DOI:
10.1039/D3TC01939J
(Paper)
J. Mater. Chem. C, 2023,
11, 10893-10904
Enabling red thermally activated delayed fluorescence by increasing the push–pull strength in naphthalimide-phenothiazine derivatives†
Received
4th June 2023
, Accepted 26th June 2023
First published on 28th June 2023
Abstract
In this study, we report the synthesis of a series of six new push–pull compounds, bearing naphthalimide as the electron acceptor and phenothiazine or phenothiazine dioxide as the electron donor, arranged in dipolar or octupolar molecular structures and showing either a single bond or a phenyl spacer as the linker between units. This synthetic effort allowed a detailed investigation of the structure–property relationships and of the key factors enabling thermally activated delayed fluorescence (TADF) in these fluorophores, performed through a joint advanced time-resolved spectroscopic and TD-DFT computational approach. Our ultrafast spectroscopic results showed larger push–pull degree for the phenothiazine compared to the phenothiazine dioxide derivatives. This resulted in negligible energy gap between the intramolecular charge transfer S1 and the naphthalimide localized T1 excited states, and thus lead to effective reverse intersystem crossing and TADF only in the case of the oxygen-free compounds. The increased conjugation in the octupolar relative to the dipolar systems as well as in the phenyl-bridged structures allowed higher fluorescence efficiencies to be achieved. The red TADF of these luminophores, less common and more difficult to achieve than blue and green TADF, was found to be enhanced in their aggregates produced both in water dispersions and in thin films. The observation of intense red TADF in the solid state makes these new all-organic materials highly promising for applications in third generation OLED devices.
Introduction
Organic light emitting diodes (OLEDs) utilizing thermally activated delayed fluorescence (TADF) active materials have attracted tremendous attention lately, more than fluorescence- and phosphorescence-based devices.1–4 In fact, according to spin statistics, electrogenerated singlet and triplet excitons are obtained in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio. Therefore, conventional fluorescent OLEDs are limited to a maximum internal quantum efficiency of 25%. On the other hand, the more efficient phosphorescent OLEDs generally employ rare noble metal complexes (e.g. Ir and Pt) with several obvious drawbacks, such as high costs and toxicity due to heavy atoms and the absence of stable blue emitters.5,6 TADF-based OLEDs, characterized by potential 100% internal quantum efficiencies and all-organic low-cost active materials, have emerged as an alternative approach for third generation devices and have aroused great interest within the last decade.
:3 ratio. Therefore, conventional fluorescent OLEDs are limited to a maximum internal quantum efficiency of 25%. On the other hand, the more efficient phosphorescent OLEDs generally employ rare noble metal complexes (e.g. Ir and Pt) with several obvious drawbacks, such as high costs and toxicity due to heavy atoms and the absence of stable blue emitters.5,6 TADF-based OLEDs, characterized by potential 100% internal quantum efficiencies and all-organic low-cost active materials, have emerged as an alternative approach for third generation devices and have aroused great interest within the last decade.
According to the El-Sayed rules, significant spin orbit coupling (SOC) in all-organic chromophores may be obtained if singlet and triplet excited states of different orbital nature are involved in the intersystem crossing (ISC). Intramolecular charge transfer interactions in molecular systems bearing electron donating and electron accepting units can not only promote SOC and hence efficient ISC but also lower the energy difference between the lowest singlet and triplet excited states.7–10 A small singlet-to triplet energy splitting (ΔEST), defined as the gap between the T1 and S1 states, is the most important criterion to achieve relatively high rates of reverse intersystem crossing (r-ISC) followed by significant delayed fluorescence. It is generally recognized that ΔEST < 0.2eV is necessary for competitive r-ISC and TADF.11
While many efficient blue and green TADF materials have been developed by suitably connecting donor and acceptor portions,5,12–14 there are currently few examples of orange-red and infrared TADF materials.15–21 Thus, an efficient molecular design strategy is required to boost the efficiency of red TADF materials. Indeed, red materials possess smaller bandgap than blue and green TADF materials. This leads to the nonradiative decay of the S1 excitons to be generally predominant in the red region due to the energy gap law. Additionally, the strong donor–acceptor character of the employed TADF fluorophores, whose excited state charge rearrangement may also be accompanied by twisting of the molecular structure, often leads to low efficiency of the radiative deactivation. An effective strategy to reduce the competition of internal conversion and vibrational relaxation processes may be to introduce more rigid and more conjugated molecular structures. On the other hand, materials with the integration of aggregation-induced emission (AIE) and TADF properties can not only effectively suppress the non-radiative pathways by restriction of intramolecular motions but also reduce the quenching of the triplet state by molecular oxygen as the aggregates may prevent the interaction.2,22 Many AIE-TADF emitters have been reported to show good performance in non-doped OLEDs.23–26
Recently, our research has been devoted to the study of several push–pull phenothiazine derivatives for their photoinduced intramolecular charge transfer (ICT) ability and non-linear optical properties.27–30 Interestingly, phenothiazine has been described in literature reports as a remarkable electron donor unit for the development of orange/red TADF luminogens.22,31–33 To this aim, we have here considered a comprehensive series of six donor–acceptor compounds (Chart S1, Scheme 1, ESI†) where the electron donor portion is phenothiazine (PTZ) or phenothiazine dioxide (PTZ(dioxy)) and the electron acceptor is naphthalimide (NPI). The donor and acceptor units were simply connected through a single bond (A–D: NPI-PTZ and NPI-PTZ(dioxy)) or through a phenyl π-bridge (A–Ph–D: NPI-Ph-PTZ and NPI-Ph-PTZ(dioxy)) in dipolar structures with lower and higher conjugation, respectively. Additionally, the molecular conjugation was also enhanced by arranging the donor and acceptor units in octupolar structures of A3–D kind (NPI(3)-PTZ and NPI(3)-PTZ(dioxy)). The present study wants to get a deep insight into the structure-TADF property relationships by a joint synthetic, spectroscopic and computational effort. State-of-the-art time resolved spectroscopies with both nanosecond and femtosecond temporal resolution, both in absorption and emission, have been employed to follow the triplet and singlet excited state dynamics of the newly synthetized compounds. Furthermore, information about the excited state energy and nature was gained by means of advanced TD-DFT calculations. The photophysics of these new fluorophores was investigated for the monomers in solution as well for the aggregates produced both in water dispersion and in the solid state.
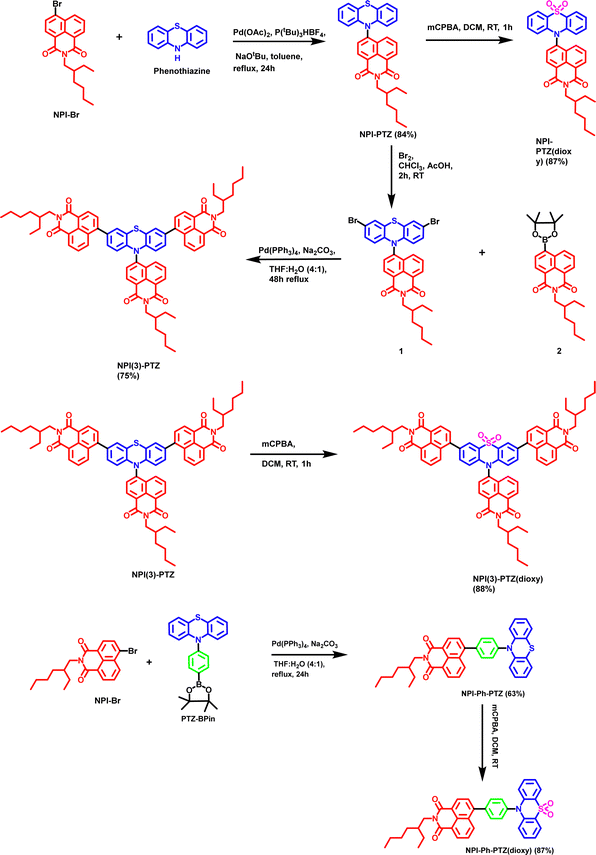 |
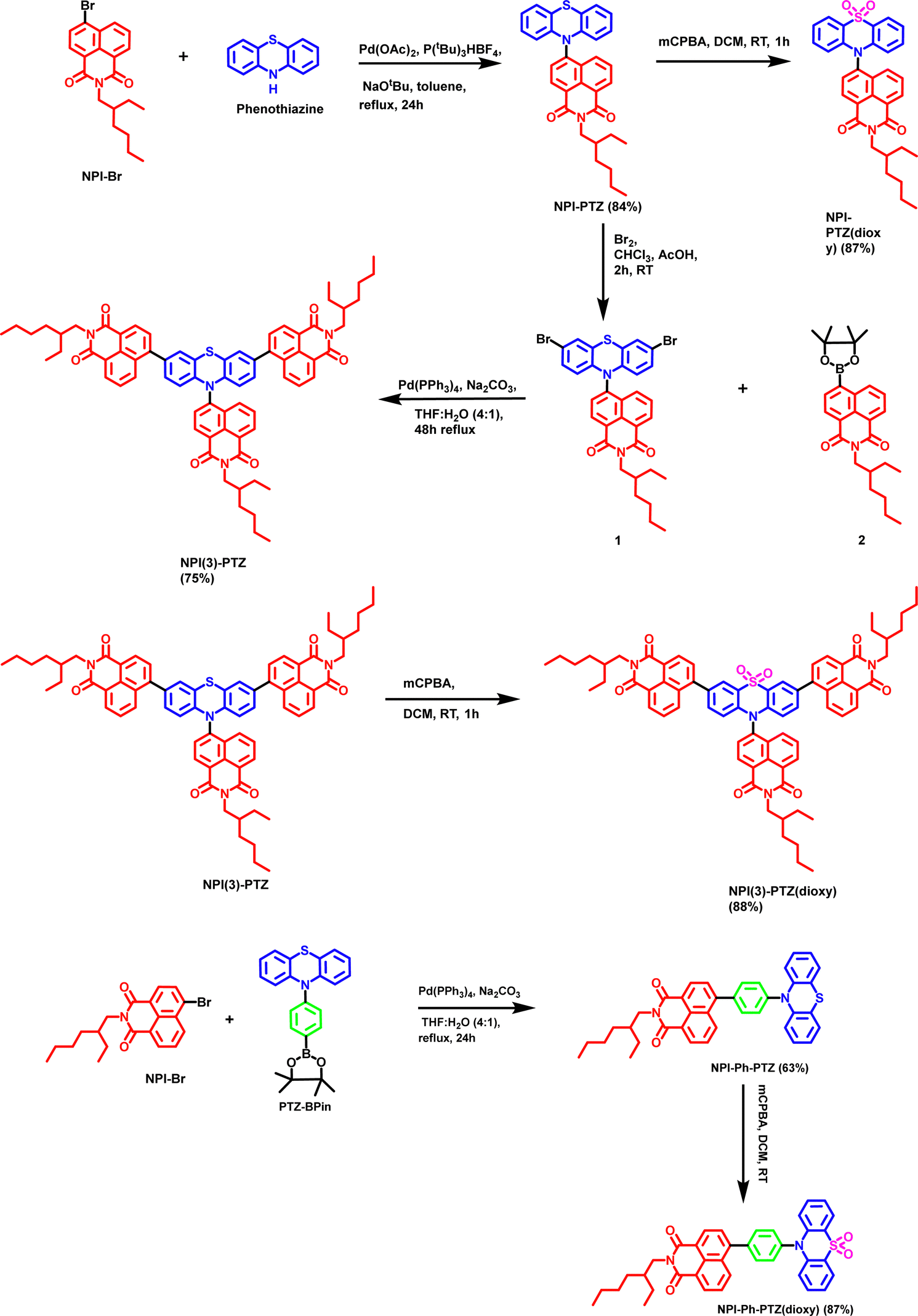
| | Scheme 1 Synthetic route to obtain NPI-PTZ, NPI-PTZ(dioxy), NPI(3)-PTZ, NPI(3)-PTZ(dioxy), NPI-Ph-PTZ and NPI-Ph-PTZ(dioxy). | |
Results and discussion
Synthesis
The synthetic procedure for the phenothiazine and phenothiazine sulfone derivatives incorporated with 1,8-napthalimide are shown in Scheme 1. The NPI-PTZ chromophore was synthesized by the Buchwald–Hartwig cross-coupling reaction of 10H-phenothiazine with one equivalent of 6-bromo-2-(2-ethylhexyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione NPI-Br in the presence of Pd(OAc)2 as the catalyst and P(tBu)3HBF4 as a ligand in 84% yield (Scheme 1). The chromophore NPI-PTZ was further reacted with 3 equiv. of 3-chloroperbenzoic acid in dichloromethane solution which resulted NPI-PTZ(dioxy) in 87% yield. The phenyl spacer containing NPI-Ph-PTZ was synthesized via the Pd-catalysed Suzuki cross-coupling of 6-bromo-2-(2-ethylhexyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione NPI-Br with one equivalent of 10-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-10H-phenothiazine PTZ-BP in 63% yield (Scheme 1). NPI-Ph-PTZ was further oxidized using three equivalents of 3-chloroperbenzoic acid in dichloromethane and resulted NPI-Ph-PTZ(dioxy) in 87% yield (Scheme 1). The star-shaped tri-1,8-napthalimde units functionalized chromophore NPI(3)-PTZ was synthesized by reacting the 6-(3,7-dibromo-10H-phenothiazin-10-yl)-2-(2-ethylhexyl)-1H-benzo[de]isoquinoline-1,3(2H)-dione 1 with 2-(2-ethylhexyl)-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-benzo[de]isoquinoline-1,3(2H)-dione 2 through the Suzuki cross-coupling reaction in 75% yield (Scheme 1). The detailed synthetic procedure of intermediates 1 and 2 is given in the ESI.† The oxidized sulfone derivative NPI(3)-PTZ(dioxy) was further synthesized from NPI(3)-PTZ by following the oxidation procedure and resulted in 88% yield (Scheme 1).
Absorption and emission properties
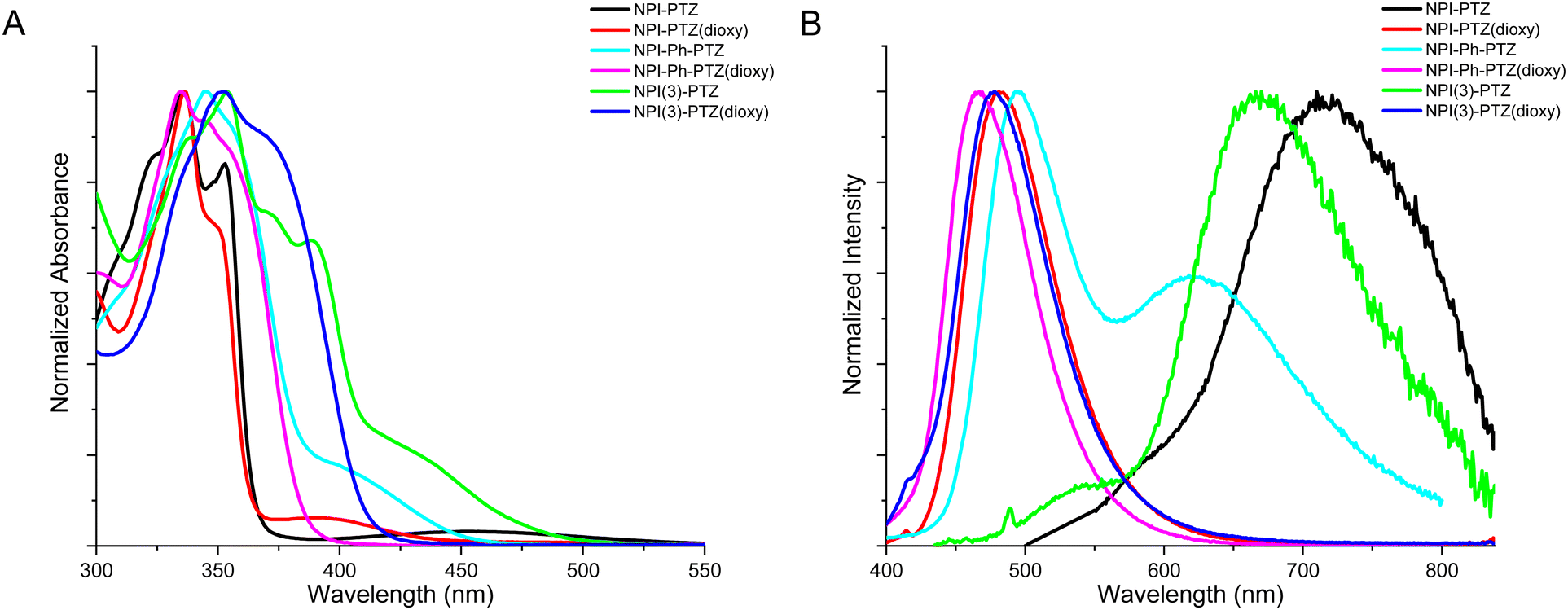
Fig. 1 shows the normalized absorption and emission spectra of the six investigated compounds in toluene. The absorption spectra (Fig. 1A) are composed by a main structured band centered at 335–355 nm depending on the molecular structure, and in most cases by a less intense bathochromic shoulder between 370 and 450 nm. The main absorption band exhibits a clear red-shift by increasing the molecular conjugation among the compounds in the series. Indeed, it is centered at 336 nm for the dipolar systems where the donor and acceptor units are connected by single bonds (D–A); it is slightly shifted toward longer wavelengths for the dipolar systems where the bridge is a phenyl ring (D–Ph–A); it is even more red-shifted for the two octupolar compounds (D–A3). The DFT computational study in toluene allowed a deep understanding of the nature of the observed absorption transitions. Fig. 2 reports the results obtained for the representative case of NPI-PTZ(dioxy). The main absorption band corresponds to the S0 → S2 transition with large oscillator strength and described by hole and electron natural transition orbitals (NTOs) localized on the naphthalimide. The bathochromic shoulder is due to the forbidden S0 → S1 described by a HOMO–LUMO transition, with the HOMO localized on the phenothiazine dioxide donor and the LUMO on the naphthalimide acceptor, thus with a clear charge transfer character. Similar findings for all the other compounds are detailed in the ESI.† The S0 → S1 transition is found at lower energies for the phenothiazine derivatives (e.g. ca. 450 nm for NPI-PTZ, ca. 400 nm for NPI-Ph-PTZ, ca. 430 nm for NPI(3)-PTZ) relative to the phenothiazine dioxide analogues (see Table S1, ESI†). This is justified by the larger ICT degree of S1 for the compounds bearing the stronger PTZ electron donor unit relative to PTZ(dioxy). This is demonstrated by the DFT simulations considering the larger charge density of the HOMO localized on the electron donor phenothiazine unit compared to that on the phenothiazine dioxide (see Fig. S40, ESI†).
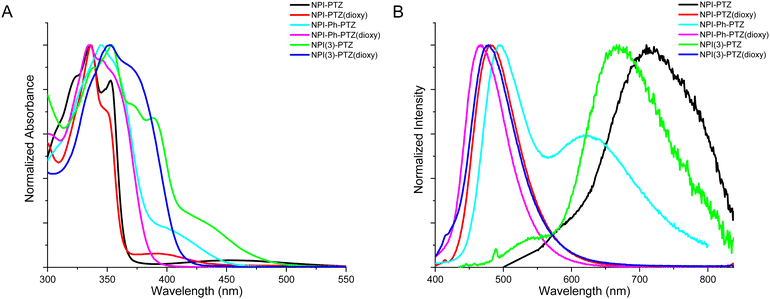 |
| | Fig. 1 Normalized absorption (A) and fluorescence (B) spectra of the investigated compounds in toluene (emission spectra were generally recorded by exciting at the lowest energetic shoulder of the absorption spectrum, with the exception of NPI-Ph-PTZ(dioxy) for which a 335 nm excitation was used). | |
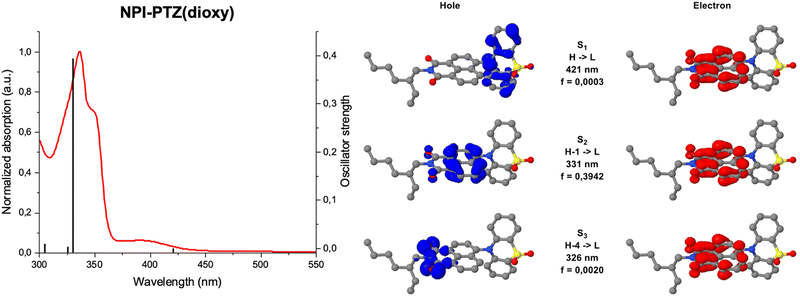 |
| | Fig. 2 Analysis of the absorption transitions for the representative case of NPI-PTZ(dioxy). | |
This computational result gives also a justification to the significant differences among the emission spectra of the six compounds (Fig. 1B). All the investigated molecules show a broad non-structured emission in toluene. The phenothiazine derivatives exhibit significantly red-shifted spectra relative to the phenothiazine dioxide analogues, particularly NPI-PTZ and NPI(3)-PTZ, in agreement with an enhanced ICT character of their lowest excited singlet state. A peculiar behavior was revealed for the case of NPI-Ph-PTZ, for which two emission bands were detected in toluene (Fig. 1B). The reasons underlying this peculiarity were further investigated through the study of the solvent effect on the spectra. For all the investigated samples, a small solvatochromism was observed on the absorption spectra: just a small red-shift on passing from the methylcyclohexane/3-methylpentane mixture to toluene and from toluene to dimethylsulphoxide (Fig. S18 and S20, ESI†). On the other hand, a large solvent effect was observed on the emission spectra, that show a significant positive fluorosolvatochromism upon increasing the solvent polarity (Fig. S19 and S20, ESI†). In the case of NPI-Ph-PTZ, the emission spectrum significantly red-shifts in the more polar solvents, with a noticeable dual emission clearly exhibited in toluene but also hinted in dimethylsulphoxide. This behavior, typical of push–pull systems,34–38 can be rationalized considering the progressive stabilization of an intramolecular charge transfer (ICT) excited state with increasing the solvent polarity as well as the presence of two relative minima on the potential energy curve of S1, that are the locally excited (LE) state populated by light absorption and the fully relaxed ICT state.
In Table 1, the fluorescence quantum yields (ϕF), lifetimes (τF) and rate constants (kF = ϕF/τF) are reported for the six fluorophores in toluene. The ϕF values are generally quite low, suggesting that non-radiative pathways may be important for the S1 deactivation of these molecules. The fluorescence efficiency is increased on passing from the phenothiazine to the phenothiazine dioxide derivatives. Moreover, a small enhancement of the radiative quantum yields and rate constants is observed on passing from the dipolar to the octupolar systems and a large enhancement by adding a phenyl bridge between the donor and the acceptor units. This finding is in agreement with the relatively more intense bathochromic shoulder observed in the absorption spectra of the octupolar and phenyl-linked derivatives (Fig. 1A), thus revealing an increased oscillator strength for the S0–S1 transition for the more conjugated compounds both in absorption and in emission. The fluorescence quantum yield is decreased while the fluorescence lifetime is generally found to be longer upon increasing the solvent polarity, with a consequent drop of the kF in the more polar solvents (Table S1, ESI†). This is a typical behavior for donor–acceptor fluorophores due to a strong competition of the ICT leading to fluorescence quenching in the most polar media.39,40
Table 1 Fluorescence properties of the investigated compounds in toluene
| Compound |
ϕ
F
|
τ
S1 (ns) |
k
F (s−1) |
|
NPI-PTZ
|
0.0015 |
2.5 |
5.9 × 105 |
|
NPI-PTZ(dioxy)
|
0.010 |
6.0 |
1.7 × 106 |
|
NPI-Ph-PTZ
|
0.076 |
9.7 |
7.8 × 106 |
|
NPI-Ph-PTZ(dioxy)
|
0.81 |
2.9 |
2.8 × 108 |
|
NPI(3)-PTZ
|
0.009 |
7.0 |
1.3 × 106 |
|
NPI(3)-PTZ(dioxy)
|
0.040 |
7.0 |
5.7 × 106 |
Ultrafast spectroscopy
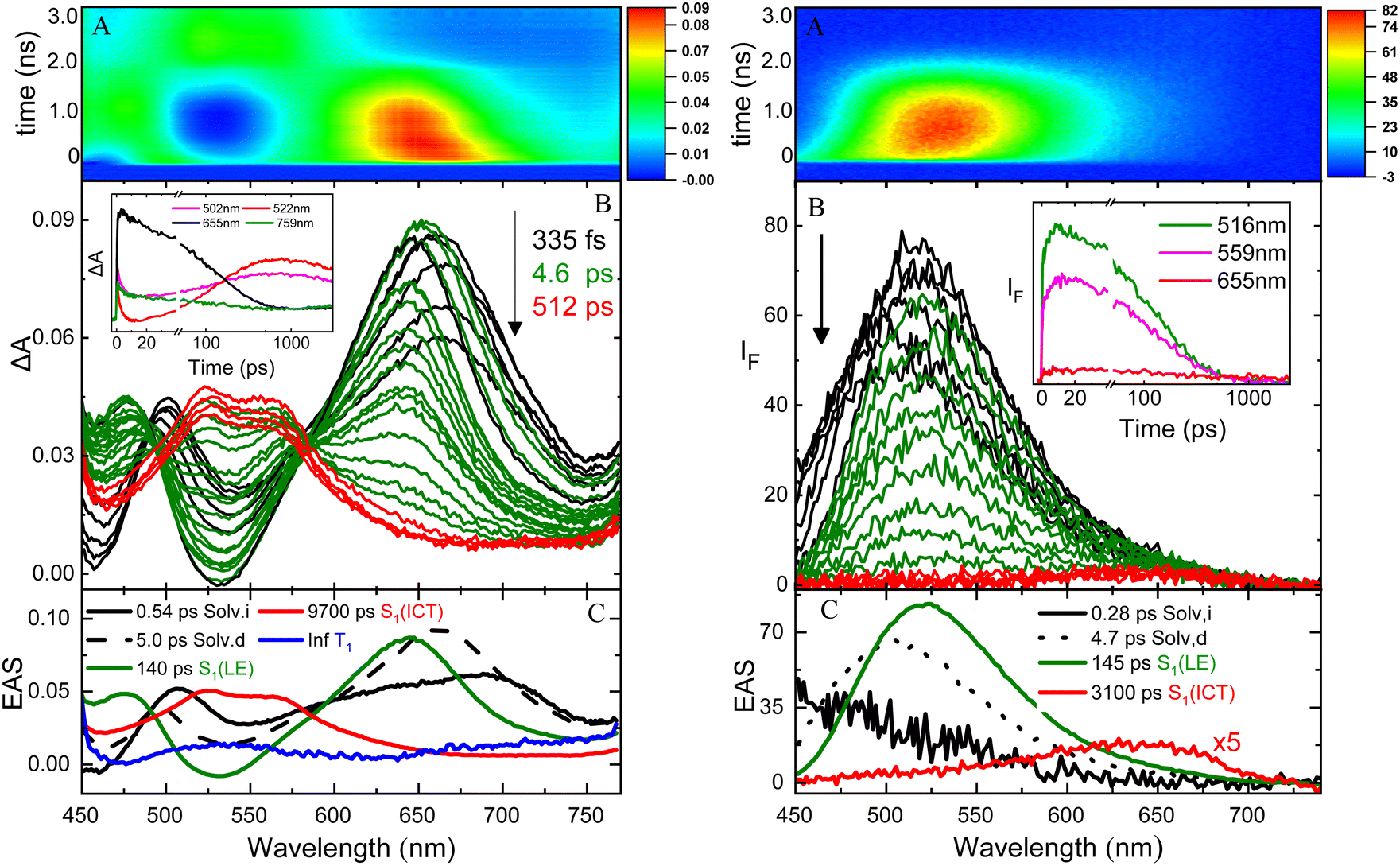
The excited state dynamics of these compounds was investigated by ultrafast spectroscopy, both in absorption and in emission, through femtosecond transient absorption (TA) and broadband fluorescence up conversion (FUC) measurements with a 400 nm laser excitation. Fig. 3 shows the results obtained for NPI-Ph-PTZ in toluene as a representative example. The TA spectra (panel B of the left graph) show a remarkable temporal evolution. The initially formed excited state absorption band (in black) exhibits a blue-shift in time after photoexcitation to give within few picoseconds another spectral shape (in green) characterized by a major peak at ca. 650 nm and a secondary band at ca. 470 nm. This transient species was found to decay in hundreds of picoseconds to give another structured excited state absorption (ESA) with two maxima at ca. 525 and 575 nm (in red). This signal decays on a longer time scale of few nanoseconds. The global analysis of the TA data revealed the presence of five components (panel C of the left graph): the first two with lifetimes of 0.54 and 5.0 ps were assigned to inertial and diffusive solvation in toluene (black); the third component with a lifetime of 140 ps and showing the 470 and 650 nm ESA peaks (green) to the LE S1 state; the forth component with a lifetime of 9700 ps and the structured 525 and 575 nm ESA (red) to the ICT state; the fifth component not decaying in the investigated time window of few nanoseconds (Inf, blue) was associated to the long-lived triplet excited state T1. Analysis of the spectral shapes of the observed transients is at the root of the LE and ICT state assignments in the TA as well as the FUC results. The broadband FUC measurements highlighted in the time-resolved emission spectra (panel B of the right graph) a small initial red-shift with time to give the main 525 nm emission band (green). This band is then found to decay and only at long delays after excitation another less intense and red-shifted emission peaked at ca. 650 nm becomes apparent (red). The global analysis of these data revealed the presence of four components, in good agreement with the TA: the first two components (0.28 and 4.7 ps, in black) associated to solvation; the third component (140 ps, in green) characterized by the intense 525 nm emission assigned to the LE state and the fourth component (3100 ps, in red) characterized by the low intensity 650 nm emission corresponding to the ICT state. These results allow a deep understanding of the steady-state dual emission exhibited by NPI-Ph-PTZ in toluene as due to the LE and ICT fluorescence. For these remarkably push–pull compounds, population of the ICT from the LE state is thus observed even in a low polar solvent, such as toluene (Table 2).
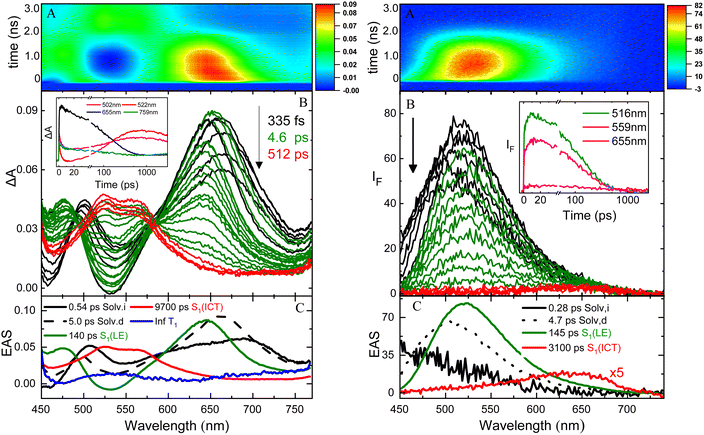 |
| | Fig. 3 Femtosecond transient absorption (left) and broadband fluorescence up conversion spectroscopy (right) of NPI-Ph-PTZ in toluene with a 400 nm laser excitation (panel A: ΔA/IFvs. wavelength and time colored map; panel B: time resolved absorption/emission spectra and kinetics at selected wavelengths (inset); panel C: Evolution Associated Spectra (EAS) and lifetimes of the components obtained through global analysis). | |
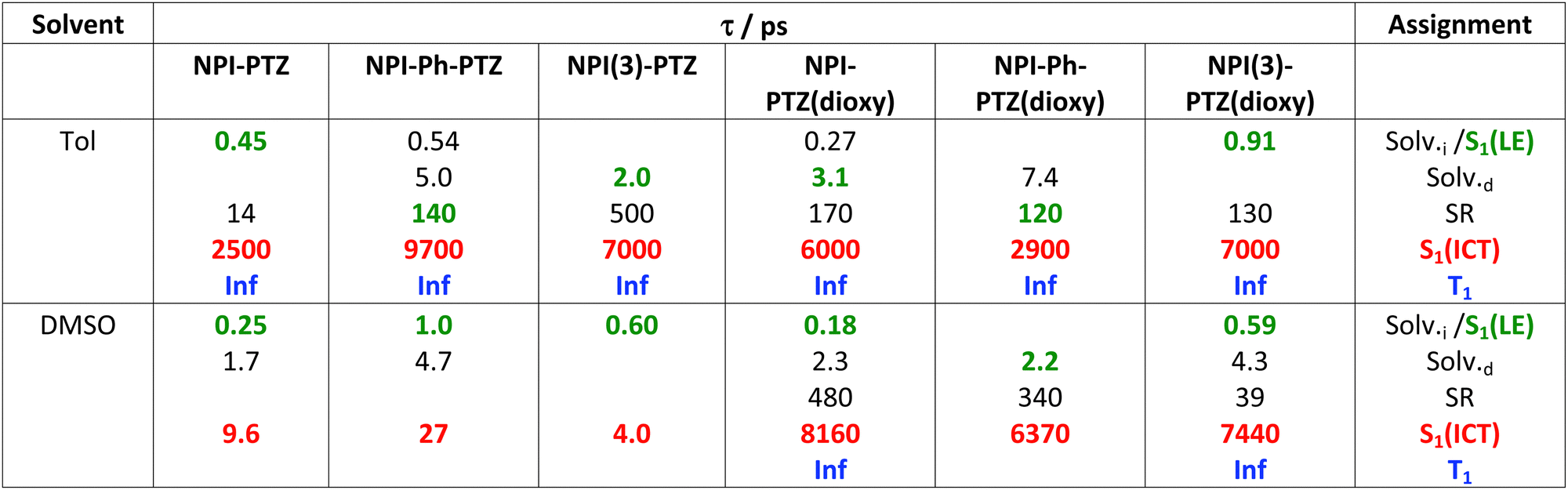
Table 2 Results of global analysis of the femtosecond transient absorption data for the investigated compounds in Tol and DMSO
| Solv.i and Solv.d: inertial and diffusive solvation; SR: structural relaxation. |
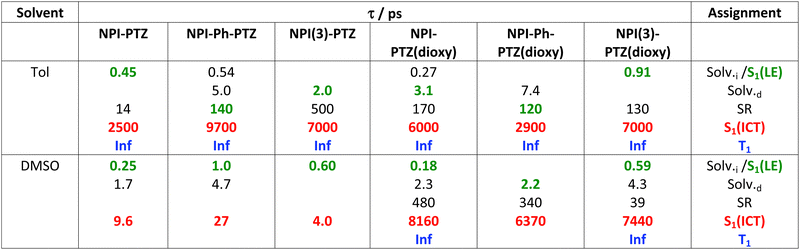
|
The LE → ICT transition in toluene takes place in ultrashort times comparable to the solvation times for the dipolar and octupolar derivatives for which the donor and acceptor units are linked through a single bond, while occurring in hundreds of picoseconds if a phenyl is inserted as π-bridge (Table 2). Indeed, increasing the distance between the donor and acceptor units through the insertion of a phenyl bridge may lead to a decrease in the rate of the ICT process. For this reason, the LE-ICT transition in NPI-Ph-PTZ appears to be slower than for the analogous compounds without the phenyl bridge (Table 2). In a more polar solvent, such as dimethylsulphoxide, the LE → ICT transition happens faster, with times comparable to the inertial solvation in all cases. It is noteworthy that for the dioxy-derivatives the ICT excited state shows a long lifetime of few nanoseconds also in the highly polar medium dimethylsulphoxide. On the other hand, for the oxygen free phenothiazine derivatives, the ICT lifetime is significantly shortened in dimethylsulphoxide, being few picoseconds for NPI-PTZ and NPI(3)-PTZ and tens of picoseconds for NPI-Ph-PTZ. This finding suggests a preferential ultrafast deactivation by internal conversion for the exceptionally stabilized ICT states of the oxygen-free phenothiazine derivatives. It is possible that population of the ICT state is accompanied by rotation around the quasi-single bonds between the donor and acceptor units in the more flexible oxygen-free derivatives (Twisted ICT states). On the other hand, for the more rigid oxygen-functionalized derivatives, ICT states characterized by a more planar geometry (Planar ICT states) are likely populated thus exibiting longer lifetimes even in highly polar solvents.29
Triplet properties
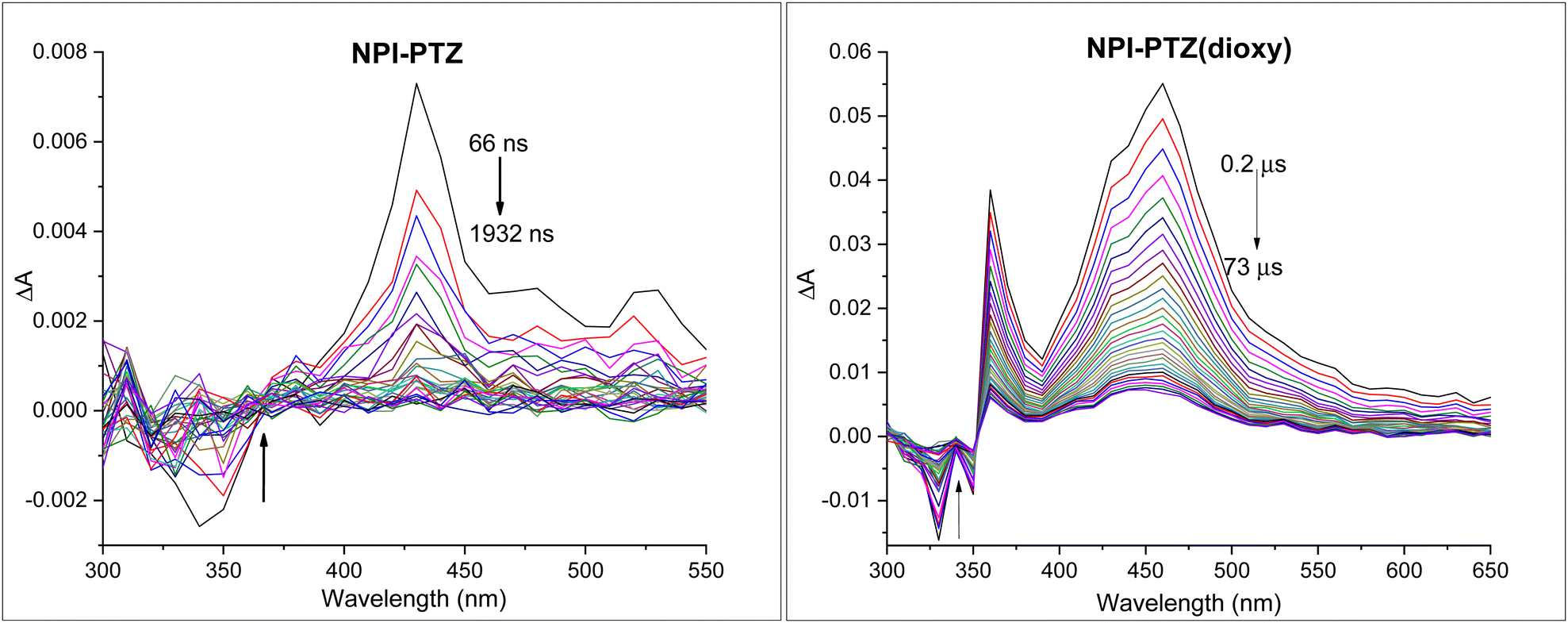
The occurrence of another non-radiative deactivation pathway, other than the ICT, such as the intersystem crossing (ISC) was investigated by means of nanosecond transient absorption measurements. Significant ESA signals were revealed for all the investigated compounds in toluene (Fig. 4 and Fig. S28, ESI†). The TA spectra in toluene showed ESA bands peaked around 400–460 nm for the dipolar systems, while being red-shifted and centred around 660–670 nm for the octupolar derivatives (Table 3). The recorded kinetics exhibited decays significantly affected by the presence of molecular oxygen in solution for all the investigated samples (Table S3, ESI†), suggesting the triplet excited state nature of the observed transient species. The triplet lifetimes were found to be longer in the phenothiazine dioxide as compared to the phenothiazine derivatives as well as in the more conjugated molecules relative to the smallest NPI-PTZ compound (Table 3). A peculiar behaviour was revealed in the decay kinetics recorded for NPI-PTZ and NPI(3)-PTZ, both in air-equilibrated and nitrogen-purged toluene. The kinetics were best fitted by using a bi-exponential decay function, with a first component showing a lifetime of few nanoseconds comparable to the ns-laser pulse duration (ca. 7 ns) and a second component showing a lifetime of tens/hundreds of nanoseconds in aerated solution which becomes significantly longer up to few microseconds in de-aereated toluene (Fig. S29 and S30, Table 3, ESI†). This latter lifetime can be unambiguously assigned to the triplet excited state. It may be possible to suggest an efficient r-ISC process which could yield a shortly delayed fluorescence in the case of these two molecules (NPI-PTZ and NPI(3)-PTZ) to justify the presence of the first short-lived component in the TA kinetics recorded by nanosecond laser flash photolysis.
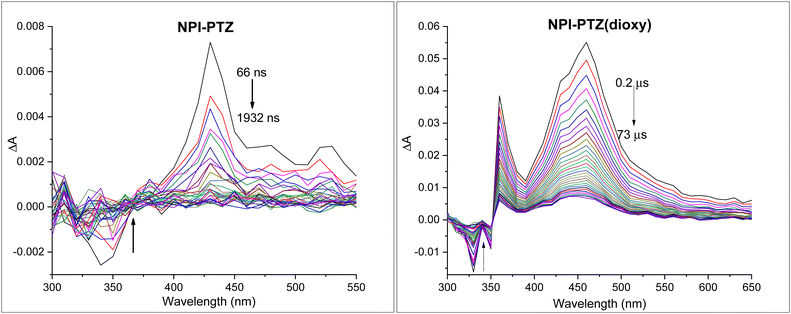 |
| | Fig. 4 Triplet absorption spectrum for NPI-PTZ and NPI-PTZ(dioxy) in nitrogen purged toluene. | |
Table 3 Triplet properties of the investigated compounds in toluene
| Compound |
λ
T (nm) |
τ
T,N2 (ns) |
ε
T (M−1 cm−1) |
ϕ
T
|
|
In acetonitrile.
In cyclohexane.
|
|
NPI-PTZ
|
430 |
6 (3%) |
7630a |
0.27 |
| 440 (97%) |
|
NPI-PTZ(dioxy)
|
460 |
44000 |
7680a |
0.88 |
|
NPI-Ph-PTZ
|
460 |
24000 |
3000a |
0.61 |
|
NPI-Ph-PTZ(dioxy)
|
400 |
12000 |
3050a |
0.19 |
|
NPI(3)-PTZ
|
660 |
19 (1%) |
7350b |
0.21 |
| 3400 (99%) |
|
NPI(3)-PTZ(dioxy)
|
670 |
45000 |
8300b |
0.93 |
Sensitization experiments allowed triplet extinction coefficients (εT) between 3000 and 8000 M−1 cm−1 to be accurately determined for the investigated molecules (Table 3). Relative actinometry measurements were carried out to obtain the triplet quantum yields in toluene (ϕT in Table 3), which were found to be significant for all the six compounds, proving ISC as an important decay pathway for their excited states. The triplet yield values were measured to be ca. 20–30% for NPI-PTZ and NPI(3)-PTZ, while being enhanced up to ca. 90% for their dioxy-analogues. A different trend was observed for the compounds showing a phenyl as π-bridge, where a more efficient ISC was revealed for the oxygen free (61%) relative to the oxygen functionalized derivative (19%). For all the phenothiazine dioxide compounds in toluene it was found that ϕF + ϕT ≈ 100%, indicating that the fluorescence and the ISC quantitively account for the S1 deactivation. The introduction of the phenyl bridge in NPI-Ph-PTZ(dioxy) has the effect of enhancing the fluorescence (81%) at the expenses of the intersystem crossing (19%) relative to the other oxygen-functionalized derivative, as expected considering the increase in conjugation. For the oxygen-free phenothiazine derivatives instead ϕF + ϕT < 100%, particularly for the case of NPI-PTZ and NPI(3)-PTZ, suggesting that an important role is played by the ICT followed by internal conversion to the ground state for these molecules. Both the fluorescence and the intersystem crossing quantum yields are enhanced in NPI-Ph-PTZ relative to NPI-PTZ and (NPI)3-PTZ for its reduced push–pull character due to the increased distance between the donor and acceptor units.
Excited state energy and nature
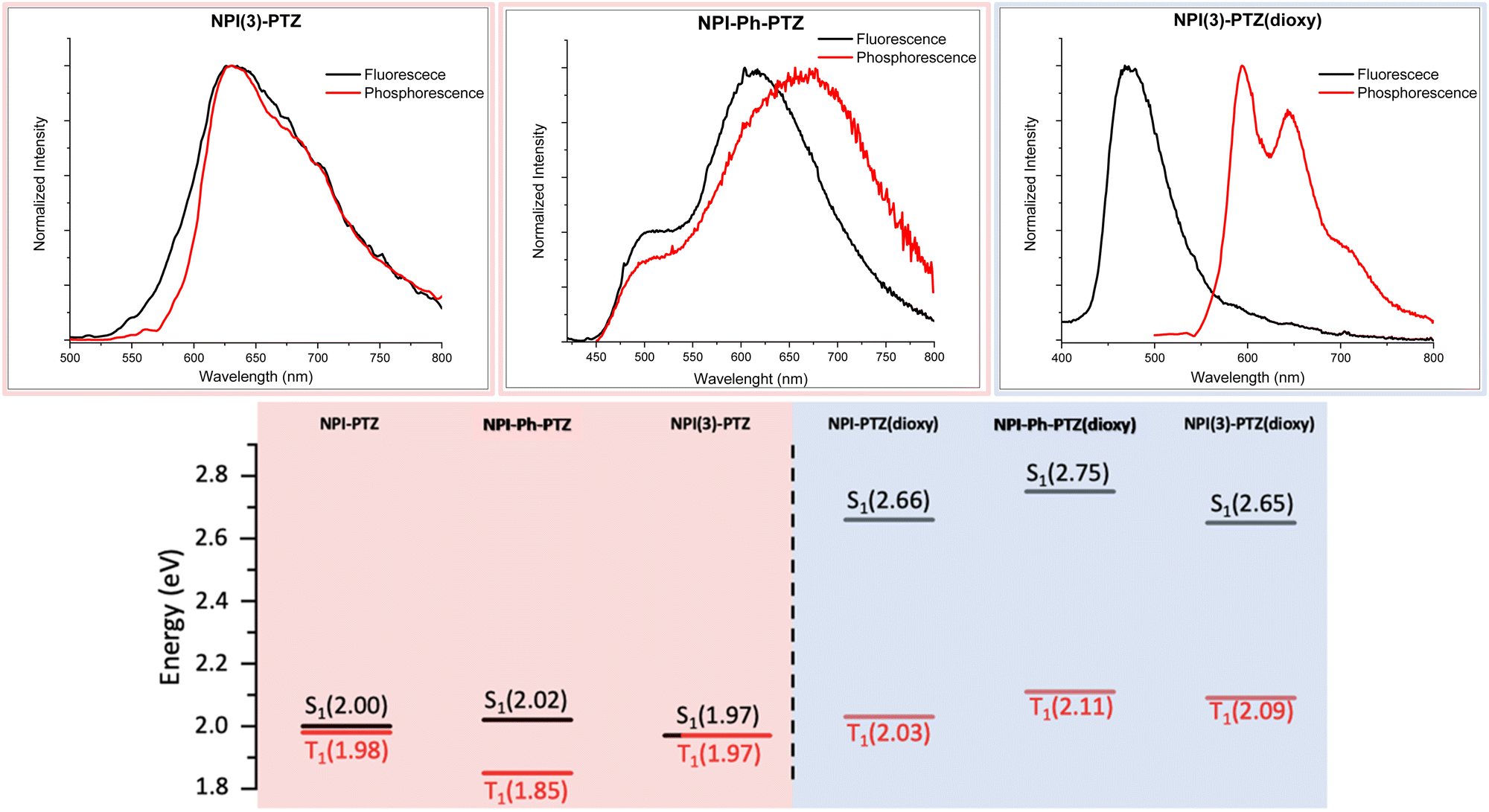
In order to investigate the possible communication between S1 and T1 suggested by the laser flash photolysis kinetics for NPI-PTZ and NPI(3)-PTZ, information about the energy of the lowest excited singlet and triplet states was gained through fluorescence and phosphorescence experiments in the methylcyclohexane/3-methylpentane mixture at 77 K. In case of the phenothiazine dioxide derivatives, the fluorescence and phosphorescence spectra were found to be well separated (Fig. 5 and Fig. S31, Table S5, ESI†) and the energy gap between the singlet and the triplet excited states resulting from these measurements was large (ΔESTca. 0.6 eV, see Table S6, ESI†). A much smaller gap was observed for NPI-Ph-PTZ, with the ΔEST being ca. 0.17 eV (see Fig. 3 and Table S6, ESI†). Surprisingly, for NPI-PTZ and NPI(3)-PTZ, roughly overlapping fluorescence and phosphorescence spectra were recorded (Fig. 3 and Fig. S31, Table S5, ESI†), suggesting a negligible ΔEST in these cases (Table S6, ESI†).
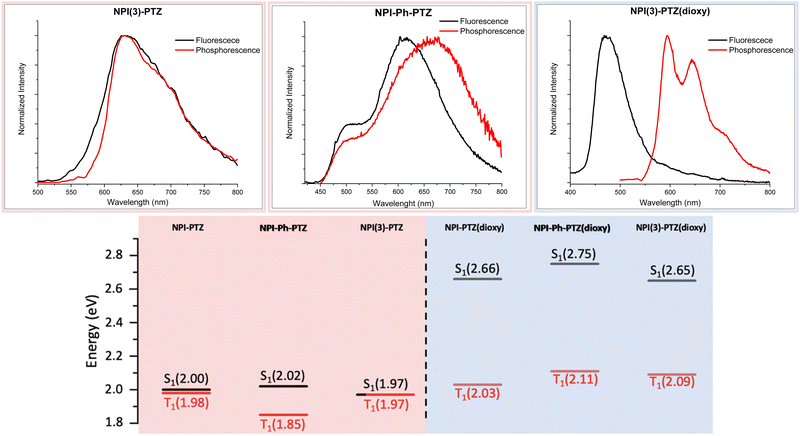 |
| | Fig. 5 Fluorescence and phosphorescence spectra in the mixture methylcyclohexane:3-methylpentane 9:1 (v/v) at 77K (upper graphs) together with the singlet and triplet energies of the investigated compounds obtained from these experiments (lower graphs). | |
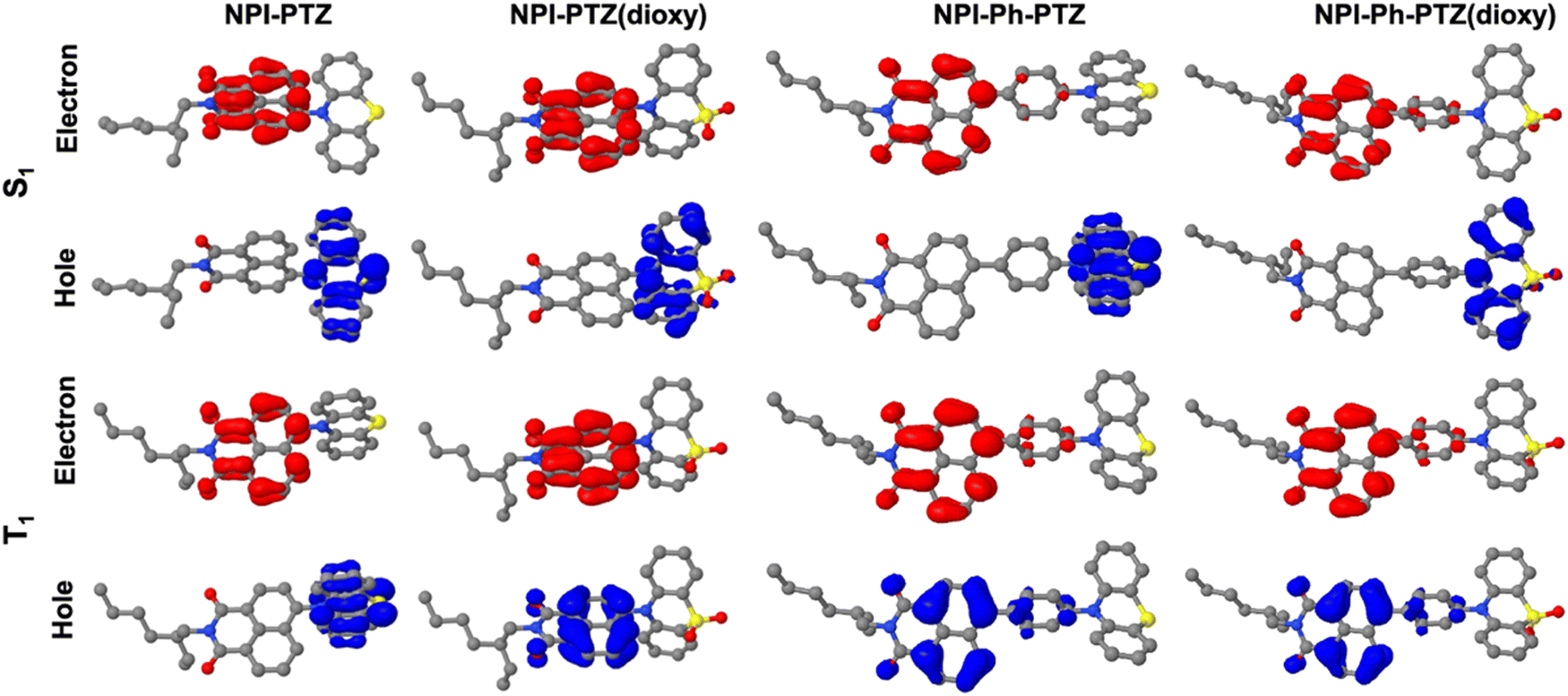
These experimental results were found to be in good agreement with the theoretical TD-DFT predictions about the energies of the S1 and T1 excited states at their respective optimized geometries. Despite the fact that the absolute values of the singlet and triplet energies are sometimes not perfectly in line with the experimental findings, the obtained trend for the ΔEST among the molecules in the series is in excellent agreement (Table 4). It is noteworthy that the triplet energy is only slightly affected by changes in the molecular structure. On the other hand, the singlet energy shows important differences among the six compounds; the energy difference with respect to the T1 state is much smaller for the oxygen-free derivatives, and particularly for NPI-PTZ and NPI(3)-PTZ, likely because of their increased ICT character (Fig. 6). The TD-DFT calculations also yield information about the structural and electronic features of the S1 and T1 states. The more significant structural rearrangement on passing from S0 to S1 for the PTZ relative to the PTZ(dioxy) derivatives (Fig. S41–S42, ESI†) is consistent with their relatively larger Stokes shift values experimentally measured (Table S1, ESI†). Additionally, while the S1 usually exhibits an ICT nature being described by a HOMO localized on the PTZ/PTZ(dioxy) and a LUMO localized on the NPI, the T1 is generally described by a configuration of NTOs localized on the NPI acceptor unit (Fig. 6 and Fig. S43, ESI†). This different orbital nature between S1 (ICT) and T1 (LE) is at the root of the significant spin-orbit coupling between the two states and thus of the efficient ISC experimentally observed for all these molecules. In summary, the ICT nature of the lowest excited singlet state S1 in these compounds makes it easily tunable in energy. This leads to the most significant energetic stabilization of the S1 in the compounds showing the largest push–pull character (NPI-PTZ and NPI(3)-PTZ), with the lowest excited singlet state becoming isoenergetic to the triplet in these cases.41
Table 4 Predicted transitions from the lowest excited states (S1 or T1) in their relaxed geometry as obtained from the TD-DFT calculations
| Molecule |
Singlet emission (eV) |
Triplet emission (eV) |
ΔEST (eV) |
|
NPI-PTZ
|
1.38 |
1.37 |
0.01 |
|
NPI-PTZ(dioxy)
|
2.43 |
1.47 |
0.96 |
|
NPI-Ph-PTZ
|
1.67 |
1.49 |
0.18 |
|
NPI-Ph-PTZ(dioxy)
|
2.79 |
1.48 |
1.31 |
|
NPI(3)-PTZ
|
1.56 |
1.46 |
0.10 |
|
NPI(3)-PTZ(dioxy)
|
2.39 |
1.48 |
0.91 |
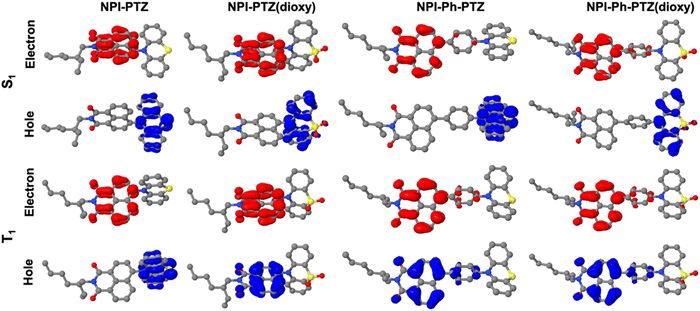 |
| | Fig. 6 Natural transition orbitals describing S1 and T1 in their optimized geometries as obtained from the TD-DFT calculations. | |
Thermally activated delayed fluorescence
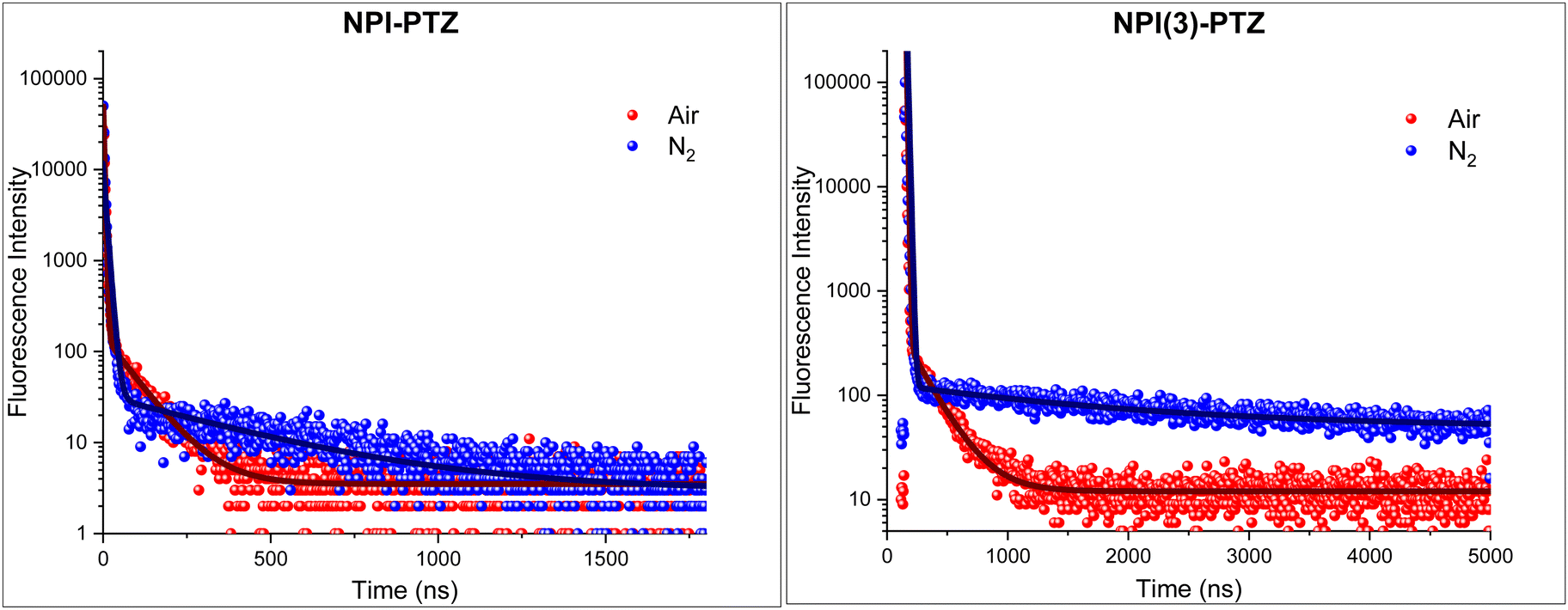
Given the negligible ΔEST demonstrated both experimentally and theoretically for NPI-PTZ and NPI(3)-PTZ, the possible occurrence of delayed fluorescence was attentively searched at room temperature for these molecules. The obtained results are reported in Fig. 7, in terms of fluorescence decay kinetics recorded by means of time correlated-single photon counting (TC-SPC) with nanosecond temporal resolution both in air-equilibrated and in nitrogen-purged toluene solution. The kinetics clearly exhibit a prompt decay component of few nanoseconds (2–8 ns), together with a delayed decay component characterized by lifetimes of tens/hundreds of nanoseconds in aerated solution which become significantly longer in de-aerated solution (Table 5). The lifetimes of this delayed emitting component are in perfect agreement with those obtained for the triplet excited state from the nanosecond transient absorption measurements (Table 5). Therefore, we have clear experimental evidence about the occurrence of delayed fluorescence for NPI-PTZ and NPI(3)-PTZ in toluene. No such evidence was found for the other investigated compounds, including NPI-Ph-PTZ, in agreement with their larger ΔEST which should disfavor the occurrence of r-ISC followed by delayed fluorescence. The determination of the fluorescence quantum yields in both air-equilibrated and nitrogen-purged conditions for NPI(3)-PTZ in toluene (Table S7, ESI†) allowed the r-ISC rate constant to be estimated according to a method of calculation reported in the literature by Adachi et al.42 A value of 1.9 × 104 s−1 was obtained for the kr-ISC of NPI(3)-PTZ in toluene (see the details in the ESI†). This value is not exceptionally high but still higher than the limit considered necessary to be overcome in the literature for a material to be considered TADF-active.43 The lower ϕF values of NPI-PTZ prevented an accurate estimate of the kr-ISC to be gained for this compound.
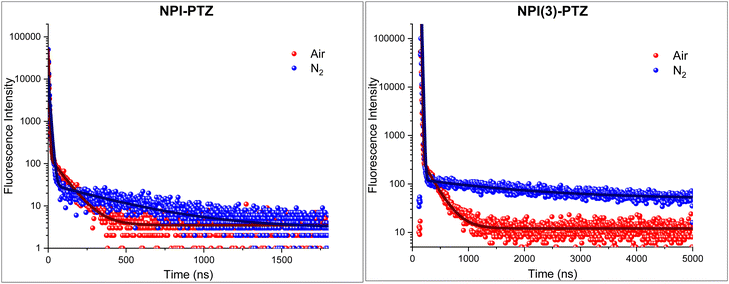 |
| | Fig. 7 Fluorescence decay kinetics of NPI-PTZ (left) and NPI(3)-PTZ (right) in air-equilibrated (red curve) and nitrogen-purged (blue curve) toluene together with their best fitting. | |
Table 5 Comparison between lifetimes of the compounds in air-equilibrated (air) and nitrogen-purged (N2) toluene, obtained from the decay kinetics recorded with nanosecond transient absorption (TA) and single photon counting (SPC) techniques
| Compound |
ns-TA |
ns-SPC |
|
τ
air (ns) |
τ
N2 (ns) |
τ
air (ns) |
τ
N2 (ns) |
|
NPI-PTZ
|
6 (5%) |
6 (3%) |
2.4 (81%) |
3.5 (88%) |
| 90 (95%) |
440 (97%) |
87 (19%) |
394 (12%) |
|
NPI(3)-PTZ
|
5 (1%) |
19 (1%) |
7.8 (89%) |
10 (87%) |
| 262 (99%) |
3400 (99%) |
215 (11%) |
1770 (13%) |
Aggregation-induced and solid-state delayed fluorescence
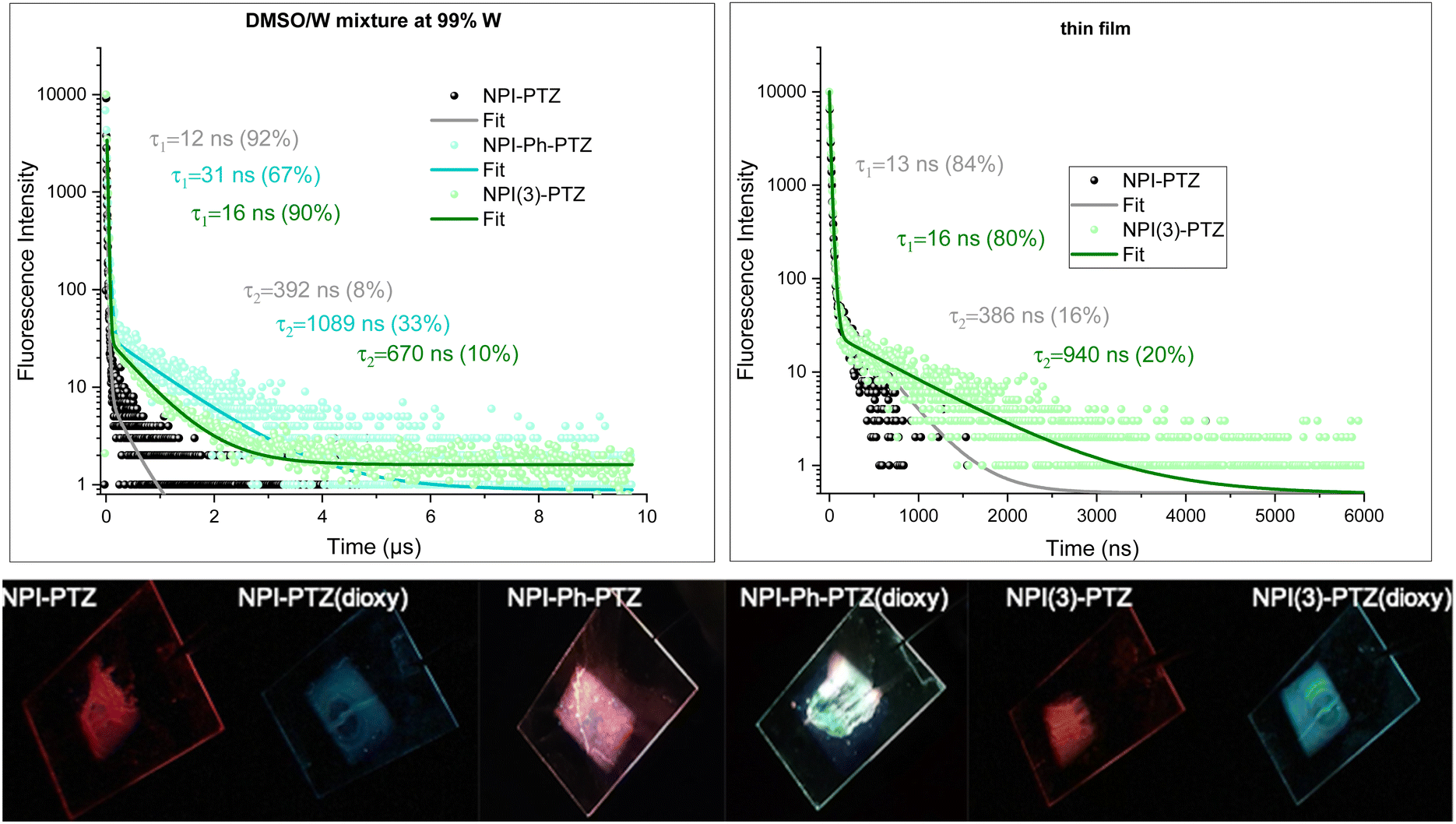
Given the low fluorescence efficiency observed for these fluorophores, particularly for the oxygen-free derivatives, and their structural flexibility as the donor and acceptor units are connected through single bonds, the possibility of an aggregation-induced emission behavior activated through restriction of intramolecular motions was considered and thoroughly investigated. To this aim, the absorption and emission properties of the compounds were studied in dimethylsulphoxide/water mixtures with different water fractions (see Fig. S32, S34 and Tables S8, S9, ESI†). Indeed, while dimethylsulphoxide is a good solvent for these organic fluorophores, water is a bad solvent and its increasing content in the mixtures leads to aggregate formation. Interestingly, aggregates were generally found to be more emissive than the corresponding monomers and were characterized by a bright blue emission for the phenothiazine dioxide derivatives and by red-emission for the phenothiazine derivatives, respectively (Fig. S33, ESI†). Strikingly, while the fluorescence decay of the monomer species took place in few/tens of nanoseconds in DMSO (see Tables S8 and S9, ESI†), the emission kinetics for the aggregates of the three oxygen-free phenothiazine derivatives in water dispersion were found to exhibit both a prompt and a delayed decay component (Fig. 8). The long emission of 392 ns, 1089 ns and 670 ns for NPI-PTZ, NPI-Ph-PTZ and NPI(3)-PTZ, respectively, is consistent with the occurrence of r-ISC from the triplet to the singlet followed by delayed fluorescence in the aggregates. We have thus evidence for aggregation-induced delayed fluorescence for these fluorophores.23,44 It is particularly appealing that the TADF is enabled in the aggregates of NPI-Ph-PTZ, for which no such behavior was revealed in solution. This might be possibly associated to the stabilization of intermolecular charge transfer states in the aggregate species, as sometime discussed in the literature.45
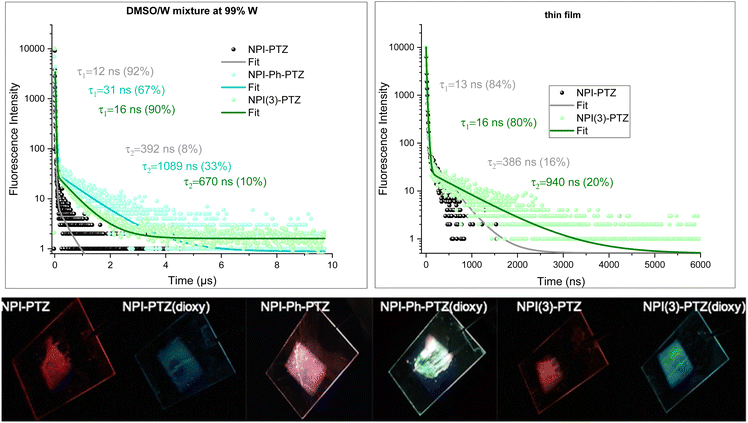 |
| | Fig. 8 (Upper panel) Fluorescence spectra decay kinetics of the investigated compounds in water dispersion (left) and in thin films (right) and relative fittings. (Lower panel) Photographs taken under UV-light of the film emission. | |
Despite the quite low fluorescence quantum yields measured for the investigated compounds in solution, significant emissions were observed in the solid state. The emission of the phenothiazine dioxide derivatives in thin films obtained by spin-coating falls in the blue, while the emission of the phenothiazine-based samples in the red (Fig. 8, Fig. S35 and Table S10, ESI†). This implies that the solid-state fluorescence of these organic materials is located in the two-extreme regions of the visible spectral range, the two regions rarely reached by employing the most common organic fluorophores. The TC-SPC measurements performed on the thin films of NPI-PTZ and NPI(3)-PTZ revealed in the fluorescence kinetics the presence of both a prompt component of tens of nanoseconds and a delayed component of hundreds of nanoseconds (Fig. 8). The observation of red TADF in the solid state for these new organic materials makes them very promising for applications in third generation OLED devices.
Conclusions
In this investigation, we report the synthesis of six new push–pull systems containing naphthalimide (NPI) as electron acceptor and phenothiazine/phenothiazine dioxide (PTZ/PTZ(dioxy)) as electron donors. The donor and acceptor units were arranged in dipolar (NPI-PTZ and NPI-PTZ(dioxy)) and octupolar (NPI(3)-PTZ and NPI(3)-PTZ(dioxy)) structures, and were connected not only by single bonds but also by a phenyl π-linker (NPI-Ph-PTZ and NPI-Ph-PTZ(dioxy)). This synthetic work was aimed at studying the effect of both tuning the push–pull strength and the molecular conjugation on the photobehaviour. The excited state dynamics, nature and energy of these chromophores were thoroughly investigated by means of an advanced joint spectroscopic and computational effort, involving time-resolved spectroscopies with both nanosecond and femtosecond temporal resolution as well as TD-DFT quantum chemical simulations. As a consequence of this mechanistic study, information about the structure-property relationships and the key factors necessary to activate red TADF in these materials was gained. Our time resolved spectroscopic and computational results showed that the increased push–pull character in the oxygen-free relative to the oxygen-functionalized phenothiazine derivatives was crucial to achieve negligible singlet-to-triplet energy splitting between the ICT S1 and the LE T1, and thus effective r-ISC and TADF. On the other hand, higher fluorescence efficiencies were obtained upon increasing the molecular conjugation by passing from dipolar to octupolar systems and particularly by introducing a phenyl spacer between the donor and the acceptor portions. Interestingly, intense red TADF was revealed and enhanced in aggregates of the phenothiazine-based fluorophores produced both in water dispersions and in thin films. The observation of red TADF in the solid state for these new organic materials makes them very promising for applications in third generation OLED devices.
Author contributions
C.M.: data curation, investigation, methodology, writing – review & editing; T.B.: data curation, investigation, methodology, writing – review & editing; M.S.: data curation, investigation, methodology, writing – review & editing; T.T.: data curation, investigation, methodology, writing – review & editing; G.C.: data curation, investigation, writing – review & editing; J.C.: investigation, supervision, writing – review & editing; R.M.: conceptualization, supervision, writing – review & editing; B.C.: conceptualization, investigation, supervision, writing – original draft, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
CM, TB, GC and BC acknowledge support by the European Union – NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 - VITALITY - CUP J97G22000170005. JC acknowledges support from Le Fonds de la Recherche Scientifique (FNRS), Belgium.
Notes and references
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 1–20 CrossRef.
- Y.-Z. Shi, H. Wu, K. Wang, J. Yu, X.-M. Ou and X.-H. Zhang, Chem. Sci., 2022, 13, 3625–3651 RSC.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef.
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS.
- X. Cai and S.-J. Su, Adv. Funct. Mater., 2018, 28, 1802558 CrossRef.
- T. Huang, X. Song, M. Cai, D. Zhang and L. Duan, Mater. Today Energy, 2021, 21, 100705 CrossRef CAS.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, Chem. Mater., 2013, 25, 3766–3771 CrossRef CAS.
- C. Chen, R. Huang, A. S. Batsanov, P. Pander, Y.-T. Hsu, Z. Chi, F. B. Dias and M. R. Bryce, Angew. Chem., 2018, 130, 16645–16649 CrossRef.
- R. Ansari, W. Shao, S.-J. Yoon, J. Kim and J. Kieffer, ACS Appl. Mater. Interfaces, 2021, 13, 28529–28537 CrossRef CAS PubMed.
- X.-K. Chen, D. Kim and J.-L. Brédas, Acc. Chem. Res., 2018, 51, 2215–2224 CrossRef CAS.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- D. H. Ahn, S. W. Kim, H. Lee, I. J. Ko, D. Karthik, J. Y. Lee and J. H. Kwon, Nat. Photonics, 2019, 13, 540–546 CrossRef CAS.
- S. Kumar, P. Tourneur, J. R. Adsetts, M. Y. Wong, P. Rajamalli, D. Chen, R. Lazzaroni, P. Viville, D. B. Cordes, A. M. Z. Slawin, Y. Olivier, J. Cornil, Z. Ding and E. Zysman-Colman, J. Mater. Chem. C, 2022, 10, 4646–4667 RSC.
- Z. Cai, X. Wu, H. Liu, J. Guo, D. Yang, D. Ma, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 23635–23640 CrossRef CAS PubMed.
- U. Balijapalli, R. Nagata, N. Yamada, H. Nakanotani, M. Tanaka, A. D’Aléo, V. Placide, M. Mamada, Y. Tsuchiya and C. Adachi, Angew. Chem., Int. Ed., 2021, 60, 8477–8482 CrossRef CAS.
- D. Karthik, Y. H. Jung, H. Lee, S. Hwang, B.-M. Seo, J.-Y. Kim, C. W. Han and J. H. Kwon, Adv. Mater., 2021, 33, 2007724 CrossRef CAS.
- S. Kothavale, W. J. Chung and J. Y. Lee, J. Mater. Chem. C, 2021, 9, 528–536 RSC.
- U. Balijapalli, Y.-T. Lee, B. S. B. Karunathilaka, G. Tumen-Ulzii, M. Auffray, Y. Tsuchiya, H. Nakanotani and C. Adachi, Angew. Chem., Int. Ed., 2021, 60, 19364–19373 CrossRef CAS PubMed.
- M. Zhao, M. Li, W. Li, S. Du, Z. Chen, M. Luo, Y. Qiu, X. Lu, S. Yang, Z. Wang, J. Zhang, S.-J. Su and Z. Ge, Angew. Chem., 2022, 134, e202210687 Search PubMed.
- Y. Liu, X. Xiao, Y. Ran, Z. Bin and J. You, Chem. Sci., 2021, 12, 9408–9412 RSC.
- S. Qi, S. Kim, V.-N. Nguyen, Y. Kim, G. Niu, G. Kim, S.-J. Kim, S. Park and J. Yoon, ACS Appl. Mater. Interfaces, 2020, 12, 51293–51301 CrossRef CAS.
- J. Guo, Z. Zhao and B. Z. Tang, Adv. Optic. Mater., 2018, 6, 1800264 CrossRef.
- R. Furue, T. Nishimoto, I. S. Park, J. Lee and T. Yasuda, Angew. Chem., Int. Ed., 2016, 55, 7171–7175 CrossRef CAS PubMed.
- X. Wang, S. Wang, J. Lv, S. Shao, L. Wang, X. Jing and F. Wang, Chem. Sci., 2019, 10, 2915–2923 RSC.
- L. Wu, K. Wang, C. Wang, X.-C. Fan, Y.-Z. Shi, X. Zhang, S.-L. Zhang, J. Ye, C.-J. Zheng, Y.-Q. Li, J. Yu, X.-M. Ou and X.-H. Zhang, Chem. Sci., 2021, 12, 1495–1502 RSC.
- M. Poddar, A. Cesaretti, E. Ferraguzzi, B. Carlotti and R. Misra, J. Phys. Chem. C, 2020, 124, 17864–17878 CrossRef CAS.
- Y. Rout, A. Cesaretti, E. Ferraguzzi, B. Carlotti and R. Misra, J. Phys. Chem. C, 2020, 124, 24631–24643 CrossRef CAS.
- Y. Rout, C. Montanari, E. Pasciucco, R. Misra and B. Carlotti, J. Am. Chem. Soc., 2021, 143, 9933–9943 CrossRef CAS.
- A. Cesaretti, T. Bianconi, M. Coccimiglio, N. Montegiove, Y. Rout, P. L. Gentili, R. Misra and B. Carlotti, J. Phys. Chem. C, 2022, 126, 10429–10440 CrossRef CAS.
- G. Tang, A. A. Sukhanov, J. Zhao, W. Yang, Z. Wang, Q. Liu, V. K. Voronkova, M. Di Donato, D. Escudero and D. Jacquemin, J. Phys. Chem. C, 2019, 123, 30171–30186 CrossRef CAS.
- M. Okazaki, Y. Takeda, P. Data, P. Pander, H. Higginbotham, A. P. Monkman and S. Minakata, Chem. Sci, 2017, 8, 2677–2686 RSC.
- R. Guo, Y. Wang, Z. Huang, Q. Zhang, S. Xiang, S. Ye, W. Liu and L. Wang, J. Mater. Chem. C, 2020, 8, 3705–3714 RSC.
- B. H. Jhun, D. Y. Jeong, S. Nah, S. Y. Park and Y. You, J. Mater. Chem. C, 2021, 9, 7083–7093 RSC.
- X. Li, S. Shen, C. Zhang, M. Liu, J. Lu and L. Zhu, Sci. China Chem., 2021, 64, 534–546 CrossRef CAS.
- B. Carlotti, A. Cesaretti, C. G. Fortuna, A. Spalletti and F. Elisei, Phys. Chem. Chem. Phys., 2015, 17, 1877–1882 RSC.
- B. Carlotti, F. Elisei, U. Mazzucato and A. Spalletti, Phys. Chem. Chem. Phys., 2015, 17, 14740–14749 RSC.
- E. Benassi, B. Carlotti, M. Segado, A. Cesaretti, A. Spalletti, F. Elisei and V. Barone, J. Phys. Chem. B, 2015, 119, 6035–6040 CrossRef CAS PubMed.
- B. Carlotti, A. Spalletti, M. Šindler-Kulyk and F. Elisei, Phys. Chem. Chem. Phys., 2011, 13, 4519 RSC.
- B. Carlotti, I. Kikaš, I. Škorić, A. Spalletti and F. Elisei, ChemPhysChem, 2013, 14, 970–981 CrossRef CAS.
- L. Fisher, R. J. Vázquez, M. Howell, A. K. Muthike, M. E. Orr, H. Jiang, B. Dodgen, D. R. Lee, J. Y. Lee, P. Zimmerman and T. Goodson, Chem. Mater., 2022, 34, 2161–2175 CrossRef.
- N. Notsuka, H. Nakanotani, H. Noda, K. Goushi and C. Adachi, J. Phys. Chem. Lett., 2020, 11, 562–566 CrossRef CAS PubMed.
- R. J. Vázquez, J. H. Yun, A. K. Muthike, M. Howell, H. Kim, I. K. Madu, T. Kim, P. Zimmerman, J. Y. Lee and T. G. Iii, J. Am. Chem. Soc., 2020, 142, 8074–8079 CrossRef.
- B. Zhang, Y. Kong, H. Liu, B. Chen, B. Zhao, Y. Luo, L. Chen, Y. Zhang, D. Han, Z. Zhao, B. Z. Tang and L. Niu, Chem. Sci., 2021, 12, 13283–13291 RSC.
- P. Tourneur, F. Lucas, C. Quinton, Y. Olivier, R. Lazzaroni, P. Viville, J. Cornil and C. Poriel, J. Mater. Chem. C, 2020, 8, 14462–14468 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section; absorption and emission properties; ultrafast spectroscopy; triplet properties; excited state energy and nature; thermally activated delayed fluorescence; aggregation induced and solid state delayed fluorescence; quantum chemical simulations. See DOI: https://doi.org/10.1039/d3tc01939j |
| ‡ Equally contributed as first authors. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Manju
Sheokand‡
b,
Titouan
Teunens
a,
Manju
Sheokand‡
b,
Titouan
Teunens