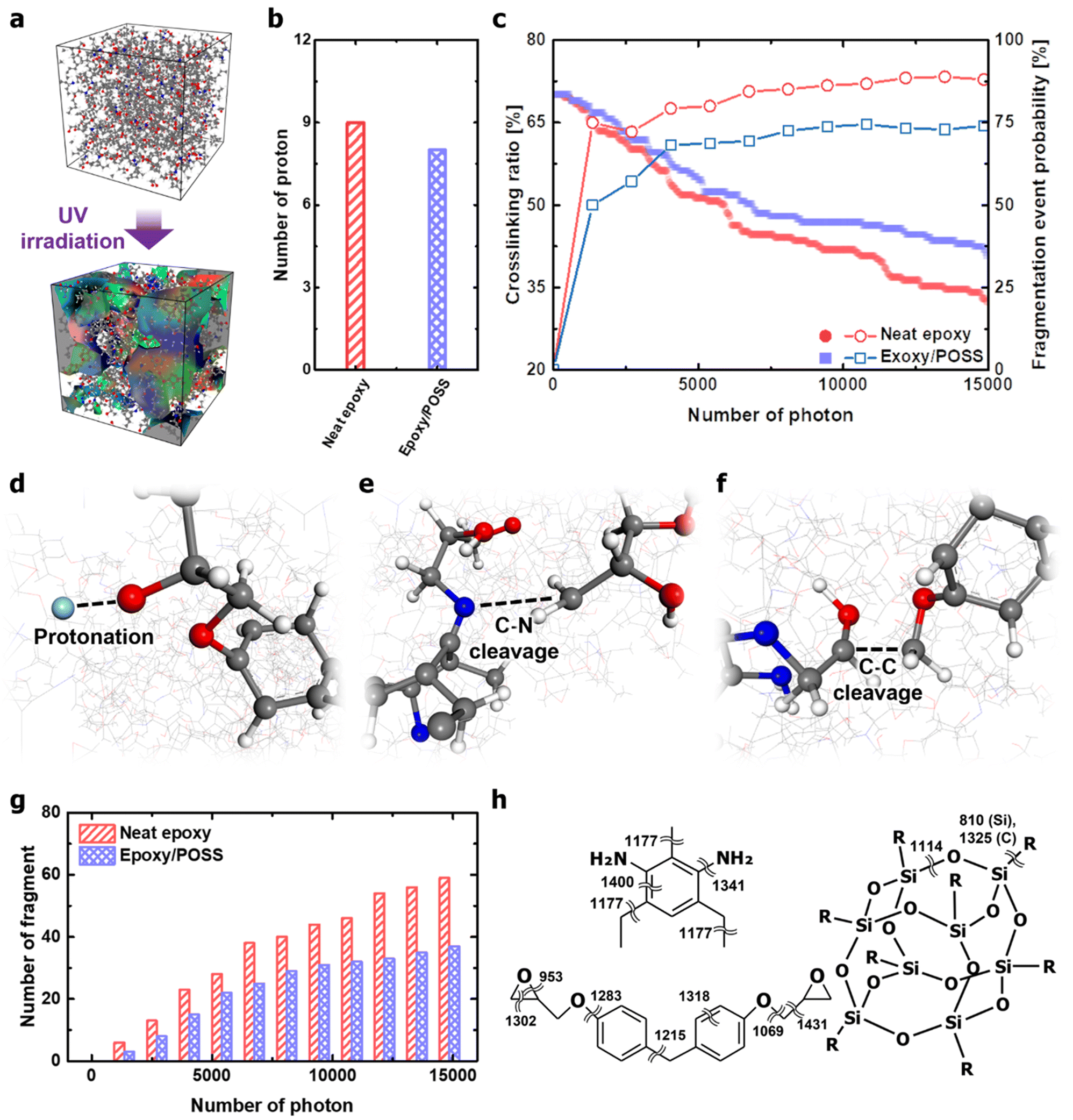 | ||
| Fig. 3 UV degradation properties of neat epoxy and epoxy/POSS nanocomposites. (a) Schematic of degradation of network structure by UV irradiation. Space occupied by fragment cleavage from the network during UV degradation is highlighted. The surface of the volume is sequentially coloured in the order of red–green–blue according to the scission order of fragments. (b) Number of protons released from the epoxy network during UV irradiation. (c) Crosslinking ratio reduction curve due to C–N cleavage under UV irradiation (filled symbols) and the corresponding fragmentation event probability (open symbols). Early stages of UV degradation of epoxy observed in MD simulations: (d) protonation, (e) C–N cleavage, and (f). C–C cleavage. (g) Number of fragments cleavage from the epoxy network during UV irradiation. (h). Distribution of stretching frequencies of the covalent bonds constituting DETDA, DGEBF, and POSS. | ||
Compared with the photodegradation in neat epoxy, the nanocomposites with intercalated POSS suppressed the decrease in the crosslinking ratio and the formation of fragments (Fig. 3c). DETDA and DGEBA exposed to high-energy photons are vibrationally excited, which is thermodynamically dissipated through intramolecular energy transfer. In the process of dissipating the provided kinetic energy, covalent bonds that are vulnerable to structural vibrations, such as stretching, scissoring, and torsion, are cleaved. The cleavage of C–N bonds was observed in both neat epoxy and epoxy/POSS nanocomposites, and the breakage of the C–C bond was identified only in the neat epoxy (Fig. 3e and f). The cleavage of the C–N bond dominantly occurs in network scission than in other types of covalent bonds, many of which lead to fragmentation. Therefore, the weakest photothermal bond in the UV-irradiated environment was C–N. This is in good agreement with the collapse of the C–N peaks of the UV-degraded epoxy reported by Liu et al.40 Note that the insertion of POSS not only directly inhibits the cleavage of the C–N bonds but also reduces the probability that the broken C–N leads to fragmentation (Fig. 3g). The fragmentation blocking behaviour is especially valid after the closed network structure is released, suggesting that the improvement in UV resistance after POSS insertion works throughout the lifespan of the material.
The enhancement of the UV resistance of the epoxy network by POSS was further interpreted as the thermodynamic dispersion of the vibration energy in the backbone chain. Molecular vibration frequencies based on classical harmonic oscillators in DETDA, DGEBF, and POSS were analysed using DFT calculations. The details of the DFT calculations are provided in Appendix C of the ESI.† The frequencies of the covalent bond stretching mode in the organic parts were mostly distributed in the range of 1200–1400 cm−1 (Fig. 3h). However, the frequency of the Si–O stretching mode constituting the inorganic cage of POSS is 1114 cm−1, which is lower than most covalent bonds in the epoxy network. The low-frequency stretching mode allows the POSS to efficiently absorb the kinetic energy inside the epoxy network. In addition, the dual frequency of Si–C due to the structural vibration of the inorganic cage improves the energy transfer kinetics from an epoxy matrix to the filler.41 The structural vibration of the inorganic cage with a low-frequency stretching mode perturbs the intrachain energy-transfer kinetics of the epoxy network, thereby inhibiting the cleavage of C–N bonds with higher stretching modes. As an exception, the C–O bond between the bisphenol A – glycidyl group with a low frequency of 1069 cm−1 did not cleave under the thermodynamic dispersion of photothermal-induced vibration. This high degree of thermodynamic robustness is due to the formation of hydrogen-bonded dampers through strong electrostatic attraction (Fig. S3, ESI†).
Effect of POSS insertion on structural deterioration by UV irradiation
The effects of POSS on morphological changes caused by network branching and fragmentation were numerically quantified using the Connolly surface analysis method.42 Although the neat epoxy experienced significant changes before and after UV irradiation, the epoxy/POSS nanocomposites showed no remarkable change in the distribution and size of the nanopores (Fig. 4a). Specifically, the neat epoxy exhibited a significant drop in the internal specific surface area with a decrease in the absolute volume fraction of pores upon exposure to UV (left column in Fig. 4b). Pore enlargement occurs because the fragments generated by C–N cleavage gather in a direction that minimises the interfacial area between the vacuum voids and the degraded epoxy. The formation of large pores is not only directly related to the degradation of mechanical properties43,44 but also causes visible cracks on the surface.44,45 In contrast, the epoxy/POSS nanocomposites show only a slight decrease in both the pore volume fraction and specific surface area during structural deterioration with UV irradiation. The structural constancy of the pores is due to the inhibition of fragment aggregation in the epoxy/POSS nanocomposites. Because the regions formed by the POSS network were sufficiently widespread within the microstructure, the structural collapse was directly prevented.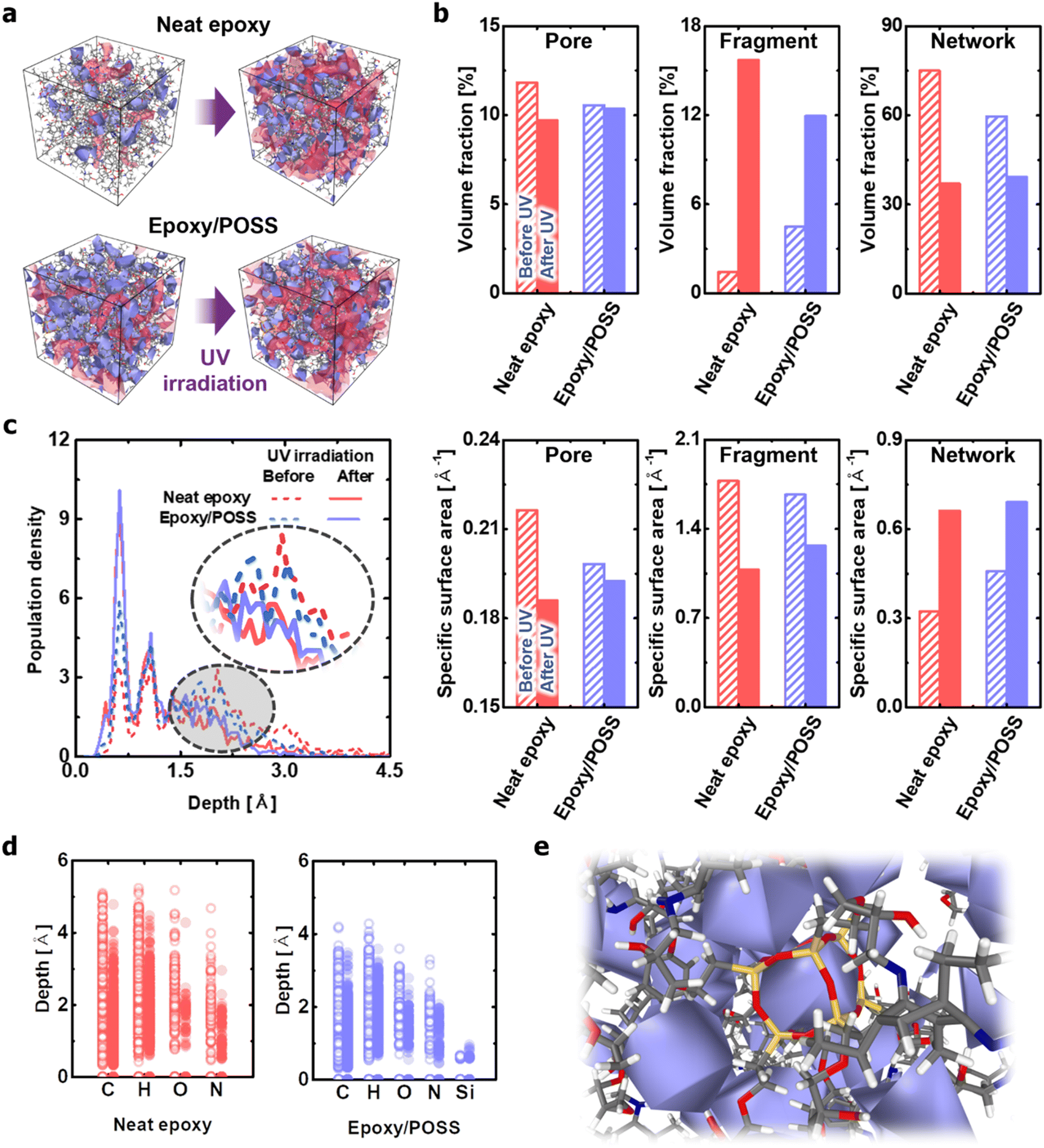 | ||
| Fig. 4 Structural deterioration of neat epoxy and epoxy/POSS nanocomposites under UV irradiation. (a) Changes in microstructure before and after UV irradiation. The occupied spaces with pores and fragments are coloured in blue and red, respectively. (b) Occupied volume and specific surface area of network, fragments, and pores in the modelled unit cells. (c) Distributions of the atoms in the network according to the distance from the epoxy chain region defined by the Connolly surface. (d) Distance distributions from the Connolly surface according to atomic species. The distance data of all atoms constituting the network structure is shown at once. (e) A representative snapshot of the microstructure showing the pore structure around the POSS network. For visibility, the area occupied by the pores is covered with a blue surface. | ||
The frequent fragmentation reactions of neat epoxy were also confirmed by the occupied volume fraction between the fragmented molecules and networks (centre and right columns in Fig. 4b). Upon UV irradiation, part of the network is changed into fragments; therefore, there is an inverse relationship between the two. The results suggest that the occupied volume expansion of fragments and shrinkage of the network within the system are greater in the neat epoxy than in the epoxy/POSS nanocomposites. This trend is attributed to the suppression of the C–N cleavage and the low fragmentation probability in epoxy/POSS nanocomposites, as previously discussed. A notable observation is a difference in the amount of change in the specific surface area; the decrease in this value for neat epoxy is greater than that for the epoxy/POSS nanocomposites. The large decrease in the specific surface area of the neat epoxy suggests that the occupied volume of the fragmented molecules expands with the growth of the aggregation sites of each fragment. The significant increase in the specific surface area of the network structure constituting the neat epoxy also suggests that a serious deterioration of the morphology occurred.
The degradation level of each microstructure was analysed by quantifying the distance distribution from the Connolly surface formed by the network to the epoxy chain (Fig. 4c). In the neat epoxy before UV irradiation, the atoms constituting the network were widely distributed along the depth, indicating a densely structured cross-linking state. However, this dense structure was degraded to a rough shape by UV irradiation, which maximised the surface area. In comparison, in the epoxy/POSS nanocomposites before UV irradiation, the corresponding atoms are mainly distributed on the surface. Therefore, the change in the distribution profile due to the structural deterioration of the POSS network is less than that of the neat epoxy. The small change in the distance distribution profiles verifies that the morphology of the networks constituting the epoxy/POSS nanocomposites was maintained during degradation, even if fragmentation occurred.
Another interesting finding is that the hydrogen and carbon atoms of the network constituting a degraded neat epoxy system exist in a deeper region of the network surface than those of the epoxy/POSS nanocomposites, even before UV irradiation (Fig. 4d). This suggests that the structurally degraded neat epoxy forms a highly aggregated network, which causes phase separation of the microstructure and weakens the mechanical and thermal load transfer of the system.46–48 Note that oxygen and nitrogen are mainly distributed on the surfaces of the degraded network owing to the exposure of hydroxyl and amine groups during fragmentation. The dominant distributions of oxygen and nitrogen on the network surface suggest that hydrophilic characteristics can be developed because of structural deterioration involving exposure to polar groups and the formation of porous structures. The feature agrees well with the hydrophilic surface manifested by exposure to the polar functional group of UV-degraded epoxy reported by Gorodki et al.49 In the epoxy/POSS nanocomposites, in contrast, the broad morphology of the POSS network with a homogeneous pore distribution prevents fragments from aggregating. The homogeneity of the pore distribution contributes to the dispersion of fragmented seeds even after UV irradiation, thereby contributing to the maintenance of a robust microstructure of the epoxy/POSS nanocomposites (Fig. 4e and Fig. S4, ESI†).
Conclusions
In this study, we computationally elucidated how POSS can effectively mitigate the photodegradation of a polymer matrix in the deep UV wavelength range. The photochemical stability of the inorganic cage was exclusively investigated using TD-DFT, and the energy release behaviour of the photothermally excited POSS networks was explored using a multiscale method that integrated DFT calculations and MD simulations. The results provide several important implications: (1) the low-frequency structural vibration of the inorganic cage derived from the shape of the POSS effectively annihilates the photothermal induced vibration mode of the neighbouring epoxy network. (2) Within the microstructure, the POSS network constitutes a unique porous structure that prevents the aggregation of degraded epoxy fragments, thereby preventing phase separation and the formation of large pores. (3) The porous structure not only inhibits the microstructure collapse of the nanocomposites but also exhibits excellent load transfer capability to efficiently convert non-bonding interaction energy into deformation energy within polymer chains.The above findings adequately explain why POSS may have superior photothermal durability properties beyond the intrinsic photochemical stability of inorganic cages, as reported experimentally. The proposed multiscale analysis regime also becomes an important link between the molecular-scale structural properties of POSS nanocomposites and the phase-separation behaviour that induces crack and pore growth. Therefore, the present study provides important insights and future development directions for a variety of engineering applications that require the design of UV-resistant materials.
The present study provides a fundamental understanding of the photothermal reaction-induced thermodynamics under an ideal configuration in which the epoxy network exists alone. However, considering the real experimental environments, all polymer materials inevitably contain various impurities such as catalysts, unreacted initiators, as well as water and intermediates.50–52 Impurities can act as catalysts, physical lubrication, and optical quenchers in the polymer matrices.29,51–53 Therefore, it is clear that the impurities contribute decisively to diversifying the decomposition pathways of epoxy systems and deserve future study.
Author contributions
Youngoh Kim: methodology, software, visualization, writing – original draft. Joonmyung Choi: conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Basic Science Research Program of the National Research Foundation of Korea, funded by the Ministry of Education (No. 2022R1F1A1063199).Notes and references
- H. Wei, J. Xia, W. Zhou, L. Zhou, G. Hussain, Q. Li and K. Ostrikov, Composites, Part B, 2020, 193, 108035 CrossRef CAS.
- J. Guo, H. Song, H. Liu, C. Luo, Y. Ren, T. Ding, M. A. Khan, D. P. Young, X. Liu, X. Zhang, J. Kong and Z. Guo, J. Mater. Chem. C, 2017, 5, 5334–5344 RSC.
- M. Cui, L. Zhang, P. Lou, X. Zhang, X. Han, Z. Zhang and S. Zhu, Polym. Degrad. Stab., 2020, 176, 109143 CrossRef CAS.
- F. L. Jin and X. Li, S. and J. Park, J. Ind. Eng. Chem., 2015, 29, 1–11 CrossRef CAS.
- W. F. Zhang, L. M. Jin, S. F. Yu, H. Zhu, S. S. Pan, Y. H. Zhao and H. Y. Yang, J. Mater. Chem. C, 2014, 2, 1525–1531 RSC.
- Y. Morita, J. Appl. Polym. Sci., 2005, 97, 1395–1400 CrossRef CAS.
- X. Fan, J.-T. Miao, L. Yuan, Q. Guan, A. Gu and G. Liang, Appl. Surf. Sci., 2018, 447, 315–324 CrossRef CAS.
- J.-Y. Bae, Y. H. Kim, H. Y. Kim, Y. B. Kim, J. Jin and B.-S. Bae, ACS Appl. Mater. Interfaces, 2015, 7, 1035–1039 CrossRef CAS PubMed.
- S. Wang, C. Yang, X. Li, H. Jia, S. Liu, X. Liu, T. Minari and Q. Sun, J. Mater. Chem. C, 2022, 10, 6196–6221 RSC.
- K.-B. Shin, C.-G. Kim, C.-S. Hong and H.-H. Lee, Composites, Part B, 2000, 31, 223–235 CrossRef.
- A. Suliga, E. M. Jakubczyk, I. Hamerton and A. Viquerat, Acta Astronaut., 2018, 142, 103–111 CrossRef CAS.
- D. W. Scott, J. Am. Chem. Soc., 1945, 68, 356–358 CrossRef.
- Y. Kim and J. Choi, Appl. Surf. Sci., 2021, 567, 150578 CrossRef CAS.
- A. S. Lee, S.-S. Choi, S. Y. Oh, H. S. Lee, B. Kim, S. S. Hwang and K.-Y. Baek, J. Mater. Chem. C, 2015, 3, 11605–11611 RSC.
- C.-G. Zhen, U. Becker and J. Kieffer, J. Phys. Chem. A, 2009, 113, 9707–9714 CrossRef CAS PubMed.
- M. Selvi, S. Devaraju, M. R. Vengatesan, J. S. Go, M. Kumard and M. Alagar, RSC Adv., 2014, 4, 8238–8244 RSC.
- Y.-Y. Deng, D. Han, D.-L. Zhou, Z.-Q. Liu, Q. Zhang, Y. Li and Q. Fu, Polymer, 2019, 178, 121609 CrossRef CAS.
- Y. Sun, A. Gu, G. Liang and L. Yuan, J. Appl. Polym. Sci., 2011, 121, 2018–2028 CrossRef CAS.
- Y. Yang, Y.-Q. Li, S.-Y. Fu and H.-M. Xiao, J. Phys. Chem. C, 2008, 112, 10553–10558 CrossRef CAS.
- Y. Cao, P. Xu, P. Lv, P. J. Lemstra, X. Cai, W. Yang, W. Dong, M. Chen, T. Liu, M. Du and P. Ma, ACS Appl. Mater. Interfaces, 2020, 12, 49090–49100 CrossRef CAS PubMed.
- B. A. M. C. Santos, A. C. P. da Silva, M. L. Bello, A. S. Gonçalves, T. A. Gouvêa, R. F. Rodrigues, L. M. Cabral and C. R. Rodrigues, J. Photochem. Photobiol., A, 2018, 356, 219–229 CrossRef CAS.
- J. Wang, H. Sun, J. Huang, Q. Li and J. Yang, J. Phys. Chem. C, 2014, 118, 7451–7457 CrossRef CAS.
- X. Sun, L. Yan, R. Xu, M. Xu and Y. Zhu, Colloids Surf., A, 2019, 570, 199–209 CrossRef CAS.
- D. Peng, W. Qin and X. Wu, Acta Astronaut., 2015, 111, 84–88 CrossRef CAS.
- A. Suliga, I. Hamerton and A. Viquerat, Composites, Part B, 2018, 138, 66–76 CrossRef CAS.
- W. Thitsartarn, X. Fan, Y. Sun, J. C. C. Yeo, D. Yuan and C. He, Compos. Sci. Technol., 2015, 118, 63–71 CrossRef CAS.
- A. Bandyopadhyay, P. K. Valavala, T. C. Clancy, K. E. Wise and G. M. Odegard, Polymer, 2011, 52, 2445–2452 CrossRef CAS.
- S. V. Kallivokas, A. P. Sgouros and D. N. Theodorou, Soft Matter, 2019, 15, 721–733 RSC.
- A. Vashisth, C. Ashraf, W. Zhang, C. E. Bakis and A. C. T. van Duin, J. Phys. Chem. A, 2018, 122, 6633–6642 CrossRef CAS PubMed.
- H. Park, B. Kim, J. Choi and M. Cho, Polymer, 2018, 136, 128–142 CrossRef CAS.
- F. Stalport, L. Rouquette, O. Poch, T. Dequaire, N. Chaouche-Mechidal, S. le Payart, C. Szopa, P. Coll, D. Chaput, M. Jaber, F. Raulin and H. Cottin, Astrobiology, 2019, 19, 1037–1052 CrossRef CAS PubMed.
- A. Usman and M. Ahmad, Chemosphere, 2017, 181, 536–543 CrossRef CAS PubMed.
- J. Franck, Trans. Faraday Soc., 1926, 21, 536 RSC.
- E. Condon, Phys. Rev., 1926, 28, 1182 CrossRef CAS.
- J. B. Coon, R. E. DeWanes and C. M. Loyd, J. Mol. Spectrosc., 1962, 8, 285–299 CrossRef CAS.
- E. V. Doktorov, I. A. Malkin and V. I. Man'ko, J. Mol. Spectrosc., 1977, 64, 1–20 CrossRef.
- R. S. C. Woo, Y. Chen, H. Zhu, J. Li, J.-K. Kim and C. K. Y. Leung, Compos. Sci. Technol., 2007, 67, 3448–3456 CrossRef CAS.
- S. Nikafshar, O. Zabihi, M. Ahmadi, A. Mirmohseni, M. Taseidifar and M. Naebe, Materials, 2017, 10, 180 CrossRef PubMed.
- M. K. Altarawneh, B. Z. Dlugogorski, E. M. Kennedy and J. C. Mackie, Combust. Flame, 2013, 160, 9–16 CrossRef CAS.
- F. Liu, M. Yin, B. Xiong, F. Zheng, W. Mao, Z. Chen, C. He, X. Zhao and P. Fang, Electrochim. Acta, 2014, 133, 283–293 CrossRef CAS.
- Y. Kim and J. Choi, Theoretical Study on the Pyrolysis Process of POSS Nanocomposites Based on the Molecular Vibration Frequency, under review.
- M. L. Connolly, Science, 1983, 221, 709–713 CrossRef CAS PubMed.
- J. Hu, X. Li, J. Gao and Q. Zhao, Mater. Des., 2009, 30, 1542–1547 CrossRef CAS.
- M. M. Khotbehsara, A. Manalo, T. Aravinthan, J. Turner, W. Ferdous and G. Hota, Polym. Degrad. Stab., 2020, 181, 109352 CrossRef CAS.
- A. Ghasemi-Kahrizsangi, H. Shariatpanahi, J. Neshati and E. Akbarinezhad, Appl. Surf. Sci., 2015, 353, 530–539 CrossRef CAS.
- C. Liu, Y. Dai, C. Wang, H. Xie, Y. Zhou, X. Lin and L. Zhang, Ind. Crops Prod., 2013, 43, 677–683 CrossRef CAS.
- D. G. Thompson, J. C. Osborn, E. M. Kober and J. R. Schoonover, Polym. Degrad. Stab., 2006, 91, 3360–3370 CrossRef CAS.
- M. Noroozi, M. Panahi-Sarmad, M. Abrisham, A. Amirkiai, N. Asghari, H. Golbaten-Mofrad, N. Karimpour-Motlagh, V. Goodarzi, A. R. Bahramian and B. Zahiri, ACS Appl. Energy Mater., 2019, 2, 5319–5349 CrossRef CAS.
- A. R. Ghohrodi, M. Ramezanzadeh and B. Ramezanzadeh, Prog. Org. Coat., 2022, 164, 106693 CrossRef CAS.
- G. Capiel, J. Uicich, D. Fasce and P. E. Montemartini, Polym. Degrad. Stab., 2018, 153, 165–171 CrossRef CAS.
- J. Pospíšil, Polym. Degrad. Stab., 1993, 39, 103–115 CrossRef.
- Y. Li, C. Liu, M. V. Lee, Y. Xu, X. Wang, Y. Shi and K. Tsukagoshi, J. Mater. Chem. C, 2013, 1, 1352–1358 RSC.
- T. Lu, M. Jiang, Z. Jiang, D. Hui, Z. Wang and Z. Zhou, Composites, Part B, 2013, 51, 28–34 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc04140e |
| This journal is © The Royal Society of Chemistry 2023 |