
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy of the NaOH(s)–CO2(g) reaction creating value for industry: green natrite production, energy, and its potential in different sustainable scenarios†
Luis
Rincón
 ab,
Claudia
Ruiz
a,
Ricardo R.
Contreras
ac and
Jorge
Almarza
*a
ab,
Claudia
Ruiz
a,
Ricardo R.
Contreras
ac and
Jorge
Almarza
*a
aEcological World for Life, Laboratorio de Transformación Química de Gases Efecto Invernadero (LabTQGEI). Fundación Gómez Pardo, Calle de Alenza, 1, 28003 Madrid, Spain. E-mail: jalmarza@ewltech.eu
bGrupo de Química Computacional y Teórica (QCT-USFQ), Departamento de Ingeniería Química, Universidad San Francisco de Quito (USFQ), Diego robles y Vía Interoceánica, Quito 17-1200-841, Ecuador
cLaboratorio de Organometálicos, Departamento de Química, Facultad de Ciencias, Universidad de Los Andes, Mérida 5101, Venezuela
First published on 8th May 2023
Abstract
In this work, we present a comprehensive analysis of a solid-based NaOH technology for CO2 capture. After CO2 capture, high purity γ-Na2CO3 synthetic natrite is formed as demonstrated through exhaustive characterization using several chemical techniques (XRD, XPS, 13C and 23Na MAS-NMR, ATR-FTIR, and TGA), and additionally a DFTB computational analysis was performed. The use of solid NaOH could offer some technical advantages such as high exothermicity (3445 MJ per tCO2) and the production of high value carbonates (natrite). Furthermore, we consider either carbonate commercialization or regeneration of NaOH in our model. This novel technology has important advantages over currently available technologies regarding efficiency in capture and potential for heat exploitation. Additionally, environmental analysis reveals a negative LCA index (life cycle assessment index) for flue gas capture and the lowest operative costs for CO2 capture. Two sustainable models are proposed that not only focus on CO2 capture, but also on the sustainability of time. One model is based on sale of natrite, because the projections in the global market for carbonates will continue to increase, due to their non-traditional applications, e.g., in molten carbonate fuel cells, batteries and other technologies for generation and storage of energy. The second model considered NaOH-raw material regeneration, and pure CO2 released can be used in obtaining of synthetic fuel, food packaging, and other industrial chemical procedures, and in this respect the current global demand for CO2 is around 140 Mt per year.
Environmental significanceCurrently, carbon capture is one of the biggest challenges facing humanity. In this sense, it is mandatory to conduct advanced research focused on reducing, capturing and storing CO2. Several methodologies are available for capturing carbon dioxide, but the great majority of them are not sustainable technologies. Consequently, it is necessary to develop CO2 capture technologies that, in addition to being viable economically and technically, are sustainable and constitute a real possibility for the industrial and manufacturing sectors. For this reason, we have proposed a new technology that has important techno-economic advantages and as we have shown it is a sustainable technology; in fact, it is simple and strictly follows green chemistry principles. The first great advantage, in comparison with state-of-the-art available methodologies, is that our technology uses “solid-based NaOH”, with the lowest associated costs and very lowest carbon footprint impact and the second advantage is that it produces high-purity synthetic natrite, a product that has several outlets on the global marketplace, as a raw material for various chemical industrial processes and for the production and storage of energy (e.g., MCFCs and batteries), stimulating a circular economy model. Here, comprehensive information about the advantages of the solid-based NaOH technology for CO2 capture is shown and an exhaustive discussion of the environmental, energy and economic impact assessments of this transformative process is presented with promising results. |
Introduction
CO2 capture has been one of the most prevalent topics for the mitigation of climate change. Nowadays, the use of amine solutions is the most popular industrial process for CO2 capture.1 Recently, different types of amine processes have been developed, including the biphasic solvent's CO2 absorption2,3 and related new technologies.4–7However, the amine process has many drawbacks such as high energy consumption for amine recovery, evaporation of the solvent and degradation of the amine or amine-based sorbents.8
In contrast with the amine solution process, the use of solid hydroxide could offer some technical advantages such as high exothermicity, large reaction velocity and the production of high value carbonates.9 Here we perform an exhaustive study of CO2 capture for a flue gas source using solid NaOH. To the best of the author's knowledge, there is no comprehensive study like this. In this introduction we start with the motivation for the present work.
Classical CO2 capture can be carried out in alkaline aqueous solutions,1 and it can be overlooked in eqn (1) in a liquid/gas medium ignoring the solid/gas possibility.
| 2NaOH(aq) + CO2(g) ⇄ Na2CO3(aq) + H2O(l) | (1) |
Eqn (1) is much more complex than is normally considered in basic chemistry,10 especially considering the equilibrium of the sodium solvated ions [Na(OH2)x+](aq).11 On the other hand, it is necessary to consider the equilibrium of CO2 in alkaline solutions (eqn (2) and (3)).1
| CO2(g) + OH−(ac) ⇄ HCO3−(ac) | (2) |
| HCO3−(ac) + OH−(ac) ⇄ CO32−(ac) + H2O(l) | (3) |
Clearly, the outcome of eqn (1) is combining the results of the carbonate equilibrium in an alkaline medium. Depending on the reactive conditions, in the solid state, it is possible to obtain not only sodium carbonate anhydrous, but other well-known forms of sodium carbonate hydrate such as thermonatrite, Na2CO3·H2O;12 natron, Na2CO3·10H2O,13 and carbonate combinations such as Wegscheider salt, Na2CO3·3NaHCO3 or trone, Na2CO3·3NaHCO3·2H2O as well.14 These products have been reported in the dry carbonation process for CO2 capture (DCP).15
At a solid/gas interface,16 the process does not involve any solvation energy. Instead, it is driven by the NaOH lattice energy UNaOH = 550 kcal mol−1.17 Consequently, the reaction with NaOH(s) is a heterogeneous process, so the reaction may present small deviations from eqn (1) stoichiometry because sodium carbonate presents a sodium deficiency, previously reported by the XPS18 study, a medium that accumulates superficially by “nucleation”. This mechanism has been previously proposed for alkali and alkaline earth metal oxides used as absorbers in CO2 capture.19 The reaction, which is spontaneous (ΔG° < 0), is also exothermic due to a favourable balance between the UNaOH and UNa2CO3 lattice energies and will continue capturing CO2(g) and generating anhydrous Na2CO3(s) until all the nucleophilic agents are consumed.
Physical adsorption of CO2 requires about 324 kJ kg−1 CO2 with equal pressure in the feed and product streams, which is by far smaller than chemisorption, that requires about 1224 kJ kg−1 CO2.20 Then, solid sorbents offer both the possibility of a lower cost, not only in energy, but also in operating costs, and application of a wide range of scales. The reaction mechanisms between a gas (CO2) and a solid (metal hydroxides and oxides) are not very easy to discern and the reaction products analysed by X-ray diffraction vary depending on the temperature and the pressure at which they occur.21
The mechanistic and kinetic studies of the transformation of carbon dioxide in water and dilute aqueous solutions of sodium hydroxide have been studied since the beginning of the 20th century.22–27 Concisely, hydroxides have been used for direct CO2 air capture with a variety of methods.28,29 Nikulshina et al.,30 reported three Na-based thermochemical cycles for capturing CO2 from a gas source of 500 ppm of CO2, including the use of solid NaOH. However, they showed that with their experimental conditions, it is very difficult for the application of Na-based solid sorbents for capturing CO2 from air. Furthermore, it is important to add that during the processes studied so far, carbonate and bicarbonate mixtures are produced.
In the present experiment, the molecular study of the reaction product between CO2(g) and NaOH(s) is performed, where an attempt is made to explain the formation of anhydrous form γ-Na2CO3(s) (synthetic natrite) with an incommensurately modulated structure and its sustainable advantages in a circular economy. The next section is divided into two parts: first, detailed mass, energy and chemical characterization of the solid-based process is presented. Second, the competitive advantages are presented at a technological level, in relation to favourable mass and energy balances that positively impact environmental (carbon footprint and LCA) and economic (techno-economic evaluation) sustainability during the CO2 capture process.
Results and discussion
The amount of CO2 captured (mean value in 28 experiments) was 1.941 ± 0.057 g of CO2 captured using 3 g of NaOH. We consider the NaOH purity to obtain an accurate ratio between CO2 moles per mol of NaOH. Based on the amount of hydrated NaOH and the scale factor (X-ray analysis) a purity of 92.5% is assumed. After considering the NaOH purity, a ratio of 0.66 CO2 moles per mole of NaOH is obtained. The large percent of CO2 capture per NaOH can be explained due to the increase in the amount of carbonate by sodium in the observed product. With this experimental evidence, the ratio between the moles of CO2 (or Na2CO3) and moles of NaOH was found to be larger than 0.50, without any evidence of NaHCO3 formation. A theoretical value of 127.6 kJ mol−1 of CO2 is predicted for the heat of reaction between solid NaOH and CO2 (eqn (4)).| 2NaOH(s) + CO2(g) ⇄ Na2CO3(s) + H2O(g) | (4) |
X-ray powder diffraction characterization
Fig. 1 shows the X-ray powder diffraction (XRD) pattern corresponding to a sample of the material obtained from the CO2 capture process using the solid-based NaOH technology.16 Clearly, the sample is mainly crystalline and there is no evidence of the presence of non-crystalline/amorphous phases, containing only the crystalline phase γ-Na2CO3 natrite,31 monoclinic space group C2/m structure. This phase is an incommensurately modulated structure.32Fig. 1a depicts the obtained X-ray powder diffraction diagram in the main angular range from 10° to 90° 2θ, with the intensity corrected to fixed 1° divergence slit values, with a main structure pattern of γ-Na2CO3 natrite (PDF#4-11-4108), with the reflections of the main structure superimposed. Fig. 1b depicts the same diagram with another pattern of γ-Na2CO3 natrite (PDF#0-61-1420), with additional satellite reflections superimposed.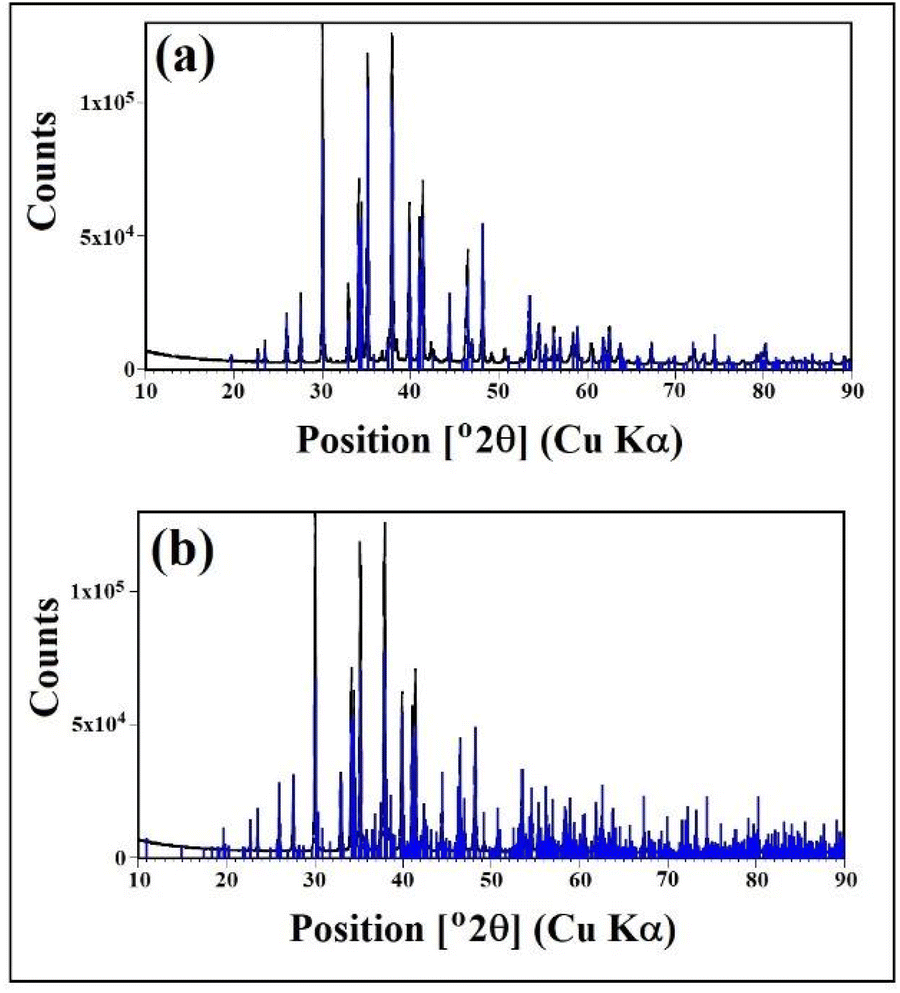 | ||
| Fig. 1 X-ray powder diffraction (XRD) pattern corresponding to a γ-Na2CO3 natrite, monoclinic space group C2/m structure, obtained by chemical transformation with solid-based NaOH technology for CO2 capture. (a) and (b) Diagram with two patterns of γ-Na2(CO3) natrite, PDF#4-11-4108 and PDF#0-61-1420 respectively. | ||
X-ray photoelectron spectroscopy analysis
The XPS spectra (see ESI, Fig. S1a†) show the expected lines for the constituent's elements in the sample previously identified by DRX as natrite, an anhydrous form of sodium carbonate, γ-Na2CO3. However, a peak corresponding to silicon was observed, a plausible impurity resulting from the material manipulation technique. The C1s spectral region (see ESI, Fig. S1b†) contains two lines at around 290 eV and 284 eV respectively. The literature reported one line centered at 289.3 eV corresponding to γ-Na2CO3.33 In this case, the second line to lower BE is discardable as an Auger peak, satellite peak, adventitious carbon34 (CxHy) or signal of the H–CO group by the sodium bicarbonate impurity, previously reported at 289.6 eV. Consequently, it is possible to propose a C1s high-resolution signal splitting into a doublet, attributable to more than one type of C–O bonding present in the crystalline structure of natrite. Multiplet splitting was observed in other carbonaceous materials as an additional sp3 C peak in the C1s XPS spectra region.35 In this instance, an extreme situation is possible for carbonate ions present in the natrite structure. First, a carbon bond with a strong sp3 component. Second, a carbon atom with a strong sp2 component, as shown in the X-ray structure.36 In a nutshell, two XPS C1s peaks are observed: an sp2 C peak with high binding energy and an sp3 C peak with low BE. The O1s spectral region for natrite (see ESI, Fig. S1c†) contains one distinguishable oxygen peak centered at 530.5 eV, previously reported at 531.8 eV.33 Additionally, we see the Auger lines NaKLL1 (a3) at 537 eV and NaKLL (a4) at 531 eV. The characteristic form of the O1s peak indicates that it can be decomposed into various components, a typical XPS signal behaviour in the presence of different oxygen bondings. This result is consistent with that observed for the C1s line, demonstrating more than one type of C–O bonding present in the incommensurately modulated X-ray structure of natrite.32 1071.5 eV BE was observed at the Na1s peak (see ESI, Fig. S1d†) corresponding to sodium atoms with octahedral coordination present in the natrite structure; however, a minoritarian signal at around 1070 eV suggests another type of Na atom too. The X-ray structure reported for natritic sodium carbonate, with graphite-like layers formed by three types of Na+ and CO32− ions37 and a distorted-octahedral coordination sphere around the sodium atom, is in accordance with the observed Na1s signal.Differential thermal and thermogravimetric analysis
Differential thermal and thermogravimetric analysis (see ESI, Fig. S2a†) carried out on the γ-Na2CO3 sample shows a loss of 0.0776% ubicated at 114.93 °C, associated with water in the lattice. Also observed were two endothermic peaks at around 232.97 °C and 337.178 °C, with losses of 0.1410% and 0.2360% respectively. These values are below the temperature range of the phase transition γ → β, but this can be attributed to carbon species (CxHy), previously reported in the TGA-DSC-MS experiment on a similar natrite sample,16 yet cannot be discarded as a polymorphism effect. The thermogram showed an endothermic steady fall from 200 °C to 700 °C, a range where we note the three phase transitions γ-, β- and α-Na2CO3.36 In fact, the γ-phase is stable until 361 °C, β-phase at around 361 °C and 489 °C, and α-Na2CO3 above 489 °C to the melting point. Finally, it was observed that the γ-Na2CO3 melting point was 851 °C, a 0.28% difference in respect to 854 °C, a value reported in the literature and corresponding to pure sodium carbonate or natrite.32 The thermogravimetric analysis result indicates that sodium carbonate obtained by chemical transformation with solid-based NaOH technology for CO2 capture has a purity level of 99.55%.ATR-FTIR analysis
The infrared spectrum with attenuated total reflectance (see ESI, Fig. S2b†) showed seven bands from 4000 to 400 cm−1, similarly, to that observed in the infrared spectrum of γ-Na2CO3 and the natrite.38 No signal in the 3500 to 3000 cm−1 region indicates an absence of water molecules, discarding the presence of thermonatrite, a typical impurity in natrite samples. In order to carry out a detailed study of the spectrum, an ideal XY3 system is considered with D3h symmetry. The application of the selection rules shows that there are four vibrations expected in these types of molecules: one nondegenerate (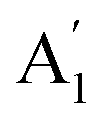 ) vibration, two doubly degenerate (E′) vibrations, and one out-of-plane (
) vibration, two doubly degenerate (E′) vibrations, and one out-of-plane (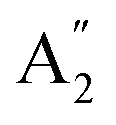 ) vibration. Consequently, the carbonate ion exhibits four normal vibrations:39ν1 (
) vibration. Consequently, the carbonate ion exhibits four normal vibrations:39ν1 (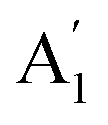 ), symmetric C–O stretching mode; ν2 (
), symmetric C–O stretching mode; ν2 (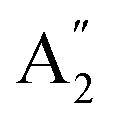 ), CO3− out-of-plane deformation mode; ν3 (E′), asymmetric C–O stretching mode and ν4 (E′), O–C–O bending in-plane deformation mode. Furthermore, in the ATR-FTIR spectrum of the sample, a weak band around the 702–695 cm−1 region was assigned to ν4 vibrational mode. The medium intensity band near the 877–854 cm−1 region must be due to ν2 normal mode. A strong broad band around the 1418–1409 cm−1 region corresponds to ν3 vibrational mode. The ν1 (
), CO3− out-of-plane deformation mode; ν3 (E′), asymmetric C–O stretching mode and ν4 (E′), O–C–O bending in-plane deformation mode. Furthermore, in the ATR-FTIR spectrum of the sample, a weak band around the 702–695 cm−1 region was assigned to ν4 vibrational mode. The medium intensity band near the 877–854 cm−1 region must be due to ν2 normal mode. A strong broad band around the 1418–1409 cm−1 region corresponds to ν3 vibrational mode. The ν1 (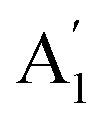 ) vibrational mode is inactive in infrared according to selection rules; however, a distortion of the punctual symmetry from carbonate ions, justified on the basis of the structure of natrite obtained by X-ray data, indicates a possible relaxation of the selection rule mechanism, and therefore now it is possible to observe ν1 (
) vibrational mode is inactive in infrared according to selection rules; however, a distortion of the punctual symmetry from carbonate ions, justified on the basis of the structure of natrite obtained by X-ray data, indicates a possible relaxation of the selection rule mechanism, and therefore now it is possible to observe ν1 (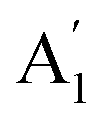 ) mode as a very weak shoulder at 1079 cm−1. Previously, this ν1 mode was reported at 1063 cm−1 and 1056 cm−1, in the γ-Na2CO3 and natrite spectrum,38,40 respectively, using a traditional infrared technique. The comparative analysis of the four vibrational modes observed in the spectrum indicates a reduction of the D3h ideal symmetry in the carbonate ion, a consequence of position modulations of carbon and oxygen atom in phase γ into an incommensurately modulated structure of natrite.37 This observation would justify the appearance of more than one component in each vibrational mode band.
) mode as a very weak shoulder at 1079 cm−1. Previously, this ν1 mode was reported at 1063 cm−1 and 1056 cm−1, in the γ-Na2CO3 and natrite spectrum,38,40 respectively, using a traditional infrared technique. The comparative analysis of the four vibrational modes observed in the spectrum indicates a reduction of the D3h ideal symmetry in the carbonate ion, a consequence of position modulations of carbon and oxygen atom in phase γ into an incommensurately modulated structure of natrite.37 This observation would justify the appearance of more than one component in each vibrational mode band.
23Na and 13C MAS-NMR analysis
The 23Na MAS-NMR spectrum (see ESI, Fig. S2c†) presented two distinct signals, at 3.15 ppm, a singlet and around −15.85 to −23.47, a broad multiplet. These signals indicate the unambiguous existence of more than one type of sodium atom, or preferably, different coordination symmetries in the lattice of the sodium atom. The X-ray structure reported for natrite showed two octahedral sodium atoms,32 Na1 and Na2, occupying interstitial sites with a highly symmetric coordination sphere in the natritic structure, a position that gives a 3.4 ppm signal. A third-sodium type (Na3) and carbonate ions form graphite-like layers, parallel to the [001] direction36 in a position with lower symmetry and surrounded by six oxygen atoms in distorted equatorial coordination with one oxygen atom above and two equidistant oxygen atoms below this plane. These types of atoms, in positions of low symmetry, appear at lower fields consequently,41,42 and the Na3 atom is observed as a broad signal below −15.85 ppm.The 13C MAS-NMR spectrum showed a singlet at 172.19 ppm, typical of the carbonyl group of the carbonate ion.43 The signal amplification into the 170–175 ppm region indicates unsymmetric slight broadening due to a variation of the CO3-group geometry coherent with X-ray details observed in the incommensurately modulated structure and with ATR-FTIR analysis which indicates a reduction of the D3h ideal symmetry of the carbonate ion in natritic chemical environments.
Clearly, the 23Na,13C MAS-NMR spectrum according to X-ray data and XPS suggests a sodium deficiency especially on the natrite surface. This means a deviation in the ideal sodium-to-carbonate ratio; in fact, this value was previously reported as 1.92 in bulk38 and 1.7533 and 1.70 on the surface.18 Normally, stoichiometric calculations that involve sodium carbonate with a value of 2 are normal, and a value between 1.92–1.70 is possible by approximation to 2, especially in fine chemicals. But, in mass balance for industrial chemistry where tons of raw materials are present, this deviation must be considered.
DFTB calculations
To compare different structures again, we generated the average values for the most important geometric parameters. From Table S1 (see ESI†), we can conclude: (1) the stability of the Na2CO3 structures increases when the C–O distance increases and the Na–C distance decreases. This keeps the number of neighbourhoods relatively constant. (2) The non-planarity of the CO32− fragment stabilizes the structure. This non-planarity is necessary for the observed decrease in the Na–C distance. (3) The charge on the sodium atom decreases as the bond is stronger. (4) These results show that possible vacancies of Na (perhaps on the surfaces) can stabilize the Na2CO3 surface.Life cycle assessment
The regeneration of NaOH is modelled according to three different methods: (i) electrolysis of sodium carbonate. The energy cost of this technology is similar to that of the industrial “chloralkali” process for the production of NaOH from aqueous NaCl solutions, that is, about 4500–5500 MJ per ton of NaOH, without considering water evaporation. In this method,44 pure CO2 is produced and sodium hydroxide is recycled back into the process through an electrolytic cell with hydrogen recycling (Fig. 2a). A theoretical maximum concentration of sodium hydroxide of 16% (w/w) is achievable due to the solubility limitations of sodium carbonate. The voltage to be applied is that of water electrolysis, slightly less than 2 V per cell. The achieved current efficiency is 85% (see Methods). If we exclude water evaporation this process is near the thermodynamics minimum. (ii) Lime cycle. Conversion of sodium carbonate into NaOH, also called “causticization”, is one of the oldest processes in the chemical industry. This process has been used by the paper industry for over a century to recover NaOH from aqueous solutions of sodium carbonate and a lignocellulosic material, known in the industry as “black liquor”. It consists of treating the sodium carbonate solution with lime (Ca(OH)2) and precipitating CaCO3. Subsequently, calcium hydroxide is regenerated by heating calcium carbonate to 900 °C. Some studies on CO2 air capture with aqueous NaOH have focused on this regeneration process.45–47 (iii) Direct causticization using titanates. Unlike the lime cycle, this process requires solid carbonate which is subsequently heated with sodium trititanate to form sodium pentatitanate and CO2. Sodium pentatitanate in water spontaneously produces NaOH and regenerates trititanate. Heating up to 900 °C of sodium trititanate and carbonate is the energy consuming step in this process. The titanate process requires 50% less energy than the lime process and 50% less water evaporation than electrolytic recovery.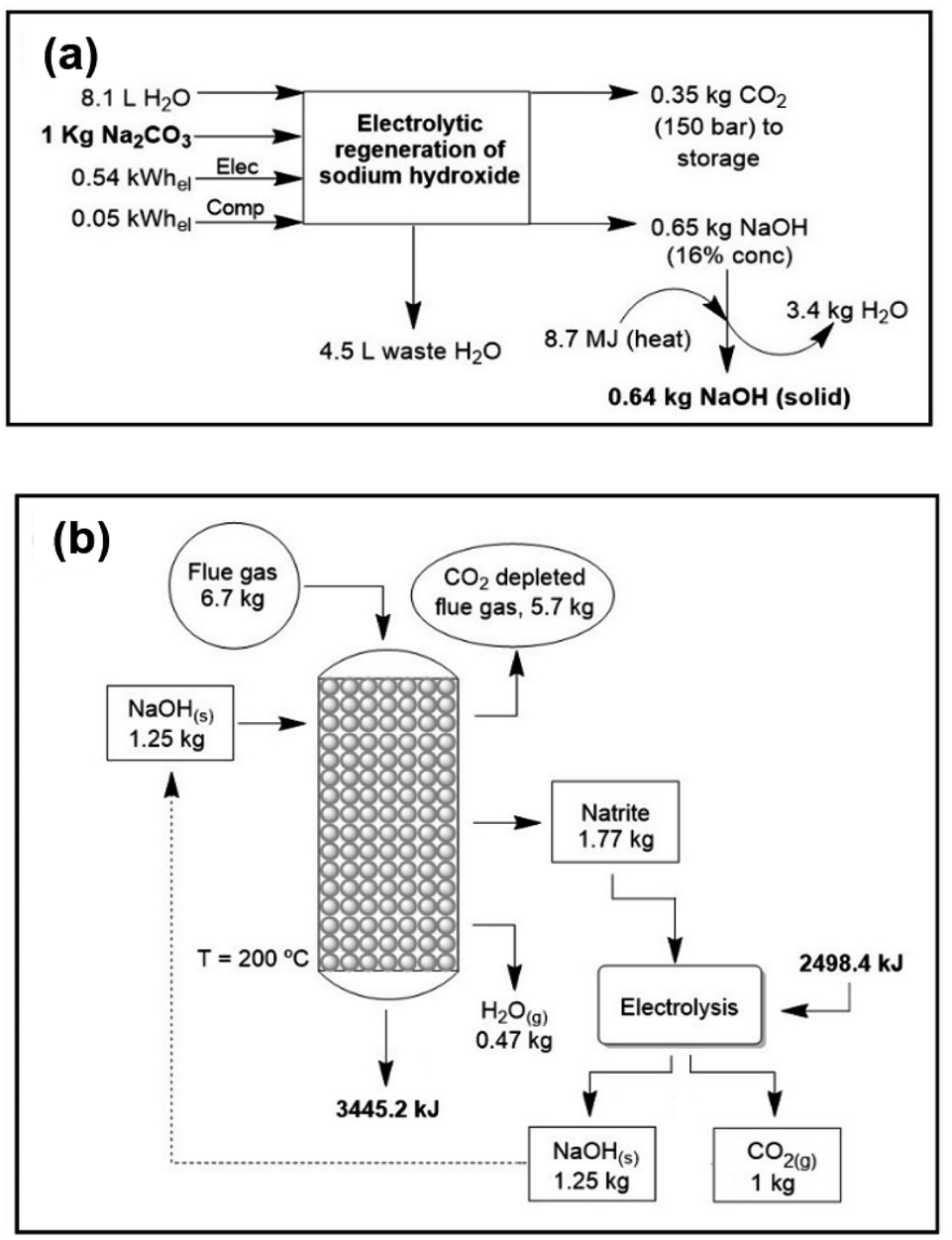 | ||
| Fig. 2 (a) Electrolytic regeneration of sodium hydroxide and pure CO2 production.44 (b) Mass and energy balance of the complete cycle with NaOH-solid technology. In this figure the complete regeneration of natrite is studied. | ||
From the regeneration point of view, the less energy intensive per CO2 ton is the electrolysis (around 40 euros per CO2 ton); however, the cost of water evaporation is high; in contrast, heating in direct caustification is costly but since the final solution is more concentrated the evaporation cost is much lower. Finally, even if costly, the lime cycle is the most mature and well-known of all technologies. Depending on water evaporation and the temperature the energetic cost of electrolysis is between 44 and 120 euros. However, due to less water evaporation the cost of using titanates can be well below that of electrolysis.
At this stage a full cost benefit analysis would be speculative due to the uncertainty in parts of the investment costs and operating costs; for this reason the analysis is focused on the energetic cost and carbon footprint.
Mass and energy balance
The final energy and mass balances of the NaOH system with flue gas are represented in Fig. 2b. Considering this, the functional unit (i.e., the unit to which the process inventory is referred to) selected was 1 kg of captured CO2 (i.e., 1 kg CO2 at the point of capture). As mentioned before, the last part (regeneration of NaOH) can be performed using an electrochemical cell. In addition to the large energy gain, solid-based processes achieve a large reaction rate when compared with the reported rate for other technologies.Carbon footprint (CF)
The CF assessment of the process not including NaOH regeneration (Fig. 3a) is −2.51 using green NaOH (see Methods) and −1.66 using the commercial one. When NaOH recovery is performed, the source of NaOH is less important, but the energy source for heating and the efficiency of the heat transfer play a major role. Assuming a conservative 50% energy recovery in the reaction the CF is estimated to be −0.57 kg CO2 for electrolytic recovery. Energy recovery in the form of electricity can be achieved using an organic Rankine cycle (ORC) using temperatures below 200 °C with high efficiency. Therefore, the assumption of a 50% heat is very reasonable in this scenario of low temperature. Under our assumptions, the CF is negative in all scenarios. Thus, there is a net decrease in greenhouse gas emissions in the atmosphere (see ESI, Table S2†).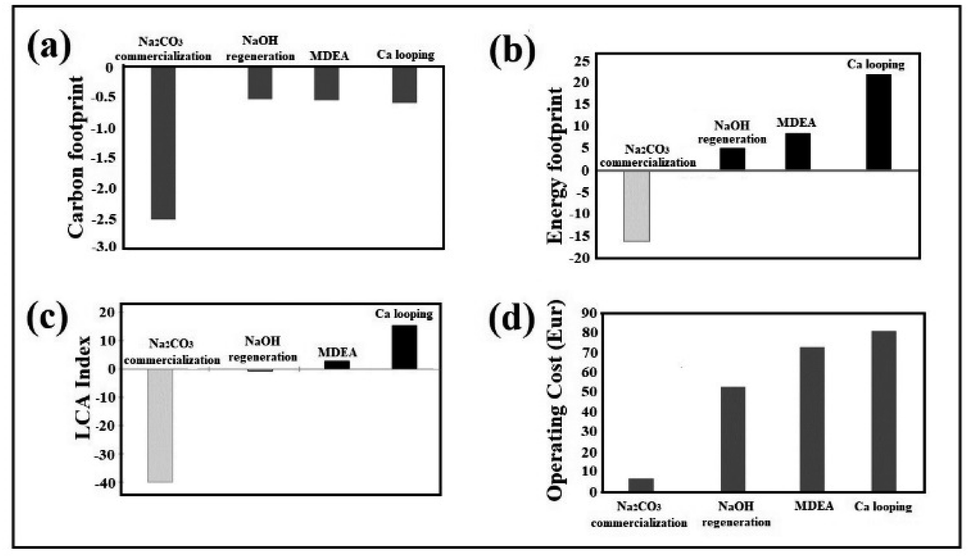 | ||
| Fig. 3 (a) Carbon footprint (kg CO2 eq. per kg CO2 captured). (b) Energy footprint (MJ per kg CO2 captured). (c) LCA index considering MDEA technologies, looping carbonate and NaOH(solid)-based solid technology. Both scenarios are taken into consideration: full Na2CO3 commercialization and full NaOH regeneration without the sale of natrite. (d) Operating cost per ton of CO2 capture. | ||
Energy footprint (EF)
This environmental category focuses on the balance of primary non-renewable energy consumption. Hence, an EF is the assessment of the energy consumption related to the whole supply chain and processing of the captured CO2. The results observed for the net balance of fossil resources are presented in Fig. 3b. The net values, in this case, are always positive, except for the scenario where CO2 is captured and sequestered in Na2CO3 for sale in the carbonate market. The other options involve an increase in the consumption of fossil fuel resources: air-based processes produce a large depletion of fossil resources, while for flue gas, our NaOH(s)-regeneration process produces the lowest impact. Indirect calcium looping increases the demand for fossil resources with respect to amines; as a matter of fact, all the alkali regeneration approaches assessed in this study (electrolytic or indirect Ca-looping) have significant energy footprints (see ESI, Table S3†).LCA index
As shown in Fig. 3c, it is possible to quantitatively appreciate the significant impact in relation to the carbon and energy footprints of the three capture technologies. Competitive advantages are not seen in a conclusive way in the previous graphs where both variables were evaluated separately.Techno-economic assessment
Fig. 3d shows the competitive advantage in relation to operating costs of the technology using NaOH(s), being the most favoured when natrite (anhydrous high purity Na2CO3) is sold at the price of conventional industrial carbonate. However, as the global carbonate market is relatively small, almost all of it must be regenerated and converted to NaOH (feedstock). 99% of raw material regeneration is considered in this study, which causes higher CO2-capture operating costs. Considering this additional process, the operating cost of the NaOH technology is still significantly lower (52 Eur per tCO2) than the operating costs of the technologies considered (≥73 Eur per tCO2) (see ESI, Table S4†).Sustainable model I
Sale of natrite and use of energy
The natrite produced with our technology represents an added value that stimulates a sustainable economy by providing value to CO2 in an economically viable way for several other industrial applications (see ESI, Fig. S3†). Using natrite would provide environmental and economic sustainability for a relatively lower cost, high purity, green carbonate industry. On the other hand, the projections in the global market for carbonates will continue to increase, among other aspects, due to their application in the technology of molten carbonate fuel cells (MCFCs),48 which require raw materials of high purity to produce clean energy.These MCFCs are a growing technology, with global potential in evaluated pilots, that meet energy needs efficiently and with great prospects in the residential, commercial, and shipping industries and capture of CO2 from industrial emissions, among other areas of energy application. Although the market for anhydrous green carbonate and molten carbonate fuel cells is not intensive, international pressures for the use of more environmentally friendly alternatives are increasingly driving these emerging markets, as is happening in maritime transport. For example, Fig. S3 (see the ESI†) shows an important release of energy (3445 MJ per tCO2 captured) which corresponds to energy released in the form of heat and can be used to feed an organic Rankine cycle (ORC) or the conventional Rankine cycle. Due to the high temperatures produced, obtaining this increase in the production of electricity or even heating water represents additional benefits to the global economic balance.
Sustainable model II
Raw material regeneration
Excess natrite can be processed to regenerate chemical converters (NaOH) which would start the capture cycle all over again with a lower carbon footprint at each regeneration step. The separated pure CO2 can be injected into geological sinks and a part could be used for enhanced oil recovery (EOR), food packaging, and synthetic fuel synthesis, among other uses. Current global demand for CO2 is around 140 Mt per year, with the oil industry as the main consumer.This regeneration has the advantage that both carbonate and solid hydroxide are stable under industrial operating conditions and easy to handle, which allows the use of existing transport infrastructure for a lower cost of transporting CO2 to regenerating locations that may be advantageous for storage or use. There may also be potential benefits to compensating the costs of the energy consumed in the raw material regeneration (NaOH) with the energy released during CO2 capture. In other words, in this model, industry has alternative evaluation: (1) if the benefits from the sale of high purity Na2CO3 are attractive, (2) which in turn would give benefits in energy savings during the regeneration process by electrolysis and (3) in turn an “additional advantage” that involves the use of energy that occurs during the capture process. These three aspects must be considered for the global economic balance of the system, which will have a direct impact on the improvement of the estimated operating costs.
In the same way, achievable savings in costs associated with CO2 allowance units (≈80 Eur per tCO2 in EU/ETS) would be an interesting incentive for industrial operations in Europe.
Conclusions
(1) The system of CO2 capture using NaOH in a solid state is a robust, efficient, and flexible technology and very environmentally friendly and additionally can be considered a green chemical technology. By means of XRD, XPS, DSC-TGA, ATR-FTIR, and 23Na and 13C MAS-NMR it was possible to confirm the sodium carbonate anhydrous production with high purity (>99.55%), present in the form of γ-Na2CO3 (synthetic natrite) with a sodium deficiency especially on the surface. It is important to highlight that in stoichiometric calculations with sodium carbonate, a value of 2 is normal and a value of 1.92–1.70 is possible to find by approximation to 2, which are valid conditions in fine chemicals, but in mass balance for industrial chemistry that involves tons of raw materials, this sodium deficiency must necessarily be considered. All results obtained in the structural characterization of natrite are consistent with those exhibited in the DFT calculations and with the X-ray results previously reported, where an incommensurately modulated structure was described.(2) The techno-economic assessment considered two sustainable models; ‘model I’ is based on sale of natrite. Furthermore, the release of energy in the form of heat during the captured process, 3445 MJ per tCO2, can be used for electric energy production. Sustainable ‘model II’ considered the NaOH-raw material regeneration and the pure CO2 obtained in the process, which can be used in other industrial applications.
(3) High exothermicity of the reaction allows for taking the energy released during the capture and making use of it in the regeneration process. This has benefits for the sustainability of the global process. Additionally, even if it is not strictly necessary for it to be a chemical conversion and regeneration, this technology is meaningfully more favorable than those offered by the technology market. Concisely, a balance that considers the sale, completed regeneration or partial regeneration of natrite, to which we may add the benefits in the carbon allowance markets, would give this technology competitive advantages over traditional CO2 capture technologies.
Methods
Gas emission concentration
The gas concentration was determined using an analyser type HORIBA Multi-Component Gas Analyzer/VA-5112G, consisting of a non-dispersive infrared method of a double beam (NDIR-nondispersive infrared) to measure gases such as SO2, CO, CO2, CH4, N2O, NO, and NH3) and the chemiluminescence method (CLA) that allows measuring NOx at low concentrations. The instrumentation bench also has a mixer Environics gas dilution system/Series 4040, a sample conditioning M&C gas conditioning unit with temperature controller/CSS Series, two flow controller systems precision gas mass flow controller/MCS-Series and M-Series, a differential pressure transducer OMEGA differential pressure transmitter/Model PX2300 and a trace detector of humidity in gases based on a P2O5 sensor DKS Aquatrace IV/Typ-R. These instruments were operated using CO2 REMO software from TCA Spain.Chemicals
Sodium hydroxide was purchased from Gie-Tec GmbH (Eiterfeld, Germany) and Merck KGaA, Darmstadt, Germany. Industrial NaOH was also purchased from Barcelona Global Chemical Solutions (Cornella de Llobregat – Spain). Sodium carbonate was obtained from PanReac AppliChem (Darmstadt, Germany). The gases CO2 and N2 were purchased from Air Liquid (Madrid, Spain). All chemicals were used directly without further purification.Experimental capture system
We employ a laboratory scale model converter to test the amount of CO2 captured using a solid NaOH chemical converter. A 50 mL Falcon conical tube was packed with three layers of solid NaOH, 1.00 g each. The system was purged using N2 to remove any traces of remaining gases. Following this 5% CO2 was captured in three steps at room temperature (≈20 °C). The solid component was recovered after each step and ground up to pack the Falcon tube with 2 layers (step 2) or 1 layer (step 3) of 1.00 g chemical converter. The captured CO2 was determined by using an instrumental method (see ESI, Fig. S4†).CO2 determination method
The amount of CO2 captured was obtained following the concentration of CO2 at the end of the reactor until the system was saturated and no more CO2 was adsorbed in each capture step. From the gas flow (1.3 L min−1), we obtained the volume of CO2 captured. From this volume, we calculated the moles of CO2. The typical error in moles of CO2 from this method is about 1%. This error was estimated assuming a 1% in the CO2 profile (see ESI, Fig. S5†).Reproducibility and repeatability of studies
For this evaluation, 4 new NaOH flasks were considered, from different provider batches (Gietec (Germany), Merck (Germany) and Barcelonesa Group (Spain)) and experiments were carried out independently for each sample for a total of 28 trials. To estimate the purity of NaOH, we use information from the X-ray analysis of fresh NaOH based on the amount of hydrated NaOH and the scale factor.The power X-ray diffraction and XPS studies
X-ray powder diffraction characterization was carried out using a PANalytical X'Pert PRO MPD apha1 powder diffractometer in Bragg–Brentano θ/2θ geometry of 240 millimeters of radius. A focalizing Ge (111) primary monochromator was used with Cu Kα1 radiation: λ = 1.5406 Å and work power: 45 kV to 40 mA. A variable divergence slit aperture was used to get an irradiated length over the sample of 10 millimeters. A mask defining the length of the beam over the sample in the axial direction of 12 millimeters was used. A diffracted beam with 0.04 radians Soller slits was used. Sample spinning was performed at 2 revolutions per second. A pixcel detector was used; active length = 3.247° θ/2θ scans from 4 to 100° 2θ with a step size of 0.026° and measuring time of 100 seconds per step (six repeated consecutive scans). Qualitative crystalline phase analysis was performed by means of the PDF (Powder Diffraction File) database, ICDD-JCPDS (International Center for Diffraction Data – Joint Committee of Powder Diffraction Standards, 2021).X-ray photoelectron spectroscopy
XPS was carried out using a Physical Electronics spectrometer, model PHI 5500. The experiment covers all elements of the periodic table, except hydrogen and helium. The depth of analysis <10 nm was performed in a chamber of ultra-high vacuum (UHV) working in the range of 1 × 10−8 to 1 × 10−9 torr (from 1.3 × 10−6 to 1.3 × 10−7 Pa) pressure. The system was calibrated in accordance with PHI procedures, with photoemission lines Eb of Ag3d5/2 = 368.3 eV and with 0.8 eV of amplitude at half peak height (FWHM), using the monochromatized aluminium source for excitation.Differential thermal and thermogravimetric analysis
DSC-TGA was performed simultaneously with an SDT Q600 V20.9 Build 20 at a heating rate of 5.00 °C min−1 in the temperature range 33–1100 °C. Approximately 15 mg of the sample was used in each run.ATR-FTIR analysis
The infrared spectrum has been obtained using a Thermo IZ10 instrument with an attenuated total reflectance accessory (ATR) of a diamond. The instrument has a DTGS detector, a KBr beam-splitter and a glow-bar source. The spectral range analysed has been 4000 to 525 cm−1 with a 4 cm−1 resolution and 32 accumulations.Nuclear magnetic resonance
Solid-state magic angle spinning (MAS) NMR spectroscopy experiments were performed on a Bruker AV 400 MHz WB (89 mm; 9.39 T) spectrometer at spinning speeds of 12 kHz at a 13C frequency of 100.61 MHz and referenced to glycine CO at 176.03 ppm and 23Na frequency of 105.84 MHz and referenced to NaCl at 7.28 ppm. The 13C direct excitation experiments were carried out with a 90° pulse with a length of 4 μs averaging 256 scans with a recycle delay of 300 s and 600 s in the MAS NMR sequence. The 23Na direct excitation experiments were performed using a 90° pulse with a length of 4 μs and a recycle delay of 5 s in the MAS NMR sequence.The DRX, XPS, TGA-DSC, IR-ATR and elemental analysis (% C and % Na) experiments were carried out in Centers Científics i Tecnològics (CCiT), Universitat de Barcelona, Spain. The NMR spectra were obtained at the Unidad de Resonancia Magnética, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, Spain.
DFTB studies
We performed non-spin polarized density functional tight binding (DFTB) calculations with the dftb+ package. The bulk Brillouin zones were sampled with 1 × 2 × 2 and gamma point Monkhorst–Pack k-point meshes. Self-consistent field calculations were converged to 1 × 10−5 eV and geometries were optimized using a force converge of 0.01 eV Å−1. We initially optimized the atomic positions and lattice constants starting from the reported γ-Na2CO3 bulk parameters (a = 8.920 Å, b = 5.245 Å, and c = 6.050 Å). The structure of Na2CO3 used as a starting point can be described as graphite-like layers formed by one Na+ for CO32− ions, which are stacked along the hexagonal axis. An additional Na+ ion is in the hexagonal inter-channels. The optimized Na2CO3 bulk structure was further revised to better represent the real material which is incommensurable. We generated 20 additional candidate starting structures by randomly perturbing the locations of Na+ ions. The 20 revised bulk structures are divided into three groups according to their relative energy differences. Group 1 contains structures that relax back to the same geometry as the initial structure. The optimized structures from these groups are all energetically more favourable than the initial structure. Group 2 consists of structures with energies lower than by about 0.5–1 kcal per formula unit. Group 3 structures are lower in energy than the initial structure by about 1–2 kcal per formula unit. The existence of several different structures that are nearly isoenergetic means that the bulk material likely consists of domains having different orientations of the carbonate ions at room temperature and above. The theoretical DFTB Na2CO3 structure results are similar to those obtained using DFT-PBE.49Mass energy balance
In this process, pure CO2 is produced and NaOH is recycled back into the process through an electrolytic cell with hydrogen recycling. This process was originally patented to produce NaOH without the production of chlorine. A theoretical maximum concentration of sodium hydroxide of 16% w/w is archivable due to the solubility limitations of sodium carbonate. The voltage to be applied is that of water electrolysis, slightly less than 2 V per cell.48 The reported current efficiency is 85%.Green sodium hydroxide assumes the use of renewable energy in the production of low-impact sodium hydroxide. The basic concept of green sodium hydroxide is the substitution of grid electricity with a renewable source (see ESI, Tables S2–S4†).
Life cycle assessment impact
In this part, two reference systems have been studied: absorption of CO2 with amines and indirect calcium looping using NaOH. Traditionally, monoethanolamine (MEA) has been the benchmark solvent for this application. Here, we consider the use of methyldiethanolamine (MDEA), which, when compared to MEA, presents a higher maximum CO2 loading capacity (1.00 mol CO2 mol−1 MDEA vs. 0.50 mol CO2 mol−1 MEA). Furthermore, thermal regeneration, which has a high energy demand and accounts for 80% of the operating cost for MEA-based capture, can be reduced if MDEA is chosen instead. The energy demand for MEA and MDEA regeneration is experimentally determined. They respectively reported 4.4 GJ per tCO2 captured and 2 GJ per tCO2 captured for MEA and MDEA.50The capture of CO2 from air is based on indirect calcium looping and causticization.9 The original publication assumed KOH for the cycle, and NaOH has been analyzed in this work. Since the process utilizes an aqueous solution of NaOH and further includes its regeneration through a thermal route using CaCO3 produced in a causticization reactor, it can be compared to other options for NaOH regeneration. In total, five scenarios for solid NaOH technology for carbon capture are considered based on the final use of the technology and the source of NaOH: (1) commercialization of Na2CO3 from commercial NaOH; (2) commercialization of Na2CO3 from green NaOH; (3) regeneration of NaOH by electrolysis and CO2 storage from green NaOH.
Carbon footprint
The CF is the total greenhouse gas emissions caused by the process, expressed as kg CO2 equivalents, characterized through the factors for chemical species of the IPCC (Intergovernmental Panel on Climate Change). If no NaOH regeneration is employed, the production of NaOH is likely to be the main contributor to the CF of the present technology. In this case, two sources of sodium hydroxide can be considered: commercial NaOH and green NaOH. For green NaOH, it was assumed that wind power is used instead of electricity from the grid. Also, the utilization of Na2CO3 based products such as sodium carbonate, reduces the carbon footprint of our technology, as it replaces the carbon footprint of current mining and chemical manufacturing processes of high-purity carbonates. For the carbon footprint, we estimated that a value of 0.50 kg CO2 eq. per kg of NaOH was used for green sodium hydroxide and 0.83 kg CO2 eq. per kg of NaOH for commercial sodium hydroxide.50 The production of green Na2CO3 has an impact of −1.01 kg of CO2 per kg of Na2CO3 produced, as it is avoided.LCA index
It is often convenient to combine both parameters, carbon and energy footprints, into a single score, in particular, if one needs a simple way to assess several CO2 capture technologies. Here, we integrated both values into a single equation (LCA index). This index allows us to appreciate, through a quantitative value, the competitive advantages of any technology in relation to the environmental impact of carbon dioxide capture. Since the energy footprint is an order of magnitude larger than the carbon footprint, the index is calculated as LCAindex = EF + 10 CF.Mass energy balance
The composition of the flue gas was 14.90% CO2, 80.50% N2, and 3.60% O2. This balance considers two steps: (1) a fluid-bed reactor of solid NaOH as the chemical sorbent of CO2, a 100% conversion reaction of NaOH can be assumed; (2) NaOH regeneration and the CO2 concentration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thanks to José Luis Gálvez (Instituto Madrileño de Estudios Avanzados, IMDEA) and Claudio Sidney (Ecological World for Life, EWL) for their valuable feedback on the manuscript.References
- S. Y. W. Chai, L. H. Ngu and B. S. How, Greenhouse Gases: Sci. Technol., 2022, 12, 394–427 CrossRef CAS.
- Y. Yu, Y. Shen, X. Zhou, F. Liu, S. Zhang, S. Lu, J. Ye, S. Li, J. Chen and W. Li, Chem. Eng. J., 2022, 428, 131241 CrossRef CAS.
- X. Zhou, Y. Shen, F. Liu, J. Ye, X. Wang, J. Zhao, S. Zhang, L. Wang, S. Li and J. Chen, Environ. Sci. Technol., 2021, 55, 15313–15322 CrossRef CAS PubMed.
- F. Huhe, J. King and S. S. C. Chuang, Res. Chem. Intermed., 2023, 49, 791–817 CrossRef CAS.
- H. Chen, H. Dong, Z. Shi and A. K. Sengupta, Sci. Adv., 2023, 9, eadg1956 CrossRef CAS PubMed.
- L. Jiang, W. Liu, R. Q. Wang, A. Gonzalez-Diaz, M. F. Rojas-Michaga, S. Michailos, M. Pourkashanian, X. J. Zhang and C. Font-Palma, Prog. Energy Combust. Sci., 2023, 95, 101069 CrossRef.
- M. Hanifa, R. Agarwal, U. Sharma, P. C. Thapliyal and L. P. Singh, J. CO2 Util., 2023, 67, 102292 CrossRef CAS.
- F. Rezaei and C. W. Jones, Ind. Eng. Chem. Res., 2013, 52, 12192 CrossRef CAS.
- D. W. Keith, G. Holmes, D. Angelo and K. Heidel, Joule, 2018, 2, 1573–1594 CrossRef CAS.
- J. Barrett, Inorganic Chemistry in Aqueous Solutions, The Royal Society of Chemistry, Cambridge, U. K., 2003, pp. 112–113 Search PubMed.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, John Wiley & Sons, New York, 6th edn, 1999, p. 102 Search PubMed.
- K. K. Wu and I. D. Brown, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 890–892 CrossRef.
- K. Mocek and D. Beruto, Mater. Chem. Phys., 1986, 14, 219–227 CrossRef CAS.
- C. E. Vanderzee and D. A. Wigg, J. Chem. Thermodyn., 1981, 13, 573–583 CrossRef CAS.
- S. Medina-Carrasco and J. M. Valverde, Cryst. Growth Des., 2018, 18, 4578–4592 CrossRef CAS.
- C. Ruiz, L. Rincón, R. R. Contreras, C. Sidney and J. Almarza, ACS Sustainable Chem. Eng., 2020, 8, 19003–19012 CrossRef CAS.
- M. C. Ball and A. H. Norbury, Physical Data for Inorganic Chemists, Longman, London, 1974, p. 96 Search PubMed.
- J. S. Hammond, J. W. Holubka, J. E. DeVries and R. A. Dickie, Corros. Sci., 1981, 21, 239–253 CrossRef CAS.
- M. T. Dunstan, F. Donat, A. H. Bork, C. P. Grey and C. R. Müller, Chem. Rev., 2022, 121, 12681–12745 CrossRef PubMed.
- A. L. Chaffee, G. P. Knowles, Z. Liang, J. Zhang, P. Xiao and P. A. Webley, Int. J. Greenhouse Gas Control, 2007, 1, 11–18 CrossRef CAS.
- R. V. Siriwardane, C. Robinson, M. Shen and T. Simonyi, Energy Fuels, 2007, 21, 2088–2097 CrossRef CAS.
- H. Hikita, S. Asai and T. Takatsuka, Chem. Eng. J., 1976, 11, 131–141 CrossRef CAS.
- C. Faurholt, J. Chim. Phys., 1924, 21, 400–455 CrossRef CAS.
- B. Pinsent and F. Roughton, Trans. Faraday Soc., 1951, 47, 263–269 RSC.
- B. Pinsent, L. Pearson and F. Roughton, Trans. Faraday Soc., 1956, 52, 1512–1520 RSC.
- D. C. Kern, J. Chem. Educ., 1960, 37, 14–23 CrossRef CAS.
- O. Levenspiel, Chemical Reaction Engineering, John Wiley & Sons, New York, 1999 Search PubMed.
- M. Mahmoudkhani and D. W. Keith, Int. J. Greenhouse Gas Control, 2009, 3, 376–384 CrossRef CAS.
- R. Sen, A. Goeppert, S. Kar and G. K. Surya Prakash, J. Am. Chem. Soc., 2020, 142, 4544–4549 CrossRef CAS PubMed.
- V. Nikulshina, N. Ayesa, M. E. Gálvez and A. Steinfeld, Chem. Eng. J., 2008, 140, 62–70 CrossRef CAS.
- N. V. Zubkova, D. Y. Pushcharovsky, G. Ivaldi, G. Ferraris, I. V. Pekov and N. V. Chukanov, Neues Jahrb. Mineral. Monatshefte, 2002, 85–96 CrossRef CAS.
- A. Arakcheeva, L. Bindi, P. Pattison, N. Meisser, G. Chapuis and I. Pekov, Am. Mineral., 2010, 95, 574–581 CrossRef CAS.
- A. Shchukarev and D. Korolkov, Open Chem., 2004, 2, 347–362 CAS.
- T. L. Barr and S. Seal, J. Vac. Sci. Technol., A, 1995, 13, 1239–1246 CrossRef CAS.
- A. Fujimoto, Y. Yamada, M. Koinuma and S. Sato, Anal. Chem., 2016, 88, 6110–6114 CrossRef CAS PubMed.
- A. Arakcheeva and G. A. Chapuis, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 601–607 CrossRef PubMed.
- M. Dusek, G. Chapuis, M. Meyer and V. Petricek, Acta Crystallogr., Sect. B: Struct. Sci., 2003, 59, 337–352 CrossRef PubMed.
- A. P. Khomyakov, Int. Geol. Rev., 1983, 25, 1111–1116 CrossRef.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley & Sons, New York, 1978, vol. 93, pp. 128–129 Search PubMed.
- P. Thirugnanasambandam and G. J. Srinivasan, J. Chem. Phys., 1969, 50, 2467–2475 CrossRef CAS.
- J. A. Iggo, NMR: Spectroscopy in Inorganic Chemistry, Oxford University Press, Oxford, 2004 Search PubMed.
- Z. E. M. Reeve, C. J. Franko, K. J. Harris, H. Yadegari, X. Sun and G. R. Goward, J. Am. Chem. Soc., 2017, 139, 595–598 CrossRef CAS PubMed.
- S. Leukel, M. Panthöver, M. Mondeshki, G. Kieslich, Y. Wu, N. Krautwurst and W. Tremel, Chem. Mater., 2018, 30, 6040–6052 CrossRef CAS.
- P. Derek, G. J. David, W. Norman and S. Ella, US Pat., 9300921-W, 1993 Search PubMed.
- R. Baciocchi, G. Storti and M. Mazzotti, Chem. Eng. Process., 2006, 45, 1047–1058 CrossRef CAS.
- J. K. Storaloff, D. W. Keith and G. V. Lowry, Environ. Sci. Technol., 2008, 42, 2728–2735 CrossRef PubMed.
- F. Zeman, Environ. Sci. Technol., 2007, 41, 7558–7563 CrossRef CAS PubMed.
- R. R. Contreras, L. Rincón and J. Almarza, Energy Sources, Part A, 2021, 1–15 Search PubMed.
- T. Cai, J. K. Johnson, Y. Wu and X. Chen, ACS Appl. Mater. Interfaces, 2019, 11(9), 9033–9041 CrossRef CAS PubMed.
- E. Medina-Martos, J. L. Gálvez-Martos, J. Almarza, C. Lirio, D. Iribarren, A. Valente and J. Dufour, J. CO2 Util., 2022, 60, 101991 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2va00281g |
| This journal is © The Royal Society of Chemistry 2023 |